Le rôle de la vasculature cérébrale et de l’immunité
innée dans la pathophysiologie de la maladie
d’Alzheimer
Thèse
Peter Thériault
Doctorat en médecine moléculaire
Philosophiae doctor (Ph. D.)
Québec, Canada
© Peter Thériault, 2017
Le rôle de la vasculature cérébrale et de l’immunité
innée dans la pathophysiologie de la maladie
d’Alzheimer
Thèse
Peter Thériault
Sous la direction de :
Résumé
La maladie d'Alzheimer (MA) constitue la cause de démence la plus répandue à travers le monde, pour laquelle il n’existe aucun traitement à ce jour. La MA est une maladie neurodégénérative associée à une détérioration progressive de la mémoire et au déclin cognitif, menant ultimement à la perte des fonctions exécutives. Cette maladie se caractérise principalement par l'accumulation et l'agrégation de la protéine neurotoxique bêta-amyloïde (Aβ) dans la neurovasculature et le parenchyme cérébral, coïncidant avec la dysfonction et la dégénérescence des neurones. Malheureusement, les causes exactes de la maladie demeurent inconnues. Toutefois, des études ont souligné le dysfonctionnement de la vasculature cérébrale et de l’immunité innée dans la pathophysiologie de la MA, soit deux systèmes biologiques cruciaux pour le maintien de l’homéostasie au cerveau.
Afin d’étudier leurs rôles dans les cascades neurodégénératives observées dans la MA, nous avons réalisé trois études distinctes visant à moduler la fonctionnalité de ces deux systèmes biologiques importants. Pour ce faire, nous avons employé diverses approches, d’abord chirurgicale pour induire la défaillance de la vasculature cérébrale, ensuite pharmacologique dans le but de réguler la réponse immunitaire au cerveau, et enfin, en modélisant deux facteurs de risques majeurs associés à la MA, soit l’âge et la diète riche en gras, afin d’identifier les mécanismes régissant la dysfonction de ces systèmes.
Les résultats de ces trois études constituant cette thèse soulignent le rôle crucial de la vasculature cérébrale et de l’immunité innée dans le maintien de l’homéostasie au cerveau, impliquant non seulement l’élimination de l’Aβ, mais également la régulation de la réponse inflammatoire, favorisant ainsi un environnement sain pour les neurones. En ce sens, nos résultats suggèrent que leur défaillance contribuent inévitablement à l’initiation des cascades neurodégénératives observées dans la MA. Les retombées de ces travaux auront sans nul doute approfondi notre compréhension de cette terrible maladie.
Abstract
Alzheimer's disease (AD) constitutes the most common cause of dementia worldwide, for which there is no curative treatment to date. AD is a neurodegenerative disease associated with progressive memory impairment and cognitive decline, which ultimately leads to the loss of executive functions. The main pathological hallmark is the accumulation and aggregation of the neurotoxic amyloid-beta (Aß) peptide within the neurovasculature and the brain parenchyma, coinciding with the dysfunction and degeneration of neurons. Unfortunately, the exact causes of AD remain unknown. However, several studies have highlighted the dysfunction of the cerebral vasculature and innate immunity in the pathophysiology of AD, which are two crucial biological systems for homeostasis in the brain.
To investigate their implications in neurodegenerative cascades observed in AD, we conducted three separate studies to modulate the function of these important biological systems. To this end, we used various approaches, first surgical to induce cerebral vasculature dysfunction, then pharmacological in order to regulate the immune response in the brain, and finally, we modeled two major risk factors associated with AD, namely age and high fat diet, in order to identify mechanisms underlying the dysfunction of these systems.
Our results obtained from these studies that constitute this thesis emphasize the crucial role of the cerebral vasculature and innate immunity in the maintenance of homeostasis in the brain, involving, on one hand, the elimination of Aß and, on the other hand, the regulation of the inflammatory response, thus promoting a healthy environment for neurons. As such, our results suggest that their dysfunction will inevitably contribute to the initiation of neurodegenerative cascades observed in AD. The impact of this work will undoubtedly deepen our understanding of this terrible disease.
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des figures ... viii
Abréviations ... ix Remerciements ... xii Avant-propos ... xiv 1. Chapitre 1 - Introduction ... 1 1.1 La maladie d’Alzheimer ... 1 1.1.1 Historique ... 2
1.1.2 Les symptômes cliniques ... 4
1.1.3 La pathophysiologie ... 4
1.1.3.1 La protéine β-amyloïde ... 6
1.1.3.2 La protéine tau ... 8
1.1.3.3 L’inflammation ... 8
1.1.4 Les hypothèses étiologiques ... 9
1.1.4.1 L’hypothèse de l’amyloïde ... 10
1.1.4.2 L’hypothèse de tau ... 12
1.1.4.3 L’hypothèse vasculaire « deux-coups » ... 13
1.1.5 Les facteurs de risque ... 15
1.1.5.1 L’âge ... 15
1.1.5.2 La diète riche en gras ... 18
1.1.5.3 La génétique ... 21
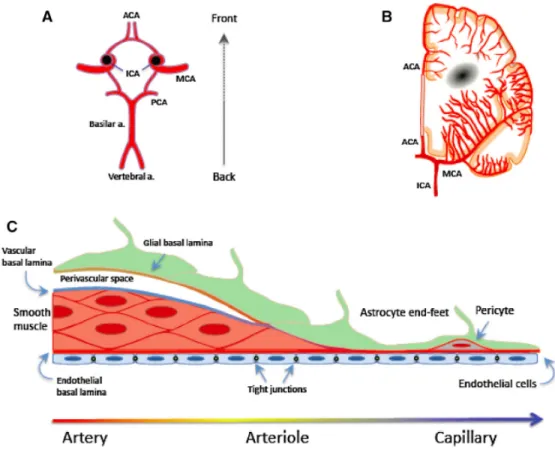
1.2 La vasculature cérébrale ... 24
1.2.1 Anatomie et fonctions ... 24
1.2.2 La barrière hémato-encéphalique ... 26
1.2.2.1 Structure et fonctions ... 26
1.2.2.2 Les échanges entre la périphérie et le SNC ... 30
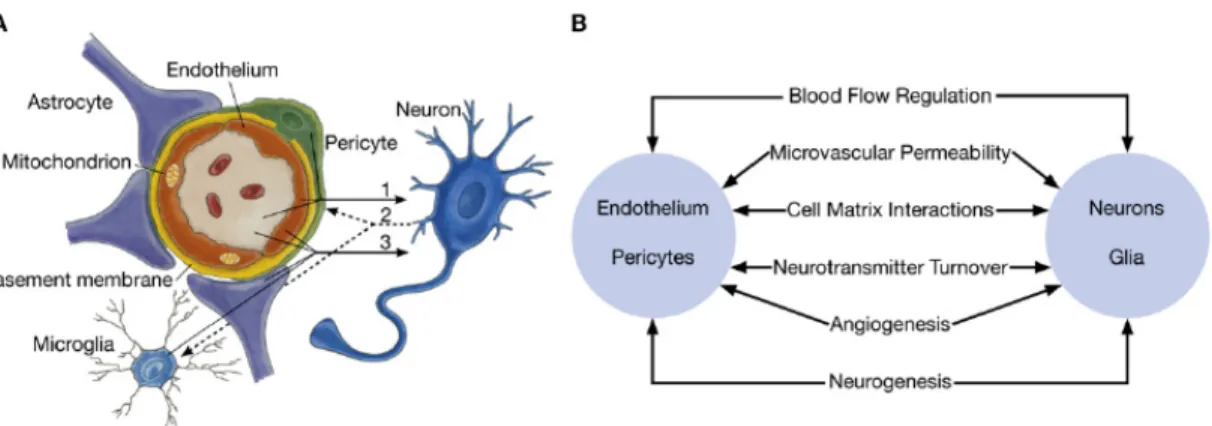
1.2.3 L’unité neurovasculaire ... 33
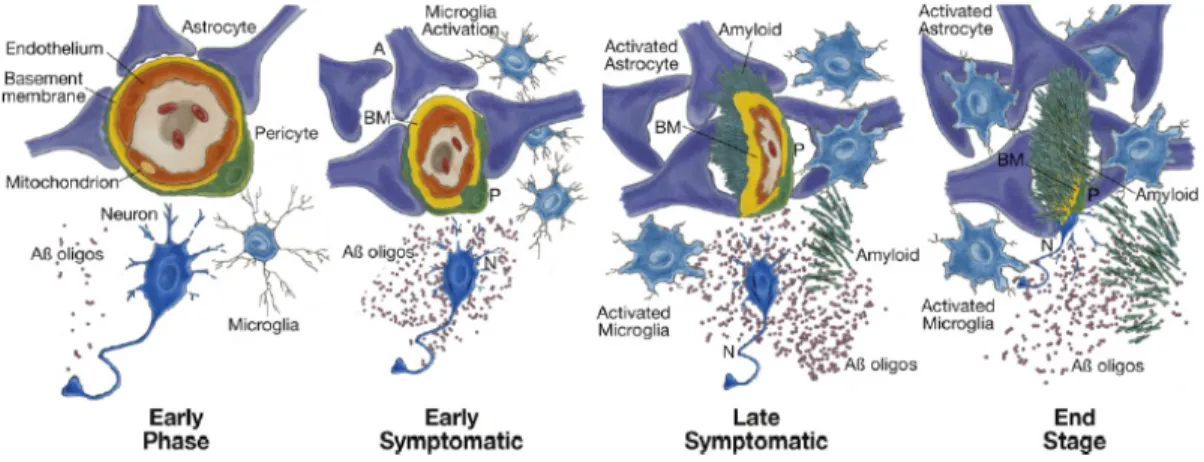
1.2.4 La vasculature cérébrale dans la maladie d’Alzheimer ... 35
1.2.4.1 L’Aβ vasculaire et le CAA ... 37
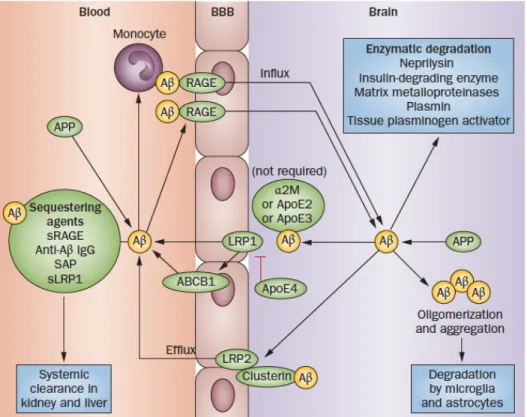
1.2.4.2 L’élimination de l’Aβ à travers la BHE ... 40
1.3 L’immunité innée ... 44
1.3.1 La réponse inflammatoire ... 45
1.3.2 Le système hématopoïétique ... 47
1.3.3 Le système phagocytaire mononucléaire ... 49
1.3.3.1 Les monocytes ... 50
1.3.3.2 Les microglies ... 53
1.3.4 L’immunité innée dans la maladie d’Alzheimer ... 55
1.3.4.1 La neuroinflammation ... 58
1.3.4.2 L’élimination de l’Aβ ... 60
L’élimination de l’Aβ par les monocytes ... 61
1.3.4.2.1 L’élimination de l’Aβ par les microglies ... 64 1.3.4.2.2
2. Chapitre 2 - Hypothèses ... 69
3. Chapitre 3 : L’hypoperfusion cérébrale chronique induit le dysfonctionnement neurovasculaire, conduisant à l’entrée et l’agrégation d’Aβ périphérique au SNC. ... 73
3.1 Note introductive ... 73
3.2 Résumé ... 73
3.3 Mild chronic cerebral hypoperfusion induces neurovascular dysfunction, triggering peripheral beta-amyloid brain entry and aggregation. ... 73
3.3.1 Abstract ... 73
3.3.2 Introduction ... 74
3.3.3 Materials and methods ... 76
3.3.4 Results ... 82 3.3.5 Discussion ... 85 3.3.6 Conclusions ... 88 3.3.7 Abbreviations ... 88 3.3.8 Competing interests ... 89 3.3.9 Authors contributions ... 89 3.3.10 Authors’ information ... 89 3.3.11 References ... 89 3.3.12 Figures ... 94 3.3.13 Supplementary material ... 101
