HAL Id: tel-02129289
https://tel.archives-ouvertes.fr/tel-02129289
Submitted on 14 May 2019HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Vers une meilleure compréhension de l’implication de
WNK1, Cullin-3 et SPAK dans l’hypertension
hyperkaliémique familiale
Chloe Rafael
To cite this version:
Chloe Rafael. Vers une meilleure compréhension de l’implication de WNK1, Cullin-3 et SPAK dans l’hypertension hyperkaliémique familiale. Cardiologie et système cardiovasculaire. Université Sor-bonne Paris Cité, 2017. Français. �NNT : 2017USPCB068�. �tel-02129289�
THESE DE DOCTORAT
DE L’UNIVERSITE PARIS DESCARTES
Présentée par
Chloé RAFAEL
Pour obtenir le grade de Docteur de l’Université Paris V
Ecole doctorale : Bio Sorbonne Paris Cité
Département : Biologie Cellulaire et Moléculaire, Physiologie, Physiopathologie (BCMPP)
Spécialité : Physiopathologie
Vers une meilleure compréhension de l’implication de
WNK1, Cullin-
3 et SPAK dans l’Hypertension
Hyperkaliémique Familiale
Dirigée par le Dr. Juliette HADCHOUEL
Présentée et soutenue publiquement le 22 novembre 2017
Devant le jury composé de :
Pr. Laurence DUBOURG : Rapporteur Dr. Frédéric JAISSER : Rapporteur
Dr. Emmanuelle VIDAL-PETIOT : Examinateur Dr. Gilles CRAMBERT : Examinateur
Pr. Xavier JEUNEMAITRE : Examinateur
Remerciements
Je souhaite tout d’abord adresser mes remerciements les plus sincères aux membres du jury qui ont accepté de prendre le temps d’évaluer ce travail malgré leurs emplois du temps chargés. Je remercie le Dr. Laurence Dubourg et le Dr. Frédéric Jaisser d’avoir accepté d’en être les rapporteurs. Je remercie également les examinateurs le Dr. Gilles Crambert, le Dr. Emmanuelle Vidal-Petiot qui m’a d’ailleurs accompagnée lors de mes premiers pas dans un laboratoire de recherche il y a 6 ans de cela maintenant, et le Pr. Xavier Jeunemaitre.
J’en profite également pour remercier le Pr. Xavier Jeunemaitre de m’avoir fait confiance en me confiant les projets sur lesquels son équipe et moi avons travaillé en collaboration au cours de ces trois dernières années. Merci également du temps que vous m’avez accordé, et ce, malgré vos contraintes d’emploi du temps !
Je tiens à exprimer ma sincère reconnaissance et gratitude au Dr. Juliette Hadchouel, ma formidable et extraordinaire directrice de thèse, sans qui ce travail de thèse n’aurait jamais vu le jour. Elle fut ma toute première encadrante et depuis, la richesse de ses connaissances, ses conseils, sa rigueur et sa confiance m’ont guidé et m’ont énormément apporté chaque jour. Je mesure la très grande chance que j’ai eu d’avoir un encadrement d’une telle qualité ! Ces années de travail à tes côtés m’ont réellement transformée et je sais maintenant grâce à toi qu’il est possible d’être de bonne humeur presque chaque jour !
Un grand merci à tous ceux que j’ai eu l’occasion de côtoyer jour après jour au premier étage, qui m’ont apporté sans le savoir un soutien moral de taille et que je continue à croiser avec le plus grand des plaisirs : Fabio, Isabelle, Angélique, Luis, Aurélie, Romuald (notre Roro national !!!!), Charlotte, Sheerazed, Chahrazade, Mélanie, Magalie, sans oublier les chefs : Maria-Christina, la grande Judith, Nabila et Christian. Egalement un grand merci à ceux qui ne travaillent pas dans les labos mais dont le rôle est tout aussi indispensable dans l’institut : Catherine, Bruno, Philippe et bien sûr Stéphanie !
Un grand merci au BDE du 1erétage qui m’accueille toujours les bras grands ouverts quand il m’arrive de repasser par le PARCC : Hélène, la petite Judith, Rami, le petit Alex et Didi. Un grand merci à l’équipe γ du PARCC avec qui j’ai travaillé une bonne partie de ma thèse et qui a continué à m’accueillir dans leur labo même une fois que Juliette et moi soyons parties du PARCC : Waed, Irmine, Ilektra, Eric et Amélie ! Encore une fois désolée pour les odeurs de souris à 9h du matin.... Merci également à Véronique pour sa collaboration et son expertise en chirurgie de souris ! Un énorme merci également à tous ceux que j’ai fréquenté dans les étages supérieurs et à l’équipe de l’animalerie avec qui cela aura été un réel plaisir de travailler.
Pour finir, il y a également celles qui n’étaient au départ que des collègues au PARCC et qui, au fil du temps, sont devenues de véritables amies ! Christelle, bien évidemment, qui a accompagné mes tout premiers pas dans un laboratoire et sur qui j’ai pu à nouveau compter lors de mon stage de Mβ et au début de ma thèse. J’en profite pour faire un petit clin d’œil à François (enfin un vrai concurrent à Mario Kart !) Bettina alias « Betty » alias « Coupinette », dès les premiers jours passés en ta compagnie j’ai su que tu serais une super coupine ! Tu as été un énorme soutien et un rayon de soleil pendant une bonne partie de ma thèse et tu continues à être une super coupine chaque jour ! Et finalement, Anne-Lyse qui a agrandi notre cercle très fermé de coupines. On se sera malgré tout quand même bien marré !
Cette thèse n’aurait jamais été la même sans le déménagement de la « WNK team » au centre de recherche de l’Hôpital Tenon. Mon intégration n’aurait pas été aussi réussie si mon bureau
ne s’était pas trouvé aussi proche du bureau des poules !! Mesdames les poules-en-chef : Sandrine alias « Sista » et Sophie. De véritables soutiens pour la vie quotidienne au laboratoire. Elles ont toujours été disponibles pour me montrer où se trouvait telle ou telle chose, pour m’expliquer le fonctionnement du matériel et elles se sont parfois même montrés très disponibles pour écouter mes blagues stupides. Sandrine, je n’oublierai également pas les carottes râpées fraiches, la quiche et toute la nourriture que tu as partagé avec moi ! Tu sais à quel point c’est important pour moi ! Et bien sûr un énorme merci à mon co-thésard et à la dernière des poulettes : j’ai nommé M. le Dr Raphaël K. ! Les heures passées à ma paillasse à Western-Blotter n’auraient jamais été les mêmes sans toi dans mon champ de vision et sans tes blagues assez nulles dans l’ensemble ! Un gros big-up à tous mes potos du premier ! Virginie pour ta gentillesse jour après jour, à Khalil, à Tiffany et Souhila pour le duo de folie qu’elles forment et leurs caractères que je peine parfois encore à décrypter ! Mais au final elles ont toujours fait preuve avec moi d’une gentillesse sans limite ! Un grand merci à mon adorable « rouya » Nadhir avec qui je partage une grande passion pour les zébrafish ! Et bien sûr, on ne peut oublier Emmanuel, le master des mots croisés, le master de la culture générale en fait ! Un grand merci à Béatrice, qui a toujours fait preuve d’une immense gentillesse à mon égard, que ce soit le matin à 8h ou au déjeuner à 12h00 (pile !!)! Il en va de même pour Chantal avec qui j’ai partagé bon nombre de mes déjeuners et une passion commune pour le tricot ! Un grand merci à Perrine pour m’avoir fait culpabiliser un certain nombre de midis où elle revenait de son sport, alors que moi je venais d’engloutir un demi kilo de pâtes aux lardons. On ne peut décemment pas remercier Perrine sans remercier son binôme de choc Dominique. Merci pour ton mauvais caractère adorable, il va me manquer !!! Je remercie également tous les grecs de cette unité, les Christos, Panos et Nikki ! Un grand merci également à Joëlle, Camille et Elie !! Je remercie également les étudiants de Juliette avec lesquels j’ai pris beaucoup de plaisir à travailler et auxquels j’ai transmis tout mon savoir (c’est dire…) : Aïssata et Samy !! Pour finir, une pensée aussi pour ceux qui sont partis peu de temps après mon arrivée à Tenon, Guillaume, Aude, Yosu et Raphaël C. Ce fut bref, mais chouette quand même !
