The Effect of Resveratrol on the Hyperproliferation of Vascular Smooth Muscle Cells from Spontaneously Hypertensive Rats: Molecular Mechanisms
Par
Sara Salem Almajdoob
Département de pharmacologie et physiologie Faculté de Médecine
Mémoire presenté à la Faculté de Médecine en vue de l’obtention du grade de
Maîtrise en Sciences (M.Sc.) en physiologie moléculaire, cellulaire et intégrative
Juin 2017
Faculté de Médecine
Ce mémoire intitulé:
The Effect of Resveratrol on the Hyperproliferation of Vascular Smooth Muscle Cells from Spontaneously Hypertensive Rats: Molecular Mechanisms
Présenté par Sara Salem Almajdoob
A été évalué par un jury composé des personnes suivantes : Dr. Réjean Couture (président-rapporteur)
Dr. Madhu B. Anand-Srivastava (directrice de recherche) Dr. Puttaswamy Manjunath (membre du jury)
Résumé:
Le remodelage vasculaire dû à l'hyperprolifération et à l'hypertrophie des cellules musculaires lisses vasculaires (VSMCs, par son abréviation en anglais) est central dans le développement de l'hypertension. Les VSMCs provenant de rats spontanément hypertendus (SHR, par son abréviation en anglais) présentent une hyperprolifération et une surexpression des protéines du cycle cellulaire. Le resvératrol est un composé polyphénolique naturel trouvé dans la peau des raisins et il est impliqué dans plusieurs effets vasoprotecteurs. Le resvératrol a également été signalé pour son effet atténuateur de la prolifération de VSMCs induite par l'angiotensine II. Cependant, il n'a pas été élucidé si le resvératrol pouvait également inhiber l'hyperprolifération des VSMCs chez les SHR. La présente étude a été entreprise pour déterminer si le resvératrol pouvait atténuer l'hyperprolifération des VSMCs et si oui, par quels mécanismes moléculaires. Méthodes: Pour cette étude, nous avons utilisé des VSMCs aortiques de rats SHR âgés de 14 semaines et de rats Wistar-Kyoto (WKY). La prolifération des VSMCs a été déterminée par la mesure de l’incorporation de la thymidine tritiée ([3H] thymidine) et l’expression des protéines a été mesurée par immunobuvardage de type Western. Résultats: Les VSMCs de SHR présentent une hyperprolifération atténuée par le resvératrol. La surexpression de la cycline D1, de la cycline E, de la kinase 4 dépendante de la cycline (CDK4), de la kinase 2 dépendante de la cycline (CDK2), de la protéine de rétinoblastome phosphorylée (pRb), de la Giα-3, de la Giα-2 et de la phosphorylation accrue de ERK1/2 (de l’anglais extracellular regulated kinase1/2) et AKT dans les VSMCs des SHR a été corrigée par le resvératrol. De plus, le resvératrol a également inhibé l'augmentation de l'anion superoxyde, l'activité de la NADPH (de l’anglais nicotinamide adenine dinucleotide phosphate) oxydase, la surexpression des protéines NADPH oxydase 2 (NOX2)/NADPH oxydase 4 (NOX4) et P47 phox, et la phosphorylation accrue de l'EGF-R (de l’anglais epidermal growth factor), IGF-1R (de l’anglais insulin-like growth factor 1) et c-Src aux niveaux témoins. Ces résultats suggèrent que le resvératrol, par l'inhibition des espèces réactives oxygénées (ROS) et des ROS qui induisent la transactivation des récepteurs des facteurs de croissance, c-Src et MAPK (de l’anglais mitogen-activated protein kinases)/PI3K (de l’anglais Phosphoinositide-3 kinase), réduit la surexpression de Giα et des protéines du cycle cellulaire et contribue à l'atténuation de l'hyperprolifération des VSMCs des SHR.
Mots clés: Resveratrol, prolifération VSMCs, protéines du cycle cellulaire, MAPK, PI3K, protéines Giα, stress oxydatif, SHR.
ABSTRACT
Vascular remodeling due to the hyperproliferation and hypertrophy of vascular smooth muscle cells (VSMCs) is central in the development of hypertension. VSMCs from spontaneously hypertensive rats (SHR) exhibit hyperproliferation and overexpression of cell cycle proteins. Resveratrol, a natural polyphenolic compound found in the skin of grapes, is implicated in several vasoprotective effects. Resveratrol has also been reported to attenuate angiotensin II-induced VSMCs proliferation. However, it was not elucidated if resveratrol could also inhibit the hyperproliferation of VSMCs from SHR. The present study was undertaken to investigate if resveratrol could attenuate the hyperproliferation of VSMCs and explore the underlying molecular mechanisms responsible for this effect. Methods: For this study, aortic VSMCs from 14-week-old SHR and Wistar-Kyoto (WKY) rats were used. The proliferation of VSMCs was determined by [3H] thymidine incorporation and the levels of proteins were determined by Western blotting techniques. Results: VSMCs from SHR exhibit a hyperproliferation which was attenuated by resveratrol. The overexpression of cyclin D1, cyclin E, cyclin dependent kinase 4 (CDK4), cyclin dependent kinase 2 (CDK2), phosphorylated retinoblastoma protein (pRb), Giα-3, Giα-2 proteins and enhanced phosphorylation of extracellular signal-regulated kinases 1/2 (ERK1/2) and AKT in VSMCs from SHR were attenuated by resveratrol. Furthermore, resveratrol also inhibited the increase of the superoxide anion,nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, overexpression of NADPH oxidase 2 (NOX2)/NADPH oxidase 4 (NOX4) and P47phox proteins, and the increased phosphorylation of epidermal growth factor receptor (EGF-R), insulin-like growth factor 1 receptor (IGF-1R), and c-Src to control levels. These results suggest that resveratrol through the inhibition of reactive oxygen species (ROS) and ROS-mediated transactivation of growth factor receptors, c-Src, mitogen-activated protein kinases (MAPK)/Phosphoinositide-3 kinase (PI3K), attenuates the overexpression of Giα and cell cycle proteins and results in the attenuation of hyperproliferation of VSMCs from SHR.
Key words: Resveratrol, VSMCs proliferation, cell cycle proteins, MAPK, PI3K, Giα proteins, oxidative stress, SHR.
Table of contents
Résumé...iii
Abstract ...v
Table of Contents ...vi
List of Figures ...ix
List of Abbreviations...xi
Dedication...xvi
Acknowledgements...xvii
CHAPTER 1- Introduction and Literature Review...1
1. Hypertension...2
1.1. Classification of Hypertension ...2
2. Blood Pressure ...3
2.1 Blood Pressure Regulation (Hemodynamics)………...3
2.2 Mechanisms of Blood Pressure Regulation ...4
2.2.1 Short Term Regulation ...4
2.2.1.1 Neural Mechanisms...5
2.2.1.2 Humoral Mechanisms ...7
2.2.2 Long Term Regulation ...7
2.2.2.1 Renin Angiotensin Aldosterone System (RAAS)………..8
2.2.2.1.1 Tissue RAAS ...8
2.3 Vascular Structure...9
3. Vascular Remodeling ...10
3.1 Vascular Remodeling in Hypertension ...11
3.1.2 SHR as a Model of Hypertension and Vascular Remodeling ...12
4. Molecular Mechanisms Implicated in Hypertension and Vascular Remodeling...13
4.1 Oxidative Stress and Reactive Oxygen Species (ROS)...13
4.1.1 Sources of ROS ……...13
4.1.2 NADPH oxidase ...14
4.1.3 The Vascular NADPH Oxidase………..…...16
4.1.4 Implication of Oxidative Stress in Hypertension………...17
4.1.5 Implication of Oxidative Stress in Cell Proliferation………....17