4. Chapitre 4 : Une forme recombinante du « tissue-plasminogen activator » diminue le développement de la pathologie associée à la maladie d’Alzheimer chez la souris APPswe/PS1. ... 103
4.1 Note introductive ... 103
4.2 Résumé ... 103
4.3 Tissue-Plasminogen Activator Attenuates Alzheimer’s Disease-Related Pathology Development in APPswe/PS1 Mice. ... 103
4.3.1 Abstract ... 103
4.3.2 Introduction ... 104
4.3.3 Materials and methods ... 106
4.3.4 Results ... 108
4.3.5 Discussion ... 113
4.3.6 Funding and disclosure ... 116
4.3.7 References ... 117
4.3.8 Acknowledgements ... 120
4.3.9 Figures ... 121
4.3.10 Supplementary information ... 127
4.3.10.1 Supplementary Material and Methods ... 127
4.3.10.2 Supplementary Figures and legends ... 140
4.3.10.3 Supplementary References ... 152
5. Chapitre 5 : La diète riche en gras exacerbe la pathologie associée à la maladie d’Alzheimer chez la souris APPswe/PS1. ... 153
5.1 Note introductive ... 153
5.2 Résumé ... 153
5.3.1 Abstract ... 153
5.3.2 Introduction ... 154
5.3.3 Results ... 156
5.3.4 Discussion ... 162
5.3.5 Materials and methods ... 167
5.3.6 Acknowledgements ... 176
5.3.7 Competing interests ... 176
5.3.8 Funding ... 176
5.3.9 References ... 177
5.3.10 Figures ... 183
5.3.11 Table and table legend ... 194
6. Chapitre 6 - Discussion et conclusion ... 195
6.1 Discussion générale ... 195
6.1.1 L’impact du dysfonctionnement neurovasculaire sur l’accumulation d’Aβ au SNC. ... 195
6.1.2 Le rôle de rt-PA dans l’amélioration de la pathologie. ... 200
6.1.3 La contribution de la diète riche en gras dans l’accélération de la pathologie. ... 203
6.2 Conclusion ... 214
Liste des figures
Figure 1. Poids économique de la MA ... 2
Figure 2. Pathophysiologie de la MA. ... 5
Figure 3. Production du peptide Aß à partir de la protéine précurseur amyloïde (APP). ... 7
Figure 4. L'hypothèse de la β-amyloïde. ... 11
Figure 5. L'hypothèse vasculaire de la MA. ... 15
Figure 6. Les caractéristiques du vieillissement. ... 17
Figure 7. Contribution du métabolisme du cholestérol et de l'apolipoprotéine E à la biogenèse, à la dégradation et l’assemblage du peptide Aβ. ... 20
Figure 8. Aperçu schématique des gènes liés à la MA. ... 22
Figure 9. Anatomie de la vasculature cérébrale. ... 25
Figure 10. Atlas moléculaire simplifié de la BHE. ... 29
Figure 11. Mécanismes de transport de la BHE. ... 32
Figure 12. Schéma de l’UNV. ... 34
Figure 13. Schéma de l'implication de l'UNV dans la pathogenèse de la MA. ... 38
Figure 14. Le transport de l’Aβ à travers la BHE. ... 42
Figure 15. Réponse inflammatoire. ... 46
Figure 16. Modèle hiérarchique d'hématopoïèse de la moelle osseuse. ... 48
Figure 17. Inflammation et rôle des monocytes sanguins. ... 51
Figure 18. Régulation de la microglie / macrophages dans le SNC. ... 55
Figure 19. Réponses immunitaires innées dans le cerveau de la MA. ... 57
Figure 20. Hypothèse de travail. ... 70
Figure 21. Illustration schématique des mécanismes potentiellement impliqués dans nos observations chez les souris APPswe/PS1 suite à la WD. ... 211
Abréviations
Aβ amyloïde-β
ABC ATP-binding cassette
ABCA7 ATP-binding cassette, sub-family A, member 7 ABCB1 ATP-binding cassette, sub-family B, member 1 ABCG2 ATP-binding cassette, sub-family G, member 2
AJ Jonctions adhérentes
APOE Apolipoprotéine E APOE4 Allèle ε4 de l'APOE APP Amyloid precursor protein ATP Adénosine triphosphate AVC Accident vasculaire cerebral
BACE1 β-sécrétase
BDNF Brain-derived neurotrophic factor BHE Barrière hémato-encéphalique CAA Angiopathie cérébrale amyloïde CBF Cerebral blood flow
CCR2 C-C chemokine receptor type 2 CCL2 Chemokine (C-C motif) ligand 2 CD Cluster of differentiation
CMoP Common monocyte progenitor CMP Common myeloid progenitor COX-2 Cyclooxygenase-2
CR Récepteur du complement
CSF Liquide cérébrospinal CVO Organe circumventriculaire CX3CR1 CX3C chemokine receptor 1
DAMP Danger-associated molecular patterns
DC Cellule dendritique
FcR Récepteur Fc (Fragment cristallisable)
GM-CSF Granulocyte macrophage colony-stimulating factor GMP Granulocyte and macrophage progenitor
GSK3β Glycogen synthase kinase 3 beta GWAS Étude d’association pangénomique HDL High density lipoprotein
HSC Cellule souche hématopoïétique IDE Enzyme de degradation de l’insuline IFN-β Interféron-β
IgG Immunoglobulines G
IκBα Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
IL Interleukin
ISF Liquide interstitiel LDL Low density lipoprotein
LDLR Low density lipoprotein receptor LPS Lipopolysaccharide
LRP-1 Low density lipoprotein receptor-related protein 1 Ly6C Lymphocyte antigen 6 complex
MA Maladie d’Alzheimer
MAC1 Macrophage-1 antigen
MAPK Mitogen-activated protein kinase
MARCO Macrophage receptor with collagenous structure MCAO Middle Cerebral Artery Occlusion
MCP-1 Monocyte chemoattractant protein-1 M-CSF Macrophage colony-stimulating factor
MDA Malondialdéhyde
MDP Macrophage and dendritic cell progenitor MHC-II Complexe majeur d’histocomptabilité II MIF Macrophage migration inhibitory factor MMP Métalloprotéinase matricielle
MO Moelle osseuse
MPL Monophosphoryl lipid A
MPS Système phagocytaire mononucléaire MSR1 Macrophage scavenger receptor 1
ND Normal diet
NEP Néprilysine
NF-κB Nuclear factor kappa-light-chain-enhancer of activated B cells NFT Neurofibrillary tangles
NOD Nucleotide-binding oligomerization domain Nr4a1 Nuclear receptor subfamily 4 group A member 1
Ox-LDL Oxydized-LDL
PAMP Pathogen-associated molecular patterns PLA-2 Phospholipase A2
PS1 Presenilin 1
PS2 Presenilin 2
RAGE Receptor for advanced glycation end products ROS Espèces réactives en oxygène
rt-PA recombinant tissue plasminogen activator sAPPα Secreted amyloid precursor protein alpha
sLRP-1 Soluble low density lipoprotein receptor-related protein 1 SNC Système nerveux central
SORL1 Sortilin-related receptor, L(DLR Class) A repeats containing SRA Scavenger receptor A
TGF-β Transforming growth factor beta
TJ Jonctions serrées
TLR Toll-like receptor
TNF-α Tumor necrosis factor-alpha t-PA tissue plasminogen activator
TREM2 Triggering receptor expressed on myeloid cells 2 UNV Unité neurovasculaire
VLDL Very low density lipoprotein VSMC Vascular smooth muscle cell
« L’imagination est plus importante que le savoir »
- Albert Einstein -Remerciements
Je voudrais tout d'abord remercier mon directeur de thèse, le Dr Serge Rivest, de m'avoir donné la chance d'évoluer au sein de son équipe. Dr Rivest est un scientifique de haut niveau dont les recherches ont un grand impact dans le domaine de la neuroinflammation, et plus particulièrement de la maladie d’Alzheimer. Sa grande générosité et disponibilité, ainsi que ses encouragements m'ont permis d'évoluer scientifiquement, mais aussi personnellement. Merci de m'avoir fait confiance!
