Rôle des microARNs dans la génétique de la maladie
d'Alzheimer.
Thèse
Emmanuelle Boscher
Doctorat en neurobiologie
Philosophiæ doctor (Ph. D.)
Québec, Canada
Rôle des microARNs dans la génétique
de la maladie d’Alzheimer
Thèse
Emmanuelle Boscher
Sous la direction de :
Dr. Sébastien Hébert, directeur de recherche
2019
Résumé
La maladie d’Alzheimer (MA) est la principale forme de démence à travers le monde (60-70%). Cette maladie neurodégénérative à progression lente est notamment caractérisée par la présence de plaques d’Amyloïde bêta (Aβ) extracellulaires ainsi que de neurodégénérescences fibrillaires intracellulaires, riches en protéine Tau. Cette maladie est associée à de nombreuses altérations cognitives et comportementales aboutissant à la perte progressive d’autonomie du patient. La part de génétique dans la MA serait majeure. Actuellement, trois gènes portent des mutations causales pour la MA : APP, PSEN1 et PSEN2. De par sa forte hétérogénéité génétique, de nombreuses voies de signalisation sont impliquées dans l’étiologie de la maladie. Les microARNs (miRs), ARN non-codants capables de réprimer plusieurs ARNm différents, ont suscité un intérêt croissant pour l’identification de nouveaux facteurs de risque. En effet, de nombreuses altérations de l’expression de miRs ont été associées à la MA. Ces données suggèrent qu’une régulation anormale miRs-ARNm pourrait influencer le développement de la MA. Ces dérégulations peuvent être dues à des mutations. Parmi ces mutations, on retrouve les variations du nombre de copies d’un gène de miR (duplication ou délétion) qui peuvent modifier le niveau d’expression du miR et les mutations ponctuelles sur des ARNm qui modifient la reconnaissance par un miR.
Notre hypothèse est que l’étude de mutations qui altèrent la régulation miR-ARNm impliquée dans la MA, pourraient permettre d’identifier de nouveaux acteurs associés à cette pathologie.
Notre 1er objectif fut d’étudier les mutations de type variation du nombre de copies d’un gène de miRs.
Les formes précoces de la MA (< 65 ans, FPMA) étant plus sévères que les formes tardives (> 65 ans), les patients atteints d’une FPMA devraient présenter des facteurs de risque plus importants. Le séquençage d’une cohorte de patients atteints de FPMA et de sujets contrôles a permit d’établir une liste de 31 variations du nombre de gènes de miRs (VNC-miRs) retrouvées uniquement chez les patients. Ces mutations auraient donc un rôle potentiel dans l’étiologie de la MA. Afin d’établir une preuve de concept, nous avons
sélectionné une mutation d’intérêt dans notre liste. Nous avons poursuivi notre étude sur la duplication du MIR-138-2, puisque ce miR serait capable d’agir sur la mémoire, l’anxiété et sur l’ARNm de FERMT2, tous associés à la MA. Nous avons, en effet, constaté qu’une surexpression de miR-138 mature, dans des souris C57bl/6, entraînerait d’une part, une altération de la mémoire, de l’apprentissage tout en favorisant l’anxiété. D’autre part, elle augmenterait Aβ42, principale composante des plaques Aβ, à la fois in vitro et in vivo, ainsi qu’Iba1, un marqueur microglial. Toutes ces altérations cognitives, comportementales et moléculaires sont retrouvées dans la MA. La duplication du MIR-138-2 pourrait donc être un facteur de risque dans cette maladie.
Notre 2ème objectif fut d’étudier des mutations ponctuelles sur des ARNm associés à la MA.
À partir de polymorphismes identifiés précédemment par notre laboratoire (Delay et collaborateurs), nous avons sélectionné trois mutations ponctuelles (MPs) d’intérêt : rs2847655C 585/MS4A 2), rs610932C 626/MS4A 6A) et rs9909G (miR-1185/NUP160). Ces mutations créeraient un site de reconnaissance pour un miR sur l’ARNm. Nous avons alors dosé les ARNm et les miRs associés à ces mutations dans une cohorte de patients atteints de la MA et de sujets sains de mêmes âges. Nous avons alors constaté que la mutation rs9909G, pouvait avoir un rôle protecteur dans la MA. Nos résultats suggèrent aussi qu’une augmentation d’expression des miR-1185 et miR-626, pourrait être impliquée dans la MA. Des études complémentaires sont nécessaires pour mieux définir le rôle des mutations rs610932C et rs2847655C.
Ainsi, ces données ont montré que l’étude de VNC-miRs et de MPs qui altèrent la régulation de miRs sur leurs cibles serait un bon outil pour identifier de nouveaux facteurs de risques potentiels (duplication du miR-138) ou protecteurs (mutation rs9909G) dans les maladies neurodégénératives, telle que la MA. Ces analyses pourront ensuite ouvrir de nouveaux champs de recherche aussi bien dans le domaine fondamental que dans la clinique (élaboration de biomarqueurs et/ou de thérapies).
Abstract
Alzheimer’s disease (AD) is the most common form of dementia characterized by cognitive and behavioral alterations leading to loss of autonomy. The two major pathological hallmarks of AD are extracellular amyloid (Aβ) plaques and intracellular neurofibrillary tangles, enriched in Tau proteins. The part of genetic is very important in AD. To date, three genes have causal mutations: APP and PSEN1 PSEN2, which are involved in Aβ production. Due to genetic heterogeneity, many different pathways are associated with AD etiology. MicroRNAs (miRs), non-coding RNAs able to repress many mRNAs, have attracted growing interest in the identification of potential new risk factors in AD. Many miRs expression are altered in AD. These data suggest that an abnormal miR regulation could trigger on AD development and/or progression. Such dysregulations could be due to mutations like copy number variations (duplication or deletion) that could alter miR expression level or single-nucleotide polymorphisms (SNPs) on miR’s recognition site in mRNA involved in AD pathogenesis.
Our hypothesis is that the study of mutations that can modify the regulation of miR on mRNA expression could be a new way to identify new risk factors in AD.
Our 1st goal was to study mutations like copy number variation in the miR gene.
The early-onset AD (< 65 years; EOAD) is a more severe form of the disease than late-onset AD (>65 years; LOAD) hence EOAD patients could have bigger risk factors than LOAD patients. We used datas from exome sequencing of a human cohort including EOAD patients and healthy individuals. We identified 31 copy number variations of miRs genes (CNV-miRs) specific to EOAD patients that suggest a potential role of these mutations in AD. To establish a proof of concept, we selected MIR-138-2 duplication in our list. MiR-138 was described to act on memory, anxiety-like behaviors and FERMT2 expression, all of which have been associated with AD. We found that the overexpression of miR-138 alters memory, learning and increase anxiety-like behaviors. Moreover, this overexpression increases Aβ42 level, the main component of Aβ plaques, in vivo and in
associated with AD. The duplication of MIR-138-2 could therefore be a new risk factor for AD.
Our 2nd goal was to study single-nucleotide polymorphisms in mRNA involved in AD.
From SNPs previously identified in our lab (Delay et al.), we selected three SNPs of interest: rs2847655C (miR-585/MS4A 2), rs610932C (miR-626/MS4A 6A) and rs9909G (miR-1185/NUP160). These mutations are predicted to create a recognition site for miRs within host mRNAs (MS4A 2, MS4A 6A, NUP160) associated to AD. We measured the expression of mRNA and miR associated to each mutation in the human cohort including AD patients and healthy age-matched individuals. We showed that the increase of miR-626 and miR-1185 expression could be associated with MA in our cohort. The rs9909G mutation could therefore be protective factor in AD. Finally, rs610932C and rs2847655C mutations need further study to elucidate their implication in AD.
Together, these results highlight that the study of CNV-miRs and SNPs that alters miR regulation can be a new way to identify new potential risk factors (like MIR-138-2
duplication) or protective factors (like rs9909G mutation) in neurodegenerative diseases,
such as AD. These investigations could lead to novel targets for fundamental and clinical researches (biomarkers and/or therapeutic treatment).
