L'UNIVERSITE TOULOUSE III - PAUL SABATIER
UFR : Physique Chime Automatique
THESE
en vue de l'obtention duDOCTORAT DE L'UNIVERSITE DE TOULOUSE délivré par l'université Toulouse III - Paul Sabatier
Ecole Doctorale de Chimie Spécialité
CHIMIE BIOLOGIE SANTE
par
Tamara DELAINE
CONCEPTION, SYNTHESE, ETUDE DE L'EQUILIBRE TAUTOMERIQUE ET EVALUATION BIOLOGIQUE DE NOUVEAUX ANALOGUES DE L'ADDUIT
ISONIAZIDE-NAD(H) COMME INHIBITEURS D'InhA DE MYCOBACTERIUM TUBERCULOSIS
Présentée et soutenue le 18 octobre 2007 Jury :
Alain BAULARD, Chargé de Recherche, INSERM, Lille Rapporteur Jean BERNADOU, Professeur, Université Paul Sabatier, Toulouse
Vania BERNARDES-GENISSON, Maître de conférence, Directeur de thèse
Université Paul Sabatier, Toulouse
Armand LATTES, Professeur Emérite, Université Paul Sabatier,
Toulouse Président
Christian MARAZANO, Directeur de Recherche, CNRS, Gif-sur-Yvette Rapporteur Bernard MEUNIER, Président de la société PALUMED, Labège
Annaïk QUÉMARD, Chargée de Recherche au CNRS, Toulouse
Stéphane QUIDEAU, Professeur, Université de Bordeaux I Examinateur
Recherches effectuées au Laboratoire de Chimie de Coordination du CNRS 205, route de Narbonne, 31077 Toulouse cedex 4
A ma famille
Le travail présenté dans ce mémoire a été réalisé au Laboratoire de Chimie de Coordination du CNRS de Toulouse dirigé par Monsieur Jean-Jacques Bonnet puis par Monsieur Bruno Chaudet. Je les remercie de m'avoir accueillie au sein de cet établissement.
Je tiens tout particulièrement à remercier Monsieur Alain Baulard, Chargé de Recherche à l'INSERM à Lille et Monsieur Christian Marazano, Directeur de Recherche au CNRS à Gif-sur-Yvette pour avoir accepter de juger ce travail en tant que rapporteurs. Leur présence à ce jury est pour moi un grand honneur.
Monsieur Stéphane Quideau, Professeur à l'Université de Bordeaux I, m'a également fait l'honneur de participer à ce jury de thèse. Je tiens à lui adresser mes remerciements pour son implication dans l'évaluation de ce travail.
J'adresse ma plus profonde gratitude à Monsieur Armand Lattes, Professeur émérite à l'Université Paul Sabatier de Toulouse, pour avoir présidé ce jury de thèse, mais aussi pour l'ensemble de ces enseignements depuis la licence. La chimie sans lui n'aurait pas été tout à fait la même.
Monsieur Bernard Meunier, PDG de la société Palumed, m'a accueilli au sein de son équipe. Je lui suis très reconnaissante de m'avoir accordée sa confiance en me proposant ce sujet captivant à l'interface de la chimie et de la biologie. Ses compétences, sa grande motivation et ses conseils ont été précieux.
Je tiens à exprimer ma profonde gratitude à Vania Bernardes-Génisson, Maître de Conférence à l'université Paul Sabatier de Toulouse, qui a assuré la direction scientifique de ce travail. Sa détermination, sa rigueur scientifique et son optimisme ont été indispensable au bon déroulement de cette thèse. Je souhaite également la remercier pour sa gentillesse, sa disponibilité et pour tout ce qu'elle m'a appris au cours de ces trois années.
Je tiens à remercier Monsieur Jean Bernadou, Professeur à l'Université Paul Sabatier de Toulouse. Sa grande culture, son optimisme, sa gentillesse et ces encouragements ont permis l'avancement de mes recherches dans la bonne humeur.
Mes chaleureux remerciements à Annaïk Quémard, Chargée de Recherche au CNRS pour m'avoir fait partager ses larges connaissances scientifiques et m'avoir initié à l'enzymologie et à la bactériologie. Sa fructueuse contribution m'a permis d'évoluer avec aisance à l'interface entre la chimie et la biologie. Je remercie également Patricia Constant pour l'ensemble de ces conseils en bactériologie et pour la réalisation des tests sur M. tuberculosis et sur les cellules eucariotes.
Je remercie Monsieur Jean-Luc Stigliani et Monsieur Philippe Arnaud pour l'ensemble des calculs de modélisation moléculaires. Merci pour votre disponibilité et votre patiente pour m'expliquer les ficelles du monde des théoriciens.
J'adresse aussi mes remerciements les plus chaleureux aux divers services grâce auxquelles ce travail a pu être réalisé dans d'excellentes conditions : le service RMN et tout spécialement Yannick Coppel, le service de diffractions aux rayons X et notament Heinz Goritzka, le service de spectrométrie de masse et le service HPLC et plus particulièrement Chantal Zedde.
Ces trois années de travail ont été facilitées par les encouragements de toutes les personnes cotoyées quotidiennement au laboratoire et que je n'oublierais pas de remercier :
Geneviève, Marguerite, Catherine, Christophe, Anne, Françoise, Isabelle et Peter merci pour vos conseils avisés et votre gentillesse.
Un grand merci à Fatima et à Maryse pour m'avoir aidé dans ce travail (et pour tout le reste biensûr) et aux thésards, post-doc et stagières avec qui la vie au laboratoire comme à l'extérieur aura été pleine de moments inoubliables : les anciens Sophie M., Céline et Sophie S. (nos dicussions interminables m'ont manqué en cette dernière années), Joel, Ludivine, Alexandrine, Guillaume, Luc, Chritophe B., Ana, Guilio, Adeline, François JBBD (vive la guiness!!!) et Charline, Christine, Vanessa (et Séb et Mathilde, courage c'est bientôt fini), François B., Jérome, Vanessa F. et la petite dernière Carmen.
Christelle et Stéphane depuis maintenant 6 ans nous avons partagé les bons moments comme les périodes difficles, merci pour votre amitiés. Merci au groupe CIES : Fanny, Flavie, Sophie, Camille et Lillian d'avoir accepté a vos repas une non monitrice, Myriam et Benoit, vous m'avez permis de décompresser dans les moment de stress.
Enfin et surtout, mes remerciements les plus forts reviennent à mes parents qui m'ont toujours encouragée et soutenue et sans qui je n'aurais pu aller au bout de mes projets. J'y associe également mon frère Hans, mes grands parents et l'ensemble de ma famille.
Pour terminer, merci à Romain qui a été à mes côtés pour partager les meilleurs comme les plus durs moments de ces trois années de thèse.
7
ABREVIATIONS... 11
INTRODUCTION GENERALE... 13
INTRODUCTION ... 17
I. La tuberculose... 19
I.1. Situation de la tuberculose dans le monde et perspectives d'évolution ... 19
I.1.1. Les causes de la recrudescence ... 20
I.1.2. Les programmes de lutte contre la tuberculose... 21
I.2. Agent pathogène et transmission de la tuberculose ... 22
I.3. Prévention et traitement ... 23
I.3.1. La vaccination... 23
I.3.2. Les antibiotiques antituberculeux et les traitements ... 24
II. Cibles thérapeutiques... 28
II.1. La paroi mycobactérienne ... 28
II.2. Structure de l'enveloppe mycobactérienne ... 29
II.2.1. Architecture de la paroi mycobactérienne ... 29
II.2.2. Les acides mycoliques ... 30
II.3. Biosynthèse des acides mycoliques... 32
III. L'isoniazide ... 33
III.1. Activation de l'INH et formation des adduits INH-NAD... 35
III.1.1. Activation enzymatique de l'INH par la catalase-peroxydase KatG ... 35
III.1.2. Activation chimique de l'INH par le pyrophosphate de MnIII... 42
III.2. Cibles moléculaires de l'INH activé ... 45
III.2.1. Inhibition de l'enzyme InhA... 45
III.2.2. Inhibition de l'enzyme MabA... 49
III.2.3. Autres cibles (KasA et DHFR) et effets pléiotropiques... 50
IV. Résistance à l'INH... 53
IV.1. Mutation dans katG... 54
IV.2. Mutation dans inhA ... 55
V. Autres énoyl-ACP-réductases et leurs inhibiteurs... 58
V.1. Inhibiteurs formant une liaison covalente avec le cofacteur NAD ... 59
V.2. Inhibiteurs formant une liaison non covalente avec le cofacteur NAD ... 62