Un grand et infini merci à Ayman (prof. ElAli!). Ayman, tu as été un véritable mentor et un ami hors pair durant tout mon cheminement au doctorat. Je te dois en majeure partie cet exploit, car tu es resté présent même dans les pires moments, et c’est ce qui a fait la différence. J'ai aimé collaborer avec toi et discuter de nos points de vue au niveau scientifique et personnel. Je me suis amusé avec toi et saches que tu m'as apporté beaucoup!
Je voudrais également remercier toutes les personnes du Team Rivest, ainsi que des laboratoires avoisinants, incluant tous les étudiants M.Sc., Ph.D., Post-docs et stagiaires que j’ai pu cotôyer. J’aimerais tout spécialement remercier David Gosselin, bientôt prof. Gosselin, un collègue exceptionnel qui a su me transmettre l’esprit scientifique. Bien sûr, je tiens aussi à remercier Giulia Cisbani notre lab manager, ainsi que les professionnels de recherche du laboratoire, Nataly Laflamme, Marie-Michèle Plante et Paul Préfontaine, qui ont été d'une grande aide technique et souvent même, une grande source de conseils de vie! Vous avez grandement contribué à ma réussite tant sur le plan professionnel que personnel! Je suis très heureux de vous avoir côtoyés.
Je tiens à remercier mes amis, mes chums, pour m'avoir changé les idées et m’avoir supporté lors de vos visites à Québec, ou lors des miennes à Sherbrooke ou Montréal. Merci à Mathieu, François, Maxime Lapointe, Antoine, Patrice, Vincent, Étienne, Guillaume Gauthier, Guillaume Fafard, Maxime Saumier, Michaël, Rémi, Alexandre, Stéphanie, etc. Un énorme merci à Pierre et Karine, mes rares amis de Québec et complices de grimpe, de vélo de montagne et surtout de bonne bouffe, vin et rhums! Vous m'avez beaucoup apporté!
Bien sûr, je voudrais remercier mes parents, Mario et Diane, mes soeurs Maggie et Audrey, ainsi que leurs conjoints respectifs Samuel et Félix pour m'avoir supporté pendant toutes ces années d'études graduées. Je vous suis grandement reconnaissant d'avoir cru en moi et en mon choix de carrière et ce, surtout quand j’étais à bout de souffle. Vous êtes tous très très importants pour moi! Je vous aime.
Maintenant, et non la moindre, j’aimerais remercier ma femme, Karine, mon amoureuse et confidente, qui pendant toutes ces longues années, jour après jour, autant pendant les moments heureux que les trop nombreux moments difficiles, a su m'épauler et me donner confiance en moi et en l'avenir. Karine, tu m’as donné un fils merveilleux, Gabriel, qui a littéralement donné un sens à ma vie et à ce que je fais. Ma femme, mon fils, votre présence et votre amour au quotidien m'ont permis de me rendre là où j'en suis aujourd’hui, pour ainsi atteindre mes objectifs de vie. Je vous dois absoluement tout, car vous êtes toute ma vie. Mes amours, je vous aime, cette thèse vous est dédiée!
Avant-propos
Cette thèse contient les résultats des principaux projets de mes études graduées et est divisée en six parties. La première partie est une introduction comprenant un survol de la littérature pertinente aux projets présentés. La seconde décrit mes hypothèses et objectifs. Ensuite, les chapitres 3, 4 et 5 constituent des articles publiés dans des journaux scientifiques révisés par des pairs et dans lesquels je suis le principal ou co- auteur, sous la tutelle du Dr Serge Rivest : 1 ElAli, A., Thériault, P., Préfontaine, P., and Rivest, S. Mild chronic cerebral hypoperfusion induces neurovascular dysfunction, triggering peripheral beta-amyloid brain entry and aggregation. Acta Neuropathologica Communications 1-75 (2013).
Contributions :
La conceptualisation et la demande de financement du projet ont été effectuées par le Dr Serge Rivest. Ce projet a été initié par le Dr Ayman ElAli, s’étant chargé du design experimental, des sacrifices et de la coupe des cerveaux. Le Dr ElAli a exécuté l’isolation des microvaisseaux, l’extraction protéique et les immunobuvardages qui en ont suivi, ainsi que les différents immunomarquages en fluorescence et quantifications en stéréologie nécessaires, incluant l’analyse des données de microscopie intravitale. Pour ma part, j’ai réalisé les ELISAs pour l’Aβ1-42 soluble incluant l’analyse des données, ainsi que les expériences de cytométrie en flux comprenant les prélèvements de sang, le marquage des cellules, suivi de l’analyse des données. L’assistance technique pour la préparation des animaux, ainsi que l’acquisition des données d’imagerie intravitale a été réalisée par M. Paul Préfontaine. Le Dr ElAli et moi-même avons tous deux contribué à l’interprétation de l’ensemble des données. La rédaction du manuscrit, l’analyse statistique, ainsi que la création des figures ont été conjointement réalisées par le Dr ElAli et moi-même. Finalement, le Dr Serge Rivest a par la suite corrigé et révisé le manuscript. Avec l’appui du Dr Serge Rivest, le Dr ElAli a été en charge de la soumission de l’article, ainsi que de la communication avec les arbitres et les éditeurs du journal. La révision du manuscrit a été conjointement réalisée par le Dr ElAli et moi-même. Je suis le 2ème de 4 auteurs sur cette étude ayant été publiée dans le journal Acta Neuropathologica Communications le 13 novembre 2013.
2 ElAli, A., Bordeleau, M., Thériault, P., Filali, M., Lampron, A., and Rivest, S. Recombinant tissue-plasminogen activator attenuates the development of Alzheimer's disease-related pathology in APPswe/PS1 mice. Neuropsychopharmacology 1-38 (2015).
Contributions :
La conceptualisation et la demande de financement du projet ont été effectuées par le Dr Serge Rivest. Ce projet a été initié par le Dr Ayman ElAli, s’étant chargé du design experimental, des sacrifices et de la coupe des cerveaux. Le Dr ElAli a exécuté l’isolation des microvaisseaux, l’extraction protéique et certains immunobuvardages qui en ont suivi, ainsi que les différents immunomarquages en fluorescence et quantifications en stéréologie nécessaires. Mlle Maude Bordeleau a exécuté les expériences in vitro incluant le maintien des cultures cellulaires, l’acquisition et l’analyse des données, ainsi que certains immunobuvardages. Pour ma part, j’ai realisé certains immunobuvardages, les ELISAs pour l’Aβ1-40/42 soluble incluant l’analyse des données, ainsi que les expériences de cytométrie en flux comprenant les prélèvements de sang, le marquage des cellules, suivi de l’analyse des données. L’assistance technique pour les tests neurocomportementaux a été réalisée par le Dr Mohammed Filali. Le Dr ElAli, Mlle Bordeleau, Dr Lampron et moi-même avons tous contribué à l’interprétation de l’ensemble des données. La rédaction du manuscrit, l’analyse statistique, ainsi que la création des figures ont été conjointement réalisées par le Dr ElAli, Mlle Bordeleau et moi-même. Finalement, le Dr Serge Rivest a par la suite corrigé et révisé le manuscript. Avec l’appui du Dr Serge Rivest, le Dr ElAli a été en charge de la soumission de l’article, ainsi que de la communication avec les arbitres et les éditeurs du journal. La révision du manuscrit a été conjointement réalisée par le Dr ElAli, Mme Bordeleau et moi-même. Je suis le 3ème de 6 auteurs sur cette étude ayant été publiée dans le journal Neuropsychopharmacology le 30 septembre 2015.
3 Thériault, P., ElAli, A., and Rivest, S. High fat diet exacerbates Alzheimer’s disease-related pathology in APPswe/PS1 mice. Oncotarget (2016).