Table des matières
Résumé ... ii
Abstract ... iv
Table des matières ... vi
Liste des figures ... ix
Liste des tableaux ... ii
Liste des abréviations ... ii
Remerciements ... vii Avant-propos ... ix Introduction ... 1 1. Maladie d’Alzheimer ... 1 1.1. Historique ... 1 1.2. Épidémiologie ... 2 1.3. Symptômes ... 2 1.4. Marqueurs pathologiques ... 3
1.4.1. Stades de Braak et Thal ... 4
1.4.2. Neurodégénérescences ... 6
1.4.3. Plaques séniles ... 7
1.4.4. Dégénérescences neurofibrillaires ... 10
1.4.5. Hypothèse de la cascade amyloïde ... 13
1.5. Formes de la maladie d’Alzheimer ... 14
1.5.1. Formes précoces ... 14 1.5.2. Formes tardives ... 17 1.6. Diagnostic et traitements ... 22 2. Les microARNs ... 25 2.1. Biogénèse ... 25 2.2. Mécanismes d’action ... 28 2.3. Rôles ... 29
2.4. Les microARNs dans les pathologies ... 31
3. Mutations génétiques ... 34
3.1. Variants nucléotidiques ... 36
3.1.1. Techniques d’identification ... 36
3.1.1.2. Technique de Sanger ... 39
3.1.1.3. SnaPshot ... 40
3.1.2. Annotations des variants ... 40
3.1.3. Implications sur l’expression de miRs. ... 41
3.2. Variation du nombre de copies d’un gène ... 44
3.2.1. Techniques d’identification ... 44
3.2.1.1. CHG-array ... 44
3.2.1.2. QMPSF ... 45
3.2.2. Implications sur la régulation exercée par un miR ... 46
Hypothèse et Objectifs ... 47
1. Objectif 1 : Identification et caractérisation de variation du nombre de copies d’un gène de microRNA dans la maladie d’Alzheimer ... 48
2. Objectif 2 : Analyse fonctionnelle de mutations ponctuelles dans la maladie d’Alzheimer ... 48
Chapitre 1 : Copy number variants in miR-138 as a potential risk factor for early-onset Alzheimer’s disease. ... 50
1.1. Résumé ... 50
1.2. Abstract ... 51
1.3. INTRODUCTION ... 52
1.4. MATERIALS AND METHODS ... 54
1.5. RESULTS ... 59
1.6. DISCUSSION ... 67
Chapitre 2 : Le miR-138 altère la mémoire, l’apprentissage et favorise un comportement de type anxieux. ... 72
Ce chapitre est rédigé sous forme d’article en vue d’une publication ultérieure. ... 72
2.1. Introduction ... 72
2.2. Matériels et méthodes ... 73
2.3. Résultats ... 79
2.4. Discussion ... 93
Chapitre 3 : La mutation rs9909G, un facteur protecteur de la maladie d’Alzheimer. ... 96
3.1. Mise en contexte ... 96
3.2. Matériels et méthodes ... 97
3.3. Résultats ... 98
3.4. Discussion ... 103
Annexes : Figures supplémentaires ... 129 Bibliographie ... 138
Liste des figures
Figure 1 : Portrait du Dr. Alois Alzheimer (A) et de sa patiente August Deter (B) ... 1
Figure 2 : Marqueurs pathologiques de la MA ... 4
Figure 3 : Propagation des dégénérescences neurofibrillaires ... 5
Figure 4 : Évolution des lésions de dépots de plaque ... 6
Figure 5 : Représentation schématique du gène de l'APP et de ces trois isoformes principales ... 7
Figure 6 : Représentation schématique de la voie amyloïdogénique de l'APP ... 8
Figure 7 : Représentation schématique de la voie non-amyloïdogénique de l'APP ... 9
Figure 8 : Isoforme de Tau ... 10
Figure 9 : Sites phosphorylables de Tau ... 12
Figure 10 : Illustration de Tau et les MTs dans un neurones sain versus un neurone qui dégénère ... 13
Figure 11 : Facteurs de risque dans la maladie d'Alzheimer ... 18
Figure 12 : Biogénèse des miRs ... 27
Figure 13 : Interaction miR-ARNm via la séquence "seed" ... 28
Figure 14 : Illustration de sites canoniques d'interaction miR-ARNm ... 29
Figure 15 : Schéma non exhaustif de voies impliquées dans la maladie d'Alzheimer et de miRs associés ... 33
Figure 16 : Nombres et types de mutations recensées chez l'Homme pouvant causer des maladies génétiques ... 36
Figure 17 : Séquençage haut débit ... 37
Figure 18 : Illustration d'un alignement de séquence pour identifier un variant ... 38
Figure 19 : Technique de Sanger ... 39
Figure 20 : Technique de SnaPshot ... 40
Figure 21 : Exemples de conséquence d’un variant sur un site de reconnaissance miR-ARNm ... 43
Figure 22 : Schéma de la technique de CGH-array ... 45
Figure 23 : Exemple de résultats de QMPSF ... 46
Figure 24 : Screening of miR-CNVs in EOAD ... 61
Figure 25 : miR-138 modulates Ab production ... 62
Figure 26 : miR-138 overexpression promotes Tau phosphorylation ... 64
Figure 27 : GSK-3β activity modulates BACE1 protein levels ... 65
Figure 28 : Model of miR-138 signaling in AD ... 66
Figure 29 : Injections intracérébroventriculaires ... 80
Figure 30 : Expression neuronale des AAV2/DJ8-CAG-eGFP ... 81
Figure 31 : Schéma du protocole de reconnaissance d'un nouvel objet ... 82
Figure 32 : Schéma du test de Barnes ... 83
Figure 33 : Altération de la mémoire et de l'apprentissage chez les souris 138 ... 85
Figure 34 : Augmentation d’un comportement anxieux chez les souris 138 ... 86
Figure 35 : Augmentation d'Aβ 42 et d'Iba1 dans l'hippocampe des souris 138 ... 88
Figure 36 : Augmentation d'Aβ42, Iba1, PSD95 et diminution de Tau totale dans le cortex frontal des souris 138 ... 91
Figure 37 : Diminution de BDNF dans le cerveau postérieur des souris 138 ... 92
Figure 39 : Variants d'intérêt ... 100 Figure 40 : Diminution de l'expression de NUP160 chez les patients atteints de la maladie d'Alzheimer ... 101 Figure 41 : Augmentation de l'expression du miR-1185 et de miR-626 chez les patients atteints de la maladie d'Alzheimer ... 102 Figure 42 : Corrélation négative entre l'expression du miR-1185 et NUP160 dans la maladie d'Alzheimer ... 103 Figure 43 : Hypothèse sur la voie de signalisation du miR-138 dans Aβ et Tau
phosphorylée ... 109 Figure 44 : Hypothèse des voies empruntées par le miR-138 pour agir sur Ab et Tau
phosphorylée ... 111 Figure 45 : Expression de la GFP est plus importante chez les souris contrôles ... 115
Supplementary Figure 1: Validation of the MIR149 duplication by QMPSF…………..…..125
Supplementary Figure 2: AD pathological markers in the EOAD patient with a MIR138-2
Liste des tableaux
Tableau 1 : Tableau récapitulatif des 3 gènes impliqués dans les FPMA-AD ... 16
Tableau 2 : Liste non-exhaustive de mutations associées à la MA ... 21
Tableau 3 : Cohorte humaine ... 97
Tableau 4 : Amorces qPCR ... 98
Tableau 5 : Récapitulatifs des mutations identifiées par Delay et collaborateurs avec leur possible conséquence sur l’expression du gène muté ... Erreur ! Signet non défini. Tableau 6 : Facteurs en commun entre voie du miR-138 et GSK3β ... 110
Supplementary Table 1: Primers used in our study……….………..……....…127
Liste des abréviations
[18F]FDG Fluorodésoxyglucose marqué au fluor-18
aa acide aminé
Aβ Amyloïd bêta
ABCA7 ATP binding cassette subfamily A member 7
AD Alzheimer's disease
AD-FPMA FPMA Autosomales Dominantes
ADAM10 Disintegrin And Metalloproteinase Domain-Containing Protein 10
ADEOAD Autosomal Dominant EOAD
Ago2 Argonaute 2
AICD APP IntraCellular Domain
AMPA Amino-3-hydroxy-5-méthylisoazol-4-propionate
APH-1 Anterior pharynx-defective 1
ApoE Apolipoprotein E
APP Amyloïd Precursor Protein
APT1 Acyl-Protein Thioesterase 1
BA39 Aire 39 de Brodmann
Bace1 Bêta-site APP cleaving enzyme1
BAM Bianry Alignment Map
BIN1 Bridging Integrator 1
BWA Burrows-wheeler alignment tool
CASS4 Cas Scaffold protein family member 4
CD2AP CD2 Associated Protein
CD33 CD33 molecule
CELF1 CUGBP Elav-like family member 1
CGH-array Comparative Genomic Hybridization-array
CLU Clusterin
CNVs Copy Number Variations
CR1 Complement C3b/C4b receptor 1
CREB1 C-AMP Response Element-binding protein
CSF CerebroCpinal fluid
DGCR8 DiGeorge Syndrome Critical Region Gene 8
DGV Database of Genomic Variants
DMEM Dulbecco’s Modified Eagle Medium
DMEM Dulbecco’s Modified Eagle Medium
EOAD Early-onset AD
EPHA1 EPH receptor A1
ExAC Exome Aggregation Consortium