V.2.1. Le triclosan, ses analogues et leurs dérivés... 62
V.2.2. Différentes structures aromatiques découvertes par criblage à haut débit .... 64
V.3. Inhibiteur n'interagissant pas avec le cofacteur NAD ... 65
OBJECTIFS... 67
RESULTATS ET DISCUSSION ... 73
CHAPITRE I : SYNTHESE D'ANALOGUES DES ADDUITS INH(BH)-NAD ... 75
I. Introduction... 77
II. Analogues des adduits BH-NAD portant le motif adénosine... 78
II.1. Analyse rétrosynthétique ... 78
II.2. Première stratégie de synthèse (voie a) ... 80 II.2.1. Préparation du sel de pyridinium avec une chaîne hydroxylée (Synthon 20)80
8
II.2.2. Préparation du sel de pyridinium phosphotriester 19 (Schéma 10) ... 82
II.2.3. Préparation des dihydropyridines ... 86
II.2.4. Réduction du sel 20 suivie de la formation des phosphotriesters 44 et 45.... 87
II.3. Seconde stratégie (voie b)... 88
II.3.1. Préparation de la chaîne phosphotriester (Synthon 23) ... 88
II.3.2. Synthèse one-pot... 88
II.4. Analogue de l'adduit BH-NAD avec un lien diphosphate... 90
II.5. Conclusion ... 92
II.6. Perspectives ... 92
III. Analogues simplifiés avec le motif isonicotinoyle ... 94
III.1. Préparation du motif hémiamidal... 95
III.2. Préparation des sels de pyridinium... 97
III.3. Préparation des dihydropyridines... 98
III.4. Article... 100
III.5. Conclusion... 105
IV. Evaluation biologique ... 105
IV.1. Inhibition de l'enzyme InhA... 105
IV.2. Inhibition de la croissance de Mycobacterium smegmatis mc2155... 106
V. Conclusion... 107
CHAPITRE II : SYNTHESE DES INHIBITEURS DE TYPE BISUBSTRAT... 109
I. Introduction... 111
II. Analyse rétrosynthétique ... 111
III. Intermédiaires fonctionnalisés... 112
III.1. Synthèse de l'hémiamidal 103 ... 112
III.2. Synthèse de l'hémiamidal 107 ... 113
IV. Molécules de type bisubstrat en série pyridine ... 113
IV.1. Famille 1 en série pyridine... 113
IV.2. Famille 2 en série pyridine... 114
IV.2.1. Tentatives de préparation des hémiamidals 115 et 116 ... 114
IV.2.2. Synthèse de l'hémaiamidal 119 ... 114
V. Molécules de type bisubstrat en série pyridinium ... 115
VI. Molécules de type bisubstrat en série 1,4-dihydropyridine ... 116
VII. Inhibition de l'enzyme InhA ... 116
VIII. Article... 119
IX. Inhibition de la croissance de mycobactéries... 119
X. Conclusion... 125
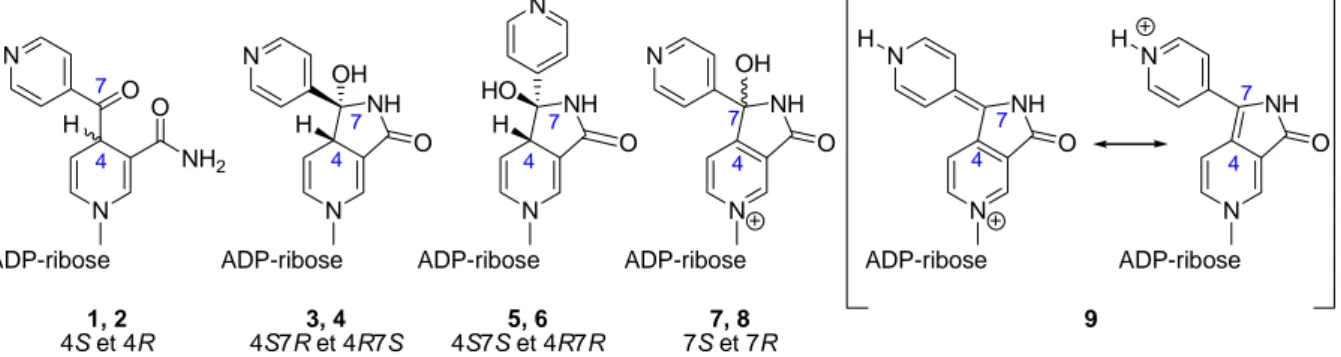
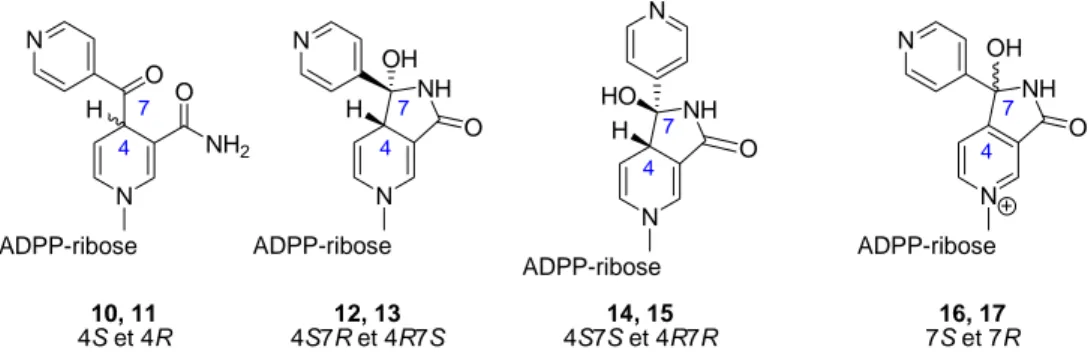
CHAPITRE III : ETUDE DE L'EQUILIBRE TAUTOMERIQUE CHAINE-CYCLE SUR DES MODELES SIMPLIFIES DES ADDUITS INH-NAD... 127
I. Introduction... 129
II. Etude expérimentale ... 129
III. Etude théorique ... 132
9
V. Conclusion... 141
CHAPITRE IV : ETUDE THEORIQUE DE L'INTERACTION DES DIFFERENTES FORMES TAUTOMERIQUES DE L'ADDUIT INH-NAD AVEC LE SITE ACTIF D' InhA... 143
I. Introduction... 145
II. Validation du modèle... 145
III. Energies d'interaction des adduits INH-NAD avec InhA... 146
III.1. Adduits ouverts 4S et 4R ... 147
III.2. Adduit BH-NAD ... 147
III.3. Adduits cycliques 4S7R et 4R7S ... 147
III.4. Adduits oxydés... 148
IV. Hypothèse d'ouverture des adduits cycliques ... 148
V. Article... 149
VI. Conclusion ... 159
CONCLUSION ET PERSPECTIVES ... 161
PARTIE EXPERIMENTALE COMPLEMENTAIRE ... 167
SYNTHESE ... 169
CHAPITRE I : SYNTHESE D'ANALOGUE DES ADDUITS INH(BH)-NAD ... 172
EVALUATION BIOLOGIQUE ... 179
I. Evaluation de l'activité inhibitrice sur InhA ... 181
I.1. Le Principe de la cinétique enzymatique sur InhA ... 181
I.2. Préparation d'InhA ... 181
I.3. Préparation et purification du substrat 2-trans-décénoyl-CoA d'InhA ... 181
I.4. Synthèse et purification des adduits INH-NAD (inhibiteurs d'InhA)... 183
I.5. Tests d'inhibition d'InhA... 184
II. Evaluation de l'activité anti-bactérienne... 189
II.1. Principe du test. ... 189
II.1. Inhibition de croissance de M. smegmatis. ... 189
II.2. Test de la spécificité des composés ... 192
II.3. Détermination des CI50 des différents composés sur M. smegmatis, E. coli et C. glutamicum. ... 195
11
ABREVIATIONS
Ac : acétyle
ACP : acyl carrier protein
ADN : acide désoxyribonucléique ADP : adénosine diphosphate AIBN : 2,2'-azabis-isobutyronitrile ARN : acide ribonucléique
BCG : Bacille de Calmette et Guérin BHI : brain heart infusion
C. : Corynebacterium
C10CoA : 2-trans-décénoyl-CoA
CcP : cytochrome c peroxydase
CI50 : concentration inhibitrice 50, concentration nécessaire à 50% d'inhibition de l'enzyme
CMI : concentration minimale inhibitrice CoA-SH : coenzyme A
DCC : dicyclohexylcarbodiimide DHFR : dihydrofolate réductase DHP : dihydropyridine
DL50 : dose létale 50, dose nécessaire pour tuer 50% des animaux soumis à l'expérimentation
DMF : diméthylformamide DMSO : diméthylsulfoxyde DO : densité optique
DOTS : directly observed therapy strategy dppf : diphénylphosphinoferrocène
E : Escherichia ETH : éthionamide Eq : équivalent molaire FAS : fatty acid synthase
Hepes : acide N-2-hydroxyéthylpipérazine-N'-2-éthanesulfonique HOMO : orbitale moléculaire la plus haute occupée
HPLC : chromatographie liquide haute performance (high pressure liquid chromatography) HPLC-SM : chromatographie liquide haute performance couplée à la spectrométrie de masse HRP : horseradish peroxydase ou peroxydase de Raifort
12
INH : isoniazide
INH-NAD : adduits covalents formés entre l'INH et le cofacteur IPTG : isopropyl β-D-thiogalactoside
IR : infra-rouge
LAM : lipoarabinomannane LB : Luria Browth Base
LDA : Diisopropylamidure de lithium
LUMO : orbitale moléculaire la plus basse inoccupée M. : Mycobacterium
mAGP : complexe petidoglycane-arabinogalactane-acide mycolique MDR : multi-drug resistances
MTT : bromure de 3-(4,5-diméthylthiazol-2-yl)-2,5-diphényl-2H-tétrazolium NAD+ : nicotinamide adénine dinucléotide (forme oxydée)
NADH : nicotinamide adénine dinucléotide (forme réduite) NADPH : nicotinamide adénine dinucléotide phosphate OMS : organisation mondiale de la santé
PBS : phosphate buffer saline
Pipes : acide pipérazine-1,4-bis(éthane-2-sulfonique) RMN : résonance magnétique nucléaire
ROE : rotating-frame overhauser effect SDS : dodécylsulfate de sodium
SIDA : sydrome d'immunodéficience acquise SM : spectrométrie de masse TB : tuberculose Tf : triflate Tr : temps de rétention THF : tétrahydrofurane UV : ultraviolet
VIH : virus d'immunodéficience humaine WT : wild type
15 Les traces les plus anciennes de la tuberculose ont été retrouvées sur des gisements osseux humains datant de la préhistoire, témoignant des ravages qu'elle causait déjà entre 3000 et 5000 ans avant JC.1 Resurgissant sporadiquement au cours de l'histoire, la tuberculose fut décrite sous diverses formes et emprunta différents noms (peste blanche, phtisie). A la fin du 18ème et au début du 19ème siècle, l'épidémie atteint son apogée en Europe et en Amérique du Nord, où la surpopulation urbaine et la dégradation des conditions d'hygiène, favorisent la contagion et donc la propagation de la maladie. A cette époque, la cure "hygiéno-diététique" et le repos dans des établissements spécialisés (sanatoriums) étaient la seule chance de récupération pour les tuberculeux. Le premier sanatorium fut ouvert en 1854 en Allemagne.2
L'épidémie de tuberculose n'a sérieusement régressé qu'à la fin des années 1960 avec l'amélioration générale du niveau de vie et des conditions d'hygiène et surtout suite à l'introduction de la vaccination par le BCG (Bacille de Calmette et Guérin) et à la mise au point de médicaments antibiotiques efficaces. C'est ainsi que l'incidence de la maladie dans le monde depuis cette date a considérablement diminué au point de considérer la tuberculose en voie d'éradication.
