Centre d’études doctorales des Sciences de la vie et de la santé FORMATION DOCTORALE : BIOLOGIE MEDICALE, PATHOLOGIE HUMAINE
ET EXPERIMENTALE ET ENVIRONNEMENT.
THESE DE DOCTORAT
L’ADRENOLEUCODYSTROPHIE LIEE A L’X :
ETUDE BIOLOGIQUE ET MOLECULAIRE
A PROPOS D’UNE SERIE MAROCAINE
Présentée et soutenue le 13 Juin 2018
Par
Mme. Fatima Zohra MADANI BENJELLOUN
JURY
Professeur El Hachmia AIT BENHADDOU Présidente
Faculté de Médecine et de Pharmacie, Université Mohammed V- Rabat
Professeur Layachi CHABRAOUI Directeur de thèse Faculté de Médecine et de Pharmacie, Université Mohammed V- Rabat
Professeur Yamna KRIOUILE Co-encadrante
Faculté de Médecine et de Pharmacie, Université Mohammed V- Rabat
Professeur Abdelhalim MESFIOUI Rapporteur
Faculté des Sciences, Université Ibn Tofail - Kénitra
Professeur Latifa CHAT Rapporteur
Faculté de Médecine et de Pharmacie, Université Mohammed V- Rabat
Professeur Ahmed BOUHOUCHE Examinateur
Faculté de Médecine et de Pharmacie, Université Mohammed V- Rabat
ANNEE 2018 THESE N° 24/17 CSVS
UNIVERSITE MOHAMMED V - RABAT FACULTE DE MEDECINE ET DE PHARMACIE
II
Au nom d’Allah, le Tout Miséricordieux, le Très Miséricordieux.
Sauf le Seigneur de l’univers,
78. qui m’a créé, et c’est Lui qui me guide ;
79. et c’est Lui qui me nourrit et me donne à boire ;
80. et quand je suis malade, c’est Lui qui me guérit,
81. et qui me fera mourir, puis me redonnera la vie,
82. et c’est de Lui que je convoite le pardon de mes fautes
le Jour de la Rétribution.
83. Seigneur, accorde-moi sagesse (et savoir)
et fais-moi rejoindre les gens de bien ;
SOURATE 26
AŠ-ŠU˒ARĀ˓ (LES POÈTES)
III
DEDICACE
A l’âme de mes frères
Oussama et Abdelmounaim,
A
Youssef mon neveu,
A ma famille,
A mon mari,
A tous les enfants marocains atteint d’X-ALD
IV
REMERCIEMENTS
Je tiens à remercier chaleureusement mon directeur de thèse Monsieur le Professeur Layachi
CHABRAOUI. Merci pour votre soutien et votre encadrement pendant ce long voyage
scientifique de sept ans. Je vous remercie pour votre savoir et votre sérieux que vous avez tentez de me transmettre, sans jamais vous fatiguer. Pour votre disponibilité dont vous avez su faire preuve à tout moment malgré vos multiples responsabilités en tant que président de nombreuses institutions scientifiques et médicales, et en tant qu’expert à l’échelle nationale et internationale. Que vous trouvez ici l’expression de mon respect, de ma reconnaissance et de mes sincères remerciements.
Je tiens à exprimer ma gratitude et ma reconnaissance pour remercier cordialement mon co-encadrante Madame le Professeur Yamna KRIOUILE, d’avoir accepté de m’encadrer et de
m’avoir accueilli dans son équipe et de m’avoir encouragé depuis le premier jour de ce projet. Je vous remercie pour votre générosité et de m’avoir ouvert toutes les voies permettant à ce travail d’aboutir. Merci pour toutes nos discussions scientifiques si intéressantes, pour tous vos conseils que j’apprécie. Vous êtes une grande dame, et un modèle à suivre.
J’adresse mes profonds remerciements au Professeur Jamal Taoufik, le Directeur du Centre d’Etudes Doctorales pour le bon suivi et la qualité de la formation au sein de l’école doctorale, ainsi que pour tout l'intérêt qu'il porte à la recherche au sein de notre faculté. Qu'il trouve ici, l'expression de mes sentiments respectueux.
Je remercie vivement le Docteur David CHEILLAN pour son aide précieuse et son implication dans la réalisation des essais biologiques au sein de son laboratoire.
De même, je remercie vivement Monsieur le Docteur Youssef IDAGHDOUR d’avoir engagé toutes les ressources nécessaires pour la réalisation du séquençage au sein de son unité et l’implication de son équipe de recherche à mettre à notre disposition tous les moyens pour faire avancer notre projet de recherche.
Je remercie particulièrement le Professeur MESFIOUI Abdelhalim, le Professeur AIT BENHADDOU El Hachmia, le Professeur CHAT Latifa ainsi que le Professeur BOUHOUCHE
V
Ahmed de nous avoir honoré et fait partie du jury de notre thèse, malgré leurs engagements, je leur exprime tout mon respect et ma gratitude.
Je tiens à présenter mes vifs remerciements au Professeur Patrick AUBOURG et son équipe,
Dr Céline BELLESMES et Mme Marie Claude BLONDEAU pour leurs apports
scientifiques et leur soutien à notre projet de recherche.
Mes remerciements vont également à tout le personnel du laboratoire de Biochimie et du laboratoire central des maladies héréditaires du métabolisme, en particulier Dr TALBAOUI
Habiba, Dr DAHRI Saloua, sans oublier Rahma pour leur aide précieuse.
Merci à Pr Ariel DANSKY, Dr Joaira BAKKACH et Mme Hikmat DAOUD-TETOUANI pour leurs apports dans les rédactions de nos articles.
VI
Je remercie du fond du cœur mon Mari Mr Mohammed FARHAOUI, pour sa présence, son soutien, son écoute et ses encouragements qu’il n’a cessé à me présenter généreusement toutes ces années de thèse. Je vous remercie pour tous les moments de joie vécus à chaque avancement de ce travail, pour toutes les larmes que vous avez essuyé à chaque blocage. Merci d’avoir vécu avec moi cette histoire, d’être toujours à mes côtés, de m’avoir compris, et d’avoir fait semblant de comprendre lorsque cela vous dépassait vous aussi. Vous étiez un élève très intelligent et vous avez bien assimilé la neurologie, la neuropathologie et en particulier l’adrénoleucodystrophie. Disons que la neurologie et l’informatique se ressemblent …
Je remercie mes parents et toute ma famille BENJELLOUN pour leur soutien et leurs prières pour que je réussisse ce grand projet de vie. Je leur remercie pour leur courage et d’avoir donné l’exemple par leur patience et leur solidarité vis – à – vis à toutes les épreuves.
Mes remerciements vont aussi à mes amies : Rajaa AGNAOU, merci pour les journées estivales passées au labo ensemble. Pour le bonheur partagé à chaque fois qu’on faisait tourner les eppendorfs pour faire apparaitre la méduse (la pelote d’ADN), quel bonheur !
Halima HAMADA, je me rappelle toujours de notre premier jour de rencontre à la bibliothèque
de la FMPR, on est allées chercher ta thèse de spécialité. Merci pour les aventures passées ensemble en apprenant la médecine chinoise et en pratiquant l’acupuncture. Merci de m’avoir fait découvrir les fleurs de Bach. Le travail de groupe lors des modules de CEDOC était un moment de plaisir partagé. Zineb ZIAN pour toutes les beaux moments passées pendant ces années lors des congrès scientifiques, et surtout pour le merveilleux séjour ensemble à Mohammedia, au parc et à casa lors de Mouqawila expo. Khawla BOUKHIMA qui était le lien entre le Maroc et Abu Dhabi en assurant le transport des prélèvements dans sa valise, merci pour le service et de m’avoir offert une merveilleuse amie MAYE. Wissal MAHER, je me rappel toujours de ta citation : « si on veut que le bon seigneur nous aide, on doit aider les autres ». Merci pour ta générosité et tes conseils. Ikram ELASRI ma conseillère, Syrine
HAMADA, Zineb TAGHDA, Khansaa JASNI, Fatima Zahraa BASRI et toutes mes amies,
merci de votre soutien, je vous souhaite tout le bonheur.
Un grand merci à mes professeurs, mes collègues et amis et tous ceux qui ont contribué de près ou de loin dans la réalisation et l’aboutissement de ce travail.
VII
Sommaire
DEDICACE ... III SOMMAIRE ... VII LISTE DES FIGURES ... XI ABREVIATIONS ... XIII RESUME ... XV ABSTRACT ... XVII صخلم ... XIX INTRODUCTION GENERALE ... 1 PARTIE THEORIQUE ... 4 1. HISTORIQUE ... 5 2. EPIDEMIOLOGIE ... 7 3. ASPECTS BIOLOGIQUES ... 8 3.1.
G
ENEABCD1
... 83.2.
L
ES ACIDES GRAS A TRES LONGUE CHAINE(AGTLC)
... 83.3.
L
A CORRELATION PHENOTYPE-
GENOTYPE ... 114. ASPECTS CLINIQUES ... 12
4.1 .
L
E SPECTRE CLINIQUE CHEZ L’
HOMME ... 124.1.1. L’insuffisance surrénalienne (Maladie d’Addison) ... 12
4.1.2. Les formes cérébrales démyélinisantes de l’enfant ... 14
a. Les formes pariéto-occipitales ... 15
b. Formes frontales ... 16
c. Formes cérébrales chroniques... 16
4.1.3. La forme du pédoncule cérébelleux moyen :... 17
4.1.4. L’adrénomyéloneuropathie (AMN) : ... 17
4.1.5. Formes cérébrales démyélinisantes de l’adulte ... 18
1.1.1. La forme asymptomatique : ... 18
1.1.2. Insuffisance gonadique ... 19
4.2.