Au cours de cette thèse j’ai également eu la chance d’être extrêmement bien entourée en dehors du labo ! Tout d’abord un énorme merci qui vient du fond du cœur à « La coloc du Bonheur », mes chers colocs avec qui j’ai vécu « au 16 » pendant mes deux premières années de thèse : Juliane, Morgane, Marine, Justine et Thomas ! Vous m’avez apporté un soutien moral sans faille quand il y en avait besoin, un vivier d’anecdotes à raconter chaque jour (de chaudières qui explosent, de fenêtres de salles de bain cassées, de cambriolage, de petites et des grosses araignées, de pulls en laine qui prennent feu quand on les approche trop près de la gazinière, de frigos qui sentent tellement mauvais qu’on doit condamner la cuisine, etc…) !! Bien que je vous aie abandonné, il se passe rarement une semaine sans que je ne pense aux moments passés à « La coloc du Bonheur » avec vous <3. Un grand merci également à Letty et Kévinou les choupinous ! C’est encore plus cool depuis qu’on a tous déménagé et que l’on est tous les 4 sur la même ligne de bus !! Bus323 Forever !! J’ai vraiment hâte que l’on reprenne nos parties endiablées de jeux de société et nos visites culturelles au musée ! Un énorme merci à mes supers copains Dévina et Vivien, toujours prêts à nous accueillir pour des week-ends à Tours et à nous proposer des activités divertissantes : Visites de châteaux médiévaux (et un peu des châteaux de la Loire aussi…), barbecue, tennis, foot, ragots, mariage, soutien de thèse, bref des supers copains comme il en existe peu… Dévina, je suis tellement heureuse que l’on ait eu à écrire nos thèses en même temps, nos messages d’encouragements mutuels chaque jour, même plusieurs fois par jour en fait, ont eu un effet on ne peut plus bénéfique sur mon moral ! Une grosse pensée pour Jacqueline et Jeannette, qui m’ont montré que l’amitié n’avait pas de limite d’âge ! Ainsi qu’un grand merci à toute
l’équipe de foot du stade Elizabeth à Porte d’Orléans et plus particulièrement à José, Pierre, Joseph, Jan, Ilya, Rémi, Aurélien, Eliott et Nikita qui, bien que sceptiques au début, on finit par accepter qu’une fille vienne jouer avec eux toutes les semaines. Soyez certains de me retrouver sur le terrain courant 2018 !!
Un énorme merci à l’ensemble de ma famille !! Evidemment une reconnaissance éternelle à mes parents qui me soutiennent dans tout ce que j’entreprends depuis toujours. Chers parents, voyez, il semblerait finalement que je les ai finis mes études !!! Je remercie également ma petite sœur Ophélia et mon petit frère Erwan sans lesquels la vie serait beaucoup moins funky. Mes grands-parents Manou et Papégé qui sont d’un soutien sans faille depuis de nombreuses années et qui, j’en suis sûre, le seront toujours dans les années à venir d’ailleurs. Une pensée à tous mes oncles, mes tantes, mes cousins et mes cousines et arrière-grands-mères qui ont suivi de près ou de loin l’avancée de cette thèse et qui m’ont témoigné tout leur soutien.
Je remercie également ma belle-famille Russe, parce qu’il est important de savoir que certaines parties de ce manuscrit ont été rédigées au beau milieu de l’Oural !!! Nina et Viktor, mes beaux-parents en or qui font toujours en sorte que je ne manque jamais de rien ! Tout comme mes belles-sœurs Vassilina et Yulia ! J’espère qu’un jour mon niveau de russe me permettra de vous dire à quel point de vous remercie de tout ce que vous faites pour moi ! En tout cas, j’y travaille sans relâche !
Finalement, je remercie Nikita qui, en bon scientifique, m’a démontré au cours de ces trois dernières années qu’il fallait parfois prendre de très grands risques pour qu’une expérience soit couronnée de succès encore plus grands.
Résumé
L’Hypertension Hyperkaliémique Familiale (FHHt) est une forme rare d’hypertension artérielle associée à une hyperkaliémie et une acidose métabolique hyperchlorémique. Ces troubles sont corrigés par les diurétiques thiazidiques qui inhibent le co-transporteur Na+-Cl -NCC exprime dans le néphron distal. Cette sensibilité aux diurétiques thiazidiques a laissé supposer que la FHHt est causée par une activation de NCC. En 2001, des mutations, de type gain-de-fonction, responsables de la FHHt ont été découvertes dans les gènes codant les sérine-thréonine kinases WNK1 et WNK4 [With No (K) lysine].
Des études in vitro ont montré que WNK1 et WNK4 stimulent NCC de façon indirecte, via la phosphorylation et l'activation de la kinase SPAK (Ste20 like Proline-Alanine rich Kinase). In vivo, l’activation de SPAK joue un rôle central dans le développement de la FHHt due aux mutations de WNK4. L'implication de SPAK n'a pas été définie dans le cas des mutations de WNK1. Nous avons donc croisé les souris WNK1+/FHHt,porteuses d'une mutation FHHt de WNK1, avec les souris SPAK243A/243A, porteuses d’une mutation abolissant l’activation de SPAK par les WNK. L’ensemble des phénotypes observés chez les souris WNK1+/FHHt est corrigé chez les souris WNK1+/FHHt:SPAK243A/243A démontrant ainsi le rôle central de SPAK dans la FHHt causée par les mutations WNK1.
En 2012, de nouvelles mutations ont été identifiées dans les gènes codant CUL3 et KLHLγ, deux composants d’un complexe ubiquitine ligase. Comme les mutations de WNK1 et WNK4, ces mutations entrainent une augmentation de l’expression de WNK1 et de WNK4. De façon inattendue, les patients FHHt porteurs d'une mutation CUL3 présentent un phénotype plus sévère que les autres. Des études suggèrent que ces mutations perturbent la fonction non seulement du néphron distal mais également des artères. Afin de vérifier cette hypothèse, nous avons généré et comparé deux modèles de souris : les souris pgk-Cul3'9, exprimant la mutation Cul3 de façon ubiquitaire, comme les patients, et les souris
sm22-Cul3'9, exprimant la mutation uniquement dans les cellules musculaires vasculaires lisses. Les deux modèles sont hypertendus, mais les souris pgk-Cul3'9 le sont significativement plus que les souris sm22-Cul3'9, ce qui prouve que l’hypertension causée par les mutations Cul3 résulte du cumul d’une atteinte rénale et vasculaire.
Récemment, de nouvelles mutations faux-sens d'un domaine dit acide de WNK1 ont été identifiées dans une petit nombre de patients atteints de FHHt. Ce domaine est nécessaire à la liaison à KLHLγ et donc à l’ubiquitination des WNK. Notre étude montre que, in vitro, ces mutations entrainent une accumulation d'une seule isoforme de WNK1. Chez la souris, la
mutation du domaine acide provoque le même phénotype que les patients, ainsi qu’une augmentation de la phosphorylation de SPAK et du co-transporteur NCC. Cette étude a donc permis de démontrer le rôle essentiel de ce domaine dans la régulation de l'abondance de WNK1 dans le néphron distal in vivo.
En conclusion, mon travail a permis une meilleure compréhension du rôle joué par SPAK, WNK1 et CUL3 dans le développement de la FHHt et, plus largement, de la physiopathologie de la FHHt.
Abstract
Familial Hyperkalemic Hypertension (FHHt) is a rare form of hypertension associated with hyperkalemia and hyperchloremic metabolic acidosis. These disorders are all corrected by thiazide diuretics that inhibit the Na+-Cl- NCC cotransporter expressed in the distal nephron. The sensitivity of FHHt patients to thiazide diuretics strongly suggested that FHHt is caused by NCC activation. In 2001, gain-of function-mutations were identified in the genes encoding the serine-threonine kinases WNK1 and WNK4 [With No (K) lysine] in a subst of FHHt patients.
In vitro studies demonstrate that WNK1 and WNK4 indirectly stimulate NCC, through the phosphorylation and activation of SPAK (Ste20 like Proline-Alanine rich Kinase). In vivo, SPAK activation plays a central role in the pathogenesis of FHHt caused by WNK4 mutations. However, the implication of SPAK has never been shown for the WNK1 mutations. Thus, we crossed WNK1+/FHHt mice, bearing the WNK1-FHHt mutation, with SPAK243A/243A mice bearing a mutation abolishing SPAK activation by the WNKs. All FHHt phenotypes observed in WNK1+/FHHt were corrected in WNK1+/FHHt:SPAK243A/243A mice demonstrating the central role of SPAK in FHHt caused by WNK1 mutations.
In 2012, new mutations have been identified in CUL3 and KLHL3 genes. The products of these genes are both part of a ubiquitin ligase complex. As WNK1 and WNK4 mutations, these mutations lead to an increased expression of L-WNK1 and/or WNK4. Surprisingly, patients with CUL3 mutations display a more severe phenotype. Previous studies have suggested that CUL3 could be involved not only in the regulation of ion transport in the distal nephron but also in the regulation of vascular tone. To verify this hypothesis, we generated and compared two mouse models: pgk-Cul3'9 mouse model bearing, as patients, an ubiquitous Cul3 mutation, and sm22-Cul3'9 mouse model that express the Cul3 mutation only in vascular smooth muscle cells. Both models are hypertensive, but pgk-Cul3'9 mice display a more severe hypertension than sm22-Cul3'9 mice. It demonstrates that the hypertension caused by Cul3 mutations results from both renal and vascular disorders.