4.2 c-Src Pathway……….18
4.3 Growth Factor Receptors Signaling……….………...19
4.3.1 Epidermal Growth Factor Receptor (EGF-R)………...……19
4.3.2 Insulin-like growth factor receptor (IGFR)………...20
4.3.3 Platelet-Derived Growth Factor Receptor (PDGFR)………21
4.4 Phosphatidylinositol-3-kinase (PI3K) Pathway………..22
4.4.1 AKT Pathway………22
4.5 MAP Kinase Pathway……….23
4.6 G-protein Signaling……….24
4.6.1 G Protein-Coupled Receptors (GPCRs)……….…...24
4.6.2 Guanine Nucleotide-Binding Proteins (G-protein)………...25
4.6.2.1 Activation of Heterotrimeric G-proteins………...26
4.6.2.2 Classification of G-proteins………..27
4.6.2.3 The Giα proteins and Hypertension ……….27
5. Cell Cycle………...28
5.1.1 Regulation of G1-Phase……….30
5.1.2 Cell Cycle Progression………...31
5.2 Signaling Pathways Involved in the Cell Cycle….……….31
5.3 Cell Cycle and Hypertension………..……32
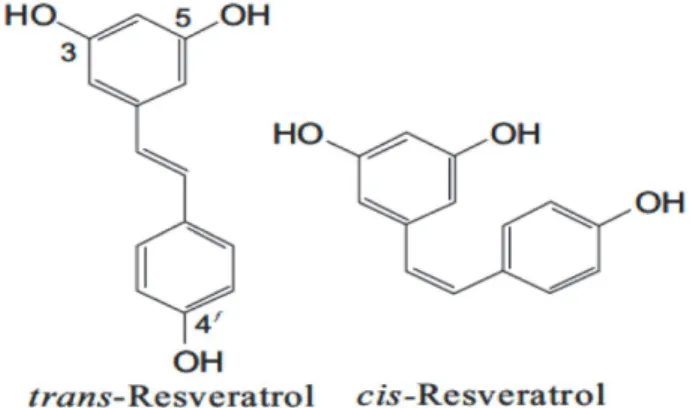
6. Resveratrol………33
6.1 Occurrence and Synthesis………. .33
6.2 Resveratrol -The French Paradox.………..34
6.3 Resveratrol Bioavailability ………....35 6.4 Resveratrol Effects …….………35 6.4.1 Antihypertensive Effects ………36 6.4.2 Antioxidant Effects ………37 6.4.3 Antihypertrophic Effects………37 6.4.4 Antiproliferative Effects ………..…..38
7. Hypothesis and Objectives……….………39
CHAPTER 2 Scientific Article ...41
CHAPTER 3 Discussion, Conclusions and Future Work ...77
8. Discussion ...78
9. Conclusions ...83
10. Future Work ...85
List of Figures Chapter 1
Figure 1: The structure of the arterial wall………10
Figure 2: Simplified scheme showing key steps in the production of reactive oxygen species (ROS)……...………..14
Figure 3: Structure of NADPH oxidase….………16
Figure 4: MAPK signaling following activation of receptor tyrosine kinase (RTKs) and G-protein coupled receptor (GPCR)………..24
Figure 5: Schematic diagram of the G-protein-coupled receptor (GPCR)…….………...25
Figure 6: G-protein-coupled receptors (GPCR)-mediated G-protein activation………...26
Figure 7: The cell cycle and its regulation by cyclin/cdk complexes….………...31
Figure 8: Chemical structures of trans-resveratrol and cis-resveratrol………..…34
Chapter 2 Figure 1: Effect of resveratrol on thymidine incorporation of VSMCs from 14-week-old SHR and age-matched WKY rats………...…68
Figure 2: Effect of resveratrol on the expression of cell cycle proteins in VSMCs from 14-week-old SHR and age-matched WKY rats………..…69
Figure 3: Effect of resveratrol on the expression of phosphorylated retinoblastoma protein (pRb) and retinoblastoma protein (Rb) in VSMCs from 14-week-old SHR and age-matched WKY rats………...70
Figure 4: Effect of resveratrol on the expression of Giα-2 and Giα-3 proteins in VSMCs from 14-week-old SHR and age-matched WKY rats………,………71
Figure 5: Effect of resveratrol on phosphorylation of ERK (A) and AKT (B) expression in VSMCs from 14-week-old SHR and age-matched WKY rats……….….72
Figure 6: Effect of resveratrol on EGF-R and IGF-1R phosphorylation and expression in VSMCs from 14-week-old SHR and age-matched WKY rats………..73
Figure 7: Effect of resveratrol on c-Src phosphorylation in VSMCs from 14-week-old SHR and age-matched WKY rats………...74
Figure 8: Effect of resveratrol on superoxide anion (O2-) production and NADPH oxidase activity in VSMCs from 14-week-old SHR and age-WKY rats………...….75 Figure 9: Effect of resveratrol on the levels of p47phox, Nox4 and Nox2 proteins in VSMCs from 14-week-old SHR and age-matched WKY rats………....76 Chapter 3
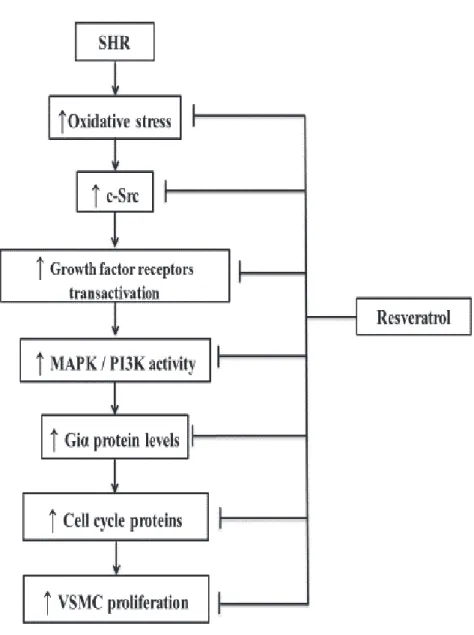
Figure 1: Schematic diagram summarizing the effect of resveratrol on hyperproliferation of VSMCs from SHR and the implicated molecular mechanisms………...………..84
List of Abbreviations -A-
AC Adenylyl cyclase AKT/PKB Protein kinase B Ang II Angiotensin II
AMPK 5' adenosine monophosphate-activated protein kinase ANP Atrial natriuretic peptide
AT1R Angiotensin II type 1 receptor ATP Adenosine triphosphate -B-
Bcl-2 B-cell lymphoma 2 BP Blood pressure -C-
Ca2+ Calcium
cAMP Cyclic adenosine monophosphate Cdk Cyclin dependent kinase
CHD Coronary heart diseases Cki Cdk inhibitor
CNP C-type natriuretic peptide CO Cardiac output
-D-
DMEM Dulbecco's Modified Eagle's medium DNA Deoxyribonucleic acid
DPI Diphenyleneiodonium -E-
EGF Epidermal growth factor
EGF-R Epidermal growth factor receptor ERK1/2 Extracellular regulated kinase 1 and 2 ET-1 Endothelin-1
-F-
FBS Fetal bovine serum -G-
GDP Guanosine diphosphate Gi Inhibitory G protein
GPCRs G protein-coupled receptors
Grb2 Growth factor receptor-bound protein 2 Gs Stimulatory G protein
GTP Guanosine triphosphate -H-
H2O2 Hydrogen peroxide -I-
IGF1 Insulin-like growth factor 1 IGF-R Insulin-like growth factor receptor -J-
JNK c-Jun-N-terminal kinase -K-
kDa Kilodalton -L-
LDL Low density lipoprotein -M-
M Molar
MAP Mean arterial pressure
MAPK Mitogen-activated protein kinases MEK MAPK/ERK kinase
MKKK Mitogen-activated protein kinases kinase kinase mmHg Millimeters mercury
mRNA Messenger ribonucleic acid -N-
NAC N-acetylcysteine
NADPH Nicotinamide adenine dinucleotide phosphate NO Nitric oxide
NOX NADPH oxidase
NPR-C Natriuretic peptide receptor-C -O-
O2- Superoxide anion -P-
PDGF Platelet-derived growth factor
PDGFR Platelet-derived growth factor receptor PGI2 Prostacyclin
p38mapk p38MAP kinase
PI3K Phosphoinositide-3 kinase PKC Protein kinase C PLC Phospholipase C PLCβ Phospholipase Cβ PLCγ Phospholipase Cγ pRb Phosphorylated Rb -R-
RAAS Renin-angiotensin-aldosterone system Raf Rapidly Accelerated Fibrosarcoma Rb Retinoblastoma protein
ROCK Rho-kinase
ROS Reactive oxygen species RTK Receptor tyrosine kinase -S-
SHR Spontaneously hypertensive rat
Sirtuin1 Silent mating type information regulation 2 homolog STATs Signal transducers and activator of transcriptions SVR Systemic Vascular Resistance
-T-
TNF-α Tumor necrosis factor-α -V-
VSMCs Vascular smooth muscle cells -W-
WKY
Wistar Kyoto rat
To my father’ soul: I am so blessed to be your daughter. To my mother: your love is beyond imagination.
Acknowledgements
First, praise be to God for everything.
I owe my deepest gratitude to my supervisor, Dr. Madhu Anand-Srivastava, for accepting me into her lab. Her constant guiding and support were very helpful and motivating. She supported me in a time that I needed it the most.
I would also like to thank the members of my jury Dr. Réjean Couture and Dr. Puttaswamy Manjunath, for accepting to evaluate my graduate research work and for investing so much time and effort to accomplish this task.
A special thanks to Dr. Yuan Li for all her teachings and helpful suggestions inside and outside the lab.
I am very thankful to my friends and colleagues in the lab: Dr. Ekhtear Hossain, Dr. Oli Sarakar, Dr. Mehdi Atef, Mr. Ashish Jain and Dr. Sofiane Rahali; every one of you added to my experience in research and in life, in general.
Heartfelt thanks go also to my brother Ahmed Almajdoob and to Adina Sigartau for their help and advice during the entire period of my master’s program.
Last but not least, I would like to thank my family for all their love, encouragement and support in all the steps of my life.