S
PECTRE CLINIQUE CHEZ LA FEMME ... 204.2.1. L’adrénomyéloneuropathie (AMN) : ... 20
4.2.2. La forme cérébrale ... 20
4.2.3. La maladie d’Addison : ... 20
4.2.4. L’atteinte ovarienne : ... 20
VIII
5.1.
D
IAGNOSTIC CLINIQUE ... 215.1.1. Les signes neurologiques ... 21
5.1.2. Les signes extra-neurologiques : ... 21
5.2.
L’
IMAGERIE:
... 235.2.1. L’imagerie par résonnance magnétique (IRM) : ... 23
5.2.2. La spectrométrie par résonnance magnétique du proton (1H-SRM) ... 24
5.3.
L
ES POTENTIELS EVOQUES:
... 255.4.
L
A PONCTION LOMBAIRE ... 255.5.
D
OSAGE DES ACIDES GRAS A TRES LONGUE CHAINES(AGTLC) :
... 255.5.1. Dosage par chromatographie en phase gazeuse (GC) : ... 26
5.5.2. La chromatographie en phase gazeuse couplée à la spectrométrie de masse (GC-MS) : ... 26
5.5.3. Electrospray ionisation – spectrométrie de masse (ESI-MS) : ... 27
5.6.
D
IAGNOSTICM
OLECULAIRE:
SEQUENÇAGE DU GENEABCD1
... 275.7.
D
IAGNOSTIC NEONATAL:
... 27 5.8.D
IAGNOSTIC PRENATAL:
... 27 5.9.D
IAGNOSTIC PREIMPLANTATOIRE:
... 28 5.10.L
ES ERREURS DE DIAGNOSTIC... 28 5.11.C
ONSEIL GENETIQUE:
... 28 6. TRAITEMENTS ... 29 6.1.T
RAITEMENTS PALLIATIFS:
... 29 6.1.1. Huile de Lorenzo : ... 296.1.2. Diète à l’acide oléique seul : ... 31
6.1.3. Traitement par immunosuppression ou par immuno-modulation... 31
6.1.4. La lovastatine... 31
6.1.5. Induction du gène ABCD2 ... 32
6.1.6. 4-phenylbutyrate (4-PBA) ... 32
6.1.7. L’acide valproïque (VPA) ... 32
6.1.8. Les stratégies antioxydants ... 32
6.2.
T
RAITEMENTS SYMPTOMATIQUES ... 326.3.
T
RAITEMENT HORMONAL DE REMPLACEMENT... 336.4.
T
RAITEMENTS CURATIFS ... 336.4.1. Greffe allogénique de cellules souches hématopoïétiques (Hematopoeitic Stem Cells Transplantation HSCT)... 33
IX
6.5.
P
ROTOCOLE DE TRAITEMENT ... 347. ROLE DES ORGANISMES OFFICIELS ET DE LA SOCIETE CIVILE ... 35
7.1.
L’
ODYSSEE DE DIAGNOSTIC ... 357.2.
L
AV
ENTUREP
HILANTHROPY ... 361.1.
S
ENSIBILISATION ... 36PARTIE PRATIQUE ... 48
CHAPITRE 1 : MISE EN PLACE D’UN PROTOCOLE DE MANAGEMENT DE L’ADRENOLEUCODYSTROPHIE LIEE A L’X AU MAROC. ... 50
R
ESUME:
... 50A
BSTRACT:
... 51M
ATERIEL ETM
ETHODES ... 53R
ESULTATS:
... 55Recrutement des patients ... 55
Protocole général du cas index : ... 56
Protocole des cas asymptomatiques ... 59
Protocole des femmes hétérozygotes ... 61
Protocole de sensibilisation ... 62
L’offre de proximité ... 62
Etat actuel au Maroc : ... 63
D
ISCUSSIONS ... 67C
ONCLUSION:
... 69CHAPITRE 2 : ETUDE MOLECULAIRE DU GENE ABCD1 ET LA DISTRIBUTION DES PHENOTYPES DANS LA POPULATION MAROCAINE : 2 NOUVELLES MUTATIONS. ... 72
R
ESUME:
... 72A
BSTRACT:
... 73 INTRODUCTION:
... 74M
ETHODES:
... 75R
ESULTATS ... 76D
ISCUSSION:
... 80C
ONCLUSION ... 82X
CHAPITRE 3 : MISE AU POINT D’UNE TECHNIQUE DE DIAGNOSTIC MOLECULAIRE POUR LA MALADIE D’X –
ALD VIA LA MISE EN EVIDENCE DE LA MUTATION C.1661G>A (P.ARG554HIS). ... 86
R
ESUME:
... 86A
BSTRACT:
... 87I
NTRODUCTION ... 87M
ATERIEL ETM
ETHODES:
... 88 1. Extraction d’ADN ... 89 2. Amplification d’ADN ... 923. Révélation sur gel d’Agarose ... 95
4. Recherche de la mutation c.1661G>A (p.Arg554His) par Polymorphisme delongueur des fragments de restriction RFLP : ... 99
R
ESULTATS:
... 100D
ISCUSSION:
... 102C
ONCLUSION:
... 103 CONCLUSION GENERALE ... 105 RECOMMANDATIONS ET PERSPECTIVES ... 109 ANNEXES ... 111ANNEXE 1 :ACCORD DU COMITE D’ETHIQUE ... 112
ANNEXE 2 :CAHIER D’OBSERVATIONS ... 114
ANNEXE 7 : FICHE D’INFORMATION SUR L’X-ALD– LANGUE FRANÇAISE ... 135
ANNEXE 8 : FICHE D’INFORMATION SUR L’X-ALD– LANGUE ARABE. ... 136
ANNEXE 9 :CELEBRATION DE LA JOURNEE MONDIALE DES MALADIES RARES DANS LE CADRE DE LA SENSIBILISATION VIS-A-VIS DE L’X-ALD, DANS UNE LUDOTHEQUE AU PROFIT DES PARENTS. ... 137
ANNEXE 10 : CREATION DE PAGES WEB DEDIEES A L’ADRENOLEUCODYSTROPHIE SUR LES RESEAUX SOCIAUX POUR UNE MEILLEURE COMMUNICATION. ... 138
ANNEXE 11 : AFFICHE DU FILM LORENZO’S OIL ... 140
ANNEXE 12 : SITES WEB INTERESSANTS ... 141
ANNEXE 13 : MODELES DE MOYENS DE SENSIBILISATION CREES PAR UNE ACTION AMIS-FAMILLE ... 142
PUBLICATIONS ... 144
ARTICLES PUBLIES DANS DES REVUES INDEXEES : ... 145
PUBLICATION D’UN LIVRE ... 162
COMMUNICATIONS ORALES : ... 163
COMMUNICATIONS AFFICHEES ... 164
XI
Liste des figures
Partie théorique :
Figure 1 : structures des AGTLC C22, C24 et C26 [33] ... 9
Figure 2: Métabolisme des AGTLC [34]. ... 10
Figure 3 : Formes cliniques de l’X-ALD [21] . ... 12
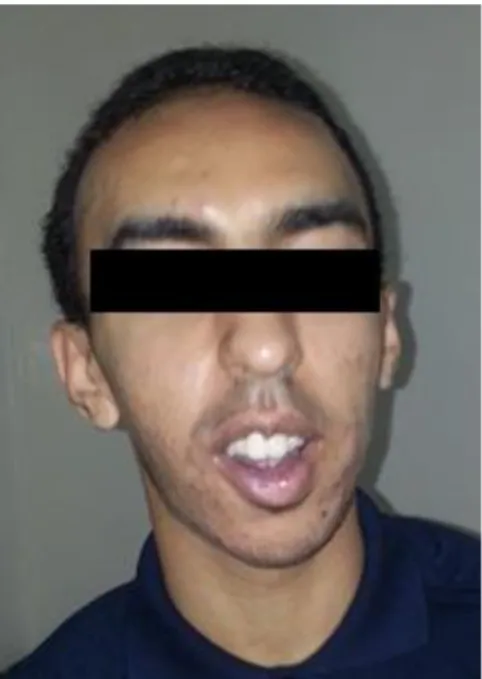
Figure 4 : Visage d’un adolescent avec mélanodermie ... 13
Figure 5 : Mélanodermie au niveau des extrémités digitées ... 14
Figure 6 : Coloration de la gencive due à une insuffisance surrénalienne chez un adolescent atteint d’X-ALD. ... 14
Figure 7 : Examen neuropathologique des hémisphères cérébraux chez un adulte avec une pure forme cérébrale de l’X-ALD. A : au niveau du striatum, B : au niveau du Thalamus, C : au niveau de la partie postérieure du lobe pariétal, D : lésions de démyélinisation au niveau de la substance blanche cérébrale [65]. ... 19
