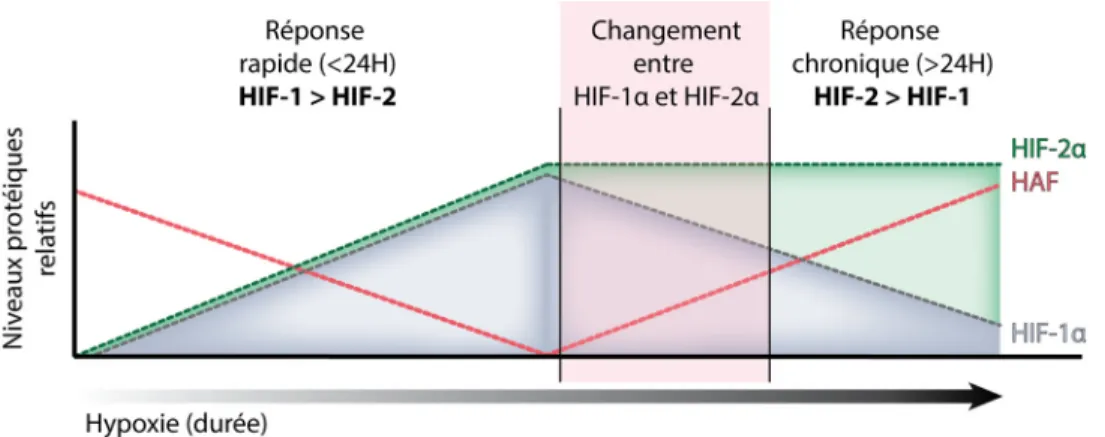
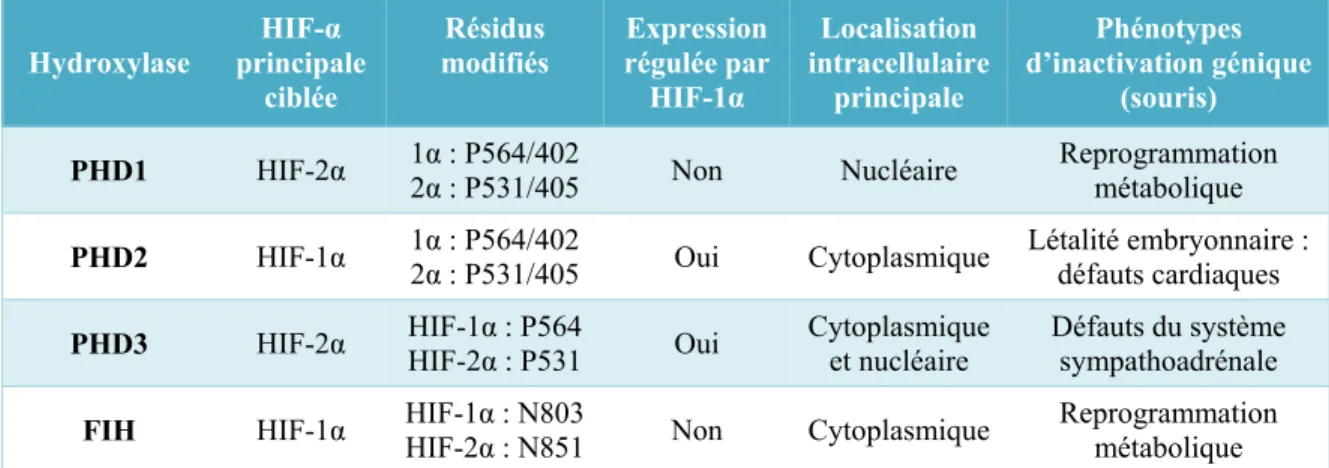
Implication de PRMT1 et de la méthylation
d’arginines dans la signalisation cellulaire et
l’expression des facteurs de transcription induits
par l’hypoxie (HIF)
Thèse
Véronique Lafleur
Doctorat en biologie cellulaire et moléculaire
Philosophiæ doctor (Ph. D.)
Québec, Canada
Implication de PRMT1 et de la méthylation
d’arginines dans la signalisation cellulaire et
l’expression des facteurs de transcription induits
par l’hypoxie (HIF)
Thèse
Véronique Lafleur
Sous la direction de :
iii
Résumé
Des conditions hypoxiques surviennent lors de situations environnementales et physiologiques spécifiques, ainsi que dans le contexte de diverses pathologies. En raison de la place cruciale de l’oxygène dans les fonctions biologiques vitales, des mécanismes complexes ont évolué afin d’assurer une adaptation cellulaire adéquate à tout stress hypoxique. Cette adaptation est induite par une réponse transcriptionnelle rapide et hautement contrôlée. L’étude des mécanismes impliqués dans cette réponse est essentielle à la compréhension de ses conséquences, notamment dans le cadre de pathologies associées à des conditions hypoxiques, tel que la progression tumorale.
Les facteurs de transcription induits par l’hypoxie (HIF ; hypoxia-inducible factors) sont les médiateurs centraux et essentiels à la réponse transcriptionnelle adaptative à l’hypoxie. Ils sont responsables de l’induction de nombreux gènes régulant les processus physiologiques, cellulaires et moléculaires nécessaires à cette adaptation. HIF-1 et HIF-2 sont les principaux membres de cette famille de facteurs de transcription, partageant des fonctions communes, distinctes et complémentaires. Leur activité dépend de leur sous-unité HIF-α respective, HIF-1α et HIF-2α. Divers mécanismes convergent afin d’assurer une modulation précise des HIF-α en fonction du niveau d’hypoxie et du contexte cellulaire. Ceci implique autant des mécanismes inducteurs que répresseurs, intervenant de manière coordonnée. L’étude de ces mécanismes est donc d’un intérêt considérable pour la compréhension de l’adaptation cellulaire en conditions hypoxiques.
Des évidences suggèrent un rôle de la méthylation d’arginines dans la réponse cellulaire à l’hypoxie. Ainsi, cette thèse se penche sur l’implication de l’arginine méthyltransférase PRMT1 dans la régulation des HIF-α. Nous caractérisons PRMT1 en tant que nouveau répresseur de la transcription des gènes HIF1A et HIF2A. Cette répression résulte de l’inhibition des facteurs de transcription Sp1/3, des régulateurs connus de ces gènes. L’étude du mécanisme impliqué nous permet alors de décrire un nouveau rôle de PRMT1 dans la cascade de signalisation ERK. En effet, nous montrons que PRMT1 interagit avec la GEF DOCK6, réprimant ainsi l’activation des kinases ERK1/2 en aval. Ceci a pour conséquence de réduire la phosphorylation et l’activité de Sp1/3. Finalement, nous révélons une dynamique hypoxique intéressante, où une répression temporellement distincte de HIF1A et HIF2A par PRMT1 survient en hypoxie.
Ainsi, cette thèse met en lumière un nouveau mécanisme de régulation des HIF-α et souligne l’importance de la répression de leur transcription dans l’adaptation cellulaire à l’hypoxie. Cette thèse contribue ainsi aux connaissances sur le contexte hautement dynamique et complexe de la régulation des facteurs de transcription HIF, des médiateurs essentiels de l’homéostasie cellulaire.
iv
Abstract
Hypoxic conditions occur under specific environmental et physiological situations, as well as in different pathological contexts. Due to the crucial requirement of oxygen for vital biological functions, complex mechanisms have evolved to ensure adequate cellular adaptation to hypoxic stress. Such adaptation is induced by a rapid and highly controlled transcriptional response. The study of this response is essential for the comprehension of its consequences, particularly in the context of pathologies associated with hypoxic conditions, such as tumor progression.
Hypoxia-inducible factors (HIFs), are central and essential mediators of the adaptive transcriptional response to hypoxic stress. They are responsible for the induction of numerous genes regulating the physiological, cellular and molecular processes necessary for this adaptation. HIF-1 and HIF-2 are the main members of this family of transcription factors and share common, as well as distinct and complementary, fonctions. Their activities are dependent on their respective HIF-α subunits, HIF-1α and HIF-2α. Diverse mechanisms converge in order to allow precise modulation of HIF-α subunits in accordance to hypoxic and cellular contexts. This implicates positive as well as negative regulators, acting in a coordinated fashion. The study of these mechanisms is of considerable interest for the comprehension of cellular adaptation to hypoxia.
Studies suggest a role for arginine methylation in the hypoxic stress reponse. Therefore, this thesis aims at evaluating the role of the protein arginine methyltransferase PRMT1 in HIF-α regulation. We characterize PRMT1 as a novel repressor of HIF1A and HIF2A gene transcription. This repression is caused by the inhibition of Sp1/3 transcription factors, known HIF1A and HIF2A gene regulators. The investigation of the underlining mechanism allowed us to describe a new role for PRMT1 in the ERK signaling cascade. Indeed, we demonstrate that PRMT1 interacts with the Rho GEF DOCK6, repressing downstream ERK1/2 kinases. This results in reduced Sp1/3 phosphorylation and activity. Finally, we reveal an interesting hypoxia-related dynamic, where a distinct temporal repression of HIF1A and HIF2A occurs under hypoxic conditions.
