HAL Id: tel-01449420
https://tel.archives-ouvertes.fr/tel-01449420
Submitted on 30 Jan 2017
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Etude du surenroulement diffusible de l’ADN
chromosomique chez la bactérie Escherichia Coli
Camille Cibot
To cite this version:
Camille Cibot. Etude du surenroulement diffusible de l’ADN chromosomique chez la bactérie Es-cherichia Coli. Biochimie, Biologie Moléculaire. Université Paris-Saclay, 2015. Français. �NNT : 2015SACLS241�. �tel-01449420�
Numéro National de Thèse : 2015SACLS241
T
HESE DE DOCTORAT
DE
L’U
NIVERSITE
P
ARIS
-S
ACLAY
PREPAREE A
L
’U
NIVERSITE
P
ARIS
-S
UD
E
COLED
OCTORALE N°577
Structure et Dynamique des Systèmes Vivants
Spécialité de doctorat :
Sciences de la Vie et de la SantéPar
Mme Camille CIBOT
Etude du surenroulement diffusible de l'ADN chromosomique chez la bactérie
Table des matières Introduction………5
Chapitre 1 L’organisation du chromosome bactérien : de la molécule d’ADN à l’orientation du nucléoïde dans la cellule ... 7 1) La structure de l’ADN ... 7 a) La forme B de l’ADN ... 11 b) La forme A de l’ADN ... 13 c) La forme Z de l’ADN ... 15 d) La forme P ... 15 2) L’ADN des procaryotes est surenroulé négativement ... 15 a) La découverte du surenroulement de l’ADN ... 17 b) De la structure secondaire au surenroulement ... 17 c) Le chromosome est surenroulé « négativement » ... 19 d) Organisation du surenroulement : les microdomaines ... 23 3) Les protéines « architectes » ... 25 a) Enroulement de l’ADN ... 29 b) Courbure de l’ADN ... 31 c) Pontage de l’ADN ... 37 d) Filaments et agrégats ... 41 4) Les macro-domaines ... 45 a) Les régions Ori et Ter ... 45 b) Des régions isolées génétiquement et spatialement : les macrodomaines ... 47 5) Orientation du chromosome dans la cellule par le processus de réplication ... Erreur ! Signet non défini. a) Organisation du chromosome en bras de réplication ... 49 b) Orientation du chromosome suivant l’axe d’élongation cellulaire ... 51 6) La cohésion des chromosomes frères ... 55 a) MukB ... 55 b) SeqA ... 57 c) Cohésion des macrodomaines Ter par MatP ... 59 Chapitre 2 Une organisation chromosomique dynamique ... 62 1) Perturbation du niveau de surenroulement ... 62 a) Les facteurs environnementaux ... 62 i) La température ... 62 ii) L’osmolarité ... 64 iii) L’anaérobiose ... 66 iv) Les lésions et modifications de l’ADN ... 66 b) Les processus cellulaires ... 68 i) La réparation ... 68 ii) La fixation de protéines ... 70 iii) L’avancée du replisome ... 72 iv) La transcription ... 72 v) Transitions de croissance ... 76 2) Maintien du niveau de surenroulement ... 76 a) Les topoisomérases de type IA ... 78 i) Topo I et Topo III ... 78 ii) L’ADN gyrase reverse ... 78 b) Les topoisomerases de type IIA ... 80 i) L’ADN gyrase ... 80 ii) La Topoisomérase IV ... 82 c) Equilibre des activités de Topo I et de l’ADN gyrase sur le surenroulement global du chromosome 82 3) Le surenroulement à deux composantes, libre et contraint ... 84
a) Les formes de surenroulement ... 84 b) La diffusion du surenroulement libre ... 86 c) Les barrières topologiques ... 88 i) Les grands domaines d’occupation protéique et les pontages ADN ... 88 ii) Les régions de haute structuration ... 90 iii) Les domaines d’interaction chromosomique (CIDs) : des régions relâchées ... 92 4) Rôle du surenroulement négatif ... 92 a) Facteur d’expression ... 94 i) Echappement de l’ARN polymérase depuis la région promotrice ... 94 1) Activité transcriptionnelle relaxation/surenroulement-dépendante ... 96 2) Contrôle de la réplication ... 98 i) L’initiation ... 98 ii) L’élongation ... 98 iii) La cohésion des chromosomes frères par les pré-caténanes ... 98 3) La ségrégation de la région de terminaison de la réplication ... 100 i) La résolution de dimère de chromosomes par la recombinase Xer au site dif ... 100 ii) La décaténation par Topo IV ... 100 4) Le stockage d’énergie ... 102 5) Système de mesure de surenroulement libre in vivo : le système res ... 104 a) La réaction de recombinaison spécifique de site du système Res ... 104 b) Le système Res in vivo ... 108 Résultats……….……..113 Chapitre 1 Le surenroulement diffusible de l’ADN chromosomique d’Escherichia coli .... 114 I. La mesure du surenroulement libre du chromosome : adaptation du système ɤδRes à la bactérie Escherichia coli ... 114 1) Deux stratégies complémentaires de mesure du surenroulement diffusible ... 114 a) Le senseur topologique ... 116 b) L’explorateur topologique ... 118 2) Le contrôle du système : induction par la température ... 120 3) Les différentes formes de la résolvase γδRes utilisées dans cette étude ... 122 4) L’expression de la résolvase ... 124 II. Mesure du surenroulement libre du chromosome par le senseur topologique ... 127 1) Variation locale du niveau de surenroulement diffusible ... 129 2) Variations de la diffusion du surenroulement suivant les conditions de croissance ... 131 a) Evolution du surenroulement diffusible avec la croissance ... 131 b) L’effet du milieu minimum sur la diffusion du surenroulement ... 135 c) L’effet de la concentration en NaCl extracellulaire sur la diffusion du surenroulement ... 135 3) Effet des protéines de fixation à l’ADN ... 137 a) NAP de phase stationnaire ... 137 b) Le système MatP/matS ... 139 III. Mesure de la diffusion du surenroulement chromosomique par l’explorateur topologique 141
1) Principe du 3C ... 161 2) Détection des jonctions par PCR ... 163 3) Détection des jonctions par qPCR ... 165 II. La localisation de MatP : microscopie à Super Résolution ... 169 1) Le PALM ... 169 a) La fusion MatP-mEos2 ... 173 b) La fusion MatP-Dronpa ... 175 2) Le BaLM ... 177 Discussion……….……179 I. Le système Res : avantages et inconvénients ... 181 1) Les avantages du système Res ... 181 2) Les inconvénients du système Res ... 182 II. De la nécessité de développer un nouvel outil senseur de la diffusion du surenroulement : définition de ses paramètres ... 183 1) La réaction de recombinaison ... 183 2) Le système d’induction ... 184 3) Un profil topologique à « haute résolution » ... 184 4) Le contrôle interne ... 185 III. L’état du surenroulement chromosomique chez E. coli ... 185 1) Les variations globales du niveau de surenroulement diffusible ... 185 a) Effet de la croissance ... 185 i) La réplication ... 185 ii) Effet de la transcription ... 187 b) Effet de la stringence ... 188 c) Effet de la résistance osmotique ... 189 2) Les variations locales du niveau de surenroulement diffusible ... 191 3) Comparaison des résultats obtenus avec d’autres études et systèmes de mesure du surenroulement diffusible ... 192 IV. Domaines et barrières topologiques ... 193 1) Comparaison du niveau de surenroulement de E. coli et de S. thyphimurium ... 193 a) Interactions à longue distance ... 193 b) La diffusion du surenroulement dans la région de terminaison de la réplication ... 197 2) Barrières topologiques induites par les protéines de fixation à l’ADN ... 198 a) Les NAPs ... 198 b) Déterminant du macrodomaine Ter : MatP ... 198 3) Comparaison avec la capture de conformation de chromosome ... 200 Table des figures.…..………203 Matériel et Méthodes………205 Bibliographie………215
Chapitre 1 L’organisation du chromosome bactérien : de la molécule d’ADN à l’orientation du nucléoïde dans la cellule L’absence de membrane nucléaire qui compacte et protège le matériel génétique est l’une des caractéristiques définissant les procaryotes. A la place du noyau, les procaryotes ont un complexe macromoléculaire appelé nucléoïde, où l’ADN et les molécules fonctionnellement associées y sont localisées. La compaction du chromosome circulaire y est imposée par une combinaison de facteurs, comprenant l’encombrement moléculaire (de Vries, 2010), la dynamique du polymère d’ADN (Pelletier et al., 2008), le surenroulement de l’ADN (Hatfield and Benham, 2002; Travers and Muskhelishvili, 2005) et l’interaction de l’ADN avec d’autres molécules telles les protéines architectes (Dillon and Dorman, 2010; Luijsterburg et al., 2008; Touzain et al., 2011). On représente souvent la force de compaction d’un système par la comparaison des dimensions de son « contenant » et de son « contenu ». Durant la phase exponentielle, la cellule de la bactérie modèle à Gram-négatif Escherichia coli mesurerait environ 2 μm de long sur 1 μm de diamètre. Par comparaison, l’opéron lac, comportant les gènes de structure lacZ, lacY et lacA (codant la β-galactosidase, la lactose perméase et la galactoside O-acétyltransférase respectivement), mesure à peu près 1,7 μm de longueur sans condensation. L’unique chromosome circulaire d’E. coli K-12 de 4,639 kb, dont on estime qu’il contient 4288 gènes codant des protéines (Blattner et al., 1997), mesure lui-même environ 1,7 mm de circonférence et s’il était complètement ouvert, aurait un diamètre de 0,5 mm, soit 500 fois celui d’une cellule d’E. coli. Cette forte compaction ne doit cependant pas entraver le déroulement correct et régulier des fonctions cellulaires ainsi que l’adaptation rapide aux conditions de l’environnement. Cette compaction fonctionnelle du nucléoïde est permise par l’organisation à différentes échelles, formant chez E. coli une structure visible à l’échelle du nanomètre et une superstructure visible à l’échelle du micromètre (Kim et al., 2004).