CHAPTER 1
1. Hypertension
Hypertension or high blood pressure (BP) is a multifactorial medical disorder defined by World Health Organization (WHO) as sustained elevated BP, systolic BP equal to or above 140 mmHg and/or a diastolic BP equal to or above 90 mmHg in adults. According to Statistics Canada 2014, 5.3 million of Canadians aged 12 and older reported being diagnosed with high BP. Hypertension is the number one risk factor for stroke and a major risk factor for heart disease, renal failure and peripheral vascular disease with their important socioeconomic burden for the community (Mulvany, 1991; Kannel, 1996; Dai et al., 2010). Most people with hypertension have no symptoms at all; 18% of Canadian adults with high BP do not even know that they have it, according to Statistic Canada 2012 to 2015. This is why hypertension is known as “the silent killer” and contributes to approximately 7.1 million deaths per year worldwide (Guilbert, 2003). In spite of its wide prevalence and intense research into its pathophysiology, the molecular mechanisms underlying hypertension are poorly understood in the majority of patients and hypertension remains inadequately managed everywhere. Understanding the molecular mechanisms underlying hypertension will probably lead to more highly targeted therapies and could significantly lower the prevalence of hypertension-related cardiovascular diseases.
1.1. Classification of Hypertension
Hypertension is classified according to etiology into two major categories: primary (essential) hypertension and secondary (nonessential) hypertension. Ninety to ninety five percent of hypertensive patients suffer from essential hypertension which is defined as chronic high BP with an unknown cause, i.e., no clear single identifiable cause is found. Many factors have been proposed to be implicated in the genesis of essential hypertension including genetic predisposition, long-term high sodium intake, sedentary lifestyle, excessive alcohol consumption, obesity, insulin resistance and inappropriate renin secretion (Lifton, Gharavi, & Geller, 2001; Geller, 2004; Reaven, Abbasi, & McLaughlin, 2004). The remaining five to ten percent of hypertension cases is categorized as secondary hypertension. This type of hypertension is linked to a diagnosed condition like chronic renal disease (Brown et al., 1976), hyperthyroidism (Prisant, Gujral, & Mulloy, 2006), aortic coarctation (Tundidor et al., 2010),
corticoadrenal disorders (Augustin et al., 1983), acromegaly (Bondanelli, Ambrosio, & degli Uberti, 2001) and many other diseases (Kaplan, 1995).
2. Blood Pressure (BP)
BP represents the force exerted on the vessel wall by the circulating blood pumped by the heart. Human's BP varies depending on age, sex, emotional status, and time of day. BP values are measured in millimeters of mercury (mmHg) and it is usually expressed in notation as a fraction of the maximum (systolic) and the minimum (diastolic) pressures of blood flow in the arteries. Systolic pressure indicates the BP that occurs on the arterial wall during heart contraction, with an average of 120 mmHg, whereas the diastolic pressure refers to the pressure inside the arteries when the heart relaxes between contractions, averages 80 mmHg. The difference between systolic and diastolic pressures is called the pulse pressure and represents the force that the heart generates each time it contracts. Another important variable for arterial BP is mean arterial pressure (MAP). MAP represents the steady state component of BP and estimated by adding one-third of the pulse pressure to the diastolic pressure. Clinical significance of the MAP comes from the fact that it represents the perfusion pressure to supply the vital organs of the body with oxygen and important nutrients (Hall & Guyton, 2011). 2.1 Blood pressure regulation (Hemodynamics)
An adequate BP is necessary for the blood to travel from the heart around the body. Arterial BP is determined physiologically mainly by the cardiac output (CO) and the vascular resistance. Blood pressure (BP) = Cardiac Output (CO) x Systemic Vascular Resistance (SVR)
CO is a term used to describe the volume of blood pumped out by each ventricle in the time interval of one minute. CO is logically equal to the product of blood volume pumped out by the left ventricle of the heart in one contraction (stroke volume) and heart rate. More is the volume of blood presents in the closed circulatory system, higher are the CO and the arterial pressure. When the heart rate increases the CO increases (to certain limit) and subsequently the arterial pressure rises.
On the other hand, SVR is described as the resistance of the systemic vasculature to blood flow. The resistance (R) to blood flow within a single vessel is determined by three different factors: length of the blood vessel (L), vessel diameter (or radius) (r) and the viscosity of the blood (η). R is directly proportional to L and η, and inversely proportional to the r to the fourth power (r4); this relation is represented by Poiseuille equation (Pfitzner, 1976)
R = 8Lη/ (πr4)
Vessel length is generally not subject to change in the body, while blood viscosity changes within a small range (when the hematocrit changes). Of these three factors, the key regulator of the vascular resistance is the vessel diameter, for example according to Poiseuille's equation when the radius of a vessel is halved, the resistance increases by 16 fold.
Smooth muscle in tunica media of a normal blood vessel exhibits contractile state (tone) that determines the diameter of the vessel. Moreover, absence of basal vascular tone in the vessel wall is rapidly incompatible with life (Lacolley et al., 2012). Vascular tone is subject to continual homeostatic changes by vasoconstrictors and vasodilators acting on the blood vessel resistance to keep BP and blood flow within normal ranges (Izzo et al., 2008). Resistance of vessels to blood flow is also related to vascular compliance which is defined as the ability of the vessel wall to expand and contract passively with changes in the pressure. Reduced arterial compliance is seen with old age, hypertension, diabetes, atherosclerosis and temporarily with contraction of smooth muscle in the arterial wall (Glasser et al., 1997; Willerson, 2007).
2.2 Mechanisms of blood pressure regulation
Maintaining near- constant BP is a necessary for blood to be delivered to all organs and tissues. BP regulation is a complex physiological system operating in both short-term and long-term reflex responses (involving hormones, local vascular factors, and neural mechanism) to return MAP to its normal value, when deviations from the norm are detected.
2.2.1 Short term regulation
The short-term regulation of BP tries within seconds to minutes to correct temporary imbalances in BP and relies mainly on neural and humoral mechanisms.
2.2.1.1 Neural mechanisms
The most rapid of the short-term regulation mechanisms is the neural mechanism. The neural control centers for the regulation of BP are often collectively referred to as the cardiovascular center, located mainly in the brain stem. This center integrates inputs and sends out impulses through the autonomic nervous system. Cardiovascular center modifies the ratio between sympathetic and parasympathetic activity in the heart and blood vessels to produce changes in the peripheral resistance and CO, which will lead to changes in the BP. This area of the brain contains three distinct regions:
I- Cardioaccelerator center: it sends stimulatory impulse over sympathetic cardiac nerves to increase CO by increasing heart rate and contractility.
II- Cardioinhibitor center: it exerts an inhibitory influence on the heart through parasympathetic vagal nerve to slow down heart rate and lower CO.
III- Vasomotor center: it contains sympathetic neurons and connects to the smooth muscle of blood vessels via efferent motor fibres, which release noradrenaline, a potent vasoconstrictor. Vasomotor tone of the blood vessels is maintained under this sympathetic control. Increased sympathetic activity produces vasoconstriction of the small arteries and arterioles with a resultant increase in peripheral resistance to blood flow, thereby increasing BP (Hall & Guyton, 2011).
Neural control of BP is mediated through multiple compensatory reflexes: the baroreceptor reflex, chemoreceptor reflex and extrinsic reflexes.
1- Baroreceptor reflex:
It is well established that the baroreceptor reflex powerfully buffer acute changes in arterial pressure, such as what occur during physical exercise, and changes in body position. Baroreceptors are sensory neurons that consistently monitor MAP and are located mainly in the carotid sinus, the aortic arch, and the right atrium. Baroreceptor reflex starts by stimulation of baroreceptors by rapid alterations of arterial BP. These baroreceptors send signals with their afferent fibres in the glossopharyngeal and vagus nerves to the cardiovascular center of brain
stem. The cardiovascular center, by way of autonomic nervous system, adjusts the MAP by managing the force and speed of the heart's contractions (CO) or blood vessel diameter (Dampney et al., 2002). Baroreceptor reflex prevents erratic fluctuations in BP; however, the baroreceptor reflex fails if the change in pressure is slow and sustained. This is because of baroreceptor resetting, wherein the baroreceptor adapts itself to new 'resting' BP. Cardiovascular diseases such as hypertension and heart failure are often accompanied by a dysfunction of baroreflex mechanisms. However, the baroreceptor reflex in general is not targeted in hypertension because, if blocked, the BP would fluctuate wildly with postural changes or Valsalva maneuver and individuals may faint (Drenjancevic et al., 2014).
2- Chemoreceptor reflex:
Chemoreceptors are chemosensitive cells that detect changes of oxygen, carbon dioxide, and pH in the arterial blood, which can indicate a change in blood flow and tissue perfusion due to BP fluctuations. They are located in the carotid bodies, which lie in the bifurcation of the two common carotids, and in the aortic bodies of the aorta and their afferent fibres, like baroreceptor afferent fibres that run in the glossopharyngeal and vagus nerves. Although the main role of chemoreceptors is to regulate respiration, they also communicate with cardiovascular centers and can induce widespread vasoconstriction. When the arterial pressure drops below a critical level, the chemoreceptors are stimulated because of diminished oxygen level, buildup of carbon dioxide and decreasing pH. As a result, chemoreceptors send impulses to the cardioacceleratory centre leading to enhanced sympathetic outflow and to increase heart rate, as well as to the vasomotor centre to cause vasoconstriction. Sympathetic vasoconstriction will tend to reduce oxygen consumption by the tissues, and thus conserve the available oxygen. Because of the hypoxemia in chronic lung disease, activation of the carotid body chemoreceptors occurs and this ultimately cause systemic and/or pulmonary hypertension (Chopra, Baby, & Jacob, 2011; Drenjancevic et al., 2014).