Contributions :
La conceptualisation et la demande de financement du projet ont été effectuées par le Dr Serge Rivest. Ce projet constitue l’étude principale de ma thèse. J’ai initié ce projet et je me suis chargé du design experimental, des sacrifices et de la coupe des cerveaux. J’ai
également effectué la majorité des immunobuvardages, les expériences in vitro, les différents immunomarquages en fluorescence, ainsi que les quantifications en stéréologie nécessaires. J’ai aussi realisé les ELISAs pour l’Aβ1-40/42 soluble incluant l’analyse des données, ainsi que les expériences de cytométrie en flux comprenant les prélèvements de sang, le marquage des cellules, suivi de l’analyse des données. Le Dr ElAli a executé l’isolation des microvaisseaux, l’extraction protéique qui en a suivi et certains immunobuvardages. L’assistance technique pour les tests neurocomportementaux a été réalisée par le Dr Mohammed Filali. Le Dr ElAli et moi-même avons tous deux contribué à l’interprétation de l’ensemble des données. La rédaction du manuscrit, l’analyse statistique, ainsi que la création des figures ont principalement été réalisées par moi-même. Finalement, le Dr Serge Rivest a par la suite corrigé et révisé le manuscript. Avec l’appui du Dr Serge Rivest, j’ai été en charge de la soumission de l’article, ainsi que de la communication avec les arbitres et les éditeurs du journal. La révision du manuscrit a été conjointement réalisée par le Dr ElAli et moi-même. Je suis le 1er de 3 auteurs sur cette étude ayant été publiée dans le journal Oncotarget le 21 septembre 2016.
Enfin, la dernière partie constitue une discussion plus approfondie sur les résultats présentés dans les chapitres 3 à 5, suivis d’une conclusion générale. De plus, sans être incluses dans cette thèse, j’ai également contribué au cours de mon doctorat à la publication d’autres articles qui ont eux aussi été publiés dans des journaux scientifiques révisés par des pairs:
• Filali, M., Lalonde, R., Thériault, P., Julien, C., Calon, F., and Planel, E. Cognitive and non-cognitive behaviors in the triple transgenic mouse model of Alzheimer's disease expressing mutated APP, PS1, and Mapt (3xTg-AD). Behavioural Brain Research 234, 334–342 (2012).
• Michaud, J.-P., Hallé, M., Lampron, A., Thériault, P., et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer's disease-related pathology. Proceedings of the National Academy of Sciences 110, 1941–1946 (2013). • ElAli, A., Thériault, P., and Rivest, S. The Role of Pericytes in Neurovascular Unit
• Thériault, P., ElAli, A., and Rivest, S. The dynamics of monocytes and microglia in Alzheimer's disease. Alzheimer's Research and Therapy 7-41 (2015).
• Thériault, P., Bordeleau, M., and Rivest, S. Isolation and purification of murine microglial cells for flow cytometry. Bio-Protocol 6-1 (2016).
• Thériault, P., Le Béhot, A., ElAli, A., and Rivest, S. Sub-acute systemic erythropoietin administration reduces ischemic brain injury in an age-dependent manner. Oncotarget (2016).
• Thériault, P., and Rivest, S. Microglia: Senescence impairs myelin debris clearance. Current Biology 16, 772-775 (2016).
Autrement, lors de mes travaux de maîtrise (M.Sc.) réalisés dans le contexte d’un partenariat industriel avec la compagnie biopharmaceutique GlaxoSmithKline (GSK), j’ai contribué à élucider un mécanisme d’action impliqué dans les effets bénéfiques du Monophosphoryl lipid A (MPL) observés chez la souris APPswe/PS1, un modèle murin de la maladie d’Alzheimer. Ces travaux ont fait l’objet d’un article intitulé : « Monophosphoryl Lipid A enhances Aβ elimination by inducing ABCB1 expression in brain microvessels », dont je suis le premier auteur. Cet article est demeuré sous forme manuscrite, puisqu’il n’a malheureusement pas pu être soumis à un journal pour publication. Par ailleurs, j’ai initié une étude visant à accroitre les connaissances concernant l’impact de l’âge et de la diète riche en gras sur la pathologie cérébrovasculaire et les fonctions cognitives en contexte non-pathologique. Cette étude en collaboration avec Dre Marie-Ève Tremblay est en cours et nous a permis d’étudier l’ultrastructure des microvaisseaux cérébraux en microscopie électronique, ainsi que d’identifier diverses molécules impliquées dans la plasticité synaptique, deux systèmes biologiques pouvant affecter les fonctions cognitives. De plus, j’ai participé à une étude décrivant les effets neuroprotecteurs et immunomodulateurs de l’érythropoïétine (EPO) chez la souris transgénique modélisant la pathologie de la maladie d’Alzheimer. En parallèle à ces études, j’ai également travaillé sur une autre étude, en collaboration avec Dr Jean Gosselin, traitant des effets protecteurs d’une molécule immunomodulatrice en contexte inflammatoire.
1. Chapitre 1 – Introduction
1.1 La maladie d’Alzheimer
En Décembre 2013, le G8 a déclaré que la recherche sur la démence constitue une priorité mondiale, soulignant du même coup l’importance de développer et de rendre disponible un traitement curatif ou préventif de cette maladie d’ici 2025 (Scheltens et al., 2016). Représentant près de 60% des cas de démence à travers le monde, la maladie d’Alzheimer (MA) ou simplement l’Alzheimer, se définissant comme une maladie neurodégénérative progressive, constitue ainsi la cause de démence la plus répandue mondialement, affectant plus de 40 millions d’individus en 2013 (Scheltens et al., 2016). Ce nombre de cas, principalement constitué d’individus âgés de 60 ans et plus, devrait doubler tous les 20 ans, du moins jusqu’en 2050 (Scheltens et al., 2016). La prévalence de la maladie coïncide avec le vieillissement de la population, celle-ci doublant à tous les 5 ans à partir de l’âge de 65 ans (Querfurth and LaFerla, 2010), pour éventuellement atteindre un individu sur deux chez les 85 ans et plus. En 2011, au Canada, près de 750 000 individus étaient touchés de la MA et autres maladies apparentées, engendrant ainsi près de 33 milliards de $ en coûts, représentant ainsi un fardeau économique important pour notre société (Duthey, 2013) (Fig.1).
http://www.brainfacts.org/diseases-disorders/degenerative-disorders/articles/2012/alzheimers-disease-tomorrow/
Figure 1. Poids économique de la MA aux États-Unis
La MA est l'une des maladies les plus coûteuses auxquelles est confronté le monde développé, en particulier puisque le nombre de personnes atteintes augmente avec le vieillissement de la population. D'ici 2050, le coût total des soins aux Américains âgés de 65 ans et plus atteint de la MA devrait quintupler, passant de 172 milliards de dollars à 1,08 trillions de dollars par an.
La forme dite « tardive » de la MA (Late-onset Alzheimer’s disease; LOAD) constitue la forme la plus répandue, soit plus de 95% des cas, dont l’âge d'apparition de la maladie est de 65 ans et plus. Autrement, la forme dite « précoce » de la MA représente entre 1 et 6% des cas, dont l’âge d’apparition se situe entre 30 et 65 ans (Piaceri et al., 2012). Du point de vue génétique, la MA se subdivise en deux formes, soit la forme dite « familiale » à caractère héréditaire et à prédominance d'apparition précoce, ainsi que la forme dite « sporadique » à caractère non-héréditaire dont l’âge d’apparition est tardive (Bertram et al., 2010). La forme sporadique constitue la majorité des cas de MA, représentant ainsi la forme la plus étudiée de par sa nature complexe et multifactorielle.
Actuellement, il n’existe aucun traitement pour la MA, puisque la pathogénèse de celle-ci demeure encore mal définie et ce, malgré de nombreux efforts investis en recherche. En ce sens, la recherche fondamentale demeure primordiale afin d’élucider les mécanismes impliqués dans le développement de la MA, visant non seulement l’identification de biomarqueurs afin de diagnostiquer plus précocement la maladie, mais aussi la découverte de cibles thérapeutiques pour ralentir ou même arrêter la progression de cette terrible maladie. 1.1.1 Historique
Historiquement, les toutes premières références à la déficience mentale associée à l’âge sont attribuées aux Pythagoras, des physiciens grecs du 7ème siècle avant J.C. qui sont à l’origine de la notion de démence (Berchtold and Cotman, 1998). En 1906, un psychiatre et neuropathologiste allemand nommé Alois Alzheimer étudia le cas d’une patiente de 51 ans du nom de Auguste Deter, qui fut admise à l’hôpital pour cause de démence. Alzheimer rapporta
des troubles de mémoire et des hallucinations chez la femme, et suite à sa mort, procéda à l’autopsie de son cerveau. À ce moment, Alzheimer décrivit deux marqueurs neuropathologiques principaux, soit les plaques séniles et les dégénérescences neurofibrillaires, constituant ainsi le tout premier cas de la MA. La découverte de la MA inclut également le travail de deux autres collaborateurs, soit le psychiatre et neuropathologiste tchèque Oskar Fischer et le médecin italien Gaetano Perusini. Ensuite, d’autres scientifiques en collaboration avec des neurologues et des psychiatres ont développé de nouveaux outils d’analyse, dont notamment la microscopie, afin de mieux décrire la MA et de la définir cliniquement (Berchtold and Cotman, 1998).
