FACULTE DES SCIENCES }^\
THESE PRESENTEE
A L’ECOLE DES GRADUES DE L'UNIVERSITE LAVAL
POUR L’OBTENTION
DU GRADE DE DOCTEUR ES SCIENCES
par
JOCELYN TREMBLAY Bachelier es Sciences de l'Université Laval
SPECTRES DE VIBRATION DES PHASES CONDENSEES DE OFg, SClg, SO2F2 et SOgClg.
JUILLET 1971 -I'
REMERCIEMENTS
En £oa£ pAemJeA ITea, je
détlAe expnùmen ma pAo^onde Aeconnalttance
au PAofietteuA PodAlgue Savoie, dont -let contellt et let encouAagementt
conttantt m'ont penmlt de meneA à bien cet tAavaux.
Amaintet AepAltet,
ton dévouement enveAt mol a dépatté let cadAet de ta fonction de dlAecteuA
de thète.
AtltAe d'exemple, je veux toullgneA ton attlôtance loAt de
ceAtalnet expénlencet qui te tant pAolongéet jutqu'à det heuAet taAdlvet.
Je
veux auttl AemeAcleA tout met con^AèAet det laboAatolAet de
tpectAotcopie pouA l'aide qu'lit m’ont fiouAnlz en plutleuAt occatlont. Je
touhalte que ce tAavail ne toit pat la filn d'mltlét tlncèAet développëet
au count de cet tnolt dennléAet annéet.
Je
AemeAcle le
Va. T.L.Weatenley, pouA let Aétultatt qu'il a bien
voulu me communlqueA à la tulte de tet tnavaux en tpectAotcopie de mlcAo-
ondet tuA le blchlonuAe de tou^Ae.
Meô
AemeAdementt t'admettent auttl au Contell National det
RecheAchet
du Canada, pouA let bouAtet [1969-1971) qui m'ont été accoAdéet.
Page
REMERCIEMENTS... i
TABLE DES MATIERES... LISTE DES FIGURES... v
LISTE DES TABLEAUX... vü 1 - INTRODUCTION GENERALE ... 1
1.1 - Effets des interactions moléculaires sur les spectres de vibration ... 2
1.2 - Vibrations dans les cristaux... 3
1.3 - Exemple d'application: SO2 cristallin ... 6
1.4 - Molécules étudiées... II 2 - PARTIE EXPERIMENTALE GENERALE... 13
2.1 - Appareils... 14 2.1.1 - Spectrophotomètre infrarouge ... 14 2.1.2 - Interféromètre Fourier ... 14 2.1.3 - Spectromètre Raman... 15 2.2 - Cellules... 16 2.2.1 - Injecteur mobile... 16
2.2.2 - Cryostat pour les études en Raman... 20 2.3 - Montages pour l'étude en infrarouge des matrices solides . 23
Ill
Page 2.3.2 - Montage pour la preparation des matrices avec
SC12...24
3 - SPECTRES RAMAN ET INFRAROUGES DE OF. EN PHASES CONDENSEES ... 30
3.1 - Introduction...31
3.2 - Partie expérimentale...32
3.3 - Résultats et discussion... 33
3.3.1 - Spectre Raman du liquide... 33
3.3.2 - Spectres Raman et infrarouge de OF2 cristallin... 35
3.3.3 - Spectres infrarouges de OF. en matrices inertes ... 41
4 - SPECTRES RAMAN ET INFRAROUGES DU BICHLORURE DE SOUFRE (SCl^ . . 49
4.1 - Introduction...50
4.2 - Partie expérimentale...51
4.3 - Résultats et discussion... 52
4.3.1 - Spectre infrarouge du gaz... 52
4.3.2 - Spectres Raman du liquide... .54
4.3.3 - Spectre infrarouge de SClg dans une matrice de Kr... 57
4.3.4 - Spectres Raman et infrarouge de SCI2 cristallin...63
Page
5 - SPECTRES RAMAN ET INFRAROUGE DE SO^ EN PHASE SOLIDE... 71
5.1 - Introduction... 72
5.2 - Partie expérimentale...73
5.3 - Résultats et discussion... 74
6 - SPECTRES RAMAN ET INFRAROUGE DE 50% Cl2 EN PHASE SOLIDE .... 85
6.1 - Introduction... 86
6.2 - Partie expérimentale... 87
6.3 - Résultats et discussion...88
RESUME... 99
V
LISTE DES FIGURES
FIGURE 1 FIGURE 2 FIGURE 3 FIGURE 4 FIGURE 5 FIGURE 6 FIGURE 7 FIGURE 8
Appareil de déposition pour 1 ' infrarouge moyen .
Appareil de déposition pour 11infrarouge lointain .
Cryostat pour les études Raman ...
Montage pour OF^ en matrices inertes ...
Montage pour matrice de SG^/Kr...
Spectre Raman de liquide, à 77°K...
Spectres infrarouge et Raman de
OF^ cristallin, à ~ 10 °K ...
Spectres infrarouges, à ~ 10 °K, de OF^ isolé dans des matrices de gaz rares dans des rapports de concentration molaire de 1:1000 ... Page 18 19 22 26 28 34 36 42
FIGURE 9 Evolution de la bande d’absorption infrarouge Vj, dans une matrice OF^rAr = 1:400
FIGURE 10 Spectre infrarouge de OF. dans des matrices de néon et d'argon dans la région de .
FIGURE 11 Spectre infrarouge de SCI2 en phase
gazeuse, à 25°C ...
45
47
53
Page FIGURE 13 Spectre infrarouge de SCI2 isolé dans une matrice
de krypton, à ~ 10 °K... 58
FIGURE 14 Spectres infrarouge et Raman de SCI2 cristallin,
à 77 °K ... 64
FIGURE 15 Spectres infrarouge et Raman de 86^2 cristallin,
à ~ 77 °K ... 75
FIGURE 16 Spectres infrarouge et Raman de SOgFg cristallin,
à ~ 77 °K... 76
FIGURE 17 Spectres infrarouge et Raman de SC^C^ cristallin,
à ~ 77 °K... 89
FIGURE 18 Spectres infrarouge et Raman de SC^Clg cristallin,
vii
LISTE DES TABLEAUX
Page
TABLEAU 1 Groupes de site et super-groupes possibles
pour me molécule de symétrie ... 5
TABLEAU 2 Diagramme de corrélation pour SCL... 7
TABLEAU 3 Quelques propriétés physiques des différents
composés étudiés... 12
TABLEAU 4 Fréquences (en an-1) des bandes observées dans les spectres de vibration du
bifluorure d’oxygène ... 37
TABLEAU 5 Diagramme de corrélation pour me structure
cristalline possible de OFg solide... 41
TABLEAU 6 Fréquences (en cm-1) des bandes observées dans les spectres infrarouges de OF. en
matrices inertes ... 43
TABLEAU 7 Constantes de force calculées pour SCI... 60
TABLEAU 8 Fréquences (en cm-1) calculées et observées
pour SCI2 en matrice solide de krypton... 61
TABLEAU 9 Contribution relative des constantes de
TABLEAU 10
Page Fréquences (en cm-1) des bandes observées dans les
spectres Raman et infrarouge du bichlorure
de soufre... 65
TABLEAU 11 Comparaison entre les rapports des intensités inté grées des bandes et de SCI. solide et le rapport
des % isotopiques de différentes espèces .... 67
TABLEAU 12 Fréquences (en cm-1) des bandes dans les spectres
de vibration de SO^F^... 77
TABLEAU 13 Fréquences (en cm-1) des bandes dans les spectres de
SECTION 1
1.1 EFFETS DES INTERACTIONS MOLECULAIRES SUR LES SPECTRES DE VIBRATION.
La théorie des groupes de symétrie simplifie considérablement 1'analyse des spectres de vibration des molécules libres. Connaissant la symétrie d'une molécule, il est facile d'en déduire le nombre de vibrations fondamentales, de même que 1'activité de celles-ci. Notons cependant que les transitions vibrationnelles dans les gaz sont généralement accompa gnées de transitions rotationnelles, ce qui complique passablement les spectres.
Lorsqu'on augmente la pression d'un gaz, les forces intermolécu laires prennent de plus en plus d'importance et, du même coup, favorisent les échanges d'énergie entre molécules voisines. Ces perturbations ont généralement pour effet de faire disparaître dans les spectres les raies individuelles associées aux transitions rotationnelles ou aux transitions de vibration-rotation. En passant à l'état liquide, on observe de plus des variations plus ou moins prononcées des fréquences de vibration et des intensités des bandes infrarouges et Raman.
Lors de la solidification, les spectres se modifient encore et se caractérisent généralement par:
3
fréquences,
ii- 11 apparition ou l'éclatement de certaines des bandes dues aux vibrations internes, et
iii- une diminution de la largeur des bandes observées.
Il est clair que l'analyse des spectres de vibration des cristaux ne peut être basée uniquement sur la symétrie de la molécule libre et que d'autres facteurs doivent être pris en considération.
