Caractérisation de la voie de dégradation de l’α-synucléine
catalysée par la Polo-Like Kinase 2
Mémoire
Manel Dahmene
Maitrise en Neurobiologie
Maître ès sciences (M.Sc.)
Québec, Canada
©Manel Dahmene, 2018
Caractérisation de la voie de dégradation de l’α-synucléine
catalysée par la Polo-Like Kinase 2
Mémoire
Manel Dahmene
Sous la direction de :
iii
Résumé
La maladie de Parkinson est une maladie neurologique chronique caractérisée par la dégénérescence progressive des neurones dopaminergiques de la substance noire pars compacta. Une deuxième caractéristique neuropathologique de cette maladie est l’accumulation des agrégats intracellulaires appelés les corps de Lewy (CLs). Ces agrégats sont majoritairement formés par une protéine pré-synaptique, α-synucléine (α-syn). Cette accumulation pathologique interfère avec les voies métaboliques vitales des neurones telles que la transmission synaptique et l’activité mitochondriale, ce qui engendre la mort cellulaire. Par conséquence, éliminer les formes toxiques, diminuer l’expression de la forme native et réduire ainsi la probabilité de la formation d’agrégats pourrait être une stratégie thérapeutique d’intérêt pour le traitement de la maladie de Parkinson et d’autres désordres qui y sont reliés.
Dans ce contexte, notre équipe a récemment décrit une nouvelle voie d’élimination de l’α-syn qui est catalysée par l’activité enzymatique de la kinase Polo-like kinase 2 (PLK2). Cependant, les mécanismes cellulaires ainsi que l’identité des molécules impliquées sont encore méconnus. Donc, mes travaux se sont concentrés sur l’étude de cette voie et ses différentes étapes qui mènent à enlever l’effet toxique de l’α-syn.
Dans ce mémoire nous montrons que, en plus de la PLK2, la PLK3, un autre membre de la famille des PLKs, est capable de phosphoryler l’α-syn au niveau du résidu S129 et induire son élimination. En plus, nous déclarons que cette action exige une interaction physique entre les 2 protéines (α-syn et PLK2) impliquant le domaine N-terminal et qu’une étape de poly-ubiquitination est essentielle pour que ce complexe protéique se dirige vers la voie de dégradation autophagique. Cette action de la PLK2 est observée également sur des formes mutées de l’α-syn tels que α-syn A30P, α-syn A53T et d’une manière plus accentuée sur la forme mutante α-syn E46K.
iv
La caractérisation de cette voie d’élimination offre de nouvelles opportunités pour le développement des traitements qui favorisent, d’une façon spécifique et sélective, la dégradation de l’α-syn et par conséquent la réduction des formes toxiques de cette dernière.
v
Abstract
Parkinson's disease is a chronic neurological disease characterized by the progressive degeneration of the dopaminergic neurons of the substantia nigra pars compacta. A second neuropathological feature of this disease is the accumulation of intracellular aggregates called Lewy bodies. These aggregates are formed by a pre-synaptic protein, α-synuclein (α-syn). This pathological accumulation interferes with the vital metabolic pathways of neurons such as synaptic transmission and mitochondrial activity, leading to cell death. Consequently, promoting the elimination of the toxic forms, reducing the expression of the native form and decreasing the probability of aggregate formation could be a therapeutic strategy of interest for the treatment of Parkinson's disease and other related disorders.
Recently, we have described a novel α-syn degradation pathway that is catalyzed by the enzymatic activity of Polo-like kinase 2 (PLK2). However, the cellular mechanism and the identity of the molecules involved are still unknown. So, my work has focused on studying this pathway and its various steps that lead to remove the toxic effect of α-syn.
In this thesis we show that, in addition to PLK2, PLK3, another member of the PLK family, is able to phosphorylate α-syn at S129 and induce its elimination. In addition, we declare that this action requires a physical interaction between the 2 proteins (α-syn and PLK2) involving the N-terminal domain and that a poly-ubiquitination step is essential for the autophagic degradation of the α-syn and PLK2 complex. This effect of PLK2 is also observed on mutated forms of α-syn such as α-syn A30P, α-syn A53T and is more pronounced in the case of the α-syn E46K mutant.
The characterization of this elimination pathway offers new opportunities for the development of treatments that allow, in a specific and selective manner, the degradation of α-syn and thus the reduction of its toxic forms.
vi
Table des matières
Résumé ... iii
Abstract ... v
Tables des matières ... vi
Liste des tableaux ... viii
Liste des figures ... ix
Liste des abréviations ... x
Remerciements ... xi
Avant-propos ... xiii
1. Chapitre I : Introduction ... 1
1.1. La maladie de Parkinson... 1
1.1.1. Historique de la maladie de Parkinson... 1
1.1.2. Épidémiologie de la maladie de Parkinson ... 3
1.1.3. Les symptômes de la maladie de Parkinson... 4
1.1.3.A. Les symptômes moteurs ... 4
1.1.3.B. Les symptômes non moteurs ... 4
1.1.4. Pathophysiologie de la maladie de Parkinson ... 5
1.1.4.A. La perte sélective des neurones dopaminergiques et son impact sur le fonctionnement des ganglions de la base ... 6
1.1.4.B. L’agrégation de l’α-synucléine et la formation des corps de Lewy ... 8
1.1.5. Causes et facteurs de risque de la maladie de Parkinson ... 9
1.1.4.A. Les risques génétiques ... 9
1.1.4.B. Les risques environnementaux ... 12
1.1.6. Diagnostique ... 13
1.1.7. Traitements pharmacologiques et chirurgicaux de la maladie de Parkinson ... 14
1.2. L’alpha-synucléine ... 16
1.2.1. Structure de l’α-synucléine ... 16
1.2.2. Localisation et fonction de l’α-synucléine ... 19
1.2.3. L’α-synucléine dans la maladie de Parkinson ... 20
1.2.3.A. Agrégation de l’α-syn ... 21
1.2.3.B. Propagation des formes toxiques d’α-synucléine et son impact sur l’évolution de la maladie de Parkinson ... 22
vii
1.2.4. Rôle des modifications post-traductionnelles dans la régulation de la toxicité de
l’α-syn : focus sur la phosphorylation ... 25
1.3. Kinases responsables de la phosphorylation de l’α-syn : Les Polo-like Kinases : ... 28
1.3.1. Structure ... 29
1.3.2. Fonction ... 30
1.3.3. Phosphorylation de l’α-syn par les PLKs... 31
2. Chapitre II : Caractérisation de la voie de dégradation de l’α-synucléine catalysée par la Polo-Like Kinase 2 ... 37 2.1. L’article scientifique ... 37 2.2. Résumé ... 38 2.3. Abstract ... 39 2.4. Introduction ... 40 2.5. Results ... 41 2.6. Discussion ... 47
2.7. Conclusions and impact for PDtherapeutic strategies ... 52
2.8. Materials and methods ... 52
3. Chapitre III : Discussion ... 69
4. Chapitre IV : Perspectives... 76
viii
Liste des tableaux
ix
Liste des figures
Figure 1: Schéma simplifié des voies striatales directes et indirectes chez un sujet sain et
un sujet parkinsonien ... 7
Figure 2: Schéma illustrant la dégénérescence des neurones dopaminergiques dans la maladie de Parkinson qui est à l’origine des troubles moteurs ... 8
Figure 3: Représentation d’un corps de Lewy ... 9
Figure 4: Représentation schématique de la structure de l’α-syn ... 18
Figure 5: Les mécanismes de neurotoxicité des agrégats de l’α-syn. ... 22
Figure 6: Les stades de Braak ... 23
Figure 7: Hypothèse de Braak ... 24
Figure 8: Mécanismes de propagation de l’α-synucléine ... 25
Figure 9: Rôle potentiel de la phosphorylation au S129 dans la régulation de l’élimination de l’α-syn, son agrégation et sa toxicité ... 28
Figure 10: Une présentation schématique de la séquence des membres de la famille des PLKs en illustrant les différences et les similitudes sur le niveau structural entre les 4 membres ... 29
Figure 11: La surexpression spécifique de la PLK2 favorise l’élimination de l’α-syn par voie autophagique in vitro. ... 34
Figure 12: La PLK2 wt induit la baisse du niveau d’α-syn et supprime sa toxicité, in vivo ...35
Figure 13: Schéma résumant le mécanisme d’élimination de l’α-syn par voie autophagique, sous l’action kinase de la PLK2 ... 36
Figure 14: Un schéma représentant les étapes de la voie de dégradation de l’α-syn sous l’action de la PLK2... 75
x
Liste des abréviations
AAV: de l’anglais «adenosine associated virus» AMS : atrophie multisystématisée
ATG5: de l’anglais «autophagy-related gene 5» ATG7: de l’anglais «autophagy-related gene 7» ATV : aire tégmentale ventrale
BHE : barrière hémato-encéphalique CKs : caséine kinases
CL : corps de Lewy
COMT: catéchol O-méthyl transférase DAergiques: neurones dopaminergiques GPe : globus pallidus externe GPi: globus pallidus interne
GRKs: de l’anglais «G-protein coupled receptor kinases» IMAO B: monoamine-oxydase B
IRM : imagerie par Résonance Magnétique LRC : liquide céphalorachidien
LRRK2: leucine-rich repeat kinase 2 MP: maladie de parkinson
NAC: de l’anglais «non-amyloidogenic component» Nh4Cl: chloride d'ammonium
PINK1: de l’anglais «PTEN-induced putative kinase 1» PLK2: polo-like-kinase 2
PLK2 wt: PLK2 forme sauvage
PLK2 KDM: de l’anglais «PLK2 kinase dead mutant» PLKs: polo-like kinases
pS129: phophorylation au S129
RBM17: de l’anglais «RNA-binding motif protein 17» SN: système nerveux
SNA: système nerveux autonome SNC: système nerveux central SNE: système nerveux entérique SNP: système nerveux périphérique SNpc: substance noire pars compacta SNpr: substance noire pars reticulata STN: noyaux sous-thalamiques TH: tyrosine hydroxylase TQ: thymoquinone
Ub-K0: de l’anglais «lysine-less ubiquitin mutant» α-syn: α-synucléine
α-syn pS129:α-syn phosphorylée au S129 β-syn: β-synucléine
xi
Remerciements
En premier lieu, j’adresse mes sincères remerciements à mon directeur de mémoire, le Dr Abid Oueslati. Merci de m’avoir accueilli dans votre laboratoire et me confier un projet de thèse si passionnant. J’apprécie énormément tout le temps que vous avez investi dans ma formation et le suivi que vous m’avez procuré pour le bon déroulement de mes travaux. Sincèrement, je dois avouer que j’ai été très chanceuse d’avoir un directeur de recherche aussi présent et disponible et privilégiée d’être votre première étudiante dans votre nouvelle équipe. Je vous suis également reconnaissante pour votre encadrement scientifique rigoureux et vos conseils précieux. Tenez en compte que vous étiez ma source d’inspiration et le stimulus de ma curiosité scientifique, tout le long de mon parcours au sein de votre laboratoire, grâce à votre passion, votre générosité et votre dévouement envers la science, et en particulier la neuroscience.