1) La structure de l’ADN
La caractérisation biochimique de l’ADN a démarré par des analyses chimiques ayant recours à diverses techniques d’hydrolyse. En 1871, Johann Friedrich Miescher isole dans des noyaux purifiés de leucocytes, une nouvelle substance composée de carbone, d’hydrogène, d’oxygène, d’azote, et de soufre, présentant un ratio phosphore/azote singulier, et correspondant à une nouvelle molécule organique très acide, contenant 22,5% d’acide
Figure 1 : représentation de l’enchaînement nucléotidiqu sous forme semi-développée composant l’ADN suivant l’orientation 5’-3’. Une base azotée est soit pyrimidique (avec un cycle azoté : cytosine ou thymine) soit purique (avec deux cycles azotés : adénine ou guanine). Le nucléoside est une base associée par une liaison N-osidique à un sucre, le désoxyribose pour l’ADN. Le nucléotide (mono-phosphate) est un nucléoside lié à un groupement phosphate au carbone 5’ du nucléoside. La liaison phosphodiester relie deux nucléotides par l’attaque nucléophile du groupement phosphate d’un premier nucléotide triphosphate, au groupement OH du carbone 3’ d’un second nucléotide.
Liaison N-osidique Liaison Phosphodiester
phosphorique. Il la nomme « nucléine » (Miescher, 1871). En 1889, Richard Altmann découvre que celle-ci est constituée d’une fraction protéique, et d’une substance acide, l’acide nucléique (Altmann, 1889). En 1896, les travaux d’Albrecht Kossel apportent des informations clés quant à la composition de l’acide nucléique dont les monomères sont caractérisés, notamment par des techniques d’hydrolyse : un sucre, un acide phosphorique et des bases azotées. Parmi ces derniers, Kossel distingue la structure des purines (adénine (A), guanine (G)), de celle des pyrimidines (thymine (T), cytosine (C), uracile (U)). Les travaux de Phoebus Levene montrent que les acides nucléiques qui diffèrent par l’exclusivité d’uracile en place de la thymine, présentent également une différence en carbohydrate. En 1909, Levene met en évidence le pentose présent dans l’acide « thymonucléique » extraits des tissus animaux (l’ARN) et qu’il appelle ribose, contraction des initiales de Rockfeller
Institute for Biochemistry et d’ose (Levene and Jacobs, 1909). Puis en 1928, grâce à un test
chimique développé par Robert Feulgen en 1922, Levene démontre que l’acide « zymonucléique » extrait des levures et des tissus végétaux (l’ADN) diffère de l’ARN par son ose, le 2-désoxyribose. En effet, l’hydrolyse acide rompt les liaisons N-osidiques entre purines et pentoses, exposant le groupe aldéhydique du désoxyribose au réactif de Schiff ou fuchsine qui, ainsi réduit, devient pourpre. Le ribose ne présentant pas cet aldéhyde, aucune coloration n’est observée. Les travaux de Phoebus Levene révèlent donc que l’acide désoxyribonucléique est constitué d’un enchainement de nucléosides reliés par des ponts phosphodiesters (Levene and London, 1928). En 1935, on parle alors d’ADN ou acide désoxyribonucléique (figure 1). Frederick Griffith avait avancé en 1928 l’existence d’un « principe transformant » transmettant la virulence d’isolats de pneumocoques à des pneumocoques non virulents, vivants dans une souris hôte (Griffith, 1928). En 1944, Oswald Avery, Colin McLeod et Maclyn McCarty extraient l’« agent transformant » et définissent le principe de « transformation » : la fonction de ce qu’on appelle désormais l’ADN, est alors mise en évidence (Avery et al., 1944). En effet, l’agent transformant isolé est un acide désoxyribonucléique ; Avery et ses collaborateurs apportent la première preuve que la séquence nucléotidique conférant à l’ADN sa structure primaire, serait le support de l’information génétique. En 1949, Erwin Chargaff démontre que la composition de l’ADN en A, T, G et C diffère d’une espèce à l’autre mais que le ratio d’adénine sur thymine et de guanine sur cytosine est toujours proche de 1.0 (Vischer and Chargaff, 1948). Il établit les règles d’équivalence de Chargaff, mettant en exergue la théorie du gène encodé d’Erwin
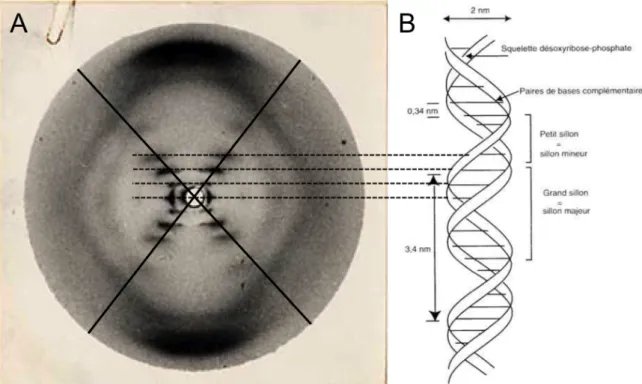
Figure 2 : structure secondaire de l’ADN bicaténaire de type B selon la théorie de Watson-Crick-Franklin. A : cliché de la diffraction de rayons X sur des cristaux d’ADN de thymus de veau, réalisé par Franklin et Gosling, le 2 mai 1952. La forme en X observée est caractéristique d’une structure hélicoïdale. B : représentation schématique de la double hélice d’ADN de type B formée par deux brins antiparallèles d’après Watson et Crick, en correspondance avec le cliché de la figure A. Les lignes en pointillées du plan à une dimension dans la figure A figurent l’espace d’empilement de deux paires de bases consécutives dans le plan à deux dimensions de la figure B.
Schrödinger (Schrödinger, 1944). Finalement, c’est le cliché de la diffraction des rayons X sur cristaux d’ADN, réalisé par Rosalind Elsie Franklin et Maurice Wilkins (Franklin and Gosling, 1953; Wilkins et al., 1953), qui amène James Watson et Francis Crick en 1953 à présenter un modèle de la structure secondaire de l’ADN (Watson and Crick, 1953): une double hélice où les deux brins s’enroulent en interagissant antiparallèlement via des ponts hydrogène entre les bases azotées (figure 2). La paire de bases G-C doit être stabilisée par trois ponts hydrogène tandis que seulement deux liaisons hydrogène stabilisent la paire de bases A-T
(figure 3). Les séquences riches en GC présentent ainsi des énergies de stabilisation et des
températures de dénaturation plus conséquentes. Au contraire, la liaison A-T offre moins de résistance aux déformations, comme la torsion individuelle des bases sur un même plan. Ainsi, la séquence d’un brin dictant celle de son complément, l’ADN double brin est souvent considéré comme une molécule unique, bien que les deux hélices ne soient pas liées de façon covalente. De surcroît, ce modèle intègre le mécanisme de réplication semi-conservative de l’ADN démontré chez E. coli (Meselson and Stahl, 1958). La double hélice d’ADN est stabilisée par des liaisons hydrogène internes mais aussi externes, variant suivant le solvant. En outre, les paramètres de l’hélice ont été analysés afin de caractériser les structures secondaires régulières des acides nucléiques (Shing and Carter, 2011). Ils regroupent principalement la répétition hélicoïdale (ou nombre de paires de bases par tour d’hélice) et le pas de l’hélice (ou distance entre nucléotides le long de l’axe de l’hélice). La répétition définit l’angle reliant chaque paire de bases le long de l’axe (tour d’hélice = 360°/répétition) tandis que le produit de la répétition et de l’élévation correspond à la hauteur (distance entre un tour complet) de l’ADN. Ces paramètres contraignent la géométrie de l’ADN qui peut donc adopter plusieurs formes, suivant la conformation de ses éléments de structures mais aussi suivant son solvant. Dans la nature, on rencontre trois formes majoritaires d’ADN, les formes A, B et Z (figure 3) dont les principales caractéristiques structurales sont décrites ci-dessous. Les caractéristiques géométriques de ces conformations en solution sont également résumées dans le tableau 1.
a) La forme B de l’ADN
C’est la forme mise en évidence par Rosalind Elsie Franklin et Maurice Wilkins. Cette conformation n’est pas définie par des paramètres géométriques stricts, mais plutôt par un ensemble de conformations apparentées, survenant à des niveaux d’hydratation élevés.