This thesis higlights a new regulatory mechanism of HIF-α subunits and underlines the importance of their transcriptional repression in cellular adaptation to hypoxia. This thesis therefore contributes to the knowlegde surrounding the highly dynamic and complex regulation of HIF transcription factors, essential mediators of cellular homeostasis.
v
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des tableaux ... viii
Liste des figures ... ix
Liste des figures supplémentaires ... xi
Liste des abréviations ... xii
Remerciements ... xvii
Avant-propos ... xix
Chapitre 1 : Introduction ... 1
1.1
L’homéostasie de l’oxygène ... 1
Le stress hyperoxique ... 2
Le stress hypoxique ... 2
1.2
Les situations de stress hypoxique et leurs implications ... 3
Situations environnementales ... 3
Situations physiologiques ... 4
Situations pathologiques ... 4
1.3
Les mécanismes d’adaptation à l’hypoxie ... 7
Les réponses physiologiques à l’hypoxie ... 7
L’adaptation cellulaire à l’hypoxie ... 10
1.4
L’orchestration de la réponse hypoxique : bases moléculaires ... 17
La réponse transcriptionnelle ... 17
La réponse épigénétique ... 21
La réponse traductionnelle ... 24
1.5
Les facteurs de transcription induits en hypoxie (HIF) ... 27
Caractéristiques générales ... 27
HIF-1 et HIF-2 ... 30
HIF-3 ... 32
HIF-1β ... 33
1.6
Les mécanismes de régulation des facteurs HIF ... 36
La régulation post-traductionnelle dépendante de l’O2 : hydroxylation ... 36
Les différentes modifications post-traductionnelles des HIF-α... 43
La régulation traductionnelle des HIF-α ... 51
La régulation transcriptionnelle des HIF-α ... 54
1.7
La méthylation d’arginines ... 61
Caractéristiques générales ... 61
Les enzymes PRMT ... 62
PRMT1, l’arginine méthyltransférase asymétrique principale ... 64
La méthylation d’arginines et la réponse hypoxique ... 67
vi
Chapitre 2: Transcriptional repression of hypoxia-inducible factor-1 (HIF-1)
by the protein arginine methyltransferase PRMT1. ... 73
2.1
Avant-propos ... 74
2.2
Résumé ... 75
2.3
Abstract ... 76
2.4
Introduction ... 77
2.5
Results ... 79
PRMT1 represses HIF-1 complex accumulation ... 79
PRMT1 does not modify HIF-1α protein stability ... 80
PRMT1 represses HIF-1α mRNA expression ... 81
PRMT1 regulates HIF-1α levels in a methyltransferase-dependent manner ... 83
PRMT1 represses HIF transcriptional activity ... 84
Sp1 and Sp3 mediate HIF-1α repression by PRMT1 ... 86
2.6
Discussion ... 89
2.7
Materials and methods ... 92
2.8
Supplementary figures ... 98
Chapitre 3 : PRMT1-mediated arginine methylation of DOCK6 represses
downstream ERK signaling and hypoxia-inducible factor (HIF) expression. . 105
3.1
Avant-propos ... 106
3.2
Résumé ... 107
3.3
Abstract ... 108
3.4
Introduction ... 109
3.5
Results ... 111
PRMT1 represses ERK1/2 signaling. ... 111
PRMT1 methylates and inhibits the RhoGEF DOCK6. ... 113
DOCK6 and PAK1 are implicated in PRMT1-dependent ERK1/2 regulation. ... 115
PRMT1 and DOCK6 regulate HIF-1α expression through ERK1/2 signaling and Sp1/3 phosphorylation. ... 117
A HIF-dependent feedback loop modulates PRMT1-mediated HIF-1α and HIF-2α regulation under prolonged hypoxic stress. ... 119
3.6
Discussion ... 122
The role of PRMT1 in ERK1/2 signaling regulation. ... 122
The implication of DOCK6 methylation in PRMT1-mediated ERK1/2 and HIF-α regulation. ... 123
The dynamic regulation of HIF-1α and HIF-2α by PRMT1-associated signaling under prolonged hypoxic stress. ... 123
vii
Chapitre 4 : Discussion ... 134
4.1
L’importance de mécanismes suppresseurs des HIF-α ... 134
Les mécanismes de résolution des HIF-α ... 135
4.2
PRMT1 : un nouveau répresseur de la signalisation hypoxique ... 135
Une répression transcriptionnelle des HIF-α sous le contrôle de PRMT1 ... 136
La modulation dynamique des gènes HIF1A et HIF2A par PRMT1 ... 138
4.3
PRMT1 : un nouveau répresseur de la signalisation ERK ... 143
DOCK6 : une nouvelle GEF activant la signalisation ERK1/2 ... 143
DOCK6 : une nouvelle cible de PRMT1 ... 144
L’impact de PRMT1 et d’ERK1/2 sur la phosphorylation de Sp1/3 ... 147
Les multiples niveaux de régulation des HIF-α par ERK1/2 ... 148
Chapitre 5 : Conclusions et Perspectives ... 149
Références ... 151
Annexe 1 ... 185
Annexe 2 ... 187
Annexe 3 ... 188
Annexe 4 ... 190
Annexe 5 ... 191
Annexe 6 ... 192
viii
Liste des tableaux
Chapitre 1 : Introduction
Tableau 1.1: Les caractéristiques des hydroxylases ciblant les HIFα. ... 39
Tableau 1.2: Les caractéristiques des ubiquitine-ligases ciblant les HIF-α... 44
Tableau 1.3: Les acétyltransférase et déacétylases ciblant HIF-α et leur régulation en hypoxie. ... 45
Tableau 1.4 : Les SUMO ligases et protéases ciblant HIF-α et leur régulation en hypoxie. ... 46
Tableau 1.5: Les kinases ciblant HIF-α et leur régulation en hypoxie. ... 47
Chapitre 2 : Transcriptional repression of hypoxia-inducible factor-1 (HIF-1) by the
protein arginine methyltransferase PRMT1
Table 2.1: Primers used for qPCR. ... 94Chapitre 3 : PRMT1-mediated arginine methylation of DOCK6 represses
downstream ERK signaling and hypoxia-inducible factor (HIF) expression
Table 3.1: Antibodies used for immunoblot analysis. ... 126ix
Liste des figures
Chapitre 1 : Introduction
Figure 1.1: Les réponses adaptatives cellulaires et physiologiques à l’hypoxie. ... 8
Figure 1.2: La reprogrammation métabolique en hypoxie médiée par le facteur HIF-1. ... 11
Figure 1.3: La modulation du cycle cellulaire en hypoxie. ... 13
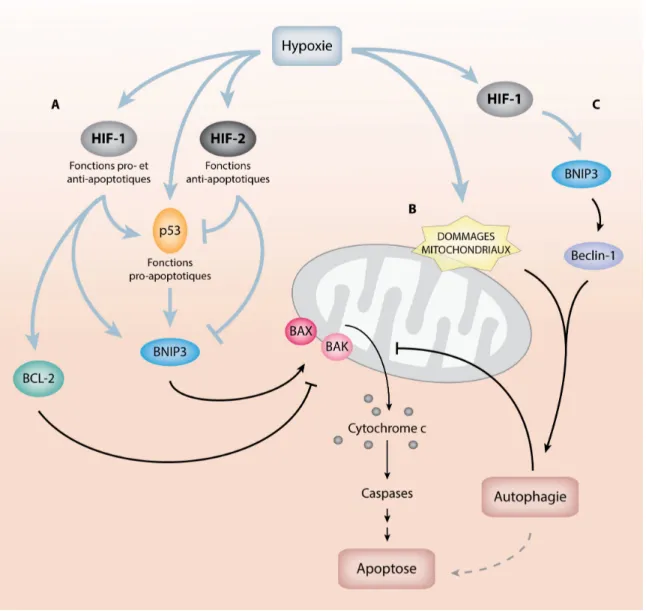
Figure 1.4: La régulation de la signalisation intrinsèque d’apoptose en hypoxie. ... 15
Figure 1.5: Les gènes cibles des HIF. ... 19
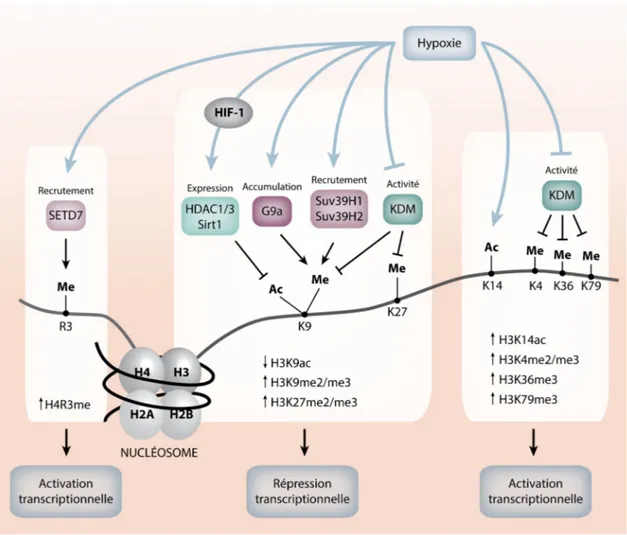
Figure 1.6: La régulation des modifications post-traductionnelles des histones en hypoxie. ... 22
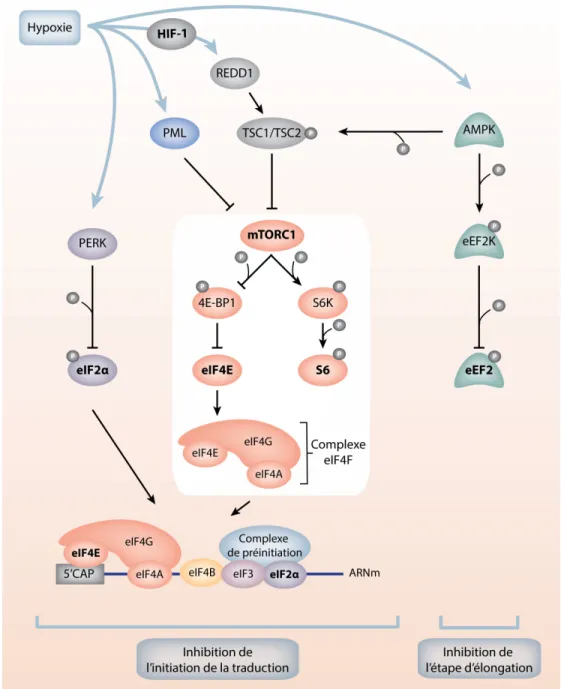
Figure 1.7: La répression globale de l’initiation de la traduction en hypoxie. ... 25
Figure 1.8: Les cinétiques de dégradation et stabilisation des HIF-α. ... 28
Figure 1.9: Les domaines fonctionnels des sous-unités HIF. ... 29
Figure 1.10: Les niveaux et l’implication respectives de HIF-1 et HIF-2 dans la réponse hypoxique. ... 32
Figure 1.11: La régulation des HIF-α dépendante de l’O2. ... 37
Figure 1.12: La sensibilité respective des PHD et de FIH face à une diminution de l’O2. ... 41
Figure 1.13: Les diverses modifications post-traductionnelles de HIF-1α et HIF-2α. ... 43
Figure 1.14: Représentation schématique de la cascade de signalisation ERK. ... 50
Figure 1.15: Les régulateurs de la traduction et de la stabilité de l’ARNm HIF1A. ... 52
Figure 1.16: Les domaines fonctionnels de Sp1 et Sp3 et leur régulation par phosphorylation. ... 56
