Je dédie ce travail
à Sophie, la femme qui partage ma vie et me supporte tous les jours,
Remerciements
Je tiens tout d’abord à remercier le Pr P. Calvas qui m’a fait l’honneur de présider le jury de cette thèse.
Je remercie également le Dr P. Desprès et le Pr L. Becquemont d’avoir accepté d’être les rapporteurs de cette thèse, ainsi que pour leurs nombreuses remarques et suggestions sur l’écriture de ce manuscrit.
Je remercier le directeur de cette thèse, le Pr D. Brassat pour m'avoir fait confiance en me laissant une grande liberté et en me déléguant plusieurs responsabilités dont j'espère avoir été à la hauteur.
Je tiens à exprimer toute ma reconnaissance au Pr R. Liblau pour m’avoir permis de faire parti de son équipe durant mon année de M2R, mais aussi pour ses nombreux conseils scientifiques durant les années qui ont suivies.
Pour sa confiance et son soutien financier durant ma thèse je remercie vivement. Pr M. Clanet.
Je remercie les Pr A. Saoudi et D. Dunia pour m’avoir initié aux Emit et aux sessions d’immunologie auxquelles ils m’ont permis d’assister et de participer
J’ai aussi une pensée particulière pour le Pr J. Zappulla qui m’a encadré durant l’année de M2R, ainsi que le Pr. E. Piaggio pour sa présence bienveillante.
Je remercie les deux femmes de mon équipe, Florence et Lise, pour leusr aides et pour leur bonne humeur.
Je remercie toutes les personnes des équipes Liblau, Saoudi et Dunia qui se reconnaitront pour tous les bons moments passés à l’heure du déjeuner et des poses.
Je remercie l’ARSEP qui a financé ma thèse et qui m’a permis d’assister à des présentations passionnantes sur la sclérose en plaques.
Afin merci à toute ma famille, mes amis, et mes compagnons de capoeira pour m’avoir permis de passer des bons moments en dehors du laboratoire durant cette thèse.
Nicolas Couturier
Etude génétique et épigénétique de la sclérose en plaques :
Susceptibilité et réponse au traitement
Directeur de thèse : Pr. David Brassat
Thèse soutenue à Toulouse, le 28 Octobre 2009
La sclérose en plaques (SEP) est une maladie auto-immune démyélinisante du système nerveux central. Cette maladie multifactorielle est la cause majeure de handicap chez le jeune adulte et affecte préférentiellement les femmes. Bien que sa pathogénie soit encore mal comprise, la communauté scientifique s’accorde à dire que la SEP se développe chez des individus génétiquement susceptibles, qui ont été en contact avec des facteurs environnementaux, qui pourraient modifier les facteurs épigénétiques. Ces derniers seraient impliqués dans deux étapes de la maladie, auxquelles je me suis intéressé pendant ma thèse: (1) la susceptibilité à la maladie et (2) la réponse au traitement.
En effet, la composante génétique influence la susceptibilité à la SEP, avec l’imputabilité de très nombreux loci ayant de faibles effets lorsqu’ils sont pris dans leur individualité. Historiquement, le complexe majeur d’histocompatibilité fut le premier locus de susceptibilité à la SEP découvert. Ce n’est qu’avec la généralisation des puces à polymorphismes et l’utilisation de larges cohortes de patients que de nouveaux gènes ont pu être découvert. Ainsi, deux gènes ont été identifiés en 2008: IL-2RA et IL-7R. Au cours de ma thèse, un polymorphisme localisé dans un nouveau gène de susceptibilité à la SEP, TYK2, a été identifié par une étude génétique dans la population française. Ce gène, codant pour une tyrosine kinase associée à de nombreux récepteurs aux cytokines telles que l’IFNβ, l’IL-6, l’IL-10, l’IL-12 et l’IL-23, présente un polymorphisme modifiant sa structure primaire et possiblement son activité enzymatique. Afin de déterminer les conséquences de ce polymorphisme sur la signalisation et la réponse immunitaire, une étude fonctionnelle a été réalisée. Par ailleurs, dans la population française, un autre gène (OAS2) serait associé à une plus grande susceptibilité à la SEP. Ce gène code pour la 2’-5’-oligoadénylate synthétase 2. Cependant, ces données restent à confirmer sur des cohortes indépendantes de tailles plus importantes à celle utilisée dans notre étude.
L’autre composante qui peut influencer la susceptibilité à la SEP est la composante épigénétique. L’épigénétique regroupe toutes les modifications transmissibles et réversibles de l'expression des gènes ne s'accompagnant pas de changements des séquences nucléotidiques. L’importance de cette composante est particulièrement marquée chez les jumeaux monozygotes, qui par définition partagent la même information génétique, mais qui peuvent être discordants dans leur statut clinique pour la SEP. Le mécanisme d’inactivation du chromosome X fait partie de cette composante épigénétique. Chez les femmes, contrairement aux hommes, les cellules possèdent deux chromosomes X. Par un mécanisme de compensation, chaque cellule va décider d’inactiver un des deux chromosomes X et conserver ce profil jusqu’à sa mort. Normalement, on retrouve dans le sang 50% de cellules exprimant le chromosome X d’origine paternelle et 50% celui d’origine maternelle. Il a été démontré que dans certaines pathologies auto-immunes, cela n’est plus le cas. En comparant une population de femmes souffrant de SEP à une population de femmes « saines », nous avons pu observer une différence dans le profil d’inactivation du chromosome X.
Une fois la maladie diagnostiquée, un traitement est proposé aux patients. La composante génétique peut alors influencer la réponse du patient au traitement. Cela a été précédemment démontré pour certains médicaments, comme la warfarine, anticoagulant utilisé dans le traitement des troubles du rythme cardiaque. Dans le cadre de la SEP, il serait important de mieux comprendre et de prédire la réponse au traitement par l’IFNβ. L’utilisation de cette molécule immunomodulatrice apporte un réel progrès dans le traitement de la SEP, mais avec l’apparition de nouveaux traitements il devient impératif pour le médecin d’avancer vers une approche thérapeutique personnalisée, à la fois plus efficace et mieux tolérée par le malade. Pour cela, nous avons constitué une cohorte européenne de patients souffrant de SEP et traités par IFNβ. Les données cliniques disponibles sur ces patients nous ont permis de les classer en deux groupes : les répondeurs et les non-répondeurs au traitement. Une analyse préliminaire des polymorphismes contenus dans des gènes de la cascade de signalisation de l’IFNβ a révélé que plusieurs polymorphismes présents dans la séquence du gène codant pour l’enzyme 2’-5’-oligoadénylate synthétase 1 (OAS1) et du gène TRAIL pourraient influencer la réponse des patients atteints de SEP à cette molécule.
Nicolas Couturier
Genetics and Epigenetics in Multiple Sclerosis:
Susceptibility and Response to Treatment
Thesis supervisor : Pr. David Brassat Toulouse, October 28th 2009
Multiple sclerosis (MS) is a demyelinating disease of the central nervous system that leads to disability in young adults. As other autoimmune diseases, MS is characterized by a striking female predominance; however its pathogenesis remains elusive. It is widely believed to occur in genetically susceptible individuals after exposure to undefined environmental factors that influence epigenetic factors. These factors may act at two different levels: (1) on the susceptibility to develop MS and (2) on response to MS treatment
Little is known about genes that are involved in MS susceptibility. Over more than 30 years, with the discovery of the association of major histocompatibility complex with MS risk and it confirmation in a wide range of population, lead us to search whether other new susceptibility genes are also involved. With the development of genome-wide association studies, two non-HLA variants (IL-2R and IL-7R variants) have been implicated with strong confidence in MS susceptibility. Recently genetic association studies have identified a polymorphism (rs34536443) in TYK2 gene associated with MS susceptibility, and we confirmed this association in the French population. Rs34536443 polymorphism localizes in the exon 21 of TYK2 gene, and codes either a proline (major allele) or an alanine at position 1104, in the tyrosine kinase. A functional consequence of this polymorphism could be to alter kinase function of this protein as suggested by two independent studies. Moreover, TYK2 deficiency revealed an important role in immunity and in lymphocyte differentiation as demonstrated in mouse model. Given these data, we investigated the potential functional consequences of the rs34536443 polymorphism on TYK2 activation, and in lymphocyte polarization. Moreover, our preliminary data suggest association between OAS2 polymorphisms and MS susceptibility. This association has to be confirmed in a new independent MS cohort.