Figure 3 : modélisation moléculaire transversale (bas) et longitudinale avec leur représentation schématique (haut) des formes majoritaires A, B et Z de la double hélice d’ADN. On distingue les atomes de phosphate en jaune, les atomes d’oxygène du sucre en rouge et les atomes d’azote des bases en bleu. La liaison A-T requiert deux liaisons hydrogène tandis que la liaison G-C en requiert trois.
A pH neutre et dans les conditions salines physiologiques, il est admis que la forme B est la forme majoritaire de l’ADN in vivo. La double hélice tourne à droite avec un pas de 3,4 nm et un diamètre de 2 nm. La distance entre deux plateaux consécutifs de paires de bases suivant l’axe de l’hélice est d’environ 0,34 nm, indiquant donc 10 paires de bases par tour d’hélice (10,4 en solution). L’axe de la double hélice passe près du centre d’appariement des bases. La double hélice montre un petit et un grand sillon de 1,2 nm et 2,2 nm de largeur respectivement. Le squelette sucre-phosphate est à l’extérieur de la double hélice et porte des charges négatives en raison des groupements phosphate. La liaison N-glycoside est en
anti et le plissement endocyclique du désoxyribose est majoritairement sous forme C2’-endo. Les paires de bases sont à l’intérieur de la double hélice, reposant
perpendiculairement au plan de l’axe de l’hélice, s’empilant ainsi parallèlement les unes sur les autres pour former un cœur hydrophobe. Chaque paire de bases tourne de 36° (34,3° en solution) autour de l’axe de l’hélice par rapport à la paire de bases suivante. Le sillon majeur est aisément accessible aux protéines. Les caractéristiques de la forme B font de celle-ci la forme la plus variable et la plus malléable. b) La forme A de l’ADN
Cette forme est observée dans des conditions plus faibles d’hydratation que celles de la forme B, c’est à dire à force ionique plus élevée ou dans un mélange éthanol-eau (Saenger et al., 1986). Cette double hélice dont l’axe est excentré du centre d’appariement des bases (non perpendiculaires à celui-ci car inclinées de 10 à 20°) est droite, mais plus courte et plus large que la forme B : son pas est de 2,8 nm pour 11 paires de bases par tour d’hélice et son diamètre est de 2,3 nm. Aussi, les plans de deux paires de bases successives sont distants d’environ 0,26 nm. La liaison N-glycoside est en anti et le plissement endocyclique du désoxyribose est majoritairement sous forme C3’-endo : la conformation des sucres apparaît ainsi moins flexible que dans la forme B. La forme A présente un petit sillon large et superficiel et un grand sillon profond où les groupes phosphate sont plus proches les uns des autres que dans la forme B, faisant apparaître un fort potentiel électronégatif dans le grand sillon, propice à l’insertion de cations. En effet, les conformations construites avec le
C3’-endo requièrent de plus fortes concentrations en sels par rapport aux conformations avec le
C2’-endo, pour contrebalancer la distance plus courte entre les groupements phosphate chargés négativement. Une transition de la configuration de type B vers celle de type A de
l’ADN matrice au site actif de l’ADN polymérase de B. subtilis serait requise pour favoriser la fidélité de la polymérase (Kiefer et al., 1998). Cette forme serait également majoritaire dans les hybrides ADN-ARN ou dans les duplexes ARN-ARN. c) La forme Z de l’ADN Son unité de répétition n’est pas un nucléotide comme dans les formes A et B, mais un di-nucléotide, faisant alterner base purique et base pyrimidique. La conformation fait donc alterner respectivement une liaison N-glycoside en syn, avec un plissement endocyclique de type C2’-exo, et une conformation du sucre en anti, avec un plissement endocyclique sous forme C2’-endo (Wang et al., 1979). Cette forme a été initialement caractérisée en conditions salines élevées, de 3M NaCl (Pohl and Jovin, 1972). La méthylation des cytosines, ainsi que la présence d’autres cations tels la spermine ou la spermidine à des concentrations millimolaires stabilisent également cette structure (Rich and Zhang, 2003). Cette double hélice dont l’axe s’écarte significativement du centre d’appariement des bases qui sont perpendiculaires à celui-ci, tourne à gauche et à l’inverse de la forme A, est plus longue et plus étroite que la forme B : elle présente un pas de 4,5 nm pour 12 paires de bases par tour d’hélice et un diamètre de 1,8 nm. Le sillon majeur ressemble plus à une surface convexe externe, tandis que le sillon mineur est profond et très étroit donc peu accessible (Wang et al., 1979). d) La forme P
Allemand et ses collègues ont mis en évidence un nouveau type d’ADN, l’ADN-P. Lorsque l’ADN-B subi une tension couplée à un surenroulement, il réalise une transition brutale où les paires de bases sont rompues et se tournent à l’extérieur de la double hélice d’ADN (Allemand et al., 1998), cachant le squelette sucre-phosphate qui devient inaccessible aux enzymes. Ce type de structure a été observé dans le bactériophage Pf1, uniquement stabilisé par l’agencement des protéines de l’enveloppe (Liu and Day, 1994).
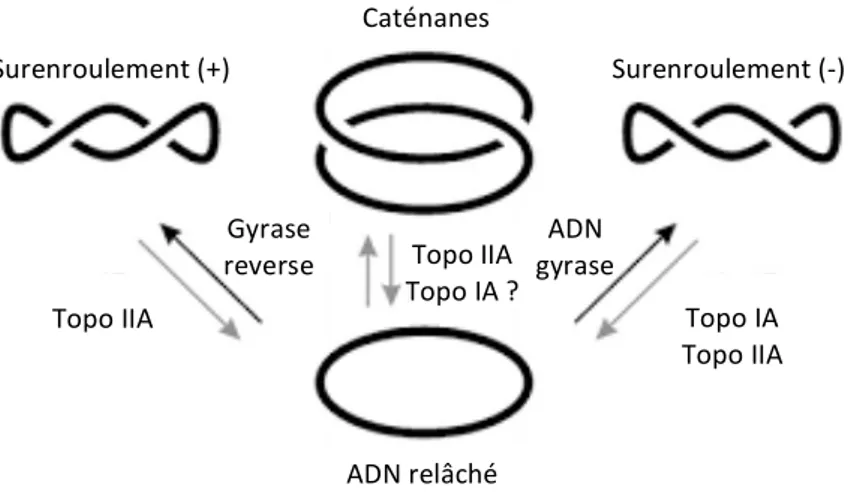
2) L’ADN des procaryotes est surenroulé négativement
Un moyen efficace de réduire le volume d’une molécule d’ADN est d’y introduire des supertours.
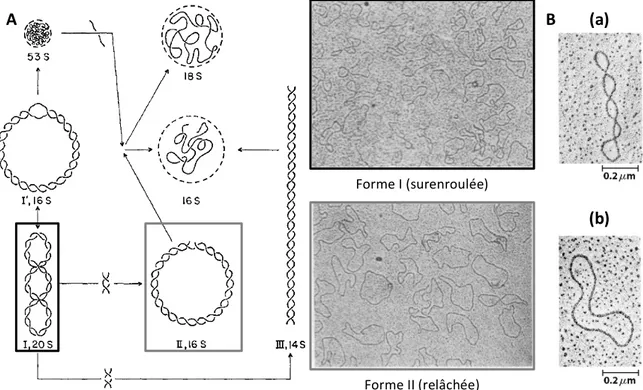
Figure 4 : l’ADN procaryote est surenroulé. A : représentation des différentes formes de l’ADN du polyomavirus (I, II et III) suivant leur coefficient de sédimentation, avec les micrographes électroniques ADN x 21000, des formes I (surenroulée, encadrée en noir) et II (relâchée, encadrée en gris) correspondants. Plus la vitesse de sédimentation est élevée, plus l’ADN est compacté donc surenroulé (d’après Vinograd et al, 1965). B : micrographes électroniques du plasmide pBR322 sous forme relâchée (a) et surenroulée (b) correspondant aux formes I et II de l’ADN du polyomavirus respectivement, observées en figure A. A Forme I (surenroulée) Forme II (relâchée) B (a) (b)
a) La découverte du surenroulement de l’ADN
La vitesse de sédimentation de l’ADN étant une propriété physique sensible à la dénaturation de celui-ci, la structure tertiaire de l’ADN a été étudiée en comparant les profils de vitesse de sédimentation de divers types d’ADN phagiques ou plasmidiques obtenus par ultracentrifugation sur gradients de sucrose. Les différentes conditions de dénaturation utilisées (Crawford and Black, 1964; Vinograd et al., 1965) montrent alors les mêmes courbes de vitesse de sédimentation-dénaturation de molécules d’ADN double brin circulaires fermées, indiquant que le degré de rotation d’un brin autour de l’autre ne varie pas. Une coupure simple brin par la DNase décompacte l’ADN extrait de la forme réplicative du bactériophage Phi X174 (Jansz and Pouwels, 1965) ou du polyomavirus, également observable en microscopie électronique comme sur la figure 4. Ces observations mettent en exergue la propriété de la topologie de l’ADN : Vinograd et ses collègues proposent que ces formes compactes sont sous forme double brin et circulaire avec une structure tertiaire enroulée gauche. Ces analyses d’ADN dites in vitro, ont ouvert la voie à l’étude du surenroulement chromosomique bactérien.