Figure 1.17: Les divers facteurs de transcription ciblant HIF-1α en réponse à différents stimuli. ... 59
Figure 1.18: La méthylation d’arginines catalysée par les PRMT. ... 62
Figure 1.19: Les domaines et motifs fonctionnels de PRMT1. ... 63
Figure 1.20: La diminution du potentiel de méthylation cellulaire en hypoxie. ... 69
Figure 1.21: L’altération du métabolisme des ADMA en hypoxie. ... 70
Figure 1.22: La modulation de HIF-1α par les PRMT. ... 71
Chapitre 2 : Transcriptional repression of hypoxia-inducible factor-1 (HIF-1) by the
protein arginine methyltransferase PRMT1
Figure 2.1: PRMT1 depletion increases HIF-1α protein accumulation. ... 79Figure 2.2: PRMT1 depletion does not modify HIF-1α protein stability. ... 81
Figure 2.3: Cellular PRMT1-depletion increases HIF-1α mRNA expression. ... 82
Figure 2.4: PRMT1’s methyltransferase activity is required for HIF-1α repression. ... 84
Figure 2.5: HIF activity is increased in PRMT1-depleted cells. ... 85
Figure 2.6: Sp1 and Sp3 transcriptional activity is increased following PRMT1-depletion and required for HIF induction. ... 87
x
Chapitre 3 : PRMT1-mediated arginine methylation of DOCK6 represses
downstream ERK signaling and hypoxia-inducible factor (HIF) expression
Figure 3.1: PRMT1-deficient cells show increased MEK1/2 and ERK1/2 phosphorylation. ... 112
Figure 3.2: DOCK6 is arginine methylated and inhibited by PRMT1. ... 114
Figure 3.3: DOCK6, as well as its downstream effector PAK1, are required for induced ERK1/2 signaling in PRMT1-deficient cells. ... 116
Figure 3.4: Induced HIF-1α expression under PRMT1 depletion or DOCK6 overexpression is mediated by ERK1/2 signaling and Sp1/3 phosphorylation. ... 118
Figure 3.5: Hypoxic conditions differentially modulate HIF-1α and HIF-2α induced expression under PRMT1 depletion, through a HIF-dependent feedback mechanism. ... 120
Figure 3.6: Schematic model of the dynamic regulation of HIF-α subunit expression by PRMT1, DOCK6 and ERK1/2 signaling. ... 125
Chapitre 4: Discussion
Figure 4.1: Représentation schématique de la répression distincte de HIF1A et HIF2A par PRMT1 en conditions hypoxiques. ... 139
Figure 4.2: Représentation schématique de la modulation différentielle de l’ARNm HIF1A et HIF2A en conditions hypoxiques, sous le contrôle de PRMT1 et d’une boucle
d’autorégulation de HIF1A. ... 140
Figure 4.3: Modèle hypothétique de régulation de HIF1A et HIF2A par PRMT1 et Sp1/3 en
hypoxie. ... 142
Figure 4.4: Représentation schématique de l’implication de PRMT1 et DOCK6 dans la régulation de la cascade ERK, de Sp1/3 et des gènes HIFA. ... 146
xi
Liste des figures supplémentaires
Chapitre 2 : Transcriptional repression of hypoxia-inducible factor-1 (HIF-1) by the
protein arginine methyltransferase PRMT1
Supplementary figure 2.1: PRMT1 depletion increases HIF-1α protein and mRNA levels at different time points under hypoxic conditions. ... 98
Supplementary figure 2.2: PRMT1 depletion increases HIF-2α accumulation. ... 99
Supplementary figure 2.3 : PRMT5 is not implicated in HIF-1α regulation. ... 100
Supplementary figure 2.4: Evaluation of actinomycin D effectiveness in blocking transcription. . 101
Supplementary figure 2.5: Asymmetric arginine methylation by wild-type PRMT1 but not a
catalytically inactive mutant. ... 102
Supplementary figure 2.6: Induction of hypoxia-associated target genes in a HIF-dependent manner. ... 103
Supplementary figure 2.7: Implication of Sp1 and Sp1 in regulation of the HIF-1α gene promoter. ... 104
xii
Liste des abréviations
ADMA de l’anglais : ω-NG,NG-asymmetric dimethylarginine
ADN Acide désoxyribonucléique
AMI de l’anglais : Arginine methyltransferase inhibitor AMP Adénosine monophosphate
AMPK de l’anglais : AMPK-activated protein kinase
AP1 de l’anglais : Activator protein 1 ARE de l’anglais : AU-rich element ARN Acide ribonucléique
ARNT de l’anglais : Aryl hydrocarbon receptor nuclear translocator
ARNTL de l’anglais : ARNT-like protein
atm Atmosphère
ATM de l’anglais : Ataxia telangiectasia mutated ATP Adénosine triphosphate
ATR de l’anglais : Ataxia telangiectasia and Rad3-related BAK de l’anglais : BCL-2 antagonist/killer
BAX de l’anglais : BCL-2-associated X
BCL-2 de l’anglais : B-cell lymphoma 2
BMP de l’anglais : Bone morphogenetic protein
BRAF de l’anglais : Serine/threonine-protein kinase B-raf
BSA de l’anglais : Bovine serum albumin CA9 de l’anglais : Carbonic anhydrase 9
CAT de l’anglais : Cationic amino acid transporter CBP de l’anglais : CEBP-binding protein
Cbx4 de l’anglais : Chromobox protein homolog 4
CDK de l’anglais : Cyclin-dependent kinase
CEBP de l’anglais : CCAAT-enhancer binding protein
CHIP de l’anglais : C-terminus of Hsc70 interacting protein
CKI/CKII de l’anglais : Casein kinase I/II
CO2 Dioxide de carbone
COX de l’anglais : Cytochrome c oxidase
CPEB de l’anglais : Cytoplasmic polyadenylation-element-binding protein
CREB de l’anglais : cAMP response element-binding
DDAH Diméthylarginine diméthylaminohydrolase
DFO Desferrioxamine
DHR de l’anglais : DOCK Homology Region
DMEM de l’anglais : Dulbecco’s modified Eagle’s medium
DMOG Diméthyloxaloylglycine
xiii DNA de l’anglais : Deoxyribonucleic acid
DNMT de l’anglais : DNA methyltransferase
DOCK de l’anglais : Dedicator of cytokinesis
DSP de l’anglais : Dithiobis(succinimidyl propionate)
DUSP de l’anglais : Dual specificity phosphatase
ECL de l’anglais : Enhanced chemiluminescence
EDTA de l’anglais : Ethylenediaminetetraacetic acid
EGF de l’anglais : Epidermal growth factor
EGFR de l’anglais : Epidermal growth factor receptor
EGLN de l’anglais : Egg-laying defective nine
EGR-1 de l’anglais : Early growth response 1
ELAVL1 de l’anglais : Embryonic lethal, abnormal vision, Drosophila-like 1
EPAS1 de l’anglais : Endothelial PAS domain protein 1
EPO Érythropoïétine
ERα de l’anglais : Estrogen receptor α
ERK de l’anglais : Extracellular signal-regulated kinase FAK de l’anglais : Focal adhesion kinase
FBS de l’anglais : Fetal bovine serum FIH de l’anglais : Factor inhibiting HIF
FOXO de l’anglais : Forkhead box O1
GAP de l’anglais : GTPase-activating protein GAR de l’anglais : Glycine- and arginine-rich motif GC de l’anglais : Glycine- and cytosin-rich motif GDP Guanosine diphosphate
GEF de l’anglais : Guanine nucleotide exchange factor
GLUT de l’anglais : Glucose transporter
GPCR de l’anglais : G-protein-coupled receptors
GSK3β de l’anglais : Glycogene synthase kinase 3β
GST Glutathione S-transférase GTP Guanosine triphosphate HA Hémagglutinine
HAF de l’anglais : Hypoxia-associated factor HAS de l’anglais : HIF-ancillary sequence HAT Histone acétyltransférase