1.2 VIBRATIONS DANS LES CRISTAUX
Une méthode introduite par Bhagavantam et Venkatarayudu (1 - 3) a permis d'établir, pour la première fois, la relation entre la structure cristalline d'un composé et le nombre de bandes de vibration présentes dans les spectres. En bref, on considère alors la maille primitive d'un cris tal comme formant un tout, et on utilise la théorie des groupes pour déter miner la symétrie des vibrations fondamentales et en prédire l'activité optique. Quoique théoriquement simple, cette méthode s'avère difficile d'ap plication dans le cas des cristaux à structure complexe.
Une théorie subséquente, élaborée par Halford (4), a considérable ment simplifié l'analyse des spectres de vibration des solides. Cet auteur a émis l'hypothèse que les mouvements d'une molécule dans un cristal étaient assujettis à la symétrie de son entourage immédiat. Dans un solide cris tallin, une molécule subit souvent des contraintes extérieures qui abais sent sa symétrie originale. Les éléments de symétrie résiduels, caracté ristiques de la symétrie du champ cristallin au niveau de la molécule, constituent alors un nouveau groupe ponctuel nommé groupe de symétrie du site. A partir des représentations irréductibles des vibrations dans ce
dernier groupe, il est possible de déterminer la symétrie de celles-ci, de même que leur activité en Raman et en infrarouge.
Cependant, on s'est vite rendu compte que l'effet du site cristal lin ne pouvait expliquer de façon satisfaisante toutes les bandes obser vées dans les spectres des solides. Homig (5) et Winston et Halford (6) ont alors proposé un modèle qui tient également compte du couplage entre
les vibrations identiques des molécules adjacentes dans les cristaux. Ainsi, pour une maille primitive comprenant N molécules, chaque mode fon damental non-dégénéré de la molécule libre peut se subdiviser en N vibra tions distinctes dans le cristal. Ce prolongement de la théorie a permis de relier la symétrie du cristal à celle de la molécule libre, par l'in termédiaire de la symétrie du site cristallin. En effet, il est toujours possible de faire le lien entre la symétrie des vibrations dans le groupe de la molécule libre, celui du site et, finalement, le groupe facteur caractéristique de la symétrie du cristal. Ceci peut se faire en compa rant les tableaux des caractères de ces divers groupes pour les opérations de symétrie communes. On peut aussi utiliser directement les corrélations qui ont été classifiées pour la plupart des groupes de symétrie (7).
Il est à noter que le groupe de symétrie du site cristallin doit toujours être un sous-groupe de celui de la molécule libre (un groupe étant également considéré comme un sous-groupe de lui-même). De même, le groupe facteur doit être un super-groupe de celui du site. Tous les sous- groupes et super-groupes des 32 groupes de symétrie ont été classifiés (8) Considérons par exemple les molécules qui font le sujet de ce travail. Toutes possèdent une symétrie moléculaire C^^. En phase solide, ces molé cules pourront occuper des sites de symétrie C« , C^, C^ ou C^. Le ta bleau 1 donne la liste des super-groupes associés à chacun de ces groupes
5
TABLEAU 1
Groupes de site et super-groupes possibles pour une molécule de symétrie Super-groupes Nombre de super-groupes C2v Oh' ?d' ?h' D6h’ °3h, C6v, 11 D4h' D2d' C4v' D2h' C2v-Cs °h» Td’ Th’ D6h' D3h’ C6v' C6h' C3h' D3d’ C3v’ D4h’ D2d' 18 C4v' C4h’ D2h’ C2v’ C2h' Cs* C2 °h’ Td' °' ?h' T' B6h' D3h’ C6v’ D6' C6h’ C6’ D3d» 25 C2h' D3' D4h’ D2d’ C4v’ D4’ C4h' S4’ C4' C2-B2h’ C2v’ D2’ C1 °h’ Td’ °' ?h' T' °6h' D3h' C6v’ D6’ C6h’ C3h’ C6’ D3d’ C3v' D3’ S6’ C3’ D4h’ 32 D2d’ C4v' D4’ C4h’ S4’ C4’ D2h’ C2v' °2’ ci> cr C2h’ Cs' C2’
de site.
1.3 EXEMPLE D'APPLICATION: 80% CRISTALLIN
L'analyse des spectres de vibration de SO- cristallin, effectuée par Anderson et Savoie (9), illustre bien l'application de la théorie des groupes dans ce domaine. La molécule SO- est de même symétrie que celles étudiées dans le présent travail, et nous nous en servirons pour expliquer l'emploi des diagrammes de corrélation. Nous en profiterons pour mettre en évidence dans les spectres de ce cristal certains aspects qui ont échappé aux auteurs ci-haut mentionnés.
Une étude par diffraction des rayons X (10) a démontré que la struc ture cristalline de SO^ était décrite par le groupe spatial C^, avec deux molécules par maille primitive, situées sur des sites de symétrie C^. La molécule libre (symétrie C^y) , possède deux modes de vibration de valence, symétrique (v-j,) et antisymétrique (v_) de symétrie A^ et respectivement, et un mode de déformation (A^). D'après le tableau des caractères du groupe ponctuel G^ , tous ces modes doivent être actifs tant en Raman qu'en infrarouge. Prenons comme exemple la vibration de valence symétrique (v^). Cette vibration est de symétrie A^ dans le groupe de symétrie de la molé cule libre (C^y) parce que les déplacements atomiques sont alors symétri ques par rapport à tous les éléments de symétrie de ce groupe (2 plans et un axe d'ordre 2). La symétrie du site dans le cristal est G-. En effet, le seul élément de symétrie ponctuelle qui passe par le centre de gravité des molécules SO- dans le solide est un axe d'ordre deux. Les déplace ments atomiques lors de la vibration v. seront, dans 1'approximation du site, symétriques par rapport à cet axe. La vibration sera donc de symétrie
7
TABLEAU 2
Diagramme de corrélation pour SCL
Modes Gaz Site Groupe facteur Activité
C2v C_2 C2v
(vx, v2, Tz) Al\.
^>A<r
Ax (z, x2, y2, z2) i.r. R.«y
/
â
R.-2
04 Bl\ ^>B<^ (x, xy) i.r. R. <v Rx3 B2^^'^\B2 (y, yx) i.r. R.
A dans le groupe du site (C2), tel qu’indiqué dans le diagramme de corréla tion (tableau 2). Ce diagramme prédit également un éclatement de l'espèce A du site en deux espèces, A^ et A^, dans le groupe facteur caractéristique de la classe cristalline (C^). Physiquement, ceci tient compte du fait que les vibrations dans les deux molécules de la maille primitive sont couplées. Les deux types de mouvements qui peuvent être excités par une radiation électromagnétique correspondent alors aux vibrations (v^) se pro duisant exactement en-phase ou hors-phase dans les deux molécules de la maille. L'effet se répercute évidemment dans tout le cristal.
Les deux composantes de dans le cristal, de symétrie et A^ dans le groupe facteur, doivent être toutes deux actives en Raman alors que seule la première doit donner lieu à une absorption dans 1'infrarouge. En effet, le tableau des caractères du groupe C2V indique qu'un vecteur déplacement z (de même qu'un vecteur Ap_) possède une symétrie A^ dans ce groupe. Inversement, une vibration de symétrie A^ doit être active en infrarouge parce qu'elle donne lieu à une variation de la composante p_ du moment dipolaire. Cette même vibration sera active en Raman par suite d'une variation des composantes o^, ot et du tenseur de polarisabi lité. Les spectres Raman (9) et infrarouge (11) de SO^ cristallin sont qualitativement en accord avec les prédictions faites ici, la vibration apparaissant sous foime de doublet à 1144/1148 cm-1 dans le spectre Raman, alors que seule la première composante a été observée en infrarouge. Les deux autres modes de vibration interne (\^ et v^) dans SCL cristallin peu vent être analysés de la même façon.
Il faut aussi inclure dans le traitement, les modes de libration des molécules dans le cristal. Les trois mouvements de rotation, R^, R et Rz ne peuvent être assimilés à des vibrations dans le cas de la molé cule libre, mais ils se transforment en rotations empêchées (librations) dans le solide, donnant lieu à des bandes dans la région des basses fré quences (région des modes de réseau). On peut prévoir d'après le dia gramme de corrélation que les librations découlant de R . par exemple, doivent donner lieu à un doublet en Raman et en infrarouge, les composantes étant de fréquences identiques dans les deux types de spectres puisqu'elles correspondent aux mêmes vibrations. Quant aux modes de translation empê chée, on les obtient de la même façon, mais il faut en éliminer les trois modes acoustiques, de fréquence nulle, qui correspondent à des
transla-9
lions de l’ensemble de la maille et, par extension, de tout le cristal. Par exemple, la translation T % donne lieu à deux types de mouvement dans le cristal. Dans le premier (symétrie A^), les deux molécules se dépla cent simultanément dans la même direction, suivant l'axe cristallographique z. Il s'agit du mode acoustique, qui ne donnera aucune bande dans les spec tres . Dans le second (symétrie A^), les deux molécules de la maille se déplacent aussi suivant l'axe z, mais leurs mouvements sont déphasés de 180°, de sorte que ces deux molécules vibrent l'une contre l'autre. Cette vibration devrait donner lieu à une bande de basse fréquence dans le spec tre Raman.