b) De la structure secondaire au surenroulement
Les deux classes les plus répandues du plissement endocyclique du sucre de l’acide nucléique décrivent chacune exclusivement, une forme d’ADN bicaténaire par leur effet sur la conformation globale de la molécule. Le type C2’-endo qui impose une distance entre phosphates de 0,7 nm, décrit un tour d’hélice de 36°, c’est à dire 10 pb/tour, correspondant à la structure de l’ADN-B. Si le sucre adopte une conformation C3’-endo, avec une distance inter-phosphates de 0,6 nm, la structure présente un tour d’hélice de 31° avec une répétition de 11 à 12 paires de bases, similaire à celle de l’ADN-A (Vargason et al., 2001). En fait, la conformation des éléments de structure de la molécule d’ADN influe profondément sur les paramètres structuraux de l’hélice mono- et bicaténaire: l’empilement des bases définit la distance entre bases, leur appariement donne le rayon de la molécule double brin et la conformation du sucre impose la distance entre les groupements phosphate d’un même brin. Ces paramètres sont étroitement liés et contribuent à l’obtention d’une torsion spécifique par la double hélice d’ADN. Aussi, dans une molécule d’ADN double brin de longueur donnée, ces paramètres définissent Tw, le nombre de tours d’hélices de l’ADN ou nombre de révolutions qu’un brin fait autour de l’autre.
A
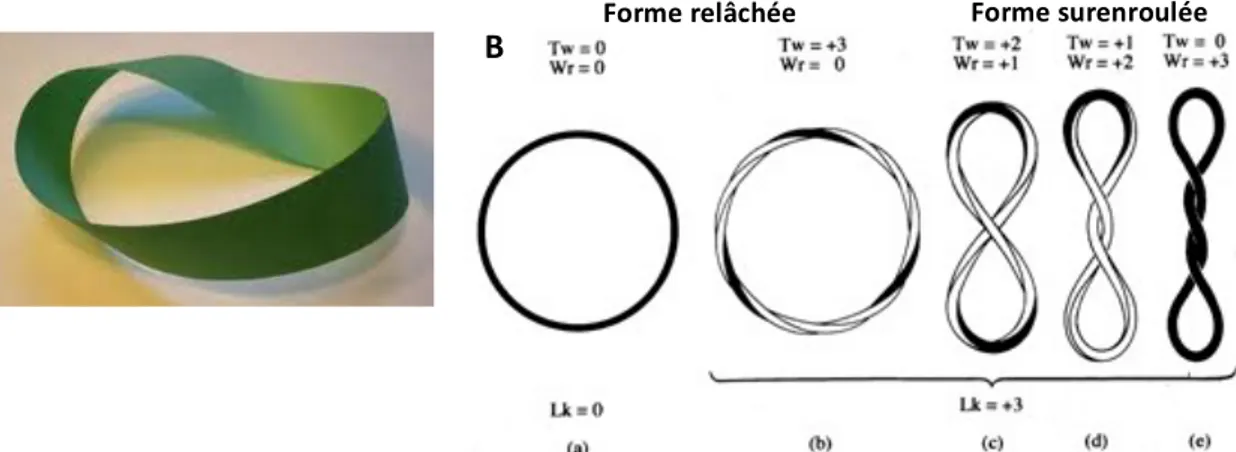
Figure 5 : la molécule d’ADN fermée est un objet topologique. A : photo représentant un ruban de Moëbius. La topologie consiste en l’étude des déformations par transformations continues. Le ruban de Moëbius est un exemple d’objet d’étude de la topologie, comme la molécule d’ADN qui est une structure fermée pouvant se déformer de façon continue sans rupture et réassemblage de brins. B : représentation schématique de la topologie de l’ADN , définie par deux paramètres, le nombre de tours d’hélices Tw et le nombre de supertours Wr, dont la somme définit le nombre total de liens topologiques Lk qui ne varie pas puisque la molécule est fermée. Les figures a et b correspondent à un état relâché puisque Wr=0. En b la molécule présente des plis blancs qui figurent trois tours d’hélices, telles deux molécules d’ADN enroulées (Tw = 3). En c, d et e, la molécule est surenroulée puisque Wr ≠ 0. Comme Lk ne varie pas, la déformation de Tw transforme Wr : alors que Tw diminue, Wr augmente. Si Tw devait augmenter, Wr diminuerait. Les plis blancs (b) ont disparu (e), mais la structure n’est pas plane comme en (a), elle est surenroulée : c’est l’effet topologique. B Forme relâchée Forme surenroulée
En effet, lorsque l’on parle d’ « un ADN », on évoque en réalité deux molécules simple brin d’ADN s’enroulant l’une autour de l’autre. De ce fait, ces brins sont liés dès que leurs extrémités sont jointes ou fixées. Un tel système peut être analysé d’un point de vue topologique. La topologie consiste en l’étude des déformations spatiales par des transformations continues, c’est à dire sans rupture et réassemblage des structures (correspondants aux brins dans notre système). L’état topologique d’une telle molécule peut être décrit par l’équation suivante (White, 1969):
Lk = Tw + Wr
où Lk est le nombre d’enlacements, c’est à dire le nombre total de fois où les deux brins du duplex d’ADN fermé se croisent. Par convention, Lk est un entier, positif dans une hélice droite, et négatif dans la configuration opposée, c’est à dire dans une hélice gauche. Lk ne varie pas tant que la structure est fermée. D’après cette formule, dans un ADN bicaténaire plan, Lk vaut le nombre de tours d’hélices Tw et Wr = 0 ; la molécule est alors dite « relâchée ». Or, si la structure n’admet pas de variation de Lk, la moindre déformation induisant une variation de Tw, crée une tension mécanique qui déplace l’équilibre. Le retour du système à l’équilibre se fait par la formation d’une structure tertiaire, une superhélice, qui possède un sens de rotation inverse à celui de la double hélice. Autrement dit, la variation de Tw est géométriquement compensée par la variation de Wr, le nombre de fois où le duplex s’enroule sur lui-même, ou nombre de supertours. Cette propriété topologique de l’ADN induit que Lk peut s’exprimer sous différentes combinaisons de Tw et de Wr et ne varie que si le duplex d’ADN subit une rupture et un réassemblage des brins. De telles réactions sont promues par les enzymes induisant des changements de topologie de l’ADN. Dans ce cas, Lk change d’un nombre entier d’unités. En 1963, Cairns apporte, par autoradiographie, la preuve que le chromosome bactérien tout entier est une molécule d’ADN double brin circulaire, donc fermée (Cairns, 1963). Dès lors, on peut parler de topologie du chromosome bactérien (figure 5). c) Le chromosome est surenroulé « négativement » D’après Vinograd, l’ADN contient des supertours d’hélice invariants gauche donc positifs car maintenus par l’équilibre des forces engendrées par la structure Watson-Crick (Vinograd et al., 1965). Dans un système « fermé », non contraint mais où un niveau standard de surenroulement
B Co ef fic ie nt d e sé di m en ta tio n (S ) [Molécule d’intercalant liée/résidu] σ, supertours/10 paires de bases τ, Supertours/ADN SV40 A BET intercalé Torsion : 10° Torsion : 36° Hélice droite Superhélice droite.