Par la suite, il a fallu attendre plus de 70 ans pour que les constituants biologiques de ces deux marqueurs neuropathologiques caractérisant la MA soient identifiés. En 1984, le pathologiste américain George Glenner parvint à mettre en évidence un constituant majeur des plaques séniles, soit la protéine bêta-amyloïde (Aβ). Plus tard, en 1985, ce fut la présence de la protéine tau hyper-phosphorylée s’accumulant dans les dégénérescences neurofibrillaires qui fut décrite par un dénommé Jean-Pierre Brion d’origine belge. Ensuite, durant les années 1990, il y a eu l’identification de plusieurs gènes responsables de la transmission de la maladie au sein de certaines familles, dont le gène APP situé sur le chromosome 21, ainsi que les gènes PS1 et PS2 situés sur les chromosomes 1 et 14 respectivement. À ce moment, il a été mis en évidence que la mutation de ces gènes menait au développement des formes précoces dites « familiales » de la MA, se manifestant avant l’âge de 60 ans. Enfin, en 1993, une étude importante a identifié le principal facteur génétique prédisposant au développement de la forme sporadique de la MA, soit l’allèle ε4 de l’apolipoprotéine (APOE). Cela dit, l’APOE4 ne demeure toutefois qu’un facteur de risque n’étant pas suffisant à lui-seul pour mener au développement de la MA (Berchtold and Cotman, 1998).
La recherche sur la MA a pris de l’expansion par la suite, procurant ainsi une meilleure compréhension de la pathophysiologie de la maladie. Ses répercussions comprennent le développement de tests diagnostiques plus performants, notamment grâce à l’identification de biomarqueurs précoces de la maladie et de nouveaux facteurs de risque. À cela s’ajoute le
développement de nouveaux modèles expérimentaux mieux adaptés, permettant ainsi de mettre en évidence les mécanismes impliqués dans la MA afin de développer de nouvelles thérapies.
1.1.2 Les symptômes cliniques
La MA se caractérise par un déclin cognitif, dont la progression et la sévérité des symptômes cliniques sont variables (Duthey, 2013). L’apparition de déficits au niveau de la cognition, se manifestant par des troubles de mémoire à court terme, des difficultés de perception du temps, d’orientation et de communication, constituent les premiers symptômes cliniques de la MA. À cela s’ajoute divers troubles comportementaux tels qu’une baisse de motivation, une hausse de l’irritabilité menant à un tempérament colérique, accompagnées de dépression. Au stade avancé de la maladie, les individus atteints de la MA perdent leur autonomie et leur indépendance, pouvant progressivement mener à une incapacité à reconnaitre leurs proches, ainsi qu’à des difficultés à exécuter les besoins primaires tels que se laver et se nourrir, de l’incontinence, une mobilité réduite, un tempérament agressif, et ultimement, à une conscience réduite voire inexistante de la réalité. Suite au diagnostic, les individus atteints de la MA survivent en moyenne 7 ans, soit 4 ans de moins qu’un individu en santé du même âge. Ce phénomène s’explique en majeure partie par des troubles découlant des symptômes cliniques, dont la perte d’autonomie au niveau des besoins primaires (ex. : hygiène, nutrition), une susceptibilité accrue aux infections (ex. : pneumonie) et aux maladies cardiovasculaires (Fitzpatrick et al., 2005; Hope et al., 2001).
1.1.3 La pathophysiologie
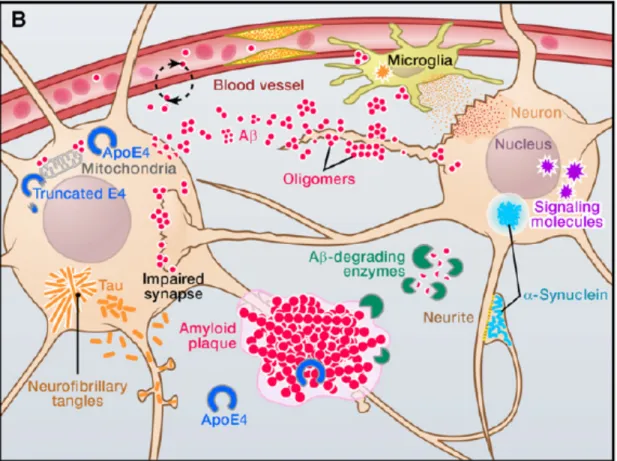
Les individus atteints de la MA présentent de nombreux caractères neuropathologiques, comprenant l’atrophie cérébrale qui résulte de la mort des neurones, autrement dit de la neurodégénérescence. Cela dit, quels sont les éléments associés à la neurodégénérescence? En fait, depuis leur identification par Alzheimer, deux marqueurs neuropathologiques principaux demeurent, soit les plaques extracellulaires de la protéine β-amyloïde (Aβ) et les enchevêtrements neurofibrillaires intraneuronaux majoritairement composés de la protéine tau
hyperphosphorylée (Querfurth and LaFerla, 2010) (Fig. 2). À cela s’ajoute un autre élément important, le phénomène d’inflammation au sein du cerveau des individus atteints de la malaide, communément appelé neuroinflammation. Les cellules de la glie, soit les microglies et astrocytes, sont associées à la neuroinflammation de part leur état d’activation et leur recrutement à proximité des plaques d’Aβ (Cameron and Landreth, 2010). En parallèle, une déficience métabolique du cerveau, c’est-à-dire une dépense énergétique en glucose réduite, a été observée chez les individus atteints de la MA (Ballard et al., 2011). Plusieurs autres caractères pathophysiologiques seraient également impliqués dans la MA, tels que la perte de plasticité synaptique, la dysfonction mitochondriale, la défaillance du transport axonal, les pathologies cérébrovasculaires, ainsi que la dérégulation du métabolisme du cholestérol (Pimentel-Coelho and Rivest, 2012; Querfurth and LaFerla, 2010).
Huang, Y. & Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204– 1222 (2012).
production d'Aβ, une diminution de sa dégradation par des enzymes protéolytiques, ou une réduction de son élimination à travers la barrière hémato-encéphalique (BHE). Les oligomères d'Aβ altèrent les fonctions synaptiques et les voies de signalisation associées, modifient l’activité neuronale et favorisent la sécrétion de médiateurs neurotoxiques par les cellules gliales. Les plaques d’Aβ fibrillaires altèrent les processus neuronaux. Tau, qui se situe normalement au niveau des axones, s’accumule au niveau du soma neuronal et des dendrites, formant ainsi des inclusions appelées enchevêtrements neurofibrillaires (NFT). Les dysfonctionnements neurovasculaires altèrent l'apport en nutriments et l'élimination des éléments métaboliques toxiques, favorisant l'activation des cellules gliales.
1.1.3.1 La protéine β-amyloïde
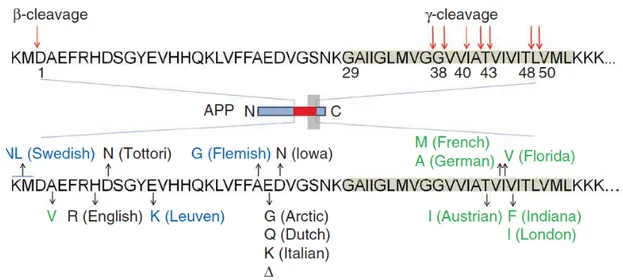
Depuis son identification il y a un siècle, l’Aβ demeure actuellement l’une des principales manifestations neuropathologiques de la MA. L’Aβ, principalement produite par les neurones, resulte d’un clivage protéolytique séquentiel de l’Amyloid precursor protein (APP) en N- et C- terminal, via les enzymes β- et γ- sécrétases, produisant ainsi des peptides monomériques d’environ 4 kDa. On retrouve diverses isoformes du peptide d’Aβ, soit allant de 37 à 43 acides aminés, de par l’action de la γ-sécrétase, ainsi que d’autres enzymes telles que les phosphatases, les glutaminyle cyclases, les isomérases et les aminopeptidases (Benilova et al., 2012; De Strooper, 2010; Hartley et al., 2008; Kumar et al., 2011) (Fig. 3). L’hétérogénéité des isoformes de peptides d’Aβ que l’on retrouve chez les patient atteints de la MA résulte non seulement de la taille de ceux-ci, mais également de leur solubilité, de leur stabilité, ainsi que de leur potentiel neurotoxique. Parmi ceux-ci, l’Aβ1-42 constitue l’isoforme ayant le plus grand potentiel neurotoxique, ainsi qu’une faible solubilité, tandis que l’Aβ1-40 représente l’isoforme la plus abondante du cerveau qui est la plus fréquemment associée à la vasculature cérébrale (Benilova et al., 2012). De plus, diverses mutations dites familliales de l’APP peuvent moduler l’affinité relative des β- et γ- sécrétases, affectant ainsi la production des différents isoformes d’Aβ (Benilova et al., 2012) (Fig. 3).
Benilova, I., Karran, E. & De Strooper, B. The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci 15, 349–357 (2012).
Figure 3. Production du peptide Aß à partir de la protéine précurseur amyloïde (APP).
Les sites de clivage médié par la ß- et γ-sécrétases sont indiqués par des flèches, et le domaine transmembranaire de l'APP est mis en évidence en gris. Le clivage via la γ-sécrétase produit un groupe de fragments Aß qui varient en longueur et en hydrophobicité. Les mutations dans la région Aβ de l'APP augmentent la production totale d’Aß (marquée en bleu), modifient les propriétés biophysiques de l'Aβ (en noir) ou affectent le spectre Aß de façon quantitative et qualitative (en vert).