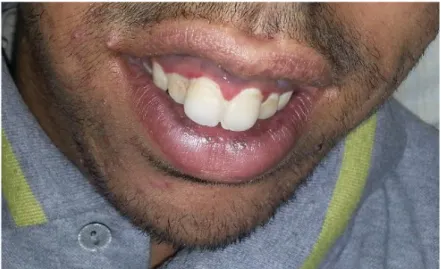
Figure 8 : Mélanodermie apparente sur le visage d’un enfant ayant une X-ALD. ... 22
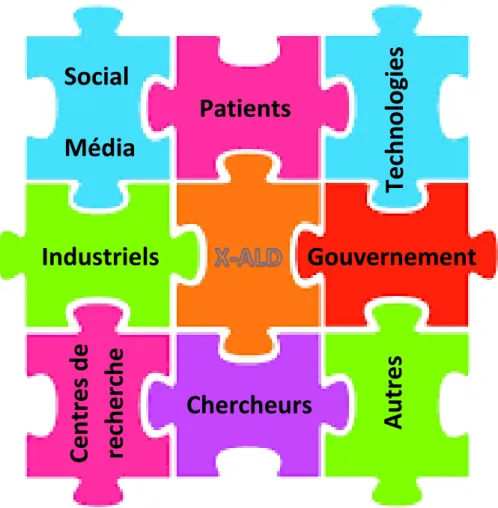
Figure 9 : Illustration des pièces de puzzle des principaux intervenants dans le développement de thérapies dans le cas de l’X-ALD. ... 37
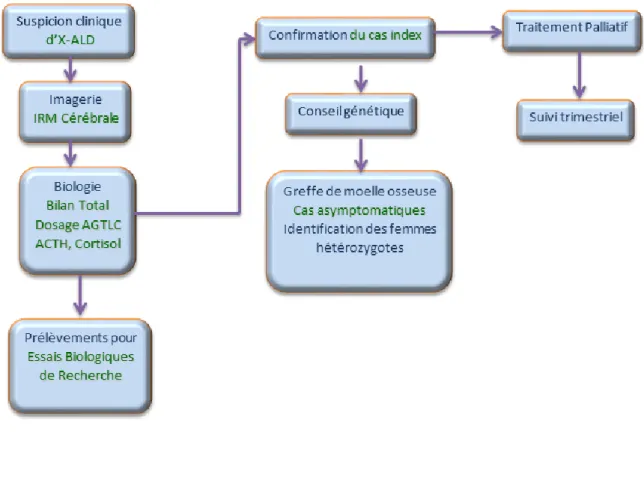
Partie pratique Chapitre 1: Figure 1 : Schéma du protocole général du cas index. ... 58
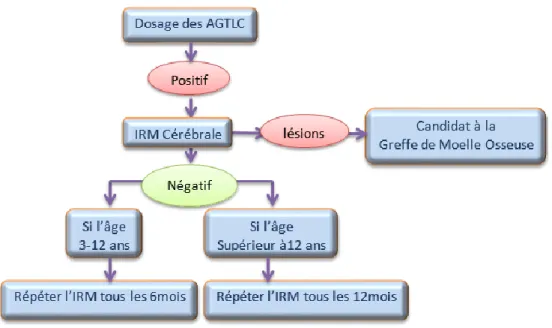
Figure 2 : Protocole des cas asymptomatiques d’X-ALD ... 60
Figure 3 : Protocole des femmes hétérozygotes porteuses d’X-ALD. ... 61
Chapitre 2 : Figure 1 : Séquence de nucléotides de la mutation p.Pro218Ala, c.652C>G au niveau du gène ABCD1 par séquençage du produit PCR. ... 76
Figure 2 : Arbre généalogique de la famille F1. ... 78
Figure 3 : Arbre généalogique de la famille F2. ... 79
Figure 4 : Arbre généalogique de la famille F3.: ... 79
Chapitre 3 : Figure 1 : Filaments d’ADN en suspension sous forme de pelote rappelant l’aspect de la méduse ... 91
Figure 2 : Thermocycleur (HVD Life sciences Qantaus®) utilisé dans nos essais. ... 93
Figure 3 : Dépôt des mélanges PCR au niveau du thermocycleur. ... 94
Figure 4 : Suivi des cycles de PCR en temps réel. ... 94
Figure 5 : Préparation de la gélose pour électrophorèse. ... 96
Figure 6 : Mise en place de la plaque à gélose dans la cuve de l’appareil de migration. ... 97
Figure 7 : Electrophorèse en cours. ... 97
Figure 8 : la plaque à gélose à la fin de la migration. ... 98
Figure 9 : Appareillage de la chambre à visualisations sous UV muni d’une caméra.: ... 98
Figure 10 : Amplification réussie du gène ABCD1 chez huit patients. ... 100
Figure 11 : Profil électrophorétique d’un échantillon muté après digestion enzymatique. ... 101
XII
Liste des tableaux
Partie pratique : Chapitre 1 :
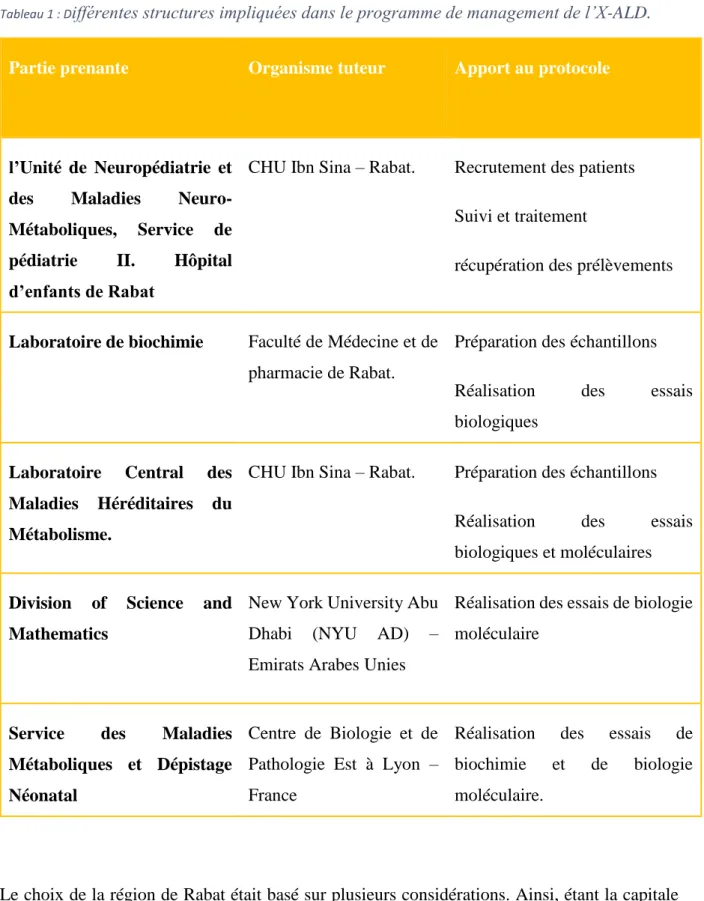
Tableau 1 : Différentes structures impliquées dans le programme de management de l’X-ALD. ... 54
Tableau 2 : Critères d’inclusion au protocole de management d’X-ALD. ... 57
Tableau 3 : Récapitulatif des cas étudiés. ... 64
Tableau 4 : Résumé des caractéristiques cliniques des cas étudiés. ... 65
Tableau 5 : Résultats des dosages des AGTLC chez les patients étudiés ... 66
Chapitre 2 : Tableau 1 : Mutations identifiées dans les 12 patients marocains étudiés ... 77
Tableau 2: Distribution des mutations du gène ABCD1 dans la population marocaine. ... 81
Chapitre 3 : Tableau1 : Protocole de préparation du tampon de protéinase K. Tableau 1 : ... 90
Tableau2 : préparation du mélange leucocytes – enzymes. ... 90
XIII
Abréviations
1H-SRM : Spectrométrie par résonnance magnétique du proton 4-PBA : 4-phenylbutyrate
ABCD1 : transporteur ATP-binding cassette (ABC) sous famille D membre 1 AGTLC : acides gras à très longue chaine
ALDP : protéine adrénoleucodystrophie AMN : adrénomyéloneuropathie BET : Bromure d’Ethidium
CCALD : children cerebral ALD / forme cérébrale de l’enfant de l’Adrénoleucodystrophie CERB : comité d’Ethique pour la Recherche Biomédicale
Cho : Choline
DPI : diagnostic préimplantatoire
EDSS : Expanded Disability Status Scale
ESI-MS : Electrospray ionisation – spectrométrie de masse GC : chromatographie en phase gazeuse
GC-MS : chromatographie en phase gazeuse couplée à la spectrométrie de masse
HPLC – ESI – MS : chromatographie en phase liquide haute performance couplée à l’électrospray d’ionisation – spectrométrie de masse
HSCT : Hematopoeitic stem cells transplantation / greffe allogénique des cellules souches hématopoïétiques
XIV
IRM : imagerie par résonnance magnétique
MDA : amplification par déplacement multiple / multiple displacement amplification MLPA : Multiple Ligation dependent Probe Amplification
NAA : N-acetylaspartate
PCR : polymerase chain reaction PEM : potentiel évoqué moteur
SCD : Siemerling-Creutzfeldt Disease (maladie de Siemerling-Creutzfeldt) VLCFA: very long chain fatty acids
VPA : acide valproïque
X-ALD : adrénoleucodystrophie liée à l’X β-IFN : interféron-β
XV
Résumé
L’adrénoleucodystrophie liée à l’X est une maladie métabolique rare neurodégénérative, due aux mutations du gène ABCD1. Elle se caractérise par plusieurs phénotypes allant de la forme asymptomatique jusqu’aux formes démyélinisantes. Nous avons mis en place un programme multidisciplinaire pour la gestion de cette maladie, en collaboration entre l’Unité de Neuropédiatrie et des Maladies Neuro-Métaboliques, Service de pédiatrie II, Hôpital d’Enfants de Rabat et le Centre des Maladies Héréditaires du Métabolisme. Nous avons travaillé ainsi sur cinq axes : l’aspect clinique, biologique, moléculaire, management de la maladie et la sensibilisation.