3- Extrinsic reflexes:
Sensors of the baroreceptor and chemoreceptor reflexes are located in the circulatory system, whereas the receptors for extrinsic reflexes are found outside the circulation. The neural pathways for these reactions include higher brain regions, such as the cerebral cortex,
hypothalamus, and the limbic system, that respond to conditions like pain, stress and cold, requiring adjustments to the BP through regulation of the cardiovascular center (Porth, 1982). 2.2.1.2 Humoral Mechanism
A number of humoral mechanisms contribute to short-term BP regulation, including these mediated by sympathetic neurotransmitters adrenaline and noradrenaline, natriuretic peptides (auricular natriuretic peptide (ANP), brain natriuretic peptide (BNP) and C-type natriuretic peptide (CNP), vasoactive peptides such as angiotensin II (Ang II), endothelin-1 (ET-1) and vasopressin, vasodilator nitric oxide (NO), leptin, adipokines, etc. They act directly on the smooth muscle, thus they instantly regulate peripheral vascular resistance. Moreover, adrenaline and noradrenaline act also on the heart and increase heart rate, cardiac contractility (adrenaline only) and subsequently BP. Some of these chemicals, like Ang II and vasopressin function in both the short- and long-term regulation of BP (Porth, 1982).
2.2.2 Long term regulation
Although, the short-term regulation mechanisms of BP act rapidly, they are unable to maintain their effectiveness. Conversely, long-term regulatory processes control BP over weeks and months through adjusting total blood volume by the kidneys and regulating water intake. Renal regulation of blood volume is achieved by the combined actions of pressure diuresis and natriuresis shifting, release of antidiuretic hormone and the renin-angiotensin-aldosterone system (RAAS) (Porth, 1982; Chopra et al., 2011; Drenjancevic et al., 2014).
Firstly, kidneys directly monitor total blood volume and arterial pressure by controlling how much fluid and salts are excreted or retained. When the arterial pressure rises as result of excess extracellular fluids in the body, the renal perfusion pressure will increase. If renal perfusion pressure increases then renal reabsorption is decreased leading to higher renal excretion of the fluid ( pressure diuresis) and sodium (pressure natriuresis) ( Hall et al., 1986).
Secondly, antidiuretic hormone, also known as vasopressin, is secreted from the posterior pituitary gland in response to decreases in blood volume and BP. The physiological effects of vasopressin are mediated by ligand binding to specific vasopressin receptors called V2 receptors which are G -protein coupled. Vasopressin has a direct vasoconstrictor effect on the systemic
circulation via V1 receptors, particularly on the vessels of the splanchnic circulation. It has been suggested that vasopressin may play a role in hypertension through its water retaining properties. It works on the kidney to increase the permeability of distal tubules and the collecting ducts, which helps increase the reabsorption of water leading to its primary physiological effects of water retention and regulation of blood osmolality (Barlow, 2002).
The third mechanism for the long-term regulation of arterial pressure involves RAAS. 2.2.2.1 Renin angiotensin aldosterone system (RAAS)
RAAS is known for its long term and consistent adjustment of arterial pressure. It has the potential to change the resistance in the arterioles of the kidney and cause the release of powerful chemical mediators to change peripheral vascular resistance. Renal afferents monitor alterations in the BP through baroreceptors. When the blood volume is low and the renal blood flow decreased, juxtaglomerular cells release the enzyme renin into the bloodstream. Plasma renin converts angiotensinogen (inactive form) produced by the liver to angiotensin I (active form). Angiotensin I is cleaved by the angiotensin-converting enzyme in the lung to form Ang II. Ang II has many effects, including increasing the BP through its vasoconstrictive properties and acting on vascular smooth muscle in arterioles both systemically and locally at the level of the kidney. Ang II also stimulates the cells of the adrenal cortex to release the hormone aldosterone. Aldosterone stimulates sodium retention and potassium excretion by the renal tubules which subsequently increases fluid retention, and indirectly, arterial pressure (Porth, 1982; Hall & Guyton, 2011).
2.2.2.1.1 Tissue RAAS
In addition to the well-known circulating RAAS, the presence of RAAS elements have been found in tissues such as heart, kidney, vasculature, adipose tissue, immune cells, and brain. It was reported that vascular smooth muscle cells (VSMCs) have a complete system to produce Ang II ( Kubo et al., 2000). RAAS in the vascular wall plays important roles not only in the regulation of vessel tone and blood flow but also in vascular proliferation and differentiation. Therefore, activated tissue RAAS has been suggested to be involved in the pathogenesis of vascular proliferative diseases such as hypertension and atherosclerosis (Hu et al., 2003).
2.3 Vascular structure
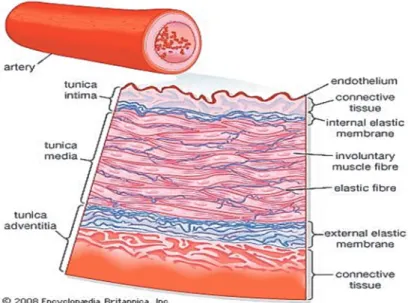
The vascular structure plays an important role in BP homeostasis. Blood vessel networks form a part of the circulatory system that carries blood away from the heart, delivers blood to all tissues, and then returns it to the heart. There are three major types of blood vessels: the arteries, the capillaries and the veins. The composition of blood vessels differs depending on their function. Arteries and veins are composed of three distinct layers, but the middle layer in the arteries is thicker than it is in the vein. The arterial wall consists of three tunicae: tunica intima, tunica media and the tunica adventitia (Figure 1). Tunica intima is the inner layer, comprised of a single layer of endothelium and a thin layer of loose connective tissue. The endothelium of tunica intima reduces friction between the vessel walls and blood. In addition, the tunica intima secretes several vasoactive substances that alter the diameter of vessels including: prostacyclin (PGI2), ET-1, NO and CNP. The tunica intima is surrounded by the internal elastic lamina which plays role in the vessel elasticity. Calcification of the internal elastic lamina contributes to the pathological process of atherosclerosis (Micheletti et al., 2008). The middle layer, the tunica media, consists mainly of VSMCs which control the caliber of the vessel and arterial tone. Relaxation and constriction of the smooth muscle layer is controlled by crosstalk between local factors and sympathetic innervation. The second and third layer is separated by the external elastic lamina. Similar to the internal elastic lamina, the external elastic lamina contributes to the elastic properties of the artery. The tunica adventitia (outer layer) consists primarily of connective tissues which help anchor the vessel to its surroundings. It is also infiltrated by nerves, blood vessels and lymphatics (Berne & Levy, 2001; Tortora & Grabowski, 2003).
Figure 1: The structure of the arterial wall. Source: https://www.britannica.com/science/renal-artery
The elasticity and muscularity of arteries influence BP homeostasis. Large conducting arteries like aorta are elastic, contain large amounts of elastin and have the property to withstand and smooth out pressure fluctuations from the heart. On the other hand, small arteries have the greatest proportion of tunica media of all the vessels making them more muscular and active in vasoconstriction, causing significant changes in total peripheral resistance. The small arteries (lumen diameters <400 μm) and arterioles are called resistance arteries because they act as the major site of vascular resistance. Any change in lumen diameter of these resistance vessels will affect BP (Intengan & Schiffrin, 2000; Berne & Levy, 2001).
Vasculature responds rapidly to changes in its biomechanical environment by functional and structural changes through autocrine and/or paracrine humoral mechanisms. Long-term hemodynamic alterations such as elevated shear stress or increased intravascular pressure may eventually lead to vascular adaptation called vascular remodeling (Willerson, 2007).
3. Vascular remodeling
Vascular remodeling is an active compensatory mechanism that occurs in response to long-term mechanical or biochemical stresses applied to the vessel walls such as increased transmural pressure and blood flow (Willerson, 2007). Vascular remodeling involves structural alterations
in at least four cellular processes: cell growth, cell death, cell migration and production or degradation of extracellular matrix. The resulting vascular remodeling may initially be adaptive, but eventually it becomes maladaptive contributing to the pathophysiology of vascular diseases and circulatory disorders such as hypertension and atherosclerosis (Korshunov, Schwartz, & Berk, 2007).
3.1 Vascular remodeling in hypertension
Hypertension is associated with structural changes, remodeling in large and resistance small arteries, impacting both the development and the complications of hypertension. Vascular remodeling of resistance small arteries in hypertension encompasses reduction in lumen diameter, increase in media thickness and an increase in wall-to-lumen ratio resulting in increase in vascular reactivity, thus enhancing peripheral resistance (Mulvany & Korsgaard, 1983; Korsgaard et al., 1993; Mulvany, 2002). Increase vascular resistance is one of multiple factors proposed in the etiology of hypertension, in both human and experimental animals (Owens & Schwartz, 1982). The thickening of arterial walls was reported even at the early or pre-hypertensive stage in animal models of essential hypertension (Mizutani, Ikeda, & Yamori, 2000). At VSMCs level, several cellular processes are involved in the vascular remodeling including VSMCs hyperplasia (increase in the VSMCs number associated with DNA synthesis) (Mulvany, Baandrup, & Gundersen, 1985), VSMCs hypertrophy (increase in the size of the VSMCs, associated with increased protein synthesis and intracellular volume), apoptosis, VSMCs elongation, and re-organization (Mulvany, Hansen, & Aalkjaer, 1978; Berk, 2001; Touyz, 2005; Renna, de Las Heras, & Miatello, 2013).