Des neuf bandes ainsi prévues dans la région des basses fréquences (librations et translations empêchées) du spectre Raman et des sept bandes prévues en infrarouge, on n'en a observé que trois (9) et sept (11) res pectivement. Ceci n'enlève rien à la valeur du modèle proposé (4 - 6). En fait, les effets de site et de couplage nous permettent de prédire le nombre maximum de bandes actives en Raman et en infrarouge, mais ne nous renseignent aucunement sur l'intensité ou la séparation des différentes bandes observables. Dans le cas de SOL cristallin, il semble que certaines bandes soient superposées ou trop faibles pour être observées.
Le spectre Raman de SOL cristallin, publié par Anderson et Savoie (9), présente certaines anomalies que ces auteurs n'avaient pu expliquer de
façon satisfaisante. En effet, chacune des fondamentales dans ce spectre est compliquée d'un épaulement du côté des hautes fréquences. Nous savons maintenant que ces épaulements s'expliquent par le fait que la fréquence d'une fondamentale Raman dans un cristal varie suivant l'angle entre la di rection du moment dipolaire induit au cours de la vibration et la direction
de propagation de l'onde vibrationnelle dans le solide. Cet effet avait été observé et expliqué théoriquement dans le cas de certains cristaux ioniques (12 - 14). On ne s'attendait cependant pas à ce que de telles anomalies se produisent dans les cristaux moléculaires. On a récemment publié une note sur ce sujet, mentionnant que cette dernière supposition était sans fondement (15) .
La théorie de ce nouvel effet, distinct des effets de site et de couplage, a été expliquée en détail ailleurs (16), et nous nous contente rons d'en indiquer ici les principales conséquences. Dans les cristaux cubiques tout d'abord, une onde vibrationnelle peut se propager de deux façons différentes, selon que la variation du moment dipolaire induit lors de la vibration est perpendiculaire ou parallèle à la direction de propa gation. On se réfère alors respectivement au mode transversal, de fré quence vt, et au mode longitudinal, de fréquence v.. La théorie prévoit dans ce cas deux bandes Raman, celle à plus basse fréquence ayant la fré quence v. et l'autre, la fréquence v^. Le problème est beaucoup plus
complexe dans le cas des cristaux uniaxes (et biaxes), alors que les bandes Raman ont des fréquences variables s'échelonnant entre vt et v^. Il est important de noter que de tels effets ne peuvent être observés dans les spectres Raman que si la vibration excitée est également active en infra rouge. Ceci est seulement possible si le cristal n'est pas centrosymétrique. En infrarouge, on peut dans certains cas observer directement le mode lon gitudinal, mais l'effet le plus commun est celui de la réflexion, qui se traduit sous forme d'un épaulement du côté des hautes fréquences des fon damentales, qui s'étend approximativement jusqu'à v».
11
1.4 MOLECULES ETUDIEES
Dans ce travail, nous analysons les spectres Raman et infrarouges des composés OF^, SClg, 5(^2 et SO2CI2 en phase solide. Notre intérêt pour ces molécules a d'abord été soulevé par l'absence complète de don nées cristallographiques sur ces composés. Même si 1'analyse des spectres de vibration d'un solide ne permet pas en général d'en déduire la struc ture cristalline exacte, il était à prévoir que les renseignements obtenus à partir des spectres Raman et infrarouges de ces composés ne seraient pas négligeables. Notons que ces solides, à cause de leur bas point de fusion, se prêtent mal à une étude par diffraction des rayons X. De plus, il
nous a semblé que l'étude en phase solide de ces composés pourrait per mettre une meilleure attribution des bandes fondamentales dans le cas de SClg, 5(^2 et SOgClg. Enfin, à notre connaissance, les spectres Raman de vibration de OFg dans ses phases condensées n'avaient jamais été étudiés.
On donne dans le tableau 3 quelques propriétés physiques de ces molécules.
Quelques propriétés physiques des différents composés étudiés Composé p.é. (°C) p.f. (°Q densité (g/cm3) couleur du solide Références °F2 -144.8 -223.8 1.90 1.52 (liq. (liq. - 223.8 °C)
- 145.3 °C) jaunâtre teinté brun 17, 18
sci2 59.6 -121 1.62 (liq. 20 °Q orange 19, 20
% -49.7 -121.4 1.7 (liq. 20 °C) blanc 21
SECTION 2
Dans cette section, nous donnerons les caractéristiques des spec- tromètres, de même qu'une description des cellules qui ont rendu possible 11 enregistrement des spectres à basse température. Nous décrirons égale ment les techniques expérimentales employées pour l'étude de OF^ et SCI2 dans des matrices inertes.
2.1 APPAREILS
2.1.1. Spectrophotometre infrarouge
Dans la région de 1'infrarouge moyen (200 - 4000 cm-1), les spectres ont été enregistrés sur un spectrophotomètre à double faisceau et à ré seaux de Perkin-Elmer, modèle 621. Dans chaque région du spectre, la fente mécanique de l'appareil et la vitesse de balayage étaient ajustées de façon à obtenir une résolution de 1 cm-1. L'appareil était calibré à partir des fréquences des bandes de rotation de gaz couramment utilisés com me étalons (23). On estime que la précision sur les fréquences citées plus
loin est de ±1 cm-1 dans le cas des bandes fines.
2.1.2. Interféromètre Fourier
15
au moyen d'un interféromètre de type Michelson, de Research and Industrial Instrument Co., modèle FS - 720. Une calculatrice (Fourier Transform
Computer, modèle FTC - 100/7), couplée à un enregistreur X-Y (modèle HR - 80), transformait les interférogrammes, et les spectres étaient directement im primés en pourcentage de transmission. Dans le domaine de fréquences de
40 - 400 cm-1, la résolution théorique maximum de cet appareil est de 2.5 cm-1, alors qu'elle passe à 1.25 cm-1 dans la région de 20 - 200 cm-1 et à 0.5 cm-1 dans la région de 10 - 100 cm-1. La précision sur la mesure des fréquences est estimée à ±1 cm-1 dans la région de 10 - 200 cm-1 et à ±2 cm-1 dans la région de 200 - 400 cm-1.
2.1.3. Spectromètre Raman
Le spectromètre utilisé pour l'enregistrement des spectres Raman a été décrit ailleurs (24). Cet appareil est muni de deux sources Laser: l'une, à 1'hélium-néon (Spectra Physics, modèle 125), d'une puissance de 75 mW à 6328.15 Â, et l'autre, à l'argon ionisé (Coherent Radiation, mo dèle 52), d'une puissance moyenne de 500 mW pour chacune des raies prin cipales à 5145.25 $ et à 4879.9 R. L'illuminateur a été construit de fa çon à pouvoir y placer les cellules à échantillons dans une position verti cale. L'analyse du faisceau Raman était effectuée par un monochromateur double (Spex, modèle 1400), alors que le détecteur était constitué par une photomultiplicatrice (Star Tracker FW - 130) refroidie à -20 °C par un courant d'azote froid. Le signal était amplifié au moyen d'un picoampère- mètre à courant continu (Victoreen, modèle 1001) et les spectres étaient reproduits sur un enregistreur Yew (modèle LER 12A).
Ce monochromateur a été fréquemment calibré au moyen d'une lampe à décharge au néon. La précision sur la lecture des fréquences est estimée à ±1 cm-1. Les fentes du monochromateur étaient ajustées au besoin, de 10 à 150 microns, ce qui permettait d'obtenir une résolution spectrale va riant entre 0.5 et 3.5 cm-1.
2.2 CELLULES
2.2.1 Injecteur mobile
Pour l'étude en infrarouge de , SCI- et SC^C^ solides, ainsi que celle en matrice inerte de OF^ et SCL^, les échantillons étaient déposés directement sur une fenêtre préalablement refroidie. Le système d'injec tion que nous avons construit, tout en limitant considérablement la perte d'échantillon par condensation sur les parties froides de la cellule, nous permettait d'obtenir des dépôts solides très uniformes.
La cellule était constituée d'une base métallique munie de deux fe nêtres extérieures (Csl,Polythene), situées dans l'axe du faisceau infra rouge. Sur le côté de cette base, un injecteur horizontal, perpendiculaire à l'axe du faisceau, était centré sur la fenêtre de déposition (Csl ou Si). L'injecteur (fig. 1) se composait d'un cône en Teflon (A) placé entre deux tubes de Pyrex concentriques (C). Une section cylindrique de fer doux (D) était fixée à l'une des extrémités du cône. L'action du champ magnétique d'une bobine d'induction (B) sur la partie métallique (D) permettait de faire glisser le cône à la position désirée. La partie supérieure de la figure 1 représente 1'injecteur en position pour déposer. Cette opération terminée, il suffisait de placer le cône en position retirée et de tourner
FIGURES 1 et 2
APPAREILS DE DEPOSITION POUR L'INFRAROUGE
Description:
A. Cône en Teflon
B. Joint verre-métal (Kovar) G. Tubes concentriques en Pyrex D. Section de fer doux
E. Bobine d'induction
F. Support métallique (cuivre) G. Fenêtre centrale (Csl ou Si) H. Base de la cellule (laiton)
I. Réservoir pour le réfrigérant liquide J. Tube de refroidissement
K. Paroi externe de la partie supérieure de la cellule L. (fig. 2) Joints d'étanchéité
M. (fig. 2) Paroi de 1'interféromètre
Note: Dans les schémas qui suivent, le faisceau infrarouge est parallèle à l'axe x.