Aussi, j’exprime ma gratitude aux membres du jury qui ont accepté d’évaluer ce travail : Dre Francesca Cicchetti et Dr Steve Lacroix.
Je voudrais également remercier toutes les personnes qui m’ont accompagné au cours de ma vie au sein du laboratoire des neurosciences qu’ils soient les membres de notre équipe, Morgan et Marilyn-2 étudiants au doctorat qui nous ont rejoints vers la fin de ma maitrise-et les stagiaires, pour leur aide appréciée. Je tiens aussi à remercier Dr Marc Morissette pour son aide précieuse avec des commentaires constructifs et des remarques amélioratrices de ce travail. Sans oublier mes voisins de paillasse et de bureau, un énorme merci, pour leur collaboration et leur amicale bienveillance.
Pour terminer, je dédie des remerciements permanents à mes parents, Awatef et Lotfi. Du fond de mon cœur, je vous remercie mes chers pour votre support constant depuis le début de mes études, vos encouragements et votre confiance illimitée en moi. C’est de vous que je tiens ma persévérance et mon ambition. Un grand merci à mes
xii
deux frères, Radhi et Ahmed yassine, qui à leur manière, ont été des grands supporteurs. Je remercie aussi mon amoureux, Achref, pour avoir partagé avec moi les moments de gloire et de découragement qu’on rencontre, bien évidemment, durant le parcours de la recherche scientifique, toujours avec autant d’amour et de soutien inestimable. Tu étais toujours mon recours et ma force chéri !
J’adresse des remerciements spécieux à tous mes amis qui m’ont aidé à décrocher lorsque j’en avais besoin, ce qui m’a permis d’être toujours si énergétique et positive dans mon travail. Je vous le dit à plein cœur que votre énorme soutien œuvrant de près ou même depuis des milliers de kilomètres, était primordial pour aboutir à cette réussite. Mes amis, ou comme je préfère vous appeler «ma 2ème famille», votre présence dans ma vie a été très marquante.
Finalement, un dernier merci pour toutes ces personnes qui, sans le savoir, m’ont permis d’avoir une expérience fabuleuse et fastidieuse dont je me souviendrai à jamais.
xiii
Avant-propos
Ce mémoire porte sur la caractérisation d’une nouvelle voie d’évacuation de l’α-synucléine sous l’action de la polo-like kinase 2 et comporte 4 chapitres. Le premier chapitre consiste en une introduction générale qui regroupe des connaissances reliées à la maladie de Parkinson, ses causes, ses traitements disponibles et surtout ses manifestations physiopathologiques en faisant appel aux travaux de recherche effectués dans ce domaine jusqu’à ce jour. Le chapitre 2 correspond à un article intitulé «Caractérisation de la voie de dégradation de l’α-synucléine catalysée par la Polo-Like Kinase 2» et publié dans le journal «Journal of Biological Chemistry» le 30 Janvier 2017 dans lequel la partie méthodes et résultats est bien détaillée. Je suis la première auteure de cet article et j’ai effectué les manipulations expérimentales, la collecte et l’analyse des données et la préparation des illustrations. Mon collègue Morgan Bérard a contribué dans l’exécution des manipulations expérimentales. La rédaction de l’article a été effectuée par Dr Oueslati, mon directeur de recherche dont la contribution fut primordiale dans la réussite du projet. Finalement, les chapitres 3 et 4, respectivement, discutent les résultats et ouvrent les portes vers des perspectives potentielles.
Étant que première étudiante de Dr Abid Oueslati, j’ai eu la chance de m’impliquer dans plusieurs projets du laboratoire qui ne sont pas inclus dans ce mémoire tels que le projet portant sur l’expression et la propagation de l’α-syn et les travaux sur la maladie d’Alzheimer et l’élimination de la protéine précurseur de l’amyloïde. De plus, c’était une expérience agréable qui m’a permis d’avoir des responsabilités plus élargies et de participer à l’organisation du milieu de travail.
1
1. Chapitre I : Introduction
1.1. La maladie de ParkinsonLa maladie de parkinson (MP) est une maladie dégénérative qui affecte le système nerveux central (SNC). Elle est caractérisée par des troubles moteurs tels que des tremblements au repos, une rigidité musculaire et une bradykinésie (akinésie)1. Plusieurs symptômes non-moteurs comme la dépression, l’anxiété et certains problèmes cognitifs peuvent également se développer durant la progression de la maladie1.
Sur le plan neuropathologique, cette maladie est caractérisée par la mort sélective des neurones dopaminergiques de la substance noire pars compacta (SNpc) et par la présence d’inclusions protéiques dans les neurones survivants2,3.
1.1.1. Historique de la maladie de Parkinson
La MP a été décrite pour la première fois par le médecin anglais James Parkinson en 1817 et documentée dans son ouvrage intitulé‘An Essay on the ShakingPalsy’ (Essai sur la paralysie agitante,1817)4. Dans cet ouvrage, il a décrit certaines caractéristiques principales de la maladie dont les tremblements au repos, les anomalies de la posture, les anomalies de la marche et la difficulté à initier des mouvements volontaires. Cet essai a été reconnu comme le véritable travail fondateur décrivant la maladie4. À cette époque, la description de James Parkinson se limite au plan moteur et n’associe pas d’autres troubles sensoriels ou mentaux. Une quarantaine d'années plus tard (en 1861), le neurologue français Jean-Martin Charcot a étudié le syndrome et a participé à l’approfondissement de la séméiologie de la maladie où il explique la différence entre rigidité musculaire, faiblesse et bradykinésie. En 1865, il a été suggéré de donner à cette maladie le nom de maladie de Parkinson pour honorer l’homme qui a décrit la pathologie pour la première fois. Cette suggestion a été proposée pour la première fois par William Sanders5 et ensuite, vulgarisée par Dr Charcot6.
2
Les recherches sur cette pathologie et ses mécanismes physiologiques et anatomiques se poursuivèrent, jusqu’en 1912, date à laquelle Fréderic Lewy a décrit la présence d’inclusions protéiques dans le cerveau des patients atteints de la MP, dénommées plus tard Corps de Lewy (CLs).