À ce titre, la souris APPswe/PS1 qui constitue l’un des modèles murins transgéniques les plus répandus relève de ce type de mutation, où l’APP portant la mutation suédoise (APPSwe) est co-exprimée avec la variante A246E de la préséniline 1 (PS1) humaine (Borchelt et al., 1996) sous le contrôle du promoteur de la protéine à prion murine. Dans ce modèle murin de la MA, les animaux produisent de l’Aβ dès leur naissance, menant à une accumulation importante de celle-ci, jusqu’à former des plaques d’Aβ insolubles aux alentours de l’âge de quatre mois. Toutefois, ce n’est que vers l’âge de six mois que ces animaux présentent des déficits cognitifs s’apparentant à la pathologie de la MA. À cet âge, on y observe des dysfonctions synaptiques et de la neuroinflammation accompagnant l’accumulation des plaques d’Aβ (Filali et al., 2009). Dans ce modèle, on retrouve un mélange des différents isoformes monomériques d’Aβ pouvant s’assembler de diverses façons de sorte à créer des oligomères de toutes tailles, des
globulomères, des amylosphéroïdes, des protofibrilles, des fibrilles, ainsi que bien sûr, des microagrégats et plaques d’Aβ (Benilova et al., 2012).
1.1.3.2 La protéine tau
Le gène de la protéine tau associée aux microtubules (MAPT) est situé sur le chromosome 17 du génome humain et exprime six isoformes de la protéine tau dans le SNC (Hanger et al., 2009). Ces isoformes proviennent de l'épissage alternatif des exons 2, 3 et 10 des 16 exons du MAPT. Les exons 2 et 3 expriment une séquence de 29 et 58 acides aminés, respectivement, et l'exon 10 exprime un domaine supplémentaire de liaison aux microtubules (Kolarova et al., 2012). En conséquence, les isoformes de la protéine tau contiennent de zéro à deux répétitions N-terminales, ainsi que trois ou quatre domaines C-terminaux de liaison aux microtubules (tau 3R ou 4R) (Kolarova et al., 2012). La phosphorylation et déphosphorylation de la protéine tau sont grandement dynamiques pendant le cycle cellulaire, puisque cette protéine doit se dissocier des microtubules pour permettre la mitose. La protéine tau est une protéine très soluble et hautement phosphorylable, de par sa conformation tri-dimensionelle accessible aux kinases (Pedersen and Sigurdsson, 2015). Près de 20%, soit 85 des résidus d'acides aminés de l'isoforme la plus longue de tau constituent des sites de phosphorylation potentiels en sérine, thréonine ou tyrosine, où environ la moitié de ceux-ci sont regroupés autour des résidus terminaux des domaines de liaison aux microtubules (Kolarova et al., 2012; Pedersen and Sigurdsson, 2015). Son rôle principal dans les neurones est d'agir à titre de stabilisateur de microtubules, permettant ainsi le transport axonal. La protéine tau ne peut que s'associer aux microtubules sous sa forme déphosphorylée, donc la phosphorylation permet de réguler directement ce système d'association-dissociation de la protéine tau aux microtubules dans le neurone (Spires-Jones and Hyman, 2014).
1.1.3.3 L’inflammation
L'inflammation au cerveau, communément nommée neuroinflammation, constitue l’un des caratères pathophysiologiques principaux associés à la MA. Plusieurs évidences ont démontré que celle-ci serait médiée par différents mécanismes, y compris l’activation de l’inflammasome,
l'activation de la microglie, la présence d’astrocytes réactifs, ainsi que le recrutement, l'infiltration et l’activation de monocytes au parenchyme cérébral (Thériault et al., 2015). En ce sens, plusieurs études ont démontré une relation étroite entre la neuroinflammation et la pathologie de la MA (Wyss-Coray and Rogers, 2012). Le système immunitaire inné joue un rôle dans le développement de la pathologie de la MA où l'exposition chronique de la microglie à l’Aβ conduit à une inflammation non contrôlée, se traduisant entre autres par la libération de radicaux libres toxiques et d’espèces réactives de l'oxygène (Cagnin et al., 2001; Uchihara et al., 1997). En outre, des études d'association pangénomique (genome-wide association study ou GWAS) sur des milliers de patients atteints de la MA ont montré que parmi les dix polymorphismes génétiques les plus étroitement liés au développement de la forme sporadique de la MA, neuf joueraient un rôle dominant dans les processus immunologiques (Moraes et al., 2012). Dans le sérum, le liquide céphalo-rachidien, et le cortex des patients atteints de la MA, des niveaux plus élevés d'IL-1β, IL-6, TNFα, IL-8, et TGFβ ont été rapportés (Akiyama et al., 2000). En ce sens, les études chez l’humain indiquent que la MA serait d'abord et avant tout une maladie immunologique.
Jusqu'à récemment, la neuroinflammation dans la MA a été exclusivement liée à l’Aβ. Cependant, des études récentes ont indiqué une contribution potentielle de l'inflammation systémique chronique dans l’initiation des cascades neurodégénératives observées dans la MA (Cunningham, 2013). Bien que le lien entre la neuroinflammation et la pathologie de la MA soit maintenant bien reconnu, comment est engendrée la réponse inflammatoire au cerveau demeure un sujet de débat, en particulier dans le contexte où la neuroinflammation serait initiée par l'inflammation systémique liée à l'âge (Pimplikar, 2014). En ce sens, ce phénomène pourrait directement agir à titre de médiateur dans l’initiation de l'inflammation cérébrale chronique qui évoluerait au fil du temps et ultimement, mènerait à la MA.
1.1.4 Les hypothèses étiologiques
Bien que la recherche sur la démence soit devenue une priorité mondiale, les causes initiant les cascades neurodégénératives observées dans la forme sporadique de la MA demeurent inconnues. Actuellement, les efforts en recherche visent à déterminer le rôle de ces
manifestations pathophysiologiques, afin de discerner celles qui constitueraient les causes de celles qui constitueraient les conséquences de la MA. Il nous est dès lors impossible d’évaluer in vivo les mécanismes impliqués dans l’initiation et l’évolution de la maladie chez un individu atteint de la MA.Toutefois, il existe des hypothèses étiologiques reconnues, étant basées sur des évidences bien établies où les mécanismes cellulaires et moléculaires sous-jacents restent à être élucidés. Parmi ces hypothèses, certains mécanismes seront, sont ou ont été des cibles thérapeutiques visant à ralentir ou même freiner la pathologie de la MA. En ce sens, les hypothèses étiologiques mentionnées et décrites ci-bas constituent celles qui sont les plus répandues et acceptées auprès de la communauté scientifique et ce, sans se limiter à leur exclusivité, puisque celles-ci peuvent co-exister de sorte à mieux expliquer le développement de la MA.
1.1.4.1 L’hypothèse de l’amyloïde
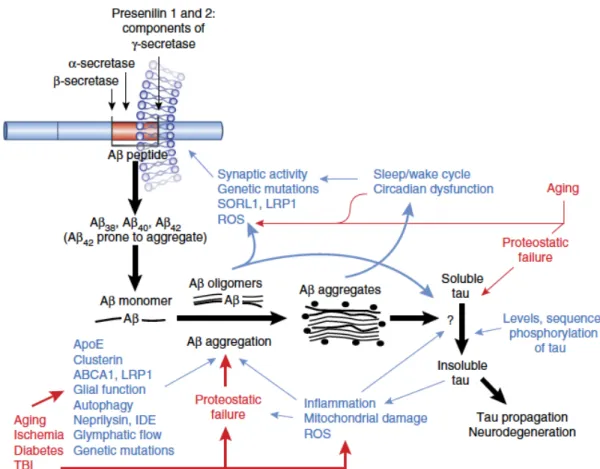
Bien qu’actuellement en cours de révision, cette hypothèse étiologique constitue l’une des plus établies et répandues au sein de la communauté scientifique, attestant que les cascades neurodégénératives menant à la MA seraient initiées par l’accumulation et l’agrégation exacerbées des peptides d’Aβ au cerveau (Benilova et al., 2012). Tel que mentionné précédemment, les différentes isoformes de l’Aβ résultent du clivage protéolytique séquentiel de l’APP via l’action de la β-sécrétase (BACE1) et la γ-sécrétase. Parmi ceux-ci, l’Aβ1-42 constitue l’isoforme ayant le plus grand potentiel neurotoxique, jouant ainsi un rôle central dans l’initiation et/ou la progression de la MA (Querfurth and LaFerla, 2010) (Fig. 4).Au cours des 20 dernières années, il a été suggéré que l’accumulation et l’agrégation des peptides d’Aβ précèderaient l’hyperphosphorylation de la protéine tau et donc, la formation d’enchevêtrements neurofibrillaires (Lewis et al., 2001; Oddo et al., 2003). En ce sens, les espèces solubles d’Aβ initieraient le dysfonctionnement des neurones au niveau des synapses, favoriseraient les dommages induits par le stress oxydatif, contribueraient à la perte d’homéostasie ionique et promouvraient l’apoptose au sein du parenchyme cérébral (Benilova et al., 2012) (Fig. 4).
Musiek, E. S. & Holtzman, D. M. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nature 18, 800–806 (2015).
Figure 4. L'hypothèse de la β- amyloïde.