HBS de l’anglais : HIF-binding site
HDAC Histone déacetyltransférase
HEK de l’anglais : Human embryonic kidney cells HGF de l’anglais : Hepatocyte growth factor HIC Hypoxie intermittente chronique HIF de l’anglais : Hypoxia-inducible factor
xiv
HIG2 de l’anglais : Hypoxia-inducible gene 2
HILPDA de l’anglais : Hypoxia-inducible lipid droplet-associated protein
bHLH de l’anglais : Basic helix-loop-helix
HNRNP de l’anglais : Heterogeneous nuclear ribonucleoprotein
HPRT Hypoxanthine phosphoribosyltransférase
HRE de l’anglais : Hypoxia-responsive element HRF de l’anglais : HIF-related factor
HRP de l’anglais : Horseradish peroxidase HuR de l’anglais : Human antigen R ID de l’anglais : Inhibitory domain IP Immunoprécipitation
IPA-3 de l’anglais : group 1 PAK inhibitor
IR de l’anglais : Insulin receptor
IRE de l’anglais : Iron-responsive element
IRES de l’anglais : Internal ribosome entry site
IRP de l’anglais : Iron regulatory protein
IRS-1 de l’anglais : Insulin receptor substrate 1
JMJD de l’anglais : JumonjiC (JmjC) domain-containing demethylase
JNK1 de l’anglais : c-Jun N-terminal kinase 1
KAT Lysine acétyltransférase
KDAC Lysine déacétylase
KDM Lysine déméthylase KMT Lysine méthyltransférase LPS Lipopolysaccharide
LZIP de l’anglais : leucine zipper
MAPK de l’anglais : Mitogen-activated protein kinase
MAT Méthionine adénosyltransférase
MCM de l’anglais : Minichromosome maintenance
MCT-4 de l’anglais : Monocarboxylate transporter 4
MEF de l’anglais : Mouse embryonic fibroblast MEK de l’anglais : MAPK/ERK kinase
MITF de l’anglais : Microphthalmia-associated transcription factor
MMA de l’anglais : ω-NG-monomethylarginine
mmHg Millimètre de mercure
MMP de l’anglais : Matrix metallopeptidase
MOP2 de l’anglais : Member of PAS superfamily
MPOC Maladie pulmonaire obstructive chronique
α-MSH de l’anglais : α-melanocyte-stimulating hormone
MYND de l’anglais : Myeloid, Nervy, DEAF1
xv
NADP Nicotinamide adénine dinucléotide phosphate
NADPH Nicotinamide adénine dinucléotide phosphate réduit
NC2 de l’anglais : Negative cofactor 2
NFĸB de l’anglais : Nuclear factor kappa light-chain-enhancer of activated B-cells
NFY de l’anglais : Nuclear transcription factor Y
NHE1 de l’anglais : Na+/H+exchanger 1
NLS de l’anglais : Nuclear localisation signal NO de l’anglais : Nitric oxide
NOS de l’anglais : Nitric oxide synthase
O2 Dioxygène
pO2 Pression partielle d’O2
ODDD de l’anglais : Oxygen-dependent degradation domain
2-OG 2-oxoglutarate
OH Hydroxyde
4-OHT 4-hydroxytamoxifène
PAK de l’anglais : p21-activated kinase
PCAF de l’anglais : p300/CBP-associated factor
PCR de l’anglais : Polymerase chain reaction
PDGF de l’anglais : Platelet-derived growth factor
PDGFR de l’anglais : Platelet-derived growth factor receptor
PDH Pyruvate déshydrogénase
PDK1 Pyruvate déhydrogénase kinase 1
PEI Polyéthylènimine
PERK de l’anglais : Protein kinase RNA-like endoplasmic reticulum kinase
PGC-1α de l’anglais : Peroxisome proliferator-activated receptor γ coactivator-1α
PHD de l’anglais : Prolyl hydroxylase domain-containing protein
PIAS de l’anglais : Protein inhibitor of activated STAT protein
PIC de l’anglais : Pre-initiation complex PKC Protéine kinase C
PMA de l’anglais : Phorbol-12-myristate-13-acetate PML de l’anglais : Promyelocytic leukemia
PRMT Protéine arginine méthyltransférase
PTB de l’anglais : Polypyrimidine tract-binding protein
PV Pervanadate
PVDF de l’anglais : Polyvinylidene difluoride membrane
RBP de l’anglais : RNA-binding protein
RBX1 de l’anglais : Ring box protein 1
REST de l’anglais : Repressor element 1-silencing transcription factor
RNA de l’anglais : Ribonucleic acid
xvi
RSUME de l’anglais : RWD-containing sumoylation enhancer
RT de l’anglais : Reverse transcription RTK Récepteur à activité tyrosine kinase SAH S-adénosyl-homocystéine
SAHH S-adénosyl-homocystéine hydrolase
SAM S-adénosyl-méthionine
SAOS Syndrome de l’apnée obstructive du sommeil
SCF SKP1/Culline1/F-box
SDMA ω-NG, N’G-symmetric dimethylarginine
SDS de l’anglais : Sodium dodecyl sulfate SEM de l’anglais : Standard error of the mean
SENP de l’anglais : Sentrin-specific protease
SETD7 de l’anglais : SET domain-containing protein 7
SFPQ de l’anglais : Splicing factor, proline- and glutamine-rich
SILAC de l’anglais : Stable-isotope labeling by amino acids in cell culture
SKP1 de l’anglais : S-phase kinase-associated protein 1
SMAD de l’anglais : Mothers against decapentaplegic
SOS de l’anglais : Son of sevenless
Sp1/3 de l’anglais : Specificity protein 1/3
STAT3 de l’anglais : Signal transducer and activator of transcription 3
SUMO de l’anglais : Small ubiquitin-related modifier
TAD de l’anglais : Transactivation domain TET de l’anglais : TET-Eleven Translocations
TFII de l’anglais : Transcription factor II
TGF-β de l’anglais : Transforming growth factor β
TIAR de l’anglais : T-cell intracellular antigen-1 (TIA-1)-related protein
TK Thymidine kinase
TNFR de l’anglais : Tumor necrosis factor receptor
TTP Tristetraproline
UTR de l’anglais : Untranslated region
VCB-CR pVHL/Élongines B,C/Culline2/RBX1
VEGF de l’anglais : Vascular endothelial growth factor
pVHL de l’anglais : von Hippel-Lindau tumor suppressor protein
VSMC de l’anglais : Vascular smooth muscle cells
WT de l’anglais : Wild-type °C Degré Celsius
xvii
Remerciements
Me penchant sur le chemin parcouru, il m’est clair à quel point la réussite associée en est une personnelle, mais également partagée. Et à partager. Peu nombreux sont les moments où l’on prend réellement le temps de se poser, de retracer nos pas et de remercier tous ceux ayant contribué à nous faire avancer. J’en prends ici l’occasion.
Les premiers pas de ce parcours au sein du laboratoire du Dr Darren Richard, je les ai faits en tant qu’étudiante au baccalauréat, impressionnée et emballée à l’idée de découvrir le monde de la recherche. Je remercie sincèrement le Dr Darren Richard, qui de par sa disponibilité et son ouverture m’a permis de tracer mon chemin dans ce milieu avec de plus en plus d’assurance. Je suis reconnaissante pour tous ses encouragements, sa patience et sa rigueur qui m’ont poussée à persévérer à travers les obstacles nombreux et à célébrer les réussites, aussi petites soient-elles. Je le remercie d’avoir façonné mon autonomie, de m’avoir accordé sa confiance et d’avoir cru en mes capacités, pour que je puisse y croire aussi. Je suis aussi très reconnaissante des opportunités qui m’ont été accordées, dont les nombreux congrès auxquels j’ai eu la chance de prendre part et à travers lesquels j’ai pu voyager tout en découvrant un monde scientifique impressionnant.