En 1919, grâce à l’examen des cerveaux des patients parkinsoniens décédés, une observation intéressante a été faite par Konstantin Tretiakoff et confirmée une vingtaine d’année plus tard (1938) par Rolf Hassler, que la région cérébrale la plus atteinte dans la MP est la substance noire. Des années plus tard, on a mis en évidence la présence d’un nouveau neurotransmetteur : la dopamine dans le striatum des patients parkinsoniens par Arvid Carlsson, un neurobiologiste suédois dont les travaux sur la dopamine ont remporté le prix Nobel de médecine (en 2000)5.
À la fin des années 60, une description symptomatique a été attribuée à la MP et depuis, elle sert comme un outil de diagnostic de la pathologie. La MP est reconnue surtout par le tremblement au repos, la rigidité musculaire, l’akinésie et la perte des réflexes posturaux.
Avec le développement des techniques biochimiques et la progression des études thérapeutiques telles que celles réalisées au Canada par Barbeau et ses collaborateurs de 1960 à 1975, par Poirier et ses collaborateurs de 1964 à 1969 et par Anden et ses collaborateurs (1964), la MP a été associée à un déficit en dopamine au niveau des noyaux caudé et putamen. Cette évidence a mené à l’apparition de traitements basés sur l’administration de la 3,4-dihydroxyphénylalanine (L-Dopa), un précurseur de la synthèse de la dopamine, qui s'imposèrent rapidement comme le traitement spécifique de la maladie et connurent une popularité fulgurante surtout au cours des années 70.
Plus tard dans les années 90, il a eu la découverte d’un gène impliqué dans l’apparition précoce de la MPavec une transmission autosomique dominante. Cette étude a été réalisée par Robert Nussbaum et ses collègues. En effet, ils ont identifié un gène (SNCA) sur le chromosome 4 (région q21-q23) responsable de la synthèse de
l’α-3
synucléine (α-syn) qui serait associé au phénotype de la MP chez plusieurs membres d’une grande famille Italo-américaine présentant une forme précoce autosomique dominante de la MP7. Le séquençage du gène de l’α-syn chez les individus affectés de cette famille a révélé que ces derniers étaient hétérozygotes relativement à une mutation faux-sens alanine→thréonine à la position 53 de la protéine7.
En août 1997, des expériences d’immunoréactivité utilisant différents anticorps dirigés contre l’α-syn chez des patients parkinsoniens ou avec une démence à corps de Lewy (DLB), effectuées par Maria Spillantini et son équipe, ont identifié l’α-syn comme le composant principal des CLs à l’origine de la pathologie7.
Depuis, des études neuropathologiques et expérimentales ont confirmé le rôle central de l’α-syn dans la mort neuronale et la pathogenèse de la MP8,9.
1.1.2. Épidémiologie de la maladie de Parkinson
La MP est la 2ème maladie neurodégénérative la plus fréquente après la maladie d’Alzheimer10. Environ 6 millions de personnes à travers le monde sont touchées par cette maladie, d’après la Fondation Nationale pour la MP. Ce chiffre continue à augmenter et à prévaloir, environ 305 000 nouveaux patients sont diagnostiqués par année dans le monde, dû principalement au vieillissement de la population11.
Généralement, cette maladie est plus répandue chez les personnes âgées de plus de 65 ans mais elle peut apparaitre chez des sujets plus jeunes, entre 25 et 30 ans11–13. De manière intéressante, son incidence est 1.5 fois plus élevée chez les hommes que chez les femmes11,13–15. Des facteurs de risque environnementaux et/ou de susceptibilités génétiques associés à l’apparition de la MP peuvent expliquer la grande variabilité de l’incidence de cette maladie entre les pays11,12.
4
1.1.3. Les symptômes de la maladie de Parkinson
Les symptômes de la MP peuvent être classés en 2 catégories: les symptômes moteurs et les symptômes non moteurs qui peuvent être soit des changements neuropsychiatriques, des changements cognitifs ou des symptômes liés à une défaillance du système nerveux autonome (SNA)1,16,17.
1.1.3.A. Les symptômes moteurs
Généralement, les symptômes observés chez la majorité des patients parkinsoniens sont une lenteur du mouvement, une bradykinésie allant jusqu’à l’akinésie dans des stades plus avancés de la maladie, un tremblement au repos, une instabilité posturale, une rigidité musculaire, de même que des difficultés d’effectuer des gestes alternatifs rapides et quotidiens tels que se brosser les dents, boutonner des vêtements, marcher ou rester debout18,1,19. Ces manifestations physiques se développent au fur et à mesure de la progression de la maladie et elles apparaissent souvent de manière asymétrique puis s’étendent aux deux côtés du corps après quelques années20. En effet, l’apparition des troubles moteurs est le résultat du manque d’un neurotransmetteur essentiel au mouvement, la dopamine. Les premiers signes cliniques de la MP apparaissent lorsque la dégénérescence des neurones dopaminergiques de la SNpc atteint environ un niveau situé entre 60-80 %21.
1.1.3.B. Les symptômes non moteurs
En plus des troubles moteurs, les patients parkinsoniens souffrent généralement des problèmes non moteurs qui précèdent les signes moteurs (phase prodromique)22. En grande partie, les symptômes non moteurs se manifestent fréquemment par des changements neuropsychiatriques caractérisés par l’apparition de la psychose chez environ 30% des patients parkinsoniens incluant des hallucinations visuelles ou auditives et des illusions23,24. De plus, ces symptômes neuropsychiques sont souvent
5
associés à la dépression22,25,26, l’anxiété et l’apathie27–29. Aussi, les personnes atteintes de la MP courent un risque plus élevé de démence, qui mène souvent à des troubles de la mémoire ou de la concentration rappelant ceux causés par la maladie d'Alzheimer30. Plus tardivement dans l’évolution de la maladie, on aperçoit chez le patient de la confusion et des troubles du sommeil31. Finalement, on note la présence de symptômes associés au SNA qui affectent significativement la qualité de vie des patients parkinsoniens. Ces symptômes comprennent la constipation, l’hyposomnie (affecte 80 à 90 % des sujets atteints), l’hypotension orthostatique, la perte de l’olfaction et la dysfonction érectile17,32,33. Également, il existe d’autres signes cliniques qui peuvent apparaitre plus tardivement dans l’évolution de la maladie comme par exemple une difficulté à avaler, une salivation excessive, la perte de dextérité, une difficulté de prononciation, une absence d’expression faciale, et une incontinence urinaire1.
Au final, l’ensemble de ces troubles affecte la qualité de la vie quotidienne, familiale et sociale des patients. Malheureusement, la majorité de ces symptômes ne sont pas corrigés par les médications dopaminergiques (se basant sur la L-Dopa et ses agonistes) ce qui complique d’autant plus le traitement de ces effets non-moteurs.
1.1.4. Pathophysiologie de la maladie de Parkinson
La MP est caractérisée par des troubles physiologiques touchant le SNC et qui se mettent en place une dizaine d’années avant l’apparition des premiers symptômes cliniques de la maladie34.
Deux conditions essentielles sont nécessaires pour l’apparition de la MP : la dégénérescence progressive des neurones dopaminergiques (DAergiques) de la SNpc innervant le striatum et la présence d’inclusions cytoplasmiques appelées CLs qui sont formées principalement d’une protéine intraneuronale, l’α-syn34.
6
1.1.4.A. La perte sélective des neurones dopaminergiques et son impact sur le fonctionnement des ganglions de la base
Malgré que la population des neurones DAergiques est très restreinte et représente moins de 1% de l’ensemble des neurones du cerveau35, elle joue le rôle d’un modulateur essentiel des fonctions motrices et psychiques. Ces neurones qui innervent plusieurs aires cérébrales, peuvent être séparés en 3 voies neuronales principales36 : 1)la voie méso-cortico-limbique dont les corps cellulaires des neurones se trouvent au niveau de l’aire tegmentale ventrale (ATV) et projettent vers les aires limbiques (incluant le striatum ventral, le septum, l’amygdale, et l’hippocampe) et le cortex frontal, 2)la voie nigro-striatale dont les prolongements neuronaux de la SN projettent vers le striatum et finalement 3)la voie tubéro-infundibulaire qui prend naissance dans l’hypothalamus et projette vers l’hypophyse via l’éminence médiane36. Le fait que ces neurones innervent des zones cérébrales variées met en évidence l’importance de la dopamine dans la régulation des différentes fonctions neurologiques.