Les flèches noires illustrent la conversion de l'APP par les ß- et γ-sécrétases pour générer le peptide Aß, qui s’accumule et s’agrège par la suite, ce qui provoque finalement l'agrégation de tau et la neurotoxicité en aval. Le texte bleu et les flèches illustrent les éléments modificateurs proposés dans la cascade de l’Aβ, et le texte rouge et les flèches montrent l'influence du vieillissement et des pathologies comorbides. Il est à noter que plusieurs inter-relations sont supposées, dont l'une impliquant un sommeil perturbé favorisant la production de l’Aß (et peut-être la clairance de l’Aß, bien que non représentée), alors que l'agrégation de l’Aß interrompt à son tour les cycles de sommeil. De multiples facteurs, du vieillissement au stress oxydatif, contribuent à l'échec protéostatique, qui à son tour favorise l'agrégation de l’Aß, de tau et probablement d'autres protéines neurotoxiques. Beaucoup des facteurs modifiant l'Aβ interagissent entre eux (comme l'inflammation modulant l'APOE), bien que cela ne soit pas représenté. IDE, enzyme dégradant l'insuline; ROS, espèces réactives de l'oxygène.
Or, le potentiel neurotoxique de l’Aβ s’illustre dans la forme dite familiale de la MA, où les individus atteints présentent des mutations dans le gène APP, promouvant ainsi la formation excessive des peptides d’Aβ. Tel que mentionné précédemment, les souris transgéniques APPswe/PS1 surexpriment l’Aβ, reproduisant ainsi plusieurs altérations neurologiques de façon similaire à celles observées chez l’humain, notamment l’accumulation et l’agrégation des peptides d’Aβ, la dysfonction des neurones, la neuroinflammation, ainsi qu’un déclin cognitif (Filali et al., 2009; McGowan et al., 2006). Toutefois, l’hypothèse de l’Aβ présente certaines limitations, découlant principalement des limitations des modèles transgéniques murins actuels. Cela se traduit par des échecs répétitifs en essais cliniques, où les traitements se basaient essentiellement sur la pathologie Aβ, suggèrant ainsi que l’Aβ initierait la maladie, mais qu’elle seule ne suffirait pas pour mener au développement de la MA (Musiek and Holtzman, 2015). En fait, les facteurs exacts qui initient l’accumulation et l’agrégation des peptides d’Aβ demeurent inconnus et ce, malgré l’identification de plusieurs facteurs de risques. Cela dit, l’impact de ces facteurs, dont l’âge, le sexe et la diète, font activement l’objet d’études afin d’élucider leur contribution dans l’initiation et la progression de la MA.
1.1.4.2 L’hypothèse de tau
Physiologiquement, la protéine tau se situe majoritairement au niveau des axones, ainsi qu’en moindre quantité au niveau des dendrites. Celle-ci joue un rôle primordial dans la régulation du transport axonal et la stabilisation des microtubules au sein des neurones (Iqbal et al., 2005). Son activité peut être modulée via des domaines de phosphorylation, pouvant ainsi avoir un effet néfaste sur sa capacité à adhérer aux microtubules. En ce sens, l’hyperphosphorylation de la protéine Tau serait responsable des dérèglements du transport axonal, ainsi que de l’instabilité des microtubules favorisant l’apoptose neuronale (Iqbal et al., 2005). Ce phénomène d’hyperphosphorylation de la protéine Tau résulterait d’une activité exacerbée des protéines kinases, favorisant l’accumulation de protéines Tau hyperphosphorylées dans les neurones, promouvant ainsi la formation d’enchevêtrements neurofibrillaires intraneuronaux et menant ultimement à la dysfonction et à la mort des neurones (Guerrero-Muñoz et al., 2015; Ittner and Götz, 2011). Cela dit la formation et l’accumulation des protéines Tau hyperphosphorylées ont également été rapportées dans d’autres contextes de stress
neurologiques, ce qui en fait un phénomène qui n’est pas exclusivement associé à la MA (Iqbal et al., 2005).
Tel que mentionné précédemment, l’Aβ et la protéine Tau hyperphosphorylée présentent toutes deux un potentiel neurotoxique. Cependant, il a été démontré que ces caractères neuropathologiques peuvent co-exister de sorte à interagir et ainsi à contribuer à la progression de la MA. De façon intéressante, dans les modèles murins transgéniques, l’accumulation et l’agrégation de l’Aβ favorise la formation de protéines Tau hyperphosphorylées, tandis que la surexpression de la protéine Tau ne favorise pas une accumulation ni une agrégation accrue d’Aβ dans le parenchyme cérébral (Lewis et al., 2001; Oddo et al., 2004). De plus, il a été démontré que la neurotoxicité des peptides d’Aβ serait médiée par la protéine Tau hyperphosphorylée, puisque chez les animaux APPtg/Tau-/-, on n’observe pas de toxicité induite par l’Aβ en l’absence de la protéine Tau (Ittner and Götz, 2011). Cela dit, les facteurs et mécanismes impliqués dans l’activation exacerbée des kinases menant à l’hyperphosphorylation de la protéine Tau restent à clarifier. Toutefois, considérant le fait que l’accumulation et l’agrégation des peptides d’Aβ précèderait l’hyperphosphorylation de la protéine Tau, il est suggéré que ce phénomène ne serait pas initiateur des cascades neurodégénératives observées dans la MA, même s’il pourrait y contribuer.
1.1.4.3 L’hypothèse vasculaire « deux-coups »
L'hypothèse vasculaire « deux-coups » (traduit de : Two-hit vascular hypothesis) constitue une alternative qui intègre la contribution cérébrovasculaire dans la pathogénèse de la MA. Cette hypothèse suggère que des dommages primaires à la vasculature cérébrale (coup #1) initierait une voie non-amyloïdogénique induisant la perte de fonction des neurones médiée par le dysfonctionnement de la barrière hémato-encéphalique (BHE) qui serait associée à une plus grande sécrétion de molécules neurotoxiques, ainsi qu’à une diminution du débit sanguin cérébral (CBF) pouvant favoriser un environement ischémique ou hypoxique (Zlokovic, 2011). En parallèle, cette hypothèse stipule que la dysfonction de la BHE conduirait à une défaillance de la clairance de l’Aβ et à une hypoperfusion cérébrale pouvant entraîner une augmentation de la production des peptides d'Aβ. Or, ces deux phénomènes contribueraient à l'accumulation
d'Aβ au cerveau (coup #2), où l’Aβ exercerait des effets vasculotoxiques et neurotoxiques, menant ainsi à la neurodégénérescence (Zlokovic, 2011; 2005) (Fig. 5). Selon cette hypothèse, la formation et l’accumulation de la protéine Tau hyperphosphorylée surviendrait suite à ces « deux coups », soit le dommage vasculaire et l’accumulation de l’Aβ.
Toutefois, l’hypothèse vasculaire « deux coups » suggère que l’initiation de la MA serait induite par la combinaison de ces deux phénomènes, où chacun d’eux ne seraient individuellement pas suffisants pour induire le développement de la maladie. Or, cette hypothèse prend en considération la dimension vasculaire, un aspect qui est de plus en plus d’intérêt actuellement, dans la pathogénèse de la MA (Fig. 5). En ce sens, il nous faut davantage porter attention aux facteurs de risques, environnementaux et génétiques, pouvant influencer la susceptibilité des individus à développer la MA avec l’âge.
Sagare, A. P., Bell, R. D. & Zlokovic, B. V. Neurovascular Dysfunction and Faulty Amyloid β-Peptide Clearance in Alzheimer Disease. Cold Spring Harb Perspect Med 2, (2012).
Figure 5. L'hypothèse vasculaire de la MA.
Les facteurs de risque vasculaires (par exemple : l'hypertension, le diabète, l'obésité, la maladie cardiaque) et/ou une lésion vasculaire initiale médiée par un trouble cérébrovasculaire (par exemple : l’ischémie, l’AVC) conduisent à l'hypoperfusion cérébrale (oligémie) et/ou une dysfonction de la barrière hémato-encéphalique (BHE) (Coup #1) associée à une diminution de l’apport sanguin au cerveau (ou hypoxie) et à l'accumulation de diverses neurotoxines dans le cerveau, qui peuvent affecter la fonction neuronale, contribuer aux processus neurodégénératifs et au déclin cognitif (lignes pleines). Parallèlement, la dysfonction de la BHE et l'hypoperfusion/hypoxie peuvent réduire la capacité de clairance vasculaire du peptide Aβ à travers la BHE et augmenter la production d'Aβ à partir de la protéine APP, causant l'accumulation d'Aβ dans le cerveau (Coup #2, lignes pointillées). L’augmentation des niveaux d'Aβ conduisent à la formation d'oligomères d’Aβ neurotoxiques, d'une part provoquant une dysfonction neuronale, et d'autre part, son auto-agrégation, ce qui conduit à l'auto-propagation et au développement de la β-amyloïdose cérébrale.
1.1.5 Les facteurs de risque
Tel que mentionné précédemment, les mécanismes initiant les cascades neurodégénératives impliquées dans la pathogénèse de la MA demeurent inconnus. Toutefois, certains facteurs de risque ont été associés à la MA, où l’étude de leur implication dans l’initiation et le développement de la maladie afin d’identifier de potentielles cibles thérapeutiques.
