© Karine Goudreau, 2019
Une étude des défauts d'homocouplage dans la
polymérisation par (hétéro)arylation directe à l'aide de
polymères modèles
Mémoire
Karine Goudreau
Maîtrise en chimie - avec mémoire
Maître ès sciences (M. Sc.)
iii
Résumé
Les polymères π-conjugués sont des matériaux à haute valeur ajoutée pour applications en électronique organique. Afin d’être utilisés dans des dispositifs électroniques, il est nécessaire d’avoir une méthode de synthèse efficace permettant d’obtenir des matériaux de bonne qualité. En effet, les polymères utilisés dans ce domaine doivent posséder une structure régulière et une masse molaire élevée. Toutefois, certains défauts peuvent se retrouver dans les chaînes polymère, ce qui diminuent leur performance en dispositif. Un des défauts possibles est l’homocouplage, soit la répétition successive d’un même motif. Ce défaut est présent peu importe la méthode de polymérisation utilisée. Dans ce projet de maîtrise, les défauts d’homocouplage dans les polymères synthétisés par poly(hétéro)arylation directe (PHAD) ont été étudiés. La PHAD est une nouvelle méthode de polymérisation novatrice permettant de diminuer le nombre d’étapes de synthèse, en plus d’être verte puisqu’elle ne produit pas de déchets organométalliques toxiques en comparaison avec les méthodes classiques de polymérisation.
Les mécanismes menant aux défauts d’homocouplage dans les polymères synthétisés par PHAD sont encore sujets à discussion. En ce sens, l’influence de la nature des monomères et des réactifs utilisés sur la régiorégularité des polymères synthétisés par PHAD a été étudiée. Pour ce faire, deux polymères, le poly(3-octyoxy-4-métylthiophène) (POMT) et le poly(1-(4-dodécylthiophèn-2-yl)-5-(2-octyldodécyl)-thiéno[3,4- c]pyrrole-4,6-dione) (P(TPD-T)), ont été choisis de manière stratégique, puisque le seul défaut possible dans leur structure est l’homocouplage. Ce défaut peut être identifié et quantifié par spectroscopie RMN 1H et ainsi permettre l’optimisation des conditions de polymérisation. De plus, les propriétés optiques et thermiques ainsi que la masse molaire des polymères ont également été analysées. Les propriétés électroniques du P(TPD-T) ont été déterminées par la mise en dispositif de transistors à effet de champs. Toutes ces caractérisations ont permis de déterminer l’influence de la présence d’homocouplage sur l’organisation du polymère à l’état solide, qui est fortement reliée aux performances en dispositifs.
v
Table des matières
Résumé………..iii
Liste des tableaux et des équations ... viii
Liste des figures ... x
Liste des abréviations et symboles ... xiv
Remerciements ... xvii
Chapitre 1 : Introduction ... 1
1.1 Les polymères π-conjugués ... 1
1.1.1 Les propriétés optoélectroniques des polymères ... 2
1.1.2 Modulation des propriétés optiques et physiques ... 4
1.2 Méthodes de synthèses des polymères π-conjugués ... 5
1.2.1 La polymérisation par polycondensation ... 5
1.2.2 La polymérisation par (hétéro)arylation directe ... 7
1.2.3 Le mécanisme et les conditions de la PHAD ... 9
1.3 Les défauts structuraux présents dans les polymères synthétisés par PHAD ... 11
1.3.1 Le poly(3-hexylthiophène) : un polymère sans défaut ... 11
1.3.2 Les défauts d’homocouplage et leur influence sur les performances en dispositifs électroniques ... 13
1.4 Objectifs du projet ... 16
Chapitre 2 : Méthodes expérimentales ... 20
2.1 Produits chimiques ... 20
2.2 Spectroscopie à résonance magnétique nucléaire (RMN) ... 20
2.3 Spectrométrie de masse à haute résolution ... 20
2.4 Spectrométrie de désorption-ionisation laser assistée par une matrice (MALDI-TOF) ... 20
2.5 Température de fusion ... 21
2.6 Spectroscopie d’absorption dans l’UV-visible ... 21
2.7 Chromatographie d’exclusion stérique ... 21
2.8 Analyse de thermogravimétrie ... 22
2.9 Analyse enthalpique différentielle à balayage ... 22
2.10 Voltampérométrie cyclique ... 22
2.11 Transistor à effet de champs ... 23
Chapitre 3 : La synthèse et l’optimisation du poly(3-octyloxy-4-méthylthiophène) (POMT) ... 25
vi
3.2 Méthode générale de polymérisation ... 27
3.3 Optimisation des conditions de polymérisation par PHAD ... 28
3.3.1 Optimisation des conditions de polymérisation pour le monomère M1 ... 29
3.3.2 Optimisation des conditions de polymérisation pour le monomère M2 ... 35
3.4 Caractérisation des polymères ... 39
3.4.1 Spectroscopie UV-visible ... 40
3.4.2 Analyse des spectres RMN 1H ... 43
3.4.3 Analyse enthalpique différentielle à balayage ... 46
3.5 Conclusion partielle ... 48
Chapitre 4 : La synthèse et l’optimisation du poly(1-(4-dodécylthiophèn-2-yl)-5-(2-octyldodécyl)-thiéno[3,4-c]pyrrole-4,6-dione) (P(TPD-T)) ... 49
4.1 Synthèse du monomère ... 49
4.1.1 Synthèse du monomère par couplage de Stille ... 49
4.1.2 Synthèse du monomère par arylation directe ... 53
4.2 Synthèse des petites molécules modèles ... 56
4.2.1 Synthèse de la molécule modèle représentant le motif régiorégulier ... 56
4.2.2 Synthèse de la molécule modèle représentant le motif d’homocouplage tête-tête ... 57
4.2.3 Synthèse de la molécule modèle représentant le motif d’homocouplage queue-queue... 58
4.3 Synthèse des polymères ... 59
4.4 Optimisation des conditions de polymérisation ... 60
4.5 Caractérisation des polymères ... 70
4.5.1 Analyse des spectres d’absorption dans l’UV-visible ... 70
4.5.2 Analyse des spectres RMN 1H ... 73
4.5.3 Analyse enthalpique différentielle à balayage ... 77
4.5.4 Mise en dispositif de transistor à effet de champs ... 79
4.6 Application du système catalytique optimisé à un copolymère ... 82
4.7 Conclusion partielle ... 84
Chapitre 5 : Conclusions et perspectives ... 86
5.1 Conclusions ... 86
5.2 Perspectives ... 89
Bibliographie ... 91
Annexe 1 : Méthodes expérimentales et caractérisations pour le poly(3-octyloxy-4-méthylthiophène) (POMT)... 93
vii
Annexe 1.2 : Méthode générale de synthèse des polymères par PHAD ... 98
Annexe 1.3 : Spectres RMN 1H des polymères POMT ... 99
Annexe 1.4 : Spectres UV-visible des polymères POMT ... 100
Annexe 2 : Méthodes expérimentales et caractérisations pour le poly(1-(4-dodécylthiophèn-2-yl)-5-(2-octyldodécyl)-thiéno[3,4-c]pyrrole-4,6-dione) (P(TPD-T)) ... 102
Annexe 2.1 : Synthèse des intermédiaires et du monomère pour le P(TPD-T) ... 102
Annexe 2.2 : Synthèse des petites molécules modèles ... 106
Annexe 2.3 : Méthode générale de synthèse des polymères par PHAD ... 111
Annexe 2.4 : Spectres RMN 1H des polymères de P(TPD-T) ... 112
Annexe 2.5 : Spectres UV-visible des P(TPD-T) ... 113
Annexe 2.6 : Voltampérométrie cyclique des polymères P(TPD-T) ... 115
viii
Liste des tableaux et des équations
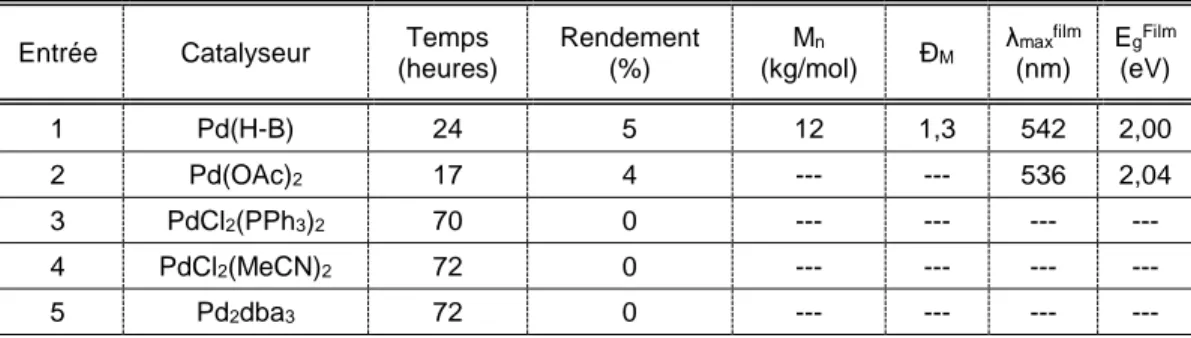
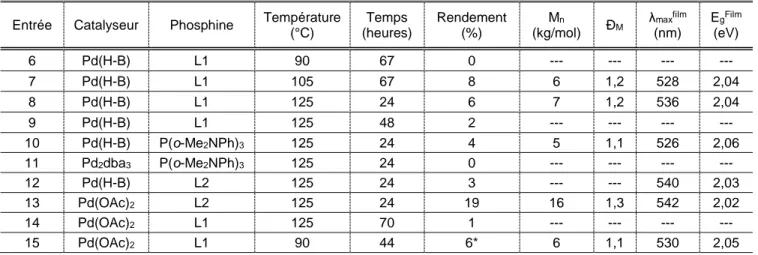