FERMT2 Fermitin Family Member 2
FPMA Formes précoces de la MA
FPMA-AD Formes autosomales dominantes de la MA
FTMA Formes tardives de la MA
GATK Genome Analysis ToolKit
GSK3β Glycogen synthase kinase 3 bêta
GWA Genome-Wide Association
GWAS Genome-wide association studies
HEK Human embryonic kindey
HGMD The Human Gene Mutation Database
HLA-DRB1 Major Histocompatibility complex Class II, DRb1
HLA-DRB5 Major Histocompatibility complex Class II, DRb 5
IDE Insuline-degrading
IGF2 Insulin Growth Factor
INESSS Institut National d’Excellence en Santé et Service Sociaux
INPP5D Inositol polyphosphate-5-phosphatase D
IRM Imagerie à résonance magnétique
LCR Liquide céphalo-rachidien
LiCl Chlorure de Lithium
LOAD Late-Onset AD
MA Maladie d'Alzheimer
MAF Minor Allelic Frequency
MAP1 Microtubule-associated protein 1
MAP2 Microtubule-associated protein 2
MAPT MT-Associated Protein Tau
MEF2C Myocyte Enhancer Factor 2C
MEM Modified eagle’s medium
micro-indels micro-insertions ou micro-délétions
miRs microARNs ou microRNAs
MMSE Mini-Mental Status Examination
MoCA Montréal Cognitive Assessment
MS4A4E Membrane-spanning 4-domains subfamily A 4E
MS4A6A Membrane-spanning 4-domains subfamily A 6A
MTs Microtubules
NMDA N-méthyl-D-aspartate
NME8 NME/NM23 Family Member 8
NOTCH3 Notch receptor 3
NUP160 Nucleoporin 160
NUP93 Nucleoporin 93
PBS Phosphate-buffered Saline
PEN2 Presenilin-enhancer 2
PHF Paires de filaments hélicoïdaux
PICALM Phosphatidylinositol Binding Clathrin Assembly Protein
PLD3 Phospholipase D Family Member 3
PSEN1 Presenilin 1
PSEN2 Presenilin 2
PTK2B Protein Tyrosine Kinase 2 beta
QMPSF Quantitative multiplex Polymerase Chain Reaction of short fluorescent
fragments
qPCR Polymérase quantitative en chaîne en temps réel
RISC miRNA-Induced Silencing Complex
SHIRPA Smith kline beecham Harwell Imperial college Royal london hospital,
Phenotype assessment
SIFT Sorting Intolerant From Tolerant
SLC12A3 Solute carrier family 12 member 3
SLC24A4 Solute Carrier Family 24 Member 4
SORL1 Sortilin related receptor 1
Souris 138 Souris injectées avec le virus AAV2/DJ8-CAG-eGFP-MIR-138-2
Souris contrôles Souris injectées avec le virus AAV2/DJ8-CAG-eGFP TEP Tomographie par Émission de Positons
TREM2 Triggering receptor expressed on myeloid cells 2
TYROBP TYRO protein tyrosine kinase binding protein
UCSC University of California, Santa Cruz
VCF Variant Call Format
VNC Variations du nombre de copies de gène
VNC-miRs VNC sur des gènes de MIRs
WES Whole exome sequencing
WT Wildtype
ZCWPW1 Zinc Finger CW-Type and PWWP domain containing 1
< À l’Univers qui aura toujours le dernier mot >
Remerciements
Tout d’abord, j’aimerais remercier mon directeur de thèse, le Dr S. Hébert, pour m’avoir accueillie dans son équipe et m’avoir permis d’assister à de nombreux congrès. Merci également pour ta confiance et la liberté que tu m’as accordé sur mes différents projets de thèse.
Je tiens à remercier également les membres de mon jury pour avoir accepté d’évaluer ma thèse. Je remercie tout particulièrement ma « best PR EVER », Claudia Goupil! Tu as été indispensable à la réalisation de cette thèse. Merci pour ta bonne humeur, tes aides précieuses ainsi que pour ta foi inébranlable. Tu as été l’âme de l’équipe pendant ces 3 années et je te souhaite de t’épanouir dans ta nouvelle équipe.
Je remercie Sara, ma labmate préférée, pour ton sourire, ta folie et ton accent italien. Merci pour toutes ces heures de fous rires qui ont ensoleillé mon doctorat et réchauffé ces longues journées glaciales d’hiver. Merci aussi d’avoir essayé de me faire aimer cuisiner, mais c’est quand même bon les sandwiCHs.
Je tiens à remercier également Léa et son frère Baya, pour avoir été là tout au long de ce doctorat, à partir en camping, regarder l’Univers marquer des points, venir squatter mon bureau et manger TOUS mes gâteaux!! Merci pour votre joie de vivre et votre soutien constant. KushoTO!!
Merci aussi à Ophélie, ma partenaire au CéNS, pour ta joie, ton humour, nos soirées Thaizone/Sense8, pour nos pauses-café et toutes les activités que tu organises pour nous faire sortir de nos cubicules riches en couleurs! Tu es un vrai rayon de soleil.
Merci à l’équipe du Dr Planel pour votre bonne humeur et votre humour jour après jour. On n’est pas officiellement de la même équipe mais bon… on partage tout!
Merci à Maude, Marie-Kim et Chloé, pour votre joie de vivre, votre humour et nos dîners au CHUL le samedi midi, sauf quand on pré-commande des ramens à la mauvaise date… Merci pour tous vos jujubes et vos chocolats bien appréciés.
Merci aux nouveaux membres de l’équipe : Serena pour ton sourire, ton aide, nos mercredi sushi et nos un ou deux chocolats chauds au Starbucks. Merci à Rémi, un grand joueur de bataille corse, pour toutes tes bêtises au labo, ton humour qui a tué plus d’un clown (paix à leur âme), pour égayer
mes journées parce que finalement même les choses floues sont sympas. Merci aussi à Andréanne, notre nouvelle PR qui va répandre son amour des fêtes de Noël dans notre coin de labo si morose. Je tiens aussi à remercier Sandrine, Françoise et Nathalie, les secondes « best PR EVER », parce qu’on ne peut pas rivaliser avec Claudia. Merci pour votre joie, votre gentillesse, vos conseils! Merci à maman et beau-papa pour votre soutien constant et votre amour. Merci d’être venus en plein mois de novembre pour assister à ma soutenance puis finalement en février pour ma date définitive de soutenance.
Merci à mes sœurs de cœur corses, vous me manquez terriblement. Merci pour votre soutien, pour votre folie, merci d’être vous. Je n’ai qu’une chose à dire, je vous aime.
Merci à ma famille de cœur parisienne : Pôpo, Nenette, Leleen, Zac et Cycy. Vous êtes des personnes merveilleuses. Merci d’être venus jusqu’ici, de votre soutien et de votre amitié. Merci pour votre humour, de m’apprendre qu’une gourde d’eau ça se lave de temps en temps et parce que la loutre ne veut pas sortir du trou!!
Merci à tous les autres qui ont animé ce coin oublié du Chul et qui ont rendu cette aventure inoubliable.
Merci enfin aux souris qui ont donné pas mal de leur personne pour l’aboutissement de ce projet de thèse.
On dit que la thèse est une période difficile mais entourée de telles personnes, tout est beaucoup plus facile.
Avant-propos
État des publications et contributions détaillées :
Le premier chapitre de cette thèse est un résumé des connaissances sur la maladie d’Alzheimer et des microARNs afin d’introduire mon sujet de doctorat.
Le chapitre 1 est une insertion d’article publié en 2019, dont les travaux ont été réalisés durant mes 3 années de doctorat dans le laboratoire de mon directeur de recherche, Dr Sébastien Hébert. La numérotation des figures a été modifiée pour s’intégrer au manuscrit de thèse.
Le chapitre 2 est présenté sous la forme d’article pour une future publication. Le chapitre 3 est un résumé de mes résultats non-publiés pour l’objectif 2.
Chapitre 1 : Copy number variants in miR-138 as a potential risk factor for early-onset Alzheimer’s disease. J Alzheimers Dis. 2019;68(3):1243-1255. doi:
10.3233/JAD-180940.
Ce chapitre a pour but de démontrer l’importance des études de variation du nombre de copies d’un gène de microARN pour identifier de nouveaux facteurs de risque dans les maladies neurodégénératives, telle que la MA.
Emmanuelle Boscher, Thomas Husson, Olivier Quenez, Annie Laquerrière, Florent Marguet, Kevin Cassinari, David Wallon, Olivier Martinaud, Camille Charbonnier, Gaël
Nicolas, Jean-François Deleuze, Anne Boland, Mark Lathrop, Thierry Frébourg, FREX
Consortium, Dominique Campion, Sébastien S. Hébert, Anne Rovelet-Lecrux.