ADN surenroulé (-) Superhélice gauche. ADN surenroulé (+) C
ADN Relâché
est maintenu, on s’intéresse à la variation du nombre d’enlacements décrit par la relation ΔLk = ΔTw + ΔWr. Lorsque Lk diminue (ΔLk < 0), l’énergie libre défavorable qui lui est associée est minimisée par la formation de surenroulement (ΔWr > 0) et/ou d’une diminution de la torsion (ΔTw < 0). Par convention, on dit que ΔTw > 0 traduit le sens de rotation de l’hélice ADN-B, c’est à dire une rotation à droite. Dans ce cas, ΔWr > 0 décrit une rotation gauche de la superhélice, et ΔWr < 0 décrit une rotation droite de la superhélice. L’utilisation d’un intercalant de l’ADN a permis de valider la prédiction du sens (ou signe) du surenroulement énoncée plus haut. Le bromure d’éthidium est une molécule qui s’intercale entre deux paires de bases successives de la molécule d’ADN, une molécule qui dénature donc la double hélice en diminuant l’angle de torsion de 26° par base occupée, ce qui augmente la distance entre les deux paires de bases de 0,34 nm (Wang, 1974) (figure 6A). L’intercalation de quantité croissante de colorant diminue donc le nombre de tours d’hélices droits (ΔTw < 0) et cette dénaturation provoque un changement continu et réversible du nombre de supertours Wr. Bauer et Vinograd proposent une équation modélisant la variation du nombre de liens en fonction de la fixation de l’intercalant sur l’ADN du virus simien 40 (SV 40) affectant sa vitesse de sédimentation (Bauer and Vinograd, 1968) (figure
6B). La variation du nombre de liens est négative ΔLk < 0. En effet, sachant que ΔTw < 0 au
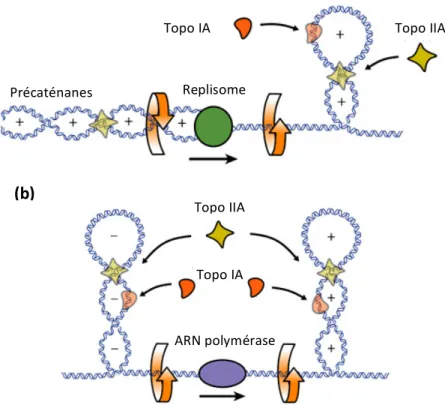
cours de l’expérience, si la saturation en intercalant mène à Wr > 0, au départ Wr < 0. Le surenroulement de l’ADN est donc négatif, ce qui va à l’encontre de la théorie de Vinograd (figure 6C). Ces résultats sont en accord avec les profils biphasiques de vitesse de sédimentation, longtemps restés inexpliqués : avec la dénaturation, la diminution de la vitesse de sédimentation indique une perte de supertours négatifs atteignant une valeur minimale qui correspond à un état non surenroulé, et suivie d’une augmentation de vitesse de sédimentation, signe d’un gain de supertours positifs. De la même façon, Wang montre que l’ADN des bactériophages λb2b5c, Phi X174, λcI857 et du plasmide 15 d’E. coli, présentent un surenroulement négatif dont il quantifie le nombre de liens (Wang, 1969a, 1969b). En 1972, Worcel et Burgi analysent de la même façon le repliement d’un nucléoïde entier isolé d’E. coli et confirment les résultats obtenus in vitro, à savoir que le chromosome bactérien montre une structure tertiaire surenroulée négativement. Bien que thermodynamiquement, l’état topologique le plus stable est le relâchement, l’ADN extrait de la plupart des organismes procaryotes présente un déficit en nombre de liens (ΔLk < 0), c’est à dire un surenroulement négatif (Wang, 1969b; Worcel and Burgi,1972).
Afin de comparer le surenroulement de molécules d’ADN de différentes tailles, une autre mesure du surenroulement a été déterminée : la densité de supertours σ de l’ADN, c’est-à-dire le nombre de superhélices par tour de double hélice d’ADN: σ = τ/β, où τ est le nombre de superhélices (nombre de rotation de l’axe de la double hélice dans l’espace ou entortillement) et β le nombre de tours d’hélice (nombre de paires de bases divisé par le pas de l’hélice) (Glaubiger and Hearst, 1967). Le changement d’état de surenroulement de l’ADN peut aussi être défini par la différence spécifique d’entrelacement ou densité de surenroulement σ, indépendante de la longueur de l’ADN, décrit par l’équation σ = ΔLk/Lk0, où Lk0 correspond au nombre d’enlacements de l’ADN lorsqu’il est relâché, c’est à dire où Wr0 = 0, donc Lk0 = Tw0 (Vologodskii et al., 1992). Si ΔTw = 0 (si le tour d’hélice ne varie pas) alors ΔLk = ΔWr et la densité de surenroulement de l’ADN est décrit par σ = ΔWr/Lk0. Chez E. coli, la valeur moyenne de la densité de surenroulement du chromosome pour 10,4 pb est de σ = - 0.06.
L’état topologique de l’ADN d’archaea thermophiles dans son ensemble n’est pas entièrement élucidé: il a été proposé qu’il soit relâché ou surenroulé positivement (Charbonnier et al., 1992; Herzel et al., 1999), alors que l’ADN d’archaea mésophiles est surenroulé négativement (Charbonnier and Forterre, 1994; Musgrave et al., 2000).
d) Organisation du surenroulement : les microdomaines
En 1971, Stonington et Pettijohn analysent, par ultracentrifugation sur gradient de sucrose, l’ADN radiomarqué natif extrait de cellules d’E. coli en phase exponentielle de croissance. L’ADN chromosomique libéré est associé à des protéines (majoritairement des ARN polymérases) et à des ARNm naissants liés à des ribosomes. La faible viscosité et la vitesse de sédimentation observées suggèrent que le chromosome possède une conformation très compacte. Cette structure est alors dépliée lorsqu’elle est chauffée et traitée à la RNase, indiquant qu’un composant ARN est impliqué dans sa stabilisation (Stonington and Pettijohn, 1971). Par un traitement à la DNase I, Worcel et Burgi estiment le nombre de coupure simple brin, capables de relâcher complètement le chromosome. Un traitement à la RNase I ne déplie quant à lui qu’une fraction du chromosome, les cellules en réplication étant plus sensibles au traitement que les cellules en carence. A partir de ces données, Worcel et Burgi construisent un modèle où le chromosome est composé d’un ensemble de boucles surenroulées, maintenues par l’interaction avec un cœur de type ribosomal (ARN
Figure 7 : le chromosome d’E. coli est organisé en domaines de surenroulement. A : micrographe électronique d’un chromosome isolé de cellules d’E. coli en phase exponentielle de croissance présentant 141±3 boucles émanant d’un noyau central (d’après Kavenoff et Bowen, 1976). B : représentation schématique de l’organisation du chromosome d’E. coli en microdomaines surenroulés (d’après Higgins et
al, 1996).
B A
polymérases), tant dans un état surenroulé que dans un état relâché (Worcel and Burgi, 1972).
Il faut attendre le travail de Richard Sinden pour obtenir une approximation in vivo du nombre de boucles de supertours d’ADN chromosomique, chez E. coli (Sinden and Pettijohn, 1981). Ces études mesurent la photo-liaison du triméthylpsoralène tritié au duplex d’ADN. Celle-ci augmentant la densité de surenroulement de 1 supertour toutes les 200 paires de bases, elle est donc directement proportionnelle au relâchement de la tension de torsion au sein de la molécule d’ADN (Sinden et al., 1980). L’analyse du nombre de coupures simple brin, introduites in vivo par irradiation aux rayons ɤ et nécessaires au relâchement complet du chromosome, indique alors la présence de 43 ± 10 domaines topologiques par équivalent-génome, c’est à dire 120 ± 30 domaines par nucléoïde. Cette estimation implique la présence de domaines d’environ 100 kb dans le chromosome des cellules d’E. coli en phase exponentielle de croissance, en milieu riche ou en milieu pauvre. Dans un milieu pauvre où le glucose a été remplacé par du succinate, le nombre de domaines chute à 25 domaines par équivalent-génome. Des cellules avec un temps de génération de 60 minutes présentent 20 domaines de moins par génome que des cellules ayant un temps de génération plus court de 25 min. Cela va à l’encontre de l’idée qu’un nombre fixe de barrières définit les domaines topologiques. Cette hypothèse est soutenue par l’observation du nucléoïde isolé d’E. coli au microscope : une structure de rosette dont les boucles, de 20 à 50 nt de long, émanent d’un « noyau » dense d’ADN (Hinnebusch and Bendich, 1997; Kavenoff and Bowen, 1976). Chaque boucle correspondrait à un seul domaine topologique (figure 7). La définition de domaine topologique correspondrait à une région relâchée par une coupure simple ou double brin de l’ADN.
3) Les protéines « architectes »
Les protéines chromosomiques architectes ont été identifiées dans les trois règnes du vivant. Ce sont généralement de petites protéines basiques interagissant avec l’ADN. Ces protéines peuvent avoir différents effets structuraux sur l’ADN, tels la courbure, le pontage ou l’enroulement, modifiant la forme locale de l’ADN pour la rendre plus compacte tout en empêchant l’agrégation. Ces effets influencent le métabolisme de l’ADN et ont été recensés chez des espèces très diverses. Chez les eucaryotes, les protéines histones et leur capacité à influencer la structure de la chromatine et les processus cellulaires sont connues en détail.
Figure 8 : représentation schématique de la présence des principales NAPs en fonction de la phase de croissance. Certaines NAPs sont spécifiques de la phase exponentielle de croissance, telles Fis, ou spécifiques de la phase stationnaire comme Dps ou CbpA. D’autres, comme H-NS ou HU sont exprimées tout au long de la croissance (d’après Sobetzko, 2012).
Phase de croissance
Abondance relative des NAPs
Exponentielle
(début) Exponentielle (milieu/fin)
Cette connaissance a influencé l’étude des protéines portant des fonctions analogues chez les bactéries, menant à les appeler protéines « histone-like » (Dorman and Deighan, 2003; Drlica and Rouviere-Yaniv, 1987). Elles sont désormais collectivement appelées Nucleoid
Associated Proteins (NAPs), reflétant leur localisation cellulaire sans impliquer de similarité
de structure avec les histones. La variabilité fonctionnelle des protéines architectes associées à l’ADN semble communément nécessaire aux procaryotes et aux eucaryotes. En effet, les eucaryotes présentent peu de variants d’histone mais ces derniers possèdent une queue sujette à de multiples combinaisons de modifications post-traductionnelles, cela leur permettant d’être fixés par des acteurs physiologiques variés. A l’inverse, les procaryotes n’ont pas de modifications post-traductionnelles mais montrent une grande diversité de NAPs. On différencie deux groupes de NAPs. Le premier groupe rassemble les NAPs au sens strict: des protéines abondantes, de faible poids moléculaire et que l’on suppose agir comme composants architecturaux au sein du chromosome bactérien alors appelé nucléoïde. De plus, ces protéines pouvant moduler l’expression du génome, elles sont impliquées dans le métabolisme (Grainger et al., 2006) et modulent souvent leur propre expression. D’après cette définition, E. coli possède au moins douze de ces NAPs (Azam and Ishihama, 1999; Dorman, 2013). Le second groupe est constitué de la grande majorité des protéines restantes, moins abondantes et qui se fixent transitoirement à l’ADN. En effet, E. coli contient au moins 240 différentes protéines qui se lient à l’ADN (Robison et al., 1998).