Dans le cas de la MP, les neurones DAergiques les plus touchés se trouvent principalement au niveau de la SNpc37, cette zone étant impliquée principalement dans le contrôle moteur. La SNpc fait partie d’un groupe de structures sous-corticales anatomiquement interconnectées appelées les ganglions de la base ou les noyaux gris centraux et connues par leur importance dans l’automatisation des mouvements et la suppression des mouvements involontaires38,39. Les ganglions de la base incluent le striatum (putamen et noyau caudé), le Globus Pallidus interne (GPi) et externe (GPe), la substance noire pars reticulata (SNpr) et pars compacta (SNpc) et le noyau sous-thalamique40. Ces structures communiquent entre elles par l’intermédiaire de 3 neurotransmetteurs principaux : l’acide -aminobutyrique (GABA), le glutamate et la dopamine. Dans la MP, la perte préférentielle des neurones DAergiques, qui partent de la SNpc vers le striatum, affecte le contrôle DAergique au niveau du striatum et par conséquent engendre un dérèglement de ses projections vers le pallidum et le STN (Figure 1). Ce dérèglement de la régulation des noyaux gris centraux est responsable de l’apparition des troubles de mouvement et la rigidité musculaire associée à la MP37.
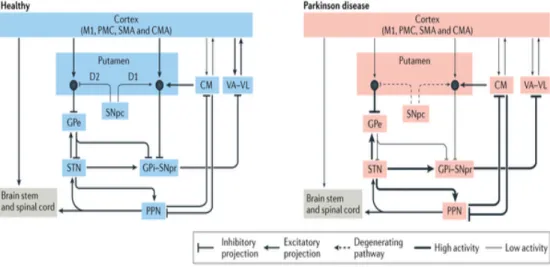
7
Figure 1: Schéma simplifié des voies striatales directes et indirectes chez un sujet sain et un sujet parkinsonien41. Dans des conditions normales, les neurones de la SNpc activent les neurones GABAergiques du striatum porteurs de récepteurs DAergiques D1 et inhibent les neurones GABAergiques qui portent les récepteurs DAergiques D2. Activés par les neurones glutamatergiques arrivant du cortex et des neurones dopaminergiques de la voie directe (D1), l’activité du complexe GPi-SNpr est inhibée. Quant aux neurones GABAergiques striataux porteurs des récepteurs D2 (voie indirecte), l’activation de ces neurones par le glutamate provenant des neurones glutamatergiques cortico-strié inhibe l’activité du GPe qui inhibe à son tour l’activité du STN et du complexe GPi-SNpr. Finalement, l’inhibition sur le thalamus moteur est réduite ce qui favorise la génération des mouvements. Dans le cas d’un patient parkinsonien, la réduction significative de l’innervation DAergique nigro-striée induit une hyperactivité de la voie indirecte qui engendre la désinhibition du GPi-SNpr via une réduction de l’activité GABAergique du GPe et par une augmentation de l’activité des neurones glutamatergiques qui projettent vers le GPE. Par conséquent, l’activation des neurones GABAergiques du complexe GPi-SNpr inhibe l’activité glutamatergique du thalamus qui projette vers les aires motrices du cortex ce qui explique en partie l’apparition des symptômes moteurs de la MP42–44. Abréviations : CM, noyau centromédian; CMA, cortex cingulaire
antérieur; GPe, globus pallidus externe; GPi, globus pallidus interne; M1, cortex moteur primaire; PMC, cortex pré-moteur; PPN, noyau pédunculopontine; SMA, aire motrice supplémentaire; SNpc, substance noire pars compacta; SNpr, substance noire pars reticulata; STN, noyau sous-thalamique; VA/VL, noyau ventral antérieur/ventral latéral.
8
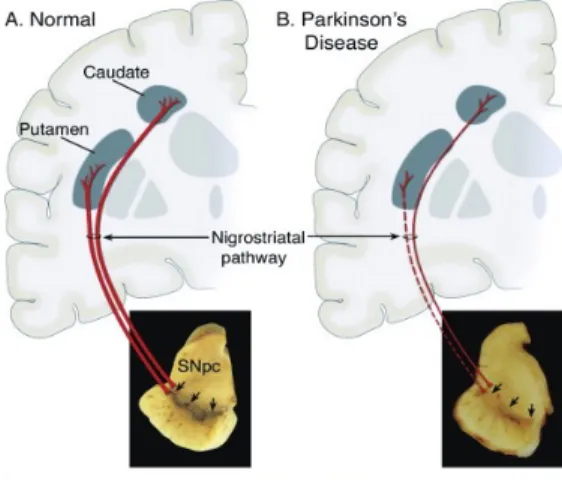
Lorsque les premiers symptômes moteurs de la maladie commencent à apparaitre, la perte neuronale des cellules dopaminergiques de la SNpc est d’environ 50% et les niveaux de la dopamine au striatum sont réduits de 80%45,46 (Figure 2).
Figure 2: Schéma illustrant la dégénérescence des neurones dopaminergiques dans la maladie de Parkinson qui est à l’origine des troubles moteurs34. (A) et (B) Schéma représentant les
projections de la voie nigro-striée à partir de la SNpc vers le striatum chez un sujet normal et un sujet parkinsonien respectivement, on note une dépigmentation de la SNpc et une dégénérescence des prolongements DAergiques dans la pathologie de la MP.
1.1.4.B. L’agrégation de l’α-synucléine et la formation des corps de Lewy
La présence des CLs, qui sont en majorité constitués des formes insolubles et agrégées de l’α-syn3,34,47–49, est une caractéristique principale de la MP (Figure 3). Ce sont des inclusions, principalement, intracytoplasmiques (aperçues parfois dans le milieu extra-neuronal)50. Ces amas se trouvent dans les corps cellulaires ou au niveau des prolongements neuronaux appelés les neurites de Lewy.
9
Figure 3: Représentation d’un corps de Lewy34. Des images résultantes d’une immunohistochimie d’un neurone dopaminergique de la SNpc montrant que les corps de Lewy sont formés de l’α-syn et de l’ubiquitine.
Ces inclusions cytoplasmiques sont éosinophiles sphériques, de 5 à 25µm de diamètre, dont les mécanismes conduisant à leur formation sont encore méconnus (voir en détails la partie 1.2.3). Les CLs ne sont pas spécifiques à la MP, on détecte également leur présence dans d’autres synucléinopathies telles que la démence à corps de Lewy51,52, la maladie d’Alzheimer53,54, la dysautonomie55, l’atrophie multisystématisée (AMS)56 et parfois même au cours du vieillissement normal57.
1.1.5. Causes et facteurs de risque de la maladie de Parkinson
Les causes de la MP sont encore peu connues. Cependant, plusieurs hypothèses associent la maladie à un ensemble des facteurs génétiques et environnementaux58. Entre autre, on considère le vieillissement comme le risque le plus pertinent pour le développement de la MP59 vu que, le plus souvent, les troubles liés à cette maladie commencent à apparaître chez des personnes âgées de plus de 50 ans15.
1.1.4.A. Les risques génétiques
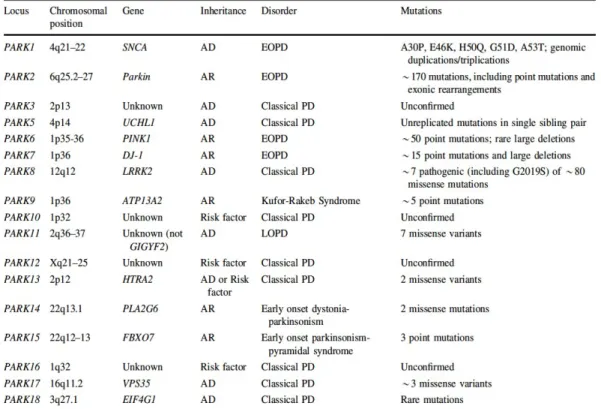
Il existe des formes familiales de la MP qui ont été mises en évidence et qui ne forment qu’environ 10% des cas. En effet, dans des cas rares, la MP peut être due à des mutations génétiques. Jusqu’à aujourd’hui, 18 loci et 11 gènes ont été identifiés
10
(Tableau 1). Généralement, ces 18 loci sont classés sous le nom de PARK 1 à 18 responsables chacun d’un phénotype différent de la MP (Tableau 1)60. Ces gènes sont capables d’induire directement la maladie ou de représenter des facteurs de risque dans certains cas sporadiques60,61.
Tableau 1: Gènes impliqués dans l’apparition de la MP. Ce tableau résume les 18 gènes et leurs mutations responsables de l’apparition des formes familiales de la MP avec les phénotypes associés à chacune des mutations.
D’ailleurs, la forme familiale de la MP est apparue avec la découverte du gène SNCA de l’α-syn en 1997. En effet, la pathologie peut être causée soit par sa duplication ou triplication62, soit par des mutations touchant ce gène et menant à des formes autosomales dominantes de la maladie63. À ce jour, cinq mutations du SNCA ont été identifiées (A53T, A30P, E46 K, H50Q et G51D)64–68.