En position pour déposer
FIGURE 1
H G F FIGURE 2 z*
X'
--- ► y D E '-O (Voir description p. 17)la partie supérieure de la cellule de façon à orienter la fenêtre centrale perpendiculairement au faisceau infrarouge. La section inférieure de la cellule (H) était indépendante, et construite de façon à pouvoir accepter soit une cellule conventionnelle pour les travaux à la température de
l'azote liquide (77 °K), soit une cellule refroidie à l'hélium liquide, qui a déjà été décrite (25). Pour les travaux dans 1'infrarouge lointain, le système de déposition a été adapté à 1'interféromètre de façon à respecter les conditions de vide nécessaires à cet appareil (voir figure 2).
2.2.2 Cryostat pour les études en Raman
Le cryostat utilisé pour nos travaux en spectroscopie Raman se dis tingue de ceux couramment employés, par la construction de la cellule à échantillon et par le mode de refroidissement de cette dernière. La par tie supérieure de ce cryostat est de type conventionnel et utilise de l'hélium liquide comme réfrigérant. Sa partie inférieure est constituée d'un tube de Pyrex de 50 mm fermé à son extrémité par une fenêtre plane du même matériel. A l'intérieur, la cellule à échantillon (en Pyrex), d'un
diamètre extérieur de 7 mm, est soudée dans un autre tube fermé (diamètre extérieur: 20 mm) qui forme l'enceinte de refroidissement. La cellule à échantillon est reliée à 1'extérieur par un tube de Pyrex, pour éviter tout contact de l'échantillon avec des substances métalliques. Le cryostat est décrit en détail dans la figure 3. Le refroidissement de la cellule à échantillon (A) était assuré par de l'hélium (liquide ou gazeux) qui péné trait dans l'enceinte de refroidissement (G) par l'intermédiaire d'un petit tube d'acier inoxydable soudé à la base du réservoir central. Après échange thermique, le gaz était évacué par un conduit métallique, au moyen d'une
FIGURE 3
CRYOSTAT POUR LES ETUDES RAMAN
Description:
A. Cellule à échantillon (tube de Pyrex de 7 mm de diamètre extérieur). B. Ecran thermique (cuivre).
C. Enceinte de refroidissement (tube de Pyrex de 20 mm de diamètre extérieur).
D. Pompe à vide mécanique. E. Robinet micrométrique.
F. Echangeur thermique (serpentin de cuivre). G. Sortie de la résistance au carbone.
IL Conduite d'entrée pour l'échantillon (tube de Pyrex de 7 mm de diamètre extérieur).
I. Support métallique (laiton). J. Résistance au carbone.
Acier inoxydable Laiton Laiton -Kovar Spectromëtre 20 mm 50 mm Faisceau Laser Kovar Pyrex FIGURE 3 (Voir description p. 21)
23
pompe à vide mécanique (D). Un robinet micrométrique (E) permettait de régler le débit d'hélium nécessaire pour maintenir la cellule à la tempé rature désirée. Une résistance au carbone (J) préalablement calibrée, située à l'intérieur de l'enceinte de refroidissement, permettait de mesu rer la température de la cellule. L'échantillon était introduit par le tube en Pyrex (H), puis condensé dans la cellule à échantillon (A). Ce système de refroidissement nous permettait de contrôler la vitesse de cristallisation des échantillons et d'obtenir ainsi des polycristaux de bonne qualité.
Au cours de nos expériences, nous avons constaté qu'il était préfé rable d'ajuster le taux de pompage de façon à ce que l'enceinte de refroi dissement ne contienne pas d'hélium liquide. Le rapport signal/bruit dans les spectres était alors environ dix fois plus grand que lorsque la cellule à échantillon beignait dans le réfrigérant liquide. Les bulles de gaz qui s'échappent de l'hélium liquide causent en effet un bruit de fond appréciable à cause des fortes réflexions qu'elles occasionnent. No tons enfin que la cellule de verre pourrait facilement être remplacée par une de quartz, qui permettrait l'étude de substances réagissant avec le verre (ex: F^).
2.3 MINTAGES POUR L'ETUDE EN INFRAROUGE DES MATRICES SOLIDES
Les propriétés corrosives de OF^ (26) et de SCl^ (27) ne nous ont pas permis d'utiliser la méthode habituellement employée pour la prépa ration des matrices solides. De plus, 1'absence de données sur la ten sion de vapeur de SCI. à différentes températures nous a obligés à uti liser une technique très spéciale dans le cas de ce composé.
2.3.1 Montage pour la préparation des matrices avec 01^.
La pression de vapeur de OF^ à 77.6 °K (P = 1.4 ran Hg) (18) est telle que notre travail sur ce composé a été considérablement simplifié. Cependant, pour éviter toute contamination du produit, le montage décrit à la figure 4 a été employé pour la préparation des différentes matrices OF./M (où M = néon, argon ou krypton).
La vapeur au-dessus de OF^ liquide à -195 °C était d'abord intro duite dans un ballon calibré de 2 litres (C) préalablement évacué. On laissait ensuite échapper ce gaz dans un ballon de 5 litres (G) également évacué. A 1'équilibre, la pression dans les deux ballons était de 0.4 mm Hg. Une jauge de Bourdon (F) placée à l'entrée du ballon de mélange (G) ser vait à mesurer la pression du gaz porteur (néon, argon ou krypton) utilisé pour la préparation des mélanges à différentes concentrations de OF./gaz
rare. Des capillaires calibrés, de différents diamètres intérieurs (H), placés à la sortie du ballon de mélange (G), servaient à maintenir le taux de déposition des mélanges à 0.5 - 1.0 millimoles de OF^/min. environ. Le temps de déposition pour les différents échantillons était d'environ une heure ; ce qui est rapide, comparativement au temps employé généralement pour la condensation des mélanges en matrice inerte. Puisque les spectres n'ont pas révélé de bandes caractéristiques d'agrégats moléculaires, nous
avons jugé préférable de hâter le temps de déposition plutôt que de risquer une décomposition du soluté. On estime que les concentrations des matri
ces obtenues avec ce système sont précises à ±10%.
2.3.2 Montage pour la préparation des matrices avec SCI2
FIGURE 4
MONTAGE POUR OF. EN MATRICES INERTES
Description:
A. Echantillon.
B. Vase de Dewar (azote liquide).
G. et D. Ballons calibrés de 2 litres.
E. Entrée du gaz vecteur (néon, argon ou krypton). F. Jauges de Bourdon.
G. Ballon calibré de 5 litres. H. Capillaires calibrés.
I. Sortie vers la cellule infrarouge. J. Conduite d'évacuation.
(Voir description p. 25)
tsj
FIGURE 5
MONTAGE POUR MATRICE DE SCWKr.
Description:
A. Entrée du gaz vecteur (krypton). B. Robinets micrométriques.
G. Ballon d'expansion. D. Manomètre à mercure.
E. Thermocouple cuivre-constantan. F. Eprouvette pour échantillon (SCl^).
G. Vase de Dewar de 5 litres (azote liquide). H. Sortie vers la cellule infrarouge.
FIGURE 5
(Voir description p. 27)
tx>
29
notamment, du Il nous a donc semblé nécessaire de déposer le mé lange gaz rare/SClg dès sa formation. Ceci a été réalisé au moyen du mon tage décrit à la figure 5. De plus, la pression de vapeur de SClg liquide n'étant pas connue, il nous a fallu procéder de façon empirique pour ob tenir la matrice désirée.
En nous basant sur 1'absorption de SCI2 liquide dans la région de 510 cm-1, nous avons calculé qu'il fallait environ 0.0035 gramme de SCI2 solide pour obtenir des bandes de 50% d'intensité environ. Pour une ma trice Kr/SClg de 700/1 (molaire), il fallait donc utiliser 2.2 grammes de krypton. En se servant des relations pour le calcul du taux d'échappement d'un gaz à travers un orifice (28), nous avons ajusté le robinet micromé trique (B) (fig. 5) de façon à ce que la quantité désirée de krypton soit déposée sur une période d'approximativement 5 heures. Il fallait égale ment que la bonne quantité de SCI2 se mélange au gaz vecteur au cours de cette même période de temps. Pour que cette condition soit remplie, nous avons déterminé par tâtonnement que le SCI2 liquide devait être maintenu à une température de -110 °C.
SPECTRES RAMAN ET INFRAROUGES DE OF_ EN PHASES CONDENSEES
3. SPECTRES RAMAN ET INFRAROUGES DE OF^ EN PHASES CONDENSEES.
3.1 INTRODUCTION
Des études par diffraction des électrons (29) et par spectroscopie de micro-ondes (30, 31), ont démontré que OF^ est une molécule non-linéaire
(symétrie ’ avec distance O-F et angle F-O-F de 1.40 % et 103° respec tivement. Le bifluorure d’oxygène possède trois modes fondamentaux de vi bration : deux modes de vibration totalement symétriques, v-^ (valence) et
\>2 (déformation), de symétrie A^, et un mode de valence antisymétrique, v?, de symétrie (ou ^ dépendant du choix des axes). L'analyse des envelop pes rotationnelles associées aux fondamentales dans le spectre infrarouge du gaz (32 - 34) a permis d’établir que cette molécule est une des rares espèces triatomiques où v, < . Jusqu’à présent, aucun spectre Raman ou infrarouge de OF^ en phases condensées n'a été publié.