Les membres du groupe des NAPs sont nombreux et diverses mais sont étudiés de façon assez inégale. La plupart des NAPs fixent la forme B de l’ADN avec une faible spécificité et la déforment localement. Le calcul de la constante de dissociation apparente (Kd) de chaque NAP pour une sonde d’ADN spécifique ou non spécifique, a permis de déterminer l’ordre d’affinité de fixation à l’ADN des différentes NAPs abordées : HU > IHF > Lrp > CbpB > Fis > H-NS > StpA > CbpA > IciA > Hfq/Dps (Azam and Ishihama, 1999). Comme les nucléosomes, il s’avère que les NAPs sont impliquées simultanément dans différents processus cellulaires, comme la transcription, la réplication, la recombinaison, la réparation, la ségrégation, etc. D’autre part, les NAPs présentent différents profils d’expression au cours des phases de croissance (Ali Azam et al., 1999) (figure 8). Cela permet à la cellule d’adapter l’organisation de son chromosome suivant la croissance et de réguler précisément l’expression de son répertoire de gènes de façon spatio-temporelle (Sobetzko et al., 2012). Il est encore difficile
Figure 9 : représentation schématique des différents effets de condensation de l’ADN par les NAPs. On peut classer les NAPs suivant leur effet de fixation l’ADN, tels l’enroulement (A) par des protéines comme Lrp, la courbure (D) par des protéines comme IHF, HU et Fis, le pontage (C) par des protéines comme H-NS, StpA, MatP et SeqA, la filamentation (B) par des protéines comme DnaA et SeqA et enfin la formation d’agrégats (E) par des protéines comme CbpA Dps et HfQ (d’après Stavans et Oppenheim, 2006).
B A
aujourd’hui d’établir le rôle précis de chaque NAP sur le repliement du génome puisque des phénotypes mutants de celles-ci sont hautement pléïotropiques, les conséquences de leur liaison étant très diverses. Néanmoins, on peut classer les NAPs suivant leur effet de fixation à l’ADN (figure 9), même si la façon dont ces protéines organisent le chromosome, au-delà de la simple distorsion locale de l’ADN, n’est pas totalement résolue. Les contacts protéine-protéine et l’espacement régulier de leur site de fixation sur l’ADN pourraient donner lieu à des supertours d’ADN ou à des boucles, telles les fibres de la chromatine eucaryote. Quelques unes de ces NAPs sont présentées ci-dessous, en fonction des conséquences locales de leur liaison à l’ADN.
a) Enroulement de l’ADN
Bien que moins fréquentes que chez les eucaryotes, les protéines architecturales qui enroulent l’ADN telles le nucléosome eucaryote sont aussi présentes chez les bactéries. Par exemple, les membres de la famille Lrp/AsnC (Leucine-responsive regulatory
protein/asparagine synthase C gene product) sont conservés chez différentes bactéries. La
protéine Lrp est un baromètre physiologique puisqu’il s’agit d’une protéine de régulation répondant à la leucine (Platko et al., 1990). Elle possède de multiples sites de fixation dont l’alignement des séquences a permis de définir un consensus dégénéré de 15 pb (Azam and Ishihama, 1999; Cui et al., 1995). La protéine Lrp s’oligomérise sous forme de disque, le site de fixation à l’ADN étant localisé dans la partie N-terminale, disposé à la surface de la protéine. Cela implique l’enroulement de l’ADN à 180° autour d’un octamère ou d’un hexadécamère, en accord avec les profils de fixation à l’ADN (de los Rios and Perona, 2007; Wang and Calvo, 1993). En absence de leucine, deux octamères de Lrp peuvent s’associer en un hexadécamère capable d’enrouler presque deux tours d’ADN (Chen and Calvo, 2002), formant ainsi un complexe qui rappelle le nucléosome eucaryote et promouvant la compaction de l’ADN. Il a ainsi été rapporté chez B. subtilis que Lrp serait capable de ponter l’ADN (Tapias et al., 2000) (figure 10). Ces propriétés influent sur la structure du chromosome mais également sur la transcription : il influence l’expression des ARN stables, telle celle de l’opéron de l’ARNr, qu’il réprime par coopération avec H-NS (Pul et al., 2007). Le gène lrp semble être positivement contrôlé par l’alarmone ppGpp à la réponse stringente, de façon indirecte (Landgraf et al., 1996). Cela est en accord avec l’abondance maximale de Lrp en fin de phase exponentielle (2500 molécules) suivi de sa diminution dès l’entrée en
phase stationnaire (Ali Azam et al., 1999). Ceci traduit une diminution du besoin en ARN stables tels les ARNr qu’il réprime, et autres composants de la machinerie de traduction. D’autre part, son niveau d’expression est deux à quatre fois plus élevé en milieu carencé qu’en milieu riche, suggérant qu’il régulerait positivement des gènes induits en cas de carence. Ce facteur régule positivement ou négativement près de 10% des gènes chez E. coli, et son activité peut être inhibée ou inaffectée par la leucine, suivant la cible régulée (Cho et al., 2008a). b) Courbure de l’ADN A l’instar des nucléosomes chez les eucaryotes, les protéines qui courbent l’ADN sont très répandues chez les procaryotes et jouent un rôle prépondérant dans l’organisation de la chromatine. Les exemples les plus étudiés sont les membres de la famille HU/IHF (Swinger and Rice, 2004).
IHF (Integrativ Host Factor) a été identifié comme le facteur de l’hôte facilitant la
recombinaison intégrative du phage lambda chez E. coli (Craig and Nash, 1984), dont l’intégration est facilitée par la formation d’une boucle d’ADN spécifique par IHF. Les gènes
ihfA et ihfB codent pour les deux sous-unités formant l’hétérodimère de IHF, α et β
respectivement (Weisberg et al., 1996). La structure cristallographique du dimère de IHF montre un corps compacte d’hélices α dont deux bras flexibles en feuillets β émanent. Une proline de chaque bras s’intercale dans le sillon mineur de la séquence d’ADN spécifique, enroulant 37 pb d’ADN autour du corps de la protéine. La fixation de IHF sur l’ADN provoque une courbure supérieure à 180° (Rice et al., 1996). Elle introduit un « U-turn » de l’ADN, centré sur le site de fixation (Azam and Ishihama, 1999; Swinger and Rice, 2004)Swinger & Rice, 2004). IHF fixe le petit sillon de l’ADN : soit une séquence nucléotidique consensus de 15 pb YAACTTNTTGATTTW (Engelhorn et al., 1995) suivant un KD= 0,3 - 2,3 nM, soit une
séquence non spécifique avec un KD= 2 μM (Murtin et al., 1998). La cellule contient environ 12000 monomères en phase exponentielle. La quantité d’IHF augmente quand la croissance des cellules diminue, atteignant 55000 molécules en début de phase stationnaire (Ali Azam et al., 1999). Ainsi, dans la transition entre la phase exponentielle et la phase stationnaire, IHF est la seconde NAP la plus abondante. Le gradient de sites ayant un Kd de 0,1 nM à 2 μM, indique une occupation graduelle des sites du chromosome au cours de la croissance
A B Figure 11 : courbure de l’ADN par la fixation de HU et Fis. A : représentation de la structure du co-cristal résolu à 2,25 Å d’un dimère de HU d’Anabena se fixant à l’ADN en induisant une courbure de 105-140° (PDB 1P78). B : représentation de la structure du co-cristal résolu à 3,11 Å d’un dimère de Fis de E. coli se fixant à l’ADN en induisant une courbure de 65° (PDB 3JRA). Les monomères sont en orange et en bleu, et le résidu intercalant lysine en jaune (d’après Gruber, 2015).
par l’augmentation de la quantité d’IHF. Une seule molécule d’IHF sur 10000 serait libre en solution, indiquant qu’IHF jouerait un rôle important dans la structure du nucléoïde et la conversion de ses fonctions (Murtin et al., 1998). Lorsqu’IHF se fixe de façon hautement spécifique en amont d’un promoteur, elle peut moduler l’initiation de la transcription en influençant la formation d’une boucle d’ADN (Dworkin et al., 1998).
HU (Histone-like protein from E. coli strain U93) présente une similarité de séquence avec
l’histone H1 eucaryote : c’est un des rares exemples de NAP qui est une protéine de type histone-like par sa séquence d’acides aminés, hautement basique (Yamazaki et al., 1984). Les gènes hupA et hupB codent pour les deux sous-unités α et β respectivement. La composition de HU varie avec la phase de croissance : homodimère α en début de phase exponentielle et β en phase stationnaire, ou hétérodimère en milieu de phase exponentielle (Claret and Rouviere-Yaniv, 1997). Néanmoins, ces trois formes ne sont pas fonctionnellement équivalentes: la forme β2, se fixe plus faiblement à de l’ADN double brin.