Un total de huit familles atteintes de la maladie a été recruté, avec seize patients (quatre femmes hétérozygotes et douze enfants). Nous avons développé trois protocoles de management de l’X-ALD : i. Protocole général du cas index, ii. ¨Protocole des cas asymptomatiques, iii. Protocole des femmes hétérozygotes. Cliniquement, les femmes hétérozygotes étaient toutes asymptomatiques. Les garçons présentaient en majorité la forme cérébrale de l’enfant. Tous les patients recrutés présentaient des taux élevés des AGTLC, ce qui confirme la maladie.
L’étude moléculaire a porté sur l’identification des variants génétiques présents au niveau de la population marocaine. Trois mutations causatives ont été découvertes incluant deux nouvelles mutations, une mutation de type faux sens c.652C>G (p.Pro218Ala), et une large délétion de l’exon 3 à l’exon 8, en plus de la mutation c.1661G>A (p.Arg554His) déjà citée au niveau de l’X-ALD Database. Pour la sensibilisation nous avons organisé et été présents dans vingt-cinq évènements scientifiques organisés dans des hôpitaux, universités, écoles et associations. Ces résultats montrent l’importance de l’X-ALD dans le contexte marocain et permettront de mieux prendre en charge les patients atteints de cette maladie et d’améliorer leur qualité de vie, ainsi que d’éviter les erreurs de diagnostic et aussi de faciliter l’accès aux traitements via le diagnostic précoce.
XVI
Mots clés : Adrénoleucodystrophie liée à l’X, acides gras à très longue chaine, gène ABCD1,
XVII
Abstract
X-linked adrenoleukodystrophy (X-ALD) is a neurodegenerative disorder caused by mutations in the ABCD1 gene. Adrenomyeloneuropathy and childhood cerebral ALD are the most common phenotypes. It is caused by mutations in the adrenoleukodystrophy protein gene
ABCD1. The principal biochemical abnormality is the accumulation of saturated very long
chain fatty acids (VLCFAs : > C22:0, mainly C26:0), which is due to the impaired capacity for β-oxidation in peroxisomes. Diagnosis is usually based on the VLCFA levels in plasma, and confirmed by molecular analysis.
Our project is a transversal descriptive study of the first program of diagnosis, treatment and follow-up of X-ALD in Morocco, with a multidisciplinary approach in collaboration between The Neuropediatric and neurometabolic service of Children's Hospital, and The Central Laboratory of Hereditary Metabolic Diseases of Rabat. This research is focuses on five axes: clinical, biological, molecular, management and awareness.
We developed three protocols of X-ALD management: general, asymptomatic and heterozygous protocol. Over a period of seven years, we recruited eight families with sixteen patients. Clinically, the presentation is primary adrenal insufficiency and behavioral changes. All patients had elevated very long fatty acids.
The goal of the molecular study was to identify genetic variants and. characterize the mutations in a Moroccan cohort. Three causative mutations were discovered including two novel mutations, missense mutation c.652C>G (p.Pro218Ala) and an hemizygous deletion mutation of 3 and 8 exons, as well as missense mutation c.1661G>A (p.Arg554His).
For awareness, we organized and were present in twenty-five events in hospitals, schools, associations and universities, including physicians, families of patients and teachers in schools.
XVIII
This is the first study of X-linked adrenoleukodystrophy in Morocco. It shows the importance of this metabolic disease. The discovery of X-ALD mutations will further help in genetic counseling and prenatal diagnosis providing a better the capacity for earlier diagnostic and therefore enhancing the chance for access to treatment.
Key words: X-linked adrenoleukodystrophy, X-ALD, neurodegenerative disorder, rare
XIX
صخلم
وأ يفروتسيدوكولونيردلأا ضرم زاهجلاب و ضيلأاب قلعتم ردان ضرم نع ةرابع وه ةيغامدلا ءاضيبلا ةداملاو رظكلا رومض .يبصعلا اذه عجريو ةثروم يف تارفط ىلإ ضرملا ABCD1 . يذلا رهظملا اهنم ةيرهاظ طامنأ ةدعب ضرملا اذه زيمتي ةيبناج ضارعأ ةيأ هيف سيل يذلا رهظملاو لكآت صخي .نيلايملا ،ضرملا اذه ةسارد لجأ نم تاصصختلا ددعتم جمانرب عضوب انمق ثحبلا اذه يف نواعتلاب كلذو نيب ضيلأا ضارمأ ةدحو ب لافطلأل ةيبصعلا لافطلأا ىفشتسم زكرمو ةقلعتملا ةيثارولا ضارملأا ضيلأاب رواحم ةسمخ ىلع انلغتشاف .طابرلاب :ىربك يريرسلا بناجلا يجولويبلاو ةرادإ اذكو يئيزجلاو ضرملا هب ةيعوتلاو . نم ةنوكم ،ضرملا اذهب ةباصم رسأ ينامث ةساردب انمق تاونس عبس تماد ةرتف للاخ رشع ةتس عضوب انمق دق و .اضيرم :ضرملا اذه ةرادلإ تلاوكوتورب ثلاث 1 ءاسنلا لوكوتوربو ،ضارعأ نودب تلاحلا لوكوتوربو ،ماعلا لوكوتوربلا . ومل تلاماحلا .ضرملا تاثر روكذلا لافطلأا ةيبلاغ ناك امنيب ،ضارعأ نودب نهلك نك ضرملل تلاماحلا ءاسنلا نأ اندجو ،يريرسلا ىوتسملا ىلع .يغامدلا فنصلاب ةقلعتم ضارعأ نورهظي ضرملل ةببسم تارفط ثلاث انفشتكاف ،ةنيعلا هذه دنع ةدجاوتملا تارفطلا ديدحتب انمق ،ةيئيزجلا ةساردلل ةبسنلاب ، يتلآاك يه و : c.652C>G (p.Pro218Ala) نوسكا نم ريبك فذح و 3 إ ل ى نوسكا 8 لا ل اذك و نيتديدج نيترفط ناربتعت نيت c.1661G>A (p.Arg554His) .ضرملاب ةقلعتملا تانايبلا ةدعاق يف اقباس اهدرس مت يتلا يف اندجاوت دقف ،ضرملاب ةيعوتلا صوصخب امأ مت ايملع اثدح نيرشع و ةسمخ ظنت سرادم و تاعماج و تايفشتسم يف اهمي .هتاقلعتم و ضرملاب فيرعتلا لجأ نم تايعمج و .برغملا يف ضرملا ةيمهأ زربت جئاتنلا هذه لك نم نكمتسو هنع جتنيس امك .هل نيلماحلا ىضرملاب ةيانعلا فورظ نيسحت نم فيفختلا صيخشتلاب ةقلعتملا ءاطخلأا لوصحلا ليهستو .ركبملا صيخشتلا قيرط نع كلذ و ةبسانملا تاجلاعلا ىلع تاملكلا :حيتافملا ةثروم ،ادج ةليوطلا ةلسلسلا تاذ ةينهذلا ضامحلاا ،ةيرظكلا ةدغلا روصق ،يفروتسيدوكولونيردأ ABCD1 .ةرفط ،1
Introduction générale
Aucune maladie n'est trop rare pour ne pas mériter attention, une citation sur la page web d’Orphanet, le portail et des maladies rares et des médicaments orphelins, qui attire l’attention sur l’importance des rare, qui sont des maladies qui existent malgré leur rareté. Ils constituent une source de souffrance pour les patients et leurs familles.
L’adrénoleucodystrophie liée à l’X fait partie de ces maladies rares métaboliques. Il s’agit d’une maladie neurodégénérative mortelle. Elle touche les deux sexes, hommes et femmes. Les femmes sont toujours conductrices et peuvent manifester une forme appelée adrénomyéloneuropathie à partir de 45 ans. Chez l’homme, plusieurs phénotypes sont possibles, ils vont de la forme asymptomatique jusqu’aux formes médullaires et cérébrales démyélinisantes selon l’âge et la topographie es lésions. Cette maladie est le résultat de mutations du gène ABCD1.
A l’échelle internationale, il existe plusieurs études sur cette maladie. En effet, de nombreux laboratoires et équipes de recherche se sont investi dans l’étude des différents aspects de cette maladie. En ce qui concerne le traitement, Plusieurs essais cliniques s’ouvrent chaque année et recrutent des patients de par le monde pour effectuer des essais in vivo. Ils s’investissent dans l’étude de traitements palliatifs tels que l’huile de Lorenzo, mais aussi dans les traitements curatifs tels que la greffe allogénique des cellules souches hématopoïétiques et la thérapie génique.
Tous ces efforts nécessitent de grands fonds pour financer ces travaux de recherches et pour permettre aux patients l’accès au diagnostic, aux traitements et aux investigations nécessaires. Ainsi, il existe plusieurs établissements gouvernementales et de la société civile qui appuient tout ce mouvement scientifique et médicale, et qui sont actifs d’une manière continue en finançant les progrès de recherches et en organisant des évènements de sensibilisation au profit des acteurs de ce domaine, et en assurant un accompagnement des familles des malades. On peut citer à titre d’exemple l’association ELA, et le projet Myéline.