Olivier Martinaud, David Wallon et le FREX Consortium ont fait le recrutement des patients et des individus contrôles. Annie Laquerrière et Florent Marguet sont les neuropathologies qui ont prélevé les échantillons humains et effectué les diagnostics cliniques. Anne Boland, Jean-François Deleuze et Mark Lathrop ont effectué le séquençage d'exome. Camille Charbonnier, Olivier Quenez et Anne Rovelet-Lecrux ont traîté les données d'exome. Kevin Cassinari et Thomas Husson ont fait les analyses d'expression du miR-138 chez le patient. Dominique Campion, Thierry Frébourg, Sébastien Hébert, Gaël Nicolas et Anne Rovelet-Lecrux ont participé à la gestion du projet et à l’écriture du manuscrit. J’ai effectué les analyses du fichier de séquençage d’exome, la génération de la liste de VNC-miRs potentiellement impliqués dans la MA, la sélection d’un VNC-miR d’intérêt pour la preuve de concept, les transfections, les immunobuvardages, les ELISA, les qPCR. J’ai également fait les analyses et les interprétations des résultats ainsi que les analyses statistiques. J’ai rédigé la première version de l’article, fait les figures puis les corrections avec l’aide de A. Rovelet-Lecrux et N. Gaël, sous la direction du Dr S. Hébert. Le papier fut publié en 2019 dans Journal of Alzheimer’s disease.
Introduction
1. Maladie d’Alzheimer
1.1.
Historique
La maladie d’Alzheimer (MA) est la principale forme de démence à travers le monde (60-70%). En 1901, le psychiatre et neurologue Alois Alzheimer examina une patiente du nom de Auguste Deter âgée de 51 ans (Figure 1). Cette patiente présentait des troubles mnésiques, de langage, ainsi que des hallucinations et de la confusion mentale. À l’époque, ces symptômes étaient caractéristiques de la démence, mais son âge étonnamment précoce suscita un intérêt particulier pour le Dr. Alzheimer. N’ayant jamais vu de cas similaire, lors du décès de A. Deter en 1906, le Dr. Alzheimer demanda l’autorisation à la famille de la patiente de pratiquer son autopsie. C’est ainsi qu’il découvrit au niveau macroscopique : la présence d’atrophies, en particulier au niveau de régions impliquées dans la mémoire et le langage, et au niveau microscopique : des dépôts anormaux, connus aujourd’hui sous le terme de dégénérescences neurofibrillaires et de plaques séniles. Le Dr A. Alzheimer
présenta ces résultats lors de la 37ème conférence de psychiatre, en Allemagne, comme une
« maladie particulière du cortex cérébral ». Ce n’est qu’en 1910, qu’elle fut baptisée maladie d’Alzheimer par le psychiatre Emil Kraepelin, considéré comme le fondateur de la psychiatrie scientifique moderne et directeur de recherche du Dr. A. Alzheimer.
Figure 1 : Portrait du Dr. Alois Alzheimer (A) et de sa patiente August Deter (B)
À l’époque, la MA était encore restreinte aux cas de démences de type précoce (avant 65 ans), alors que les troubles démentiels des personnes âgées étaient classés parmi les pathologies de type vasculaire ou considérés comme des conséquences naturelles du vieillissement. Ce n’est qu’en 1980, que les cas tardifs de démences furent retirés des causes normales du vieillissement pour être regroupés avec les cas précoces de la MA. Dès lors, les découvertes scientifiques sur cette pathologie ont connu un fort essor, avec l’identification de marqueurs pathologiques.
1.2.
Épidémiologie
En 2000, la MA et les démences associées furent classées à la 14ème place des causes
de mortalité chez l’Homme à travers le monde. En 2016, elles ont atteint la 5ème position.
Ces chiffres risquent malheureusement de doubler d’ici 2030 si rien n’est fait (selon l’Organisation mondial de la santé). C’est plus de 45 millions d’individus dans le monde, dont 564 000 personnes au Canada (CIUSSS centre-Ouest-de-l’Île-de-Montréal), qui sont diagnostiquées pour la MA. Sa prévalence augmente avec l’âge : 0,8% chez les individus âgés de 65 ans ou moins, 6% pour ceux âgés de 75 ans et 25% pour ceux ayant 85 ans ou plus, d’après l’agence de la santé publique du Canada. Les femmes sont d’ailleurs plus souvent atteintes (près de deux tierds des américains atteints de la MA sont des femmes)
que les hommes probablement dû à leur plus grande espérance de vie1. De nombreux essais
cliniques ont vu le jour, avec plus de 800 milliards de dollars investis dans le monde en
20152, cependant aucun traitement ou biomarqueur, permettent de diagnostiquer
précocement, retarder ou prévenir la pathologie2.
1.3.
Symptômes
Les symptômes ont été décrits il y a déjà plusieurs années, sous le terme « d’altération de la raison liée à l’âge ». Cependant, cette pathologie n’est pas une suite logique du vieillissement. La plasticité cérébrale réussirait à compenser les lésions occasionnées par la pathologie durant une vingtaine d’année. Cette compensation finirait cependant par ne plus être suffisante et les premiers symptômes apparaîtraient.
Il existe plusieurs stades cliniques dans la MA qui peuvent être regroupés selon la sévérité :
Stades précoces: déficits cognitifs légers. L’amnésie (altération de la mémoire), en particulier celle à court terme, est souvent le premier symptôme décelé.
Stades modérés: aggravation de l’amnésie, apparition d’aphasie (altération du langage), d’agnosie (trouble de la reconnaissance d’objet ou de proche) et/ou d’apraxie (trouble de l’exécution des tâches de la vie quotidienne). Le patient développe également des troubles du comportement (dépression, colère inexpliquée, anxiété) avec un changement de personnalité (méfiance, hallucinations, troubles obsessionnels compulsifs,
etc.)1. Son autonomie diminue progressivement, s’accompagnant de pertes de notion du
temps et d’espace et il peut également avoir des difficultés à réaliser des tâches de la vie quotidienne (comme manger et/ou s’habiller).
Stades sévères: Le patient a besoin d’aide pour les activités les plus basiques. Il ne s’exprime plus qu’avec des mots ou phrases simples, ses capacités physiques et ses réflexes sont amoindris l’obligeant à rester alité1. À ce stade, tout le cerveau est atteint par la pathologie. Le patient devient vulnérable aux thromboses, aux infections et aux sepsis,
pouvant entraîner des défaillances d’organes1. Les troubles de la déglutition rendent
difficile le simple fait de manger ou de boire et peuvent induire des infections pulmonaires,
telle que la pneumonie, l’une des principales causes de mortalité dans la MA1.
1.4.
Marqueurs pathologiques
La MA est une maladie neurodégénérative caractérisée au niveau macroscopique par des atrophies cérébrales et des dilatations ventriculaires. Au niveau microscopique, cette maladie présente des plaques séniles extracellulaires ainsi que des dégénérescences
Figure 2 : Marqueurs pathologiques de la MA
Niveau macroscopique : atrophies cérébrales et dilatations ventriculaires. Coupe coronale d’un cerveau d’un sujet sain (à gauche) versus celui d’un patient atteint de la MA (à droite) (A). Niveau microscopique, plaque sénile (flèche verte) et dégénérescence neurofibrillaire (flèche bleue) (B) (Adapté de Bird, 2008).
1.4.1. Stades de Braak et Thal
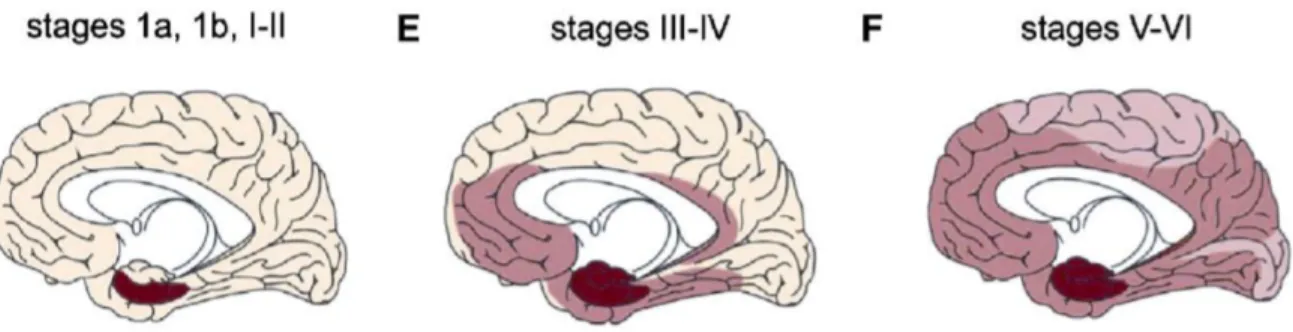
Les études de Braak et collaborateurs, ont permis de diviser la MA en 6 stades selon
son évolution (Figure 3)3. Les premières dégénérescences neurofibrillaires apparaissent
dans le locus coeruleus, noyau situé dans le tronc cérébral, définissant les stades I et II de la MA. Aux stades III et IV, les lésions s’étendent dans les aires entorhinale et
hippocampale. L’atrophie de l’hippocampe est d’ailleurs l’un des marqueurs les plus
précoces de la MA, ce qui expliquerait que l’amnésie soit l’un des premiers symptômes3.
Aux stades V et VI, les lésions continuent de s’étendre dans l’isocortex, en atteignant les
aires associatives puis les aires sensorielles primaires. Ces atteintes entraîneraient
l’apraxie, l’aphasie, l’agnosie et/ou les troubles comportementaux observés plus tard dans la pathologie4.