3.1.1 Hyperplasia of vascular smooth muscle cells (VSMCs)
VSMCs, the main cellular component of the medial layer of the vascular wall, are essential in regulating BP and flow by controlling the diameter of blood vessels. They are involved in the pathological changes taking place in vascular diseases such as atherosclerosis and hypertension (Lacolley et al., 2012). In intact arteries, VSMCs are contractile and exhibit extremely low rates of proliferation. The increase in proliferation results from an imbalance between the factors that stimulate proliferation and those that inhibit it. VSMCs lose their contractile function and convert to a highly proliferative state by mechanical injury, oxidative
stress, and humoral factors (Yuan, 2011; Salabei & Hill, 2013). Smooth muscle cell proliferation contributes significantly to the process of vascular remodeling associated with hypertension (Owens & Schwartz, 1982). The intracellular mechanisms implicated in VSMCs hyperplasia are complex. VSMCs hyperplasia occurs in response to vasoactive agents (Ang II, ET-1, catecholamine, cytokines, and vasopressin) that stimulate G protein-coupled receptors (GPCRs) or Tyrosine Kinase Receptors (RTKs) (Touyz, 2000; Berk, 2001; Ljuca & Drevensek, 2010). However, Ang II appears to be one of the most important in the hypertension (Touyz, Tabet, & Schiffrin, 2003). Ang II, via the angiotensin II type 1 receptor (AT1Rs), stimulates cell growth through diverse signaling pathways including stimulation of tyrosine kinases, MAPK, mobilization of intracellular calcium (Ca2+) and generation of reactive oxygen species (ROS) through activation of vascular NAD(P)H oxidase (Berk, 2001; Filipeanu et al., 2001; Touyz, 2005). Targeting some of these signaling pathways with novel therapeutic strategies could prevent or induce regression of arterial remodeling thereby ameliorating the development of hypertension and other forms of cardiovascular disease. The spontaneously hypertensive rat model (SHR) is particularly suitable for the study of the molecular mechanisms behind the increased proliferation of VSMCs. It is known that VSMCs from SHR have an intrinsic ability to proliferate more rapidly than those from normotensive control Wistar Kyoto rat (WKY) (Scott-Burden et al., 1989).
3.1.2 SHR as a Model of Hypertension and vascular remodeling
SHR is a genetic model of essential hypertension. It is also used to study various cardiovascular diseases, often with the WKY as the normotensive control (Pinto, Paul, & Ganten, 1998). The SHR strain was developed by Okamoto and colleagues during the 1960s, by selective inbreeding of rats from Wistar strain having a high BP, obtaining SHR in which the incidence of spontaneous hypertension is 100 percent (Okamoto & Aoki, 1963). The increased BP develops over the first 12 weeks and increases with age (Okamoto & Aoki, 1963).The SHR model has been shown to display most of the characteristics of human essential hypertension (Yamori & Okamoto, 1974). Both in essential hypertension and in SHR, similar changes in the structure of small arteries (reduced lumen, increased media to lumen ratio) have been observed pari passu with the increase in BP (Heagerty et al., 1993). Therefore, this model allows
researchers to study the mechanisms behind vascular remodeling including VSMCs hyperplasia, as well as possible therapeutic approaches.
4. Molecular mechanisms implicated in hypertension and vascular remodeling 4.1 Oxidative Stress and Reactive Oxygen Species (ROS)
Oxidative stress reflects an imbalance between the production of ROS and their elimination by antioxidant, in which oxidant overproduction overwhelms the cellular antioxidant capacity (Lyle & Griendling, 2006). ROS are a family of highly reactive oxygen-containing molecules (due to the presence of unpaired valence shell electrons) such as superoxide anion (O2-), hydrogen peroxide (H2O2) and hydroxyl radical (OH-) that act as oxidants (Touyz & Schiffrin, 2004). ROS are normally generated by the cells during reduction-oxidation (Redox) reactions leading from molecular oxygen (O2) to water (H2O) (Fridovich, 1997; Thannickal & Fanburg, 2000) Physiologically, ROS are produced in a controlled manner at low concentrations and have important roles in cell signaling and homeostasis, with scavenging mechanisms protecting against the toxic effects of excess ROS (Touyz, Tabet, & Schiffrin, 2003; Touyz, 2004b). However, under pathological conditions, excessive endogenous formation of ROS overcomes cellular antioxidant defense mechanisms. Accumulation of ROS results in the stimulation of an enzymatic cascade leading to pathological changes in the cell (ROS-initiated modification of lipids, proteins, carbohydrates and DNA) (Thannickal & Fanburg, 2000; Lassegue & Griendling, 2004).
4.1.1 Sources of ROS
Cellular production of ROS comes from enzymatic and nonenzymatic sources (Touyz & Schiffrin, 2004). The major intracellular source of ROS is the mitochondria, which convert 1-2% of consumed molecular oxygen into superoxide anion. Mitochondria generate ROS as by-products during Adenosine triphosphate (ATP) production via electron transfer through cytochrome c oxidases (Boveris & Chance, 1973). ROS can also be produced by NAD(P)H oxidase, xanthine oxidase, cyclooxygenase, lipoxygenase, heme oxygenases, peroxidases, as well as hemoproteins (Touyz, 2004a). Interestingly, all vascular cell types have the ability to generate ROS in a controlled and regulated manner (Channon & Guzik, 2002). In the
vasculature, ROS are produced to varying degrees and by several enzyme systems, including NADPH oxidases, endothelial NO synthases, xanthine oxidase and the mitochondrial electron transport system (Lee & Griendling, 2008). Their production is regulated by anti-oxidant enzymes such as catalase, superoxide dismutase, thioredoxin, and glutathione (Thannickal & Fanburg, 2000). Autooxidation of small molecules such as dopamine, epinephrine, flavins and hydroquinones is responsible for the nonenzymatic production of ROS (Freeman & Crapo, 1982).
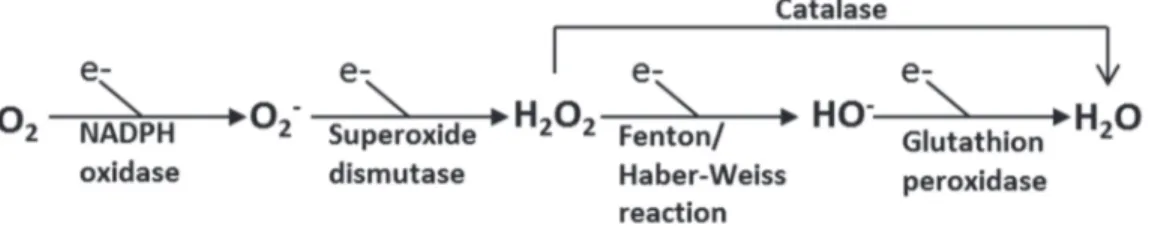
The primary form of most ROS is superoxide anion (Turrens, 2003). Superoxide anion is produced by the univalent reduction of oxygen using NADPH as an electron donor (Figure 2) (Taniyama & Griendling, 2003). Superoxide anion converts to H2O2 either spontaneously or by the action of superoxide dismutase (SOD) (Fridovich, 1997). Unlike O2-, H2O2 is not a free radical and is a much more stable molecule (Thannickal & Fanburg, 2000). H2O2 is reduced to H2O by catalase or glutathion peroxidase. H2O2 can also react with reduced transition metals (Fenton/ Haber-Weiss reaction) to be converted to the highly reactive hydroxyl radical (OH-) (Figure 2) (Blanc, Pandey, & Srivastava, 2003). The reaction of O2- with nitric NO, inactivates NO and generates peroxynitrite, a potentially deleterious ROS (Lassegue & Griendling, 2004). NADPH oxidase is responsible for the majority of the superoxide produced in the vasculature (Griendling, Sorescu, & Ushio-Fukai, 2000).
Figure 2: Simplified scheme showing key steps in the production of reactive oxygen species (ROS). Source: (Blanc, Pandey, & Srivastava, 2003).