Recently, new missense mutations have been identified in WNK1 acidic motif in a small number of FHHt patients. This acidic motif is necessary for the liaison to KLHL3 and therefore for WNK ubiquitination. Our study shows that, in vitro, these mutations lead to the accumulation of only one isoform of WNK1. In mice, the mutation of WNK1 acidic motif leads to an increased phosphorylation of SPAK and NCC. Therefore, it demonstrates the
essential role of the acidic motif in the regulation of WNK1 abundance in vivo in the distal nephron.
This work contributes to a better comprehension of the role played by SPAK, WNK1 and CUL3 in FHHt and more generally in the regulation of blood pressure and Na+/K+ homeostasis.
Table des matières
Liste des abréviations 1
INTRODUCTION GENERALE : De l’hypertension Hyperkaliémique Familiale à la
découverte de régulateurs de la pression artérielle 3
1. REIN ET HYPERTENSION ARTERIELLE 4
1.1 Le rôle du sodium et potassium dans l’hypertension artérielle essentielle 4
1.2 Structure du néphron 5
1.3 Les mécanismes de transport ionique le long du néphron distal 7
1.3.1 Les mécanismes du transport dans le DCT 7
1.3.2 Les mécanismes de transport dans le CNT et le CD 8
1.3.2a Les mécanismes de transport dans les CP 8
1.3.2b Les mécanismes de transport dans les CI 10
2. DE L'HYPERTENSION HYPERKALIEMIQUE FAMILIALE À LA
DÉCOUVERTE D'UNE NOUVELLE VOIE DE RÉGULATION DU TRANSPORT
IONIQUE DANS LE NÉPHRON DISTAL 13
2.1 Aspects cliniques de l'Hypertension Hyperkalémique Familiale (FHHt) 13 2.2 Quatre nouveaux gènes pour une pathologie très rare 14 2.3 Gènes impliqués dans la FHHt et hypertension artérielle essentielle 16
2.4 Les WNK, des kinases particulières 17
2.4.1. Structure 17
2.4.2. Profil d'expression 19
2.4.3 Le gène WNK1 donne naissance à plusieurs variants exprimés de façon
tissu-spécifique. 20
2.4.4 Partenaires et substrats de WNK1 23
2.4.4a. WNK1 et molécules impliquées dans l'exocytose 23
2.4.4b. WNK1 et voie du TGF-β 24
2.4.4c SPAK et OSR1 25
3. RÉGULATION DU TRANSPORT IONIQUE PAR WNK1 ET WNK4 25
3.1 La voie WNK-SPAK/OSR1 25
3.1.1 Les kinases SPAK et OSR1 : des régulateurs des co-transporteurs cation-chlorure
CCC en aval des WNK 25
3.1.1a SPAK/OSR1 et les CCC 25
3.1.1b Activation de SPAK et OSR1 par les WNK 26
3.1.2 Régulation de NCC par les WNK 28
3.1.2a SPAK est essentiel à la phosphorylation et à l’activité de NCC 28
3.1.2b L-WNK1 active SPAK indépendamment de WNK4 30
3.1.2c WNK4 : inhibiteur ou activateur de NCC ? 33
3.1.3 Rôles extra-rénaux de la voie WNK-SPAK/OSR1 35
3.1.3a Régulation du développement et du tonus vasculaires par L-WNK1, SPAK et OSR1 35
3.1.3b La voie de signalisation WNK-SPAK/OSR1 dans les neurones 37
3.2. Rôles de WNK1 et WNK4 indépendants de SPAK et OSR1 38 3.2.1 Régulation de la réabsorption de sodium par WNK1 et WNK4 indépendante de
SPAK et OSR1 39
3.2.1a Régulation de NCC par les WNK indépendante de SPAK et OSR1 39
3.2.1b Régulation de ENaC par WNK1 et WNK4 39
3.2.2 Régulation de la sécrétion de potassium par WNK1 et WNK4 42
3.2.2b Régulation de BKCa par WNK1 et WNK4 44 3.2.3 Régulation du transport de Cl- par WNK1 et WNK4 44
3.2.4 Régulation de la réabsorption de Ca2+ par WNK1 et WNK4 45 4. RÉGULATION DE L'EXPRESSION ET DE L'ACTIVITÉ DE WNK1 ET WNK4 45
4.1 Régulation par les apports sodés et le système Rénine-Angiotensine-Aldostérone 46
4.2 Régulation par le potassium extracellulaire 49
4.3. Autres modes de régulation hormonale 51
4.3.1. Par l'insuline 51
4.3.2 Par la vasopressine 52
4.4 Régulation de l'abondance protéique par CUL3 et KLHL3 52
4.4.1 Le complexe CUL3/KLHL3 53
4.4.2 WNK1 et WNK4 sont ubiquitinés par le complexe CUL3/KLHL3 55 4.4.3 Implication du complexe CUL3-ubiquitine ligase dans la régulation physiologique de
WNK1 et WNK4 57
4.4.3a Par le système rénine-angiotensine-aldostérone 57
4.4.3b En réponse à l'insuline et à la vasopressine 58
4.4.3c Dans les cellules musculaires lisses vasculaires 58
5. WNK1, WNK4, CUL3, KLHL3 ET PHYSIOPATHOLOGIE DE LA FHHt 59
5.1 Les apports de la FHHt dans la compréhension du paradoxe de l’aldostérone 59 5.2 Plusieurs gènes et mutations mais des conséquences communes ? 62 5.2.1 Les mutations de CUL3 perturbent l'activité ubiquitine ligase du complexe 63 5.2.2 Les mutations de KLHL3 empêchent le recrutement du substrat ou la liaison à
Cullin-3 65
5.2.3 Les mutations de WNK4 empêchent son interaction avec KLHL3 66 5.3.4 Les patients CUL3 souffrent d'une forme plus sévère de FHHt 70
6. CONCLUSIONS ET OBJECTIFS DU TRAVAIL DE THESE 71
PREMIERE PARTIE : Implication de la kinase SPAK dans l’Hypertension
Hyperkaliémique Familiale causée par les mutations introniques de WNK1 74
1. INTRODUCTION 75
1.1 Implication de SPAK dans la FHHt : état des connaissances 75 1.1.1 SPAK est impliqué dans la FHHt causée par les mutations de WNK4 75 1.1.2 Implication de SPAK dans la FHHt causée par les mutations WNK1? 76
1.2 Objectifs de l’étude 77
1.3 Résumé de l’article 77
2. ARTICLE : Consequences of SPAK inactivation on Hyperkalemic Hypertension caused by WNK1 mutations: evidence for differential roles of WNK1 and WNK4 78
3. CONCLUSIONS ET PERSPECTIVES 97
DEUXIEME PARTIE : Etude de la sévérité du phénotype causé par les mutations de Cullin-3 grâce à la caractérisation de deux modèles murins porteurs de la mutation
Cul3'9 100
1. INTRODUCTION 101
1.1 Cullin-3 régule RhoA dans les cellules musculaires lisses vasculaires 101 1.2 La mutation de Cul3 entraine un phénotype vasculaire 102
1.2 Résumé de l’article 103
2. ARTICLE : Cullin-3 mutations cause a more severe form of hypertension by
3. NOUVEAUX RESULTATS : L’inactivation de WNK4 prévient la FHHt causée par la
mutation Cul∆9 134
4. CONCLUSIONS ET PERSPECTIVES 137
TROISIEME PARTIE : 140
Caractérisation d’un modèle murin porteur d’une nouvelle mutation de WNK1
responsable d’une forme particulière d’Hypertension Hyperkaliémique Familiale 140
1. RESUME DE L’ARTICLE 141
2. ARTICLE : Mutations in the acidic motif of WNK1 cause normotensive hyperkalemic
hyperchloremic acidosis 143
3. CONCLUSIONS ET PERSPECTIVES 195
DISCUSSION GENERALE 198
L-WNK1 et WNK4 : même "combat" ? 199
Quels sont les mécanismes responsables de l'hyperkaliémie dans la FHHt ? 201 Inhibition de SPAK et/ou WNK1/WNK4 : une stratégie anti-hypertensive ? 203
REFERENCES BIBLIOGRAPHIQUES 206
Liste des abréviations
ADSN: néphron distal sensible à l’aldostérone AngII : angiotensine II
ARH : autosomal recessive hypercholesterolemia BKCa: Ca2+-dependent K+ channel
Cab39 : Ca2+-binding protein 39 CCD : coiled-coil domain CCD : canal collecteur cortical CI : cellule intercalaire
CP : cellule principale CNT : tubule connecteur CUL3 : cullin-3
cTAL : branche ascendante large corticale DCT : tubule contourné distal
ENaC : canal sodium épithélial