Ainsi, c’est le cortex temporal qui est affecté en premier, puis le cortex préfrontal
et pariétal pour finir par les noyaux sous-corticaux3.
Figure 3 : Propagation des dégénérescences neurofibrillaires
Aux stades I et II de la maladie d’Alzheimer, les premières dégénérescences neurofibrillaires surviennent dans le locus coeruleus. Aux stades III et IV, les lésions s’étendent dans les aires entorhinale et hippocampale. Aux stades V et VI, les lésions continuent de s’étendre dans l’isocortex, en atteignant les aires associatives puis les aires sensorielles primaires. (Source : Braak et al., 2011).
Les études de Thal et collaborateurs, se sont interessées à la propagation des plaques séniles et proposent cinq phases (Figure 4)5. Les premières lésions, phase 1, surviennent dans le néocortex avec une réapartition plutôt étendue. Elles continueraient ensuite, en phase 2 et 3, dans des régions plus internes tels que les noyaux du diencéphale et le striatum. À la phase 4, ces lésions se retrouvent dans les noyaux du tronc cérébral comme la substance noire, les colliculus et les noyaux gris centraux. Le cervelet est touché durant la dernière phase (phase 5) ainsi que d’autres noyaux du tronc cérébral (tels que le locus
coeruleus, noyaux du pont et noyaux tegmental ventral)5. Cependant, cette charge cérébrale
de dépots ne serait pas corrélée avec les altérations cognitives retrouvées dans la MA. L’hypothèse sugérée serait que les plaques séniles se formeraient avant l’apparition des
Figure 4 : Évolution des lésions de dépots de plaque
En phase 1 : les premières lésions surviennent dans le néocortex. Elles continuent de s’étendre, en phase 2 et 3, dans des régions plus internes tels que les noyaux du diencéphale et le striatum. À la phase 4, ces lésions se retrouvent dans les noyaux du tronc cérébral. Puis en phase 5, le cervelet est touché avec d’autres noyaux du tronc. (Source : © BIOPRO Baden-Württemberg GmbH)
Ces atteintes hiérarchiques pourraient être dues à plusieurs facteurs comme : la
localisation et/ou le type de neurones, l’activité neuronale (neurone plus ou moins actif),
la plasticité propre à chaque région ou encore les connexions qui pourraient « propager » la pathologie4.
1.4.2. Neurodégénérescences
Les atrophies sont dues à une perte neuronale majeure, dont la plupart (90%) sont observées dans l’hippocampe et le cortex entorhinal dans la MA7–9. Elles sont d’ailleurs accompagnées de dilatations ventriculaires et corticales. Un patient atteint par la MA peut perdre entre 8 à 10% du poids de son cerveau en 10 ans versus 2% en moyenne chez un
1.4.3. Plaques séniles
En 1984, le pathologiste américain George Glenner identifia l’Amyloïde bêta (Aβ)
comme la composante majeure des plaques séniles11.
Cette Aβ est issue du clivage de l’Amyloïd Precursor Protein (APP), dont le gène est situé sur le chromosome 21 chez l’Homme. Dans le cerveau, l’épissage alternatif de l’APP résulte en 3 isoformes majoritaires qui se distinguent par la présence ou non des exons 7, 8 et 15 (Figure 5) : APP 695 (-7; -8; +15) majoritairement neuronale, APP 751 (+7; +8; -15)
et APP 770 (+7; +8; -15) sont exprimées surtout par les cellules gliales12–14.
Figure 5 : Représentation schématique du gène de l'APP et de ces trois isoformes principales
Les exons 7, 8 et 15 peuvent être épissés, donnant différents isoformes dont les 3 majoritaires possèdent 770, 751 ou 695 acides aminés (aa). Illustration faite à l’aide d’images issues de « Servier Medical Art ».
L’APP est une protéine transmembranaire avec un long domaine N-terminal
extracellulaire, un domaine transmembranaire et un domaine C-terminal cytosolique15. Elle
est impliquée dans de nombreux mécanismes, telles que l’activité neuronale, la synaptogenèse, l’adhésion cellulaire, la neurogenèse mais aussi l’apoptose17–20,21. Durant son transport à la membrane, elle peut emprunter deux voies: la voie amyloïdogénique ou la
voie non-amyloïdogénique15.
Dans la voie amyloïdogénique (Figure 6) : l’APP est clivée par la β-sécrétase APP 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 Ab Gène APP 695 APP 751 APP 770NH2- -COOH NH2- -COOH NH2- -COOH cytoplasmique extracellulaire Principales isoformes
fragment APPs-β extracellulaire. Le peptide C99 est ensuite coupé par une γ-sécrétase formée de : PSEN1 ou 2 (Presenilin 1 ou 2), APH-1 (Anterior pharynx-defective 1), PEN2
(Presenilin-enhancer 2) et Nicastrine. Cette coupure génère un fragment AICD (APP
IntraCellular Domain) intracellulaire et le peptide Aβ12, 19. L’Aβ est retrouvée chez
l’Homme, le singe, la souris mais aussi chez le chien, suggérant un rôle crucial dans l’évolution.
L’Aβ produit peut-être de différentes longueurs (37 à 49 aa), où Aβ40 et Aβ42 sont les
deux principales formes22. En effet, Aβ40 constituerait environ 90% des formes produites
contre moins de 10% pour Aβ4223,24. Cependant la longueur de ce peptide influencerait sa
capacité à s’auto-agréger25,26. L’Aβ42 serait plus encline à l’agrégation et constituerait le
cœur des plaques séniles, alors que l’Aβ40 pourrait avoir des rôles neuroprotecteurs25–28. Le
rôle de l’Aβ est encore mal connu, mais elle pourrait agir comme facteur neurotrophique,
antimicrobien ou encore dans le transport du cholestérol29–33. Ses actions peuvent d’ailleurs
se faire à de très faibles concentrations, entre 10-11M et 10-10M 29.
Figure 6 : Représentation schématique de la voie amyloïdogénique de l'APP
L’APP est coupée par la β-sécrétase et la γ-sécrétase pour générer les fragments APPs-β, Aβ et AICD, durant la voie amyloïdogénique.
Dans la seconde voie, appelée voie non-amyloïdogénique (Figure 7) : l’APP membranaire est la cible d’une α-sécrétase, qui se trouve être principalement l’ADAM10 (Disintegrin And Metalloproteinase Domain-Containing Protein 10). Cette sécrétase coupe dans la séquence de l’Aβ, empêchant ainsi la production de ce peptide15. Cette coupure libère un fragment APPs-α extracellulaire et un fragment C83 cytoplasmique. Le peptide
Voie amyloïdogénique Ab b-sécrétase g-sécrétase APP AICD APPs-b
C83 est ensuite coupé via la γ-sécrétase produisant un fragment p3 extracellulaire et un
fragment AICD cytoplasmique16.
Cette voie empêche ainsi la production d’Aβ et permet la production d’APPs-α qui serait impliquée dans la neurogenèse (en particulier à l’âge adulte), la synaptogenèse, l’activité de kinases ou encore dans la mémoire18,20,34,35,36.
Figure 7 : Représentation schématique de la voie non-amyloïdogénique de l'APP
L’APP est coupée par une α-sécrétase et une γ-sécrétase permettant la libération de fragement APP-α, p3 et AICD, durant la voie non-amyloïdogénique.
L’Aβ aurait donc un rôle important au sein du cerveau, mais une mauvaise
élimination ou une augmentation de sa production/agrégation pourrait être neurotoxique37.
Bien que ce peptide soit majoritairement extracellulaire, l’Aβ intracellulaire aurait des effets neurotoxiques qui précèderaient celui de l’Aβ extracellulaire38,39. L’accumulation
intracellulaire entraînerait la mort neuronale et la libération extracellulaire de l’Aβ40. Ses
monomères solubles s’agrègeraient alors en oligomères puis en protofibrilles de moins en
moins solubles, avant de former des plaques plus larges de feuillets-bêta insolubles41,40. La
neurotoxicité de l’Aβ extracellulaire proviendrait de ses pré-plaques de monomères ou de ses petits oligomères, alors que les plaques plus larges seraient un mécanisme de défense
pour séquestrer les monomères/petits oligomères d’Aβ42,43. Cette neurotoxicité proviendrait
de l’action de l’Aβ sur la taille, la densité, l’activité mais aussi la composition des synapses43–47 et/ou via les cellules gliales48. Cependant, selon l’hypothèse de la cascade amyloïde, l’Aβ seul ne serait pas suffisant pour induire de la neurotoxicité. Celle-ci passerait par le second marqueur pathologique de la MA situé, quant à lui, dans les
Voie non-amyloïdogénique APP g-sécrétase a-secretase AICD p3 APPs-a
1.4.4. Dégénérescences neurofibrillaires
C’est en 1985, que Jean-Pierre Brion et collaborateurs, constatèrent la présence de
Tau hyper- ou anormalement phosphorylée dans les dégénérescences neurofibrillaires49.