Lorsque nous avons entrepris l'étude de cette molécule, notre prin cipal objectif était d'obtenir des informations sur la structure cristal line de ce composé, pour lequel on ne possède encore aucune donnée cristal lographique. L'analyse des spectres de vibration de OF^ nous apparaissait d'autant plus intéressante que cette molécule ne possède aucun isotope sus ceptible de perturber appréciablement les spectres du solide. En effet, 1'espèce isotopique la plus abondante, ^OF^, n'est présente qu'en concen tration négligeable (0.2%). De plus, au cours de ce travail, nous avons
pensé que ce genre de molécule se prêterait bien à l'étude des différents sites de piégeage dans les matrices solides de gaz rares.
3.2 PARTIE EXPERIMENTALE
La méthode employée pour la préparation de OF. a été sensiblement la même que celle décrite par Lebeau et Damiens (35). Le produit a été syn thétisé par la réaction du fluor gazeux (pureté de 98%, Matheson Co. Ltd) avec une solution aqueuse de NaOH (3%) continuellement régénérée. Le gaz produit était ensuite purifié du fluor excédentaire par barbotage dans de
l'eau distillée, et débarrassé de 1’oxygène dissous, par pompage après avoir été condensé dans un piège à 77 °K. La coloration d'une solution de Kl par les bulles d'oxygène dégagé permettait de déterminer l'instant où ce gaz était complètement éliminé. Finalement, le produit résiduel était purifié par distillations successives dans des pièges refroidis à la tempé rature de l'azote liquide. Les impuretés les plus probables, CF. et SiF^, n'ont jamais été détectées dans les spectres infrarouges et Raman des échan tillons que nous avons préparés.
Pour les spectres infrarouges du solide pur et de OF^ en matrice inerte, les échantillons étaient condensés sur une fenêtre en Csl (ou en silicium pour 1'infrarouge lointain) refroidie par un courant d'hélium li quide. Les appareils décrits à la section 2.2.1 (voir figures 1 et 2) ont servi à la déposition des différents échantillons. Les films de solide pur ont été obtenus par évaporation du liquide contenu dans un piège à la tem pérature de l'azote liquide (la pression de vapeur de OF^ à 77 °K est de 1.4 mm Hg (18)). Les dépôts pour les matrices inertes ont été préparés en utilisant le montage décrit à la section 2.3.1 (figure 4). Il s'est
33
avéré difficile d'obtenir les spectres Raman de OF^ solide car ce composé est coloré en phases condensées. Nous n'avons pu obtenir de spectre en uti lisant la raie à 6328 $ d'un laser He-Ne à une puissance de ~ 70 mW. Les raies principales à 5145 Â et 4880 Â d'un laser à l'argon ionisé ont suffi à produire une diffusion Raman relativement faible, d'autant plus que la puissance de la source devait être réduite à ~ 100 mW pour éviter un réchauf fement local de l'échantillon, causé par l'absorption d'une partie de la lumière incidente. Les spectres du solide ont été enregistrés avec la cel lule décrite à la section 2.2.2 (voir figure 3).
3.3 RESULTATS ET DISCUSSION
'f* 3.3.1 Spectre Raman du liquide
Le spectre Raman de OF^ liquide est reproduit à la figure 6 et les fréquences observées, mesurées au maximum d'intensité des bandes, sont énu mérées dans le tableau 4. Pour obtenir ce spectre, un volume de 0.5 ml de liquide était condensé dans une cellule en Pyrex, de 6 mm de diamètre, immergée dans de l'azote liquide dans un vase de Dewar conventionnel (Pyrex) muni de fenêtres planes- à son extrémité. Au cours de nos expériences, nous avons remarqué que le liquide (d'une coloration jaunâtre) devenait légère ment teinté de brun après avoir été soumis à une exposition prolongée au faisceau laser. Cependant, même dans ce cas, nous n'avons pu détecter dans le spectre de bandes caractéristiques d'impuretcs.
^ Au cours de la rédaction de cette thèse, le spectre Raman de OFg liquide a été publié par D.J. Gardiner et J.J. Turner. J. Mol. Spectry. 38, 428 (1971). Leurs résultats, de même que les conclusions qu'ils en tirent, sont pratiquement identiques aux nôtres.
450 460 470 820 830 920 930
FREQUENCE (cm"')
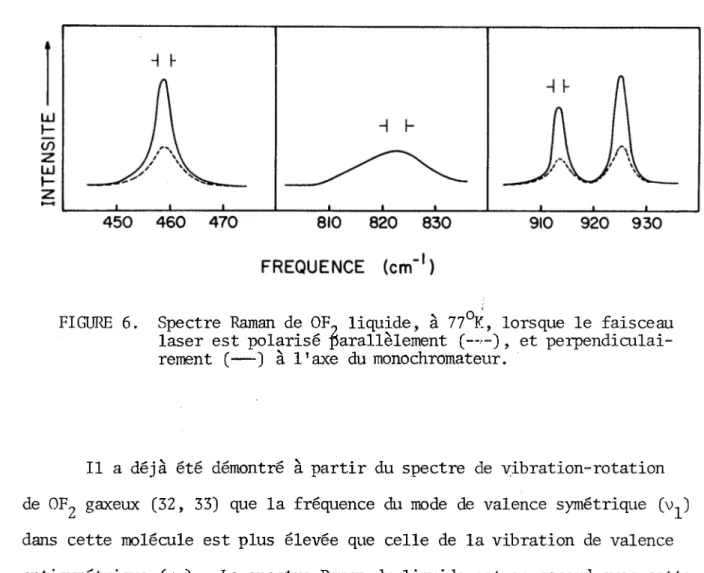
FIGURE 6. Spectre Raman de 0F„ liquide, à 77°K, lorsque le faisceau laser est polarisé parallèlement (--■—) , et perpendiculai rement (—) à l'axe du monochromateur.
Il a déjà été démontré à partir du spectre de vibration-rotation de OFg gaxeux (32, 33) que la fréquence du mode de valence symétrique (\u) dans cette molécule est plus élevée que celle de la vibration de valence antisymétrique (v_). Le spectre Raman du liquide est en accord avec cette attribution: le facteur de dépolarisation mesuré pour la bande à 458 cm-1 (p = 0.48) et le doublet à 913/925 cm-1 (p = 0.35) confirme que ces bandes sont dues à des modes totalement symétriques (A^). Malheureusement, la faible intensité de la bande large à 823 cm-1, attribuée au mode , ne nous a pas permis d'en mesurer la polarisation. Une autre caractéristique in téressante dans ce spectre est le doublet dans la région de v^, causé par résonance de Fermi entre et 2Vg. Cet effet est moins apparent dans le spectre infrarouge du gaz (34, 36), à cause de la présence des bandes dues aux transitions rotationnelles associées à ces deux vibrations.
35
3.3.2 Spectres Raman et infrarouge de OF^ cristallin
Les spectres Raman et infrarouge de OF^ cristallin à ~ 10 °K, sont reproduits à la figure 7 et les fréquences des bandes observées sont énu mérées dans le tableau 4. Des modifications importantes se sont produites dans le spectre infrarouge lorsque les échantillons ont été recuits en éle vant la température aussi près que possible du point de fusion (49.4 °K). Ce phénomène était particulièrement évident dans la région de où la ban de à 823 cm-1 disparaissait complètement, pour faire place à un pic très fin à 812 cm-1 et à une bande très large s'étendant de 815 à 850 cm-1. Dans la région de le processus de la cristallisation se traduisait par un dédoublement de chacune des composantes dues à la résonance de Fermi. Dans le cas des spectres Raman, où les solides polycristallins étaient pro duits par refroidissement contrôlé, nous n'avons observé que des variations mineures de l'intensité des bandes à la suite de recristallisations suc cessives des échantillons.
La caractéristique intéressante du spectre infrarouge de 0F_ cris tallin est la bande très large du côté des hautes fréquences de Notant que l'intensité relative de cette bande augmentait lorsque le faisceau infra rouge pénétrait l'échantillon à un angle différent de 90°, nous avons pensé qu'elle pouvait être due à de la réflexion. Pour vérifier cette hypothèse, nous avons enregistré le spectre de réflexion infrarouge d'un film épais de OF2 solide, en utilisant un montage optique semblable à celui décrit par Bessette et Cabana (37). Nous avons effectivement observé une forte bande de réflexion dans la région de 815 à 850 cm-1 du spectre infrarouge. Dans
IN T E N S IT E
----►
-l
o
g
I/
20 40 60 80 KX) 450 460 800 810 820 830 840 850 900 910 920 930 FREQUENCE (cm-1)Spectres infrarouge (A) et Raman (B) de OFo cristallin à ~10 °K. La ligne pointillée représente le spectre d'un film amorphe.