Cependant, la tuberculose connait aujourd'hui une recrudescence inquiétante aussi bien dans les pays en voie de développement que dans les pays industrialisés. Ceci est dû d'une part à la synergie prononcée entre le virus d'immunodéficience humaine (VIH) et la tuberculose et d'autre part à l'apparition de souches résistantes aux antibiotiques spécifiques de la mycobactérie (comme par exemple l'isoniazide). Ainsi, la résurgence de la tuberculose est considérée aujourd'hui par l'Organisation Mondiale de la Santé (OMS) comme un problème majeur de santé publique. En effet, le nombre de décès est de plus de 2 millions par an dans le monde.
La recrudescence de la maladie est associée pour une part importante à l'accroissement des résistances des microorganismes à de nombreux antibiotiques, en effet aucun nouveau médicament de première ligne n'a été introduit en thérapeutique depuis près de quarante ans. Ceci rend nécessaire le développement de nouvelles stratégies thérapeutiques, basées notamment sur une meilleure connaissance du mécanisme d'action des médicaments existants. Une meilleure compréhension du mécanisme d'action moléculaire de l'isoniazide (un des médicaments le plus utilisé dans le traitement de la tuberculose) permettrait de concevoir de nouvelles molécules capables d'agir comme antituberculeux. Depuis plusieurs années, le
16
groupe du Professeur J. Bernadou collabore pour la meilleure compréhension de ce mécanisme d'action.
Ainsi, nous proposons d'approfondir la compréhension du mécanisme d'action de l'isoniazide et de synthétiser de nouveaux composés à activité antimycobactérienne potentielle basés sur les connaissances de ce mécanisme.
19
I. La tuberculose
La tuberculose, première cause de mortalité mondiale due à un agent infectieux unique, tue plus de 2 millions de personnes chaque année. Depuis une vingtaine d'années, en dépit des traitements antibiotiques et du vaccin existant, on assiste à une recrudescence mondiale de cette maladie qui semble avoir été un peu trop vite considérée comme éradiquée dans les pays industrialisés.
I.1. Situation de la tuberculose dans le monde et perspectives d'évolution
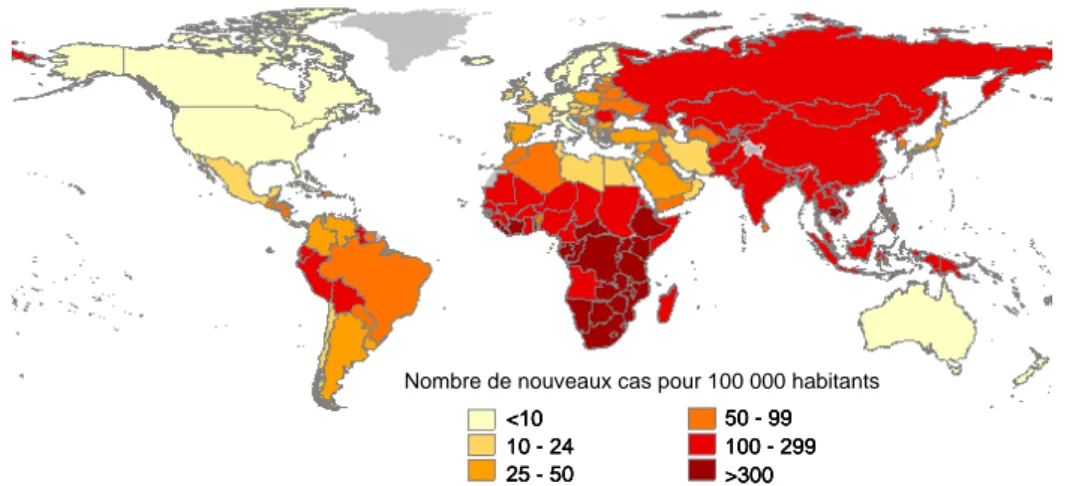
Aujourd'hui, la tuberculose reste un problème majeur de santé publique : chaque année, 8 à 10 millions de personnes contractent la maladie et plus de 2 millions en meurent, soit un décès toute les quinze secondes.3 Selon l'OMS, la tuberculose est l'une des principales causes de mortalité d'origine infectieuse avec le SIDA (syndrome d'immunodéficience acquise) et le paludisme. Elle touche essentiellement les pays en voie de développement qui totalisent 95% des cas et 98% des décès, mais aussi les régions industrialisées comme les Etats-Unis ou l'Europe. Trois continents sont principalement concernés : l'Afrique, l'Asie et l'Amérique Latine (Figure 1) où la surpopulation, les conditions de vie insalubres, l'alimentation déficiente et le manque de moyens des autorités sanitaires sont propices à la propagation de l'infection.4 <10 10 - 24 25 - 50 <10 10 - 24 25 - 50 <10 10 - 24 25 - 50 50 - 99 100 - 299 >300 50 - 99 100 - 299 >300 50 - 99 100 - 299 >300
Nombre de nouveaux cas pour 100 000 habitants
Figure 1 : Estimation de l'incidence de la tuberculose dans le monde en 2005.
(Source : The Global Plan to Stop TB http://www.stoptb.org/globalplan/)
A la fin des années 1970, avec la mise en place d'un traitement basé sur une polychimiothérapie obligatoire d'une durée de six mois, l'éradication était envisagée pour les années 2005-2010 dans les pays développés. Cependant, la tuberculose est aujourd'hui en recrudescence. En effet, ces dix dernières années, le nombre de tuberculeux dans le monde a augmenté de 20% et l'OMS estime que le nombre de morts imputé à la tuberculose va croître
20
pour atteindre 5 millions en l'an 2050.5 De problème majeur de santé publique, la tuberculose est maintenant devenue une urgence mondiale.
I.1.1. Les causes de la recrudescence
Plusieurs facteurs peuvent être évoqués pour expliquer cette recrudescence :5
Les changements démographiques
Les régions du globe les plus touchées sont également celles où la population croît le plus rapidement. Ces changements démographiques compteront pour 75% dans l'augmentation du nombre de cas dans la décennie à venir.
La co-infection VIH / Mycobacterium tuberculosis
Le VIH et l'agent pathogène de la tuberculose, qui accélèrent mutuellement leur progression, forment une association meurtrière. La tuberculose est responsable de 13% des décès chez les malades atteints du SIDA dans le monde. En particulier en Afrique, le VIH est la principale raison de la hausse de l'incidence de la tuberculose ces dix dernières années.
L'appauvrissement de certaines populations
L'association entre pauvreté et tuberculose est bien établie. La plus grande majorité des personnes souffrant de tuberculose disposent des services médicaux les plus pauvres. Il en résulte un mauvais suivi des traitements par les malades, donc des risques de rechute et d'émergence de bacilles résistants. Même dans les pays industrialisés, les taux les plus élevés de malades se retrouvent dans les couches les plus pauvres de la population.
Les mouvements de population
Dans les pays développés où l'incidence de la tuberculose est faible, l'immigration est le facteur qui contribue le plus à l'augmentation du nombre de cas. Ceci est dû à l'importation de la maladie depuis des pays fortement touchés.
La résistance multi-drogues
L'une des causes majeures de cette nouvelle avancée de l'épidémie de tuberculose est l'émergence de souches multirésistantes de Mycobacterium tuberculosis. L'acquisition d'une souche résistante peut s'effectuer de deux manières :
Multirésistance primaire : à la suite d'une infection par un bacille de Koch d'emblée multirésistant chez un patient n'ayant jamais reçu d'antibiotique auparavant.
21 Multirésistance acquise ou secondaire : lorsqu'un traitement inadéquat ou mal pris,
entraîne chez un patient initialement infecté par une souche sensible, une sélection de mutants résistants.
La multirésistance secondaire résulte souvent d'un traitement suivi de façon irrégulière ou partielle, les malades omettant de prendre régulièrement tous leurs médicaments jusqu'à la fin de la période prescrite parce qu'ils commencent à se sentir mieux, ou encore lorsque l'approvisionnement en médicaments n'est pas fiable. La résistance acquise, induite par un traitement inadéquat, est la marque d'une déficience des programmes de lutte contre la tuberculose.
I.1.2. Les programmes de lutte contre la tuberculose
C'est dans ce contexte que l'OMS a développé de nouvelles stratégies ainsi que des programmes nationaux et internationaux de recherche visant à améliorer les outils pour combattre avec plus d'efficacité la recrudescence de cette pandémie mondiale.
Le programme DOTS (Directly Observed Therapy Strategy) mis en place par l'OMS depuis 1995 a fourni des lignes directrices afin de lutter contre la tuberculose. Cette stratégie repose sur cinq points essentiels :6
l'engagement des gouvernements apportant un support financier durable,
la détection rapide des cas de tuberculose reposant sur le renforcement des équipements (radiographie) dans les laboratoires et sur un personnel formé,
un traitement standardisé sous surveillance directe (aider les patients à prendre leurs médicaments régulièrement et à achever leur traitement) et l'amélioration de l'accès au traitement dans les populations les plus pauvres et les plus vulnérables,
un système d'approvisionnement en médicament efficace et régulier, un système d'analyse de l'efficacité de ce programme.
Le programme DOTS a récemment été étendu.7,8 Il s'inscrit dans une stratégie globale appelée "Halte à la tuberculose" qui prend en compte de nouveaux objectifs :
la lutte contre la co-infection VIH/M. tuberculosis, le renforcement des systèmes de santé,
22
donner aux personnes atteintes de tuberculose et aux communautés les capacités d'agir,
favoriser et promouvoir la recherche.