Tout au long de mes années au laboratoire, plusieurs personnes ont été présentes et ont contribué à l’avancement de mon parcours. Je suis extrêmement reconnaissante pour tout ce que chacune d’elles a pu m’apporter, tant du point de vue scientifique que personnel. Je débute par souligner le groupe qui fut là à mon premier été au laboratoire : Marc-André, Marie-Claude, Maude, David, Wagner, Françoise et Cristina. L’ambiance amicale et collaboratrice régnant dans le laboratoire grâce à eux contribua à ma motivation et à mon envie de poursuivre dans cette voie. Un merci tout particulier à Marc-André, qui m’a pris sous son aile et a su me transmettre de nombreux outils et connaissances nécessaires à ma réussite future. Je souhaite ensuite remercier Geneviève, qui fut d’une aide indispensable à travers ma maîtrise. J’ai appris énormément à travers elle, tant sa disponibilité, sa gentillesse et sa patience furent infinies. Je garde en bon souvenir tous nos diners en tête-à tête et notre escapade en congrès sous le soleil de Los Angeles. Je remercie également Laurent, présent à travers mon doctorat, pour ses conseils, son soutien et ses encouragements, mais également pour son calme et sa bonne humeur contagieuse. Nos partages musicaux et discussions de toutes sortes ont su apporter une atmosphère de travail agréable.
Mon cheminement a reçu un vent de motivation à l’arrivée de celles qui ont été mes partenaires et complices à travers l’aventure du doctorat, Sophie et Maroua. J’ai découvert en elles des amies, des confidentes et des personnes encourageantes. Je les remercie énormément d’avoir partagé les hauts et les bas du doctorat avec moi. Leur présence fut non seulement rassurante, mais
xviii
stimulante, et a su grandement embellir mon quotidien. Sophie : pour son écoute, son dynamisme, sa motivation contagieuse et pour les nombreuses expériences partagées en voyage et en congrès. Maroua : pour ses mots d’encouragements, son calme, sa détermination et pour les nombreux rires et partages culturels. Je garderai en mémoire l’environnement d’entraide que nous avons pu créer.
Au fil des années, plusieurs personnes ont également apporté leur énergie au laboratoire, le temps de leur passage. Je remercie notamment Noémie, que j’ai eu le plaisir de superviser et qui a su mettre sa motivation à contribution de mon projet. Sans oublier Bruna, avec toute sa délicatesse et sa minutie, et Ronald, avec toute son énergie et sa confiance. Enfin, Catherine, faisant maintenant le pont vers la troisième génération du laboratoire avec assurance et habileté et envers qui je suis très reconnaissante pour l’aide précieuse en cette fin de parcours.
Je souhaite remercier les membres du jury qui ont accepté d’évaluer ma thèse et de m’en faire leurs commentaires. Je me dois également de souligner les organismes subventionnaires qui m’ont octroyé des bourses d’études tout au long de ma maitrise et de mon doctorat : le Fonds de recherche du Québec - Nature et technologies (FQRNT), le Fonds de recherche du Québec - Santé (FRQS), les Instituts de recherche en santé du Canada (IRSC) et la Fondation du CHU de Québec.
À l’extérieur du cadre scientifique, je tiens à remercier ceux et celles qui ont fait de mon passage à Québec une étape remplie de rencontres et d’expériences enrichissantes. Je pense notamment à Niraj, pour tous ces moments partagés autour d’un café, d’un repas et lors de diverses soirées. Je pense à mes nombreux colocs, qui ont partagé mon quotidien à différentes périodes, y apportant une touche de réconfort et de distraction. Une mention toute spéciale aussi à mes amies de longue date, qui ont fait la route à plusieurs reprises pour venir me visiter, me ramenant un peu de par chez moi avec elles, tout en me faisant sortir de ma bulle.
Je termine par remercier sincèrement les personnes m’ayant soutenu inconditionnellement à travers ce parcours. Mes parents : toujours présents avec leurs paroles encourageantes ; toujours disponibles pour me soutenir dans mes déceptions et pour célébrer mes réussites. Mon frère : me transmettant toute sa fierté et son optimisme ; me démontrant à quel point la persévérance et le travail nous mènent loin dans la poursuite de nos objectifs. Finalement, Anthony : qui fait ce parcours tumultueux à mes côtés depuis les trois dernières années ; dont les encouragements ont su me relever à de nombreux moments et ont su me pousser à me surpasser ; et dont la force et la générosité sont continuellement sources d’inspirations.
Arrivée au bout de ces longues années d’études, j’ai le sentiment d’avoir énormément grandi et appris. Les défis surmontés me permettent maintenant de me tourner vers la suite avec espoir et confiance, gardant en poche tous les conseils et outils obtenus en cours de route grâce à la présence de chacune de ces personnes. Bonne lecture!
xix
Avant-propos
L’intérêt global du laboratoire du Dr Darren Richard consiste en la compréhension de la régulation des facteurs de transcription induits en hypoxie, HIF. Des résultats préliminaires effectués au laboratoire suggéraient que la sous-unité HIF-1α pouvait être modifiée par méthylation d’arginines, une modification post-traductionnelle majeure. L’enzyme responsable restait toutefois inconnue. À mes débuts au laboratoire, j’ai alors entrepris d’investiguer l’implication de l’arginine méthyltransférase PRMT1 dans cette potentielle modification de HIF-1α. Mes travaux initiaux lors d’un stage estival m’ont permis de montrer que PRMT1 ne semblait pas être impliquée dans une méthylation directe de HIF-1α, mais modulait néanmoins l’activité de HIF-1α. Ceci m’amena, au cours de ma maitrise et mon doctorat, à me pencher sur le rôle précis de PRMT1 dans la régulation indirecte des facteurs HIF.
Le Chapitre 1 de cette thèse consiste en un aperçu du rôle des facteurs de transcription HIF au sein de la réponse cellulaire à l’hypoxie, mais aussi en une description des différents membres de cette famille et des divers facteurs impliqués dans leur régulation. Ce chapitre permet également de décrire le processus de méthylation d’arginines et de faire état des évidences sur ses liens avec la signalisation hypoxique.
Au Chapitre 2 sont présentés mes travaux traitant de l’impact de PRMT1 sur l’expression et l’activité des sous-unités HIF-α. Ces travaux intitulés : Transcriptional repression of hypoxia-inducible factor-1 (HIF-1) by the protein arginine methyltransferase PRMT1 (Lafleur VN, Richard S et Richard DE), furent l’objet d’une publication dans le journal Molecular Biology of the Cell en janvier 2014.
Le Chapitre 3 de cette thèse consiste en la poursuite de cette étude, où j’ai pu caractériser le
mécanisme par lequel PRMT1 régule l’expression des HIF-α. Ceci me permit de mettre en évidence une signalisation impliquant les kinases ERK1/2, des acteurs importants dans la signalisation hypoxique. Ces travaux intitulés : PRMT1-mediated arginine methylation of DOCK6 represses downstream ERK signaling and hypoxia-inducible factor (HIF) expression (Lafleur VN, Turgeon C, Couturier A, Lamalice L et Richard DE), sont en actuellement en finalisation en vue d’une soumission pour publication.
À travers mon parcours, j’ai eu l’opportunité de prendre part à d’autres projets du laboratoire, ayant tous comme objectif d’approfondir la compréhension des mécanismes régulant les HIF, mais déviant du sujet précis de cette thèse. Les résumés et hyperliens de ces articles publiés sont présentés en annexe de cette thèse.
xx
Ainsi, j’ai participé à des travaux sur la régulation des HIF par l’isomérase Pin1, suite à leur phosphorylation par les kinases ERK1/2. Ces travaux intitulés : The prolyl isomerase Pin1 regulates hypoxia-inducible transcription factor (HIF) activity (Jalouli M, Déry MA, Lafleur VN, Lamalice L, Zhou XZ, Lu KP et Richard DE), furent publiés en aout 2014 dans le journal Cellular Signalling (Annexe 4).
J’ai également contribué à deux études démontrant le rôle des ROS mitochondriaux dans l’induction de HIF-1α dans deux différents contextes :
- L’implication de l’hélicase/exonucléase Werner dans la régulation de HIF-1α a fait l’objet d’une publication en aout 2012 dans le journal Experimental Cell Research : The Werner syndrome gene product (WRN) : a repressor of hypoxia-inducible factor-1 activity (Labbé A, Lafleur VN, Patten DA, Robitaille GA, Garand C, Lamalice L, Lebel M et Richard DE) (Annexe 5).
- L’étude du rôle des ROS mitochondriaux dans la régulation de HIF-1α par l’hormone vasoactive angiotensine II fut publiée en septembre 2010 dans le journal Molecular Biology of the Cell : Hypoxia-inducible factor-1 activation in nonhypoxic conditions : the essential role of mitochondrial-derived reactive oxygen species (Patten DA, Lafleur VN, Robitaille GA, Chan DA, Giaccia AJ et Richard DE) (Annexe 6).