11
D’autres gènes sont associés aux formes familiales soit le leucine-rich repeat kinase 2 (LRRK2)69,70. La protéine exprimée par ce gène se trouve en grande partie soit dans le cytoplasme ou associée aux membranes mitochondriales. Ceci met en évidence la liaison importante entre la MP et les mitochondries. D’ailleurs, une mutation à l’intérieur de ce gène est capable d’induire les mêmes signes rencontrés chez un patient parkinsonien71 et responsable de l’apparition de la forme autosomale dominante de la maladie72.
Des mutations de certains gènes impliqués dans le système ubiquitine-protéasomique (protéolyse intracellulaire), tels que PARK 2 codant pour la protéine Parkine et PARK 5 codant pour la protéine ubiquitine carboxy-terminale hydrolase L1 (UCH-L1), peuvent provoquer le dysfonctionnement de ce système et causer des formes rares de la MP à transmission héréditaire73. Ceci suggère que la dysfonction de la voie ubiquitine-protéasomique est impliquée dans l’apparition de la pathologie de la MP.
En plus, on mentionne des mutations dans les gènes PTEN-induced putative kinase 1 (PINK1) et parkine, des gènes liés à la mitophagie74, seraient à l’origine de la mise en place d’une forme autosomale récessive de la maladie73,75.
Publiée dans Nature Genetics en juillet 2014, une étude réalisée dans le but de définir le profil génétique de la MP a mis à jour 6 nouveaux facteurs de risque génétique impliqués dans la maladie qui s’ajoutent aux 24 déjà existants76.Ces facteurs de risques comportent 9 gènes qui sont déjà associés à des formes rares familiales de la MP et d’autres qui sont impliqués dans la signalisation de la dopamine et de l'α-syn. Ceci fut démontré grâce à une méta-analyse réalisée à partir des données de plus de 100 000 individus, dont 13 708 individus atteints de la MP et 95 282 individus sains provenant d'études d’association pangénomique (GWAS). Donc, plus un individu était porteur de ces variations génétiques ou de leur combinaison, plus le risque qu’il développe la MP était grand.
12
1.1.4.B. Les risques environnementaux
En plus des facteurs de risque génétique, certains facteurs environnementaux seraient également associés à l’augmentation du risque de développer la MP77. De nombreuses études évoquent la possibilité d’une intoxication (exposition aux pesticides, aux neurotoxines comme le 1-méthyl-4-phényl-1,2,3,6-tétrahydropyridine (MPTP), aux agents pathogènes de l’environnement,…). En 2004, des chercheurs ont mis en lumière la capacité de certains pesticides d’induire des symptômes de la maladie chez les animaux similaires à ceux observés chez les patients parkinsoniens78. De plus, en 2013, d’autres ont démontré que l’exposition environnementale aux pesticides et autres solvants toxiques augmente de 2 fois le risque de contracter la MP79, malgré que le plus souvent ces expositions sont à des concentrations assez faibles mais soutenues sur de longues périodes80. Cet effet nocif est causé par leur haute capacité d’être absorbés par la peau jusqu’à atteindre, dans certains cas, le système nerveux (SN) et altérer son fonctionnement79–81.
Cette maladie multifactorielle peut aussi être causée par le MPTP, un sous-produit de la synthèse d’une drogue illicite, le 1-methyl-4-phenyl-4-propionoxypiperidine (MPPP), qui peut causer de manière soudaine une forme grave et irréversible de la MP. De nos jours, le MPTP est utilisé chez les animaux de laboratoire, généralement les primates et les rongeurs, comme un inducteur irréversible du syndrome parkinsonien82 et ainsi permettre l’élaboration des modèles d’animaux parkinsoniens pour la science.
D’autres causes ont été mises en évidences pour expliquer la différence de la répartition de la maladie dans le monde, comme l’exposition à des solvants hydrocarbonés tels que le monoxyde de carbone ou le manganèse83, l’utilisation de l’eau des puits et la consommation des fruits exotiques84.
En résumé, la MP doit son apparition à une combinaison de prédispositions génétiques dues à la mutation de certains gènes et à une exposition à certains composés chimiques et produits à risque.
13
1.1.6. Diagnostique
Puisque la MP met plusieurs années à se développer avant l’apparition de ses premiers symptômes, il est essentiel de diagnostiquer la maladie le plus tôt possible afin de pouvoir agir plus rapidement. Plusieurs projets de recherche, qui requièrent des équipes d’experts, se sont concentrés sur l’identification des biomarqueurs capables de détecter la maladie avec une haute sensibilité et le développement des outils destinés à l’identification spécifique de cette atteinte. Mais malheureusement, jusqu’à ce jour, aucun test pré-clinique fiable n’a été identifié pour cette pathologie. C’est grâce à l’apparition de ses signes moteurs les plus caractéristiques, tels que le tremblement au repos, la rigidité et l’instabilité posturale, qu’elle est diagnostiquée. Ces signes moteurs progressent graduellement au cours du temps, ce qui rend le diagnostic plus évident.
Comme ces symptômes sont aussi bien présents dans le cas d’autres maladies neurodégénératives, comme par exemple l’AMS où la réponse du patient à un traitement avec la L-Dopa aide à différencier la MP des autres désordres.
Plusieurs méthodes de diagnostic et pronostic ont émergées au cours du temps telles que l’imagerie par résonance magnétique (IRM), la tomographie par émission des positrons (en anglais ‘Positron emission temography scan’ ou ‘PET scan’) et l’ultrasonographie. Ainsi, la maladie peut être diagnostiquée, le traitement convenable peut être prescrit avec des doses qui peuvent être ajustées au fur et à mesure de l’évolution de la maladie.
De nombreux laboratoires concentrent leurs efforts sur l’étude de la protéine α-syn, ses formes modifiées et sa propension à former des agrégats en liaison avec l’apparition et l’évolution de la maladie. Dans ce cas, les modifications post-traductionnelles de la protéine α-syn, telles que sa phosphorylation, peuvent s'avérer utiles en tant que biomarqueur de la MP. C’est en 2013 que la détection des formes de phosphorylation de l'α-syn dans le liquide céphalorachidien (LRC) et le plasma a été
14
développée grâce à la chromatographie en phase liquide couplée à la spectroscopie de masse.
En raison de l’urgente nécessité d’identifier un biomarqueur spécifique pour la MP et en s’intéressant toujours aux modifications subies par l’α-syn, de nombreuses équipes de partout dans le monde s’unissent et collaborent au développement d’un moyen de diagnostique pertinent par l’étude des modifications subies par l’α-syn. Par exemple, la compagnie ‘Covance Antibody Products’ en collaboration avec l'Université d'Ottawa et la Fondation Michael J. Fox a développé un kit ultra-sensible ELISA qui permet de détecter les modifications post-traductionnelles de la protéine α-syn, incluant la phosphorylation et la troncation.
Malheureusement, l’absence d’un biomarqueur capable de détecter la MP avec une haute sensibilité et l’absence d’outils de diagnostic beaucoup plus précis permettant un dépistage plus simple et précoce des patients demeurent des préoccupations encore actuelles.
1.1.7. Traitements pharmacologiques et chirurgicaux de la maladie de Parkinson
Divers médicaments sont disponibles et visent à réduire les symptômes de la maladie. Généralement, ces médicaments sont attribués à la thérapie dopaminergique et tendent à remplacer la dopamine dans le cerveau85,86.
Un traitement révolutionnaire : la L-Dopa ou la lévodopa, un précurseur de la synthèse de la dopamine, capable de traverser la barrière hémato-encéphalique (BHE), est aujourd’hui la thérapie ‘gold standard’ pour la symptomatologie de la MP77. En effet, il est le plus souvent prescrit afin de pallier le manque en dopamine et pouvoir atténuer les symptômes moteurs86. Ce traitement est attribué au patient sous forme des comprimés renfermant deux molécules : la lévodopa et la carbidopa87. L’introduction de la molécule carbidopa associée à la lévodopa est essentielle pour empêcher la
15
conversion de la lévodopa en dopamine dans la périphérie88. Par contre l’utilisation de ce traitement prolongée génère chez la vaste majorité des patients parkinsoniens des effets indésirables souvent plus néfastes que la maladie elle-même. Ces effets néfastes sont généralement le développement de dyskinésies (mouvements anormaux involontaires incessants au cours de l'éveil et s'arrêtant lors du sommeil caractérisés par une motricité lente et stéréotypée affectant préférentiellement le visage mais qui peut également concerner le tronc et les membres), une réduction de l’efficacité thérapeutique de la L-Dopa en fin de dose (akinésie de fin de dose, wearing-off) et finalement l’apparition de phénomène on-off (fluctuations motrices imprévisibles caractérisées par une alternance aléatoire d'un état de parkinsonisme à un état où les symptômes disparaissent avec parfois l’apparition de dyskinésies)89.