Les objectifs de ces programmes étaient de parvenir à dépister 70% des nouveaux cas de tuberculose infectieuse et en guérir au moins 85% en 2005, le but final étant de maîtriser la tuberculose d'ici 2015 et d'observer une inversion de la tendance actuelle. Les résultats sont en dessous des objectifs concernant le dépistage avec une estimation de 53% en 2004. Mais les taux de succès thérapeutiques étaient de 82% la même année, soit très proches de l'objectif des 85%.7 La poursuite des efforts est encore indispensable et dépendra en grande partie du soutien financier des gouvernements.
I.2. Agent pathogène et transmission de la tuberculose
Peu de temps après la mise en évidence du caractère infectieux et contagieux de la tuberculose par le médecin français Jean-Antoine Villepain, le microbiologiste allemand Robert Koch, en 1882, observa et décrivit pour la première fois le germe pathogène responsable de la tuberculose : Mycobacterium tuberculosis également appelé bacille de Koch. En 1905, il est récompensé par le Prix Nobel de médecine et de physiologie pour cette découverte.
La tuberculose est donc due à une bactérie acido-alcoolo-résistante, aérobie stricte, communément appelé bacille (bactérie en forme de bâtonnet) tuberculeux. L'espèce la plus répandue est représentée par le bacille de type humain, Mycobacterium tuberculosis (M. tuberculosis) (99% des cas). Dans les régions d'élevage, les bovidés peuvent être infectés par une autre espèce du bacille, M. bovis qui est transmissible à l'homme par le lait non pasteurisé (1% des cas). En Afrique, un autre bacille a été identifié chez l'homme, M. africanum, dont la pathogénicité est la même que celle de M. tuberculosis. Ces trois espèces de mycobactérie ainsi que M. microti font partie du complexe M. tuberculosis. Les trois premières bactéries sont les plus dangereuses en ce qui concerne l'infection des sujets humains. Parmi les espèces non pathogènes, M. smegmatis et M. aurum sont particulièrement étudiées en laboratoire.
La transmission de la maladie se fait essentiellement par voie aérienne (toux, éternuement, expectoration). L'homme constitue à la fois le réservoir et l'agent de
23 transmission (ou vecteur) de la maladie.9 Bien que 2 milliards de personnes soient infectées par le bacille tuberculeux, seulement 10% d'entre elles développent la maladie. En effet, un porteur de bacilles peut rester en bonne santé toute sa vie (la maladie est dite latente) ou bien développer une tuberculose active lorsque son équilibre immunitaire est bouleversé, par exemple s'il est affaibli à la suite de malnutrition ou s'il est infecté par d'autres microorganismes comme le VIH. De ce fait, la tuberculose est connue comme étant une "maladie des pauvres" ou "maladie opportuniste".
Lorsque la tuberculose est déclarée, elle affecte le plus souvent les poumons (80% des cas, tuberculose pulmonaire). Les symptômes sont des toux persistantes, des douleurs thoraciques, du sang dans les expectorations et de la fièvre. La tuberculose se caractérise aussi par une perte de poids et d'appétit et de la fatigue. Si elle n'est pas traitée, une tuberculose active tue plus de la moitié de ses victimes. L'infection peut également toucher n'importe quel autre organe (tuberculose extra-pulmonaire). Mais seule la tuberculose pulmonaire est responsable de la transmission du bacille.
I.3. Prévention et traitement
I.3.1. La vaccination
Ce sont les bactériologistes français Albert Calmette et Camille Guérin qui mirent au point un vaccin, connu sous le nom de BCG, préparé à partir d'une souche atténuée de Mycobacterium bovis. Ce vaccin fut utilisé pour la première fois chez l'homme en 1921 en France, mais ce n'est qu'à partir de la seconde guerre mondiale que la vaccination au BCG s'est répandue à travers le monde.
Le BCG est toujours le seul vaccin utilisé de nos jours. Environ 115 millions de vaccins sont distribués chaque année et on estime que 80% des enfants le reçoivent à travers le monde. Plusieurs études montrent que l'efficacité de ce vaccin est de 80% chez les enfants, uniquement pour les formes les plus dangereuses de la maladie (par exemple la tuberculose méningée).10 Cependant, une étude réalisée à grande échelle en Inde a montré que son efficacité pour prévenir les tuberculoses pulmonaires est très variable surtout chez les adultes qui ne seraient protégés que dans un cas sur deux.11 Ce vaccin ne permet donc pas d'empêcher la transmission de la maladie ni d'enrayer l'épidémie mondiale. De nouveaux vaccins sont actuellement en cours de développement.12
24
I.3.2. Les antibiotiques antituberculeux et les traitements
Les antibiotiques13,14
C'est en 1944, que le microbiologiste américain Selman A. Walksman, découvrit le premier antibiotique actif contre la tuberculose : la streptomycine. Les résultats furent tout de suite impressionnants : arrêt immédiat de la progression de la maladie et rétablissement complet des malades. Dans les années qui ont suivi, de nombreux antituberculeux ont été développés.
La plupart des médicaments utilisés de nos jours sont encore d'anciennes molécules, découvertes il y a plus de 30 ans, grâce a un effort de recherche clinique entrepris entre 1945 et 1970. Le succès de l'antibiothérapie a ainsi conduit à un abandon quasi-total des recherches. En effet, aucun autre antituberculeux de première ligne n'a été introduit en thérapeutique durant ces quarante dernières années. C'est ainsi que l'isoniazide (INH) est et demeure depuis 1952, l'antituberculeux le plus puissant de l'arsenal actuel d'antibiotiques, tout en étant le moins cher. Aujourd'hui, il reste le médicament le plus utilisé dans le traitement de la tuberculose mais aussi dans sa prophylaxie.
D'une façon générale, les médicaments antituberculeux sont divisés en deux catégories : Les composés à large spectre (non spécifiques) qui possèdent une activité contre
plusieurs espèces de bactéries, y compris les mycobactéries. Ils agissent sur des cibles moléculaires présentes dans une grande variété de bactéries. Ils bloquent la transcription (rifampicine, fluoroquinolone), la réplication de l'ADN (fluoroquinolone), la traduction (streptomycine) ou la synthèse d'autres macromolécules (acide gras : thiolactomycine ; peptidoglycane : D-cyclosérine).
Les composés dont l'activité est spécifique des mycobactéries, voire une seule espèce de mycobactérie. Ce sont les antibiotiques les plus sélectifs et ils interfèrent en général avec la biosynthèse de la paroi mycobactérienne, une structure propre des mycobactéries. C'est le cas de l'isoniazide, le pyrazinamide, l'éthionamide et l'ethambutol. Les trois premiers sont des prodrogues nécessitant une étape d'activation à l'intérieur de la mycobactérie. Ceci contribue à la sélectivité de ces médicaments.
Au niveau clinique, les molécules utilisées dans le traitement contre la tuberculose peuvent également être classées en deux catégories :
Les antibiotiques de "première ligne"
Les antibiotiques de "première ligne" sont des médicaments utilisés durant la phase initiale de traitement de la tuberculose. Il s'agit d'une période de traitement intensif, durant
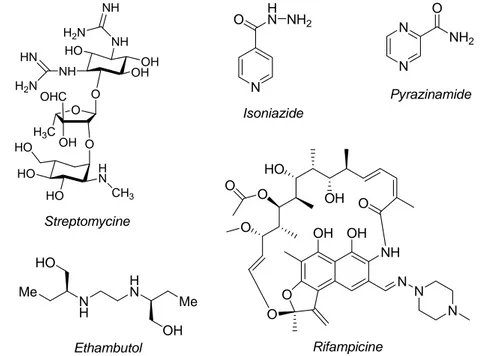
25 laquelle une combinaison de quatre antituberculeux, parmi les cinq que nous allons présenter ici (Figure 2), est prescrite au malade.
La streptomycine a été isolée de Streptomyces griseus et fut le premier antibiotique
réellement efficace contre la tuberculose. Elle possède une concentration minimale d'inhibition (CMI) de 1 µg/mL. Elle pénètre dans la membrane interne de M. tuberculosis et inhibe la biosynthèse des protéines en se liant de manière irréversible à la petite sous unité des ribosomes.
L'isoniazide (INH) est très actif vis-à-vis de plusieurs agents pathogènes du genre
mycobactérien en particulier sur les bacilles en multiplication et possède des CMI allant de 0,02 à 0,2 µg/mL. Cette molécule est une prodrogue nécessitant une activation in vivo (à l'intérieur de la bactérie) pour former le véritable principe actif. Il inhibe la biosynthèse des acides mycoliques qui sont des constituants essentiels de la paroi mycobactérienne. Nous reviendrons en détail sur le mécanisme d'action de l'INH par la suite.
N O HN NH2 Isoniazide HO H N O HO O HO CH3 OH H3C O OHC OHOH NH HO NH HN H2N NH H2N Streptomycine N N NH2 O N H H N Me Me HO OH Ethambutol Pyrazinamide O N N N NH OH OH O O OH HO O O O Rifampicine
Figure 2 : Antituberculeux de première ligne.
Le pyrazinamide est un analogue de l'isoniazide. Il s'agit également d'une prodrogue dont
l'activité dépendrait d'une amidase bactérienne. L'acide pyrazinoïque serait en fait la molécule active. Son activité est très spécifique de M. tuberculosis et agit principalement sur les bacilles quiescents ("dormants"). Ce composé présente un fort effet de synergie avec l'INH et le pyrazinamide et a permis le raccourcissement du traitement de 12 à 6 mois.
La rifampicine est un composé naturel isolé de Streptomyces mediterranei, introduit
26
agissant sur l'ARN polymérase ADN-dépendante et n'a aucun effet sur les enzymes présentes chez les mammifères. La CMI de cet antibiotique est de 0,2 µg/mL. Avec l'INH, la rifampicine constitue la base de la chimiothérapie antituberculeuse.