Tableau 1: Optimisation des conditions de polymérisation de M1 selon différents catalyseurs dans le THF ... 29 Tableau 2: Optimisation des conditions de polymérisation de M1 selon différents catalyseurs et phosphines dans le THF ... 30 Tableau 3: Optimisation des conditions de polymérisation de M1 selon différentes phosphines en présence d’acide pivalique dans le THF ... 31 Tableau 4: Optimisation des conditions de polymérisation de M1 selon différents catalyseurs dans le toluène ... 32 Tableau 5: Optimisation des conditions de polymérisation de M1 selon différents catalyseurs dans le toluène avec un large excès de K2CO3 ... 32 Tableau 6: Optimisation des conditions de polymérisation de M1 selon différents catalyseurs et différentes concentrations dans le toluène ... 33 Tableau 7: Optimisation des conditions de polymérisation de M1 selon différents nombres d'équivalents de réactifs dans le toluène ... 34 Tableau 8: Optimisation des conditions de polymérisation de M2 selon différents ligands dans le THF ... 36 Tableau 9: : Optimisation des conditions de polymérisation de M2 selon différents ligands et différentes températures dans le THF ... 36 Tableau 10: Optimisation des conditions de polymérisation de M2 à différentes températures dans le toluène ... 38 Tableau 11: Réplicats du polymère P2-6 ... 39 Tableau 12: Les polymères POMT qui ont fait l’objet d’une caractérisation détaillée ... 40 Tableau 13: Les déplacements chimiques en ppm des différents protons aliphatiques du POMT permettant d’en déterminer la régiorégularité... 45 Tableau 14: Quantification de la régiorégularité des différents POMT par spectroscopie RMN 1H ... 45 Tableau 15: Mesures thermiques des différents POMT par analyse enthalpique différentielle à balayage ... 47 Tableau 16: Optimisation des conditions de couplage par arylation directe pour obtenir le TPD-T ... 55 Tableau 17: Optimisation des conditions de polymérisation du P(TPD-T) selon différents solvants et différentes concentrations ... 61 Tableau 18: Optimisation des conditions de polymérisation du P(TPD-T) selon différents catalyseurs ... 62 Tableau 19: Optimisation des conditions de polymérisation du P(TPD-T) selon différents assemblages de catalyseurs et de ligands ... 63 Tableau 20: Optimisation des conditions de polymérisation du P(TPD-T) à une plus faible concentration selon différents catalyseurs et ligands ... 64
ix
Tableau 21: Optimisation des conditions de polymérisation du P(TPD-T) selon différentes concentrations et températures ... 65 Tableau 22: Optimisation des conditions de polymérisation du P(TPD-T) selon différentes concentrations de toluène ... 66 Tableau 23: Optimisation des conditions de polymérisation du P(TPD-T) avec les concentrations d’acide et de base augmentées selon différentes températures ... 67 Tableau 24: Optimisation des conditions de polymérisation du P(TPD-T) selon différents ligands ... 68 Tableau 25: Réplicats des polymères P3-23 et P3-24 ... 69 Tableau 26: Les polymères P(TPD-T) qui ont fait l’objet d’une caractérisation détaillée ... 70 Tableau 27: Déplacements chimiques en ppm des différents protons du P(TPD-T) permettant le calcul de la régiorégularité... 75 Tableau 28: Détermination de la régiorégularité des différents P3 à l'aide des spectres RMN 1H ... 76 Tableau 29: Mesures thermiques des polymères P3 par analyse enthalpique différentielle à balayage ... 78 Tableau 30: Valeurs des niveaux d’énergie HOMO et LUMO et de la bande interdite des différents P3 ... 79 Tableau 31: Paramètres électroniques des différents polymères P3 mis en dispositif de transistor à effet de champs de type n ... 80 Tableau 32: Paramètres électroniques des différents polymères P3 mis en dispositif de transistor à effet de champs de type p ... 81 Tableau 33: Paramètres électroniques bruts des différents polymères P3 mis en dispositif de transistor à effet de champs de type n ... 115 Tableau 34: Paramètres électroniques bruts des différents polymères P3 mis en dispositif de transistor à effet de champs de type p ... 117
Équation 1: Équation générale de Carothers ... 6 Équation 2: Équation de Carothers pour un monomère ... 7
x
Liste des figures
Figure 1: Structure du poly(acétylène) ... 1
Figure 2: Structure du polythiophène, polypyrolle et polyaniline ... 1
Figure 3: Structure anti-coplanaire ... 1
Figure 4: Théorie des bandes; comparaison des matériaux isolants, semi-conducteurs et conducteurs ... 2
Figure 5: Hybridation des orbitales dans une série d’oligothiophènes ... 3
Figure 6: a) structure benzenoïde b) structure quinoïde ... 4
Figure 7: Hybridation des orbitales dans un complexe donneur-accepteur (D-A) ... 5
Figure 8: Arrangement des chaînes alkyle dans le P3HT ... 5
Figure 9: Représentation de l'équation de Carothers ... 6
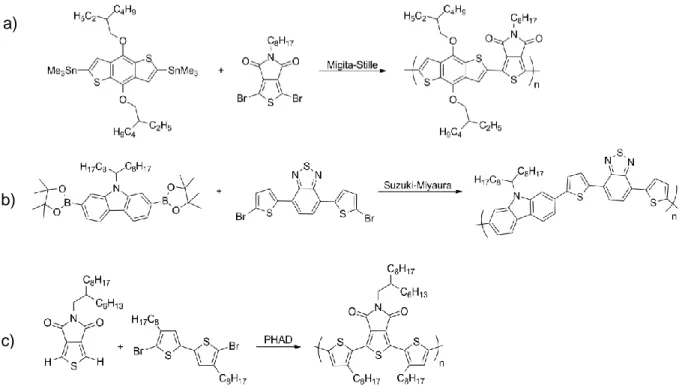
Figure 10: Exemples de polymères synthétisés par couplage de a) Migita-Stille, b) Suzuki-Miyaura et par c) PHAD ... 7
Figure 11: Mécanisme proposé de la PHAD ... 9
Figure 12: Conditions générales de PHAD a) d’un homopolymère b) d’un copolymère .... 10
Figure 13: a) Deux monomères possibles afin de synthétiser le P3HT b) défauts d’homocouplage c) défaut en β ... 11
Figure 14: Influence de l’homocouplage sur les propriétés optiques et sur l’efficacité de conversion énergétique (ECE) en cellule solaire ... 13
Figure 15: Mécanisme de réduction du Pd(II) en Pd(0) ... 14
Figure 16: Influence de la déhalogénation sur l'homocouplage ... 14
Figure 17: Mécanisme trans menant à la déhalogénation et à l’homocouplage C-H/C-H . 15 Figure 18: Structure du POMT et du P(TPD-T) ... 16
Figure 19: Polymérisation par (hétéro)arylation directe des deux monomères du POMT . 17 Figure 20: Polymérisation par (hétéro)arylation directe du P(TPD-T) ... 18
Figure 21: Synthèse du PTPD2T(C8) par PHAD ... 19
Figure 22: Synthèse des monomères M1 et M2 ... 25
Figure 23: Mécanisme de couplage d'Ullmann ... 26
Figure 24: Schéma général de polymérisation par PHAD pour obtenir le POMT ... 27
Figure 25: Les différents motifs de répétition possibles dans la structure du POMT ... 28
Figure 26: Structure de a) Pd(Herrmann-Beller) et b) de différentes phosphines ... 28
Figure 27: Synthèse des polymères P1-1 à P1-5 ... 29
Figure 28: Synthèse des polymères P1-6 à P1-15 ... 30
Figure 29: Synthèse des polymères P1-16 et P1-17 ... 31
Figure 30: Synthèse des polymères P1-18 à P1-22 ... 32
xi
Figure 32: Synthèse des polymères P1-26 à P1-31 ... 33
Figure 33: Synthèse des polymères P1-32 à P1-35 ... 34
Figure 34: Synthèse et caractérisation du polymère P1-36 ... 35
Figure 35: Synthèse des polymères P2-1 à P2-3 ... 36
Figure 36: Synthèse des polymères P2-4 à P2-7 ... 36
Figure 37: Synthèse des polymères P2-8 et P2-9 ... 38
Figure 38: Synthèse du polymère P2-6 ... 39
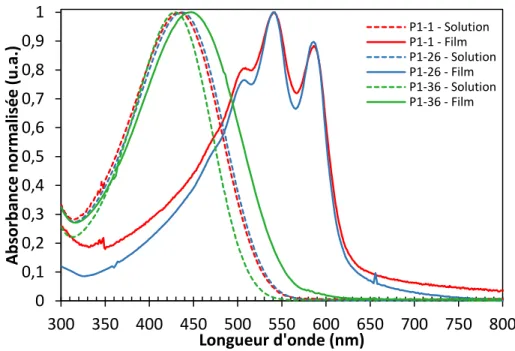
Figure 39: Spectres UV-visible des polymères P1 en solution dans le CHCl3 et en film ... 40
Figure 40: Structure tordue du POMT obtenue par sa solubilisation ou par chauffage du film ... 41
Figure 41: Spectres UV-visible des polymères P2 en solution dans le CHCl3 et en film ... 42
Figure 42: Spectre RMN 1H du POMT (500 MHz, 50°C, CDCl3) ... 44
Figure 43: Spectres RMN 1H des polymères POMT (500 MHz, 50°C, CDCl3) ... 44
Figure 44: Analyse enthalpique différentielle à balayage des polymères POMT ... 47
Figure 45: Synthèse du P(TPD-T) par (hétéro)arylation directe ... 49
Figure 46: Schéma de synthèse du 1-iodo-5-(2-octyldodécyl)-4H-thiéno[3,4-c]pyrrole-4,6-dione ... 49
Figure 47: Intermédiaire de réaction pour la synthèse du 3-éthyl-4-méthyl 2-aminothiophène-3,4-dicarboxylate ... 50
Figure 48: Intermédiaires de réaction pour la synthèse du 1-iodo-5-(2-octyldodécyl)-4H-thiéno[3,4-c]pyrrole-4,6(5H)-dione ... 50