4.1.2 NADPH oxidase
The NADPH oxidases are membrane-bound enzymes that catalyze the production of superoxide anion from oxygen and NADPH. They are present in phagocytic cells (neutrophils,
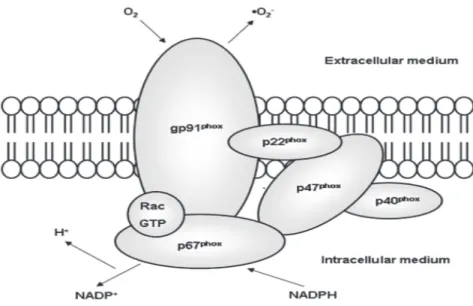
macrophages, and eosinophils) and vascular tissue (Griendling, Sorescu, & Ushio-Fukai, 2000; Babior, Lambeth, & Nauseef, 2002; Vignais, 2002). The NADPH oxidase is a multimeric protein (Figure 3) and consists mainly of four major subunits: two cell membrane components gp91phox/NOX2 and p22phox which together form a heterodimer, called flavocytochrome b558, as well as two cytosolic components, p47phox and p67phox (Griendling, Sorescu, & Ushio-Fukai, 2000). A low molecular weight G protein, rac, participates in the assembly of the cytoplasmic units and the activation of the enzyme (Vignais, 2002). Cytochrome b558 is the crucial catalytic component responsible for the transport of electrons across biological membranes to reduce oxygen to superoxide (Koshkin & Pick, 1993). Gp91phox is an essential component of the NAD(P)H oxidase which binds to the electrons and carries components of oxidase such as flavine adenine dinucleotide, and a pair of hemes molecules (Brandes & Kreuzer, 2005). Over the last years, six homologs of gp91phox/ Nox2 were found: NOX1, NOX3, NOX4, NOX5, DUOX1 and DUOX2. Together with gp91phox/NOX2 they form a family called NOX (for NADPH oxidase) (Bedard & Krause, 2007). On the other hand, p47phox, p67phox and Rac translocate from the cytosol to the membrane during NADPH oxidase assembly (Heyworth et al., 1991). P47phox is the protein that carries the cytosolic subunits to the membrane subunits to assemble the active oxidase. P67phox contains two Src homology 3 (SH3) domains, one in the middle of the protein, and one near the carboxyl terminus. The SH3 domains interact with p22phox to activate the enzyme. P22phox is located in the membrane, along with gp91phox and has a tail in the cytosol. When p22phox is phosphorylated, it binds to p47phox; essential an interaction in enzyme activation (Brandes & Kreuzer, 2005).
Figure 3: Structure of NADPH oxidase. Source: (Rabelo et al., 2010)
4.1.3 The Vascular NADPH Oxidase
Several features differentiate the vascular NAD(P)H oxidase from the phagocytic NAD(P)H oxidase (Lassegue & Clempus, 2003). Vascular NAD(P)H oxidase is regulated by Ang II, ET-1, platelet derived growth factors (PDGF), thrombin and tumor growth factor-α (Duerrschmidt et al., 2000; Zalba et al., 2001; Rueckschloss et al., 2002). Superoxide anion produced by vascular NAD(P)H oxidase participates in cellular signaling, whereas phagocytes produce superoxide anions playing role in their microbicidal functions (Szasz et al., 2007). Production level of superoxide anion by blood vessel cells is also different from that of phagocytes. The level of O2- generated by vascular NAD(P)H oxidase is significantly lower compared to that produced by the phagocytic enzyme (Hohler, Holzapfel, & Kummer, 2000). NAD(P)H oxidase is found in the different layers of vascular wall: the intima (Muzaffar et al., 2003), the media (Berry et al., 2000) and the adventitia (Rey et al., 2002). Only p47phox and p22phox in VSMCs appear to be expressed consistently (Lassegue & Clempus, 2003). Nox4 is highly expressed in all cardiovascular cells, higher than Nox1 and Nox2 (Griendling, 2004). Aortic smooth muscle cells express Nox1 and Nox4 in rodents, and also Nox5 in humans (Lyle & Griendling, 2006). VSMCs from the rat resistance arteries express all subunits, including gp91phox (Azumi et al., 1999; Touyz et al., 2002).
4.1.4 Implication of oxidative stress in hypertension
A considerable number of studies have demonstrated that hypertension is associated with increased oxidative stress and also with an impairment of endogenous antioxidant defense mechanisms (Lassegue & Griendling, 2004; Khullar, Relan, & Sehrawat, 2004). The levels of ROS have been shown to be increased in vascular tissue of SHR, and several models of experimental hypertension as well as in patients with various hypertensive disorders (Zalba et al., 2000; Higashi et al., 2002; Touyz, 2004b). This is associated in SHR with the overexpression of different subunits of NADPH oxidase such as p47phox and Nox4 in VSMCs, which appear to be responsible for the enhanced activity of NADPH oxidase and ROS production exhibited by these cells (Saha, Li, & Srivastava, 2008; Saha, Li, Lappas, et al., 2008; Anand-Srivastava, 2010). ROS mediate their effects through redox-sensitive signaling pathways including Ang II signaling (Touyz, Tabet, & Schiffrin, 2003). The role of Ang II in NADPH subunits expression and activation has been reported by several studies (Griendling et al., 1994; Touyz, Cruzado, et al., 2003; Hossain & Anand-Srivastava, 2017). In fact, our lab group has reported that enhanced levels of O2- in VSMCs from SHR was attenuated by AT1R antagonist, losartan, supporting the implication of Ang II in ROS production (Anand-Srivastava, 2010). The involvement of oxidative stress in the pathogenesis of hypertension is also mediated by the inactivation of NO by O2- in the vasculature and kidney as well as by ROS-induced vascular remodeling (Taniyama & Griendling, 2003; Hsieh et al., 2014). Exogenous administration of antioxidants has been shown to attenuate elevated BP in DOCA-salt hypertensive rats, SHR and mild hypertensive patients (Nakazono et al., 1991; Beswick et al., 2001; Boshtam et al., 2002). Nonetheless, the relationship between oxidative stress and hypertension as well as the effect of antioxidant treatment of hypertension are still being studied.
4.1.5 Implication of oxidative stress in cell proliferation
Recent evidence has shown that ROS are also implicated in endothelial dysfunction, VSMCs migration, growth and apoptosis, inflammation and increased depositions of extracellular matrix proteins, which are important factors in arterial remodeling during cardiovascular disease (Griendling, Sorescu, Lassegue, & Ushio-Fukai, 2000; Taniyama & Griendling, 2003). Previous study by Rao and colleagues has reported that H2O2 stimulates DNA synthesis of VSMCs (Rao
& Berk, 1992). Furthermore, our research group demonstrated that the hyperproliferation of VSMCs from SHR was attenuated by the antioxidants diphenyleneiodonium (DPI) and N-acetylcysteine (NAC), suggesting the involvement of oxidative stress in enhanced DNA synthesis of VSMCs from SHR (Li, Levesque, & Anand-Srivastava, 2010). At molecular level, oxidative stress contributes in the cellular signaling pathways involved in the cell growth and proliferation (Cheng et al., 1999; Cheng et al., 2003). For example, ROS generation was shown to contribute in the activation of mitogenic MAPK pathway by Ang II and ET-1 in VSMCs (Touyz et al., 2004).
4.2 c-Src Pathway
C-Src kinase is an important member of a family of non-receptor tyrosine kinases called Src family kinases (SFK). SFK consists of 9 members, although only c-Src, Fyn and Yes are ubiquitously expressed in all cells, and c-Src is highly expressed in VSMCs (Wheeler, Iida, & Dunn, 2009). C-Src kinase is composed of two binding domains, SH2 and SH3, a kinase domain, a non-catalytic domain in the C-terminal and a myristoylation sequence in the N-terminal. SFK plays a vital role in cell differentiation, proliferation and survival signaling mechanisms (Roskoski, 2004). Recent studies have highlighted the contribution of c-Src in molecular and cellular processes underlying Ang II-induced hypertension (Qin & Zhou, 2015). Accumulating evidence suggests that c-Src activation is implicated in various Ang II vascular responses, such as cell growth (Touyz, He, Wu, et al., 2001), cell migration (Mugabe et al., 2010) and contraction (Touyz, Wu, et al., 2001). Src family is a key mediator of Ang II-induced VSMCs proliferation through multiple intracellular signaling pathways including the Shc/ growth factor receptor-bound protein 2 (Grb2)/ ERK2 pathway (Sayeski & Ali, 2003), the signal transducers and activators of transcription (STATs) (Bromberg et al., 1998) and the PI3K signaling pathway (Fincham, Brunton, & Frame, 2000). In VSMCs, a feed-forward mechanism was suggested, in which low levels of H2O2 activate c-Src, which in turn initiates a signaling cascade leading to NAD(P)H oxidase activation, generation of additional ROS, further activation of Src, and the amplification of oxidase activity (Seshiah et al., 2002; Touyz, Yao, & Schiffrin, 2003). Our lab group has showed that PP2, a c-Src inhibitor, attenuates the enhanced activation of growth factor receptors as well as the enhanced DNA synthesis of VSMCs from SHR, suggesting its implication in the transactivation of growth factor receptors and the resultant hyperproliferation
of VSMCs from SHR (Li, Levesque, & Anand-Srivastava, 2010; Atef & Anand-Srivastava, 2016).