(a) et (b) identifient un échantillon placé à incidences nor male (90°) et oblique (60°) respectivement. (X5) indique une expansion correspondante de l'intensité de la bande.
37
TABLEAU 4
Fréquences (en cm-1) des bandes observées dans les spectres de vibration du bifluorure d'oxygène.
Gaz Liquide Solide Attribution
Infrarouge Raman Infrarouge Raman
Réf. 34 77 °K ~ 10 °K ~ 10 °K 48 47 56? 54 59 58 Vibrations 66 — de réseau 71 73 77 77 — 82 86 85 456 — 461 458 461 462
Z
812 812" v-7 transversal-
820 831 823 (815-850) 845 V3 longitudinal Réflexion 914 912 ' 913 916 1 et (922) (922) 925 925 2 v-928 925 927Z
sauf pour ce qui est d'une bande très fine à 845 cm-1, dont la fréquence coïncide approximativement avec la limite supérieure de la bande de ré flexion en infrarouge. La grande séparation entre le doublet principal de Vg à 812/820 cm""1 dans le spectre Raman et la bande à 845 cm-1 élimine la possibilité que cette dernière résulte d'un éclatement du mode antisymétrique par effet de couplage. On attribue cette bande à 845 cm-1 au mode longitu dinal associé à la vibration transversale responsable de la bande à 812 cm-1. Savoie et Pézolet (15) ont déjà indiqué que les fréquences longitudinales et transversales pouvaient être séparées de façon appréciable dans les cris taux moléculaires piézoélectriques. La présence d'un mode longitudinal dans le spectre Raman de OF^ solide est une indication certaine que ce cristal est non-centrosymétrique (38). De plus, il semble bien que le so lide soit, par raison de symétrie ou par accident, complètement ou presque complètement isotrope au point de vue optique, puisque la fréquence du mode longitudinal a toujours été observée à 845 cm-1 dans nos spectres Raman, indépendamment des échantillons étudiés. Ceci suggère que OF^ cristallise dans le système cubique. Une telle conclusion est d'autant plus acceptable que les échantillons étudiés étaient tous polycristallins, les microcris taux étant sans doute orientés de façon quelconque par rapport au faisceau laser. Dans ces conditions, si OF_ solide possédait une structure cris talline uni axe (ou biaxe) , on aurait dû observer en Raman une large bande de diffusion entre 812 et 845 cm"1.
Les spectres de vibration de OF_ cristallin donnent aussi des indi cations quant au nombre de molécules par maille primitive dans le cristal. On remarque que toutes les fondamentales présentent au moins deux compo santes , soit dans le spectre infrarouge (v, et v.), ou bien dans le spectre Raman (v_). Un tel dédoublement ne peut se produire qu'au niveau du groupe
39
facteur, puisque la molécule à l'état libre ne possède aucun mode fonda mental dégénéré. Déjà, on peut conclure qu'il y a au moins deux molécules par maille primitive dans le crystal de 0¥^.
Dans la région de 1'infrarouge lointain, le spectre de OF^ cristal lin révèle sept bandes situées entre 40 et 90 cm-1. Chacune de ces bandes correspond à une bande en Raman, de fréquence identique, compte tenu de la précision sur les mesures des fréquences. Bien que le nombre des modes de réseau observés soit inférieur au nombre maximum de neuf prévus (pour des transitions à k=0) si la maille primitive du cristal contient deux molé cules, nous croyons qu'en réalité, ce nombre est encore plus élevé. Par expérience, on sait que tous les modes externes prévus pour une structure cristalline donnée sont rarement observés dans les spectres Raman et infra rouges, sauf lorsque le nombre de bandes prédites est très réduit. Dans le cas présent, par exemple, puisque l'un des moments d'inertie de OF^ est d'environ six fois plus petit que les deux autres (29 - 31), on devrait observer dans le spectre Raman et (ou) dans le spectre infrarouge au moins une bande à ~ 200 cm-1 due au mode de libration de la molécule par rapport à l'axe du plus petit moment d'inertie. Aucune bande n'a été décelée dans cette région. En outre, la présence de sept modes de réseau actifs dans les spectres de vibration d'un cristal avec deux molécules par maille pri mitive serait caractéristique d'une structure cristalline de basse symétrie Or ceci est incompatible avec les conclusions tirées plus haut à partir du mode longitudinal associé à la vibration fondamentale v_.
L'analyse des spectres de vibration d'un composé à l'état cristallin permet généralement d'éliminer un certain nombre de structures cristallines possibles pour ce solide (5, 6). Dans le cas de OF2, nous savons au départ
que les spectres Raman et infrarouge du solide peuvent s'expliquer par au moins 86 combinaisons possibles de groupes de site et de groupes facteurs
(voir section 1.2, tableau 1). La symétrie de site peut être éli minée ici, car, pour tous les super-groupes de ce dernier on prédit un singulet pour les modes de symétrie et Bp tant en Raman qu'en infra rouge. Or on observe un doublet pour les modes v, et Vg en infrarouge et pour le mode en Raman. De même, les molécules ne peuvent occuper un
site de symétrie ^ dans ce cristal, car pour toutes les corrélations avec les super-groupes de ce site on prévoit un singulet en infrarouge pour
et \)_. De la meme façon, on peut déduire que la structure cristalline de OFg ne peut être monoclinique, triclinique ou trigonale si la symétrie du site est C , et qu'elle ne peut être triclinique si le site est de symétrie Cp
Il nous est impossible de poursuivre plus loin notre analyse à par tir de ces seules données expérimentales. Cependant, si le cristal possède une structure cubique, comme le suggère la présence d'un mode lon gitudinal dans le spectre Raman, la combinaison d'un site de symétrie et d'un groupe-facteur pourrait très bien expliquer les spectres de vibration de OF^ cristallin. Une telle combinaison (voir tableau 5) prédit dans le spectre infrarouge huit modes de réseau, des doublets pour
et Vp et un singulet ou un doublet pour (dépendant avec quel plan de symétrie de la molécule, coincide celui du site). Ce type de struc ture, avec 12 molécules par maille primitive (8), devrait cependant se caractériser par un spectre Raman assez complexe : 17 bandes sont prévues dans la région des basses fréquences, alors que 4 bandes devraient être
41
TABLEAU 5
Diagramme de corrélation pour une structure cristalline possible de OF. solide.
Mode Gaz Site G. Facteur Activité
*
\>2 peut aussi être de symétrie B^.
observées pour les modes et , et un doublet ou un quadruplet dans la région de excluant les modes longitudinaux possibles.
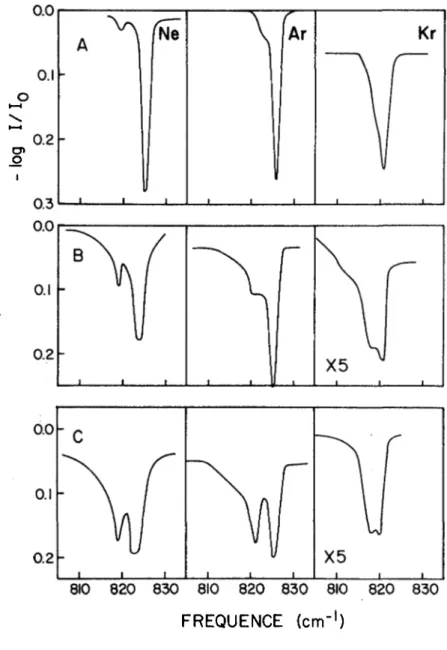
3.3.3 Spectres infrarouges de OF. en matrices inertes.
Los spectres infrarouges, dans la région de , de OF. isole dans des matrices de Ne, Ar et Kr, dans un rapport de concentration de 1:1000, sont reproduits à la figure 8. Les fréquences observées après plusieurs cycles de recuits , sont données au tableau 6. On constate que la vibration
-lo g
I/
810 820 830 810 820 830 810 820 830 FREQUENCE (cm"1)FIGURE 8. Spectres infrarouges, à ~10°K, de 0F2 isolé dans des matrices de gaz rares dans aes rapports de concentration molaire de 1:1000,
(A) avant, et (B,C) après recuits (X5) indique une expansion correspondante de l'intensité de la bande.
43
TABLEAU 6
Fréquences (en cm x) des bandes observées dans les spectres infrarouges de 0F_ en matrices inertes.
Mode OF /Ne 1/1000 OF2/Ar i/iûoo OTkT OF2/Kr 1/1000 Vv 819 821 820 818 O 823 825 825 820 (900) 911 (910) v. 919
_
1 924 922de valence antisymétrique v_, a un comportement à peu près identique dans chacune de ces matrices. Un dépôt brut du mélange gazeux à ~ 10 °K pro
duit d'abord un singulet (à 825 cm-1 dans le néon et l'argon, et à ~ 820 cm-1 dans le krypton), avec des épaulements à ~ 821 et ~ 818 cm-1 dans les ma trices d'Ar et de Kr respectivement et une bande faible, mais bien résolue, à 819 cm-1 dans le cas du néon. Sous l'effet de recuits successifs de ces matrices par élévation de la température à ~ 20 °K pour le Ne, ~ 35 °K pour l'argon, et ~ 50 °K pour le krypton et par refroidissement subséquent à ~ 10 °K, on remarque que la bande de basse fréquence croît graduellement, en intensité pour devenir presque aussi intense que le pic principal. Ces transformations s'effectuent sans variation de fréquences, sauf dans le cas de la matrice de néon où la fréquence du pic principal est abaissée
d'en-viron 2 cm 1.