Figure 49: Synthèse du 2-tributylstannyl-4-dodécylthiophène ... 51
Figure 50: Schéma de synthèse du monomère M3 par couplage de Stille ... 52
Figure 51: Schéma de synthèse du (2-octyldodécyl)-thiéno[3,4-c]pyrrole-4,6-dione) ... 53
Figure 52: Schéma de synthèse du 2-bromo-4-docécylthiophène ... 53
Figure 53: Schéma de synthèse du TPD-T par arylation directe ... 55
Figure 54: Synthèse de la petite molécule régiorégulière (TPD-T-TPD-T) ... 56
Figure 55: Synthèse de la petite molécule d'homocouplage tête-tête (T-TPD-TPD-T) ... 57
Figure 56: Synthèse de la petite molécule d'homocouplage queue-queue (TPD-T-T-TPD) ... 58
Figure 57: Schéma de synthèse générale de la PHAD pour obtenir P3 ... 59
Figure 58: Structures de différents catalyseurs de palladium et de différentes phosphines ... 60
Figure 59: Synthèse des polymères P3-1 à P3-5 ... 61
Figure 60: Synthèse des polymères P3-6 à P3-10 ... 62
Figure 61: Synthèse des polymères P3-11 à P3-13 ... 63
xii
Figure 63: Synthèse des polymères P3-17 et P3-18 ... 65
Figure 64: Synthèse des polymères P3-19 à P3-21 ... 66
Figure 65: Synthèse des polymères P3-22 et P3-23 ... 67
Figure 66: Synthèse des polymères P3-24 à P3-26 ... 68
Figure 67: Synthèse des polymères P3-23 et P3-24 ... 69
Figure 68: Spectres UV-visible des polymères de P(TPD-T) à l'état solide ... 71
Figure 69: Spectres UV-visible a) de P3-14 en film et en solution dans le CHCl3 et b) dans l'oDCB en fonction de la température ... 72
Figure 70: Spectres RMN 1H des petites molécules modèles permettant l'assignation des signaux d'homocouplage ... 74
Figure 71: Spectre RMN 1H du P(TPD-T) (500 MHz, 90°C, TCE-d2) ... 74
Figure 72: Assignation des défauts d'homocouplage et des bouts de chaîne sur les spectres RMN 1H des polymères P3 ... 75
Figure 73: Analyses enthalpiques différentielle à balayage des différents P3 ... 78
Figure 74: a) Spectres UV-visible du PTPD2T(C8) en solution dans le CHCl3 et sous forme de film et b) analyse enthalpique différentielle à balayage du PTPD2T(C8) ... 82
Figure 75: Synthèse du PTPD2T(C8) par la méthode optimisée de PHAD ... 82
Figure 76: Spectres RMN 1H du PTPD2T(C8) et l'assignation des bouts de chaînes et défauts d'homocouplage ... 83
Figure 77: Structures des polymères POMT et P(TPD-T) ... 86
Figure 78: Structures du POMT et des isomères pour l'étude de l'influence de l'oxygène 89 Figure 79: Structure des polymères a) PTPD2TF b) PTPDBDT ... 89
Figure 80: Structures des polymères a) PCDTBT et b) PiIEDOT ... 90
Figure 81: Spectre RMN 13C du monomère M1 ... 94
Figure 82: Spectre RMN 1H du monomère M1 ... 94
Figure 83: Spectre RMN 1H du composé 4 ... 95
Figure 84: Spectre RMN 13C du composé 4 ... 96
Figure 85: Spectre RMN 13C du monomère M2 ... 97
Figure 86: Spectre RMN 1H du monomère M2 ... 97
Figure 87: Spectres RMN 1H des différents POMT (500 MHz, 50 °C, CDCl3)... 99
Figure 88: Spectres UV-visible des différents POMT en solution dans le CHCl3 et en film ... 101
Figure 89: Spectre RMN 1H du composé 9 ... 103
Figure 90: Spectre RMN 13C du composé 9 ... 103
Figure 91: Spectre RMN 1H du monomère M3 ... 104
Figure 92: Spectre RMN 13C du monomère M3 ... 105
xiii
Figure 94: Spectre RMN 13C de la petite molécule régiorégulière ... 107
Figure 95: Spectre RMN 1H de la petite molécule d'homocouplage T-T ... 108
Figure 96: Spectre RMN 13C de la petite molécule d'homocouplage T-T ... 109
Figure 97: Spectre RMN 13C de la petite molécule d'homocouplage Q-Q ... 110
Figure 98: Spectre RMN 1H de la petite molécule d'homocouplage Q-Q ... 110
Figure 99: Spectres RMN 1H des différents P(TPD-T) (500 MHz, 90 °C, TCE-d2) ... 112
Figure 100: Spectres UV-visible des polymères P(TPD-T) en solution dans le CHCl3 et en film ... 114
xiv
Liste des abréviations et symboles
AcOEt Acétate d’éthyle
BHT Hydroxytoluène butylé
Bu3SnCl Chlorure de tributylétain
CDCl3 Chloroforme deutérié
CMD Métallation-déprotonation concertée
Cs2CO3 Carbonate de césium
CV Voltampérométrie cyclique d Doublet dd Doublet de doublet ÐM Indice de polymolécularité DMAc Diméthylacétamide DMF Diméthylformamide DPP Dicétopyrrolopyrrole
DSC Analyse enthalpique différentielle à balayage ECE Efficacité de conversion énergétique
Egel Valeur de bande interdite électronique
EgFilm Valeur de bande interdite optique du polymère sous forme de film
Et2O Éther éthylique
eV Électronvolt
Hexane Mélange d’isomères d’hexanes
HOMO Orbitale la plus haute en énergie occupée Iouvert/Ifermé Ratio de courant de circuit ouvert et fermé
J Constante de couplage
K2CO3 Carbonate de potassium
LUMO Orbitale la plus basse en énergie non-occupée
m Multiplet
Me3SnCl Chlorure de triméthylétain
MeOH Méthanol
MgSO4 Sulfate de magnésium
mmol Milimoles
Mn Masse molaire moyenne en nombre
xv NBS N-bromosuccinimide n-BuLi n-butyllithium NMP N-méthyl-2-pyrrolidone oDCB o-dichlorobenzène P(o-anisyle)3 Tris(o-méthoxyphényle)phosphine P(o-Me2NPh)3 Tris[o-(N,N-diméthyleaniline)]phosphine P(TPD-T) Poly(1-(4-dodécylthiophèn-2-yl)-5-(2-octyldodécyl)-thiéno[3,4- c]pyrrole-4,6-dione) P3HT Poly(3-hexylthiophène)
PCy3HBF4 Tricyclohexylphosphine tétrafluoroborate
Pd(Herrmann-Beller) Trans-bis(acétato)bis[2-[bis(2-méthylphényl)phosphino]benzyl] dipalladium(II)
Pd(PCy3)2 Bis(tricyclohexylphosphine)palladium(0) Pd2dba3 Tris(dibenzylidèneacétone) dipalladium(0) PdCl2(MeCN)2 Bis(acétonitrile)dichloropalladium(II)
PdCl2(PCy3)2 Dichlorobis(tricyclohexylphosphine)palladium(II) PdCl2(PPh3)2 Bis(triphénylephosphine)dichloropalladium(II) PHAD Poly(hétéro)arylation directe
PivOH Acide pivalique
POMT Poly (3-octyloxy-4-méthylthiophène)
ppm Partie par million
PTPD2T(C8) Poly(1-(4,4'-dioctyl-[2,2'-bithiophèn]-5-yl)-5-(2-hexyldécyl)-thiéno[3,4-c]pyrrole-4,6-dione) q Quadruplet Q-Q Homocouplage queue-queue quint Quintuplet R Rendement
RMN 13C Résonance magnétique nucléaire du carbone RMN 1H Résonance magnétique nucléaire du proton
RR Régiorégularité
s Singulet
SCE Électrode saturé en calomel
t Triplet
T Température
t.a. Température ambiante
xvi TCB 1,2,4-trichlorobenzène TCE-d2 1,1,2,2-tétrachloroéthane-d2 Tf Température de fusion THF Tétrahydrofurane TMEDA N,N,N',N'-tétraméthyléthylènediamine TMS Tétraméthylsilane TPD (2-octyldodécyl)-4H-thiéno[3,4-c]pyrrole-4,6(5H)-dione) T-T Homocouplage tête-tête Vth Voltage seuil
%m/m Pourcentage en rapport massique
δ Déplacement chimique
ΔHc Enthalpie de cristallisation
ΔHf Enthalpie de fusion
µe Mobilité d’électrons
µh Mobilité de trous d’électrons
λmaxfilm Longueur d’onde maximale d’absorption du polymère sous forme de film
xvii
Remerciements
J’aimerais tout d’abord remercier mon directeur de recherche, le Pr Mario Leclerc, de m’avoir permis d’effectuer un stage et une maîtrise dans son laboratoire. Ce laboratoire m’a permis de découvrir la chimie des polymères conjugués et m’a fait changer d’idée sur les études graduées. Merci M. Leclerc pour votre présence ainsi que pour votre aide et vos idées. Merci également pour votre confiance qui m’a permis de démarrer plusieurs nouveaux projets dans le laboratoire. Cette expérience a été très instructive pour moi autant au niveau professionnel que personnel. Merci également pour vos encouragements lorsque je vous écrivais pour vous faire part des derniers résultats prometteurs qui arrivaient toujours le vendredi soir.