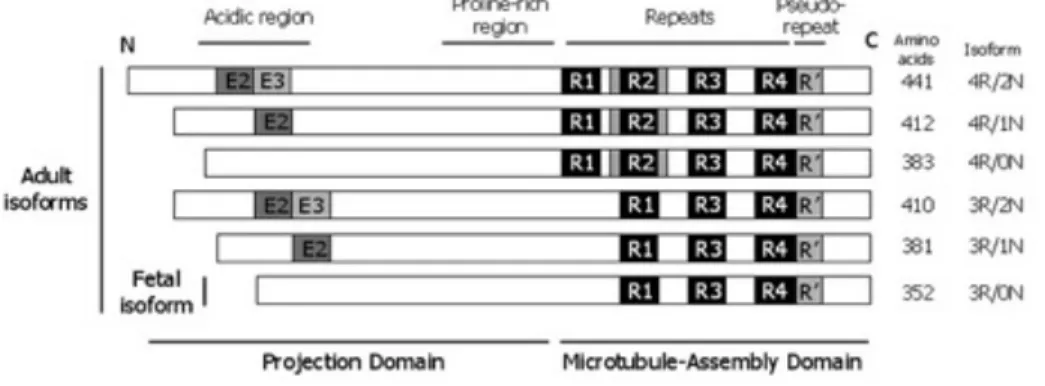
Tau est une protéine associée aux microtubules (MTs)50. Elle est codée par le gène MAPT
(MT-Associated Protein Tau), situé sur le chromosome 17 chez l’Homme. L’épissage alternatif de ce gène produit 6 isoformes. L’excision des exons 2 et/ou 3, en N-terminal, donne les isoformes : 0N (-2; -3), 1N (+2; -3) ou 2N (+2; +3). Parallèlement, l’excision ou non de l’exon 10, en C-terminal, donne les isoformes : 3R (-10) ou 4R (+10) (Figure 8). Ainsi, les 6 isoformes de Tau sont : 0N3R, 1N3R, 2N3R et 0N4R, 1N4R ou 2N4R. Le transcrit 1N est plus abondant que le 0N, lui-même plus abondant que le 2N (1N>0N>2N)51. Il est intéressant de noter que l’isoforme 0N3R est la seul retrouvée à l’état
fœtal chez l’Homme, alors qu’à l’âge adulte toutes les isoformes sont exprimées52–54.
Figure 8 : Isoforme de Tau
L’excision des exons 2 et/ou 3 en N-terminal donne les isoformes : 0N (-2; -3), 1N (+2; -3) ou 2N (+2; +3). L’excision ou non de l’exon 10, en C-terminal, donne les isoformes : 3R (-10) ou 4R (+10). Seul le 0N3R est trouvé chez le fœtus. À l’âge adulte toutes les isoformes de Tau sont retrouvées. (Source : Chun and Johnson, 2007)
Dans le cerveau, Tau est majoritairement neuronale, avec une localisation plutôt
axonale que dendritique55,56. Grâce à ses quatre domaines de liaisons aux MTs, dont un sur
l’exon 10 épissable, elle peut interagir avec les dimères d’α- et β- tubuline qui constituent les MTs. Cette interaction va influencer la polymérisation, la stabilité, et l’espacement des
synaptique et le maintien de l’intégrité ADN/ARN en condition physiologique et de stress61–68. Tau a ainsi divers rôles au sein de la cellule69.
Ses rôles peuvent être régulés par différentes modifications post-traductionnelles comme la phosphorylation, l’acétylation, la glycosylation ou encore la troncation. Or, l’altération de son état de phosphorylation est associée à une vingtaine de maladies neurodégénératives qui sont regroupées sous le nom de tauopathies, dont la MA. Cette protéine posséderait près de 85 sites de phosphorylation possibles (Figure 9), dont la majorité se situe sur ses domaines de liaisons aux MTs70–72. La phosphorylation de Tau
peut influencer sa fixation aux MTs (Figure 10)73,74. Sous forme non phosphorylée, Tau se
lie aux MTs et favorise leur stabilité. Lors de stress cellulaire, Tau déphosphorylée se délocalise dans le noyau pour protéger l’ADN de dommages oxydatifs. Sous forme phosphorylée, Tau est majoritairement somato-dendritique et se détache des MTs, qui vont
alors se déstabiliser et se dépolymériser75,76. Il y aurait environs 40 épitopes anormalement
ou hyper-phosphorylés dans la MA:
Tau hyperphosphorylée se caractérise par une augmentation de la quantité de
protéines Tau phosphorylées et/ou de groupements phosphates par épitope (3 à 8 fois plus
de qu’à l’état physiologique qui compte 2 à 3 groupements par épitope)77–79.
Tau anormalement phosphorylée se caractérise par la phosphorylation d’épitope
Figure 9 : Sites phosphorylables de Tau
Sites phosphorylés chez des patients atteints de la MA (rouge), chez des sujets sains (vert), chez les deux (bleu). Sites potentiels de phosphorylation non démontrés in vivo ou in vitro (noir). Épitopes reconnus par des anticorps anti-Tau (flèches jaunes). Les résidus sont numérotés selon la longueur de l’isoforme de Tau (Source : JC. Luna-Munoz and B. Floran, 2013).
Tau soluble va s’auto-assembler en oligomères puis former des paires de filaments hélicoïdaux (PHF) insolubles qui s’organisent ensuite en neurofibrilles (NFT). Cette
agrégation entraînerait une perte de fonction de la protéine Tau soluble83–85 et un gain de
fonction toxique via la séquestration d’autres protéines associées aux MTs (telles que
Microtubule-associated protein 1/2, MAP1 et MAP2), par exemple86–88. Ces protéines séquestrées ne pourraient alors plus assurer leurs les rôles physiologiques et/ou assurer une compensation physiologique.
Figure 10 : Illustration de Tau et les MTs dans un neurones sain versus un neurone qui dégénère
Dans un neurone sain, Tau normalement phosphorylée va permettre de stabiliser les MTs. Dans un neurone qui dégénère, la protéine Tau anormalement phosphorylée va favoriser la dépolymérisation des MTs et s’auto-agréger. (Source : National Institute on Aging/U.S. National Institutes of Health).
1.4.5. Hypothèse de la cascade amyloïde
Aβ et Tau seraient donc les deux principaux marqueurs pathologiques de la MA. La question est de savoir s’ils interagissent entre eux et si oui, par quel mécanisme? En 1991,
J. Hardy et D. Allsop, proposent l’hypothèse de la cascade amyloïde89. La MA
commencerait par l’accumulation d’Aβ, via l’altération de sa production et/ou de son élimination, ce qui induirait une réponse inflammatoire et des dommages oxydatifs. Ce stress induirait ensuite, l’agrégation de Tau et la formation des neurofibrilles à l’origine de la dégénérescence neurofibrillaire.
Cette hypothèse a été confortée par différentes études qui ont montré: qu’une mutation ou une augmentation d’APP entraînerait une augmentation de Tau intracellulaire90–92. Les dimères d’Aβ, quant à eux, favoriseraient l’hyperphosphorylation de Tau ainsi que la dépolymérisation du cytosquelette pour, in fine, entraîner une
dégénérescence neuronale93. Inversement, des neurones qui n’expriment pas de Tau
favoriser l’activité de kinases ciblant Tau, (telle que Glycogen synthase kinase 3 bêta, GSK3β) et induire une neuroinflammation, où les cytokines pro-inflammatoires favoriseraient l’hyperphosphorylation de Tau. Ils ont montré aussi que l’Aβ pouvait diminuer la capacité du protéasome et donc la dégradation de Tau ainsi qu’altérer le transport axonal et donc la localisation protéique et nucléique de Tau, entre autres94,95.
Ainsi l’Aβ serait l’élément déclencheur et Tau serait l’effecteur de la mort neuronale, dans la MA, selon l’hypothèse de la cascade amyloïde. Elle est cependant remise en cause, principalement depuis que des études ont constaté une absence de corrélation entre les dynamiques spatiales et temporale de la pathologie Aβ avec la sévérité du déclin cognitif de la MA96–101. L’absence de résultats encourageants des essais cliniques qui ciblent la voie Aβ ne fait que renforcer cette remise en question102–105.
1.5.
Formes de la maladie d’Alzheimer
La MA peut se distinguer selon l’âge d’apparition des symptômes : les formes
précoces (FPMA), plus sévères, qui apparaissent avant 65 ans mais peu fréquentes (environ
5%) et les formes tardives (FTMA) qui sont majoritaires et apparaissent après 65 ans (environ 95%).