ERK : extracellular signal-regulated kinase FHHt : Hypertension Hyperkaliémique Familiale GRE : Elément de réponse aux glucocorticoïdes HCTZ : hydrochlorothiazide
HEK : human embryonic kidney
HSN2 : Hereditary Sensory Neuropathy type 2 HTA : hypertension artérielle
KCC: co-transporteur potassium-chlore KIR5.1: inwardly rectifying K+ channel KLHL3 : kelch-like 3
KS-WNK1 : kidney-specific WNK1 L-WNK1 : long-WNK1
MEK : MAP/extracellular signal regulated protein kinase (ERK) kinase MAPK : Mitogen-Activated Protein Kinase
NCC : co-transporteur sodium-chlore
NDCBE : sodium-driven chloride/bicarbonate exchanger
NKCC1 : co-transporteur Na, K, 2Cl de type 1 NKCC2 : co-transporteur Na, K, 2Cl de type 2 OMIM: online mendelian inheritance in man OSR1 : oxidative stress-responsive kinase-1 PAS : pression artérielle Systolique
PF2 : PASK/FRAY homology 2
PHA2 : Pseudohypoaldostéronisme de type 2
PPARJ : peroxisome proliferator-activated receptor-J RAAS : Système rénine-angiotensine-aldostérone ROMK : Renal outer medullary potassium channel SGK1 : serum-glucocorticoid regulated kinase 1 SNP : Single-nucleotide polymorphism
SPAK : sterile 20/SPS21-related proline/alanine-rich kinase SYT2 : Synaptotagmin 2
TGF-E : Transforming Growth Factor beta
TRPV5: transient receptor potential vanilloid 5 Ca2+ channel V2R : récepteur à la vasopressine de type II
WNK : With No lysine (K) HEK : human embryonic kidney MO25 : Mouse protein-25
INTRODUCTION GENERALE :
De l’hypertension Hyperkaliémique Familiale
à la découverte de régulateurs de la pression
1. REIN ET HYPERTENSION ARTERIELLE
1.1 Le rôle du sodium et potassium dans l’hypertension artérielle
essentielle
L’hypertension artérielle est une maladie complexe causée par différents facteurs génétiques et environnementaux. De nombreuses études ont montré qu’il existait une corrélation entre la consommation de sel et la prévalence de l’hypertension artérielle. Chez le chimpanzé, un régime riche en sel pendant 20 mois entraine une augmentation de la pression artérielle de 110 à 140mmHg. Ces valeurs reviennent à la normale dès lors que l'animal reçoit un régime normosodé (Denton et al., 1995). Chez l’Homme, cette corrélation est également retrouvée, et ce même dans les populations les plus disparates (Dahl, 2005). De la même façon, une restriction sodée entraîne une diminution de la pression artérielle, surtout chez des personnes hypertendues (2002). Depuis, de nombreuses études ont montré que ce n’était pas l’apport alimentaire en sodium seul qui influençait la pression artérielle, mais l’apport en NaCl (Schmidlin et al., 2005; Whitescarver et al., 1984).
Très récemment, une étude a recensé l’ensemble des analyses portant sur la consommation de potassium et la pression artérielle et a mis en évidence une association entre une consommation importante de potassium et une diminution de la pression artérielle (McDonough et al., 2017).
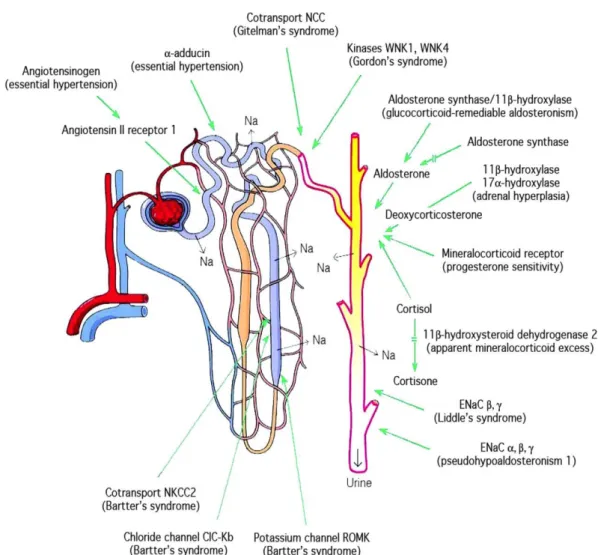
Dans des conditions environnementales normales, le maintien de la balance du sodium, du chlorure et du potassium se fait majoritairement par les urines. A la fin des années 60, Guyton et Coleman développent un modèle mathématique intégrant tous les facteurs pouvant avoir un impact sur la régulation de la pression artérielle et concluent que toutes les formes d’hypertension chroniques impliquent un défaut d’excrétion de sodium par le rein (Guyton and Coleman, 1968). L'étude de Guyton a depuis été confirmée par l'analyse génétique de formes rares et Mendéliennes qui affectent la pression artérielle (Lifton et al., 2001). Malgré la variété de systèmes physiologiques affectant la pression artérielle, les mécanismes moléculaires identifiés dans tous ces syndromes mendéliens sont une dérégulation des transporteurs ou canaux ioniques dans le néphron distal (Lifton et al., 2001). En d’autres termes, tous ces gènes codent des molécules qui sont capables de contrôler la capacité du rein à maintenir la balance sodée et potassique, ce qui confirme l’importance de ce système dans la régulation de la pression artérielle (Meneton et al., 2005) (Figure 1). Ces troubles mendéliens ont notamment permis de confirmer le rôle central qu’occupent le canal épithélial sodique
ENaC [Epithelial sodium (Na) Channel] et le co-transporteur Na+-Cl- NCC dans la régulation de la pression artérielle.
Figure 1. Mutations et polymorphismes affectant les niveaux de pression artérielle chez l’Homme.
La plupart des gènes a été mise en évidence par l’étude des formes mendéliennes d’hypertension et d’hypotension. La majorité de ces gènes sont impliqués directement ou indirectement dans la réabsorption du sel. D’après Meneton et al., β005.
1.2 Structure du néphron
Le rein est donc l’organe responsable de la régulation de la balance du sodium, du chlorure et du potassium et, en accord avec l’hypothèse de Guyton, nombres d’études montrent que le rein joue un rôle prépondérant dans la régulation de la pression artérielle. Il joue un rôle central dans l’homéostasie du milieu intérieur grâce à la filtration du plasma et à la sécrétion et/ou la réabsorption d’eau et de solutés ainsi que de la rénine. Chez l’Homme, le rein est compose d’environ un million de néphrons, son unité fonctionnelle. Chaque néphron est composé d’un glomérule qui assure l’ultrafiltration du plasma suivi d’un tubule rénal assurant
le transport des solutés et de l’eau. Chez l’Homme, jusqu’à 180 litres de plasma peuvent être ultrafiltrés chaque jour par les glomérules. L’ultrafiltrat s’écoule le long de la lumière du tubule rénal et sa composition est modifiée par des mécanismes de sécrétion et/ou de réabsorption d’eau et de solutés de façon à ce que soit excrétée dans l’urine la même quantité d’eau et d’ions que celle qui est ingérée quotidiennement. Les apports en eau et en sodium variant considérablement d’un individu a l’autre et d'un jour à l'autre, ces mécanismes de réabsorption et de sécrétion sont, de ce fait, finement régulés et contrôlés.
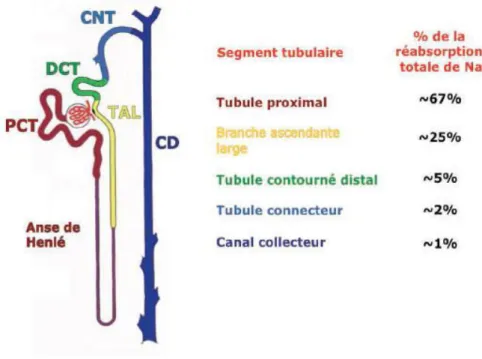
Le tubule rénal est une succession de segments tubulaires morphologiquement différents contenant un ou plusieurs types cellulaires et possédant des propriétés de transport et de régulation spécifiques. Les segments initiaux du néphron assurent la majeure partie de la réabsorption d’eau et de solutés. Le tubule proximal qui fait directement suite au glomérule assure la réabsorption massive d’eau et de Na+. A l’inverse, les segments terminaux, eux, sont l’objet de régulations permettant d’ajuster finement la balance entre les apports en eau et en Na+, Cl- et K+ et leur excrétion. Le néphron distal, qui comprend le tubule contourné distal (DCT), le tubule connecteur (CNT) et le canal collecteur (CD), assure l’ajustement final de l’excrétion de ces ions et d’eau (Figure β).