Un merci spécial à Serge Beaupré qui a été une ressource incroyable et qui a su répondre à mes nombreuses questions. Merci Serge pour la musique latino lors des tempêtes de neige et aussi pour toutes les blagues partagées lors des deux dernières années.
Je tiens également à remercier François Grenier de m’avoir formée lors de mon stage, ceci m’a permis d’être autonome dès le début de ma maîtrise. Un énorme merci à Amélie Robitaille qui a été une grande aide au laboratoire et avec qui j’ai pu partager les bons et les mauvais moments autant au laboratoire qu’en voyage. Merci Amélie d’avoir crié avec moi « C’est mauve »! Je souhaite également remercier les deux stagiaires qui ont travaillé sur ce projet : Joëlle Perron et Valérie Gauthier. Merci également à tous les autres membres du laboratoire pour l’aide et pour la merveilleuse ambiance: Philippe, Jean-Rémi, Carl, Nicolas, Maxime G., Pierre-Olivier, Terry, Thomas, Eliane, Mathieu, Catherine et Josyane. Finalement, un merci spécial à toute ma famille, mais plus particulièrement à mes parents, Henriette et Gervais, de m’avoir encouragée et d’avoir cru en moi tout au long de mes études. C’est en très grande partie grâce à vous que je me suis rendue aussi loin et je vous en remercie énormément. Je tiens également à remercier mon conjoint, Maxime Daigle, pour sa présence au quotidien et son support. Un immense merci d’avoir toujours cru en moi et de m’avoir encouragée, cela a été très important pour moi surtout dans les journées plus difficiles. Merci beaucoup!
1
Chapitre 1 : Introduction
1.1 Les polymères π-conjugués
L’engouement pour les polymères conjugués a débuté vers la fin des années 70 lors de la publication d’un poly(acétylène) conducteur (Figure 1). En effet, Alan J. Heeger, Alan MacDiarmid et Hideki Shirakawa ont publié conjointement qu’en présence d’un oxydant ou d’un réducteur, ce polymère devient sous forme dopée et possède les propriétés d’un conducteur. Ces trois chercheurs recevront en 2000 le prix Nobel de chimie.1
Le poly(acétylène) est toutefois instable à l’air et ne peut être mise en œuvre facilement, c’est pourquoi la recherche sur les polymères conducteurs s’est rapidement tournée vers des polymères possédant un cœur aromatique, tel que le thiophène2, le pyrrole3 et l’aniline4 (Figure 2). Beaucoup plus stables, les composés aromatiques peuvent être polymérisés par électrochimie. Cependant, les polymères obtenus sont insolubles et donc difficiles à mettre en œuvre.
C’est alors qu’une deuxième génération de polymères conjugués fut développée. Cette nouvelle gamme de polymères possède des chaînes alkyle sur les unités aromatiques. Le premier exemple de cette nouvelle génération est la famille des poly(3-alkylthiophène)s (P3AT) synthétisée par polymérisation oxydative en 1986-1987.5,6 Cette nouvelle famille de matériaux attire l’attention tout d’abord par sa solubilité qui facilite sa mise en œuvre, mais également par sa structure anti-coplanaire à l’état solide (Figure 3).
Figure 1: Structure du poly(acétylène)
Figure 2: Structure du polythiophène, polypyrolle et polyaniline
2
Cette structure permet de réduire considérablement l’encombrement stérique entre les unités de répétition et d’obtenir un meilleur empilement des chaînes polymère à l’état solide par les interactions entre les chaînes aliphatiques ainsi que par des interactions π-π.
Suite à ces travaux, le domaine des polymères conjugués s’est ensuite dirigé vers les polymères conjugués semi-conducteurs. Le principal intérêt des polymères π-conjugués comme semi-conducteurs est dû à leurs propriétés optoélectroniques modulables. En effet, la variété de matériaux accessibles est quasi-infinie. Cette caractéristique fait des polymères conjugués des matériaux intéressants pour des applications en électronique organique. Il est alors possible de concevoir un polymère selon les propriétés désirées pour une application bien précise, comme un transistor à effet de champs,7 une cellule photovoltaïque8,9 ou une diode électroluminescente.10 Contrairement au silicium utilisé actuellement dans ces dispositifs, les polymères conjugués permettent la fabrication de dispositifs minces et flexibles. Soluble dans les solvants usuels, le polymère est mis sous forme d’encre pour ensuite être imprimé sur un substrat flexible par des méthodes déjà existantes, comme l’impression en continu ou l’impression à jet d’encre. Ceci permet donc de produire des dispositifs électroniques à l’échelle industrielle avec un faible coût.
1.1.1 Les propriétés optoélectroniques des polymères
La théorie des bandes permet de déterminer si un matériau est un isolant, un semi-conducteur ou un semi-conducteur selon la valeur de bande interdite (Eg), soit la différence d’énergie entre la bande de valence et la bande de conduction. La bande de valence, similaire à l’orbitale la plus haute en énergie occupée (HOMO) d’une molécule, est constituée d’orbitales moléculaires toutes occupées par des électrons. La bande de conduction, quant à elle, réfère aux orbitales moléculaires non occupées, soit l’analogue
Figure 4: Théorie des bandes; comparaison des matériaux isolants, semi-conducteurs et conducteurs
3
de l’orbitale non occupée de plus faible énergie (LUMO) d’une molécule simple. En ce sens, un matériau semi-conducteur est un matériau auquel un stimulus doit être appliqué afin d’exciter un électron de sa bande de valence à sa bande de conduction. L’électron peut alors être délocalisé tout au long du squelette conjugué. Selon la théorie des bandes (Figure 4), un semi-conducteur possède une valeur de bande interdite (Eg) non-nulle inférieure à 3 eV. Lorsque la valeur de Eg est nulle, le matériau est un conducteur, c’est-à-dire que les électrons se déplacent librement aux conditions ambiantes entre la bande de valence et la bande de conduction. La conduction est alors très élevée, similaire aux métaux. À l’opposé, lorsque la valeur de bande interdite est supérieure à 3 eV, on parle alors d’un isolant. Un électron ne peut donc pas être excité jusqu’à la bande de conduction due à la trop grande différence d’énergie entre celle-ci et la bande de valence. La conduction dans ce type de matériau est pratiquement nulle.