À des stades avancés de la maladie, il est possible d'offrir des options chirurgicales aux personnes qui ne répondent plus au traitement ou à celles qui subissent des dyskinésies invalidantes provoquées par les médicaments. Un exemple de ces interventions chirurgicales est la stimulation cérébrale profonde (SCP) qui consiste à l’implantation d’électrodes dans certaines parties spécifiques du cerveau (le thalamus, le globus pallidus interne ou externe, le noyau sous-thalamique (STN), le noyau pédonculopontin). Un stimulateur envoie ensuite des impulsions électriques permettant de réduire les mouvements involontaires et les tremblements90. Tout dépendamment du positionnement des électrodes, l’effet du SCP pourrait être soit inhibiteur via la modulation de la libération de GABA, soit excitateur via la libération du glutamate, ou soit les deux quand les électrodes sont dans le STN. Généralement, les aspects des comportements moteurs sont améliorés avec cette approche thérapeutique91.
Aujourd’hui de nouveaux traitements novateurs sont en cours d’étude comme la greffe des cellules dopaminergiques fœtales, la thérapie génique, la neuroprotection qui vise à protéger les neurones dopaminergiques et la chirurgie fonctionnelle telle que l’ablation chirurgicale de certaines régions du cerveau qui a l'avantage de permettre une diminution de la quantité de médicaments basés sur la L-Dopa et réduire ainsi l’apparition des dyskinésies.
16
Jusqu’à ce jour, les traitements existants sont seulement capables de réduire les symptômes. Or, il est primordial de développer un ou des traitements pré-symptomatiques afin d’arrêter ou du moins de ralentir la progression de la maladie.
1.2. L’alpha-synucléine
L’α-syn est une protéine appartenant à la famille des synucléines qui est très abondante dans l’organisme humain et surtout dans le cerveau46. Cette famille comprend en plus de l’α-syn, la β- et la γ-syn92,93. L’α-syn est plus susceptible de s’agréger que la β- et la γ-syn 94. Grâce à cette capacité, elle est le constituant principal des CLs impliqués dans l’apparition de la MP95. Ainsi, une meilleure connaissance de sa structure, de ses fonctions physiologiques et de ses interactions moléculaires sont donc essentielles afin d’élucider son implication dans la pathogenèse de la MP et des autres synucléinopathies.
1.2.1. Structure de l’α-synucléine
L’α-syn est codée par le gène SNCA localisé sur le chromosome 4q22.1 (http://www.ncbi.nlm.nih.gov/gene/6622). Cette protéine possède une masse moléculaire de 14 kDa et comprend 140 acides aminés avec un pKa de 4.7 96. Elle peut exister sous d’autres isoformes comme l’α-syn 126, 112, ou 9897.
Depuis les années 90, il a été démontré que l’α-syn, extraite avec d’autres protéines, se retrouve sous une forme monomérique98,99 et demeure stable même après être exposée à des conditions qui perturbent la structure des protéines en général, suggérant qu’elle ne présente pas une structure secondaire ordonnée et qu’elle est plutôt dépliée et sans conformation organisée98,100. Dû à ses propriétés structurales, l’α-syn a été incluse dans la famille des protéines intrinsèquement désordonnées (ou en anglais: intrinsically-disordered protein, IDP)95. Grâce à sa conformation flexible, elle est capable d’interagir avec d’autres protéines et de se lier à des membranes
17
biologiques101,102. Cette association lui permet de se manifester sous plusieurs conformations, de former différents complexes protéiques et d’acquérir des rôles variés dans différentes conditions physiologiques101,102. Ainsi, on l’aperçoit sous la forme des hélices-alpha dans le cas où elle est liée aux lipides membranaires103–105 ou formant des feuillets bêta en s’agrégeant avec d’autres protéines.
Des études récentes ont postulé une nouvelle hypothèse concernant la structure de l’α-syn, notamment les travaux de Bartels et ses collègues, qui proposent que l’α-syn se trouve sous la forme d’un tétramère stable avec des hélices alpha dont la masse moléculaire est d’environ 58-60 kDa106.
Au niveau structural, l’α-syn comprend 3 domaines :
-le domaine N-terminal : ce domaine est composé de 60 acides aminés (acides aminés 1-60) riche en résidus lysine et contient une séquence répétitive de 11 résidus. Ce motif est conservé entre les espèces et au sein de la famille des synucléines. De plus, il lui est unique et spécifique. Cette séquence répétitive, rappelant le domaine de liaison lipidique des lipoprotéines, est responsable de la formation des hélice alpha avec les phospholipides membranaires, in vitro et in vivo103,104,107. En effet, une mutation touchant ce site, dans le cas du mutant A30P par exemple, est capable d’affecter la liaison de l’α-syn aux membranes in vitro108 et nuire à son fonctionnement. Fait intéressant, les mutations associées à l’apparition des formes familiales de la MP, A53T, A30P, E46K en plus de G51D et H50Q récemment décrites65–67,109–111, affectent ce domaine.
-Une partie central hydrophobe (acides aminés 61-95) appelée NAC (non-amyloidogenic component)96,112 responsable de l’agrégation de la protéine α-syn113–115et de la formation des feuillets béta avec d’autres α-syn96,112. Ce fragment a été décrit pour la première fois comme un constituant principal des plaques séniles caractéristiques de la maladie d’Alzheimer96.
18
-Le domaine C-terminal (acides aminés 96-140)116 qui est reconnu par sa grande variabilité, vu qu’il ne présente pas une structure secondaire particulière au sein de la famille des synucléines et de même qu’entre les espèces93. Ce domaine, polaire et riche en résidus chargés117, contrôle la localisation cellulaire de l’α-syn et est important dans son interaction avec les autres protéines, les petites molécules et les métaux99,118. Ce domaine contient plusieurs sites potentiels de modifications post-traductionnelles tels que la phosphorylation (Y125, S129, Y133 et Y136)119,120, la troncation (D115, D119, P120, E130, et D135), la nitration (au Tyrosine125, Tyr133 et Tyr136)119,121et l’ubiquitination (K96)119,122 (Figure 6). Ceci suggère que ce domaine protéique joue un rôle important dans la régulation de la conformation, de l’oligomérisation et ainsi de la fonction de la protéine121,123.Contrairement au NAC, le domaine C-terminal semble avoir un effet préventif contre l’agrégation. En effet, une étude in vitro utilisant l’α-syn humaine recombinante a démontré que la troncation du domaine C-terminal (α-syn 1-120) favorise l’assemblage en filament124.
Figure 4: Représentation schématique de la structure de l’α-syn125. L’α-syn comprend 3
domaines : un domaine N-terminal (en bleu), une région NAC (en orange) et la partie C-terminal (en rouge). De plus, ce schéma présente (en haut) les différents sites de liaison aux métaux et les sites potentiels pour les modifications post-traductionnelles au niveau du domaine C-terminal (en bas).
19
1.2.2. Localisation et fonction de l’α-synucléine
L’α-syn est exprimée, surtout, par les différentes populations neuronales du SNC et SNP46,126. D’ailleurs, elle peut se trouver dans divers parties du cerveau tels que le bulbe olfactif, le cortex frontal, le striatum, l’hippocampe, l’hypothalamus, le thalamus, le mésencéphale, le cervelet et la médula127–129. Dans la cellule, l’α-syn peut se trouver dans différents compartiments, soit au niveau du corps cellulaire, dans le cytoplasme ou tout le long des prolongements axonaux et dendritiques126,130. D’une manière préférentielle, cette dernière s’accumule dans la zone pré-synaptique92, évoquant ainsi ses rôles variés dans chaque compartiment.
La localisation subcellulaire variée de l’α-syn est sous le contrôle de certains facteurs tels que sa séquence protéinique, surtout celle du domaine N-terminal131,132, les ligands auxquels elle se trouve associée132, ses mutations131 et ses modifications post-traductionnelles133.