Figure 2. Représentation des différents segments tubulaires du néphron et leur réabsorption respective de sodium.
1.3 Les mécanismes de transport ionique le long du néphron distal
1.3.1 Les mécanismes du transport dans le DCT
Le tubule contourné distal (DCT) est localisé dans le cortex rénal et réabsorbe 5 à 10% du Na+ filtré. Chez la souris et le rat, le DCT est divisé en deux parties (DCT1 et DCT2), chacune exprimant différents transporteurs apicaux de Na+. Le DCT humain est quant à lui uniforme et présente le même profil que le DCT1 murin (Subramanya and Ellison, 2014) (Figure 3).
Dans la partie initiale du DCT (DCT1), le cotransporteur Na+-Cl- NCC localisé à la membrane apicale permet l’entrée de NaCl dans la cellule. La réabsorption de sodium via NCC est électroneutre et sensible aux diurétiques thiazidiques tels que l’hydrochlorothiazide (HCTZ). La partie terminale du DCT (DCT2) se caractérise par la présence à la fois du cotransporteur NCC et du canal sodique épithélial ENaC à la membrane apicale (Reilly and Ellison, 2000; Subramanya and Ellison, 2014). La réabsorption de sodium via ENaC est électrogénique et sensible à l’amiloride.
Dans l'ensemble du DCT, sont présents à la face basolatérale :
- la Na+/K+-ATPase qui maintient un sortant de sodium favorisant la réabsorption de sodium par le cotransporteur NCC ;
- le canal CLCK2/ClC-Kb, qui assure l'extrusion du chlorure en fonction du potentiel de membrane ;
- les canaux potassiques Kir4.1 (KCNJ10) et Kir5.1 (KCNJ16), qui forment un hétérodimère permettant le recyclage et la sortie de K+de la cellule vers l’interstitium.
Chez l’Homme, les mutations perte-de-fonction de NCC sont responsables du syndrome de Gitelman, une maladie autosomique récessive rare caractérisé par une hypotension, une hypokaliémie, une alcalose métabolique hypochlorémique, une hypomagnésémie et une hypocalciurie (Gitelman et al., 1966; Simon et al., 1996a). Plus récemment, deux équipes ont montré que des mutations perte-de-fonction du canal Kir4.1 sont responsables du syndrome EAST (Epilepsy Ataxia Sensorineural-deafness Tubulopathy), affectant plusieurs organes mais dont le phénotype rénal est proche de celui du syndrome de Gitelman (Bockenhauer et al., 2009; Scholl et al., 2009).
Si le récepteur aux minéralocorticoïdes (MR) est exprimé tout du long du DCT, seul le DCT2 est probablement sensible aux actions de l’aldostérone, qui se fixe et active ce récepteur. En effet, le DCTβ exprime l’enzyme 11-β hydroxysteroid dehydrogenase 2 (11-βHSD2), qui
métabolise le cortisol en cortisone, un métabolite inactif, empêchant ainsi la fixation des glucocorticoïdes circulants au MR (Bostanjoglo et al., 1998). Les récepteurs minéralocorticoïdes dans le DCTβ sont donc exclusivement occupés par l’aldostérone. Le DCT2 appartient ainsi au néphron distal sensible à l'aldostérone (ASDN), avec le tubule connecteur (CNT) et le canal collecteur cortical (CCD). L’angiotensine II (AngII), un autre acteur du système Rénine Angiotensine Aldostérone (RAAS), sécrété en situation d’hypovolémie est également capable de réguler le transport de NaCl et d’activer NCC dans le DCT. Je reviendrai en détail sur la régulation de l'activité du néphron distal par l'angII et
l'aldostérone dans la partie 4.
Figure 3. Modèle cellulaire du transport de sodium, chlorure et potassium dans les cellules du DCT1.
1.3.2 Les mécanismes de transport dans le CNT et le CD
A l’inverse du DCT, les épithélia du CNT et du CD sont composés de deux types cellulaires, les cellules principales (CP) et les cellules intercalaires (CI), avec des fonctions différentes et spécifiques.
1.3.2a Les mécanismes de transport dans les CP
Les CP représentent environ 60% des cellules du CNT/CD et expriment ENaC à leur membrane apicale. La réabsorption de Na+ à travers les CP est électrogénique : elle est couplée à la sécrétion de K+ via le canal potassique ROMK (Renal Outer Medullary-K) (Frindt and Palmer, 1989). L’importance fonctionnelle de ROMK est illustrée par le fait que les mutations pertes de fonction du canal sont responsables du syndrome de Bartter
caractérisé par une perte de sel, une alcalose hypokaliémique, une hypercalciurie et une diminution de la pression artérielle (Simon et al., 1996b). Comme NCC, l’activité des canaux ROMK et de ENaC est directement liée à l’activité de la pompe Na+/K+-ATPase située au pôle basolatéral.
Les mutations gain-de-fonction de ENaC causent le syndrome de Liddle qui associe une sévère hypertension et une hypokaliémie. Ces mutations induisent une augmentation du nombre de canaux présents à la membrane apicale des CP, ce qui entraîne une réabsorption de Na+ dans le CNT et le CD, couplée à une sécrétion excessive de K+ par ROMK (Warnock, 2001). A l’inverse, une perte de fonction de la sous-unité D d’ENaC résulte en un pseudo-hypoaldosteronisme de type I, caractérisé par une perte de Na+ urinaire, une diminution de la pression artérielle et une hyperkaliémie.
Pour finir, les cellules principales réabsorbent de l’eau par les canaux à eau AQPβ (Aquaporine 2) exprimés au pôle apical (Fushimi et al., 1993), et AQP3 et AQP4 présents au pôle basolateral (Ecelbarger et al., 1995; Terris et al., 1996). Chez l’Homme, des mutations perte-de-fonction du gène AQP2 sont responsables de l’apparition de diabète insipide néphrogénique (Mulders et al., 1997).
Le transport de NaCl dans les CP est principalement sous contrôle de l’aldostérone et de l’AngII, qui stimulent la réabsorption de NaCl dans les CP. Ce signal permet en effet d’augmenter dans un temps très court le nombre de canaux ENaC à la membrane apicale des cellules ainsi que de la Na+/ K+-ATPase à la membrane basolatérale (Loffing et al., 2001; Mamenko et al., 2012). L’action de l’aldostérone régule également l’expression des gènes codant la sous-unité D du canal ENaC et la sous-unité D1 de la Na+/K+-ATPase (Féraille and Doucet, 2001).
Figure 4. Modèle cellulaire du transport de sodium et de potassium dans les cellules principales.
1.3.2b Les mécanismes de transport dans les CI
Les CI peuvent être de trois types: type A (A-CI ou D-CI), type B (B-CI ou E-CI) ou non-A non-B (Teng-umnuay et al., 1996). Les cellules intercalaires sont traditionnellement impliquées dans la régulation de la balance acide/base. Néanmoins, des résultats récents ont montré que les E-CIs jouent également un rôle dans la réabsorption de Na+ et Cl-.
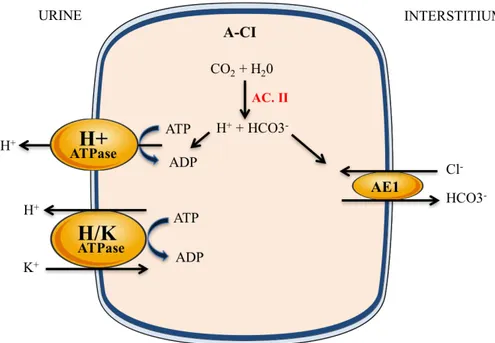
Les D-CI sécrètent des protons dans la lumière tubulaire et expriment l’anhydrase carbonique (AC). Les AC II et IV sont prédominantes dans le rein chez l’homme et le rongeur (Purkerson and Schwartz, 2007). Dans les D-CI, l’ACII génère un ion bicarbonate et un proton à partir de CO2 et d’eau. Les protons générés par la réaction de l’ACII sont transportés à travers la
membrane apicale dans la lumière tubulaire via la pompe à protons H+-ATPase tandis que les ions bicarbonates sont transportés à travers la membrane basolatérale via l’échangeur chlore/bicarbonate AE1 (Figure 5). Une acidose métabolique entraine une augmentation de la sécrétion de protons par la H+-ATPase, limitant ainsi l’acidose (Bastani et al., 1991). Les mutations du gène SLC4A1, qui code AE1, et les mutations inactivatrices des sous-unités de la H+-ATPase sont responsables d’une acidose tubulaire rénale caractérisée par un défaut d’excrétion des protons ou une perte de bicarbonates.,
Figure 5. Modèle cellulaire du transport de sodium, potassium, chlore et bicarbonates dans les cellules intercalaires de type A.