Afin d’être un semi-conducteur et ainsi posséder une faible valeur de bande interdite, un polymère conjugué doit posséder certaines caractéristiques clés comme une structure plane, une longueur de conjugaison élevée ainsi qu’une forme quinoïde favorisée par rapport à la forme benzenoïde.11
4
Tout d’abord, la structure plane d’un polymère est nécessaire à l’hybridation des orbitales π et à la délocalisation des électrons sur l’ensemble du corps conjugué. Cette hybridation des orbitales permet de diminuer la largeur de la bande interdite, tel que démontré à la Figure 5. Ainsi, plus le squelette conjugué est étendu, plus la valeur de la bande interdite diminue. Une structure plane est favorisée par le biais d’interactions électrostatiques entre deux hétéroatomes, par exemple entre le soufre et l’oxygène, ce qui rigidifie la structure. Les chaînes alkyle, quant à elles, créent de l’encombrement stérique, ce qui peut mener à la torsion du polymère. Il est donc essentiel de trouver un équilibre entre la solubilité et l’encombrement stérique. De plus, c’est la forme quinoïde, Figure 6, d’une chaîne polymère qui assure la délocalisation des électrons sur tout le squelette conjugué. En effet, la forme benzenoïde de l’unité de répétition est en compétition avec la forme quinoïde. Afin de favoriser cette dernière, il est nécessaire d’avoir une structure coplanaire, ce qui mène également à la diminution de la valeur de bande interdite.11
1.1.2 Modulation des propriétés optiques et physiques
Tel que mentionné précédemment, la modulation des propriétés optoélectroniques des polymères π-conjugués est une des principales caractéristiques de ces matériaux. En effet, il existe quelques méthodes afin de moduler la valeur de bande interdite d’un polymère. La méthode la plus répandue est l’introduction d’une combinaison donneur-accepteur (D-A). Comme illustré à la Figure , l’unité riche en électrons, le donneur, possède des orbitales frontières plus élevées en énergie que l’unité pauvre en électrons, l’accepteur. Lors de l’hybridation des orbitales moléculaires, le donneur dicte à la hausse la HOMO du complexe alors que l’accepteur abaisse la LUMO. Cette alternance d’unités riches et pauvres en électrons permet donc de diminuer la valeur de bande interdite finale du polymère.12,13
5
En plus des propriétés optiques, Il est possible de moduler la cristallinité du polymère par le choix des chaînes alkyle. Une chaîne droite favorise davantage la cristallinité alors qu’une chaîne ramifiée assure une solubilité accrue du polymère. Les chaînes alkyle s’assemblent de façon à maximiser les interactions de Van der Waals, ainsi les chaînes linéaires s’organisent tel qu’illustré à la Figure 8. Cet arrangement favorise une structure définie, ce qui augmente la cristallinité du polymère à l’état solide. Alors que les chaînes ramifiées, ne permettant pas un tel arrangement, facilitent la solubilisation par l’insertion de molécules de solvants entre celles-ci.
1.2 Méthodes de synthèses des polymères π-conjugués
1.2.1 La polymérisation par polycondensation
Afin d’obtenir des polymères performants en électronique organique, il est nécessaire d’obtenir une haute masse molaire. Pour ce faire, le degré de polymérisation doit donc être élevé. Pour les polymères synthétisés par des méthodes de polycondensation, l’équation de Carothers indique que des polymères possédant une haute masse molaire sont obtenus seulement lorsque le degré d’avancement de la réaction est près de l’unité. En
Figure 7: Hybridation des orbitales dans un complexe donneur-accepteur (D-A)
6
effet, l’augmentation de la masse molaire est exponentielle lorsque le degré d’avancement de la réaction est près de 1 (Figure 9).14 Au début de la réaction, seuls les monomères réagissent entre eux pour former des oligomères. Lorsque la quantité de monomères est très faible, il y a alors couplage d’oligomères entre eux pour ainsi former des chaînes polymère de plus en plus longues.
L’équation de Carothers14 décrivant cette relation est la suivante :
𝜒𝑛 =
1 + 𝑟 1 + 𝑟 − 2𝑝𝑟
Équation 1: Équation générale de Carothers
où
χ
n est le degré de polymérisation, p est le taux de conversion (lorsque p = 1, la réaction est complète) et r est le rapport stœchiométrique entre les deux monomères.Pour un copolymère, le rapport stœchiométrique entre les monomères utilisés est un paramètre très important. En effet, un faible débalancement de la stœchiométrie mène à un polymère de faible masse tel qu’illustré à la Figure 9. Afin d’assurer un rapport stœchiométrique près de l’unité, il est nécessaire d’avoir des monomères très purs ainsi qu’une pesée très précise. De plus, une méthode de polymérisation efficace est essentielle pour assurer un degré d’avancement élevé et donc une haute masse molaire.
Pour un homopolymère, c’est-à-dire lorsqu’un seul monomère est polymérisé sur lui-même, le rapport stœchiométrique (r) est nécessairement égal à 1. L’équation de Carothers peut donc être réduite à l’Équation 2 :
0 50 100 150 200 250 0,8 0,82 0,84 0,86 0,88 0,9 0,92 0,94 0,96 0,98 1
X
np
r = 1 r = 0,99 r = 0,987 𝜒𝑛=
1 1 − 𝑝
Équation 2: Équation de Carothers pour un monomère
Toutefois, il est important de noter que pour avoir un degré de polymérisation élevé, le monomère doit posséder une pureté très élevée. Une méthode de polymérisation efficace est également nécessaire afin d’obtenir un degré d’avancement de polymérisation élevé. En effet, plusieurs méthodes de polymérisation efficaces ont été développées au cours des années. Quelques exemples de ces méthodes de polymérisation sont présentés à la section suivante.
1.2.2 La polymérisation par (hétéro)arylation directe
Les méthodes classiques de polymérisation utilisées dans le domaine des polymères π-conjugués sont les réactions de couplage de Migita-Stille et de Suzuki-Miyaura (Figure 10 a et b).15,16 Ces polymérisations utilisent des monomères portant des halogènes et des groupements organométalliques d’étain et de bore, respectivement. Les monomères sont ensuite polymérisés à l’aide d’un catalyseur de Pd(0) pour former les liens C-C désirés. Toutefois, ces méthodes de polymérisation engendrent des coûts élevés pour la synthèse
a)
b)
c)
Figure 10: Exemples de polymères synthétisés par couplage de a) Migita-Stille, b) Suzuki-Miyaura et par c) PHAD
8
des monomères en plus de rejeter une quantité stœchiométrique de déchets organométalliques toxiques, comme les organoétains.17,18
Depuis quelques années, une nouvelle méthode de polymérisation est utilisée pour la synthèse de polymères π-conjugués; la poly(hétéro)arylation directe (PHAD). En comparaison avec les méthodes classiques de couplage, la PHAD ne nécessite aucun intermédiaire métallique sur les monomères tel qu’illustré à la Figure 10c.13,19 En effet, la PHAD repose sur l’activation de lien C-H aromatique par le biais d’un catalyseur de Pd(0) et d’une base. Cette nouvelle méthode de polymérisation diminue le coût de production des polymères par un nombre inférieur d’étapes de synthèse et par l’absence de groupements fonctionnels dispendieux comme les esters boroniques. De plus, le seul sous-produit rejeté en quantité stœchiométrique est l’acide bromique qui est neutralisé in
situ par la présence d’une base. La PHAD est donc une méthode plus verte que les
méthodes classiques de polymérisation. De plus, la poly(hétéro)arylation directe a démontré son efficacité par le fait qu’il est possible d’obtenir des polymères possédant des masses molaires et des rendements similaires et parfois même supérieurs à ceux obtenus par polymérisation de Suzuki ou de Stille.20-22 Un autre avantage de cette nouvelle méthode est l’accessibilité à de nouveaux polymères qui étaient difficiles, voire impossibles, à obtenir avec les méthodes traditionnelles de polymérisation.
Toutefois, cette nouvelle méthode de synthèse de polymères comporte son nombre de défis. Comme dans le cas de couplage par Stille, la PHAD peut générer certains défauts d’homocouplage dans la structure du polymère final.23 L’homocouplage est la répétition subséquente d’une même unité. De plus, un défaut spécifique à la PHAD s’ajoute à l’homocouplage, soit les défauts de branchement en β. Étant donné que la réaction repose sur l’activation de liaisons C-H aromatiques et qu’il est possible de retrouver plusieurs atomes d’hydrogène accessibles sur un monomère, il peut donc y avoir activation d’un lien C-H non désiré. Ces défauts seront présentés plus en détail à la section 1.3. Une optimisation des conditions de réactions est nécessaire afin d’obtenir un polymère comportant peu de défauts et une masse molaire élevée. Tous les paramètres sont sujets à optimisation, comme le choix du catalyseur et du ligand, la température, la position de l’atome du brome, le solvant, la concentration, etc. Afin de comprendre davantage l’effet de chaque réactif dans la réaction de PHAD, il est nécessaire de comprendre le cycle catalytique.