In biology, the term epigenetics refers to changes in gene expression caused by mechanisms other than modifications in the underlying DNA sequence. These changes may remain through cell divisions. These epigenetic modifications could be implicated in MS susceptibility too. Monozygotic twins share the same genetic information but two third of identical twins are discordant for MS status. In this case, the importance in MS susceptiblity of epigenetic factors is clearly visible. X-chromosome inactivation is an epigenetic mechanism occurring only in females. In female mammalian cells, one of the two X-chromosome is inactivated in early embryonic life. Thus, females are mocaics for two cell populations, cells with either the paternal or the maternal X in the active form. X-choice is assumed to be random, and the result is generally 50% of cells expressed the paternal and the remaining 50% expressed the maternal genes. Studies reported for some autoimmune disease (lupus, autoimmune thyroid diseases and rheumatoid arthritis) an association with a deviation from this ratio 50:50%. In MS, ours results revealed a difference in degree of skewing in MS patients compare to controls.
After MS development, genetic factors may influence patient response to MS treatment. Pharmacogenetics is a science that provides some clue on how to define best responders. For instance, variants in the VKORC1 gene could explain why efficient anticoagulant warfarin dose is variable between patients. Ten years ago, interferon beta (IFNβ) was the first therapy proposed with a demonstrated efficacy in MS. This immunomodulatory drug is a major advancement in MS therapy as it allows a 30% decrease in the relapse rates, in disability progression and in lesion load, and an increase in patient’s quality of life. But with the development of new drugs, it will be of importance to personalise medicine. Hence, exploring the degree of variability in candidate genes for direct association with treatment response represents a promising approach to improve the overall efficacy of the treatment. For this study, naïve patients to immunotherapy and starting IFNβ treatment were recruited in the European community. We categorized MS patients on clinical criteria in two groupes: responders and non-responders to IFNβ treatment. In a preliminary study, significant associations with OAS1 and TRAIL polymorphisms were found suggesting that genetic variants in these two genes may be of clinical interest in MS as predictors of the efficacy to IFNβ therapy.
ABREVIATION ET ANGLICISMES
P1LISTE DES FIGURES
P3INTRODUCTION
P7I
I
.
.
L
L
a
a
s
s
c
c
l
l
é
é
r
r
o
o
s
s
e
e
e
e
n
n
p
p
l
l
a
a
q
q
u
u
e
e
s
s
P9I.1. Les différentes formes cliniques de sclérose en plaques et évolution de la
maladie P10
I.2. Conséquences anatomiques de la maladie P13
I.3. Pathogénie de la sclérose en plaques P13
I.4 Critères de diagnostic de la sclérose en plaques ou critères de McDonald P15
I.5. Les méthodes d’investigation paracliniques P16 I.5.1. Imagerie par résonnance magnétique
I.5.2. Analyse du liquide céphalo-rachidien I.5.3. Examen des potentiels évoqués visuels
I.6. Mesure de la progression du handicap P19
I
I
I
I
.
.
L
L
e
e
s
s
f
f
a
a
c
c
t
t
e
e
u
u
r
r
s
s
d
d
e
e
s
s
u
u
s
s
c
c
e
e
p
p
t
t
i
i
b
b
i
i
l
l
i
i
t
t
é
é
à
à
l
l
a
a
s
s
c
c
l
l
é
é
r
r
o
o
s
s
e
e
e
e
n
n
p
p
l
l
a
a
q
q
u
u
e
e
s
s
P23II.1. Les facteurs génétiques P23
II.1.1. Les éléments soulignant l’importance de la génétique dans la maladie II.1.2. Etiologie génétique de la sclérose en plaques
II.1.3. Les différentes classes de polymorphismes génétiques chez l’Homme
1.3.1. Les polymorphismes d’un seul nucléotide 1.3.2. Les polymorphismes structuraux
II.1.4. La notion de déséquilibre de liaison entre plusieurs polymorphismes II.1.5. Les moyens mis en œuvre dans l’identification des gènes de susceptibilité à la sclérose en plaques
1.5.1. L’approche gène candidat vs l’approche sans à priori a. Approche gène candidat
b. Approche sans à priori par GWAS
1.5.2. Configuration des cohortes utilisées en association génétique
II.1.6. Importance du complexe majeur d’histocompatibilité dans la susceptibilité à la sclérose en plaques
1.6.2. Implication du CMH-I dans la susceptibilité à la sclérose en plaques
II.1.7. Les autres gènes associés à la susceptibilité à la SEP
1.7.1. Le récepteur à l’IL-7 1.7.2. Le récepteur à l’IL-2 1.7.3. Le CD58
1.7.4. La tyrosine kinase 2
II.2. Les facteurs environnementaux P51
II.2.1. Les risques micro-environnementaux II.2.2. Les risques environnementaux
2.2.1. Les agents infectieux a. Le virus d’Epstein-Barr
b. Les autres pathogènes suspectés
2.2.2. L’exposition lumineuse et la vitamine D 2.2.3. La cigarette
II.3. Les facteurs épigénétiques P60
II.3.1. Influence de l’épigénétique dans la susceptibilité aux maladies auto- immunes
II.3.2. Comment l’épigénétique pourrait modifier la susceptibilité à la sclérose en plaques ?
3.2.1. Modification épigénétique du locus PAD2
3.2.2. Impact de l’épigénétique sur la différenciation cellulaire 3.2.3. L’inactivation du chromosome X
a. Mécanismes d’inactivation du chromosome X
b. Implication de l’inactivation du chromosome X dans l’expression phénotypique c. Causes du biais dans l’inactivation du chromosome X
d. Conséquences de l’inactivation du chromosome X e. Monosomie ou perte du chromosome X
I
I
I
I
I
I
.
.