1
1
Chapitre 1 : Introduction
1.1 L’homéostasie de l’oxygène
L’oxygène, deuxième élément le plus abondant sur terre, est retrouvé dans la majorité des molécules biologiques nécessaires à la vie. Lorsque retrouvé sous sa forme de molécule de dioxygène (O2), il est indispensable aux organismes aérobiques, pour lesquels il représente le comburant
essentiel au métabolisme cellulaire. Dans ces organismes, le processus de respiration mitochondriale hautement sophistiqué exploite les propriétés de la molécule d’O2 pour la synthèse d’ATP
(adénosine-5’-triphosphosphate). L’hydrolyse de l’ATP fournit l’énergie nécessaire aux réactions chimiques des cellules. Ainsi, un équilibre précis entre l’apport en O2 et sa consommation, assurant son homéostasie,
doit être maintenu au sein des organismes et leurs tissus.
La quantité d’O2 dans le sang, les tissus et organes est déterminée sous forme de pression
partielle d’oxygène (pO2), principalement exprimée en millimètre de mercure (mmHg) ou en
pourcentage (%). La pression partielle d’un gaz se définit comme la pression de ce gaz au sein d’un mélange gazeux, mais est également utilisée pour décrire la pression d’un gaz dissout dans le sang.
Sur terre, la pression absolue (ou atmosphérique, atm) au niveau de la mer est de 1 atm (760 mmHg). Ainsi, l’O2 qui représente 20,9 % du mélange total a une pression de 0,21 atm ou
160 mmHg. Cette pO2 diminue graduellement à mesure qu’elle passe des poumons (110-150 mmHg ;
15-20 %) et diffuse du système artériel (100 mmHg ; 13 %) vers les différents tissus et organes périphériques [1]. Chaque organe possède une pO2 distincte et optimale (en majorité située entre
10-70 mmHg ; 1-10 %) correspondant à sa vascularisation, ses fonctions et ses besoins métaboliques. Par exemple, le cerveau, le foie, le cœur, les reins et la rétine qui sont parmi les organes les plus actifs au niveau métabolique ont une demande particulièrement élevée en O2. Une altération dans la
disponibilité d’O2 peut avoir des conséquences néfastes pour les fonctions de ces organes et mener à
divers troubles pathologiques.
La pO2 optimale, propre à chaque tissu ou organe, se définit comme un état de normoxie.
Aussi bien un apport insuffisant qu’un apport excessif en O2 peut être néfaste pour l’organisme. On
parle alors respectivement d’hypoxie (du grec hupo : sous, et de oxus : oxygène) et d’hyperoxie (du grec huper : au-dessus). Il est à noter qu’en plus des besoins relatifs de chaque tissu et organe, des processus physiologiques et pathologiques peuvent influencer les besoins en oxygène à un moment précis, par exemple lors du développement, et ainsi faire varier l’état considéré comme normoxique, en un état hypoxique ou hyperoxique.
2
Le stress hyperoxique
L’hyperoxie survient principalement en conditions cliniques, lors de l’administration d’O2 à
un patient par ventilation mécanique (ventilateur/respirateur artificiel). Une carence en O2 est un des
problèmes les plus réguliers dans le contexte de soins intensifs, faisant de l’O2 un des agents
thérapeutiques les plus utilisés [2, 3]. Ainsi, l’administration de concentrations élevées en O2 (de
20-100 %) est routinière pour contrer l’hypoxémie (diminution de la pO2 artérielle) causée par une
oxygénation ou ventilation inadéquate. Ceci peut être nécessaire lors de différentes pathologies, dont quelques-unes seront présentées à la section 1.2.3.
Néanmoins, une hyperoxie prolongée ou trop prononcée peut entrainer des dommages cellulaires importants, menant à diverses conséquences non négligeables sur les organes [4]. Le système pulmonaire peut être particulièrement affecté, de même que le système oculaire (rétinopathie), le système nerveux central (convulsions et crises) et le système sanguin (érythrocytose) [5].
Le stress hypoxique
L’hypoxie survient lorsque la quantité d’O2 disponible descend sous un seuil critique,
empêchant le maintien d’une production d’énergie optimale pour combler les besoins cellulaires. Chez les mammifères, l’apport en O2 aux tissus est principalement dépendant de la pO2 artérielle et
du débit sanguin. Toute situation affectant ces paramètres peut donc être associée à un stress hypoxique : une diminution de la concentration d’O2 inspirée et atteignant le sang (hypoxémie), une
diminution de la capacité du sang à transporter l’O2, une altération dans la perfusion des tissus
(ischémie), une altération de la l’utilisation de l’O2 par les cellules.
Une carence en O2, même pour de courtes périodes, peut être néfaste pour les processus
biologiques. Ainsi, quelle qu’en soit la cause, un apport d’O2 insuffisant résulte en divers processus
de compensation, locaux ou systémiques, permettant à l’organisme ou aux organes de maintenir leurs activités vitales. Ces processus sont abordés plus loin dans ce chapitre.
3
1.2 Les situations de stress hypoxique et leurs implications
Cette section fait état des situations d’hypoxie les plus communes chez l’humain et présente certaines implications physiologiques et/ou physiopathologiques associées. Ceci, dans le but de souligner l’importance et l’intérêt derrière la compréhension des divers mécanismes d’adaptation à l’hypoxie.
Il existe différents contextes pouvant être à l’origine d’un stress hypoxique chez un individu. Ceux-ci peuvent être environnementaux, physiologiques ou pathologiques, et peuvent provoquer un stress local, systémique, aigu ou chronique.
Situations environnementales
Tout individu peut être exposé à des conditions extérieures menant à de l’hypoxémie et de l’hypoxie systémique. Les exemples principaux sont la montée en altitude et la plongée en apnée, où une diminution de l’O2 atteignant le sang survient.
1.2.1.1 L’altitude
La pression atmosphérique décroit lorsque l’altitude augmente, entrainant la diminution de la pO2 de manière proportionnelle. En altitude (généralement > 1500 m), la quantité d’O2 atteignant le
sang et les tissus est donc inadéquate et se définit en tant qu’hypoxie hypobare. La pO2 sanguine la
plus faible chez l’humain fut enregistrée près du sommet du mont Everest, à 8400 mètres d’altitude [6]. Les alpinistes présentaient alors une pO2 artérielle moyenne de 25 mmHg, soit quatre fois
inférieure à la pO2 artérielle normale.
En réponse à l’altitude, le corps enclenche diverses réponses physiologiques, principalement une augmentation des activités cardiorespiratoires (fréquence respiratoire et rythme cardiaque élevés) et des réponses hématologiques (production accrue d’érythrocytes), pour contrer les déficits en O2.
Par contre, une mauvaise acclimatation à l’altitude ou une expositition à des altitudes extrêmes provoque le syndrome du mal aigüe de l’altitude, comprenant une variété de symptômes, et entraine un risque de complications sévères et mortelles [7, 8].
1.2.1.2 La plongée en apnée
La plongée en apnée (ou plongée libre) désigne la plongée sans ventilation au cours de laquelle la quantité d’O2 atteignant le sang et les organes diminue. Les réponses physiologiques
enclenchées lors de cette plongée sont regroupées sous le terme de réflexe d’immersion (diving response). Ce réflexe inné agirait en mécanisme de défense, permettant de conserver les réserves d’O2
et de retarder l’apparition de conséquences hypoxiques sérieuses [9]. Ceci est accompagné d’une augmentation dramatique de la pression sanguine artérielle (hypertension) et une diminution du
4
métabolisme de l’O2. Une exposition prolongée à ces conditions hypoxiques systémiques provoque
néanmoins le développement de troubles physiopathologiques, dont l’arythmie et l’asphyxie [9].
Situations physiologiques
Des situations physiologiques altérant l’homéostasie de l’O2, tel que l’exercice d’endurance,
peuvent provoquer des contextes hypoxiques localisés. Il est également à noter que le développement embryonnaire et fœtal normal représente un contexte hypoxique majeur, où l’hypoxie représente un stimulus physiologique favorable.
1.2.2.1 L’exercice physique
Lors d’efforts physiques, les cellules des muscles squelettiques requièrent un apport énergétique accru. L’augmentation conséquente de la consommation d’O2 musculaire, accompagnée
d’une hypoxémie découlant des limitations respiratoires, crée alors des stress hypoxiques au niveau des muscles squelettiques [10, 11]. Ceci est donc associé à une inaptitude à soutenir les demandes énergétiques et à maintenir l’effort physique, en plus de résulter en un apport d’O2 cérébral insuffisant
[12]. Ceci, malgré les réponses physiologiques mises en place pour tenter de maintenir un apport d’O2
adéquat aux tissus musculaires engagés, notamment par une élévation des activités cardiorespiratoires et du flot sanguin.
1.2.2.2 Le développement embryonnaire
Le développement embryonnaire s’effectue dans un microenvironnement hypoxique par rapport au niveau d’oxygénation de l’adulte, en raison du manque d’accès à la circulation maternelle. Cet état d’hypoxie est régulé de manière spatiotemporelle et est vital pour la formation du système vasculaire et cardiaque et pour le développement des organes [13, 14]. Dans ce cas, l’hypoxie agit en tant que stimulus pour la formation (vasculogenèse) et le développement (angiogenèse) des vaisseaux sanguins, grâce à la production de facteurs proangiogéniques. Ceci sera discuté plus en détail dans la section 1.3.1.2. Cet état hypoxique persiste au cours du développement embryonnaire et le développement fœtal se poursuit également dans un état relatif d’hypoxie favorable.