9
1.2.3 Le mécanisme et les conditions de la PHAD
Comme illustré à la Figure 11, la première étape (a) consiste à une addition oxydante du lien C-halogène, ici l’atome de brome. Ainsi le catalyseur de Pd(0) passe à l’état d’oxydation Pd(II). Cette étape est suivie d’un échange de ligand (b); de la phosphine pour une base conjuguée de carboxylate bidentate. Ce ligand bidentate s’ouvre pour laisser place à l’unité portant le lien C-H à activer, le thiophène (c). L’étape suivante induit généralement la sélectivité du mécanisme, soit l’état de transition de métallation-déprotonation concertée (CMD). Il y a alors formation du lien C-Pd simultanément au bris du lien C-H. Le thiophène est déprotoné par le groupement carboxylate adjacent pour former un acide carboxylique. Lors de cet état de transition, il est possible de séparer l’énergie d’activation en deux : soit en énergie de distorsion défavorable et en énergie d’interaction favorable à la CMD. D’un côté, l’énergie de distorsion correspond à la fois à l’énergie de distorsion nécessaire au complexe de Pd afin d’ouvrir le ligand bidentate ainsi qu’à la difficulté du lien C-H à se tordre pour adopter la géométrie nécessaire à l’état de transition. La distorsion du complexe de Pd est intrinsèque au système catalytique utilisé. Alors que, pour l’unité thiophène, plus le lien C-H est fort, plus il est difficile d’induire une torsion et de briser le lien en question, l’énergie d’activation de la CMD est alors augmentée. En ce sens, cette énergie de distorsion est réduite dans les composés pauvres
a b c f e d
Figure 11: Mécanisme proposé de la PHAD
10
en électrons puisque le lien C-H est plus faible. D’un autre côté, le facteur favorisant la CMD consiste en l’énergie d’interaction entre l’atome de carbone portant l’atome d’hydrogène et le centre de Pd. Ce dernier devient déficient en électrons lorsque le ligand bidentate s’ouvre. Un carbone riche en électrons favorise donc la formation du lien C-Pd afin de combler ce manque d’électrons du Pd. Une fois la CMD complétée (d), l’unité thiophène est liée au Pd. Par la suite, un deuxième échange de ligand prend place afin de relarguer l’acide carboxylique formé (e). Ce dernier est déprotoné par une base, généralement le carbonate de césium, pour être régénéré. La dernière étape (f) consiste à une élimination réductrice des deux unités aromatiques adjacentes formant ainsi le lien C-C désiré tout en régénérant le catalyseur de Pd(0) de départ. Lors d’une polymérisation, l’unité formée possède les deux groupements réactionnels nécessaires (l’halogène et l’atome d’hydrogène) pour retourner dans le cycle catalytique afin d’effectuer d’autres couplages.24-26 Dans le mécanisme de la PHAD, il est également possible que le ligand bidentate carboxylate soit remplacé par la base de carbonate27. Celui-ci joue le même rôle, mais doit être présent en quantité stœchiométrique puisqu’il ne peut pas être régénéré.
Les conditions réactionnelles de la PHAD peuvent être résumées telles que retrouvées à la Figure 12. Il est possible de polymériser un seul monomère portant à la fois un halogène et un atome d’hydrogène actif en arylation directe ou de copolymériser deux monomères, un portant deux halogènes et l’autre deux atomes d’hydrogène actifs. Les catalyseurs utilisés en arylation directe sont très variés. Généralement, des précatalyseurs de Pd(II) sont utilisés dus à leur stabilité supérieure à l’air en comparaison avec les catalyseurs de Pd(0). Les précatalyseurs sont réduits en Pd(0) in situ par l’intermédiaire d’une oxydation de la phosphine présente dans le système.28,29 Les phosphines utilisées pour la PHAD sont des phosphines riches en électrons et très encombrées qui se lient fortement au catalyseur ralentissant ainsi la vitesse de réaction et assurant une meilleure sélectivité du mécanisme.20,27 Pour ce qui est de l’additif, un acide carboxylique encombré comme l’acide
11
pivalique (PivOH) ou l’acide néo-décanoïque (NDA) permet d’augmenter la sélectivité de la CMD.26,30 Le dernier réactif est la base, les carbonates de césium (Cs2CO3) et de potassium (K2CO3) sont les plus utilisés. Pour ce qui est du choix de solvant, deux grandes familles de systèmes réactionnels sont connues dans la littérature, soit les systèmes polaires et apolaires. L’utilisation d’un solvant polaire comme le DMAc ou le DMF permet l’élimination de la phosphine du système réactionnel puisque ces solvants jouent le rôle de ligand. Toutefois, les polymères comportant de longues chaînes alkyle possèdent une faible solubilité dans ces solvants, ce qui peut limiter les masses molaires. Les solvants peu polaires ou apolaires comme le THF et le toluène assurent une meilleure solubilité des polymères en croissance permettant ainsi d’augmenter les masses molaires.31 L’influence des réactifs sur la quantité de défauts structuraux retrouvés dans un polymère sera discutée dans la prochaine section.
1.3 Les défauts structuraux présents dans les polymères synthétisés
par PHAD
1.3.1 Le poly(3-hexylthiophène) : un polymère sans défaut
Le poly(3-hexylthiophène) (P3HT) est de loin le polymère le plus étudié jusqu’à ce jour en électronique organique. Tel qu’illustré à la Figure 13a, deux monomères peuvent mener au P3HT par PHAD. Toutefois, les deux monomères mènent à un P3HT possédant des caractéristiques différentes. En effet, le monomère 1 produit un polymère avec une masse molaire plus élevée et une meilleure régularité structurale.32
Dans le cas du P3HT, il est possible d’observer à la fois les défauts d’homocouplage et de défauts en β. L’homocouplage est induit par un couplage entre deux atomes de carbone Figure 13: a) Deux monomères possibles afin de synthétiser le P3HT b) défauts d’homocouplage c) défaut en β
12
portant la même fonctionnalité, soit C-H/C-H ou C-Br/C-Br. Ce défaut affecte les propriétés optiques et électroniques des polymères menant ainsi à une diminution des performances en dispositifs électroniques. Les défauts en β spécifiques à la PHAD sont dus à l’activation d’un lien C-H en β de la molécule. Ceci peut soit briser la conjugaison au sein du polymère ou créer un branchement, c’est-à-dire que deux chaînes polymère partent d’une même unité. Les défauts de branchement en β affectent l’organisation à l’état solide des polymères, ce qui mène à une diminution des performances en dispositifs électroniques.33 Ce défaut est responsable de la différence de réactivité entre les deux monomères du P3HT. En effet, le monomère 1 portant l’atome de brome sous la chaîne alkyle assure une meilleure sélectivité que son homologue, le monomère 2. Dans ce dernier, l’atome de brome ayant un effet inductif électroattracteur diminue l’énergie d’activation du lien C-H adjacent, ce qui mène à une plus faible sélectivité entre les deux liens C-H présents sur le monomère.
Il est possible de quantifier le pourcentage de couplages réguliers par rapport aux couplages menant à des défauts. On parle alors de régiorégularité (RR). Celle-ci est déterminée à l’aide de la spectroscopie RMN 1H d’un polymère par l’intégration des signaux de protons réguliers et ceux correspondant à des défauts. Ainsi, plus un polymère possède un ratio élevé entre le signal régiorégulier et celui des défauts, plus la régiorégularité est élevée. Cette caractéristique structurale est généralement proportionnelle à la performance en dispositif électronique. Il est donc nécessaire d’optimiser les conditions réactionnelles afin d’obtenir une régiorégularité élevée.
Suite à plusieurs années d’études sur ce polymère, des conditions réactionnelles de PHAD menant à un P3HT régiorégulier (RR > 99 %) ont été découvertes et publiées par Ozawa et al.27 La polymérisation a été effectuée en présence du précatalyseur Pd(Herrmann-Beller), du ligand P(o-Me2NPh)3 et de la base Cs2CO3 dans le THF. Il a été démontré que la phosphine, ayant un fort caractère électrodonneur et un encombrement stériquement important, permettait d’assurer une sélectivité élevée dans ce système autant sur les défauts d’homocouplage que de branchement en β. Une étude de ce polymère a également été effectuée dans un solvant polaire, le DMAc, en présence du précatalyseur Pd(OAc)2, d’acide néo-décanoïque et de K2CO3.34 Cette étude a mené à un P3HT moins régiorégulier (RR = 93,5 %) que celui obtenu dans le THF. Ces études démontrent l’influence du choix de solvant utilisé. En effet, à la suite de ces études, les conditions
13
apolaires ont été davantage utilisées pour la PHAD afin d’obtenir des polymères de haute régiorégularité.