4.3 Growth factor receptors signaling
Many growth factors, such as epidermal growth factor (EGF), platelet-derived growth factor (PDGF) and insulin-like growth factor 1 (IGF 1), mediate their diverse biologic responses by binding to and activating transmembrane receptors with tyrosine kinase activity, designated receptor tyrosine kinases (RTK) (Schlessinger, 2014). RTK have risen as key regulators of different cellular processes including proliferation and differentiation, cell survival, cell migration and cell cycle control (Prenzel et al., 2001). Aberrations in the activation or signaling of RTKs have been linked to cancer, severe bone disorders and cardiovascular disease (Lemmon & Schlessinger, 2010). All RTKs consist of a ligand-binding extracellular N-terminal domain, an intracellular C-terminal domain responsible for (RTK) activity and a single transmembrane helix (Schlessinger, 2014). The binding of the ligand to its receptor causes two receptor monomers to form a dimer and this dimerization induces the autophosphorylation of the tyrosine residues. Once the receptor is activated and dimerized, it recruits SH-containing domain proteins, which trigger downstream events (Lemmon & Schlessinger, 2010). RTKs can also undergo phosphorylation in a ligand-independent manner by a process called transactivation and mediate the response of GPCRs agonist, such as Ang II and ET-1, thrombin, lysophosphatidic acid, ROS (Touyz, Cruzado, et al., 2003) and others (Daub et al., 1996; Hackel et al., 1999; Eguchi and Inagami 2000; Li, Levesque, & Anand-Srivastava, 2010). Among the RTKs, a special attention is paid to the transactivation of epidermal growth factor receptor (EGF-R), platelet-derived growth factor receptor (PDGFR) and insulin-like growth factor receptor (IGF-R), and subsequent signaling cascades involving the MAPK, PI3K -AKT and c-Src pathways.
4.3.1 Epidermal Growth Factor Receptor (EGF-R)
EGF-R/ErbB1 belongs to the EGF-R (ErbB/HER) subfamily of RTK containing three other members (ErbB2, ErbB3 and ErbB4) (Prenzel et al., 2001). EGF-R is increasingly recognized as a vital factor in the control of normal cell proliferation, growth and cellular survival (Prigent & Lemoine, 1992). Binding of EGF-R with its ligand, EGF, results in the homodimerization of
the EGF-R receptor or the heterodimerization of the EGF-R receptor with the other members of the ErbB family (Xian, 2007). The phosphorylation of EGF-R on Tyr1068, as a result of ligand binding-induced dimerization, recruits the adaptor protein Grb2, leading to the activation of Ras/ERK1/2 pathway. Multiple studies have shown that GPCRs agonist, such as Ang II, ET-1 (Gomez Sandoval & Anand-Srivastava, 2011), as well as intracellular pathways, involving c-Src (Biscardi et al., 2000) or ROS can cause the activation and subsequent phosphorylation of EGF-R through receptor transactivation phenomenon (Touyz, Cruzado, et al., 2003; Li, Levesque, & Anand-Srivastava, 2010 ). ET-1 and Ang II-induced EGF-R transactivation has been demonstrated in multiple cell types, including VSMCs (Eguchi, Iwasaki, Inagami, et al., 1999; Iwasaki et al., 1999), cardiomyocytes (Kodama et al., 2002) and pancreatic stellate cells (Hama et al., 2004). An increasing body of evidence suggests that the transactivation of EGF-R plays a critical role in vasoactive peptide-induced physiological responses linked to MAPK, c-Src and Akt/PKB signaling, such as growth, hypertrophy and proliferation in VSMCs (Eguchi et al., 1998; Bokemeyer, Schmitz, & Kramer, 2000; Prenzel et al., 2001; Ohtsu et al., 2006). Recent reports by our laboratory showed that EGF-R, AT1R, ET-A and B receptor inhibition decrease exaggerated ERK1/2 phosphorylation and hyperproliferation of VSMCs from SHR to levels found in VSMCs from WKY. These results are suggesting that the transactivation of growth factor receptor by endogenous vasoactive peptides through activation of MAPK contributes to the enhanced cell growth of VSMCs in SHR ( Li, Levesque, & Anand-Srivastava, 2010).
4.3.2 Insulin-like growth factor receptor (IGF-R)
IGF-R is a RTK that shares structural and functional homology with the insulin receptor and is abundantly expressed in VSMCs. Its structure consists of two extracellular α-chains and two intracellular β-chains (Adams et al., 2000). The binding of IGF1 or insulin (at very high, unphysiological concentrations) to the α-subunit stimulates a conformational change and induces the activation of tyrosine kinase domain of the IGF-1Rβ subunit leading to autophosphorylation in multiple tyrosine residues and the induction of the RTK catalytic activity (Arnqvist et al., 1995). The activated IGFR not only recruits the Grb2/Sos complex and activates the Ras/rapidly Accelerated Fibrosarcoma (Raf)1/MEK/ERK pathway (Radhakrishnan et al., 2008) but also triggers the PI3-K and its downstream targets AKT/PKB and p7OS6k (Zheng &
Clemmons, 1998). Beside activation by the ligand, the IGF-R is also activated by Ang II, ROS, Ca+2 and c-Src (Touyz, Cruzado, et al., 2003; Tu et al., 2010) (Oligny-Longpre et al., 2012). Bouallegue et al. have demonstrated that IGF-1R transactivation plays a role in ET-1 and Ang II-induced PKB phosphorylation and hyperproliferative responses in A10 vascular smooth muscle cells with c-Src being upstream to IGF-1R in this signaling cascade (Bouallegue, Vardatsikos, & Srivastava, 2009). Moreover, the fact that the dominant negative of IGF-1R is able to suppress VSMCs proliferation and migration, and to induce apoptosis that leads to the reduction of neointima formation in an injured carotid artery rat model supports a possible pathogenic role of upregulated IGF-1R signaling in vascular remodeling (Lim et al., 2004). 4.3.3 Platelet-Derived Growth Factor Receptor (PDGFR)
PDGFR is a membrane protein-tyrosine kinase consisting of an intracellular split kinase domain and an extracellular immunoglobulin-like domain (Heldin & Lennartsson, 2013). It exists in two isoforms: PDGFR-α and PDGFR-β (Andrae, Gallini, & Betsholtz, 2008) and is expressed in many cell types including VSMCs with the expression of PDGFR‐β being higher in VSMCs (Kitami et al., 1995; Kiyan et al., 2005). PDGFR-α is activated by A, PDGF-B and PDGF-C, while PDGF-PDGF-BPDGF-B and PDGF-DD bind and activate PDGFR-β (PDGF-Board & Jayson, 2005). Several reports have shown that PDGFR undergoes tyrosine phosphorylation in response to Ang II (Gao et al., 2006), ET-1 (Gomez Sandoval & Anand-Srivastava, 2011) and ROS (Saito et al., 2002). In vitro and in vivo studies have suggested that PDGF receptors are implicated in several well-characterized signaling pathways. These include c-Src, PI3K, PLCγ, Ras-MAPK and Giα proteins pathways which are known to be involved in cell growth and proliferation (Andrae, Gallini, & Betsholtz, 2008; Gomez Sandoval & Anand-Srivastava, 2011; Heldin & Lennartsson, 2013). PDGFR signaling is implicated in the pathogenesis of a variety of diseases such as atherosclerosis, pulmonary hypertension and leukemias (Andrae, Gallini, & Betsholtz, 2008). In addition, PDGF receptor is implicated in vascular remodeling through the modulation of VSMCs migration and proliferation (Koyama et al., 1992; Sano et al., 2001). Both α and β isoforms of PDGFR are expressed at high levels in cultured VSMCs from SHR. However, in VSMCs from WKY, PDGFR-α is suppressed almost completely (Inui et al., 1994).
4.4 Phosphatidylinositol-3-kinase (PI3K) pathway
PI3K pathway is another significant cellular signaling pathway that plays an important role in cell growth, survival, proliferation and gene expression. The PI3Ks are a group of lipids and protein kinases, divided into three classes according to their structure and mechanism of regulation namely class I, class II and class III (Rameh & Cantley, 1999). Class I PI3Ks are heterodimeric proteins consisting of a catalytic and a regulatory (accessory) subunit and represent the dominant form of PI3Ks in cardiovascular tissues (Oudit et al., 2004). Class I PI3Ks are subdivided further into class IA and IB, where class IA PI3Ks are activated by RTK, while class IB is activated by GPCR (Leevers, Vanhaesebroeck, & Waterfield, 1999). Activated PI3Ks catalyze the transfer of phosphate from ATP to the 3’position of the inositol ring of the membrane localized phosphoinositides such as phosphatidylinositol (Ptdln), phosphatidylinositol4-phosphate (Ptdlns4P) and phosphatidylinositol4,5-bisphosphate (PtdIns(4,5)P2). Furthermore it promotes the generation of phosphatidylinositol 3-phosphate (PtdIns3P), phosphatidylinositol (3,4)-bisphosphate (PtdIns(3,4)P2) and phosphatidylinositol (3,4,5)-trisphosphate (PtdIns(3,4,5)P3), respectively. These phospholipids act as second messengers to activate several proteins like phosphoinositide dependent kinase 1 (PDK1), Akt and p7OS6K (Kandel & Hay, 1999; Katso et al., 2001).