Dans les médias, l’X-ALD a fait l’objet d’un film dramatique « Lorenzo's Oil », qui raconte l’histoire d’un enfant Lorenzo atteint d’une X-ALD, basé sur la véritable histoire d'Augusto et
2
Michaela Odone, deux parents dans une lutte acharnée pour chercher un traitement pour leur fils Lorenzo, atteint d'X-ALD. Augusto Odone est le fondateur du Projet Myéline International. Il est aussi, avec son épouse Michaela, à l’origine de la mise au point de l’huile de Lorenzo. Malgré le progrès qu’a connu la recherche scientifique liée à cette pathologie, plusieurs aspects restent encore méconnus et non compris. Et on avance toujours des hypothèses qui méritent d’être vérifiées.
Il existe plusieurs problématiques liées à l’X-ALD :
La maladie se manifeste sous plusieurs phénotypes et il n’existe pas de corrélation phénotype-génotype, ce qui donne lieu généralement à un tableau clinique non spécifique qui ressemble à de nombreuses autres pathologies, ce qui donne lieu à des erreurs de diagnostic.
L’importance et la difficulté du diagnostic précoce. En effet, le patient ne peut avoir accès au traitement que pendant le stade asymptomatique ; une fois les lésions de démyélinisation ont lieu, l’accès à la greffe de moelle osseuse et à la thérapie génique n’est pas possible.
La fenêtre thérapeutique très étroite entre le moment d’apparition des premières lésions de démyélinisation et le moment optimal de traitement par greffe.
A l’échelle nationale, Au Maroc, avant de démarrer ce programme, il n’y avait aucune étude structurée sur la maladie, et à cause de son aspect rare, les erreurs de diagnostic font que les patients restent perdus entre les services, et le diagnostic spécifique n’a lieu que dans de rares cas. Il en résulte que les patients n’ont pas accès aux traitements palliatifs ni au traitement de substitution par hormonothérapie de l’insuffisance surrénalienne. Aussi il n’existe pas d’association dédiée à accompagner ces patients.
Vu l’aspect couteux des investigations et en l’absence de soutien des établissements de la société civile, plusieurs patients abandonnent le suivi. Pour en résumer il n’y avait pas de structure organisée dédiée à cette maladie qui s’intéresse à ses différents aspects dans une optique globale et multidisciplinaire.
Dans ce contexte nous avons senti le besoin de participer dans le changement de la situation par la mise en place d’une ébauche scientifique qui s’intéresse à l’étude de la maladie dans sa globalité. Ainsi, et après une bonne réflexion et discussion avec les Professeurs Chabraoui et
3
Pr Kriouile, nous avons réalisé le design de cette étude qui touche tous les aspects de la maladie, à savoir l’aspect clinique, biologique et moléculaire, et qui essaye de répondre aux questions suivantes :
Quelle est la situation de l’X-ALD au Maroc ?
Quel est le profil génétique de l’X-ALD chez les patients marocains ? Comment assurer un diagnostic précoce dans le contexte marocain ? Comment prévenir la maladie au Maroc ?
En tant que première étude, elle a nécessité un temps important de sept ans, et une collaboration entre plusieurs unités scientifiques, et un grand effort pour créer des partenariats, chercher les familles, les intégrer au programme, les accompagner, trouver des solutions pour les problèmes financiers liés à la bonne marche du programme et à la participation des familles aux investigations couteuses et à assurer les thérapies adaptées.
Nous avons mis en place un programme multidisciplinaire pour la gestion de cette maladie, en collaboration entre l’Unité de Neuropédiatrie et des Maladies Neuro-Métaboliques, Service de pédiatrie II, Hôpital d’Enfants de Rabat, le Centre des Maladies Héréditaires du Métabolisme, the Division of Science and Mathematics - New York University Abu Dhabi (NYU AD) – Emirats Arabes Unies et le service des Maladies Métaboliques et Dépistage Néonatal du Centre de Biologie et de Pathologie Est à Lyon – France. Nous avons travaillé ainsi sur cinq axes : l’aspect clinique, biologique, moléculaire, management de la maladie et la sensibilisation vis-à-vis de la maladie.
Cette thèse comporte deux parties principales ; une partie théorique qui donne une image sur les différents aspects de la maladie, et une partie pratique subdivisée en trois grands chapitres. Dans le premier chapitre nous avons travaillé sur le management de l’X-ALD. Dans le deuxième nous avons étudié le gène ABCD1, et la distribution des phénotypes, et nous avons mis en évidence deux nouvelles mutations chez des patients marocains ; et le dans le troisième chapitre nous avons mis au point une technique de diagnostic de l’X-ALD, via la recherche de la mutation c.1661G>A (p.Arg554His).
4
5
1. Historique
Le premier cas d’X-ALD a été décrit et publié en 1910 par Haberfeld et Spieler [1]. Trois ans plus tard, le premier cas mort probablement de l’X-ALD était le frère du premier cas publié et a été décrit dans une publication de Paul Schilder en 1913 [2]. La maladie est appelée depuis la maladie de Schilder « Schilder’s Disease ».
Depuis 1923, la maladie devient « Siemerling-Creutzfeldt Disease » (SCD) en rapport avec les deux chercheurs Siemerling et Creutzfeldt qui ont décrit la maladie chez deux garçons avec un syndrome qui consiste en une leucodystrophie, une atrophie surrénale et une hyperpigmentation associés à une infiltration leucocytaire. Leur papier est considéré comme étant le premier rapport détaillé et complet sur la maladie [3]. Plus de 30 cas de SCD ont été rapporté depuis [4].
En 1963, on commence à parler de la liaison à l’X (X-Linkage) en se basant sur l’analyse de pédigrées [5]. La maladie est appelée ainsi « Sex-linked Schilder’s disease » [6]. le nom « adrénoleucodystrophie liée à l’X » (X-linked adrenoleukodystrophy, X-ALD) n’a pris place qu’en 1970 avec Michael Blow [7].
En ce moment, la maladie a été classée en tant que maladie métabolique dont le diagnostic repose sur la biopsie des glandes surrénales. [8] [9].
En 1976, le premier cas de la forme adulte d’X-ALD a été décrit par Herbert Budka et al. à Vienne [10]. Cette forme a été nommée Adrénomyéloneuropathie (AMN) par Griffin et al. en 1977 [11]. Tandis que le premier cas d’X-ALD chez la femme n’a été publié qu’en 2012[12]. La démonstration de l’accumulation des AGTLC, caractéristique de l’X-ALD est due à Powers et al. en 1976, ce qui a permis le développement d’essais de diagnostic via la détection de l’accumulation des AGTLC dans les cellules et les fluides de l’organisme, et de lancer des protocoles de diagnostic prénatal par la suite [13]. Un biomarqueur pour le diagnostic de la maladie devint par la suite disponible, et la signature biochimique était l’accumulation anormale de l’acide hexacosanoique (C26:0) [14] [15].
Les publications des années 70, ont été en accord sur les trois principales caractéristiques de la maladies : la leucodystrophie, l’atrophie des glandes surrénales et l’hyperpigmentation de la peau [8].
6
Dans les années 70 et 80 , l’équipe de Hugo Moser à l’Institut Kennedy Krieger de Baltimore, ont apporté une grande contribution à la caractérisation clinique de l’X-ALD, et ont créé une prise de conscience de la maladie auprès des médecins [16] [17].
Les années 90, ont été marquées par la découverte de l’huile de Lorenzo, qui a été le fruit d’un travail de recherche des parents de Lorenzo, enfant atteint d’X-ALD [18], [19]. Cette découverte avait une contribution significative dans le développement de nouvelles approches thérapeutiques. Cette trouvaille a fait l’objet d’un film intitulé « Lorenzo’s Oil » qui a joué un rôle important dans la sensibilisation vis à vis des maladies rares et métaboliques, et a fait l’objet d’un long débat [20].
A l’aube du 21e siècle, les travaux réalisés portent sur l’amélioration des moyens de diagnostic
et la compréhension de la physiopathogénie de la maladie, dans l’optique de développer de nouvelles thérapies.
Nous avons présenté ici une brève revue sur les principales découvertes de la maladie et l’évolution de son appellation. Cependant Kemp et al. expriment que l’appellation « adrénoleucodystrophie » est erronée, car toutes les personnes atteintes ne développent pas forcement une atteinte neurologique ou surrénalienne, telles que les femmes. Il propose en effet, une nouvelle appellation « le déficit d’ABCD1 » (ABCD1 deficiency), qui sera la mieux adaptée. Toutefois, le changement de terminologie prend des décennies avant d’être adopté [21].
7
2. Epidémiologie
Actuellement nous disposons de données relatives à l’incidence de l’X-ALD en Europe, en Amérique du nord et en Amérique du sud, en Asie et en Australie [22]. Elle va de 1/16000 aux états unis d’Amérique [23] jusqu’à 1/100000 en France et en Hollande [24], [25]
Il n’est facile de déterminer la prévalence réelle et l’incidence de l’X-ALD, vu la nature de la maladie et sa complexité, due aux caractéristiques suivantes :
L’hétérogénéité clinique : l’X-ALD ressemble cliniquement à plusieurs maladies neurologiques te métaboliques.
Le phénotype asymptomatique : Les cas asymptomatiques et les femmes hétérozygotes ne montrent aucun symptôme cliniques et ne sont pas reconnus à l’examen cliniques et ne peuvent être diagnostiqués que lors du conseil génétique.