Un tel comportement peut s'expliquer soit par la formation d'agrégats moléculaires, soit par l'existence de différents sites de piégeage dans ces matrices. Nous avons opté pour la dernière hypothèse puisque, tant sur le plan théorique qu'expérimental, l'agglomération des molécules de 0F_ dans de telles matrices est improbable. Pimentel et Charles (39) estiment que 1'aggregation en quantité appréciable des molécules de soluté dans les ma trices solides est possible pour des rapports de concentrations (molaires) inférieur à 1:300. On peut exclure un tel effet dans le cas présent (rap port OFg: matrice = 1:1000), d'autant plus que les molécules de OF^ ne donnent probablement pas lieu à de fortes interactions moléculaires, con trairement au cas de molécules telles que NO^, qui peuvent facilement for mer des dimères (40). De plus, si la composante du côté des basses fré quences du doublet observé pour était due à un phénomène d'aggloméra tion, on devrait s'attendre à ce que cette bande soit plus intense à des concentrations plus élevées de OF^. En fait, dans le spectre d'une matrice de 0F_ dans de l'argon à une concentration de 1:400 (fig. 9), la bande à 821 cm-1 est plus intense que celle à 825 cm-1. Cependant, lorsque la matrice est soumise à des recuits successifs, la bande à 825 cm-1 ga gne de l'intensité au dépens de celle à plus basse fréquence, jusqu'à ce que les deux composantes possèdent approximativement la meme intensité. Ceci suggère l'existence de différents sites de piégeage dans l'argon so
lide (de meme que dans le néon et le krypton), les molécules de OF^ ten dant à se distribuer dans ces différents sites lors du recuit de la matrice.
Dans plusieurs occasions, on a fait appel à l'existence de différents sites de piégeage pour expliquer le dédoublement de certaines bandes dans
45
FREQUENCE (cm-1)
FIGURE 9. Evolution de la bande d'absorption infrarouge dans une matrice OF2:Ar = 1:400, à la suite de recuits successifs.
les spectres infrarouges de molécules isolées dans des solides. Un exemple typique est celui de CIF^ isolé dans une matrice solide d'argon dans un rapport de concentration de 1:2000 (41), alors que la vibration v., des
deux espèces ^CIF^ et 37C1F^ donne lieu à un doublet dans chaque cas. Des études par résonnance de spin électronique de certaines espèces isolées, telles NO2 (42) et PF
g
(43) , ont également démontré 11 existence de plusieurs sites de piégeage dans les matrices de gaz rares. Cependant, la nature exacte de ces différents sites est difficile à concevoir. En première approxima tion, vu l'empilement moléculaire dans les cristaux de gaz rares (44) et les rayons de Van der Waals de ces molécules, on peut calculer qu'une molécule de OF2 peut s'insérer assez facilement, du moins pour l'argon et le krypton, dans des sites de substitution dans ces matrices. Quant à l'autre (ou aux autres) type de site, il peut être interstitiel, quoiqu'on peut alors s'at tendre à une déformation locale considérable de la matrice.Il est improbable que le dédoublement de v_ dans nos spectres résulte de la rotation, soit complètement libre, soit restreinte à des mouvements autour de l'axe du plus petit moment d'inertie, des molécules de OFg dans ces matrices. La rotation libre serait facilement identifiable par la for me caractéristique des bandes de vibration-rotation dans les spectres.
D'autre part, il est tout à fait possible qu'une rotation de la molécule au tour de l'axe du plus petit moment d'inertie puisse se produire dans les matrices d'argon et de krypton, quoique ceci ne devrait pas causer de dé doublement aussi important que celui que nous avons observé pour \)_. Une telle rotation restreinte explique apparemment la présence d'une bande, dont l'intensité varie avec la température, à environ 0.8 cm-1 du pic prin cipal de Vj dans les spectres de SO2 et NOg isolés dans des matrices de Kr
(45). Soulignons que cette bande ne peut être isolée que sous une haute résolution (0.3 cm-1). Il est possible qu'un phénomène semblable se pro duise dans une solution de OF2 en matrice solide, mais qu'il n'ait pas été décelé par manque d'une résolution spectrale adéquate.
47
900
910
920
900 S
FREQUENCE (cm"1)
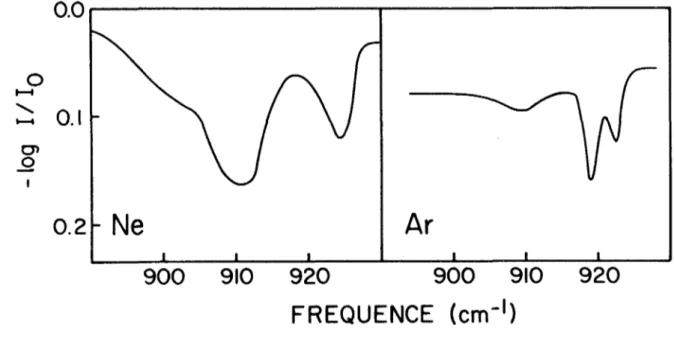
FIGURE 10. Spectre infrarouge d'une matrice recuite de OF2 :Ne = 1:1000, et C^rAr = 1:400 dans la région de v^.
Même avec des films très épais, aucune absorption appréciable n'a été décelée dans la région du mode de déformation (x^) de OF^ dans les ma trices de Kr et Ar à une concentration molaire de 1:1000. De même, nous n'avons pu observer de bande correspondant au mode v^. Il faut dire ce pendant qu'avec les films très épais, 1'opacité de la matrice, augmentait
considérablement le temps de réponse de l'appareil. Cependant, dans une matrice de néon à la même concentration, nous avons observé deux bandes assez larges à 911 et 924 cm-1 (fig. 10). Ce dédoublement est sans doute causé par la résonance de Fermi entre et 2 v?, comme dans les spectres du compose pur. Un tel effet n'a pas etc observé dans une matrice d'argon, à une concentration de 1:400, (fig. 10), à cause probablement d'un déplace ment de la fréquence de v. dans cette matrice. Notons que, dans ce dernier
les deux composantes de v^, lorsque la matrice est recuite.
Notre spectre de OFg isolé dans l'argon diffère passablement de celui que Arkell et ses collaborateurs (46) ont obtenu, à une concentra tion molaire de 1:40, dans leur étude visant à isoler le radical GF. Leur spectre révèle un doublet bien résolu à 915/925 cm-1, lequel est tout à fait semblable à celui que nous avons observé dans le cas de films amorphes de GF2 solide. Il est donc très probable qu'une agglomération appréciable des molécules OF^ se soit produite au cours de leurs expériences, bien que ceci ne signifie pas nécessairement que la bande attribuée au radical OF soit due à autre chose.
SECTION 4
SPECTRES RAMAN ET INFRAROUGES DU BICTILORURE DE SOUFRE (SC12)
4.1 INTRODUCTION
Des etudes par diffraction des electrons (47, 48) et par spectrosco- pie Raman (49) et infrarouge (50) ont démontré que la molécule de bichlo- rure de soufre est de symétrie C^v- Récemment, une étude par micro-ondes
(51) a permis de spécifier les paramètres géométriques de la molécule: les distances S-Cl sont de 2.014 ± 0.004 Â, et l'angle Cl-S-Cl est de 102.8° ± 0.2°. A notre connaissance, aucune étude par spectroscopie ou par dif fraction des rayons X n'a été faite sur SClg en phase solide.
Au début, notre principal objectif était d'obtenir des informations sur la structure cristalline de SCl^, afin de déterminer si cette structure est semblable à celle de OF. cristallin. Nous avons cependant constaté en étudiant les travaux déjà publiés sur les phases vapeur et liquide de ce
composé, qu'il existait encore une certaine ambiguïté dans l'attribution des fondamentales et v^. Les résultats obtenus par spectroscopie infrarouge
(50) laissaient supposer que SC^ pouvait appartenir à cette catégorie de molécules triatomiques pour lesquelles (ex. 0?, OF.). Nous avons donc jugé nécessaire de reprendre le spectre infrarouge du gaz et celui en Raman du liquide, à plus haute résolution, et de compléter nos travaux par l'étude de SCI. isolé dans une matrice inerte. A maintes occasions, cette
dernière technique s'est avérée efficace pour séparer des bandes qui sont superposées en phases gazeuse et liquide.
51
4.2 PARTIE EXPERIMENTALE
La méthode employée pour la purification du bichlorure de soufre, était essentiellement la même que celle suggérée par Rosser et Whitt (52). Environ 200 ml de SC^ (Technical Grade, de Matheson, Colemann and Bell) étaient distillés à reflux en présence de 4 ml de PCl^ pendant environ 6 heures, avec une colonne (en Pyrex) de 60 cm de long, remplie de billes de verre. Une première fraction du distillât était recueillie dans un ballon contenant quelques gouttes de PCl^, et on répétait le processus de distil lation jusqu'à ce que le point d'ébullition du liquide demeure constant à ~ 59.5°C. Le produit était ensuite recueilli dans un piège préalablement évacué et refroidi à la température de l'azote liquide, afin d'éviter toute dissociation du SCl^. Chaque échantillon étudié était préalablement distil lé sous vide et recueilli directement dans la cellule appropriée, en main tenant le piège principal à ~ 170°K. Nous estimons que la concentration des impuretés dans le produit final ne dépassait pas, dans chaque cas, 1%.