Les ß-CI présentent une polarité opposée à celle des cellules α-CI avec l’échangeur Cl -/HCO3- localisé du coté apical et la pompe à proton H+-ATPase du côté basolateral. L’échangeur Ae4 exprimé à la membrane basolatérale des E-CI (Hentschke et al., 2009) assure le transport du sodium et du bicarbonate à travers la membrane basolatérale de la cellule vers l’interstitium (Chambrey et al., 2013)
Les E-CI sont connues pour sécréter du bicarbonate en réponse à l’alcalose métabolique. Cette sécrétion est assurée par la pendrine, un échangeur Cl-/HCO3- apical. L’expression de la pendrine est régulée par les modifications de l’état acido-basique. Lors d’une acidose métabolique ou respiratoire, son expression diminue (Seigneux et al., 2007). A l’inverse, une charge alcaline entraine une augmentation de son expression (Wagner et al., 2002). Leviel et
al. ont montré en 2010 que E-CIs réabsorbent également du sodium et du chlorure de façon électroneutre et sensible aux diurétiques thiazidiques. Le chlorure est réabsorbé dans les E-CI grâce à la pendrine, tandis que le sodium est réabsorbé par NDCBE (aussi appelé SCL4A8), un cotransporteur Cl-/HCO3- dépendant du Na+ (Leviel et al., 2010) (Figure 6). L’inactivation du gène codant la pendrine abolit l’absorption transépithéliale de chlorure dans des CCD isolés et microperfusés (Wall et al., 2004). A l’inverse, la réabsorption de chlorure augmente dans le canal collecteur de souris surexprimant la pendrine dans les cellules intercalaires (Jacques et al., 2013). La réabsorption de NaCl par le système NDCBE/pendrine est dépendante de la pompe H+-ATPase basolatérale (Chambrey et al., 2013). Comme NCC, le cotransporteur NDCBE est une cible des diurétiques thiazidiques tels que l’hydrochlorothiazide (HCTZ). De par ce couplage, la pendrine joue un rôle non seulement dans la régulation de l'équilibre acido-basique mais également dans celle de la pression artérielle. L’ablation génétique de la pendrine chez la souris entraine une diminution de la pression artérielle (Trepiccione et al., 2017). Bien que la pendrine joue un rôle essentiel dans le rein, les mutations inactivatrices de son gène sont responsables d’une pathologie associant troubles de la thyroïde et de l’oreille interne sans phénotype rénal particulier (Maciaszczyk and Lewiński, β008).
Dans les ß-CI, l’expression de la pendrine et de la H+-ATPase apicale est stimulée par l'angiotensine II et l'aldostérone. Cependant, un régime riche en potassium cause l’effet inverse malgré des niveaux élevés d’aldostérone. L’identification récente de la phosphorylation du récepteur minéralocorticoïde sur le résidu Ser843empêchant l’aldostérone de se lier à son récepteur dans les ß-CI a fourni un début d’explication (Shibata et al., 2013a). En situation de déplétion volémique, l’angiotensine II favorise la déphosphorylation du
récepteur, permettant ainsi à l’aldostérone de se fixer sur son récepteur et de stimuler l’expression de la pendrine et de la H+-ATPase (Shibata et al., 2013a). A l’inverse, un régime riche en potassium favorise la phosphorylation du récepteur et empêche l’action de l’aldostérone via son récepteur. Jusqu’à maintenant, la phosphorylation MRS843-P n’a été retrouvée que dans les ß-CI et permet d’expliquer comment l’aldostérone est capable de produire deux réponses distinctes à l’hypovolémie et à l’hyperkaliémie dans ces cellules (Shibata et al., 2013a).
Figure 6. Modèle cellulaire du transport de sodium, chlore et bicarbonates dans les cellules intercalaires de type B.
L’ensemble de ces observations montre que l’aldostérone joue un rôle important dans la régulation de la réabsorption du NaCl et de la sécrétion de K+ dans le néphron distal. L’hypovolémie et l’hyperkaliémie sont les deux stimuli de la sécrétion d’aldostérone. Pour maintenir l’homéostasie, l’aldostérone doit être capable soit de maximiser la réabsorption de Na+ sans modifier la sécrétion de K+pendant l’hypovolémie, soit de maximiser la sécrétion de K+ sans modifier la réabsorption de Na+en réponse à l’hyperkaliémie. Le fait qu’une seule et même hormone puisse avoir des effets différents sur le transport de Na+ et K+ en fonction de la situation physiologique a été nommé le « paradoxe de l’aldostérone ».
L’étude de l’Hypertension Hyperkaliémique Familiale (FHHt) s’est révélée particulièrement pertinente à la compréhension du paradoxe de l’aldostérone, puisque chez les patients FHHt la réabsorption accrue de Na+ est associée à une inhibition de la sécrétion de K+, entrainant hypertension et hyperkaliémie (Kahle et al., 2003; Wilson et al., 2003). La compréhension des mécanismes impliqués dans la FHHt a permis de mieux comprendre comment la sécrétion de
potassium et la réabsorption de sodium dans le néphron distal sont découplées en réponse à l’aldostérone.
2. DE L'HYPERTENSION HYPERKALIEMIQUE FAMILIALE À LA
DÉCOUVERTE D'UNE NOUVELLE VOIE DE RÉGULATION DU
TRANSPORT IONIQUE DANS LE NÉPHRON DISTAL
2.1 Aspects cliniques de l'Hypertension Hyperkalémique Familiale
(FHHt)
L’Hypertension Hyperkaliémique Familiale (FHHt), également appelée Syndrome de Gordon ou Pseudohypoaldostéronisme de type II (PHA2), est une forme très rare d’hypertension artérielle transmise majoritairement selon un mode autosomique dominant. Cette pathologie est caractérisée par une élévation de la pression artérielle et des anomalies métaboliques, à savoir une hyperkaliémie, une acidose métabolique hyperchlorémique et une hypercalciurie. L'ensemble de ces symptômes est intégralement corrigé par l’administration de diurétiques thiazidiques (Gordon and Hodsman, 1986). En France, on dénombre 100 cas authentiquement diagnostiqués. Gordon et ses collègues ont rapporté leur premier cas en 1970 (Gordon et al., 1970) mais les caractéristiques cliniques et biologiques de la maladie ont été décrites pour la première fois en 1964 (Paver and Pauline, 1964). Il s’agit du cas d’un jeune homme de 15 ans souffrant d’une hypertension sévère (180/1β0 mmHg) associée à une hyperkaliémie majeure (8,2 mEq/L), une acidose avec une fonction rénale normale et une excrétion tubulaire de potassium altérée. La description de son cas a été complétée quelques années plus tard (Arnold and Healy, 1969). Son aldostéronémie était normale avec une aldostérone urinaire légèrement élevée. L’administration de diurétiques thiazidiques a permis la normalisation de l’hypertension artérielle et de l’hyperkaliémie. Cette sensibilité aux diurétiques thiazidiques a par ailleurs été rapportée chez d’autres sujets (Stokes et al., 1968). Le terme de « pseudohypoaldostéronisme de type 2 » sera proposé en 1981 (Schambelan et al., 1981), en raison de l’hyperkaliémie résistante aux minéralocorticoïdes, et par opposition au pseudohypoaldostéronisme de type 1 dans lequel l’hyperkaliémie est associée à une perte de sel et une hypotension. Cette dénomination contestée (Gordon, 1986) reste encore fréquemment utilisée de nos jours.
Parmi les nombreux cas décrits dans la littérature, il existe une grande variabilité d'âge de découverte de la maladie et de phénotypes (Gordon, 1995; Achard et al., 2001). Deux jeunes
filles ont développé la maladie durant leurs deux premières semaines de vie (Gereda et al., 1996). Des détections du syndrome chez des sujets plus âgés ont également été rapportées. Brautbar et al. rapportent le cas un homme de 52 ans souffrant d'hypertension et d’hyperkaliémie (Brautbar et al., 1978). Son identification a d’ailleurs permis de diagnostiquer quatre autres membres atteints de la même famille (Brautbar et al., 1978). Grâce à l’étude de ces différents cas, la grande sensibilité aux diurétiques thiazidiques a été identifiée comme étant une caractéristique de la pathologie (Lee et al., 1979; Mayan et al., 2005). Le traitement par un diurétique de l’anse de Henlé, le furosémide, a également été rapporté comme efficace chez plusieurs groupes de patients (Lee et al., 1979; Achard et al., 2001). L’ensemble de ces observations a laissé penser que la FHHt pouvait être due à une dérégulation fonctionnelle des voies de signalisation de l’homéostasie des ions au niveau du rein, et plus précisément du néphron distal.