Situations pathologiques
Des conditions d’hypoxémie et d’hypoxie sont très communes dans le contexte de divers troubles ou pathologies, d’où l’intérêt et l’importance de bien comprendre les réponses associées. De l’hypoxie systémique peut être reliée à divers troubles pulmonaires et cardiovasculaires, tandis que de l’hypoxie sévère et aigüe est communément associée à des troubles ischémiques, notamment au niveau du cœur et du cerveau. L’hypoxie peut participer à certains processus pathologiques, en
5
favorisant la progression de la maladie en question ou l’apparition d’autres troubles. Quelques exemples de situations pathologiques associées à l’hypoxie sont présentés ici.
1.2.3.1 Les troubles pulmonaires
La maladie pulmonaire obstructive chronique (MPOC) et le syndrome de l’apnée obstructive du sommeil (SAOS) sont parmi les troubles respiratoires chroniques les plus courants.
La MPOC, dont la première cause est le tabagisme, est une pathologie progressive, englobant la bronchite chronique (voies respiratoires irritées, enflées et présence de mucus) et l’emphysème (alvéoles endommagées), pouvant engendrer une hypoxémie chronique [15]. Cette dernière joue un rôle majeur dans le développement de processus pathologiques supplémentaires, dont l’hypertension artérielle pulmonaire (due à un épaississement des parois vasculaires) et la polycythémie (augmentation de la quantité d’érythrocytes). Un apport thérapeutique d’O2 à long terme serait en fait
une des rares interventions démontrant un impact sur la durée de vie des patients MPOC hypoxémiques [16].
Le SAOS est caractérisé par des épisodes récurrents d’effondrement partiel ou complet des voies respiratoires supérieures pendant le sommeil [17, 18]. Ceci provoque une respiration peu profonde, un rythme respiratoire anormalement lent ou un arrêt respiratoire complet. L’hypoxie intermittente chronique (HIC) qui en résulte est donc la caractéristique physiopathologique principale de cette pathologie, détectée par une chute notable de la saturation artérielle en O2. Le SAOS est un
syndrome majeur présentant une forte prévalence clinique et des impacts à long terme sur la santé, puisque associé au développement de pathologies secondaires [19].
1.2.3.2 Les maladies cardiovasculaires
La maladie des artères coronaires (ou insuffisance coronarienne ou cardiopathie ischémique) est la première cause mondiale d’insuffisance cardiaque [20]. Elle est la conséquence du développement d’artériosclérose (durcissement et épaississement des artères) ou d’athérosclérose (accumulation de dépôts lipidiques à la surface interne des artères) au niveau des artères coronaires, entrainant une réduction de l’apport d’O2 au cœur (myocarde). Cette ischémie peut alors mener au
développement d’angines de poitrine, soit des douleurs thoraciques résultant de l’effort supplémentaire devant être fourni par le cœur en carence d’O2 [21]. L’obstruction complète des artères
coronaires par la rupture des plaques d’athérosclérose peut quant à elle mener à un infarctus du myocarde (crise cardiaque) [22].
L’insuffisance cardiaque est la conséquence finale de nombreuses maladies cardiovasculaires, telle que la maladie des artères coronaires, mais peut également être causée par une variété de troubles ou pathologies sous-jacentes [23]. L’insuffisance cardiaque consiste en
6
l’incapacité du cœur à procurer assez de sang aux tissus pour répondre aux demandes énergétiques. Ceci peut provoquer de l’hypoxie systémique et/ou des mécanismes physiologiques ou pathologiques compensatoires.
1.2.3.3 Le développement tumoral
L’hypoxie est une caractéristique commune du microenvironnement des tumeurs [24]. Celles-ci contiennent des régions hypoxiques dès les premières étapes de leur développement, en raison de leur état avasculaire et de la limite de diffusion de l’O2. Même lors de la formation de leur
réseau vasculaire, stimulée par l’état hypoxique lui-même, les cellules tumorales demeurent partiellement en carence d’O2. Ceci est notamment dû à leurs vaisseaux anormaux, tortueux et
présentant des fuites. Une forte hétérogénéité dans le microenvironnement tumoral est présente. Des régions d’hypoxie sévère (< 0.02 % d’O2) à modérée (0.5 à 1 % d’O2), de même que des régions
anoxiques (0 %) sont retrouvées au sein d’une même masse tumorale.
L’état hypoxique tumoral favorise l’émergence de mécanismes adaptatifs promouvant la survie cellulaire, notamment par une reprogrammation métabolique et une résistance à l’apoptose [25]. Ces éléments, ainsi que leur régulation, seront abordés plus en détail dans la prochaine section. L’hypoxie tumorale soutient également des réponses contribuant à la vascularisation, la croissance, l’invasion et la formation de métastases [25, 26].
En radiothérapie, une résistance importante des tumeurs hypoxiques est fréquemment observée [27]. Ce traitement a un effet plus grand envers les régions oxygénées des tumeurs que les régions dépourvues d’oxygène. La molécule d’O2 est en fait considérée en tant que puissant élément
sensibilisant les cellules aux radiations [28]. Depuis les premières observations, il y a près d’un siècle, d’un rôle critique de l’apport sanguin et de l’O2 dans la réponse des cellules à l’irradiation, de
nombreuses avancées furent effectuées afin de développer des traitements favorisant l’oxygénation tumorale, et donc la radiosensibilité des cellules [29]. De nouvelles technologies, particulièrement dans la détection de l’hypoxie tumorale et ses changements en cours de traitements, et de meilleures connaissances quant aux propriétés des vaisseaux tumoraux offrent un potentiel prometteur de nouvelles approches ciblées [30-32].
7
1.3 Les mécanismes d’adaptation à l’hypoxie
La présente section se penche sur les principaux mécanismes de l’adaptation à l’hypoxie, impliquant des processus physiologiques et cellulaires. La régulation de ces processus est également abordée, permettant de souligner le rôle central des facteurs HIF, soit HIF-1 et HIF-2. Les particularités de ces facteurs seront détaillées dans les sections subséquentes.
En raison de la place cruciale de l’O2 dans les fonctions biologiques vitales, des systèmes
complexes ont évolué chez les organismes eucaryotes afin de « sentir », réguler et s’ajuster à tout changement d’O2. Les processus impliqués doivent être finement contrôlés et être appropriés face aux
changements de pO2 pour permettre une réponse rapide et prolongée. Ceci, dans le but de maintenir
un niveau d’homéostasie métabolique adéquat et de minimiser les dommages cellulaires et tissulaires. L’activation de telles réponses est dépendante de « senseurs » hautement modulables. C’est ici qu’interviennent les facteurs de transcription induits par l’hypoxie (HIF ; hypoxia-inducible factors), des médiateurs centraux et essentiels à la réponse à l’hypoxie.
Les réponses physiologiques à l’hypoxie
Les réponses physiologiques à l’hypoxie peuvent être de type cardiovasculaire (augmentation du rythme cardiaque, modification du flot sanguin, constriction des vaisseaux périphériques), ventilatoire (augmentation du rythme respiratoire), hématologique (augmentation du niveau d’hémoglobine) et même morphologique (développement pulmonaire, augmentation de la vascularisation tissulaire) (Figure 1.1).
1.3.1.1 Les fonctions cardiovasculaires et ventilatoires (cardiorespiratoires)
Certains réflexes cardiorespiratoires sont impliqués dans la réponse très rapide (de secondes à minutes) à des changements d’O2. Comment une modulation du niveau d’O2 peut-elle être « sentie »
si rapidement ? Comment ces réflexes sont-ils engendrés ? C’est à ce niveau qu’agissent des chémorécepteurs artériels, les cellules de glomus, qui, en quelques secondes, induisent de l’hyperventilation et une activation du système nerveux sympathique menant à des modifications cardiovasculaires (Figure 1.1) [33]. Les cellules de glomus sont localisées dans le corpuscule carotidien (ou glomus carotidien), un organe à la bifurcation des carotides artérielles au niveau du cou. Elles y sont en contact avec des capillaires leur permettant de détecter les niveaux d’O2 artériel
et sont innervées par des fibres sensorielles reliées au centre de contrôle cérébral des fonctions respiratoires [34, 35]. Bien que l’implication de canaux potassiques dans la fonction de ces cellules soit claire, via une dépolarisation membranaire menant au relâchement de neurotransmetteurs, les mécanismes par lesquels ils sont rapidement régulés restent globalement indéfinis.
8
Figure 1.1: Les réponses adaptatives cellulaires et physiologiques à l’hypoxie.
Le corpuscule carotidien et les facteurs de transcription HIF agissent en tant que senseurs et médiateurs des différentes réponses biologiques au stress hypoxique. Voir le texte pour plus de détails.
Illustration par V.N. Lafleur.