L'éthambutol est un aminoalcool synthétique décrit en 1961. Il agit comme agent
bactériostatique avec une CMI de 8 µg/mL et il est actif contre la plupart des espèces du genre Mycobacterium. Il inhibe la biosynthèse des arabinanes qui entrent dans la composition de la membrane mycobactérienne.
Les antibiotiques de "seconde ligne"
D'autres composés sont occasionnellement utilisés pour traiter la tuberculose (Figure 3), tels l'éthionamide, la cyclosérine, l'acide para-aminosalicylique ou des fluoroquinolones comme la lévofloxacine et l'ofloxacine, dans le cas de tuberculoses résistantes à plusieurs antituberculeux de première ligne.
N NH2 S Ethionamide OH COOH H2N Acide para-aminosalicilique (PAS) O N H O NH2 Cyclosérine N O O COOH N F N Ofloxacine N O COOH F N NH Moxifloxacine
Figure 3 : Quelques antibiotiques de seconde ligne.
Cependant, la faible disponibilité et le coût élevé de certaines de ces molécules les rendent inabordables pour la majeure partie des malades. De plus, à l'exception des fluoroquinolones, l'utilisation de ces composés est délicate à cause de leur faible activité par rapport aux antituberculeux de première ligne et à cause de plus grands risques d'effets secondaires. Les fluoroquinolones sont extrêmement actives contre M. tuberculosis et sont souvent choisies pour traiter les cas de tuberculose multirésistante, malgrès les effets secondaires.
27
Les traitements13,14
Sans traitement la tuberculose est souvent fatale, environ un tiers des patients atteint de la maladie décèdent au cours de la première année suivant l'inoculation et la moitié dans les cinq ans.
Il fut évident, dès le début de l'utilisation des antituberculeux, qu'une monothérapie serait responsable d'échecs et de sélection de mutants résistants. Le phénomène de résistance fut à l'origine de la recommandation d'une polychimiothérapie obligatoire de 6 mois. Elle permet d'augmenter et de diversifier le nombre de cibles biologiques en additionnant les effets propres à chaque antibiotique. La polychimiothérapie, lorsqu'elle est convenablement appliquée permet un taux de guérison de 95%. Ce traitement est nécessairement long (6 à 18 mois selon le régime thérapeutique) à cause de la lente croissance des bacilles tuberculeux (une division toutes les 20 heures, contre 20 minutes à 1 heure pour la majorité des autres bactéries) et de la présence de bacilles quiescents ("dormants") à métabolisme réduit et donc moins sensibles aux antibiotiques.
A l'heure actuelle, la thérapie standard pour les tuberculoses actives se fait en deux phases : une période de traitement intensif de deux mois avec quatre antibiotiques : l'INH, la rifampicine, la pyrazinamide et l'éthambutol ou la streptomycine, suivie d'une seconde période de quatre mois durant laquelle le malade continue à prendre deux d'entre eux : l'INH et la rifampicine. Bien que la plupart des patients n'aient plus de germe pathogène dans leurs crachats après les deux premiers mois de traitement, la prolongation du traitement est requise pour empêcher les rechutes et l'apparition de bacilles résistants.
Dans le cas d'infections par des bacilles de Koch résistants à l'INH, des traitements plus longs arrivent généralement à bout de l'infection. Les personnes atteintes de tuberculose multirésistante (la résistance multi-"drogue" étant définie comme une résistance à l'INH et à la rifampicine) peuvent également être soignées, cependant la chimiothérapie requise est encore plus longue (jusqu'à deux ans) et plus toxique pour les patients. Pour les cas les plus délicats, les antituberculeux sont administrés aux doses maximales tolérées et les traitements peuvent faire appel à des antituberculeux encore en cours de développement.
L'ensemble de ces faits accentue le besoin de stratégies alternatives dans le traitement de la tuberculose, basées notamment sur une meilleure connaissance du mécanisme d'action des médicaments existants.
28
II. Cibles thérapeutiques
Généralement les composés antituberculeux ciblent la biosynthèse de macromolécules (protéines, acides nucléiques, polysaccharides ou lipides de l'enveloppe cellulaire) essentielles à la survie de la mycobactérie. Il est important d'une part que ces cibles n'aient pas d'équivalents chez le mammifère pour limiter les effets secondaires et d'autre part qu'elles soient spécifiques à M. tuberculosis pour éviter l'apparition de résistances communes à plusieurs types de bactéries.
Dans le cadre de cette thèse, consacrée à l'étude du mécanisme d'action et à la synthèse d'analogues de l'INH, nous allons décrire plus particulièrement la paroi mycobactérienne qui est la cible privilégiée de cet antibiotique.
II.1. La paroi mycobactérienne
L'enveloppe cellulaire des mycobactéries est essentielle pour la croissance et leur survie chez l'hôte. Elle permet à la mycobactérie de résister, lors de la contamination, à un environnement très agressif, comme par exemple l'intérieur des macrophages. En effet, dès leur entrée dans les bronches et les alvéoles pulmonaires, les bacilles sont confrontés aux défenses immunitaires de l'hôte. Il arrive que le système immunitaire parvienne à éliminer complètement l'infection. Mais il est également possible que les mycobactéries résistent à la dégradation macrophagique. Dans ce cas, ils peuvent rester dans un état de latence, ou se multiplier en entraînant la lyse des macrophages ce qui conduit au développement de l'infection.14
L'enveloppe mycobactérienne possède une structure unique qui la distingue des autres bactéries : sa forte teneur en lipides la rend particulièrement imperméable et lui confère une résistance à la plupart des antibiotiques et agents thérapeutiques courants. Ces caractéristiques font de l'enveloppe mycobactérienne une excellente cible pour le développement de nouveaux antituberculeux. Il est donc indispensable d'avoir une bonne connaissance de la structure de cette enveloppe et d'identifier les enzymes responsables de sa biosynthèse.
Dans ce paragraphe, nous décrirons l'architecture de l'enveloppe mycobactérienne ainsi que la structure et la biosynthèse des acides mycoliques qui la constitue (en relation avec le mécanisme d'action proposé pour l'INH, objet de ce travail).
29
II.2. Structure de l'enveloppe mycobactérienne
La structure de cette enveloppe est aujourd'hui bien connue, notamment grâce aux travaux des équipes de P.J. Brennan,15 M. Daffé et P. Draper16 et de D.E. Minnikin.17
II.2.1. Architecture de la paroi mycobactérienne
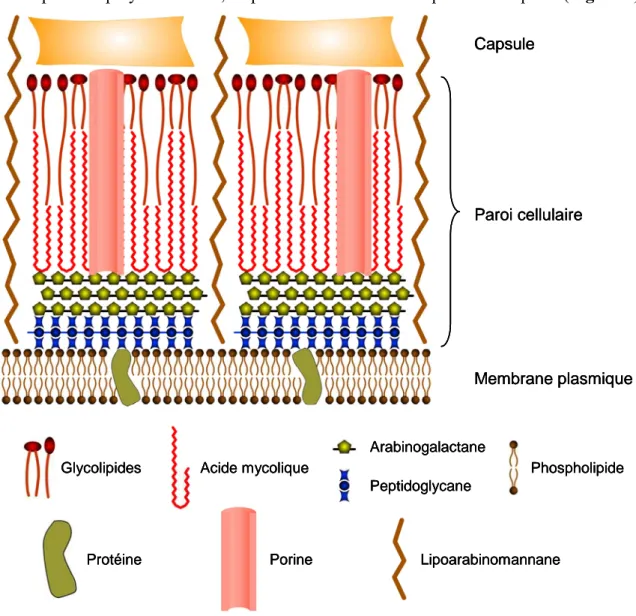
L'enveloppe mycobactérienne est constituée de trois éléments majeurs : la membrane plasmique (assemblage de lipides associés à des protéines pour former une bicouche lipidique asymétrique) entourée d'une paroi cellulaire riche en lipides et en sucres, elle-même encerclée d'une capsule de polysaccharides, de protéines et d'une faible quantité de lipides (Figure 4).
Acide mycolique Peptidoglycane Arabinogalactane Phospholipide Protéine Glycolipides Lipoarabinomannane Porine Capsule Paroi cellulaire Membrane plasmique Acide mycolique Acide mycolique Peptidoglycane Arabinogalactane Peptidoglycane Arabinogalactane Phospholipide Phospholipide Protéine Protéine Glycolipides Glycolipides Lipoarabinomannane Lipoarabinomannane Porine Porine Capsule Paroi cellulaire Membrane plasmique Capsule Paroi cellulaire Membrane plasmique
Figure 4 : Représentation schématique de l'enveloppe des mycobactéries.
(Adapté des travaux de Daffé et Draper16)
La paroi cellulaire est constituée de l'association de trois polymères (peptidoglycanes, arabinogalactanes et acides mycoliques) liés entre eux de façon covalente pour former le complexe mAGP (complexe peptidoglycane-arabinogalactane-acide mycolique).
30
Du cytoplasme vers l'extérieur de la bactérie, on distingue :
La couche de peptidoglycane constituée par des chaînes linéaires de polysaccharides (N-acétylglucosamine et acide muramique) reliées entre elles par de courts tétrapeptides.
Le peptidoglycane est relié à l'arabinogalactane (arabinose et galactose) par un pont diglycosyl-phosphodiester.
Les acides mycoliques, branchés sur un motif penta-arabinose, sont les molécules les plus abondantes (en poids) de la paroi mycobactérienne et leur structure varie en fonction de l'espèce bactérienne considérée.
Les acides mycoliques forment en surface une seconde bicouche par interaction hydrophobe avec la partie cireuse de glycolipides et glycopeptidolipides.
Une autre macromolécule est présente dans la paroi des mycobactéries : le lipoarabinomannane (LAM). Sa localisation et la façon dont il est associé à la paroi ne sont pas encore bien établies mais il semblerait qu'il traverse la paroi sur toute son épaisseur.