HU montre une structure similaire à celle de IHF mais le site de fixation est aspécifique et sa taille très variable. Comme pour IHF, la proline de chacun des deux bras en feuillet β s’intercalant dans le petit sillon de l’ADN, la distance entre les deux prolines est de 9 pb d’ADN. Le corps du dimère ayant des contacts variables avec des sites ADN supplémentaires, le site de fixation pourrait s’étendre à 19 pb et diminue le tour d’hélice moyen d’ADN à 31° par paire de bases (Swinger et al., 2003). HU se lierait préférentiellement à l’ADN présentant des déformations structurales, telles que des coupures ou une jonction (Balandina et al., 2002; Pontiggia et al., 1993). Elle se fixe soit avec un KD de 200 à 2500 nM pour l’ADN non
déformé (Pinson et al., 1999) ou à 2 nM pour les substrats déformés (Kamashev and Rouviere-Yaniv, 2000). La fixation à de telles déformations suggère un rôle de HU dans la réparation de l’ADN par recombinaison homologue (Li and Waters, 1998). La fixation aspécifique de HU induit et/ou stabilise une grande variété de séquences d’ADN courbés, variant de 105 à 140° (Swinger et al., 2003) (figure 11 A). Cette flexibilité du complexe HU-ADN, facilite la formation de boucles sur de courtes distances et à faible concentration, permettant la régulation de l’expression des gènes ou la condensation de la région cible (Becker et al., 2007). Très récemment, une étude utilisant une fusion HupA-mCherry a été réalisée afin de visualiser le chromosome d’E. coli à haute résolution spatiale et temporelle (Fisher et al., 2013). Les propriétés d‘interaction de HU avec l’ensemble du nucléoïde ont
permis de révéler la structure globale de type hélicoïdale et la nature dynamique du nucléoïde d’E. coli. Le nombre maximal de protéines HU présentes dans la cellule d’Escherichia coli a été estimé à près de 30 000 en phase exponentielle, permettant de recouvrir et de plier 10 % de l’ADN chromosomique en se fixant à tous les 300 à 400 paires de bases environ (Ali Azam et al., 1999).
Fis (Factor for inversion stimulation) a d’abord été identifié comme un facteur impliqué dans
la recombinaison spécifique de site. Cette protéine est conservée dans la plupart des bactéries à Gram négatif. Elle se lie en homodimère à une séquence consensus de 17 pb, riche en AT, exceptée pour les positions 2 et 16, où des résidus G et C sont communément trouvés (Azam and Ishihama, 1999; Cho et al., 2008b). L’homodimère présente un corps hydrophobe d’où émanent deux motifs de fixation à l’ADN, une par monomère, de type hélice-tour-hélice (Pan et al., 1996). Les deux hélices de reconnaissance sont séparées de 2,5 nm et l’insertion dans les deux petits sillons adjacents séparés de 3,4 nm induit la courbure de l’ADN au niveau du site de liaison, jusqu’à 50° (Skoko et al., 2006). Une courbure additionnelle est générée par les interactions entre l’ADN flanquant le site de fixation de Fis et les côtés du dimère de Fis, résultant en une courbure jusqu’à 90°. La courbure moyenne est de 65° et le site de fixation est d’environ 27 pb (Stella et al., 2010) (figure 11B). La fixation de Fis conduit à une compaction considérable de l’ADN, sans doute due à de la fixation aspécifique et à un repliement concomitant (Skoko et al., 2005), similaire au mécanisme suggéré pour HU (van Noort et al., 2004). Fis contribue à de nombreuses activités cellulaires impliquant l’ADN, notamment la transcription, la réparation et la recombinaison (Dorman, 2009). Un analyse bio-informatique et des expériences d’Immunoprécipitation de Chromatine (Chip) chez E. coli semblent montrer que Fis se lie préférentiellement en dehors des régions codantes de gènes, tous les 200 à 300 paires de bases en moyenne le long du chromosome (Grainger et al., 2006). Fis a un impact majeur sur les transcriptomes de E. coli et de S. thyphimurium (Bradley et al., 2007; Kelly et al., 2004) en régulant l’expression de nombreux gènes impliqués dans la croissance tels ceux qui codent pour les ARNr, les ARNt ou pour d’autres NAPs. Le promoteur de Fis présente six sites de fixation permettant une auto-régulation négative de son expression (Ball et al., 1992). Fis serait capable de faire des ponts ADN-protéine-ADN (Schneider et al., 2001). Environ 50 000 protéines de Fis s’accumulent en phase exponentielle précoce puis leur nombre décroît
Figure 12 : courbure de l’ADN par IciA (ou ArgP). Modèle de fixation du tétramère du régulateur transcriptionnel IciA d’après la résolution à 2,7 Å de la structure du cristal de IciA de Mycobacterium
tuberculosis. Le premier dimère se fixerait avec une haute affinité au RBS (Recognition Binding Sequence)
libre (a), puis la fixation d’un deuxième dimère serait facilitée par la reconnaissance de leur domain régulatoire C-terminal et induirait une courbure de l’ADN (b) (d’après Zhou et al, 2010). Dimère I Dimère II se lie à l’ADN Dimère II Dimère I se lie à l’ADN ADN A C D
jusqu’à des niveaux indétectables en phase stationnaire (Ali Azam et al., 1999).
IciA (Inhibitor of chromosome initiation A) se fixe sous forme d’homodimère sur de l’ADN
courbé riche en AT au niveau de l’origine de réplication (Hwang and Kornberg, 1992; Wei and Bernander, 1996) (figure 12). Elle se fixerait à trois séquences répétées de 13 pb situés à côté d’oriC (Hwang and Kornberg, 1990). De cette façon, elle inhibe l’initiation de la réplication de l’ADN in vitro en bloquant l’ouverture de la région oriC promue par l’initiateur DnaA. IciA est également appelé Arp et fait partie de la famille des régulateurs transcriptionnels de type Lys (Thöny et al., 1991) et à ce titre est essentielle pour la transcription de Argol, la protéine d’export de la L-Arginine (Nandineni et al., 2004). Arp se fixe au promoteur de dnaA et contrairement à SeqA, active sa transcription (Lee et al., 1997). La cellule compte environ 800 molécules d’IciA (ou 400 dimères) en phase exponentielle puis 500 molécules en phase stationnaire (Ali Azam et al., 1999).
c) Pontage de l’ADN
Les NAPs qui pontent l’ADN créent des connections dynamiques entre duplexes d’ADN, formant des boucles pouvant s’étendre sur plusieurs kilobases. La formation de ponts se prête au contrôle de la transcription, puisque ceux-ci peuvent potentiellement capturer l’ARN polymérase ou l’exclure des promoteurs, réprimant les gènes concernés (Becker et al., 2007; Grainger et al., 2006). Néanmoins, ce type de pontage s’opposerait à un large spectre de mécanismes de fixation de NAPs sur l’ADN, tels la torsion et l’enroulement (Luijsterburg et al., 2008; Stoebel et al., 2008), comme le mécanisme de compaction de Dps qui inhibe la formation de boucles par Fis (Sato et al., 2013).
H-NS (Histone-like Nucleoid Structuring protein) se fixe en homodimère avec une haute
affinité à des séquences d’ADN double brin non spécifiques mais riches en AT et courbées (Rimsky et al., 2001; Yamada et al., 1990). La protéine H-NS se fixe au sillon mineur de l’ADN par son domaine C-terminal (Shindo et al., 1995), et s’oligomérise par le domaine N-terminal via deux sites séparés (Bloch et al., 2003). En l’absence de son domaine de fixation à l’ADN, elle peut former une hélice qui compacte la double hélice ADN par des contacts de dimères tête-tête et queue-queue (Arold et al., 2010) et semble ponter une même molécule d’ADN en présence de 2 mM de MgCl2 (Lim et al., 2012) (figure 13). Caractérisée comme un
Figure 14 : pontage de l’ADN par MatP. Représentation de la structure du co-cristal d’un tétramère de MatP lié à deux sites matS de E. coli résolu à 3,55 Å (PDB 4D8J). Les monomères sont en orange et en bleu. Chaque dimère se fixe à l’ADN en induisant une courbure de 8° et un changement conformationnel qui leur permet de se tétramériser (d’après Dupaigne et al, 2012).
répresseur global de la transcription, elle affecte plus de 35 gènes ou opérons chez E. coli (Blot et al., 2006), notamment ceux dont l’initiation nécessite le facteur RpoS par séquestration de ce dernier dans une boucle (Shin et al., 2005). HN-S auto-réprime son expression en occupant son promoteur (Falconi et al., 1993). La quantité de cette protéine atteint un niveau maximum en phase exponentielle à 20000 protéines puis décroît jusqu’à 40% de ce niveau en fin de phase stationnaire (Ali Azam et al., 1999).