T
T
r
r
a
a
i
i
t
t
e
e
m
m
e
e
n
n
t
t
e
e
t
t
p
p
h
h
a
a
r
r
m
m
a
a
c
c
o
o
g
g
é
é
n
n
é
é
t
t
i
i
q
q
u
u
e
e
d
d
e
e
l
l
a
a
s
s
c
c
l
l
é
é
r
r
o
o
s
s
e
e
e
e
n
n
p
p
l
l
a
a
q
q
u
u
e
e
s
s
P77III.1. Les traitements de fond disponibles en 2009 P77 III.1.1. L’interféron bêta
III.1.2. L’acétate de glatiramère
III.1.3. L’anticorps monoclonal natalizumab III.1.4. La mitoxantrone
III.3. Optimisation des traitements existants P86
III.4. Les nouveaux traitements en cours de développement P88
III.5. Définition de la réponse au traitement dans la sclérose en plaques P93 III.5.1. Traitement de la sclérose en plaques par une approche d’escalade
thérapeutique
III.5.2. Prédiction de la réponse au traitement des patients sclérose en plaques en vue d’une médecine personnalisée
5.2.1. Recherche de bio-marqueurs prédictifs de la réponse au traitement 5.2.2. Anticiper la réponse au traitement par analyse de l’expression génique 5.2.3. Recherche de marqueurs génétiques prédictifs de la réponse au traitement
IV. Les
i
i
n
n
t
t
e
e
r
r
f
f
é
é
r
r
o
o
n
n
s
s
d
d
e
e
t
t
y
y
p
p
e
e
1
1
P99IV.1. La voie de signalisation des interférons de type 1 P99
IV.2. Les protéines antivirales induites par les interférons de type 1 P100 IV.2.1. La protéine MxA
IV.2.2. La voie des OAS et de la RNaseL IV.2.3. La protéine kinase PKR
IV.2.4. Le facteur ISG15
IV.3. Interférons de type 1 et sclérose en plaques P105 IV.3.1. Evidences de l’importance des interférons de type 1 dans la sclérose
en plaques
IV.3.2. Mécanisme d’action de l’IFNβ dans le traitement de la sclérose en plaques
3.2.1. Effet de l’IFNβ sur la migration des cellules immunes a. Adhésion à la barrière hémato-encéphalique
b. Migration à travers l’espace péri-vasculaire
3.2.2. Effet de l’IFNβ sur l’activation et la polarisation des cellules immunes 3.2.3. Effet de l’IFNβ sur l’apoptose des cellules immunes
MATERIELS ET METHODES
P113RESULTATS
P125Facteurs génétique et susceptibilité à la SEP P127
Facteurs épigénétiques et susceptibilité à la SEP P153
DISCUSSION ET PERSPECTIVES
P175ANNEXES
P191ABREVIATIONS ET ANGLICISMES
APC Cellules présentatrices de l’antigène Mx Myxovirus-Resistance-Protein
ARNdb ARNs doubles brins MZ Jumeaux monozygotes
BBB Barrière hémato-encéphalique Nabs Anticorps neutralisants
BDNF Brain Derived Neurotrophic Factor NO Oxyde nitrique
CMH Complexe Majeur d’Histocompatibilité OAS 2’-5’OligoAdénylate Synthétase
CNV Variation du nombre de copies OPC Oligodendrocyte Precursor Cell
DZ EAE Jumeaux dizygotes Encéphalomyélite Auto-immune PKR PLP
Protéine Kinase Régulée par les ARNs Protéine protéolipide de la myéline
EDSS
Expérimentale
Echelle élargie de progression du
PP-MS PEV
SEP progressive primaire Potentiels Evoqués Visuels handicap PF Paramètres Fonctionnels
EBV Virus d’Epstein-Barr RR-MS SEP rémittente-récurrente
GA Acétate de glatiramère SNC Système Nerveux Central
GWAS Genome-Wide Analysis Study SEP Sclérose En Plaques
HHV6 Virus herpétique humain 6 SNP Single Nucleotide Polymorphism
IFN Interféron SP-MS SEP progressive secondaire
IgG Immunoglobuline d’isotype G S1P Sphingosine-1-Phosphate
IL Interleukine TDT Test de Déséquilibre de Transmission
ILxR Récepteur à l’interleukine x Thx Lymphocytes T-helper x
IRM Imagerie par Résonnance Magnétique TYK2 Tyrosine Kinase 2
ISGF3 IFN-Stimulated Gene Factor 3 VCAM-1 Vascular Cell Adhesion Molecule-1
ISRE IFN-Stimulated Response Element VD Vitamine D
JAK KIR
Janus Kinase
Killer cell Immunoglobulin-Like
VLA-4 Xce
Very Late Activating Antigen-4 X-Controlling Element
Receptor XCI Inactivation du chromosome X
LCR Liquide Céphalo-Rachidien Xic X Inactivation Center
LD LEMP Déséquilibre de liaison LeucoEncéphalopathie Multifocale Xist XITE
X -Inactive Specific Transcript X-Inactivation Intergenic Transciption
MAF
Progressive
Fréquence de l’allèle mineur d’un Xpr
Elements
X-Pairing Region polymorphisme
MBP Protéine basique de la myéline
MMP Métallo-protéases
MSSS Score de sévérité de la sclérose en plaques
LISTE DES FIGURES
Figure 1 : Schéma des principaux symptômes rencontrés dans la SEP. Figure 2 : Description schématique de l’évolution clinique de la SEP.
Figure 3 : Présence de bandes oligoclonales d’IgG retrouvées uniquement dans le LCR d’un
individu souffrant de SEP.
Figure 4 : Comparaison d’un PEV enregistré chez un individu souffrant de SEP avec celui
d’un individu sain.
Figure 5 : Echelle EDSS simplifiée, ne présentant que les scores principaux de la SEP. Figure 6 : Score de sévérité de la SEP (MSSS) global.
Figure 7 : Les différentes voies pouvant conduire au développement de la SEP. Figure 8 : Risque de récurrence intrafamiliale pour la SEP.
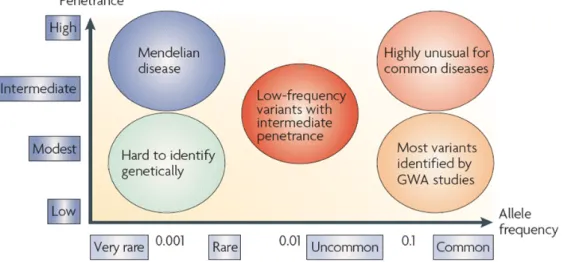
Figure 9 : Les variants de faible fréquence et la susceptibilité aux maladies.
Figure 10 : Les différentes catégories de polymorphismes génétiques chez l’Homme.
Figure 11 : Nombre de polymorphismes détectés dans le génome de 4 individus de
différentes ethnies.
Figure 12 : Nombre et caractéristiques des variations structurales détectées dans le génome
d’un individu d’origine caucasienne.
Figure 13 : Les déséquilibres de liaison des variants communs du génome humain diffèrent
entre les populations.
Figure 14 : Association des polymorphismes par une approche directe ou par une approche en
tagSNP.
Figure 15 : Recouvrement des loci contenant les facteurs de risque génétique aux maladies
communes humaines.
Figure 16 : Approche GWAS en plusieurs étapes afin de réduire la taille des échantillons. Figure 17 : Allèles de l’haplotype du HLA-DR2 associés à la sclérose en plaques
(l’implication des marqueurs entre parenthèse semble due à un déséquilibre de liaison).
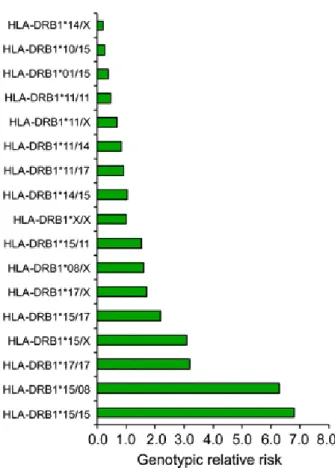
Figure 18 : Risque génotypique relatif pour la sclérose en plaques en fonction des
Figure 19 : Association du HLA avec la sclérose en plaques.
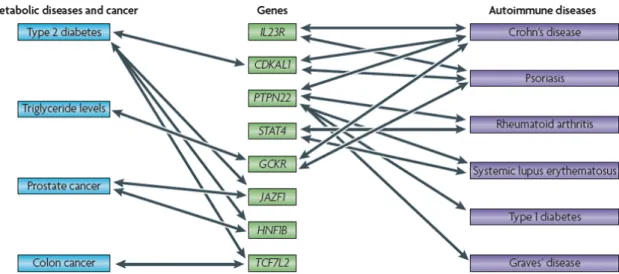
Figure 20 : Les gènes associés au risque de développer une sclérose en plaques.