II.2.2. Les acides mycoliques
Fonctions
Les fonctions des acides mycoliques sont variées et peuvent se classer en cinq catégories. Structurale
Les acides mycoliques sont impliqués dans le maintien de la forme et de la rigidité du bacille. La stabilité exceptionnelle de ces acides est illustrée par la possibilité de l'étude de la tuberculose en paléo-épidémiologie dans l'antiquité.
Protection
Les acides mycoliques contribuent à la formation d'un bouclier hydrophobe imperméable à de nombreux composés tels que les antibiotiques classiques ou les radicaux exogènes.
Taxonomie
La nature et la variété des acides mycoliques synthétisés par les différentes espèces bactériennes revêtent une grande importance, dans la mesure où ils constituent des éléments spécifiques et donc des critères majeurs de différenciation et de classement.
31 Immunitaire
Les acides mycoliques constituent antigènes susceptibles d'être reconnus par les cellules immunitaires (lymphocytes T) de l'organisme.18,19
Cible
Une des voies d'investigation pour la recherche actuelle est le ciblage de la paroi cellulaire qui constitue un élément hautement spécifique des mycobactéries. En effet, les enzymes impliquées dans cette synthèse représentent des cibles idéales pour des médicaments potentiels et ainsi plusieurs médicaments fonctionnent par interruption de cette biosynthèse.20 C'est ainsi que l'INH et l'éthionamide inhibent la biosynthèse des acides mycoliques.
Structure
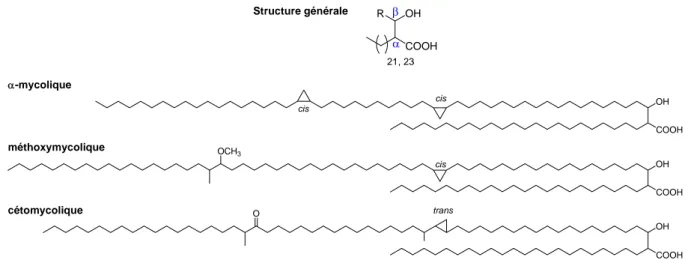
Les acides mycoliques sont des acides gras complexes, à longue chaîne de 60 à 90 atomes de carbone, α-ramifiés et β-hydroxylés. La structure générale de ces acides gras de haut poids moléculaire a été élucidée en 1950 par Asselineau (Figure 5).21
Le degré de complexité et la diversité des structures des acides mycoliques sont surtout liés à la chaîne principale R ou chaîne méromycolique et, dans une moindre mesure, à la longueur de la chaîne en α.15 La chaîne méromycolique peut comporter des groupes
fonctionnels variés tels qu'une double liaison, un cyclopropane ou une ramification méthyle, ainsi que des motifs carbonyle, époxyde, méthoxy ou alkoxy carbonyle. Trois types d'acides mycoliques ont été caractérisés chez M. tuberculosis : les acides α-mycoliques, les acides méthoxymycoliques et les acides cétomycoliques (Figure 5).
OH R COOH 21, 23 α β COOH OH cis cis COOH OH cis OCH3 COOH OH O trans Structure générale α-mycolique méthoxymycolique cétomycolique
32
II.3. Biosynthèse des acides mycoliques
Plusieurs étapes de la voie de biosynthèse des acides mycoliques sont connues, cependant, le schéma de biosynthèse aboutissant à la formation des acides mycoliques reste encore incomplet. La biosynthèse de ces molécules complexes comprend 4 étapes :15
La synthèse des acides gras saturés de C24-C26 (chaîne α).
La synthèse de la chaîne méromycolique (C40-C60).
La modification de cette chaîne par l'introduction des groupements fonctionnels. La condensation de type Claisen de la chaîne α et de la chaîne méromycolique.
La biosynthèse des acides mycoliques fait intervenir plusieurs systèmes de synthèse d'acide gras. Un complexe enzymatique multifonctionnel appelé "Fatty Acid Synthase" FAS-II est chargé de produire les chaînes méromycoliques par élongation des acyl-CoA C16-C18
produits par le système FAS-I (enzyme multifonctionnelle de sept domaines).22,23
Le cycle d'élongation comprend quatre activités enzymatiques indépendantes (Schéma
1) : deux activités réductases, l'une spécifique d'une double liaison en β ou 2-trans-énoyl-ACP
(InhA), l'autre spécifique d'une fonction cétone en β ou β-cétoacyl-ACP réductase (MabA), une activité β-cétoacyl synthase (KasA) qui introduit 2 atomes de carbone supplémentaires et enfin une activité β-hydroxyacyl-ACP déshydratase qui permet d'éliminer une molécule d'eau.24 Système FAS-I S O n CoA acyl-CoA O S O O AcpM malonyl-ACP S O O AcpM n Kas A KasB S O AcpM n S OH O AcpM n 3-cétoacyl-ACP acyl-ACP 3-hydroxyacyl-ACP 2-trans-énoyl-ACP S O AcpM n MabA Dehydratase InhA Système FAS II NADPH, H+ NADP+ NADH, H+ NAD+ KasIII H2O Système FAS-I S O n CoA acyl-CoA O S O O AcpM malonyl-ACP S O O AcpM n Kas A KasB S O AcpM n S OH O AcpM n 3-cétoacyl-ACP acyl-ACP 3-hydroxyacyl-ACP 2-trans-énoyl-ACP S O AcpM n MabA Dehydratase InhA Système FAS II NADPH, H+ NADP+ NADH, H+ NAD+ KasIII H2O
33 Le mécanisme d'introduction des groupements fonctionnels sur la chaîne méromycolique n'est pas encore parfaitement élucidé, mais la première étape correspondrait à l'introduction d'une double liaison cis.25
L'étape finale de la biosynthèse des acides mycoliques est la condensation de type Claisen entre la chaîne méromycolique et la chaîne α. L'enzyme impliquée dans cette étape de condensation correspond à la protéine Pks13, protéine multifonctionnelle contenant cinq domaines catalytiques, qui est un membre des polykétides synthases (Pks) de type I.26,27
Après la condensation, l'intermédiaire β-céto-ester-3-oxomycolique devra être réduit par une réductase CmrA, conduisant à l'acide mycolique (Schéma 2).
FAS-II FAS-I R1 S R 2 O O X CoA Acyl-CoA (C16-C18) activation carboxylation R1 O AMP S R 2 O CoA COOH R1 X R2 O O Réductase Condensase Pks13 R1 X R2 OH O Mycolate β-céto-ester mycolique R2 : C 22-C26 R1 : C40-C60 X : transporteur CmrA
Schéma 2 : Modèle proposé pour l'étape finale de la biosynthèse des acides mycoliques.
(Adapté de Portevin et al).28
III. L'isoniazide
La découverte en 1945 par Chorine29 de l'effet antibactérien du nicotinamide sur M. tuberculosis stimula la recherche de nouveaux médicaments contre la tuberculose, parmi les dérivés de la pyridine. Ainsi, en 1952, Fox entrepris la synthèse de la thiosemicarbazone du pyridyl-4-carboxaldéhyde (Schéma 3). L'étude fortuite des intermédiaires de cette synthèse mit en évidence l'activité antituberculeuse de l'hydrazide de l'acide isonicotinique ou isoniazide,30,31 bien supérieure à celle des substances jusqu'alors utilisées. Ceci conduisit rapidement à des applications thérapeutiques. L'INH est une vieille molécule, synthétisée dès 1912 par Meyer et Mally, mais qui n'avait jamais été testée comme antituberculeux.
34 N O O R N O HN NH2 N O HN HN SO2 Ph N H N N H S NH2 Ester d'acide isonicotinique Isoniazide Benzènesulfonyl-isonicotinoylhydrazine Thiosemicarbazone du pyridyl-4-carboxaldéhyde Schéma 3.
In vivo, l'expérimentation entreprise sur les animaux a entièrement confirmé les remarquables propriétés antituberculeuses de l'INH.30 Les premiers résultats sur l'homme ont été publiés par Elmendorf et al32 en 1952 et les propriétés préventives et curatives de l'INH ont rapidement été démontrées.
L'INH est un antibiotique à spectre d'action étroit, hautement spécifique des mycobactéries et plus particulièrement de celles du complexe M. tuberculosis. Ces dernières sont extrêmement sensibles à l'INH dans une gamme de CMI (concentration minimale inhibitrice) de 0,02-0,20 µg/mL (soit 0,15-1,50 µM), alors que les autres espèces de mycobactéries sont moins sensibles et requièrent des concentrations de l'ordre de 1-10 µg/mL pour M. kansaii et 100 µg/mL pour M. phlei. L'INH possède une activité antibactérienne très faible voire nulle en dehors du genre Mycobacterium : les autres bactéries telles que Escherichia coli sont capables de croître en présence de concentrations en INH supérieures à 1 µg/mL. Même administré de façon prolongée (6-18 mois), le traitement des patients n'occasionne qu'un faible risque d'effets secondaires nocifs. En effet, l'INH est remarquablement peu toxique et la dose létale chez l'animal (DL50 ~ 160 mg/kg chez la souris)
est très supérieure à la dose active (0,25 à 5 mg/kg/jour).33,34
On a longtemps cru que l'INH était rapidement intégré dans les bacilles par un processus oxygène-dépendant de transport actif inhibé par addition de cyanure ou par compétition avec d'autres dérivés hydrazides.35 Cependant des études plus récentes contestent ce processus
d'internalisation et suggèrent une diffusion passive de l'INH.36 Son action bactéricide est
extrêmement efficace sur les bacilles tuberculeux à multiplication rapide et poussant dans des conditions de cultures optimales. Par contre, elle est moins efficace dans des conditions qui ralentissent la croissance des bacilles telles que l'absence de nutriments préférentiels ou la sous-oxygénation.35 L'effet de l'INH devient irréversible au bout de quelques heures, mais la croissance même des bacilles n'est définitivement arrêtée qu'une génération après le début du traitement.
35 Des auteurs continuent à l'heure actuelle de travailler sur divers hydrazides analogues de l'INH, dans le but de trouver de nouvelles molécules plus efficaces (notamment sur les souches résistantes à l'INH).37 Cependant, aucun médicament n'est arrivé en clinique.