1.5.1. Formes précoces
Les FPMA se distinguent des FTMA pas seulement par l’âge d’apparition des symptômes mais aussi par certains aspects comme une atrophie plus marquée dans le cortex
pariétal et temporal plutôt que l’hippocampale observée chez les FTMA106,107. Elles
présentent des symptômes de dysfonctionnement visuo-spatial ainsi que des apraxies, plutôt
que l’amnésie chez les FTMA106,107. Les fonctions exécutives, les tâches visuo-spatiales et
les capacités motrices sont également plus atteintes. Environ 22-64% des formes précoces
seraient d’ailleurs non-amnésiques106,107. Enfin, la région hippocampale serait plus
différences soient notables, la pathologie reste identique du point de vue de la présence de plaques d’Aβ et des dégénérescences neurofibrillaires106,107.
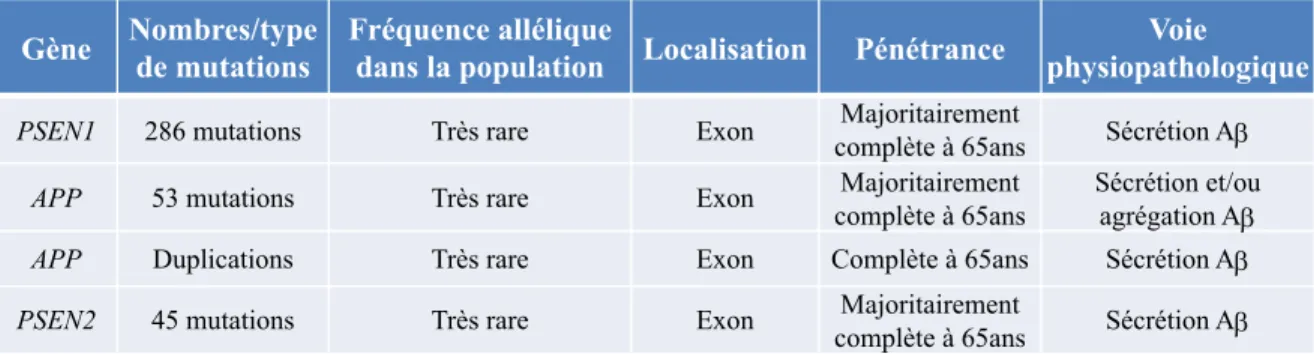
Les FPMA peuvent être soit familiales, soit sporadiques. Trois gènes, PSEN1, APP et PSEN2, ont des mutations causales dans les formes familiales (Tableau 1) et sont autosomales dominantes (FPMA-AD):
PSEN1, (environ 80% des cas FPMA-AD) située sur le chromosome 14q24.2 chez
l’Homme108. Les patients porteurs d’une mutation sur le gène de la Psen1 ont des
symptômes plus précocement (vers 43 ans) que ceux porteur d’une mutation APP (vers 51
ans) ou PSEN2 (vers 57 ans)108. Certains patients montrent même une pathologie vraiment
très précoce (vers 35 ans). À ce jour, plus de 200 mutations ont été rapportées (Alzheimer
Disease ans Frontotemporal Dementia Mutation Database) dont la majorité sont situées
sur les exons 5 à 8 du gène de la PSEN1 et augmentent le ratio Aβ42/Aβ40109,110.
L’augmentation de ce ratio suggère qu’il y a une hausse de la forme pro-fibrille d’Aβ dans le cerveau.
APP, (environ 15% des cas FPMA-AD) situé sur le chromosome 21q21.3 chez l’Homme.
Environ 50 mutations sur le gène de l’APP ont été répertoriées (Alzheimer Disease and
Frontotemporal Dementia Mutation databse) dont la plupart altèrent la protéolyse de l’APP
et donc la quantité d’Aβ produite. Certaines mutations peuvent modifier la coupure effectuée par la β-sécrétase, telles que D7H, E682K ou K16N. Ces mutations entraînent
une augmentation de la quantité d’Aβ40 et d’Aβ42111–113. D’autres mutations, comme
T714I, V715M ou encore V715A, sont retrouvées sur le site de coupure de la γ-sécrétase
entraînant une augmentation du ratio Aβ42/Aβ40114. La mutation V717I, quant à elle, va
affecter le site de coupure de la β- et de la γ-sécrétase augmentant la production d’Aβ38 et
d’Aβ42115. Mais des mutations récessives sont également identifiées, telle que E693Delta
qui induit une résistance à la dégradation protéolytique et favorise l’accumulation d’Aβ105.
La mutation A673V est également une mutation récessive mais, de manière étonnante, à l’état hétérozygote elle serait anti-amyloïdogénique, alors qu’à l’état homozygote elle aurait
un effet plutôt pro-amyloïdogénique116. De plus, si l’adénosine 673 (A673) est remplacée,
un effet protecteur vis-à-vis de la MA, en diminuant d’environ 40% la production d’Aβ106. Enfin, des duplications du gène de l’APP ont aussi été associées aux formes familiales des
FPMA, avec une pénétrance quasi complète à l’âge de 65 ans117,118.
PSEN2, (environ 5% des cas de FPMA-AD) situé sur le chromosome 1q42.13 chez
l’Homme. Les mutations sur la PSEN2 sont bien plus rares et ne sont pas toutes causales pour la MA. En effet, « seulement » 17 mutations sur les 45 répertoriées (Alzheimer
Disease and Frontotemporal Dementia Mutation Database; Alzforum) seraient causales.
Dix autres mutations ne seraient pas pathologiques. Quant aux dernières mutations, leurs implications sont encore à l’étude. La plupart de ses mutations pathologiques élèvent le
ratio d’Aβ42/40 telles que T122P, N141I et M239V119.
Tableau 1 : Tableau récapitulatif des 3 gènes impliqués dans les FPMA-AD
Liste du nombres/types de mutations identifiées sur les gènes PSEN1/2 et APP, leur pénétrance, leur fréquence allélique dans la population ainsi que la localisation des gènes et leur voie physiopathologique.
Ces trois gènes mutés conduisent généralement à des formes amnésiques typiques de la MA. Cependant, elles ne représentent qu’une faible portion des FPMA (5-10%). Plus de 50% des formes mendéliennes et la majorité des formes sporadiques sont encore sans causes identifiées. Des mutations sur TYROBP (TYRO protein tyrosine kinase binding
protein) et NOTCH3 (Notch receptor 3) pourraient être des facteurs de risque pour les
FPMA mais des études sur des cohortes plus larges sont encore nécessaires pour mieux définir leurs contributions120,121,106.
Gène Nombres/type de mutations Fréquence allélique dans la population Localisation Pénétrance physiopathologiqueVoie
PSEN1 286 mutations Très rare Exon complète à 65ansMajoritairement Sécrétion Ab
APP 53 mutations Très rare Exon Majoritairement complète à 65ans
Sécrétion et/ou agrégation Ab
APP Duplications Très rare Exon Complète à 65ans Sécrétion Ab
PSEN2 45 mutations Très rare Exon Majoritairement
1.5.2. Formes tardives
Les FTMA dépendraient à 70% de facteurs génétiques à transmission non-dominante
et à 30% de facteurs environnementaux122. Le principal facteur de risque est l’âge, environ
2 à 4% des patients sont diagnostiqués à 65 ans, 15% après 80 ans et 30% après 85 ans123.
L’historique familial est aussi un fort facteur de risque, puisque si un des deux parents est
atteint, le risque pour l’enfant est multiplié par 1,5 et si les deux le sont, le risque est
multiplié par 2123. Enfin, l’allèle ε4 de l’ApoE, une apolipoprotéine, est fortement associé à
la MA124,125. ApoE possède trois allèles différents, ε2, ε3 et ε4, dont la fréquence varie au
sein de la population. L’allèle ε2 a une fréquence d’environ 5 à 10%, l’allèle ε3 d’environ 65 à 70% et 15 à 20% pour l’allèle ε4. À l’état hétérozygote, l’allèle ε4 augmente de 3 fois le risque de développer la MA et à l’état homozygote il l’augmente de 12 fois. À l’inverse, l’allèle ε2 serait un facteur protecteur1,126.
Côté environnemental, les facteurs comme l’alimentation, l’exercice physique ou l’état cardio-vasculaires sont associés à la MA. Le diabète, l’hypertension ou encore le tabagisme sont des facteurs de risque alors que l’exercice physique et une alimentation
riche en oméga 3 seraient neuroprotecteurs127.