4.4.1 AKT pathway
The most widely studied downstream target of PI3K pathway is protein kinase B (PKB), also known as Akt (a product of akt proto-oncogene). Akt is a serine/threonine kinase whose activation depends on the generation of 3-phosphorylated phosphoinositides by class I PI3K. It exists as three isoforms in the mammalian system: PKBα/Akt1, PKBβ/Akt2 and PKBγ/Akt3 (Nakatani et al., 1999). PI3K/AKT pathway is triggered in response to insulin (van der Heide et al., 2005), Ang II (Saward & Zahradka, 1997), ET-1 (Daou & Srivastava, 2004) and many other growth factors (Duan, Bauchat, & Hsieh, 2000). Moreover, ROS play an important role in mediating Akt activation by Ang II (Ushio-Fukai et al., 1999) and ET-1 (Daou & Srivastava, 2004) in VSMCs. Once activated, Akt modulates function of several intracellular substrates such as p70S6-kinase (Eguchi, Iwasaki, Ueno, et al., 1999), c-Myc , B-cell lymphoma 2 (Bcl-2) and caspases (Coffer, Jin, & Woodgett, 1998) resulting in regulation of glycogen synthesis,
cell cycle regulation, cell growth, cell survival, and protein synthesis (Marte & Downward, 1997). Dysregulation of the PI3K/Akt pathway is implicated in a number of human diseases including cardiovascular disease. Contribution of AKT in vascular disease was suggested from studies in which angiotensin-induced hypertension in New Zealand White rats was associated with an elevated PI3K/AKT activity (Ljuca et al., 2001).
4.5 Mitogen-activated protein kinases (MAPK) pathway
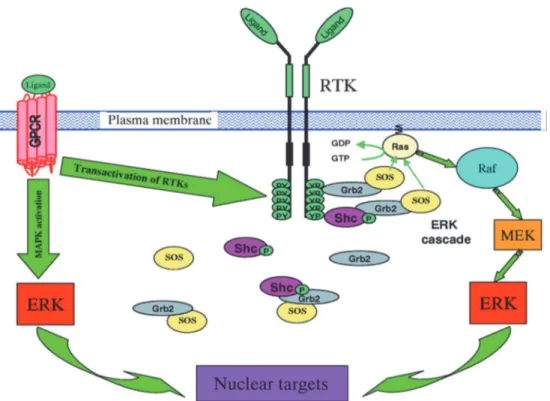
MAPKs are a family of ubiquitous serine/threonine protein kinases (Pearson et al., 2001). ERK1/2, p38MAP kinase (p38mapk), c-Jun-N-terminal kinases (JNK) and ERK5 are the main well characterized groups of MAPKs (Cargnello & Roux, 2011). ERK1/2 is a major growth signaling kinase, whereas p38mapk and JNK influence cell survival, apoptosis, differentiation and inflammation (Pearson et al., 2001). ERK5 is involved in protein synthesis, cell cycle progression and cell proliferation (Abe et al., 1996; Nicol et al., 2001). A variety of stimuli such as growth factors, vasoactive peptides (Ang II and ET-1) and ROS can activate MAPK pathway (Seger & Krebs, 1995). Signals from activated RTK or GPCR to ERK1/2 are transmitted via Ras, a small membrane-bound GTP-binding protein. Once Ras is activated, it recruits Raf, also known as (MAPK) kinase kinase (MKKK). Raf phosphorylates MEK, MKK or MAPK kinase at specific serine/threonine residues, which in turn, phosphorylates MAPKs, such as ERK1/2 on threonine and tyrosine residues, which activate a number of transcription factors involved in gene activation (Figure 4) (Robinson & Cobb, 1997). Much evidence supports that an aberrant activation of the MAPK is often associated with vascular remodeling in cardiovascular diseases (Muslin, 2008). For example, the enhanced activation of vascular MAPKs has been demonstrated in various models of hypertension such as SHR (Xu et al., 1996; Touyz, Deschepper, et al., 2002). Moreover, the activation of MAPK by vasoactive peptides in VSMCs was shown to be involved in vascular changes associated with hypertension ( Touyz, He, El Mabrouk, et al., 2001; Kubo et al., 2002). Moreover, the fact that the pharmacological inhibition of the enhanced activity of MAPK in VSMCs from SHR by the MEK inhibitor, PD98059, inhibits the overexpression of Giα proteins and restores the enhanced proliferation of VSMCs from SHR to control level supports the notion of the implication of MAPK pathway in the hyperproliferation of VSMCs through enhancing expression of Giα proteins (Bou Daou, Li, & Anand-Srivastava, 2016).
Figure 4: MAPK signaling following activation of receptor tyrosine kinase (RTKs) and G-protein coupled receptor (GPCR). Source: (Kholodenko, 2003)
4.6 G-protein signaling
4.6.1 G Protein-Coupled Receptors (GPCRs)
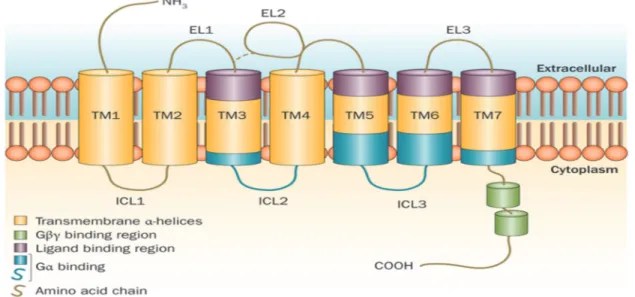
GPCRs are among the most abundant transmembrane receptors that transduce extracellular stimuli into the interior of the cells through interaction with various G proteins leading to a physiological response (Wheatley et al., 2007). The cyclic adenosine monophosphate (cAMP) signal pathway and the phosphatidylinositol signal pathway are the two principal signal transduction pathways of the G protein–coupled receptors (Gilman, 1987). G protein–coupled receptors are implicated in many major diseases such as hypertension, cardiac dysfunction and depression, making them the target of more than 50% of therapeutic drugs (Klabunde & Hessler, 2002; Becker et al., 2004). Catecholamines, peptides, lipids, proteins and glycoprotein hormones are examples of the ligands capable of activating GPCRs. The common structural characteristic of all GPCRs is the presence of 7 transmembrane helices. These transmembrane helices interconnected by three extracellular loops (EL1, EL2, EL3) containing the ligand-binding domain and three alternating intracellular loops (ICL1, ICL2, ICL3) provide ligand-binding
sites for intracellular signaling proteins (Figure 5). They also have an extracellular N-terminus and an intracellular C-terminal tail. The binding of the ligand to the extracellular domain of GPCRs disturbs non-covalent interactions between the transmembrane α helices, thus causing the receptor to take an active conformation which is translated by a rotation or movement of these helices and a change in the conformation of the cytoplasmic loops. This in turn leads to an increase in the affinity of the receptor for a G protein and cause its activation by exchanging the guanosine diphosphate (GDP) bound to the G protein for a guanosine triphosphate (GTP) (Gilman, 1987).
Figure 5: Schematic diagram of a GPCR. Source: (Neumann, Khawaja, & Muller-Ladner, 2014)
4.6.2 Guanine nucleotide-binding proteins (G- protein)
G-proteins are heterotrimeric and membrane-bound proteins that are coupled to GPCRs. They are a large family of guanine nucleotides (GDP or GTP) binding proteins that transduce the signal to intracellular effector molecules. All members of the G proteins family share a common structural core, composed of three distinct subunits: α, β, and γ. There are at least 20 different genes for α-subunits, 5 for β-subunits and 12 for γ-subunits in mammals (Fleming, Wisler, & Watanabe, 1992; Premont, Inglese, & Lefkowitz, 1995). The α subunits is the component that binds guanine nucleotides and confers specificity in receptor and effector interactions (Gilman, 1984).
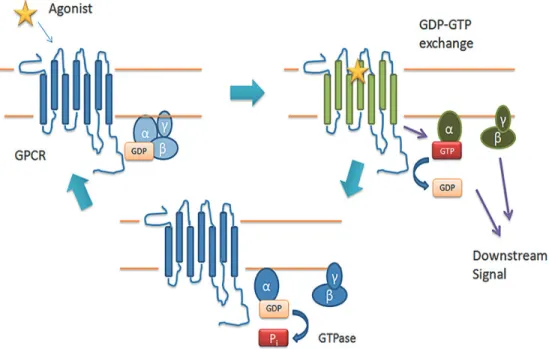
4.6.2.1 Activation of Heterotrimeric G-proteins
In its inactivated state, a G protein maintains its heterotrimeric state and its Gα subunit binds GDP. Upon ligand binding and receptor activation, the G protein is turned on by the interaction with an activated receptor (GPCRs) which replaces GDP by GTP on the guanine nucleotide binding site of the Gα subunit (Fleming, Wisler, & Watanabe, 1992). Binding of GTP to Gα induces a conformational change and promotes the dissociation of G-protein into Gα and Gβγ (Figure 6). After dissociation of the Gα subunit from the Gβγ, Gα subunit binds to an effector such as adenylyl cyclase (AC), while the Gβγ dimer binds to effectors such as ions channels, activate phospholipase Cβ (PLCβ) and phospholipase A. All α-subunits possess intrinsic GTPase activity and hydrolyze guanosine GTP to GDP; by that, the Gα subunit subsequently dissociates from the effector and reassociates with the Gβγ subunit to reform the inactive heterotrimeric G protein (Neer, 1995; Sprang, 1997). Regulators of G protein signaling also known as GTPase- activating proteins play a crucial role in controlling the activity of G proteins by accelerating the rate of GTPase activity ( Ross & Wilkie, 2000).
Figure 6: G-protein-coupled receptor (GPCR)-mediated G-protein activation. Source: (Smith, Sim-Selley, & Selley, 2010)