Les spectres Raman du liquide et du solide ont été enregistrés à l'aide du spectromètre décrit à la section 2.1.3. L'effet Raman a dû être excité à partir de la raie à 6328 % d'un laser He-Ne, car le bichlorure de soufre est coloré (le liquide est rouge foncé) et ne devient complètement transparent qu'au-dessus de 7000 X (49). Le cryostat employé pour les étu des à basse température était le même que celui décrit par Savoie et
Pézolct (53). Le vase de Dcwar contenant la cellule à échantillon était place verticalement, étant donné la géométrie de la chambre à illumination
à 1’entrée du spectromètre.
Pour le spectre infrarouge du gaz, nous avons utilise une cellule conventionnelle de 10 cm de parcours optique, munie de fenêtres en chlo rure d'argent. Les films du solide pur ont été déposés, à l'aide du mon tage décrit à la section 2.2.1 (voir figures 1 et 2), sur une fenêtre en AgCl ou en silicium refroidie par un courant d'azote liquide. Tous les échantillons étudiés ont été recuits en élevant la température aussi près que possible du point de fusion. Enfin, le montage pour l'étude de SCl^ isolé dans une matrice solide de krypton a été décrit à la section 2.3.2 (fig. 5), alors que la cellule utilisée était la même que celle employée dans nos travaux sur OF^.
4.3 RESULTATS ET DISCUSSION
4.3.1 Spectre infrarouge du gaz.
Les constantes rotationnelles de 32S35Cl2 (A = 0.4866 cm""1,
B = 0.0973 cm-1 et G = 0.0811 cm-1) et le paramètre d'asymétrie k = -0.9201, calculés à partir des spectres micro-ondes (51), indiquent qu'en première approximation la molécule de bichlorure de soufre est une toupie symétrique allongée. Dans ces conditions, le spectre de SC^ à l'état de vapeur de vrait se caractériser par des bandes de vibration-rotation de types paral lèle pour v_ (type A) et perpendiculaire pour et v^ (type B). Sous fai ble résolution, les spectres infrarouges de SCI. gazeux et liquide (50) présentent une bande de forte absorption à 525 et 514 cm""1 respectivement. En se basant sur le spectre Raman du liquide (49), Barrow (50) attribue ces bandes au mode de valence symétrique v^.
-L
o
g
(
I
/
I
0
) 53FREQUENCE (cm'1)
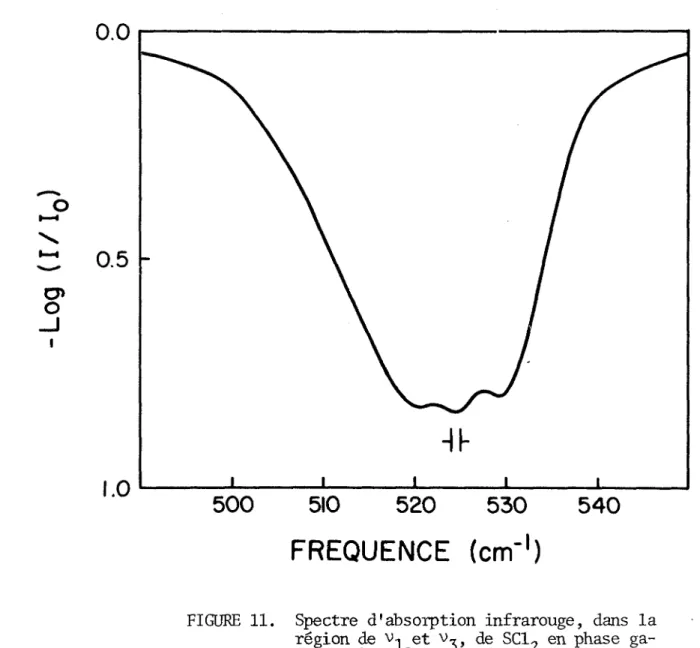
FIGURE 11. Spectre d'absorption infrarouge, dans la région de Vi et v~ de SCI- en phase ga zeuse, à 25°C.
Généralement, en phase vapeur, le mode antisymétrique des molécules triatomiques (0^, NO-, OF^, H^O, Cl^O, SO^) se caractérise par une absorption infrarouge beaucoup plus intense que celle due au mode de valence symétrique (54). Le spectre infrarouge d'un échantillon gazeux de SCI- fraîchement distillé, enregistré avec une résolution de 1 cm-1, est représenté à la fi gure 11. A première vue, ce spectre semble révéler la présence des branches
P, Q et R de à 520, 525 et 530 cm-1 respectivement. La séparation des branches P-R (10 cm-1) serait cependant en désaccord avec la séparation mi nimale (15.3 cm-1) calculée à partir des moments d'inertie (55) d'après la relation
1/2 Av = S(|3)[kT/B' ] /TT
où B' est la moyenne des constantes rotationnelles B et C, et la fonction S(g) est prise à sa valeur minimale de 1.0. Il semble que nous soyons plutôt en présence d'une bande hybride ayant pour caractère dominant celui d'une bande parallèle (type A). La proximité de et v_ favorise le recouvrement des branches P et R associées au mode de valence symétrique et des branches P, Q et R caractéristiques du mode antisymétrique, de sorte qu'il est im possible de séparer les contours particuliers de chacune de ces fondamentales.
4.3.2 Spectres Raman du liquide
Nous avons enregistré le spectre Raman de SCI2 liquide à diverses températures (25°C (fig. 12), -20°C, -50°C et -90°C), dans l'espoir de pou voir séparer les modes v^ et Vy Abstraction faite de la diminution de la largeur des bandes lors de l'abaissement de la température, les formes de celles-ci sont demeurées essentiellement identiques à celles à 25°C.
L'asymétrie du côté des basses fréquences de la bande à 519 cm-1 peut être causée soit par la superposition do et Vy soit par les vibra tions Vj des différentes espèces isotopiques. Dans la première hypothèse, on devrait observer dans le spectre Raman en lumière polarisée du liquide un déplacement apparent de la fréquence au maximum d'intensité, puisque les deux modes fondamentaux, de symétries distinctes (v^ (A^), v, (B^)),
de-55
FREQUENCE (cm-1)
FIGURE 12. Spectre Raman de SCI2 liquide, à 25°C, avec le faisceau laser polarisé parallèlement (—), et perpendiculairement (--- ) à l'axe du monochromateur.
vraient alors exhiber un comportement différent. Lorsqu'on change le plan de polarisation du faisceau laser, le maximum de la bande se déplace effec tivement de 519 à 516 cm-1, ce qui pourrait laisser supposer que > v,. Ce décalage très faible ne constitue pas une preuve de la présence de deux bandes dans cette région. La seconde hypothèse semble plus réaliste pour deux raisons: (i) la bande observée en lumière polarisée est également asy métrique, et (ii) le facteur de dépolarisation mesurée (p = 0.17) est ce lui attendu pour une vibration totalement symétrique.
En résumé, la bande Raman vers 519 cm-1 est due en presque totalité à la vibration et son asymétrie est causée par des espèces isotopiques, notamment 32S35C137C1. La bande due à également présente dans cette région est trop faible pour être isolée. Il est même possible que ces deux vibrations soient mélangées dans le liquide à cause de 1'anisotropie loca le dans ce milieu. Mentionnons que, même dans les meilleures conditions, il nous a été impossible de déceler la bande observée à 535 cm-1 par Stammreich et ses collaborateurs (49) et attribuée à v_. Il est probable que cette bande résulte de la présence de S^Clg dans le liquide suite à une décomposition partielle du SClg.
Au cours de notre étude, nous avons porté une attention spéciale à un phénomène assez curieux qui se produit lorsqu'on abaisse graduellement la température de SCIg liquide. A ~ -20°C, on constate en effet que le li quide se trouble, prenant l'aspect d'une suspension de couleur orange qui devient de plus en plus dense lorsque la température est abaissée jusqu'au point de solidification. Beckman (56) a expliqué ce comportement par une polymérisation ou par la formation de SCl^. Mondain-Monval (27) l'explique soit par des impuretés dans SC^, soit par une décomposition spontanée de ce dernier en monochlorure et en chlore. Enfin, plus récemment, Whiting (20), à la suite d'une étude systématique sur les points de fusion de différents mélanges de SCI2 et S2CI2, propose la formation d'un composé intermédiaire de formule chimique S^Cl^. Nos spectres Raman de SCI2 liquide entre 25°C et -90°C ne présentent aucune nouvelle bande qui pourrait laisser supposer la formation de polymères ou autres espèces chimiques. En particulier, la bande la plus intense de S^Clg (Vj) et celle caractéristique de Clg n'ont pu être décelées dans nos spectres. Il semble donc que la phase d'aspect