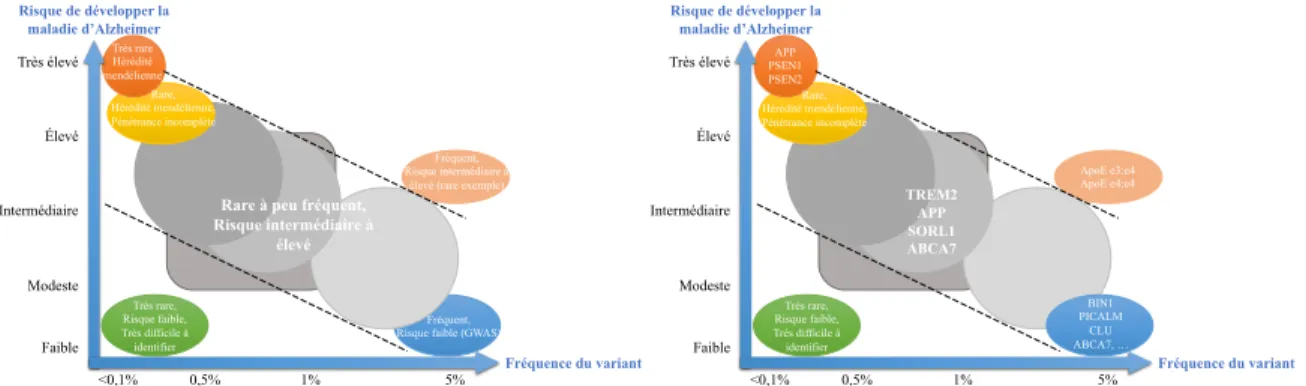
La part de la génétique est prépondérante dans la MA, de nombreuses méta-analyses et nombreux séquençages du génome ont permis d’identifier de nouvelles causes/facteurs de risque qui ont permis de grandes avancées dans la compréhension de la maladie (Tableau 2). Le « genome-wide association studies » (GWAS) a permis d’identifier 11 locis de susceptibilités : CR1, BIN1, CLU, CD2AP, EPHA1, PICALM, MS4A6A/4E, ABCA7, DSG2 et APOE. Puis, une étude de méta-analyse a ajouté 11 nouveaux locis: la région HLA-DRB5-DRB1, SORL1, PTK2B, SLC24A4, ZCWPW1, CELF1, CASS4, FERMT2, INPP5D, MEF2C et NME8128,129. Les gènes PLD3 et TREM2 ont été ensuite
ajoutés à cette liste130,131. La susceptibilité que confère chacun des variants des gènes
identifiés varient de l’un à l’autre tout comme leur fréquence dans la population. ApoE est le facteur de risque le plus important avec une fréquence proche des 5%. La majorité des autres variants sont associés à des risques faibles mais certains auront une fréquence élevée
Figure 11 : Facteurs de risque dans la maladie d'Alzheimer
(Source : Modifiée de Le Guennec)
Ces gènes sont impliqués dans de nombreuses voies différentes, qui ont ainsi pu être associées à la MA, telles que :
Le métabolisme lipidique, avec les gènes de l’ApoE, ABCA7, SORL1 et CLU, entre autre. Il s’avère que le cholestérol est essentiel à la formation de la gaine de myéline et des membranes cellulaires. Une mauvaise redistribution de cholestérol peut affecter la plasticité
synaptique, la mémoire et l’activité neuronale et être un facteur de risque pour la MA133,134.
En effet, l’APP est retrouvée dans les radeaux lipidiques et l’Aβ peut être liée par le cholestérol où une élévation de la concentration du cholestérol intracellulaire corrèle avec le processus amyloïdogénique de l’APP30,135. ApoE serait capable d’éliminer les plaques d’Aβ avec une efficacité différente en fonction de son isoforme (ε2 > ε3 > ε4). L’isoforme
ε4 aurait une efficacité d’élimination insuffisante136,37,16. SORL1 participerait au recyclage
de l’APP membranaire vers l’appareil de Golgi. Cette protéine aurait donc un effet protecteur dans la MA en diminuant la production d’Aβ, ainsi des mutations de SORL1
seraient associées aux FPMA ou aux FTMA137. CLU est une apolipoprotéine (ou ApoJ)
impliquée dans le transport du cholestérol, des phospholipides et l’élimination d’Aβ via son endocytose et/ou son transport au travers de la barrière hémato-encéphalique. La diminution de CLU réduirait la formation de plaque amyloïde et la neurotoxicité associée
aux dépôts d’Aβ138,139. CLU serait d’ailleurs augmentée dans le LCR de patients atteints de
la MA140. Un niveau élevé de CLU plasmatique est associé à une atrophie du cerveau ainsi
qu’à la sévérité et la prévalence de la MA141,142. CLU joue aussi un rôle dans le système du
complément en inhibant la réponse immunitaire143.
Faible Modeste Intermédiaire Élevé Très élevé Risque de développer la maladie d’Alzheimer Fréquence du variant <0,1% 0,5% 1% 5% Très rare Hérédité mendélienne Rare, Hérédité mendélienne, Pénétrance incomplète
Rare à peu fréquent, Risque intermédiaire à élevé Très rare, Risque faible, Très difficile à identifier Fréquent, Risque intermédiaire à
élevé (rare exemple)
Fréquent, Risque faible (GWAS)
Faible Modeste Intermédiaire Élevé Très élevé Risque de développer la maladie d’Alzheimer Fréquence du variant <0,1% 0,5% 1% 5% APP PSEN1 PSEN2 Rare, Hérédité mendélienne, Pénétrance incomplète Très rare, Risque faible, Très difficile à identifier ApoE e3:e4 ApoE e4:e4 TREM2 APP SORL1 ABCA7 BIN1 PICALM CLU ABCA7, …
La réponse immunitaire, où les gènes tels que CLU, CR1, CD33,
HLA-DRB5-DRB1, MS4A et TREM2 ont été associés à la MA. Il s’avère qu’une forte activation
microgliale dans les aires lésées a été décrite chez les patients atteints par la MA37. De la
microglie est retrouvée autour et au sein des plaques amyloïdes et est fortement associée aux altérations neurofibrillaires37,144. Le système du complément est lui aussi observé au niveau des plaques amyloïdes ainsi que dans les neurofibrilles et serait sur-régulé dans la MA145,37. CR1 code pour un récepteur érythrocytaire qui peut être lié par l’Aβ oligomérique afin d’exporter cette Aβ hors de la circulation sanguine145,146. TREM2 est
retrouvée au niveau des membranes microgliales et serait impliquée dans la phagocytose ainsi que dans l’élimination des débrits neuronaux. Sa mutation R47H pourrait d’ailleurs
augmenter le risque de développer la MA par trois 147,148. CD33, un récepteur de cellules
myéloïdes et microgliales principalement, est impliqué dans la recapture d’Aβ extracellulaire149. Il serait augmenté chez des patients atteints de la MA et son variant
rs3865444 serait un facteur protecteur dans la pathologie149. De plus, CD33 est aussi
impliqué dans des processus d’endocytose.
L’endocytose et les fonctions synaptiques avec des gènes tels que PICALM, BIN1,
CD2AP, FERMT2 et EPHA1. PICALM assure l’endocytose dépendante de la clathrine, la
libération de neurotransmetteurs et agit dans la formation de la mémoire. Or, cette protéine colocalise avec l’APP et sa sous-expression altère le transport de l’APP, alors que sa surexpression favoriserait la formation de plaques amyloïdes dans la MA150. CD2AP est une protéine adaptatrice impliquée dans le transport membranaire. Elle favoriserait le
transport de l’APP de l’endosome précoce vers la voie de dégradation lysosomale151.
FERMT2 (ou Kindlin2) est une co-activatrice d’intégrine impliquée dans l’adhésion
cellule-matrice extracellulaire et influencerait le recyclage de l’APP, via les endosomes
Rab4A-positif152,153. Une sous-expression de FERMT2 entraînerait une augmentation
d’APP à la membrane cellulaire et favoriserait la production d’Aβ153. Elle serait également
impliquée dans la toxicité de Tau où la diminution de FERMT2 entraînerait une neurodégénérescence oculaire chez la drosophile liée à Tau154. Enfin, BIN1 est proposé
comme le facteur de risque le plus important après ApoE. Cette protéine est impliquée dans
L’homéostasie du calcium est une voie bien connue pour être impliquée dans les maladies neurodégénératives, telle que la MA, avec des gènes comme BIN1, PTK2B et
SLC24A4. Sa dérégulation peut induire de la mort neuronale et un déclin cognitif. Cette signalisation est mise en jeu dans de nombreux processus intra- et extra- cellulaires allant de l’activité synaptique à la communication cellulaire, en passant par l’adhésion cellulaire. Dans le cerveau, l’ion calcique est fondamental pour le contrôle de l’activité synaptique et
l’élaboration de la mémoire157. Le contrôle de cette homéostasie est donc primordial pour
l’état physiologique du cerveau mais aussi pour le maintien de l’intégrité neuronale et la survie à long terme des cellules. La dérégulation de son homéostasie peut expliquer la vulnérabilité de certaines populations de neurones dans certaines maladies. Or, il se trouve que l’Aβ serait étroitement liée à cette homéostasie, notamment via l’excitotoxicité où elle stimulerait l’activité des récepteurs glutamatergiques de type NMDA
(N-méthyl-D-aspartate)158. L’Aβ serait capable de former des pores calciques159,160 entraînant un influx
d’ion calcium et la modification du potentiel membranaire. Le niveau de calcium intracellulaire agirait sur la production d’Aβ ainsi que sur le processus de l’APP161,162,