Depuis sa découverte, la compréhension du mode de fonctionnement de l'INH a fait l'objet de nombreux travaux. Ainsi au fur et à mesure de l'avancement des connaissances en particulier sur la génétique et la biochimie des mycobactéries, plusieurs hypothèses ou modèles ont été proposés pour tenter d'expliquer le mode d'action de cette molécule. Au-delà des effets pleiotropiques de l'INH chez le bacille, l'hypothèse la plus fréquemment avancée pour expliquer son effet bactéricide est son action sur l'intégrité de la paroi bactérienne en inhibant la biosynthèse des acides mycoliques. Ces derniers étant confinés essentiellement à la paroi des mycobactéries, ils représentent donc des cibles spécifiques.
Bien que son mécanisme d'action au niveau moléculaire ne soit pas encore complètement élucidé, il est maintenant largement admis que l'INH agit principalement par un processus impliquant deux étapes intracellulaires, chacune d'elles faisant intervenir une ou plusieurs enzymes de M. tuberculosis :
Une première étape d'activation de l'INH par la catalase peroxydase KatG, cette étape sera détaillée dans le paragraphe III.1. de cette introduction.
Une seconde étape d'inhibition cellulaire constituée par l'interférence de la forme activée de l'INH avec la biosynthèse des acides mycoliques. Cette étape sera décrite dans le paragraphe III.2. de cette introduction.
III.1. Activation de l'INH et formation des adduits INH-NAD
III.1.1. Activation enzymatique de l'INH par la catalase-peroxydase KatG
La protéine KatG
Le rôle essentiel de la protéine KatG dans l'action de l'INH contre M. tuberculosis a été partiellement révélé peu de temps après l'introduction de l'INH dans les thérapies antituberculeuses, lors des phénomènes de résistance. Lorsque que Middlebrook a isolé les premiers mutants de M. tuberculosis résistants à l'INH à partir de patients traités avec de l'INH en monothérapie, il a observé que la plupart des mutants hautement résistants avaient perdu leur activité catalase-peroxydase.38
Ce n'est que 40 ans plus tard, en 1992, que la base moléculaire de ces observations a été comprise. La découverte du gène katG qui code l'enzyme KatG de M. tuberculosis a permis à Zang et al39 d'expliquer la relation entre résistance à l'INH et perte de l'activité
catalase-36
peroxydase. Ils ont démontré que la sensibilité à l'INH pouvait être restaurée par introduction du gène katG sauvage dans une souche INH-résistante, catalase-peroxydase-déficiente. Ainsi la protéine KatG est indispensable à la sensibilité à l'INH qu'elle permet d'activer, cette étape d'activation étant obligatoire pour provoquer la mort bactérienne.
Structure
Dans sa forme native, la protéine KatG de M. tuberculosis est, en solution aqueuse, sous forme d'un dimère constitué de deux sous-unités identiques de 82 kDa chacune, soit un enchaînement de 744 acides aminés. Elle appartient à la classe des hydroperoxydases I ou catalase-peroxydase40 qui possède au sein de la même enzyme les activités catalase et peroxydase. Ce type d'enzyme est distinct des catalases et peroxydases dites typiques qui ne possèdent qu'une seule activité catalytique par enzyme.
La catalase-peroxydase n'est pas spécifique aux mycobactéries et existe dans de nombreux microorganismes procaryotes sans qu'ils soient spécialement sensibles à l'INH.
Les propriétés catalytiques de ces enzymes sont liées à la présence du motif hème dans le site actif : un ion fer(III) est complexé par un macrocycle tétrapyrrolique, la protoporphyrine IX. Cette hémoprotéine fait intervenir une espèce fer-oxo de haut degré d'oxydation dans son cycle catalytique. De plus, il a été montré que l'hème est à cheval sur les deux sous-unités protéiques et que le fer possède une histidine comme ligand axial.41
Fonction
Cette enzyme joue un rôle déterminant dans la réponse au stress oxydatif. Ce stress peut résulter de la génération continue d'espèces réactives de l'oxygène et/ou de l'azote par la bactérie elle-même et/ou par l'environnement dans lequel vit la bactérie. Chez M. tuberculosis la protéine KatG revêt une importance particulière car elle protège le bacille contre les effets délétères des espèces réactives produites par l'hôte. Elle permet alors la survie du bacille parasite en dégradant ces espèces nocives. Il a en effet été montré que KatG protège M. tuberculosis aussi bien contre H2O2 que contre les peroxydes organiques.41
KatG peut fonctionner selon les deux modes catalase et peroxydase qui sont détaillés dans le Schéma 4. Nous utiliserons les notations classiques de composés I et II pour décrire les états successifs de l'enzyme.
Dans le mode catalase (Schéma 4 : voie 1 (R = H) et 2), KatG possède une forte activité catalytique41 et joue un rôle classique mais néanmoins clé dans le contrôle de la concentration de l'eau oxygénée dans les compartiments cellulaires. Son action permet d'éliminer H2O2 par
37 réaction de dismutation en O2 et en H2O, là où elle n'est pas nécessaire ou bien est en excès.
M. tuberculosis est une bactérie aérobie stricte et a besoin de H2O2 comme cofacteur des
réactions catalysées par les peroxydases.
L'eau oxygénée est obtenue par réduction d'oxygène moléculaire prélevé dans le milieu environnant. Le transfert d'un électron vers l'oxygène moléculaire forme l'anion superoxyde O2•- qui est rapidement dismuté en O2 et H2O2. La réduction à un électron de H2O2 peut
conduire à la formation du radical hydroxyle HO• extrêmement réactif et très toxique (Réaction de Fenton). Ces espèces radicalaires peuvent s'additionner sur des biomolécules comme l'ADN ou les protéines et entraîner des dégâts irréversibles sur le métabolisme de la mycobactérie. De plus c'est aussi par ce mécanisme via H2O2 que les bacilles phagocytés à
l'intérieur des macrophages sont détruits. Le contrôle de la quantité de H2O2 est donc
nécessaire pour la survie du bacille dans les macrophages.
N N N N FeIII His KatG au repos N N N N FeIV His O N N N N FeIV His O + Composé I Composé II KatG au repos KatG au repos ROOH ROH 1 2 4 3 H2O2 H2O + O2 AH H+ + A H2O + A AH + H+
Schéma 4 : Cycle catalytique physiologique de KatG avec AH comme substrat ; mode
catalase 1 et 2 avec R = H ; mode peroxydase 1, 3 et 4.
Dans le mode peroxydase (Schéma 4 : voie 1, 3 et 4), KatG fonctionne comme une peroxydase à large spécificité,41 capable d'accepter les électrons d'une grande variété de donneurs tels que l'o-diasinidine ou le NAD(P)H et les utilise pour réduire H2O2 en H2O.42 Par
ce biais, la catalase-peroxydase joue un rôle de régulateur métabolique dans de nombreux systèmes enzymatiques NAD(P)H-dépendants en contrôlant le rapport du cofacteur réduit/oxydé.43 Enfin l'activité peroxydase de KatG est nécessaire pour activer l'INH, probablement en radical isonicotinoyle et par conséquent pour inhiber la synthèse de la paroi bactérienne (comme nous le verrons plus tard).
KatG est donc une enzyme bifonctionnelle avec un seul site actif, capable de catalyser de manière sélective deux réactions très différentes (effet catalase ou oxydation peroxydasique).
38
Les mécanismes d'oxydations de l'INH
Il est maintenant bien établi que l'INH est une prodrogue et requiert une activation intracellulaire pour former une espèce active avant d'exercer un effet toxique sur les bacilles tuberculeux. C'est la catalase peroxydase KatG qui est responsable de cette oxydation. Cette particularité permet un ciblage spécifique des microorganismes à détruire, car eux seuls possèdent le matériel enzymatique nécessaire à l'activation de l'INH. Ceci permet de minimiser les effets secondaires potentiels dus aux dommages engendrés sur les cellules de l'hôte.
Le mécanisme de l'activation oxydante de l'INH par KatG demeure encore un sujet à débat du fait de la multiplicité des activités catalytiques de l'enzyme.
KatG est une enzyme multifonctionnelle et polyvalente capable, en plus de sa fonction primaire de détoxification, d'activer l'INH selon plusieurs modes. Toutefois, malgré de nombreux travaux, les données actuelles de la littérature ne permettent pas de désigner avec certitude la voie d'activation préférentielle suivie par M. tuberculosis pour oxyder l'INH. En effet, selon les conditions expérimentales in vitro, KatG semble adapter son activité et les résultats obtenus ne sont pas toujours comparables. Le point sur lequel tout le monde s'accorde est qu'in vivo, la bactérie est placée dans un environnement complexe, comportant différentes variables (pH, présence de métaux, …), susceptible d'influencer directement le mode de fonctionnement de KatG.
Les différentes voies proposées pour décrire l'oxydation de l'INH par KatG sont les suivantes :
La voie catalase-peroxydase.41,44 La voie cytochrome "P450 like".43,45 La voie superoxyde-dépendante.46,47 La voie manganèse-peroxydase.48-50
Il faut noter que de nombreux système enzymatiques ont été utilisés comme modèle d'étude du mécanisme d'oxydation de l'INH pour suppléer à la disponibilité problématique de la protéine KatG : le cytochrome P450,51 la prostaglandine synthase,52 la myéloperoxydase,53 la peroxydase de raifort (HRP).52 Enfin, des systèmes non enzymatiques basés sur l'oxydation de l'INH catalysée par des métaux (MnII le plus souvent) en présence d'oxygène moléculaire ont également été utilisés,45,48,49 ainsi que, au laboratoire, l'oxydation par un complexe de MnIII mimant l'activité manganèse-peroxydase de KatG (cf. paragraphe III.1.2.)
39
Les produits d'oxydation de l'INH
Les espèces intermédiaires réactives
L'activation de l'INH conduit à la formation d'espèces activées responsables des effets bactéricides.