L’hypothèse physiopathologique d’une hyperactivation d’un co-transporteur sodium-chlore comme anomalie primitive de la maladie est clairement énoncée en 1991 (Take et al., 1991). Les auteurs soumettent un père et ses enfants atteints à des explorations fonctionnelles, dont la perfusion de chlorure de sodium, de sulfate de sodium et de bicarbonate de sodium. L’excrétion urinaire de potassium augmente dans tous les cas chez les sujets contrôles et seulement dans les deux derniers cas chez les patients, c'est-à-dire qu’elle n’augmente pas en cas de perfusion de chlorure de sodium. De plus, un traitement avec de l’hydrochlorothiazide à la dose de β5mg/j pendant γ semaines corrige entièrement les anomalies. L’hypothèse est donc que les patients souffrent d’une réabsorption augmentée de sodium et de chlorure dans le segment tubulaire sensible aux thiazidiques, ce qui entraîne un apport diminué de sodium dans les tubules en aval et donc une moindre excrétion potassique, d’où l’hyperkaliémie.
2.2 Quatre nouveaux gènes pour une pathologie très rare
Les diurétiques thiazidiques utilisés pour contrôler les symptômes de la FHHt agissent en bloquant la réabsorption de sodium et de chlorure par le co-transporteur sodium-chlorure NCC dans le rein. Ce dernier point, associé au fait que cette maladie se comporte comme le miroir du syndrome de Gitelman, pathologie autosomique récessive résultant de mutations inactivatrices du gène SLC12A3 (Simon et al., 1996a) codant NCC, suggère fortement que la FHHt est une anomalie du tubule distal rénal. Cependant, aucune mutation de la séquence codante de SLC12A3 n’a été identifiée chez les patients FHHt. Trois loci différents associés à la FHHt ont initialement été localisés sur les chromosomes 1q31-42, 12p13 et 17p11-q21 (Mansfield et al., 1997; Disse-Nicodème et al., 2000). L’étude de trois nouvelles familles et le
fait que deux d’entre elles n’étaient liées à aucun des trois loci précédemment identifiés a suggéré une hétérogénéité génétique encore plus grande, avec au moins un quatrième locus (Disse-Nicodeme et al., 2001).
Une collaboration entre les équipes du Pr. Lifton et du Pr X. Jeunemaitre a permis de découvrir que les deux gènes sur les chromosomes 12 et 17 sont respectivement WNK1 et WNK4 (Wilson et al., 2001). Ces gènes codent des membres d’une famille de sérine-thréonine kinases, les With No lysine (K) kinases, composée de quatre membres WNK1, WNK2, WNK3 et WNK4.
Cependant, les mutations retrouvées dans les gènes WNK1 et WNK4 n’expliquaient qu’une partie minoritaire des cas décrits (6,5% de la cohorte française). Un ou plusieurs autres gène(s) devai(en)t donc être responsable(s) de cette pathologie. En 2012, deux équipes ont utilisé les outils modernes de la génétique humaine, en particulier des études de séquençage d’exome entier, à la recherche de mutations codantes dans la FHHt. Ces recherches ont abouti à la découverte simultanée par les deux équipes de mutations dans les gènes KLHL3 (Kelch-like 3) et Cullin-3 (CUL3) responsables de la FHHt (Boyden et al., 2012; Louis-Dit-Picard et al., 2012). Depuis, il a été établi que 29% des patients FHHt sont porteurs de mutations WNK1 ou WNK4 et 47% des patients de mutations KLHL3 ou CUL3.
L’implication de WNK1, WNK4, CULγ et KLHLγ dans la régulation du transport ionique rénal et de la pression artérielle était jusqu’alors insoupçonnée. WNK1 est identifiée en β000 (Xu et al., 2000) par clonage à partir d’une banque d’ADNc de cerveau de rat avec des sondes présentant des séquences communes aux membres de la famille des MEK (MAP/extracellular signal regulated protein kinase (ERK) kinase) dans le but d’identifier de nouveaux régulateurs des MAP kinases. La kinase WNK1 humaine est ensuite isolée à partir de lignées cellulaires humaines de cancer colorectal (Verissimo and Jordan, 2001). L’identification initiale de WNK1 a très rapidement été suivie par l’identification de trois membres supplémentaires de la famille : WNK2, WNK3 et WNK4 (Verissimo and Jordan, 2001). Le gène CUL3 code la cullin3 (CUL3). CUL3 appartient à la famille des Cullins composée de 7 protéines (CUL1, -2, -3, -4a, -4b, -5 et -7) impliquées dans une multitude de processus biologiques comme le contrôle du cycle cellulaire, la réplication de l’ADN et le développement (Petroski and Deshaies, 2005). Toutes ces protéines sont impliquées dans l’ubiquitination des protéines intracellulaires de par leur capacité à former des complexes ubiquitine-ligases (Genschik et al., 2013). Protéines d'échafaudage, les cullins permettent la mise en contact du substrat avec l’enzyme ubiquitine-ligase E2. En ce qui concerne les rôles connus de CUL3 lors de son
implication dans la FHHt, des études avaient notamment démontré son implication dans l’activation de caspases lors de la différentiation spermatique chez la Drosophile. KLHL3fait partie de la famille des protéines Kelch-like composée de 49 membres (Dhanoa et al., 2013) Ces protéines sont les homologues de la protéine Kelch de Drosophile melanogaster. Kelch participe à la mise en place de l’ovogénèse chez la Drosophile et fonctionne comme un stabilisateur en interconnectant les filaments d’actine entre eux (Kelso et al., 2002; Tilney et al., 1996). Lors de la découverte des mutations chez les patients atteints de FHHt, peu de chose sont connues à propos de KLHL3. Les protéines KLHL jouent le rôle d’adaptateur de substrat, qui recrute les protéines afin qu’elles soient ubiquitinées par le complexe ubiquitine ligase E3. Je décrirai les mutations identifiées chez les patients atteints de FHHt et leurs conséquences un peu plus loin dans le manuscrit.
2.3 Gènes impliqués dans la FHHt et hypertension artérielle
essentielle
WNK1, WNK4, CUL3 et KLHL3 étant mutés dans une forme rare et mendélienne d’hypertension, plusieurs équipes ont fait l’hypothèse que des variants génétiques affectant ces gènes et leurs voies de signalisation pourraient être à l’origine de formes plus communes d’hypertension artérielle essentielle.
Kokubo et al ont montré l’association entre une augmentation de la pression systolique et un polymorphisme dans un des introns de WNK4 dans une population japonaise (Kokubo et al., 2004). Un autre polymorphisme localisé dans l’intron 10 de WNK4 a été associé à une hypertension dans une population blanche américaine (Erlich et al., 2003). Cependant, cette association n’a pas été retrouvée dans une population anglo-australienne (Speirs and Morris, 2004)
La recherche d’association entre des variants de WNK1 et l’hypertension artérielle essentielle chez des patients de la cohorte BRIGHT (British Genetics of Hypertension) a permis d’identifier le polymorphisme rs1468γβ6 comme étant associé à la sévérité de l’hypertension artérielle (Newhouse et al., 2005). Ce polymorphisme, localisé à proximité du promoteur de WNK1, est également associé au à un risque d’hypertension artérielle au sein de deux populations chinoises indépendantes (Han et al., 2011). D’autres analyses menées sur la cohorte BRIGHT ont montré que le polymorphisme rs765β50, localisé dans l’intron 1, est associé à une augmentation de la pression artérielle systolique (PAS), à la pression artérielle diastolique et à l’hypertension artérielle. Ce variant a été retrouvé dans 6 populations
indépendantes (Newhouse et al., 2009). Il est également fortement associé au niveau de PAS nocturne dans une étude conduite chez des familles caucasiennes européennes (Tobin et al., 2005). D’autres polymorphismes de WNK1 ont été associés au niveau de réponse à un traitement par hydrochlorothiazide chez des populations africaines et caucasiennes non hispaniques (Turner et al., 2005), ou encore à l’influence du régime alimentaire sur le niveau de pression artérielle dans une population japonaise d’hommes sains (Osada et al., 2009). Enfin, différents variants et haplotypes de WNK1 ont été associés à des variations de pression artérielle, d’hypertension artérielle essentielle et d’excrétion urinaire de potassium (Newhouse et al., 2009). La majorité des études suggère donc que des variants communs de WNK1 interviennent dans la régulation de la pression artérielle au sein de la population générale.
Les tests d’association entre 794 SNPs présents au locus KLHL3 et la pression artérielle n’ont montré aucune association significative avec la pression artérielle systolique ou diastolique ou les deux combinées. Les variations communes dans KLHL3 ne semblent donc pas avoir d’impact sur la régulation de la pression artérielle au sein de populations saines (Louis-Dit-Picard et al., 2012).