Figure 21 : Récepteurs aux cytokines qui utilisent TYK2 dans leur voie de signalisation. Figure 22 : Prévalence de la SEP dans le monde.
Figure 23 : Représentation schématique de l’incidence de la SEP en fonction de l’infection
par le virus d’Epstein-Barr.
Figure 24 : Métabolisme de la vitamine D.
Figure 25 : Modèle pour la transmission d’un phénotype en l’absence de mutation du gène
Kit proposé par Rassoulzadegan et al.
Figure 26 : Mécanisme proposé dans le développement de la SEP suite à une dérégulation
épigénétique du gène PAD2 conduisant à la surexpression de la protéine dans les oligodendrocytes.
Figure 27 : Modèle possible d’inactivation du chromosome X.
Figure 28 : Mécanisme expliquant le biais dans l’inactivation du chromosome X.
Figure 29 : Représentation schématique du mécanisme proposé pour expliquer l’effet
immuno-modulateur de l’acétate de glatiramère.
Figure 30 : Les différents types d’anticorps monoclonaux utilisés en thérapie.
Figures 31 : Les cibles thérapeutiques possibles dans le traitement de la sclérose en plaques. Figures 32 : La voie de signalisation des interférons de type 1.
Figure 33 : Mécanisme d’action de la protéine MxA. Figure 34 : La voie antivirale OAS1-RNase L. Figure 35 : Mécanisme d’action de la PKR. Figure 36 : Mécanisme de l’ISGylation.
Figure 37 : Le polymorphisme rs34536443 ne modifie pas l’expression protéique de TYK2. Figure 38 : Le polymorphisme rs34536443 modifie le niveau d’activation de TYK2.
Figure 39 : Le polymorphisme rs34536443 modifie le niveau d’activation de la voie de
Figure 40 : Le polymorphisme rs34536443 contrôle le niveau d’expression des gènes induits
par l’IFNβ.
Figure 41 : Le polymorphisme rs34536443 influence l’expression des facteurs nucléaires
impliqués dans la polarisation lymphocytaire.
Figure 42 : L’expression des facteurs nucléaires impliqués dans polarisation lymphocytaire
permet de classer les individus en fonction de leur génotype pour le polymorphisme rs34536443.
Figure 43 : Le polymorphisme rs34536443 influence la sécrétion de cytokines par les
lymphocytes T.
Figure 44 : Etude par cytométrie de l’effet du polymorphisme rs34536443 sur la production
de cytokines par les lymphocytes.
Figure 45 : Comparaison de l’expression des gènes de la voie des IFNs de type 1 chez des
patients SEP par rapport à des témoins.
Figure 46 : Le gène OASL n’est pas associé avec la susceptibilité à la SEP dans les familles
trio françaises.
Figure 47 : Le gène OAS2 est associé avec la susceptibilité à la SEP dans les familles trio
françaises.
Figure 48 : Protocole expérimental permettant de mesurer le profil d’inactivation du
chromosome X chez une femme.
Figure 49 : L’immortalisation des cellules immunitaires par l’EBV modifie leur profil de
XCI.
Figure 50 : Le profil du XCI diffère entre la population de patientes SEP et la population de
témoins.
Figure 51 : Comparaison du coefficient de corrélation pour le profil du XCI au sein de
cohortes de jumelles MZ, en fonction du statut clinique pour la SEP.
Figure 52 : Monosomie du chromosome X au sein de paires de jumelles MZ discordantes
pour la SEP.
Figure 53 : Association du polymorphisme rs1131532 du gène TRAIL avec la réponse au
Figure 54 : Association du polymorphisme rs2660 du gène OAS1 avec la réponse au
traitement de la SEP par l’IFNβ.
Figure 55 : Invalidation de l’association du polymorphisme rs1131532 du gène TRAIL avec
la réponse au traitement de la SEP par l’IFNβ.
Figure 56 : Invalidation de l’association du polymorphisme rs2660 du gène OAS1 avec la
réponse au traitement de la SEP par l’IFNβ.
Figure 57 : Le polymorphisme rs34536443 contrôle le niveau d’expression des facteurs
nucléaires de polarisation lymphocytaire en présence d’IFNβ.
Tableau I : Critères de McDonald révisés (2005) pour le diagnostic de la SEP.
Tableau II : Analyse d’association du polymorphisme rs34536443 de TYK2 dans la SEP par
une approche en cas-témoins.
Tableau III : Analyse TDT du polymorphisme rs34536443 de TYK2 dans la SEP sur 640
familles trio françaises.
Tableau IV : Analyse d’association du polymorphisme rs12815666 d’OAS2 dans la SEP par
une approche en cas-témoins.
Tableau V : Analyse d’association du polymorphisme rs1298301 d’OAS2 dans la SEP par
une approche en cas-témoins.
Tableau VI : Analyse d’association du polymorphisme rs34536443 de TYK2 avec la réponse
au traitement de la SEP par l’IFNβ.
Tableau S1 : Liste et localisation sur la plaque de PCRarray des gènes amplifiés.
Tableau S2 : Analyse TDT des polymorphismes rs3741981 et rs10774671 d’OAS1 dans la
SEP sur 591 familles trio françaises.
Tableau S3 : Analyse TDT des haplotypes formés par des polymorphismes rs3741981 et
I
I. La sclérose en plaques
La sclérose en plaques (SEP) est une maladie auto-immune inflammatoire chronique touchant le système nerveux central (SNC), c'est-à-dire le cerveau ainsi que la moëlle épinière. Elle affecte environ 2,5 millions de personnes dans le monde (80 000 personnes en France) et en moyenne 120 personnes pour 100 000 habitants sont nouvellement diagnostiquées chaque année [Compston et al., 2002]. 70% des nouveaux patients sont des sujets jeunes, ayant entre 20 et 40 ans. La SEP représente la cause majeure de handicap dans cette tranche d’âge. La prévalence de la maladie dans la population générale est variable en fonction des régions du monde étudiées, avec 60-200 patients atteints de SEP pour 100 000 personnes en Europe et en Amérique du nord, alors que dans les zones de faible prévalence comme le Japon, la prévalence est environ 10 fois moins importante. De façon similaire aux autres maladies auto-immunes humaines, les femmes sont plus fréquemment touchées que les hommes, avec un ratio homme : femme de 1:3. De plus, plusieurs études à travers le monde suggèrent que, durant les 50 dernières années, l’incidence de la maladie a augmenté [Barnett et al., 2003], et que cette augmentation est plus rapide chez les femmes que chez les hommes, modifiant encore plus le ratio homme : femme [Wallin et al., 2004; Orton et al., 2006].
Chez la majorité des patients, les manifestations cliniques apparaissent dès le début de la maladie et indiquent l’implication du système nerveux moteur, sensoriel, visuel et autonome (Figure 1). Par ailleurs, il existent d’autres symptômes ou signes moins perceptibles (dépression, fatigue, etc…) qui peuvent passer inaperçus et qui ne seront diagnostiqués que de manière rétrospective [Compston et al., 2002].
Figure 1 : Schéma des principaux symptômes rencontrés dans la SEP.