1.1.5.1 L’âge
L’âge, responsable du phénomène du vieillissement, constitue le facteur de risque primaire associé à la forme sporadique de la MA, puisque l’âge d’apparition de la maladie se situe vers 65 ans. Or, la prédisposition à développer la MA tend à augmenter exponentiellement et ce, à près de 15 fois entre l’âge de 60 ans et 85 ans (Evans et al., 1989). Par définition, le vieillissement se caractérise par une perte progressive de l'intégrité physiologique, conduisant à l’altération de la fonction de l’organisme et à une vulnérabilité accrue à la mort. Cette détérioration représente le principal facteur de risque pour les principales pathologies
humaines, y compris le cancer, le diabète, les troubles cardio-vasculaires et les maladies neurodégénératives, dont la MA (López-Otín et al., 2013). Durant les dernières années, la recherche sur le vieillissment a permis de déterminer que le taux, ou la sévérité, du vieillissement serait contrôlée par des voies génétiques et processus biochimiques ayant persisté lors de l'évolution. En fait, le vieillissement découle de plusieurs phénomènes en simultané, dont l'instabilité génomique, le raccourcissement des télomères, les altérations épigénétiques, la perte de protéostasie, la dysfonction mitochondriale, la sénescence cellulaire, ainsi que l’altération de la communication intercellulaire, constituant tous des mécanismes communs dans divers organismes (López-Otín et al., 2013) (Fig. 6). Ces phénomènes peuvent mener à la dysfonction de différents organes en altérant le fonctionnement de plusieurs systèmes biologiques importants au sein de l’organisme.
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Figure 6. Les caractéristiques du vieillissement.
L'instabilité génomique, l'attrition des télomères, les altérations épigénétiques, la perte de protéostase, la sensibilité réduite aux nutriments, la dysfonction mitochondriale, la sénescence cellulaire, l'épuisement des cellules souches et la communication intercellulaire altérée.
Nul besoin de mentionner le rôle primordial qu’occupe le système nerveux central (SNC), composé du cerveau et de la moelle épinière, au sein de notre organisme. En ce sens, notre compréhension de l’effet du vieillissement sur le cerveau demeure cruciale afin d’identifier les mécanismes responsables des cascades neurodégénératives impliquées dans la MA. En ce qui a trait au déclin cognitif observé chez les individus atteints de la MA, on rapporte peu de changements dans la quantitié ou la densité neuronale (Riddle et al., 2003) . Toutefois, il a été démontré que le taux de remplacement des neurones ou synapses est réduit, entraînant ainsi une perte de la plasticité neuronale menant ultimement à la sénescence (Grill and Riddle, 2002). De façon plus subtile, le vieillissement peut également mener à la dysfonction des neurones sans induire de dommages perceptibles par le biais d’études histologiques. Étant donné que le bon fonctionnement du cerveau dépend de plusieurs systèmes biologiques importants, dont l’immunité innée et la vasculature cérébrale, le dysfonctionnement de ceux-ci peut engendrer une plus grande vulnérabilité du cerveau avec l’âge.
En ce sens, concernant le système immunitaire inné, le vieillissement contribue au phénomène appelé « inflamma-aging », qui se définit par une dérégulation induite par l'âge du système immunitaire inné, favorisant ainsi une inflammation systémique chronique (López-Otín et al., 2013). En fait, les cellules immunitaires sénescentes sécrètent des niveaux élevés de molécules pro-inflammatoires dans la circulation sanguine (van Deursen, 2014), dont les espèces réactives de l'oxygène (ROS) qui contribuent au stress oxydatif (Nathan and Cunningham-Bussel, 2013). Par ailleurs, le vieillissement altère également la fonction de la microvasculature cérébrale, dont le cerveau est dépendant afin d’assurer le bon fonctionnement des neurones (Zlokovic, 2008). En fait, il a été démontré chez divers modèles murins transgéniques de la MA (ex. : APPswe/PS1) que de nombreuses altérations au niveau de la vasculature cérébrale pouvaient précéder la neurodégénérescence, ainsi que le déclin cognitif (Pimentel-Coelho and Rivest, 2012). En ce sens, il a été proposé qu’avec le
vieillissement survient une perte de densité des vaisseaux sanguins dans certaines régions du cerveau, accompagnée de changements dans la structure des vaisseaux persistants (Riddle et al., 2003). Ces phénomènes ont pour effet de contribuer à la réduction du CBF, diminuant ainsi l’apport en éléments nutritifs et l’élimination des éléments toxiques, rendant ainsi les neurones plus vulnérables aux dommages avec le vieillissement (Iadecola, 2013). En parallèle, le vieillissement peut également avoir des effets indirects sur la vasculature cérébrale, entre autre en affectant le remodelage neurovasculaire, se traduisant par une capacité réduite des vaisseaux sanguins du cerveau à répondre efficacement aux changements de la demande métabolique des neurones, rejoignant ainsi le concept de couplage neurovasculaire (Zlokovic, 2008).
1.1.5.2 La diète riche en gras
L’augmentation de la prévalence de l'obésité au Canada et aux États-Unis a suscitée une attention accrue en recherche et ce, principalement à cause des effets néfastes qui en découlent sur la santé, incluant les maladies cardiovasculaires, l’hypertension et le diabète de type II, augmentant ainsi de 10% à 25% le risque de développer la MA (Barnes and Yaffe, 2011). De plus, des études ont mis en évidence la contribution de l'obésité dans le déclin cognitif, tel qu’observé dans la MA, ainsi démontré par de fortes corrélations entre l'indice de masse corporelle (IMC) et le développement de la MA et ce, dans divers groupes d'âge (Hsu and Kanoski, 2014; León and Mitchell, 2013). En ce sens, l’obésité résultant d’une malnutrition constitue donc un facteur de risque dans le développement de la MA, dont les effets sur le cerveau font l’objet d’un nombre grandissant d’études depuis les dix dernières années (Freeman et al., 2014).
En fait, un régime alimentaire occidental peut engendrer une carence en éléments nutritifs, ainsi qu’une inflammation qui pourrait directement avoir un impact sur les fonctions cognitives. Or, il a été rapporté que la prévalence de la MA est plus grande dans les pays où la diète est élevée en calories, tandis qu’elle est plus faible chez ceux qui consomment principalement des aliments pauvres en gras (Barnes and Yaffe, 2011). De plus, des études épidémiologiques suggèrent que la diète riche gras constitue un facteur de risque majeur pour le développement
de la MA (Freeman et al., 2014), et ce risque est encore plus élevé chez les individus présentant l'APOE4 (Hsu and Kanoski, 2014). La diète riche en gras contient généralement des hauts niveaux d'hydrates de carbone simples, d’acides gras saturés et de cholestérol, induisant ainsi des perturbations métaboliques telles que l'hyperglycémie et l'hyperlipidémie (Hsu and Kanoski, 2014).
Or, la diète riche en gras exacerberait le phénomène du vieillissement, en altérant davantage la fonctionnalité du système immunitaire inné et de la vasculature cérébrale. En ce qui a trait au système immunitaire, il a été démontré qu’une hyperlipidémie induite par une diète riche en gras favorise l’inflammation systémique, entre autre en stimulant la production de leucocytes circulants dans le sang (Soehnlein and Swirski, 2013). Or, l’augmentation de la réponse immunitaire, comme l'inflammation, constituerait l'une des principales conséquences d'une diète riche en gras. En ce sens, plusieurs études suggèrent que l'augmentation de la réponse inflammatoire au cerveau (ou neuroinflammation) serait corrélée avec la perte de mémoire chez les patients atteints de diverses maladies, y compris la MA (Heneka et al., 2015). Chez la souris, la consommation d'une diète riche en gras engendre une neuroinflammation et un déclin cognitif (Julien et al., 2010), ce qui suggère que la diète riche en gras affecterait la progression de la MA en engendrant une réponse inflammatoire au cerveau.
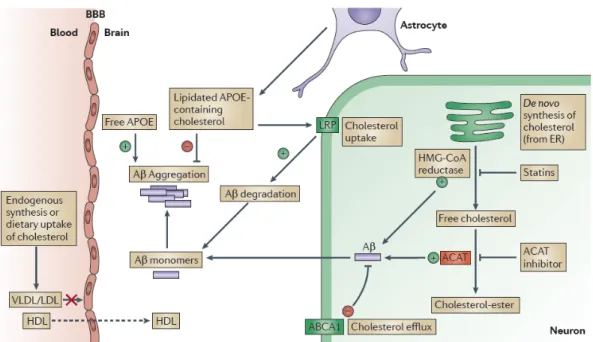
Au niveau de la vasculature cérébrale, il a été démontré que la diète riche en gras a pour effet d’augmenter les niveaux de lipoprotéines de faible densité (LDL) dans la circulation sanguine, étant très susceptibles à l’oxydation. Ceci promouvoit la formation de LDL-oxydés (ox-LDL), soit une molécule hautement réactive associée à l’inflammation systémique et aux dommages/pathologies vasculaires, tel que l'athérosclérose (Franciosi et al., 2009; Hansson and Libby, 2006). En ce sens, la diète riche en gras augmente le risque d’accident vasculaire cérébral (AVC), augmentant ainsi de 50% le risque de développer la MA chez ces personnes (Iadecola, 2013). De façon intéressante, une diète riche en gras pourrait également affecter le métabolisme du cholestérol au cerveau, qui serait impliqué directement ou indirectement (ex. : via APOE) dans la production, l’agrégation et le transport de l’Aβ à travers la BHE (Di Paolo and Kim, 2011) (Fig. 7). En ce sens, la diète riche en gras constitue un facteur de risque majeur associé à la MA, pouvant affecter divers aspects de l’homéostasie au cerveau et donc,