1.3.2 Les défauts d’homocouplage et leur influence sur les performances en
dispositifs électroniques
Tel que mentionné précédemment, l’homocouplage diminue l’organisation du polymère à l’état solide et affecte donc ces propriétés optiques et électroniques. En effet, la présence d’homocouplage peut être déterminée par spectroscopie d’absorption UV-visible ainsi que par spectroscopie RMN 1H. Il est également possible d’étudier son influence sur les propriétés électroniques par la mise en dispositif du polymère. En spectroscopie d’absorption UV-visible, la présence de ce défaut se traduit par une diminution de la longueur d’onde maximale d’absorption du polymère à l’état solide et, par conséquent, par une augmentation de la valeur de bande interdite. Cette méthode permet donc de comparer de façon qualitative une série de polymères. Par la suite, la spectroscopie RMN 1H permet de calculer le pourcentage de régiorégularité du polymère, c’est-à-dire le pourcentage de couplages régioréguliers par rapport à ceux d’homocouplage. La mise en dispositif, tels que les cellules solaires, permet d’analyser l’influence de la présence de l’homocouplage sur les performances électroniques. À la Figure 14, il est possible de voir l’influence du taux d’homocouplage sur les propriétés optiques et sur l’efficacité de conversion énergétique en cellules solaires (ECE) pour le PDPPTPT. Les auteurs ont inséré volontairement des défauts d’homocouplage de l’unité DPP dans ce polymère synthétisé par couplage de Suzuki-Miyaura. Une faible augmentation du taux d’homocouplage induit une diminution de la longueur d’onde maximale d’absorption et mène à une chute importante des performances de ce polymère en cellule solaire.35
Figure 14: Influence de l’homocouplage sur les propriétés optiques et sur l’efficacité de conversion énergétique (ECE) en cellule solaire
14
Les mécanismes menant à la présence de ce défaut au sein de polymère synthétisé par poly(hétéro)arylation directe sont encore sujets à discussion. Jusqu’à maintenant, trois explications sont présentes dans la littérature. La première explication implique l’utilisation de précatalyseur de Pd(II) qui pourrait être réduit en Pd(0) par l’intermédiaire d’un couplage oxydatif menant à de l’homocouplage de type C-H/C-H, Figure 15.28
La deuxième proposition est la déhalogénation, c’est-à-dire le remplacement d’un halogène par un hydrogène. Comme illustré à la Figure 16a, l’atome d’hydrogène inséré pour former le benzène n’est pas réactif et ne pourra réagir davantage. Dans le cas représenté en b, l’atome d’hydrogène substitué sur le thiophène est réactif en arylation directe, il s’insèrera dans la structure et créera un motif d’homocouplage C-Br/C-Br.25
La dernière explication de l’homocouplage est un mécanisme proposé dans lequel le thiophène s’insère en position trans de l’aryle, Figure 17. Cette conformation trans ne permet pas l’élimination réductrice et mène plutôt à un échange du proton de l’unité thiophène vers l’aryle expliquant ainsi la débromation de l’aryle en question. Par la suite, un autre thiophène effectue la CMD et s’additionne sur le catalyseur en cis. L’élimination réductrice est maintenant possible, ce qui mène à la production d’une unité bithiophène et donc d’un motif d’homocouplage de type C-H/C-H.36
Figure 16: Influence de la déhalogénation sur l'homocouplage Figure 15: Mécanisme de réduction du Pd(II) en Pd(0)
15
Plusieurs facteurs sont connus pour influencer la présence de défauts d’homocouplage dans les polymères synthétisés par PHAD. Tout d’abord, le choix de solvant est primordial. En effet, les solvants polaires mènent généralement à une présence accrue d’homocouplage, alors que les solvants apolaires sont reconnus pour limiter ces défauts.18 De plus, l’utilisation de phosphines encombrées et très riches en électrons, la diminution de la quantité de catalyseur ainsi qu’une diminution de la température permettent généralement d’augmenter la régiorégularité.31 Ces facteurs ralentissent la vitesse de réaction afin de favoriser le produit thermodynamique, le couplage régiorégulier. Malgré la prise en compte de ces paramètres lors de la conception du système catalytique, il est possible de retrouver des défauts d’homocouplage au sein de la structure polymérique. C’est pourquoi ce projet de maîtrise porte principalement sur l’étude de ce défaut.
16
1.4 Objectifs du projet
L’objectif principal de ce projet est d’effectuer une étude des défauts d’homocouplage au sein de polymères synthétisés par poly(hétéro)arylation directe. Pour ce faire, deux polymères modèles ont été sélectionnés, le poly (3-octyloxy-4-méthylthiophène) (POMT) et le poly(1-(4-dodécylthiophèn-2-yl)-5-(2-octyldodécyl)-thiéno[3,4-c]pyrrole-4,6-dione) (P(TPD-T)), Figure 18.
Le choix de ces polymères est basé sur le fait que les monomères soient bloqués en position β, c’est-à-dire qu’aucun hydrogène en β n’est présent, comme dans le cas du POMT, ou qu’il est trop encombré stériquement pour être activé en PHAD, comme le P(TPD-T). Ceci permet donc d’éliminer toute possibilité de défaut en β et de seulement se concentrer sur les défauts d’homocouplage. Dans les deux cas, les polymères sont en fait des homopolymères, c’est-à-dire qu’un seul monomère est polymérisé sur lui-même. Ceci permet d’éviter tout débalancement stœchiométrique du système qui limiterait les masses molaires et qui, en plus, pourrait induire de l’homocouplage. Tel que mentionné précédemment dans la description de l’équation de Carothers, il est nécessaire d’avoir un ratio parfait des monomères afin d’obtenir de hautes masses molaires. Ce facteur est donc évité en utilisant un homopolymère. Afin d’étudier l’homocouplage à l’aide d’homopolymère, il est nécessaire d’utiliser des monomères asymétriques pour en déterminer la régiorégularité par spectroscopie RMN 1H. Le POMT et le P(TPD-T) répondent donc à tous ces critères.
Le projet est divisé en deux sections : la première étant l’optimisation des conditions de polymérisation et la caractérisation du POMT alors que la deuxième consiste aux mêmes aspects pour le P(TPD-T). Tout d’abord, le POMT est un polymère riche en électrons qui a déjà été synthétisé dans la littérature par couplage oxydatif.37,38 Ce polymère a été choisi puisque les signaux des protons sur le groupement méthyle ainsi que les premiers atomes d’hydrogène de la chaîne alkoxy sont très distincts en spectroscopie RMN 1H. Une faible
POMT
P(TPD-T)
17
variation de leur environnement chimique influence leur déplacement chimique, ce qui permet de quantifier la régiorégularité du polymère. De plus, cet homopolymère permettra d’étudier l’influence de la position de l’atome de brome sur le monomère de départ. Le monomère M1 possède l’atome de brome sous la fonction éther alors que le monomère
M2 porte le même halogène sous le groupement méthyle, Figure 19.
Deux aspects seront étudiés, soit l’influence de l’encombrement stérique ainsi que l’influence électronique du groupement adjacent aux atomes de brome et d’hydrogène. L’atome d’oxygène sert d’espaceur à la chaîne carbonée, ce qui diminue l’encombrement stérique autour du carbone 2 en plus d’être un fort donneur d’électrons par effet mésomère. Par ailleurs, le groupement méthyl est un groupement plus encombré stériquement et moins électrodonneur que le groupement alkoxy puisqu’il est un donneur par effet inductif. De plus, on retrouve seulement dans la littérature des exemples de poly(3,4-dialkoxythiophène) synthétisé par PHAD, ce polymère est donc le premier exemple de 3-alkoxythiophène polymérisé par cette méthode. Afin d’effectuer l’optimisation des conditions de polymérisation du POMT, tous les polymères synthétisés seront analysés par spectroscopie d’absorption UV-visible afin de déterminer la longueur maximale d’absorption ainsi que la largeur de bande interdite. En effet, tel que mentionné précédemment, la présence d’homocouplage au sein d’un polymère influence ces propriétés optiques. Cette analyse simple et rapide permettra de comparer les polymères entre eux et de déterminer les polymères possédant les meilleures propriétés optiques et donc une régiorégularité plus élevée. Suite à l’optimisation des conditions de polymérisation, les polymères les plus prometteurs seront analysés en détails par spectroscopie RMN 1H afin de déterminer la régiorégularité puis par analyse enthalpique différentielle à balayage (DSC) afin de déterminer l’influence de l’homocouplage sur les propriétés thermiques des différents POMT.