I.1. Les différentes formes cliniques de sclérose en plaques et évolution de la
maladie
Dès le 19ème siècle, trois signes cliniques maintenant connus comme associés à la SEP - dysarthrie (troubles de l’articulation), ataxie (trouble de l’équilibre) et tremblements - étaient déjà décrits par les médecins. C’est le neurologue français, Jean-Martin Charcot, qui en 1868 associa tous ces symptômes à une seule pathologie qu’il nomma la « sclérose en plaques »
[Charcot et al., 1868]. Un siècle plus tard, Schumacher et al. définirent le terme de poussées dans la SEP. Ils les qualifièrent comme la dysfonction localisée d’une fonction, affectant la substance blanche, et dont l’effet doit perdurer durant au moins 24 heures tout en étant précédé d’au moins 30 jours de stabilité clinique [Schumacher et al., 1968]. Les poussées de SEP sont le reflet clinique de la présence de foyers inflammatoires actifs au niveau du SNC. Ces zones inflammatoires conduisent à des dommages au niveau des fibres myélinisées d’axones et des neurones, dont la conséquence est d’entraîner des défauts dans la conduction des signaux neurologiques. Cependant, cette définition originale de poussées n’est pas totalement vraie. En effet, la substance grise du SNC peut elle aussi être affectée au cours de la maladie, comme cela est visible en imagerie par résonnance magnétique (IRM) par une diminution du volume total de la matière grise [Sanfilipo et al., 2006] et par la présence de lésions [Bo et al., 2003]. Par ailleurs, les 30 jours d’intervalle de stabilité clinique entre les poussées furent arbitrairement choisis, sans aucune correspondance avec les connaissances actuelles sur la
biologie de la maladie. On sait qu’un même patient présente de nombreuses lésions dans le SNC, à des stades d’évolution différents, qui progressent de manière indépendante, et qui sont toutes susceptibles d’induire des poussées. Enfin, il existe des pseudo-poussées, qui sont rapidement réversibles et se déclarent en présence d’un stress physiologique. Ces pseudo-poussées ne sont pas dues à l’apparition de nouveaux foyers inflammatoires, mais sont plutôt la conséquence des dommages présents dans d’anciennes lésions et qui perturbent la conduction du signal en présence d’un stress.
La SEP n’est pas une maladie homogène quant à l’expression des symptômes. En effet, il est possible de distinguer deux formes cliniques majeures de SEP : les SEP à forme
rémittente-récurrente (RR-MS) et les SEP progressives primaires (PP-MS). Ces deux
formes peuvent être considérées comme deux maladies à part entière car très hétérogènes sur le plan radiologique [McFarland et al., 1999 ; Rovaris et al., 2003], histologique [Lucchinetti et al., 2000] et clinique [Sospedra et al., 2005] (Figure 2). La forme la plus fréquente (85% des cas) est la forme RR-MS qui va présenter une évolution par poussées, tandis que les 15 autres pourcents des cas sont des formes PP-MS présentant une évolution plus linéaire de la maladie
[Vollmer et al., 2007].
- La forme RR-MS se caractérise donc par des poussées. Une poussée est l’apparition d’un signe clinique qui généralement perdure dans le temps (entre une semaine et un mois). Ces poussées sont associées à l’apparition de lésions inflammatoires dans le SNC, visibles en IRM. Au début de l’évolution de la RR-MS, la majorité des patients, récupèrent complètement de leur handicap après ces épisodes aigüs de la maladie. On assiste alors à une alternance dans le temps de poussées suivies de périodes de récupération plus ou moins longues. Puis avec le temps, les poussées deviennent plus fréquentes et le patient commence à ne récupérer que partiellement de son handicap ce qui conduit à l’accumulation de dommages neurologiques [Lublin et al., 1996 ; Thompson et al., 1990]. Les poussées sont des événements qu’il est toujours difficile de prédire, cependant des changements environnementaux peuvent influencer le risque de de survenue d’une poussée. Les changements hormonaux durant la grossesse et après l’accouchement sont aussi connus pour affecter la fréquence des poussées. Le nombre moyen de poussées annuelles diminue progressivement durant les 3 trimestres de grossesse (passant de 0,7 avant la grossesse, à 0,6, puis 0,5 et enfin 0,2 aux 1er, 2ème et 3ème trimestres respectivement). Puis, rapidement après l’accouchement, le nombre moyen de poussées annuelles augmente fortement (taux annuel moyen : 1,2) pour
retrouver progressivement une valeur normale un an après l’accouchement [Confavreux et al., 1998]. Ainsi, bien que cette période dans la vie d’une femme soit susceptible d’influencer le nombre de poussées, si on prend l’ensemble de la période (grossesse et post-accouchement) le risque global de progression du handicap n’est pas modifié. Plusieurs études suggèrent également que les médiateurs inflammatoires associés à une infection aigüe pourraient précéder le début d’une poussée dans 20 à 30% des cas. Suite à cette période de RR-MS, environ 65% des patients entrent dans une nouvelle phase de la maladie, appelée SEP progressive secondaire (SP-MS). A ce stade, la maladie n’évolue plus par poussées, mais le handicap progresse de manière continue. - La forme PP-MS se caractérise, dès le début de la maladie, par une lente aggravation du handicap sans qu’il n’y ait de rémission constatée. Cette forme de SEP se manifeste souvent comme une atteinte de la moelle épinière même si une atteinte du cerveau peut aussi arriver. Comparé à la forme RR-MS, le handicap évolue généralement plus rapidement chez les patients souffrant de cette forme de la maladie.
Il est important de noter que le temps moyen entre le diagnostic de SEP et le décès du patient est de 30 années environ, ce qui représente une réduction de l’espérance de vie de 5 à 10 ans
[Bronnum-Hansen et al, 2004]. Les patients souffrant de SEP ont aussi un risque accru de suicide, reflétant une augmentation des périodes de dépression au cours de leur vie [Minden et al., 1990]. Par ailleurs, chez des patients présentant des troubles neurologiques importants, le décès peut dans 2/3 des cas être attribuable à la maladie, à une augmentation de la susceptibilité aux pathogènes, ou à un risque de complications sévères de maladies induites par les infections.
Figure 2 : Description schématique de l’évolution clinique de la SEP. D’après Vollmer et al., J. Neurol. Sci.,
I.2. Conséquences anatomiques de la maladie
La principale cellule cible du système immunitaire est l’oligodendrocyte. Cette cellule du SNC synthétise et maintient une gaine de myéline autour de courts segments de 20 à 40 axones avoisinants. La gaine de myéline consiste en une membrane condensée, spiralée autour de l’axone, et qui forme une gaine isolante segmentée nécessaire à la conduction saltatoire de l’influx nerveux axonal. Ainsi, on retrouve tout au long de l’axone une alternance de segments myélinisés et de segments non-myélinisés ou nœuds de Ranvier qui regroupent des canaux sodium voltage-dépendants. Le potentiel d’action, lorsqu’il se propage le long de l’axone, passe d’un nœud de Ranvier à un autre et de manière passive dans les segments myélinisés du nerf (d’où l’utilisation du terme de conduction saltatoire).
La démyélinisation des axones explique en grande partie les signes cliniques que l’on observe chez un patient souffrant de SEP. La destructuration de la segmentation de l’axone empêche la conduction saltatoire, ce qui ralentit la vitesse de conduction de l’influx nerveux. Par ailleurs, les axones démyélinisés peuvent spontanément déclencher des potentiels d’action. Cela peut se traduire chez le patient par la sensation de décharges électriques ou la visualisation de flashs lumineux. Enfin, si des axones démyélinisés sont proches, le signal peut se propager d’un axone vers un autre ce qui entraîne de nombreux signes cliniques (douleurs, tétanie…).
La caractéristique majeure de la SEP est l’apparition de lésions (ou plaques) dans le SNC du patient. Ces lésions sont le stade final d’un procédé impliquant l’inflammation, la succession de démyélinisation et de remyélinisation, la déplétion des oligodendrocytes, l’astrocytose et la dégénérescence axonale et neuronale [Compston et al., 2008].