Les cas présentant des symptômes non spécifiques. Les cas faux diagnostiqués pour d’autres maladies. Les faux négatifs au dosage des AGTLC [26], [27].
Le manque de coordination entre les structures médicales pour l’enregistrement des patients atteints de la maladie dans certains pays.
Or, la collaboration entre les centres de recherche augmente le potentiel d’identification d’une large proportion de cas d’X-ALD [23]. Les études ont montré que seuls 33% des cas sont testés dans un pédigrée atteint d’X-ALD, ce qui renforce la théorie que l’incidence de cette maladie est sous-estimée. L’étape la plus importante pour palier à ce problème est de sensibiliser les professionnels de santé et les familles atteintes de l’importance potentielle de s’inscrire aux programmes d’étude de cette maladie [23].
En Afrique, aucune donnée sur la prévalence de l’X-ALD n’est publiée pour les pays africains. Mais, il parait que la prévalence est approximativement la même dans tous les groupes ethniques [28].
8
3. Aspects biologiques
3.1. Gène
ABCD1
L’X-ALD est une maladie métabolique due aux mutations du gène ABCD1 (transporteur
ATP-binding cassette (ABC) sous famille D membre 1), localisé au niveau du chromosome Xq28
[29]. Le gène ABCD1 code pour le ABC hémi-transporteur peroxisomal (anciennement appelé la protéine adrénoleucodystrophie, ALDP).
L’X- ALD est une maladie lié à l’X. Les garçons ne reçoivent jamais le chromosome X de leurs pères, ils le reçoivent toujours de leur mamans et expriment la maladie selon les phénotypes décrits dans le chapitre aspects cliniques.
Les femmes reçoivent un chromosome X de la maman et un chromosome X du papa, elles sont toujours conductrices. La probabilité de transmission du gène affecté est de 50% [30].
A cause de la localisation du gène ABCD1 sur le chromosome X, un seul allèle affecté peut entrainer la maladie. Par conséquent, un énorme nombre de différentes mutations pathogènes ont été décrites dans le cas de l’X-ALD. Ces mutations sont décrites dans la base de données X-ALD (http://www.x-ald.nl) [31].
Bien que la majorité des patients héritent généralement l'allèle ABCD1 défectueux de l’un des parents, entre 4,1% et 19% des cas X-ALD ont été signalés porteurs de mutations acquises de novo [22].
3.2. Les acides gras à très longue chaine (AGTLC)
Les AGTLC sont des acides gras saturés non ramifiés, avec une chaine qui dépasse 20 atomes de carbones [32], à savoir l’acide docosanique C22 :0, l’acide tétracosanoique C24 :0 et l’acide héxacosanoique C26 :0 (figure 1) [33]. Ils sont présents chez les mammifères en tant que constituants des sphingolipides [33]. Ils proviennent de l'alimentation et de l’élongation des
9
acides gras à longue chaine qui a lieu au niveau du réticulum endoplasmique, et qui est la principale source de l’accumulation des AGTLC en cas d’X-ALD [34], [35].
Figure 1 : structures des AGTLC C22, C24 et C26 [33]
L’élongation des acides gras à longue chaine s’effectue par les élongases ELOVL6 pour les acides gras C18:0 et C22:0 et par les élongases ELOVL1 pour les acides gras C24:0 et C26:0. La protéine ALDP permet le transport des C24:0/C26:0-CoA à travers la membrane peroxysomale en cas d’X-ALD, la protéine ALPD est complètement absente ou déficitaire ce qui en résulte un déficit de la β-oxydation peroxysomale pour la dégradation des AGTLC, ce qui augmente les niveau cytosoliques des AGTLC (figure 2) [34].
Les AGTLC sont activés dans le cytosol par l'acyl-CoA synthase en CoA-thioesters. Les peroxysomes des mammifères sont équipés de trois transporteurs ABC : ABCD1, ABCD2 et ABCD3, dont le rôle est la transportation d’une manière ATP-dépendante d’un large spectre d’acyl-CoA-thioesters à savoir les AGTLC, à travers la membrane peroxysomale [36]. En effet, les AGTLC sont transportés principalement par le transporteur ABCD1. En cas de déficience dans l’expression de l’ABCD1, les transporteurs ABCD1 et ABCD2 ne peuvent compenser que très partiellement cette fonction [37], d’où l’accumulation de ces AGTLC.
10
Figure 2: Métabolisme des AGTLC [34].
Les AGTLC sont dégradés en coopération entre les voies de la β-oxydation peroxysomale et mitochondriale.
Les peroxysomes sont des organites essentiels du cytosol chez les cellules eucaryotes supérieures. Ils sont siège de de multiples fonctions métaboliques, y compris la voie peroxysomale de la β-oxydation [36], [38].
Chez les patients atteints d’X-ALD, les AGTLC, en particulier C26, s’accumulent dans les tissus et les fluides du corps, et servent ainsi comme biomarqueurs pour le diagnostic de la maladie [28]. Les AGTLC ont un effet cytotoxique sur la cellule nerveuse [39]. L’élévation de leurs taux induit la mort cellulaire chez les oligodendrocytes et les astrocytes, et une dérégulation de l’hémostase intracellulaire du calcium [40].
11
3.3. La corrélation phénotype-génotype
Il est devenue une évidence qu’il n’existe pas une corrélation phénotype – génotype pour la maladie d’X-ALD [32]. En effet, la même mutation peut donner lieu à tous les phénotypes possibles de l’X-ALD [41]. Ceci peut avoir lieu dans la même famille avec la même mutation et entre les vrais jumeaux [42]. Aussi il n’existe pas une relation entre la localisation de la mutation et le degré de sévérité de la maladie ou son évolution [31], [43], [44].
La grande variabilité dans l’expression phénotypique du même gène peut être due à des facteurs épigénétiques et stochastiques ou à la présence de gènes modificateurs [45]–[50].
12
4. Aspects cliniques
L’X-ALD est une maladie progressive qui se présente sous un large spectre de formes cliniques, de ce fait elle peut être diagnostiquée et confuse à d’autres maladies. De plus, son aspect rare, rend son étude encore plus difficile.
En se basant sur les données publiées, on peut raisonnablement estimer que tous les patients atteints d’X-ALD sont nés en un état présymptomatique [21]. Le plus jeune patient symptomatique reporté avait 3 ans [34]. La figure 1, résume les formes cliniques de l’X-ALD.
Figure 3 : Formes cliniques de l’X-ALD [21] .
4.1. Le spectre clinique chez l’homme
4.1.1. L’insuffisance surrénalienne (Maladie d’Addison)
L’insuffisance surrénalienne primaire ou maladie d’Addison, peut être le seul symptôme chez les garçons et les hommes pendant des années voir des décennies avant l’apparition de signes neurologiques [51], comme elle peut rester la seule manifestation clinique de l’X-ALD pendant
0 0.5 3 < 20
Age (années)
La forme cérébrale
Interaction entre les variantes génétiques Facteurs environnementaux (Traumatisme crânien, inflammation, inconnu) Facteurs modificateurs La Myéloneuropathie L’insuffisance surrénalienne P rés ym p to m atiq u e
13
toute leur vie [52]. Environ 70 % des patients atteints d’ALD finissent par développer à un moment de leur vie une insuffisance surrénalienne. L’ALD est la cause la plus fréquente d’insuffisance surrénale chez le garçon après l’âge de quatre ans. Chez l’adulte, elle est la deuxième cause d’insuffisance surrénale après une origine auto-immune [53]–[55]. Il est donc important de suspecter l’X-ALD chez tout patient de sexe masculin présentant une insuffisance surrénalienne [12]. L’insuffisance surrénalienne affecte initialement la fonction glucocorticoide de la glande. Toutefois, la fonction minéralocorticoide est déficiente approximativement chez la moitié des patients atteints d’X-ALD [53].
Les signes cliniques sont évolutifs et comportent une mélanodermie puis des crises d’insuffisance surrénale aiguë avec son cortège habituel (douleurs abdominales, vomissements, hypotension, hypoglycémie, et plus ou moins perte urinaire de sel). [55].
L’insuffisance surrénalienne peut coexister avec toutes les formes d’X-ALD chez l’homme [56]. Chez la femme, elle est exceptionnelle.
14
Figure 5 : Mélanodermie au niveau des extrémités digitées
Figure 6 : Coloration de la gencive due à une insuffisance surrénalienne chez un adolescent atteint d’X-ALD.
4.1.2. Les formes cérébrales démyélinisantes de l’enfant
Elles touchent les garçons entre 5 et 12 ans et évoluent en trois phases : [57]–[59]
une phase latente sans signe clinique, même cognitif, avec l’apparition de lésions de démyélinisation à l’IRM cérébrale qui évoluent lentement ;
une deuxième phase où apparaissent des signes cliniques contemporains d’une progression importante et rapide des lésions de démyélinisation qui deviennent inflammatoires (prise de gadolinium visible sur les séquences T1) ;
15
une phase de stabilisation avec des séquelles motrices, sensorielles et cognitives majeures (état grabataire) pouvant conduire au décès.
Les symptômes cliniques dépendent de la localisation des lésions de démyélinisation. Celles-ci débutent toujours dans des régions spéCelles-cifiques [57] :
Splénium du corps calleux (avec ensuite extension dans la substance blanche pariéto-occipitale);
Genou du corps calleux (avec extension dans la substance blanche frontale) ;
Voies pyramidales au niveau du tronc, des capsules internes de manière uni- ou bilatérale (avec extension vers les centres ovales de la substance blanche pariéto-frontale) ;
Voies auditives au niveau du tronc (avec extension vers les lobes temporaux et circonvolutions de Heschl).