Lors de son activation, l'INH passe par des intermédiaires réactionnels radicalaires et ioniques avant de conduire à des produits d'oxydation stables. Ces produits, bien identifiés (acide isonicotinique, isonicotinamide, isonicotynaldéhyde (Schéma 5) et 4-pyridylméthanol), ne présentent pas, à concentration physiologique, de toxicité pour M. tuberculosis ni d'activité inhibitrice de la synthèse des acides mycoliques (cible majeur de l'INH). Par contre les espèces intermédiaires radicalaires et ioniques sont toxiques et non spécifiques dans la mesure ou elles sont capables d'alkyler des macromolécules (protéines, saccharides, lipides ou acides nucléiques).35,44
La formation d'un radical isonicotinoyle a été mise en évidence comme espèce réactive dans l'action bactéricide de l'INH (Schéma 5).44 Ce radical apparaît comme l'intermédiaire principal de l'oxydation de l'INH et est très probablement impliqué dans la formation de(s) l'espèce(s) responsable(s) des effets bactéricides ou bactériostatiques.54
N O HN NH2 Isoniazide N O Radical isonicotinoyle + N O X
-OH ac. isonocontinoique -NH2 isonicotinamide
-H isonicotinaldéhyde X =
Oxydation par KatG
Espèces non bactéricides
N O HN NH
Schéma 5 : Produits et intermédiaires clés de l'oxydation de l'INH.
Formation des adduits INH-NAD
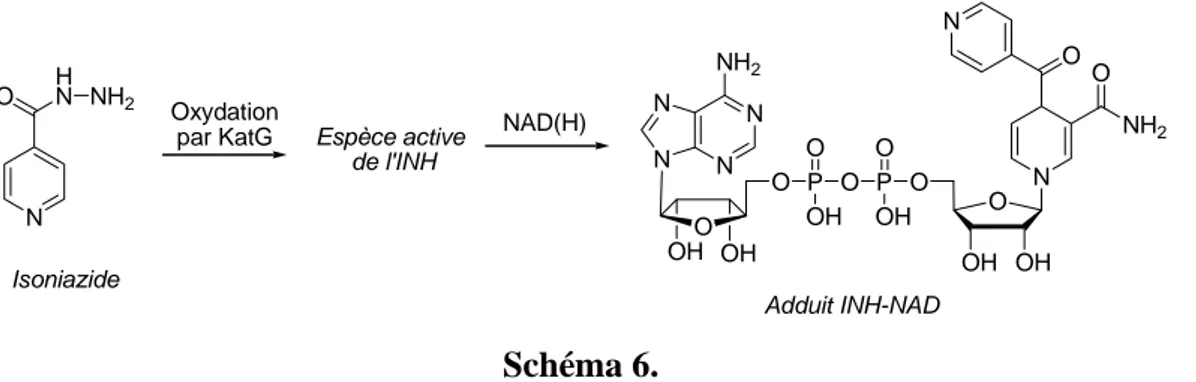
La proposition de mécanisme par lequel l'INH inhibe probablement InhA est "indirecte". Après activation par KatG, la forme activée de l'INH se lie de façon covalente au NAD(H) et un composé d'addition l'adduit INH-NAD est formé (Schéma 6).
40 O O N N N N NH2 O P O P O N O NH2 O N OH OH OH OH O O OH OH N O HN NH2 Isoniazide Espèce active de l'INH Oxydation par KatG Adduit INH-NAD NAD(H) Schéma 6.
La prodrogue n'interagit pas directement avec l'enzyme, mais seulement à travers cet adduit covalent qui serait le véritable responsable de l'inhibition compétitive d'InhA. La démonstration de l'existence d'un complexe InhA-adduit INH-NAD a été obtenue par résolution de la structure cristalline aux rayons X et études en spectrométrie de masse d'un tel complexe (Figure 6).55
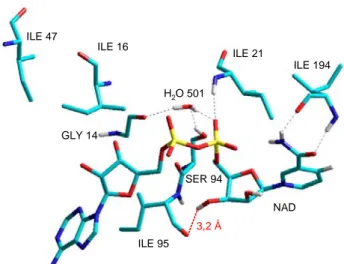
Figure 6 : Contacts moléculaires entre le site actif d'InhA et l'adduit INH-NAD(H) d'après
Sacchettini.55 Les chiffres représentent la distance (en angströms) entre les atomes sélectionnés (Ph = atome de phosphore).
En raison de sa localisation et de son orientation le motif isonicotinoyle interagit bien avec les chaînes latérales des acides aminés du site actif. En effet, la chaîne latérale de la Phe149, par une rotation de 90°, forme un stacking de type π-π avec le cycle pyridine de l'adduit INH-NAD. Ce nouvel arrangement structural du site actif augmente l'affinité d'InhA pour l'adduit INH-NAD (estimée à 100 nM56 ou 0,5 nM57 selon les auteurs; cet écart a été
41 expliqué par un mécanisme de type slow tight binding,24 comme nous le verrons dans le paragraphe III.2.1) par rapport au NADH (KmNADH = 8 µM58 ou 66 µM24).
Les données de la spectrométrie de masse de cet adduit sont en accord avec les résultats cristallographiques puisqu'ils font état d'un incrément supplémentaire de masse de 106 (m/zadduit = 770 et m/zNADH = 664) correspondant au motif isonicotinoyle ajouté.55-57
Le complexe stœchiométrique InhA-adduit INH-NAD montre un pic d'absorption à 278 nm et un épaulement vers 326 nm, avec un rapport caractéristique A326/A278 de 0,16.57
Dans l'adduit INH-NAD, la configuration du carbone en position C4 du motif nicotinamide est 4S ; c'est-à-dire que le motif isonicotinoyle occupe la même face que l'hydrure transféré lors la réduction du substrat (trans-énoyl-ACP) par le NADH.22,55,56
L'inhibiteur INH-NAD libre possède un pic d'absorption à 260 nm (
ε
260 = 27 000 M-1.cm-1) caractéristique des groupements aromatiques (adénine, pyridine) et un pic compris entre
326 et 333 nm (
ε
326 = 9600 M-1. cm-1) caractéristique du motif dihydropyridine.56,57 Mécanisme de formation des adduits INH-NAD
Il y a quelques années, trois hypothèses ont été envisagées dans la littérature pour expliquer la formation de ces adduits INH-NAD : la première voie impliquait une réaction d'addition de type ionique entre le dérivé NAD+ et le carbanion acyle de l'INH, la deuxième supposait une réaction d'addition radicalaire entre le NAD• et le radical isonicotinoyle alors que la troisième mettait en jeu une réaction d'addition "hybride" entre le cation NAD+ et le radical isonicotinoyle suivie d'une étape de réduction à un électron pour conduire aux adduits INH-NAD.
Parmi les trois scénarios proposés, pour la formation de ces adduits, ce sont les voies impliquant le radical isonicotinoyle qui sont les plus admises aujourd'hui.
En 1998, Sacchettini est le premier à soutenir la deuxième voie.55 Pour cela, il se base sur des études montrant que l'inhibition INH-dépendante d'InhA est plus rapide en présence de NADH que de NAD+.59,60 De plus, il avance l'hypothèse que la formation du radical NAD• peut être catalysée par la protéine InhA ou par le MnII/O2, ce dernier étant capable d'effectuer
une telle réaction avec l'INH.
Un an plus tard, Wilming et Johnsson56 proposent que l'addition du radical isonicotinoyle se fasse directement sur la forme oxydée du cofacteur (NAD+) et soit suivie d'une réduction du radical cation pour donner les adduits INH-NAD (Schéma 7). Des expériences de formation des adduits INH-NAD en absence d'InhA ont conduit à l'observation de quatre
42
isomères de même masse 770 par chromatographie liquide de haute performance couplée à la spectrométrie de masse (HPLC-SM). Deux isomères peuvent être logiquement attendus correspondant à l'attaque du radical isonicotinoyle sur l'atome C4 du NAD+ en solution sur les faces re et si du noyau nicotinamide. Pour expliquer la formation des autres produits, Wilming et Johnsson ont proposé l'hypothèse d'un stacking entre la purine et la partie isonicotinoyle ou d'une hydratation de la fonction cétone. Nous verrons dans le paragraphe suivant une explication proposée dans notre équipe mettant en jeu une cyclisation intramoléculaire. O O N N N N NH2 O P O P O N O NH2 O N OH OH OH OH O O OH OH N O HN NH2 Isoniazide N O Radical isonicotinoyl Oxydation par KatG ou MnII / O 2 1) NAD+ 2) +1e -Adduits INH-NAD H 4
Schéma 7 : Mécanisme de formation des adduits INH-NAD proposé par Wilming et
Johnsson.56
III.1.2. Activation chimique de l'INH par le pyrophosphate de MnIII
Oxydation biomimétique de l'INH par le pyrophosphate de MnIII
Les travaux effectués au laboratoire par Michel Nguyen au cours de sa thèse ont permis de mettre au point un système d'oxydation in vitro de l'INH biomimétique de la catalase-peroxydase KatG. Celui-ci utilise un complexe de MnIII pour mimer l'activité manganèse-peroxydase de KatG. La réaction de l'INH avec le pyrophosphate de MnIII en présence de cofacteur NAD+ ou NADH permet d'obtenir des adduits INH-NAD présentant les mêmes données spectroscopiques (masse, UV)61,62 que ceux obtenus par réaction avec KatG.55-57 De plus ces adduits sont aussi des inhibiteurs compétitifs de l'enzyme InhA.61,63
Le rendement de la réaction de formation de ces adduits est bien meilleur avec ce système d'oxydation biomimétique qu'avec le système MnII/O2 et est comparable à celui obtenu avec
l'enzyme KatG (41%).56,57 Le pyrophosphate de MnIII présente deux intérêts par rapport au système enzymatique : la réaction est considérablement accélérée (15 minutes au lieu de plusieurs heures) et il permet d'éviter d'utiliser KatG dont la disponibilité est fluctuante. Ce système chimique simple permet donc d'obtenir des adduits INH-NAD rapidement et avec un bon rendement.