I.3. Pathogénie de la sclérose en plaques
Il est généralement admis que la SEP débute par une infiltration de lymphocytes auto-réactifs, passant du sang vers le SNC à travers la barrière hémato-encéphalique (BBB). Cette transition d’un état de surveillance physiologique vers une cascade pathologique se produirait suite à un défaut de régulation. C’est ce défaut de régulation qui autoriserait aux cellules immunitaires de monter une réponse inflammatoire dans le SNC. En effet, il a été démontré que les cellules régulatrices de patients souffrant de SEP sont moins efficaces dans la suppression d’une réponse effectrice [Viglietta et al., 2004]. Par ailleurs, les cellules T CD4+
auto-réactives de ces patients présenteraient une sensibilité moindre à l’apoptose via la surexpression d’une molécule de signalisation : l’arrestine bêta 1 [Shi et al., 2007]. Ce serait aussi la défaillance de mécanismes de régulation locaux du SNC qui serait responsable de l’accumulation périvasculaire de lymphocytes T CD8+ retrouvés à proximité (ou au contact) d’oligodendrocytes et d’axones démyélinisés [Neumann et al., 2002]. L’importance du rôle historiquement assigné aux lymphocytes T-helper 1 (Th1) par l’étude chez animal de l’encéphalomyélite auto-immune expérimentale (EAE), modèle murin de la SEP, est aujourd’hui remise en cause [Sospedra et al., 2005]. L’inflammation serait plutôt montée par un
nouveau sous-type de lymphocytes T capables de sécréter de l’interleukine 17 sous le contrôle de l’interleukine 23 [Langrish et al., 2005]. Deux interleukines (IL) sécrétées par les lymphocytes Th17, l’IL-17 et l’IL-22, seraient impliquées dans l’ouverture de la BBB [Kebir et al., 2007], permettant l’entrée de ce sous-type de lymphocytes dans le SNC [Tzartos et al., 2008]. Les lymphocytes Th17 pourraient alors directement tuer les neurones ou recruter d’autres cellules impliquées dans la réponse inflammatoire conduisant à la destruction de la myéline. Enfin, le système immunitaire inné (microglie, mastocytes…) pourrait aussi jouer un rôle dans la progression de l’inflammation via la production de composés réactifs de l’oxygène
[Sospedra et al., 2005] ou l’implication de récepteurs membranaires de mort cellulaire [Zajicek et al., 1992].
La question du ou des antigène(s) spécifique(s) de cette réponse immune n’a pas encore trouvé de réponse, principalement car des lymphocytes auto-réactifs sont naturellement présents chez des personnes saines. Historiquement, les protéines composant la myéline était considérées comme les cibles uniques de la réponse immunitaire à l’initiation de la SEP. Cependant d’autres composants du SNC, comme la crystalline αB [Ousman et al., 2007]
ou la neurofascine [Mathey et al., 2007] semblent aussi être la cible de la réponse immunitaire. Les lésions débuteraient par des points focaux d’inflammation du SNC qui, par la suite, donneraient naissance à des plaques de démyélinisation présentant une expansion radiale progressant à travers la substance blanche d’apparence normale [Kutzelnigg et al., 2005].
Les phases de rémission dans la SEP sont le reflet d’un mécanisme au cours duquel les gaines de myéline sont reformées et la conduction saltatoire restaurée : ce phénomène est appelé remyélinisation [Franklin et al., 2008]. Cette remyélinisation se traduit en IRM par l’apparition de plaques de remyélinisation dites « fantômes ». Ce phénomène, qui est généralement plus actif dans les formes RR-MS, peut cependant avoir lieu durant la phase progressive de la maladie. Par ailleurs, une différence inter-individuelle dans l’efficacité de la
remyélinisation existe [Patrikios et al., 2006]. Les débris de myéline sont alors phagocytés et des
précurseurs d’oligodendrocytes (OPC), naturellement présents dans le SNC mature [Horner et al., 2000], peuvent migrer en réponse à des chimiokines telles que les sémaphorines 3A et 3F
[Williams et al., 2007]. Des OPC ont été retrouvés au niveau des lésions de SEP [Scolding et al., 1998] et pourraient constituer les cellules capables de remyéliniser les axones nus [Chadran et al., 2008]. Cependant, comme le reflète l’accumulation du handicap tout au long de l’avancée de la maladie, la succession de cycles de démyélinisation et de remyélinisation semble progressivement épuiser le système de réparation du tissu. Même si plusieurs hypothèses peuvent expliquer cet épuisement, comme la présence de facteurs inhibiteurs (ou l’absence de facteurs activateurs) de la différenciation des OPC en oligodendrocytes ou encore un épuisement du nombre d’OPC [Franklin et al., 2002 ; Penderis et al., 2003], aucun mécanisme n’a été clairement démontré.
I.4. Critères de diagnostic de la sclérose en plaques ou critères de McDonald
Il est important de rappeler qu’aujourd’hui encore aucun test reposant uniquement sur l’analyse d’un seul critère clinique ou biologique ne permet de diagnostiquer de manière fiable un début de SEP. A l’origine, le diagnostic de la maladie reposait sur des critères cliniques et parfois paracliniques. En 2001, un regroupement international de neurologues proposa un nouveau consensus pour le diagnostic de la SEP basé sur des critères cliniques, paracliniques et IRM. Ce nouveau consensus fut appelé « les critères de McDonald »
[McDonald et al., 2001]. Les critères de McDonald reposent sur le principe que les lésions doivent être disséminées dans l’espace mais aussi dans le temps afin que le diagnostic de SEP soit posé de manière non ambiguë. Chose nouvelle, les critères de McDonald incluent aussi un schéma permettant de diagnostiquer les formes PP-MS qui sont caractérisées, dès l’apparition de la maladie, par l’absence d’une succession de poussées et de rémissions. A la suite du diagnostic, le neurologue peut classer les patients en 3 catégories : patients souffrant de SEP, patients ne souffrant pas de SEP et patients souffrant possiblement de SEP. Dans ce dernier cas, des examens complémentaires (analyse du liquide céphalo-rachidien, examen des potentiels évoqués visuels) doivent être pratiqués, et le patient doit être réévalué par la suite. Ces critères sont internationalement reconnus et ont été rapidement adoptés par l’ensemble de la communauté de neurologues. En 2005, une révision des critères de McDonald fut apportée afin de simplifier et de raccourcir le temps de diagnostic de la SEP, tout en maintenant une
bonne sensitivité et spécificité [Polman et al., 2005]. Comme représenté dans le tableau I, le neurologue s’appuie sur des informations cliniques qui sont complétées par des informations IRM ou parfois paracliniques.
Tableau I : Critères de McDonald révisés (2005) pour le diagnostic de la SEP. D’après Polman et al., Ann. Neurol., 2005.
I.5. Les méthodes d’investigation paracliniques
Les différentes méthodes d’investigation paracliniques permettent d’aider le neurologue dans son diagnostic en renforçant les observations cliniques lorsqu’elles ne sont pas suffisantes.
I.5.1. Imagerie par résonnance magnétique
L’IRM conventionnelle est un outil paraclinique précieux pour le clinicien à la fois dans le diagnostic d’un début de SEP et dans l’évaluation de la progression et de l’activité de la maladie. Les lésions apparaissent sous forme de taches hypo- ou hyper-intenses reflétant une inflammation locale ou une destructuration de l’intégrité du tissu. La prise d’images du SNC se fait en deux temps afin de scanner le cerveau puis la moëlle épinière du patient. Les données IRM peuvent présenter certains avantages par rapport à une approche purement
clinique. D’abord, ces données sont beaucoup moins subjectives. De plus, la technique est très sensible aux changements induits par l’avancée de la SEP [Rovaris et al. 1999]. En effet, les événements inflammatoires visibles en IRM sont de 5 à 10 fois plus fréquents que les poussées cliniques [Miller et al., 1996]. Cependant, la corrélation entre les mesures IRM, l’activité de la maladie, et les manifestations cliniques sont faibles. Cette mauvaise corrélation pourrait résulter en partie de l’incapacité à quantifier par l’approche IRM l’étendue de la lésion, mais aussi la nature du tissu atteint.