Dans le cadre de la MP, on se concentre sur l’étude de l’α-syn dans les neurones DAergiques de la SNpc puisque ce sont les cellules les plus touchées et responsables de l’apparition des premiers symptômes de la pathologie37. Une fois produite dans le soma, l’α-syn est transportée à travers les prolongements axonaux vers les terminaisons synaptiques126,134,135. Rendue à ce niveau, l’α-syn peut s’associer aux vésicules synaptiques136. Ce qui suggère un rôle de cette dernière dans la régulation du trafic vésiculaire et de la transmission synaptique. Un ensemble croissant d’études, effectuées in vitro sur des cellules PC12 qui surexpriment l’α-syn d’une manière stable137 et in vivo dans des modèles animaux parkinsoniens138–142, ont montré qu’un niveau élevé de l’α-syn l’α-synaptique réduit l’exocytose et donc la libération du neurotransmetteur140,141. En plus, cette surexpression affecte le processus de recyclage des vésicules synaptiques, leur trafic intersynaptique143 et diminue l’efficacité de la réincorporation de la dopamine se trouvant dans les fentes synaptiques142. Aussi, grâce à sa capacité à interagir avec certaines protéines synaptiques, l’α-syn se comporte comme une protéine chaperonne dans la modulation de la formation, la maintenance et la distribution du complexe
20
protéique pré-synaptique SNARE (syntaxine, SNAP-25 et synaptobrévine), ce qui lui confère un rôle indirecte dans la libération de la dopamine144. Par contre, ce rôle ne s’avère pas essentiel la libération du neurotransmetteur puisque l’invalidation du gène produisant l’α-syn n’affecte pas les processus d’exocytose et de la relâche de la dopamine145,146.
Dans le cas de dommages au niveau des terminaisons neuronales, les travaux de Chandra et son équipe ont révélé un rôle important de l’α-syn contre la neurodégénérescence. En effet, cette action essentielle pour la survie neuronale exige son association avec la cysteine-string protein-alpha (CSPα) et le complexe protéique SNARE146. D’autres études ont démontré que l’α-syn ne vient s’installer au niveau synaptique qu’après la formation de cette dernière (synapse)135, suggérant que cette protéine n’est pas impliquée dans le développement de la synapse mais plutôt dans le maintient de l’homéostasie synaptique. D’après ces observations, l’α-syn semble avoir un rôle régulateur de l’activité fonctionnelle pré-synaptique.
1.2.3. L’α-synucléine dans la maladie de Parkinson
Certaines évidences ont mis en lumière un lien causal entre la protéine α-syn et la MP. En effet, l’α-syn est soupçonnée d’être impliquée dans l’apparition de la pathologie de Parkinson pour 2 raisons. Premièrement, il a été démontré qu’une mutation faux-sens, sur le chromosome 4 touchant le gène exprimant cette protéine, SNCA, peut être à l’origine de l’apparition des formes familiales de la maladie65,66,110. Deuxièmement, il s’est avéré qu’elle constitue le principal composant des CLs 147,148,où elle se trouve en grande quantité sous sa forme agrégée. Ces agrégats sont également observés dans diverses maladies neurodégénératives comme l’AMS ou la démence à corps de Lewy149.
D’autres expériences ont montré que l’introduction des formes agrégées de l’α-syn est suffisante pour déclencher des symptômes semblables à ceux observés dans la MP. En effet, l’injection d’agrégats de l’α-syn prélevés de cerveaux de patients décédés
21
au niveau du striatum des souris et des macaques induit une pathologie s’apparentant à la MP150. De plus, il a été démontré in vitro sur des cultures cellulaires que les agrégats de l’α-syn peuvent se propager aux cellules avoisinantes115. Cependant, les mécanismes pathophysiologiques de la MP, médiés par l’α-syn, conduisant à la formation progressive des amas insolubles de l’α-syn et aboutissant à la neurodégénérescence demeurent encore mal connus151.
1.2.3.A. Agrégation de l’α-syn
L’α-syn monomérique est capable de se polymériser et s’agréger formant des structures variées de fibrilles. Cette agrégation peut être le résultat soit de l’instabilité de sa structure native ou déclenchée par le stress environnemental, tel que le stress oxydatif et les modifications post-traductionnelles, ou due à une cause génétique soit par des mutations de certains gènes qui augmentent la cytotoxicité et qui favorisent l’agrégation152,153 ou à la multiplication du gène codant pour l’α-syn (une duplication154 ou triplication62). Dans ces conditions pathologiques, l’α-syn a tendance à adopter un état replié et s’agréger en feuillets bêta pour donner naissance aux CLs 155. La formation des agrégats d’α-syn se fait selon différents mécanismes et implique plusieurs intermédiaires moléculaires. Un ensemble d’études récentes appuie le fait que l’agrégation de l’α-syn et la formation des formes fibrillaires induisent une toxicité plus sévère pendant la progression de la neurodégénérescence156,157, tandis que son blocage induit une réduction significative du nombre d’agrégats avec moins de perte de neurones DAergiques chez des rats parkinsoniens158.
En effet, des études in vitro et in vivo ont montré qu’au niveau cellulaire les formes agrégées de l’α-syn (pré-fibrilles, fibrilles et oligomères) affectent la neurotransmission en réduisant la libération de la dopamine dans le cerveau qui résulte en l’apparition des difficultés motrices rencontrées dans la MP159,160. De plus, ces agrégats sont capables d’une part d’augmenter la perméabilité membranaire aux ions calcium provoquant un influx vers le milieu intérieur de la cellule qui induit à son tour la mort neuronale161. D’autre part, ils peuvent causer des dommages aux
22
mitochondries162, induire une fuite des lysosomes163 et perturber le transport axonal des protéines synaptiques164 aboutissant à des synapses dysfonctionnelles et une éventuelle neurodégénérescence139 (Figure 5).
Figure 5: Les mécanismes de neurotoxicité des agrégats de l’α-syn8. Les formes agrégées de
l’α-syn (oligomères et fibrilles) sont toxiques pour la vie neuronale. Elles altèrent la fonction des mitochondries, de l’appareil de Golgi et du réticulum endoplasmique, dérégulent la dégradation des protéines et affectent la transmission synaptique, ce qui provoque la mort neuronale.
1.2.3.B. Propagation des formes toxiques d’α-synucléine et son impact sur l’évolution de la maladie de Parkinson
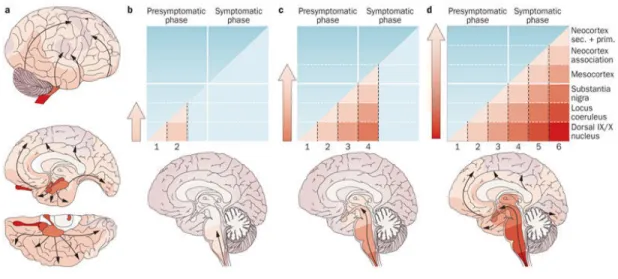
Dans des conditions pathologiques, les formes toxiques d’α-syn sont capables de se propager d’une cellule à une autre165–167 et peuvent infecter, ainsi, différentes régions du SNC. En effet, leur localisation reflète la propagation de la maladie et sa progression d’où il est possible de distinguer 6 différents stades168,169. En premier lieu, les agrégats s’accumulent au niveau du bulbe olfactif et le noyau dorsal moteur du nerf vague (stade 1). Ensuite, les CLs se propagent vers le bulbe rachidien (stade 2). Le 3ième stade de la MP est marqué par l’apparition des symptômes moteurs de la maladie suite à la destruction massive des neurones de l’amygdale et la substance noire. La pathologie
23
s’aggrave touchant le cortex temporal (stade 4). Finalement, des corps et neurites de Lewy apparaissent au niveau du néocortex, ce sont les stades 5 et 6170,171 (Figure 6).
Figure 6: Les stades de Braak 169. Images illustrant la dispersion des CLs selon les stades de Braak, au cours de la progression de la pathologie de Parkinson. Au cours des stades 1 et 2, les CLs se trouvent dans le tronc cérébral, c’est la phase pré-symptomatique. Ensuite, les CLs colonisent d’autres régions cérébrales (stades 3 et 4) pour finalement atteindre le cortex (stades 5 et 6).
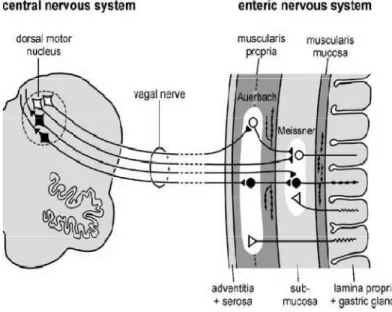
Dans le cadre de l’étude de la progression spatio-temporelle des agrégats de l’α-syn chez des patients parkinsoniens, un chercheur allemand, Dr Heiko Braak, a détecté la présence de ces agrégats dans des neurones autres que les neurones DAergiques, avant même l’atteinte de ces derniers. La présence des CLs en périphérie du SNC, dans les neurones des parois gastriques et intestinales172–175, est l’une des découvertes majeures de Dr. Braak et ses collaborateurs. En lien avec celle-ci, il a développé une hypothèse (hypothèse de Braak) qui explique l’origine de la maladie en se basant sur l’interconnexion entre le nerf X et le système nerveux entérique (SNE) et suppose qu’un agent pathogène est capable de traverser la muqueuse intestinale et entrer en contact avec les neurones du SNE pour remonter via un transport rétrograde à travers le nerf
24
vague vers le SNC (Figure 7)168,176. Ainsi, la prévention des lésions intestinales pourrait être un moyen de protection contre la MP.