L’implication des protéines HIF dans la régulation des fonctions de cet organe sensoriel fut soulevée. En effet, une déficience en HIF-1 atténue les fonctions de détection d’O2 du corpuscule
carotidien, tandis qu’une carence en HIF-2 les augmente [36]. Il en résulte dans les deux cas des perturbations néfastes dans l’homéostasie cardiorespiratoire en réponse à l’hypoxie. Un antagonisme réciproque entre HIF-1 et HIF-2 a été démontré dans ce contexte, soulignant l’importance d’un équilibre entre les niveaux respectifs de ces facteurs pour la sensibilité adéquate du corpuscule carotidien envers l’O2 [37].
Le corpuscule carotidien n’est pas requis seulement pour la réponse rapide à l’hypoxie, il l’est également pour l’acclimatation à long terme à l’hypoxémie [38]. Le maintien de l’hyperventilation lors d’hypoxie chronique est permis par le développement d’une hypersensibilité des chémorécepteurs [39]. De plus, des conditions d’hypoxie chronique mènent à une augmentation de la croissance même du corpuscule carotidien, à partir d’une population de cellules souches [40, 41]. Ceci permet une activation marquée, due à l’augmentation du nombre de signaux afférents.
9
1.3.1.2 Les changements morphologiques et hématologiques : angiogenèse et érythropoïèse En plus des cellules chémoréceptrices agissant en tant que « senseurs » des niveaux d’O2
sanguin, toutes les cellules nucléées de l’organisme ont la capacité de détecter des altérations des niveaux d’O2 et d’engendrer des réponses précises, grâce aux facteurs de transcription HIF. L’hypoxie
est un signal proangiogénique majeur. L’angiogenèse, un processus à multiples étapes, consiste en la formation des vaisseaux sanguins à partir de vaisseaux préexistants. Ceci entraine un remodelage des vaisseaux pour élargir les réseaux d’artères, capillaires et veines.
Les facteurs de transcription HIF sont les régulateurs centraux du processus d’angiogenèse en réponse à l’hypoxie, autant en contexte physiologique que pathologique [42]. Leur rôle est particulièrement étudié dans le cadre de l’angiogenèse tumorale [43]. Les HIF assurent l’orchestration des différentes étapes d’angiogenèse. Ils augmentent l’expression de plusieurs facteurs et récepteurs angiogéniques : le facteur de croissance de l’endothélium vasculaire (VEGF ; vascular endothelial growth factor), le principal facteur proangiogénique ; le facteur de croissance placentaire (PlGF ; placental growth factor), le facteur de croissance dérivé des plaquettes (PDGF ; platelet-derived growth factor) et les angiopoïétines 1 et 2 (ANGPT1 et ANGPT2) [42]. Ceci entraine ce qui est appelé le switch (ou changement) angiogénique. Les HIF facilitent ensuite le recrutement des cellules endothéliales progénitrices via la régulation de chémokines et leurs récepteurs, mais aussi via l’expression de métalloprotéases (MMP) modifiant la matrice extracellulaire pour faciliter la migration de ces cellules [44]. Ceci est accompagné d’une augmentation de la prolifération de ces cellules pour former les nouveaux vaisseaux.
L’érythropoïèse est un autre processus majeur ciblé en hypoxie afin d’augmenter l’apport d’O2 aux cellules et tissus. Celui-ci consiste en la différenciation des cellules souches
hématopoïétiques en érythrocytes hautement spécialisées, à travers plusieurs étapes complexes se déroulant au sein de la moelle osseuse [45]. L’hypoxie entraine l’induction de l’expression de l’érythropoïétine (EPO), un facteur sérique requis pour la prolifération et différenciation des précurseurs des érythrocytes et donc essentiel à la production d’érythrocytes matures [46]. La génération d’EPO est sous le contrôle direct du facteur de transcription HIF-2 et se produit majoritairement, mais non exclusivement, au sein du rein chez l’adulte [47]. D’ailleurs, les facteurs HIF et leur induction en hypoxie furent initialement découverts grâce à l’analyse de l’expression de l’EPO en hypoxie. Une augmentation, même faible et transitoire, de l’EPO a des effets systémiques et persistants sur l’hématocrite (% du volume sanguin occupé par les érythrocytes).
Les évènements d’angiogenèse et d’érythropoïèse consistent donc en des réponses durables pour contrer une carence systémique ou localisée d’O2, particulièrement lors d’hypoxie prolongée ou
10
L’adaptation cellulaire à l’hypoxie
Une réponse complexe et finement orchestrée est mise en place par les cellules lors d’une carence en O2. Cette réponse implique des changements majeurs dans différents processus cellulaires,
notamment le métabolisme, la prolifération et la survie cellulaire (Figure 1.1). 1.3.2.1 L’adaptation hypoxique et la reprogrammation métabolique
La dépendance envers l’O2 pour une production efficace d’ATP par les mitochondries signifie
que les processus métaboliques sont particulièrement affectés lors de conditions hypoxiques. Une activation marquée de la glycolyse anaérobique survient donc, dans un effort de maintien de la production d’ATP. Les processus exigeant une forte consommation d’ATP sont également réduits. Cette reprogrammation métabolique majeure constitue la réponse de survie principale à l’hypoxie et est sous le contrôle direct et majeur du facteur de transcription HIF-1 [48].
La glycolyse consiste en la conversion du glucose, macronutriment essentiel, en deux molécules de pyruvate. L’oxydation des liens carbone du glucose permet alors de capter de l’énergie sous forme d’ATP. En présence d’O2, le pyruvate est ensuite converti en acétyl-CoA pour être
davantage oxydé au sein des mitochondries par le cycle de Krebs (ou cycle d’acide tricarboxylique, ou cycle de l’acide citrique) et la chaine de transport des électrons, produisant ainsi jusqu’à 38 molécules d’ATP. Toutefois, en carence d’O2, le pyruvate est plutôt réduit en lactate, permettant ainsi
l’oxydation du NADH et le maintien du processus de glycolyse (Figure 1.2) [49]. La glycolyse anaérobique ne produit toutefois que deux molécules d’ATP par molécule de glucose. Étant un processus inefficace, mais plus rapide, sa forte activation est donc primordiale en hypoxie.
La majorité des enzymes impliquées dans les réactions successives de la glycolyse voient leur expression accrue en hypoxie par l’action de HIF-1 (Figure 1.2) [50, 51]. Il en est de même pour les transporteurs de glucose 1 et 3 (GLUT1/3 ; glucose transporters 1/3). La réduction du pyruvate en lactate est aussi favorisée en hypoxie par l’induction de la lactate déshydrogénase (LDH) [52]. Ce processus n’est toutefois pas sans conséquence, puisque le lactate et les ions hydrogène (H+) produits peuvent provoquer une acidification dommageable du milieu intracellulaire. L’exportation du lactate est donc augmentée sous le contrôle de HIF-1 par la surexpression du transporteur de monocarboxylate 4 (MCT-4 ; monocarboxylate transporter 4) (Figure 1.2) [53]. Ceci est accompagné d’une surexpression de l’anhydrase carbonique 9 (CA9 ; carbonic anhydrase 9) et de l’échangeur 1 de Na+/H+ (NHE1 ; Na+/H+exchanger 1), contribuant à l’excrétion de H+ et au maintien d’un milieu intracellulaire alcalin favorable [54, 55].
11
Figure 1.2: La reprogrammation métabolique en hypoxie médiée par le facteur HIF-1.
En hypoxie, HIF-1 est centrale dans l’activation de la glycolyse anaérobique et l’inhibition de la respiration mitochondriale. Cette régulation métabolique est essentielle au maintien des réserves énergétiques en conditions de stress hypoxique. Voir le texte pour plus de détails. PDK1 : pyruvate dehydrogenase kinase 1; PDH : pyruvate déshydrogénase; LONP1 : lon peptidase 1, mitochondrial; COX1 : cytochrome c oxidase
4-2/4-1. Illustration par V.N. Lafleur.
Afin de favoriser davantage la glycolyse anaérobique en hypoxie, le cycle de Krebs et la phosphorylation oxydative sont activement réprimés [56]. Il y a d’abord une réduction de l’activité de la pyruvate déshydrogénase (PDH) qui convertit le pyruvate en acétyl-CoA, limitant ainsi son entrée dans le cycle de Krebs (Figure 1.2). Ceci est causé par l’augmentation de sa kinase PDK1 (pyruvate dehydrogenase kinase 1) par HIF-1 [57, 58]. La phosphorylation oxydative est modulée en hypoxie grâce à la modification de l’enzyme oxydase du cytochrome c (COX; cytochrome c oxidase). Ce complexe terminal de la chaine respiratoire mitochondriale voit en effet sa sous-unité régulatrice COX4-1 remplacée par la sous-unité alternative COX4-2 [59]. Ce changement, permis par la dégradation de COX4-1 et l’expression accrue de COX4-2 par HIF-1, semble optimiser l’activité du complexe en fonction des conditions hypoxiques.
Cette reprogrammation métabolique est essentielle à la survie des cellules exposées à un stress hypoxique. Néanmoins, ce processus peut être détourné par les cellules cancéreuses même en présence d’O2 et de mitochondries fonctionnelles. Ceci, qui correspond à de la glycolyse aérobique,