On peut globalement dire qu’il existe principalement trois séquences d’acquisition des images en IRM conventionnelle: la séquence T1, la séquence T1 associée à la prise de contraste au gadolinium, et enfin la séquence T2.
- La méthode la plus sensible pour la détection de l’ensemble des lésions de SEP est l’acquisition d’images en séquence T2. Cette sensibilité est très utile pour le diagnostic de la maladie [McDonald et al., 2001] mais aussi pour suivre son évolution (en comptant le nombre de nouvelles lésions, ou en observant l’extension des anciennes). Les lésions apparaissent sous la forme de taches hyper-intenses (blanches).
- La méthode donnant des images en séquence T1 est beaucoup moins sensible que la précédente, et certaines lésions n’y sont pas visibles. Néanmoins, elle présente l’avantage d’autoriser la visualisation de lésions hypo-intenses (noires) qui ont été appelées « trous noirs ». Cependant, la définition d’un trou noir n’est pas arbitraire et dépend fortement de l’opérateur qui analyse les clichés d’IRM. Ces trous noirs sont des lésions hypo-intenses chroniques qui ont été reportées comme étant des zones où une très forte démyélinisation et une perte axonale ont eu lieu [Van Walderveen et al., 1998]. Ce sont les séquelles de lésions anciennes associées à la présence d’une atrophie locale.
- La séquence T1 associée à l’injection intraveineuse d’un composé à prise de contraste, le gadolinium, avant l’acquisition d’images permet de distinguer les lésions actives des lésions inactives. En effet, le gadolinium ne peut normalement pas diffuser dans le SNC. Une prise de contraste n’est alors possible que dans les zones où la perméabilité de la BBB est augmentée, révélant ainsi les lésions où l’inflammation a encore lieu. Toutes ces approches permettent aussi de mesurer l’atrophie qui a lieu au niveau du cerveau et de la moëlle épinière du patient, ce qui permet au neurologue d’estimer l’étendue de la perte totale de tissu [Filippi et al., 2002].
En plus de ces approches conventionnelles, des variations dans les protocoles d’imagerie IRM permettent aujourd’hui de distinguer de plus en plus de composants impliqués dans la pathologie de la SEP : l’inflammation, les dommages axonaux, la démyélinisation, et l’astrocytose par exemple [Compston et al., 2002].
I.5.2. Analyse du liquide céphalo-rachidien
Dans 90% des cas de SEP, l’électrophorèse des protéines contenues dans le liquide
céphalo-rachidien (LCR) révèle un profil de bandes oligoclonales d’immunoglobulines d’isotype G (IgG) (Figure 3). Une anormalité du LCR se traduit par la présence de ces
bandes oligoclonales d’IgG qui ne sont pas présentes dans le sérum du même patient. Leur présence indique une production locale ce qui suppose la présence de lésions immunes et inflammatoires dans le SNC. Cette pratique fait partie du diagnostic courant de la SEP car il permet d’éliminer les pathologies aux symptômes proches de la SEP (lupus, autres maladies auto-immunes systémiques).
Figure 3 : Présence de bandes oligoclonales d’IgG retrouvées uniquement dans le LCR d’un individu souffrant
de SEP.
I.5.3. Examen des potentiels évoqués visuels
Les potentiels évoqués visuels (PEV) désignent un signal électrique produit par le système nerveux en réponse à une stimulation externe de nature visuelle (flashs lumineux, motifs à damiers). Chez une personne souffrant de SEP, le PEV enregistré présente une forme typique : la forme en vague est conservée mais retardée dans le temps (Figure 4). Ils
permettent de rechercher une atteinte spécifique du nerf optique qui serait passée inaperçue et de poser le diagnostic de SEP si les critères cliniques et IRM sont insuffisants.
Figure 4 : Comparaison d’un PEV enregistré chez un individu souffrant de SEP avec celui d’un individu sain.
I.6. Mesure de la progression du handicap
La progression de la maladie chez un patient peut être quantifiée en se reportant à une échelle de mesure : l’échelle élargie de progression du handicap (EDSS) [Kurtzke et al., 1955]. Bien qu’il existe d’autres échelles pour évaluer le handicap, l’échelle EDSS est l’un des instruments les plus anciens et les plus utilisés par les neurologues pour mesurer le degré d’invalidité du patient [Sharrack et al., 1996]. L’échelle EDSS se répartit en 20 niveaux (répartis de 0 à 10 avec des demi-points) [Kurtzke et al., 1983]. Elle débute avec un score de 0 pour un examen neurologique normal, et finit avec un score de 10 qui correspond au décès de l’individu comme conséquence de la SEP (Figure 5). Les scores les plus bas évaluent surtout des limitations fonctionnelles peu visibles, tandis que les scores les plus élevés mesurent davantage l’invalidité. Cette échelle prend en compte 8 paramètres fonctionnels (PF) du SNC ainsi que la capacité du patient à marcher. Les 8 PF testés (les 4 premiers sont majeurs alors que les 4 derniers sont plus mineurs) sont mutuellement exclusifs : (1) fonction pyramidale, (2) fonction cérébelleuse, (3) fonction du tronc cérébral, (4) fonction sensitive, (5) transit intestinal et fonction urinaire, (6) fonction visuelle, (7) fonction cérébrale et (8) autres fonctions [Kurtzke et al., 1984]. L’ensemble des PF couvre la totalité des déficits que l’on peut retrouver dans la SEP. Chaque PF présente des grades ordonnés de 0 à 5 (ou 6). Jusqu'au score EDSS de niveau 3,5 ce sont les scores obtenus dans chaque PF qui déterminent automatiquement le score EDSS. Puis, à partir d’un score EDSS de 4, la définition de chaque niveau est aussi complétée par la capacité de marche du patient [Sharrack et al., 1996].
présente plusieurs imperfections. Tout d’abord, cette échelle ne repose pas uniquement sur des mesures objectives, mais fait appel à des mesures subjectives rendant parfois difficile l’allocation d’un score EDSS à un patient. De plus, comme cette échelle repose sur une combinaison de mesures de PF et de mesures ambulatoires, un patient semblant présenter une maladie plus sévère qu’un autre peut cependant se voir attribuer un score EDSS identique. En effet, si le score EDSS est validé pour suivre l’évolution de la maladie chez un patient, il l’est beaucoup moins pour la comparaison de la maladie entre plusieurs patients. Enfin, il est important de garder en mémoire que l’échelle EDSS n’est pas une mesure linéaire. En effet, les patients progressent plus rapidement entre les scores 1 à 5 qu’entre les scores 5 à 7 [Myers et al., 1992].