Une atteinte cérébrale démyélinisante peut apparaître quelques années après l’apparition d’une insuffisance surrénale (souvent non diagnostiquée et limitée à une mélanodermie qui apparaît vers l’âge de trois ou quatre ans).
a. Les formes pariéto-occipitales
Ce sont les plus fréquentes (début dans le splénium du corps calleux puis extension dans la substance blanche pariéto-occipitale en regard du cuneus). Les premiers symptômes consistent en des troubles cognitifs au niveau visuomoteur et visuospatial, un déficit de la mémoire immédiate et parfois une atteinte frontale par déconnection des lobes frontaux avec les lobes occipitaux. Ces déficits cognitifs ne sont mis en évidence que par une évaluation neuropsychologique approfondie.
Apparaissent ensuite, des troubles du comportement, consécutifs aux déficits cognitifs. Ils se manifestent généralement par un repliement sur soi-même, des troubles de l’attention, parfois de l’agressivité et des troubles émotionnels. Puis rapidement on assiste à des troubles sensoriels (amputation du champ visuel, puis diminution de l’acuité visuelle ; surdité verbale), des troubles moteurs (syndrome pyramidal des deux membres inférieurs, troubles de la marche, ataxie cérébelleuse, hémiparésie) et parfois des crises convulsives (tonicocloniques bilatérales ou latéralisées), des céphalées, voire même des signes d’hypertension intracrânienne (avec œdème au fond de l’œil).
16
Une fois que les symptômes cliniques sont évidents, la maladie évolue très rapidement de semaine en semaine, et de mois en mois, avec la perte quasi complète de toutes les fonctions cognitives, une cécité corticale et une tétraplégie. Les enfants peuvent se stabiliser à ce stade avec une survie qui peut atteindre deux à trois décennies avec les conditions de prise en charge palliative actuelles [57]. Ces formes représentent environ deux tiers des formes cérébrales d’ALD.
b. Formes frontales
Les formes frontales débutent dans le genou du corps calleux. Elles se traduisent essentiellement par un syndrome frontal (troubles de l’attention, troubles des fonctions exécutives) puis une hémiparésie. Elles apparaissent habituellement après l’âge de dix ans et évoluent plus lentement au début. Ces formes représentent 20 % des cas cliniques [57].
c. Formes cérébrales chroniques
Moins de 5 % des formes cérébrales démyélinisantes n’évoluent pas vers un stade inflammatoire. Elles sont essentiellement observées vers l’âge de dix ans. Les lésions de démyélinisation évoluent très progressivement, aboutissant à une destruction de la myéline dans les régions pariéto-occipitales ou frontales.
Ces formes se révèlent souvent par des troubles cognitifs qui ne sont pas diagnostiqués en tant que tels, mais amènent souvent une prise en charge psychologique chez l’enfant, psychiatrique chez l’adulte.
Bien que les patients présentent à l’évidence un syndrome frontal ou un syndrome pariétal, ce n’est souvent qu’à l’occasion d’une crise convulsive généralisée que le diagnostic est évoqué devant des anomalies en neuro-imagerie. Même en présence de lésions pariéto-occipitales importantes, les patients ne développent pas de troubles sensoriels visuels. Ils ne présentent pas non plus de troubles moteurs sauf lorsqu’à l’âge adulte une AMN apparaît [28], [57].
D’une manière générale, la maladie évolue d’autant plus vite qu’elle débute tôt, les formes pariéto-occipitales ayant une évolution plus rapide que les formes frontales. En majorité, les formes cérébrales de l’enfant ont un pic d’apparition de signes cliniques entre six et sept ans, les premières lésions apparaissant en fait vers l’âge de quatre ans et demi à cinq ans.
17
4.1.3. La forme du pédoncule cérébelleux moyen :
Elle est rapportée chez l’adulte, et se caractérise par des anomalies progressives du comportement, des difficultés de marche avec un déficit d’attention, associés à des signes d’une atteinte pyramidale et cérébelleuse [60].
4.1.4. L’adrénomyéloneuropathie (AMN) :
Décrite pour la première fois en 1981, ce phénotype est le plus fréquent chez l’adulte, atteignant 60 % des hommes porteurs d’une mutation du gène ABCD1 [28], [57], [61]. Les symptômes de l’AMN ne sont pas spécifiques [21].
L’AMN se caractérise par l’apparition d’une paraparésie spastique progressive associée à des troubles de l’équilibre (par atteinte des cordons postérieurs de la moelle) et des troubles urinaires (dysurie, envies pressantes). Il s’y associe parfois des signes cliniques de neuropathie périphérique (démyélinisante, axonale ou les deux) chez 60 % des hommes atteints d’AMN. Les premiers symptômes d’AMN apparaissent entre l’âge de 20 et 50 ans avec un pic entre 20 et 30 ans chez l’homme.
Dans un tiers des cas, la paraparésie spastique évolue rapidement en cinq ans vers un handicap moteur significatif (marche avec une ou deux cannes) ; dans les deux tiers des cas restants, l’évolution est progressive sur 15 à 20 ans. Le diagnostic est facilement évoqué devant une paraparésie spastique progressive associée à des troubles de la paresthésie et des troubles urinaires et fécaux.
Trente-cinq pour cent des hommes atteints d’AMN développent dans un second temps une atteinte cérébrale démyélinisante [62].
L’AMN chez l’homme peut être associée à des troubles du comportement, tel que la perte du contrôle de la vie quotidienne pour la gestion du foyer et des finances [56], la manie, l’hypersexualité et la préservation [63]. Avec le temps, des symptômes de dépression, des troubles émotionnels et une démence peuvent avoir lieu [63] [64].
Il n’existe cependant pas encore de marqueur biologique permettant de prédire l’évolutivité sur le plan individuel, aussi bien chez les hommes que chez les femmes [57].
18
4.1.5. Formes cérébrales démyélinisantes de l’adulte
Elles ont une évolution tout à fait identique aux formes cérébrales de l’enfant avec l’existence de formes occipitales et de formes frontales. La seule différence majeure est que la période initiale pendant laquelle les lésions de démyélinisation évoluent lentement sans manifestations évidentes, dure beaucoup plus longtemps, en règle générale entre cinq à dix ans. Cependant, une fois que les premiers symptômes cliniques deviennent évidents, contemporains de l’apparition de prise de gadolinium à l’IRM cérébrale, l’évolution est aussi rapide que chez l’enfant [28], [57].
Une atteinte cérébrale démyélinisante peut être diagnostiquée chez l’adulte avant que ne soit diagnostiquée une AMN. Tous les adultes avec une atteinte cérébrale démyélinisante présentent cependant des signes cliniques et électro-physiologiques d’une atteinte médullaire. Comme indiqué précédemment, cette atteinte cérébrale peut survenir dans un second temps, après la survenue de signes cliniques d’AMN [57]. La figure 7 montre des lésions cérébrales post mortem chez un adulte avec une pure forme cérébrale démyélinisante.
1.1.1. La forme asymptomatique :
Les formes débutant au niveau des voies pyramidales restent longtemps asymptomatiques jusqu’au moment où apparaît une hémiparésie ou une diplégie, ensuite compliquée de troubles pseudobulbaires, puis d’une atteinte cognitive lorsque les lésions atteignent la substance blanche pariéto-frontale [57]. De même pour les formes débutant au niveau des voies auditives, jusqu’à l’apparition de lésions dans les lobes temporaux ; en fait, elles deviennent le plus souvent symptomatiques quand d’autres lésions de démyélinisation, occipitales ou frontales, apparaissent [57]. Ces formes ne sont diagnostiquées que chez les enfants appartenant à des familles ALD connues. Ces enfants sont alors identifiés lors du conseil génétique.
Une faible proportion d’adultes se présentent à l’âge de 50 ans avec des signes très modérés de paraparésie spastique et sont pratiquement asymptomatiques sur le plan fonctionnel [57].
19
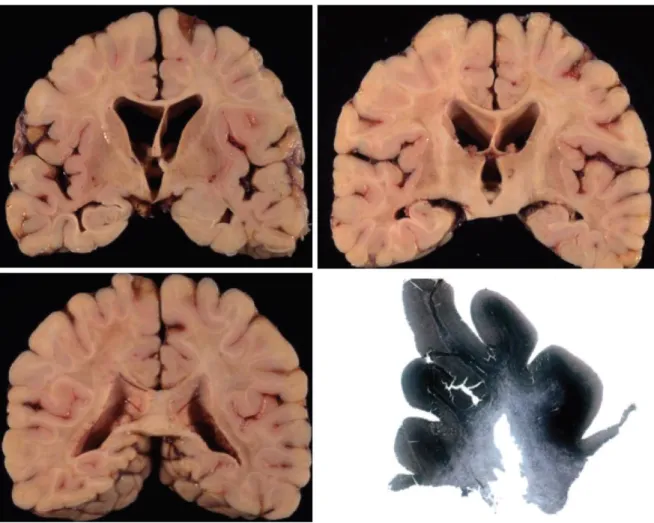
Figure 7 : Examen neuropathologique des hémisphères cérébraux chez un adulte avec une pure
forme cérébrale de l’X-ALD. A : au niveau du striatum, B : au niveau du Thalamus, C : au niveau de la partie postérieure du lobe pariétal, D : lésions de démyélinisation au niveau de la substance blanche cérébrale [65].