StpA (Suppressor of td mutant phenotype A) partage 58% d’identité dans sa séquence en
acides aminés avec H-NS (Zhang and Belfort, 1992). StpA semble avoir un mode d’action et un profil d’expression identique à celle-ci (Ali Azam et al., 1999; Azam and Ishihama, 1999; Zhang et al., 1996). Elle fixe préférentiellement l’ADN surenroulé négativement (Keatch et al., 2005). Par multimérisation, elle forme un filament rigide de protéines sur l’ADN, qui est alors protégé des dommages, et peut ponter ce filament avec un ADN nu, ou condenser des filaments par pontage en présence de magnésium (Lim et al., 2012). Cependant, StpA se lie à l’ADN avec une affinité quatre à six fois supérieure à celle d’H-NS (Sonnenfield et al., 2001) et sa fixation est insensible à l’effet du pH, de la température ou de la concentration en sels. Elle présente également une activité de chaperonne ARN in vitro (Zhang et al., 1995).
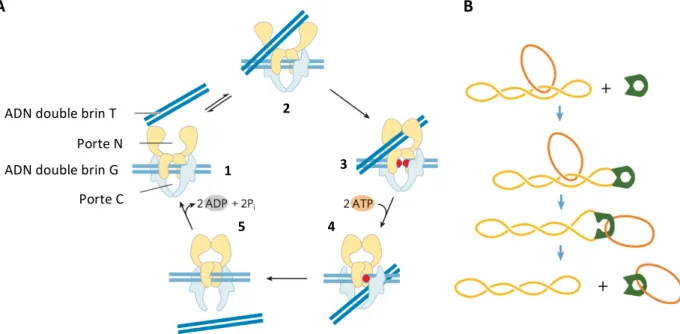
MatP (Macrodomain ter Protein) se lie à 22 sites matS au sein de la région terminale de
réplication, suivant un KD= 2-5 nM. Une analyse structure-fonction, basée sur une
expérience de double hybride bactérien, a été menée parallèlement à la résolution du cristal de la protéine MatP. Elle présente 3 domaines distincts: un domaine de fixation à matS, un domaine de dimérisation (motif Ribbon-Helix-Helix), et un domaine coiled-coil dans la région C-terminale (Dupaigne et al., 2012). Ce dernier est impliqué dans la formation de tétramères de MatP et dans l’interaction de MatP avec la machinerie de division (Espéli et al., 2012). MatP se fixe en dimère au site matS et plie l’ADN suivant un angle de 8°. Des analyses de diffraction de rayon X aux petits angles (SAXS) montrent également que cette fixation induit un changement de conformation du dimère permettant la tétramérisation par les domaines C-terminaux coiled-coil des dimères. Des analyses de microscopie à force atomique montrent que MatP est capable de former des boucles en fixant deux sites matS éloignés, excepté dans le mutant de tétramérisation (Dupaigne et al., 2012). On suppose donc que MatP est capable de ponter deux sites matS par tétramérisation (figure 14).
Figure 15 : pontage de sites GATC hémiméthylés par SeqA. A : représentation de la structure du co-cristal d’un dimère de SeqA lié par coopération à deux sites GATC hémiméthylés de E. coli résolu à 2,98 Å (PDB 3FMT). Les monomères sont en vert et en bleu. B : modèle de pontage d’après l’analyse des différents mutants de SeqA à oriC et au delà de oriC (d’après Chung et al, 2009).
A oriC
Au-delà d’oriC
SeqA a initialement été identifiée comme un facteur séquestrant l’origine de réplication oriC
chez E. coli, en empêchant la formation du complexe ouvert de réplication et l’expression du facteur d’initiation de la réplication DnaA. La protéine SeqA contribue à réguler la réplication du chromosome en empêchant la ré-initiation aux origines nouvellement répliquées pendant environ un tiers cycle (Lu et al., 1994). Elle s’associe au grand sillon des séquences GATC double brin qui sont hémi-méthylées sur le A après réplication mais aussi l’ADN complètement méthylé à oriC qui présente 11 sites GATC (Guarné et al., 2002; Slater et al., 1995). SeqA et la Dam méthytransférase seraient en compétition pour la fixation à l’ADN (Waldminghaus et al., 2012). SeqA se fixe stablement en dimère à deux séquences GATC par sa partie C-terminale, de façon coopérative, en se multimérisant par sa partie N-terminale (Guarné et al., 2005). La fixation d’une région de plus de 100 kb, à une distance de 300 à 400 kb de la fourche (Helgesen et al., 2015). Aussi, il est supposé que SeqA promeut le pontage intermoléculaire de l’ADN, à la manière d’une cohésine, en maintenant assemblées les molécules filles nouvellement répliquées in vitro (Fossum et al., 2007) et in vivo (Joshi et al., 2013) (figure 15). Environ 1000 molécules de SeqA sont présentes dans la cellule en phase exponentielle (Slater et al., 1995). d) Filaments et agrégats Les protéines qui polymérisent sous forme de filaments sur l’ADN ou qui s’y s’agrègent, ont généralement un rôle protecteur : elles plus spécifiquement exprimées dans des conditions de létalité cellulaire.
DnaA (DNA-binding protein A) liée à l’ATP se fixe de façon coopérative à des boîtes R ou DnaA de 9 pb 5'-TTAA/TNCACA suivant un KD de 4 à 20 nM dans la région d’oriC et d’origines
de réplication de plasmides tels pSC101, pBR322 et ColE1 (Fuller et al., 1984; Schaper and Messer, 1995). Pour initier la réplication, elle s’oligomérise par son domaine N-terminal de façon ATP-dépendante (Sekimizu et al., 1987; Simmons et al., 2003) stimulée par le facteur DiaA (Keyamura et al., 2007) et ouvre la double hélice d’ADN en l’étirant, s’associant à l’ADN simple brin (Duderstadt et al., 2011) (figure 16). Cette protéine joue aussi un rôle d’activateur ou de répresseur de sa transcription et de celle de nombreux autres gènes, y compris dans la région Ori (Atlung et al., 1985; Braun et al., 1985). La quantité intracellulaire de DnaA est de 1000 à 2000 molécules par cellule (Sekimizu et al., 1988), atteignant son
Figure 17 : agrégation de l’ADN par Dps. A : représentation de la structure du cristal d’un homo-dodécamère de Dps de E. coli résolu à 2,60 Å (PDB 1F33) (Luo et al, unpublished). B : micrographes de force atomique montrant l’auto-agrégation de Dps condensant simultanément l’ADN (d’après Ceci et al, 2004).
A B
maximum en milieu de phase exponentielle (Ali Azam et al., 1999).
CbpA (Curved DNA binding protein A) est une co-chaperonne conservée dans la plupart des
γ-protéobactéries (Yamashino et al., 1994). La protéine CbpA se fixe de façon préférentielle à de l’ADN courbé et riche en AT sous forme d’homodimère fonctionnnel (Azam and Ishihama, 1999; Yamada et al., 1990). Elle fixe le petit sillon de l’ADN et forme des agrégats (Chintakayala et al., 2015). CbpA contribue à la croissance à basse et à haute température. Elle forme un complexe avec la protéine de modulation de chaperonne CbpM qui inhibe alors son activité de co-chaperonne en bloquant sa dimérisation ainsi que sa fixation à l’ADN (Bird et al., 2006). La transcription de CbpA est sous contrôle de Lrp et RpoS (Chenoweth and Wickner, 2008) et réprimée par Fis (Chintakayala et al., 2013). Elle s’accumule jusqu’à plus de 2500 copies en fin de phase stationnaire (Ali Azam et al., 1999). Dps (DNA-binding protein from starved cells) est une protéine qui protège l’ADN en s’y fixant
de façon aspécifique en cas de carence, de stress nutritionnel ou de stress oxydatif, donc notamment en phase stationnaire (Almirón et al., 1992). Cette protéine de type ferritine s’oligomérise sous forme de dodécamère (Azam and Ishihama, 1999). Le chromosome est protégé des dommages oxydatifs par un co-cristal de dodécamère de Dps et d’ADN, hautement stable et ordonné par la séquestration des ions Fe2+ du milieu intracellulaire et empêchant la production de radicaux oxygénés (Grant et al., 1998; Wolf et al., 1999) (figure 17 A). Dps protège également du stress oxydatif en phase exponentielle, des dommages UV et des irradiations, de la toxicité du cuivre, du fer et du stress thermique (Nair and Finkel, 2004). Elle condense l’ADN via sa région N-terminale en formant des agrégats par diminution du pH (Ceci et al., 2004) (figure 17 B). La transcription de dps en phase stationnaire requiert le facteur RpoS de la transcription et IHF (Almirón et al., 1992) alors qu’elle est réprimée par Fis et H-NS (Grainger et al., 2008). Cette protéine est donc présente en une faible quantité en phase exponentielle (6000) qui augmente progressivement pour atteindre un niveau maximal en fin de phase stationnaire (180000) (Ali Azam et al., 1999).
Hfq (Host Factor for phage Qbeta) a été identifiée comme le facteur de l’hôte requis pour la
réplication du phage ARN Qβ sous la forme d’un hexamère à de l’ADN simple ou double brin mais plus spécifiquement à de l’ARN (Franze de Fernandez et al., 1972). L’inactivation de Hfq