Par ailleurs, la vitesse d’évolution clinique de la SEP est très variable d’un patient à un autre. Or, en s’appuyant uniquement sur un score EDSS (qui n’est qu’une image de la maladie au temps T), on ne prend pas en compte cette notion de vitesse d’évolution. La radiologie et le nombre de poussées annuelles (fréquemment utilisés pour mesurer l’activité de la maladie) n’y font pas référence eux aussi. Pourtant la durée d’évolution de la maladie est un facteur important dans l’accumulation des dommages dans le SNC et la sommation des handicaps physiques. En 2005, une approche basée sur un algorithme fut développée afin de prendre en compte « l’agressivité » de la maladie : il fut nommé score de sévérité de la SEP (MSSS)
[Roxburgh et al., 2005]. En regardant la grille de MSSS (Figure 6), on se rend compte par
exemple, qu’un score élevé de MSSS peut être aussi bien assigné à un patient ayant rapidement démontré un score EDSS intermédiaire, ou alors à un patient présentant un handicap sévère mais après une évolution moyennement longue de la maladie. Il semblerait que chez un même patient, le score MSSS soit relativement stable sur de longues périodes, même si certains patients peuvent avoir des variations plus ou moins importantes [Pachner et al., 2009]. Cette méthode pourrait ainsi être un outil utile pour prédire rapidement la sévérité de la SEP chez un patient, c'est-à-dire son évolution au cours du temps.
II. Les facteurs de susceptibilité à la sclérose en plaques
La communauté scientifique s’accorde à penser que le développement de la SEP n’est pas sous la dépendance d’un seul facteur, mais résulte plutôt de l’action globale de plusieurs facteurs : des facteurs de susceptibilité génétique, des facteurs environnementaux et enfin des facteurs épigénétiques qui sont à l’interface des deux premiers facteurs cités. La maladie se développerait chez des personnes génétiquement susceptibles qui auraient été mises en présence de facteurs environnementaux dont une conséquence serait de modifier le profil épigénétique du génome (Figure 7).
MS MS MS MS O1 O1 O1 O1 EBV EBV VD VD G G G G Pathway #1 Pathway #2 Pathway #3 Pathway #4 MS MS MS MS O1 O1 O1 O1 EBV EBV VD VD G G G G Pathway #1 Pathway #2 Pathway #3 Pathway #4
Figure 7 : Les différentes voies pouvant conduire au développement de la SEP. G : facteurs génétiques, VD :
carence en vitamine D, EBV : infection par le virus d’Epstein-Barr, O1-4 : autres facteurs environnementaux inconnus. D’après Goodin, PLoS ONE, 2009.
II.1. Les facteurs génétiques
II.1.1. Les éléments soulignant l’importance de la génétique dans la maladie
L’étiologie génétique de la SEP est suggérée par l’augmentation du risque pour un patient de développer une SEP si un des membres de sa famille en est atteint (Figure 8). Un outil de mesure de cette agrégation familiale est le λs. Ce paramètre est défini comme le ratio du risque de développer une SEP chez une personne apparentée au patient SEP (Ks) sur la prévalence de la maladie dans la population générale (K = 0,1%-0,2%) : (λs = Ks/K)
[Oksenberg et al., 2001]. Ainsi, une valeur de λs égale à 1 indiquerait l’absence d’agrégation familiale d’une maladie. Dans la SEP, cette valeur de λs varie en général entre 20 et 40 pour
d’épidémiologie génétique standard et d’une correction du facteur âge a démontré que les personnes liées aux 1er, 2ème et 3ème degrés à une personne atteinte de SEP présentent un risque augmenté de développer la maladie par rapport à la population générale. Le risque passe ainsi de 0,1% à 3% pour une personne apparentée au 1er degré (5% pour les frères et sœurs, 2% pour les parents et 2% pour les enfants), soit un λs de l’ordre de 15 à 30. Pour les individus apparentés au 2ème et 3ème degrés, ce risque est moins élevé (proche de 1%) mais reste qu’en même supérieur à celui de la population générale [Robertson et al., 1996]. Cependant ces données sont insuffisantes puisqu’elles ne permettent pas de faire la part entre le poids de la génétique et celui de l’environnement familial [Dyment et al., 2004]. Ce sont des travaux réalisés chez des enfants adoptés et chez des demi-frères/sœurs qui supportent le concept que les facteurs génétiques sont majoritairement responsables de l’agrégation familiale de la maladie
[Oksenberg et al., 2001]. Les résultats de l’étude menée sur l’adoption révélèrent que, bien que des enfants adoptés aient vécu depuis leur enfance avec une personne souffrant de SEP, ils ne présentaient pas plus de risque de développer une SEP que la population générale (λs = 1)
[Ebers et al., 1995]. Des études furent aussi menées sur les demi-frères et sœurs, ce qui permit de tester l’effet du partage du patrimoine génétique sur le risque de développer la maladie (les demi-frères et sœurs partagent seulement 25% de leur information génétique, alors que les enfants partageant les mêmes parents ont 50% de leur information génétique en commun). Il fut montré que les demi-frères/sœurs d’un enfant atteint de SEP présentaient un risque significativement plus faible que celui des « vrais » frères et sœurs (1,3% vs 3,5%, p < 0,001)
[Sadovnick et al., 1996]. Par ailleurs, ce travail démontra que le sexe du parent commun aux deux enfants n’influençait pas le risque de SEP puisque les demi-frères/sœurs d’origine maternelle et paternelle partageaient un risque comparable (risques respectifs de 1,4% et 1,2%) [Sadovnick et al., 1996]. Enfin, des études réalisées sur des familles dont les deux parents souffraient de SEP démontrèrent que les enfants issus de ces couples présentaient un risque significativement augmenté par rapport à des enfants provenant de familles dont un seul parent était atteint [Roberston et al., 1997 ; Ebers et al., 2000]. De plus, les auteurs de ce travail confirmèrent le rôle majeur de la génétique sur l’environnement en démontrant que les épouses de personnes souffrant de SEP présentaient un risque de développer la maladie comparable à celui de la population générale [Ebers et al., 2000]. Enfin, des études réalisées sur des jumeaux apportèrent la preuve du rôle majeur de la génétique dans la maladie. Alors que des jumeaux monozygotes (MZ) partagent 100% de leur information génétique, des
« singuliers ». La concordance observée chez les jumeaux MZ était significativement supérieure à celle observée chez des jumeaux DZ ou frères/sœurs singuliers (concordances équivalentes à 25% pour les jumeaux MZ, 5% pour les jumeaux DZ et 3% les frères/soeurs)
[Willer et al., 2003]. Ainsi, chez des jumeaux MZ, le risque de récurrence est d’environ 34% ce qui confère une augmentation du risque (λs) de 170 fois [Dyment et al., 2004]. De plus, l’importance du sexe des jumeaux fut soulignée puisque les auteurs démontrèrent que la différence de risque observée entre des jumeaux MZ et des jumeaux DZ n’était pas retrouvée chez des jumeaux de sexe masculin [Willer et al., 2003]. Cependant, malgré une information
génétique identique la majorité des jumeaux MZ sont discordants pour la SEP (environ 75% sont discordants) ce qui suggère malgré tout l’importance de facteurs non-génétiques intervenant dans l’étiologie de la maladie (nous reviendrons par la suite sur les facteurs non-génétiques impliqués) [Willer et al., 2003].
Figure 8 : Risque de récurrence intrafamiliale pour la SEP. D’après Compston et al., Lancet, 2008.
II.1.2. Etiologie génétique de la sclérose en plaques
La SEP fait partie de la grande famille des maladies complexes. Il est important de comprendre la différence qu’il existe entre les maladies génétiques mendéliennes et les maladies complexes (Figure 9). Dans les maladies génétiques à transmission mendélienne, ce sont généralement des mutations dans un ou quelques gène(s) qui sont responsables de l’apparition de la maladie. Par ailleurs, la présence de la mutation est associée à une très forte pénétrance de la maladie qui est généralement égale à 100% [Prichard et al., 2002]. De plus, la fréquence des mutations conduisant à l’apparition de la maladie est généralement faible