1.1.2. Insuffisance gonadique
Les signes cliniques de la dysfonction gonadique se manifestent par une diminution de la libido (46%), une dysfonction érectile (58%), et une cryptorchidie (15%). L’examen physique révèle une diminution de la pilosité pubienne (50%), des testicules de petite taille (12%) et une gynécomastie (35%). Les examens biologiques montrent une baisse du taux de testostérone total plasmatique (12%) [66]. Il existe sur le plan histologique une atteinte des cellules de Leydig et de Sertoli [57].
Parfois, c’est devant des signes cliniques d’insuffisance gonadique que le diagnostic de l’X-ALD est évoqué. Les patients ayant en règle toujours des signes d’AMN associés même mineurs [57].
20
4.2. Spectre clinique chez la femme
4.2.1. L’adrénomyéloneuropathie (AMN) :
Elle touche 50 % des femmes conductrices hétérozygotes [28], [57], [61]. Les premiers symptômes apparaissent habituellement après l’âge de 40 ans. D’une manière générale, une femme conductrice hétérozygote avec des signes d’AMN a une forme évolutive de la maladie de moindre gravité que chez l’homme ; la neuropathie périphérique est moins fréquente aussi, mais les douleurs neurogènes sont plus fréquentes et plus sévères.
4.2.2. La forme cérébrale
La femme n’a jamais la forme cérébrale. Celle-ci survient exceptionnellement chez des femmes porteuses d’une mutation du gène ABCD1 sur un chromosome X et d’un réarrangement ou une délétion du deuxième chromosome X intéressant la région Xq28 où se trouve le gène ABCD1 [57].
4.2.3. La maladie d’Addison :
l’existence d’une insuffisance surrénale biologique avec ou sans signes cliniques est exceptionnelle chez la femme [67].
4.2.4. L’atteinte ovarienne :
Si chez les hommes la fonction gonadique est atteintes, les femmes conductrices hétérozygotes ne présentent aucune anomalie de la fonction ovarienne [57].
21
5. Diagnostic
5.1. Diagnostic clinique
Les symptômes cliniques liés à l’X-ALD sont très hétérogènes. On distingue des symptômes neurologiques et extra-neurologiques, qu’on peut qualifier de signes alarmants et en leur présence on peut suspecter une atteinte par la maladie [68] :
5.1.1. Les signes neurologiques
Les patients présentent souvent des signes moteurs. On assiste alors à une stagnation ou une régression du développement moteur. Chez l’enfant les premiers signes sont généralement une démarche maladroite, une détérioration des compétences fonctionnelles telles que les activités sportives [68], avec une baisse des performances intellectuelles scolaires [57]. Les patients peuvent avoir une implication précoce des voies cortico-spinales, ce qui en résulte des signes de motoneurones supérieurs tels que la quadri-parésie spastique ou la paraparésie spastique. Ces signes moteurs sont souvent associés à des symptômes psychiatriques [68].
L’échelle EDSS (Expanded Disability Status Scale) , bien que critiquée, reste un bon outil de cotation clinique pour juger l’évolution des patients atteints d’X-ALD [69].
5.1.2. Les signes extra-neurologiques :
a. L’insuffisance surrénalienne : qui se caractérise souvent par une mélanodermie, une
hyponatrémie, des symptômes digestifs tels que des nausées et des vomissements [70], et rarement une hypoglycémie. Les patients peuvent montrer une récupération prolongée de l'anesthésie générale comme première indication de l'insuffisance surrénale [68].
b. Troubles endocriniens : liés à l’hypogonadisme chez l’homme. En effet, Les adultes
22
c. Troubles ophtalmologiques : le strabisme, ce qui donne un facies caractéristique de la
maladie chez les patients atteints.
d. Symptômes cutanés : l’hyperpigmentation de la peau (la mélanodermie) et des
gencives liée à la maladie d’Addison [70].
Figure 8 : Mélanodermie apparente sur le visage d’un enfant ayant une X-ALD.
e. Symptômes digestifs : liés à la maladie d’Addison aussi, tels que les vomissements et
la diarrhée [70].
Le diagnostic clinique doit prendre en considération toutes les informations disponibles à savoir l’âge au premier symptôme, l’historique familial, l’ensemble des signes neurologiques et extra-neurologiques décrits ci-dessus.
23
5.2. L’imagerie :
5.2.1. L’imagerie par résonnance magnétique (IRM) :
L’IRM cérébrale dans le cas de l’X-ALD est un élément de très grande importance, car elle intervient dans les différentes phases de la maladie, depuis la suspicion jusqu’au suivi post traitement par greffe de cellules souches hématopoïétique. Ainsi, lors de la suspicion de la maladie, l’IRM permet la confirmation de l’X-ALD chez le cas index en repérant les lésions de démyélinisation. Chez les cas asymptomatiques, elle permet d’évaluer la susceptibilité au traitement par greffe HSCT [71], [72]. Après la greffe, elle permet de contrôler l’efficacité du traitement et l’évolution de l’état du patient [73].
L’X-ALD est une maladie non prédictible, l’IRM cérébrale étant un moyen de diagnostic très important, elle peut être aussi un élément de pronostic qui prédit l’expression et l’évolution de la maladie, selon la topographie des lésions [57], [73].
Il existe deux variantes majeures en IRM cérébrale chez les patients à X-ALD. La variante avec une prédominance postérieure, la plus courante et qui représente 80% des cas. Elle touche la région pariéto-occipitale, le splénium du corps calleux avec une atteinte fréquente des voies auditives et des voies visuelles. La variante avec une prédominance antérieure, qui représente entre 10 à 15% des cas d’X-ALD, elle couvre la substance blanche de la région frontale, le genou du corps calleux avec occasionnellement une atteinte cérébelleuse. A côté de ces deux variantes majeures, 5% des cas peuvent manifester des formes atypiques, avec des anomalies isolées [74]. Le score moyen de sévérité de la variante postérieure est de 9 pour une série allant de 0.5 à 25, alors que celui de la variante antérieure est de 10 pour une série allant de 2 à 18. Il a été démontré qu’il existe une corrélation entre l’âge du patient de la sévérité de la maladie. En effet, la présence de l’une des variantes antérieure ou postérieure à jeune âge est associée avec une progression rapide de la maladie. Le score de progression à l’IRM est aussi fortement corrélé avec la survie du patient [74].
Le changement de signal en IRM est dû au processus inflammatoire de démyélinisation, qui est à l’origine d’une prolongation du temps de relaxation de T1 et de T2.
24
Cinq modèles d’atteinte cérébrale ont été identifiés en prenant en considération l’âge du patient, le score de gravité initial à l’IRM selon les scores de l’échelle de Loes, et la localisation anatomique des lésions.
5.2.2. La spectrométrie par résonnance magnétique du proton (1H-SRM)
Cette technique consiste à analyser les ratios des métabolites NAA/Cr, Cho/Cr, Glu/Cr et MI/Cr dans la substance blanche et la substance grise du patient. Etant donné que les changements biochimiques du métabolisme membranaire ont lieu avant les changements structurels, il faut prendre en considération qu’un phénotype avec une apparence normale chez un adulte ayant une X-ALD peut donner lieu à des changements précoces en son neurochimie. Ces changements précoces peuvent servir comme marqueurs de l’X-ALD chez l’Adulte [69].Actuellement l’utilisation du 7 Tesla Scanner permet de distinguer les changements neurochimiques précoces chez l’adulte. Il a été démontré qu’il existe une corrélation entre l’importance des changements neurochimiques et la sévérité des symptômes chez l’adulte [69]. Ainsi, des essais menés chez des adultes atteints d’X-ALD avec un phénotype d’apparence normale a conclu qu’il existe une diminution des ratios NAA/Cr, Cho/Cr, Glu/Cr et MI/Cr en les comparant aux patients non atteints d’X-ALD [69].
Dans le cas de l’AMN, on assiste à une diminution de la N-acetylaspartate (NAA) au niveau de la substance blanche, qui reflète un dysfonctionnement ou une perte axonale [75]. Dans la forme cérébrale de l’adulte, la démyélinisation peut induire une élévation de la Choline (Cho) qui est un marqueur de la rupture membranaire [76]
Il existe aussi des différences de valeurs des ratios entre les différents phénotypes chez l’adulte. Ainsi, il a été démontré que ces ratios sont plus élevés dans le cas de la forme cérébrale par rapport à l’AMN, mais aucune différence significative entre l’AMN chez l’homme et la femme hétérozygote n’a été retrouvée [69].
Il est à noter que la méthode traditionnelle basée sur les trouvailles cliniques et biochimiques, combinées aux résultats d’IRM à 1.5T, demeure incapable d’expliquer les différences dramatiques en terme de sévérité des symptômes entre les différents phénotypes d’X-ALD. La SRM à 7T permet de mieux visualiser l’architecture des lésions et de détecter les changements liés aux métabolites, qui peuvent servir de marqueurs neurochimiques de la maladie.
![Figure 2 : Métabolisme des AGTLC [34].](https://thumb-eu.123doks.com/thumbv2/123doknet/15061718.698968/29.892.114.713.193.559/figure-métabolisme-des-agtlc.webp)
![Figure 3 : Formes cliniques de l’X-ALD [21] .](https://thumb-eu.123doks.com/thumbv2/123doknet/15061718.698968/31.892.147.668.503.850/figure-formes-cliniques-l-ald.webp)