Figure 7: Hypothèse de Braak176. Schéma mettant en évidence les connexions entre le SNE et le
SNC via le nerf vague. Des informations provenant du réseau neuronal entérique remontent par le nerf vague vers le SNC en traversant le noyau moteur dorsal du bulbe rachidien.
De plus, des agrégats de l’α-syn ont été détectés dans des neurones impliqués dans les voies olfactives, dans la sécrétion des glandes salivaires ou dans d’autres structures essentielles au maintien de l’équilibre et la posture175.
Sur le plan cellulaire et moléculaire, la libération des oligomères de l’α-syn peut se faire soit par simple exocytose105, dans des vésicules177 ou dans des exosomes166,178. Dans le milieu extérieur, ces oligomères se transfèrent vers la cellule hôte soit par une pénétration directe179, par endocytose180, par dissémination trans-synaptique166 ou par l’intermédiaire d’un récepteur membranaire177 (Figure 8). Une fois dans la cellule réceptrice, ces formes toxiques continuent à s’agréger et incorporent des monomères d’α-syn endogène de la cellule hôte. Ce mécanisme a été observé par Luk et ses collègues dans un modèle cellulaire de la MP avec lequel ils ont réussi à mieux décrire
25
la propagation des formes toxiques de la maladie. En effet, l’α-syn produite par ces neurones sains s’associe et s’agrège avec les protéines de l’hôte, ce qui met en évidence la capacité de l’α-syn pathologique à quitter les neurones et se déplacer vers d’autres pour envahir des zones variées du SN. L’α-syn pathogénique peut se transmettre d’un neurone à un autre neurone ou à une cellule gliale177 par propagation prionique165 (Figure 8).
Figure 8: Mécanismes de propagation de l’α-synucléine 8. L’α-syn sous sa forme monomérique
ou agrégée se transfère d’un neurone à un autre par des multiples façons : l’exocytose, la pénétration directe, la transmission trans-synaptique ou par l’intermédiaire de récepteurs membranaires. De telle manière, la maladie se propage dans les différentes régions cérébrales.
1.2.4. Rôle des modifications post-traductionnelles dans la régulation de la toxicité de l’α-syn : focus sur la phosphorylation
La MP se caractérise notamment par l’accumulation de l’α-syn dans les neurones dopaminergiques. Cette protéine s’agrège, dans le cas où elle est produite en trop grande quantité, mal éliminée ou modifiée sous certaines formes, formant des agrégats toxiques intra-neuronales, les CLs, menant ainsi à la neurodégénérescence. Il y a plusieurs années, les scientifiques ont découvert que ces protéines agrégées dans le cerveau avaient subi plusieurs transformations post-traductionnelles telles que la
26
phosphorylation, l’ubiquitination, la troncation et la nitration qui, en modifiant leurs propriétés biochimiques, jouent un rôle important dans la régulation de l’agrégation et la toxicité de l’α-syn in vivo119. Ils ont rapporté que la phosphorylation au résidu Sérine 129 (pS129) est la plus abondante dans les cerveaux des patients parkinsoniens, de même que chez les modèles animaux de la MP121,181. Cette phosphorylation touche 90% de la fraction totale de la protéine α-syn dans les inclusions neuronales. Cependant, chez un sujet sain, les niveaux d’α-syn phosphorylée au résidu S129 (α-syn pS129) sont inférieurs à 4 %121,181. Ceci suggère que l’α-syn pS129 est en corrélation avec la génération des CLs et pourrait jouer un rôle important dans la destruction neuronale121. La compréhension de l’effet de cette modification sur les propriétés structurales de l’α-syn, la modulation de sa fonction physiologique, son agrégation et sa toxicité sera essentielle pour le développement d’un éventuel moyen de diagnostic ou de traitement de la MP.
Parmi les kinases responsables de la phosphorylation de l’α-syn au S129, on cite les caséines kinases (CKs)182, les GRKs (G-protein coupled receptor kinases)183, la LRRK2 (leucine rich repeat kinase 2)184 et les PLKs (polo-like kinases)108,185.
En ce qui concerne le rôle de la pS129 et son implication dans la pathologie de Parkinson, les études sont controversées. Un ensemble d’études confèrent à l’α-syn pS129 un certain caractère de toxicité. En effet, une analyse des cerveaux post-mortem de patients parkinsoniens a montré une accumulation de l’α-syn insoluble et phosphorylée186. De plus, il a été démontré que la phosphorylation de l’α-syn sur le résidu S129 pourrait changer la conformation de l’α-syn native et compromettre son habilité à se lier aux membranes cellulaires (membranes cytoplasmique, membrane mitochondriale, membrane des vésicules synaptiques,…)157,183,187–189. Ces évidences démontrent que cette transformation post-traductionnelle de l’α-syn serait à l’origine de son agrégation et ainsi la mort neuronale. Cependant, la contribution de la pS129 dans la toxicité de l’α-syn reste encore mal connue. Afin de mieux explorer cette possible contribution, des modèles de cultures cellulaires et d’animaux surexprimants l’α-syn avec une kinase qui est capable d’induire sa phosphorylation sur le site S129, ont été
27
développés. Deux expérimentations utilisant ces modèles décrits, une chez la mouche190 et l’autre chez le rat191, ont été étudiées. Ainsi, il a été démontré chez la drosophile (mouche du vinaigre) 190 et chez le rat191 que la surexpression de l’α-syn et des kinases GRK2 et GRK6 résulte à une élévation des niveaux de pS129 accompagnée d’une perte neuronale190,191. Ceci suggère que les GRKs en phosphorylant l’α-syn augmentent sa toxicité et favorisent la neurodégénérescence. Globalement, ces études affirment que la pS129 est importante pour la formation des inclusions pathologiques dans la MP190,192,193.
Des résultats récents de notre laboratoire ont démontré qu’une polo-like kinase, la PLK2, est capable d’induire efficacement la phosphorylation d’α-syn au S129. Notamment une surexpression de l’α-syn avec la PLK2 favorise son élimination par voie autophagique et, d’une manière intéressante, la suppression de la mort neuronale194. D’autre part, le Dr Luk et son équipe ont montré que la pS129 n’est pas essentielle pour la formation des CLs en induisant la surexpression dans des cultures cellulaires soit de l’α-syn S129A ou l’α-syn tronquée (1-120), 2 formes de l’α-syn incapables d’être phosphorylées au S129. De plus, il a été montré que la pS129 ralentit le processus de fibrillation in vitro suggérant son rôle dans la réduction de la formation des inclusions cytoplasmiques de l’α-syn193,195. Cette action est due grâce à certaines kinases, particulièrement les PLKs, qui sont capables d’agir aussi bien sur les formes solubles de l’α-syn que les formes agrégées108,193,196,197.
28
Figure 9: Rôle potentiel de la phosphorylation au S129 dans la régulation de l’élimination de l’α-syn, son agrégation et sa toxicité 125. La PLK2 interagie et phosphoryle l’α-syn au S129
favorisant sa dégradation par voie autophagique, d’une manière à réduire sa toxicité in vivo194. D’un autre côté, la pS129 est déclarée comme capable d’inhiber la genèse des formes agrégées de l’α-syn198. De plus l’α-syn sous sa forme monomérique ou agrégée peut être phosphorylée au
S129, ce qui inhibe et déstabilise son agrégation. Ensemble ces études appuient l’effet neuroprotecteur de la pS129.
L’α-syn pS129 se trouve dans plusieurs zones du corps humain autres que le SNC, tout dépendamment du stade de l’évolution de la pathologie. En effet, des études de la composition du sang ou des fluides cérébrospinaux de patients parkinsoniens en α-syn pS129 ont montré un niveau élevé de pS129 chez les patients parkinsoniens en comparaison avec les sujets contrôles sains199. À part son accumulation pathologique dans les liquides corporels (sang et liquide céphalo-rachidien), l’α-syn pS129 a été détectée dans certaines structures du SNP, dans les fibres nerveuses cutanées200,201 et le système gastrique, où elle s’installe plusieurs années avant l’apparition des premiers symptômes moteurs. Ceci suggère que l’α-syn pS129 pourrait représenter un marqueur spécifique pour le diagnostic précoce de la MP.