18
Par la suite, l’optimisation et la caractérisation du P(TPD-T), comme présenté à la Figure 20, seront décrites dans le chapitre 4. Ce polymère permettra d’analyser l’homocouplage au sein d’un polymère de type donneur-accepteur. En effet, l’unité thiénopyrroledione possède un caractère électroattracteur très important dû à la fonction imide, alors que l’unité thiophène est électrodonneur par la seule présence d’une chaîne alkyle. Dans ce cas-ci, l’influence de la position de l’atome de brome ne peut être étudiée, puisque l’insertion d’un atome de brome seulement sur l’unité TPD représente un défi synthétique complexe, voire impossible à faire. Par ailleurs, l’utilisation de TPD portant des atomes d’hydrogène est beaucoup plus efficace en PHAD que la même unité portant des halogènes. De plus, le P(TPD-T) est un nouveau polymère, il est toutefois analogue à certains polymères à base de TPD et de thiophène retrouvés dans la littérature.13,19
Ce polymère a été conçu tout d’abord pour l’unité TPD, une unité très connue, facile à synthétiser et qui démontre d’excellentes performances en transistor à effet de champs ainsi qu’en cellule solaire.12,15 Ensuite, l’unité thiophène a été choisie comme co-unité pour sa simplicité de synthèse et pour son caractère donneur d’électrons. De plus, des petites molécules modèles représentant les motifs de répétition pouvant être présents dans la chaîne polymère seront synthétisés. Les spectres RMN 1H de ces molécules modèles permettront de déterminer les signaux correspondant aux motifs régiorégulier et d’homocouplage, de transposer cette assignation aux signaux présents dans les spectres RMN 1H des polymères et ainsi d’en déterminer la régiorégularité.
Comme dans le cas du POMT, l’optimisation des conditions de polymérisation afin d’obtenir un P(TPD-T) hautement régiorégulier sera basée sur la longueur maximale d’absorption ainsi que sur la valeur de bande interdite des polymères. Une fois l’optimisation des conditions de polymérisation effectuée, les polymères possédant les propriétés optiques les plus prometteuses seront caractérisés par spectroscopie RMN 1H
19
et par DSC afin de déterminer respectivement la régiorégularité et les propriétés thermiques. De plus, ces polymères seront mis en dispositif de transistor à effet de champs afin de déterminer l’influence de l’homocouplage sur les propriétés électroniques. Finalement, les conditions réactionnelles ayant mené au P(TPD-T) le plus régiorégulier seront utilisées pour la synthèse d’un copolymère analogue, le PTPD2T(C8) (Figure 21). Ce polymère a déjà été synthétisé par PHAD, le polymère obtenu sera donc comparé à la littérature.19
20
Chapitre 2 : Méthodes expérimentales
2.1 Produits chimiques
Les produits de départ utilisés ont été achetés chez Combi-Blocks, Sigma-Aldrich, TCI, Oakwood Chemical, Matrix Scientific, Frontier Scientific ou Fisher Scientific. Aucune purification supplémentaire n’a été effectuée sur ces composés, ils ont été utilisés tels que reçus.
2.2 Spectroscopie à résonance magnétique nucléaire (RMN)
Les spectres RMN 1H et 13C ont été enregistrés sur un spectromètre Agilent DD2 500 MHz. Toutes les analyses ont été faites à température ambiante sauf si spécifié. Les spectres RMN ont été effectués dans le chloroforme deutérié (CDCl3), dans le 1,1,2,2-tétrachloroéthane-d2 (TCE) ou tout autre solvant deutérié tel que spécifié. Les solvants deutériés ont été achetés chez Sigma-Aldrich. Les déplacements chimiques (δ) sont rapportés en partie par million (ppm) relativement à un standard interne de tétraméthylsilane (TMS). Les symboles utilisés pour décrire les signaux sont : s = singulet, d = doublet, t = triplet, q = quadruplet, quint = quintuplet, dd = doublet de doublet, m = multiplet. Le temps de relaxation entre chaque impulsion pour les spectres RMN 1H est de 10 secondes pour tous les composés aromatiques.
2.3 Spectrométrie de masse à haute résolution
L’appareil utilisé pour enregistrer les spectres de masse haute résolution est un LC/MS-TOF Agilent 6210 à l’aide du mode d’ionisation par électronébulisation (ESI).
2.4 Spectrométrie de désorption-ionisation laser assistée par une
matrice (MALDI-TOF)
La masse moléculaire de certains composés a été déterminée à l’aide d’un MALDI-TOF Axima Assurance de la compagnie Mandel Scientific. Une solution de polyéthylène glycol et de polypropylène glycol ayant des masses molaires respectives de 400 kg/mol et 2000
21
kg/mol a été utilisée afin de calibrer l’appareil. Une solution de dithranol est utilisée comme matrice. L’échantillon est mis dans le CHCl3 de grade HPLC à une concentration de 10-3 mol/L. Le même volume de matrice et d’échantillon sont mélangés afin d’obtenir le mélange ionisable utilisé pour l’analyse.
2.5 Température de fusion
L’appareil Stanford Research Systems Optimelt Automated Melting Point System a permis de déterminer le point de fusion de certains composés à un taux de chauffe de 2 °C/minute.
2.6 Spectroscopie d’absorption dans l’UV-visible
L’appareil Hewlett Packard 8452A a été utilisé pour effectuer les spectres d’absorption UV-visible des polymères. Pour les mesures en solution dans le chloroforme des polymères, une cuvette de quartz de 1 cm de parcours optique a été utilisée. Des lamelles de microscope en verre ont été utilisées pour y déposer par enduction centrifuge une solution de polymère dans le chloroforme (5 mg/mL). Le spectre du polymère à l’état solide est ensuite utilisé afin de calculer la largeur de bande interdite optique (Egopt). Cette dernière est calculée suite à l’obtention du point d’inflexion de la courbe trouvé à l’aide de tangentes.
2.7 Chromatographie d’exclusion stérique
Les masses molaires des polymères, masses molaires moyennes en nombre (Mn) et moyennes en masse (Mp), ont été déterminées à l’aide de l’appareil Varian Instrument PL120 équipé d'un détecteur à indice de réfraction et d'un viscosimètre PL BV400 HT. Cet appareil est équipé de deux colonnes, une de PLgel Mixed C (300 x 7,5 mm) et une colonne de garde PLgel Mixed C. L’éluant utilisé est le 1,2,4-trichlorobenzène (TCB) avec un stabilisant d’hydroxytoluène butylé (BHT) à 0,0125 %p/v. Le tout est préalablement filtré sur un filtre de fibre de verre d’une porosité de 0,45 μm. L’éluant passe dans l’appareil à un débit de 1,0 mL/minute à une température de 110 °C. L’appareil est calibré à l’aide de la méthode standard au polystyrène avec des étalons de polystyrène Easi-Vials PS-M de la compagnie Varian Polymer Laboratories. Pour la préparation des échantillons, le polymère est solubilisé dans du TCB chaud à une concentration de 2,5 mg/mL à l’aide
22
d’un Varian Polymer Laboratories PL-SP 260VC. Les échantillons sont chauffés et agités 20 minutes avant d’être filtrés sur un filtre de fibre de verre d’une porosité de 0,45 μm.
2.8 Analyse de thermogravimétrie
L’appareil Mettler-Toledo TGA/SDTA 851e a permis de déterminer la température de dégradation des polymères grâce à la thermogravimétrie (TGA). Les échantillons sont chauffés à une vitesse de 20°C/min de 50°C à 700°C sous atmosphère d’argon. La température de dégradation est déterminée à la suite d’une perte massique de 5%.
2.9 Analyse enthalpique différentielle à balayage
L’analyse enthalpique différentielle à balayage (DSC) permet de déterminer les températures de fusion (Tf) ainsi que de cristallisation (Tc) des polymères. Les analyses sont effectuées à l’aide de l’appareil Perkin-Elmer DSC823e. L’appareil est d’abord calibré avec un étalon d’indium ultra-pur. Une quantité entre 5 et 10 mg de polymère est pesée et mise dans une capsule en aluminium afin d’effectuer les analyses. L’échantillon subit une première chauffe à 20°C/min afin d’éliminer l’histoire thermique. Il est ensuite refroidi à une vitesse de 10°C/min et chauffé à une vitesse de 20°C/min afin d’observer les températures de cristallisation et de fusion respectivement. Deux intervalles de température ont été utilisés, le premier pour le poly(3-octyloxy-4-méthylthiophène) est de 50°C à 200°C et le deuxième pour le P(TPD-T) est de 50°C à 350°C.
2.10 Voltampérométrie cyclique
L’appareil utilisé afin de déterminer les niveaux énergétiques des polymères est un Solartron 1287. L’électrolyte utilisé est une solution à 0,1 mol/L de tétrabutylammonium tétrafluoroborate (Bu4NBF4) dans l’acétonitrile anhydre (CH3CN). Le sel de Bu4NBF4 est préalablement recristallisé trois fois dans un mélange 1 :1 eau:MeOH et séché à 70°C sous vide pendant 24 heures. Les cyclovoltamogrammes (CV) ainsi que les potentiostats sont effectués à une vitesse de 50 mV/s. L’électrode de référence est une électrode Ag/Ag+ avec une concentration de 0,01 mol/L d’AgNO3. Les bornes de courant utilisées sont de -0,4 à 1,8 V ainsi que -2,0 à -0,3 V pour la mesure d’oxydation et de réduction respectivement. Un film de polymère est appliqué sur une électrode de platine à partir