Une seule étude suggère que le polymorphisme rs17479770 du gène CUL3 pourrait être un facteur protecteur dans la pathogénèse de l’hypertension artérielle essentielle. En effet, la présence de ce variant est associée à une diminution du risque d’hypertension artérielle essentielle dans une population chinoise (Li et al., 2017).
2.4 Les WNK, des kinases particulières
2.4.1. Structure
Les With No lysine (K) kinases WNK doivent leur nom au fait que la lysine présente au sein du sous-domaine catalytique II de la grande majorité des kinases est remplacée par une cystéine. La lysine catalytique (lysine 233) est déplacée dans le sous-domaine I (Xu et al., 2000). Cette lysine remplace une glycine dans la chaîne de glycines du sous-domaine I et est indispensable à l’activité kinase de WNK1 (Xu et al., 2000).
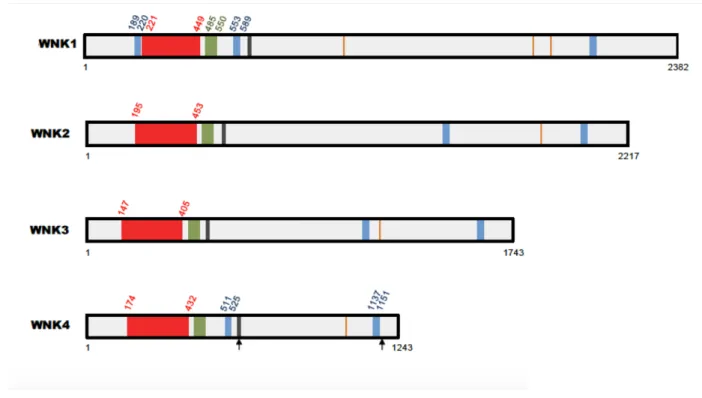
Le domaine kinase est la région la plus hautement conservée entre les membres de la famille WNK (Figure 7). In vitro, le domaine kinase de WNK1 est capable d’autophosphorylation et de phosphoryler un substrat exogène (Xu et al., 2000). L’activité kinase de WNK1 dépend de la phosphorylation de deux sérines, les sérines 378 et 382 (Xu et al., 2002). La mutation ponctuelle de la sérine γ78 entraîne une diminution de 50% de l’activité kinase. La mutation
ponctuelle de la sérine γ8β ou des deux résidus entraîne la perte de la totalité de l’activité kinase (Zagórska et al., 2007).
Les protéines WNK possèdent deux à trois domaines coiled-coil (CCD) et trois domaines riches en proline (Verissimo and Jordan, 2001) (Figure 7). Les domaines coiled-coil jouent un rôle majeur dans les interactions protéiques (Woolfson, 2005). Dans le cas des WNK, le domaine coiled-coil C-terminal permet la formation d'hétéro- ou d'homo-mères de WNKs, requise pour l'activité de ces kinases (Thastrup et al., 2012). Il est important de noter que les WNKs ne semblent pas être présents sous forme de monomères dans la cellule mais uniquement sous forme de complexes de haut poids moléculaire (Thastrup et al., 2012). L'étude de la conservation de la séquence de ce CCD entre les quatre WNKs et entre espèces, suivie d'une "alanine-scan" a permis de mettre en évidence un petit motif HQ (HIQEVVSLQT) essentiel pour la formation de ces complexes. L’analyse de la séquence de WNK1 révèle également des motifs RFXV permettant l’interaction de WNK1 avec ses substrats et notamment avec la kinase SPAK (Verissimo and Jordan, 2001; Vitari et al., 2006). Ces motifs sont présents dans WNK2, WNK3 et WNK4.
Les kinases WNK possèdent également un domaine très conservé appelé « motif acide » (Figure 7). On parle de motif acide en raison des 5 résidus acides localisés dans une petite portion de 10 acides aminés (Holden et al., 2004; McCormick and Ellison, 2011). La plupart des mutations de WNK4 responsables de la FHHt est localisée dans ce motif et nous verrons plus loin dans le manuscrit qu'elles affectent l'interaction de WNK4 avec KLHL3.
Les kinases WNK possèdent également un domaine initialement identifié comme étant un domaine auto-inhibiteur de l’activité kinase (WNK1-AI) (Xu et al., 2000). Depuis, une recherche NCBI-BLAST a identifié le domaine WNK1-AI comme étant homologue au domaine PF2 (PASK/FRAY homology 2) de SPAK et OSR1. Le domaine OSR-PF2 est capable de se lier aux motifs RFXV et de la même façon, le domaine WNK1-AI interagit avec les peptides contenant les motifs RFXV (Moon et al., 2013). Cela suggère que le domaine WNK1-AI n’est pas un domaine auto-inhibiteur, mais qu’il intervient dans les interactions protéines-protéines.
Figure 7. Représentations schématiques des kinases WNK1-2-3-4.
Toutes les kinases WNK ont en commun un domaine kinase (en rouge), deux à trois domaines coiled-coil (en bleu), un motif acide (en noir), le domaine WNK1-AI (en vert) et des motifs RFXV de liaison à SPAK (orange). Les flèches indiquent les deux sites des mutations FHHt de WNK4. Les localisations des domaines et les longueurs des protéines sont données pour les séquences humaines. Illustration tirée du manuscrit de thèse du Docteur Emmanuelle Vidal-Petiot.
2.4.2. Profil d'expression
WNK2 est le seul membre de la famille WNK à ne pas être exprimé dans le rein (Delaloy et al., 2003). Il est principalement exprimé dans le cœur, le cerveau et le colon (Verissimo and Jordan, 2001). Les transcrits WNK3 sont détectés par RT-PCR dans l’intégralité des tissus étudiés chez l’Homme (rein, cerveau, poumon, placenta, foie et rate), excepté dans le muscle squelettique (Holden et al., 2004). WNK3 est particulièrement exprimé dans le système nerveux central (hippocampe, cervelet et cortex cérébral). On le retrouve également dans les cellules épithéliales du système digestif (estomac, intestin, pancréas, voies biliaires) (Kahle et al., 2005). Au niveau rénal, WNK3 est exprimé dans tous les segments du néphron, mais son niveau d’expression relatif dans chaque segment n’a pas été mesuré (Oi et al., 2012). Si WNK3 est capable d'activer fortement le co-transporteur NCC in vitro (Rinehart et al., 2005), ce rôle n'a pas été confirmé in vivo (Oi et al., 2012). Je n'en parlerai donc plus dans le reste du manuscrit.
Chez l'Homme et la souris, WNK4 est exprimé dans de nombreux tissus (rein, testicules, cœur, foie, cerveau, poumon, rate et colon), mais pas dans le muscle squelettique. WNK4 est également retrouvée au niveau des jonctions serrées et des membranes latérales des épithélia polarisés impliqués dans le transport du chlorure tels que le colon, les glandes sudoripares, les glandes salivaires ou la peau (Choate et al., 2003; Kahle et al., 2004). L’immunohistochimie a également permis de mettre en évidence WNK4 au niveau de l’endothélium de la barrière hémato-encéphalique (Kahle et al., 2004). Au niveau rénal, WNK4 est exprimé dans le glomérule et dans l’ensemble du tubule avec une expression plus importante dans le DCT puis dans la branche ascendante de l’Anse de Henlé et le tubule connecteur (Ohno et al., 2011). De manière générale, les sites d’expression de WNK1 et WNK4 se superposent grandement, toutefois, le profil d'expression de WNK1 est plus complexe car ce gène possède plusieurs promoteurs et est soumis à des phénomènes d'épissage alternatif.
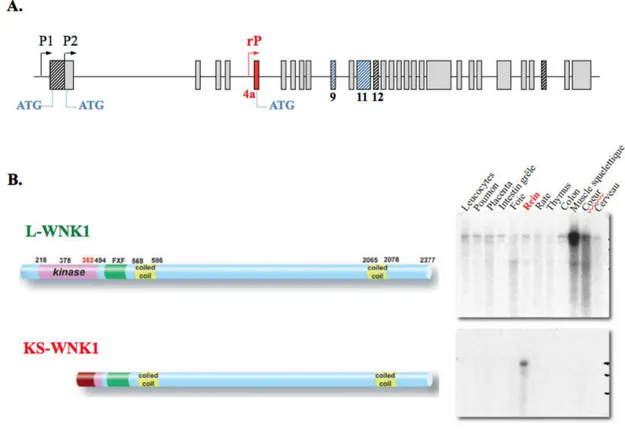
2.4.3 Le gène WNK1 donne naissance à plusieurs variants exprimés de façon tissu-spécifique.
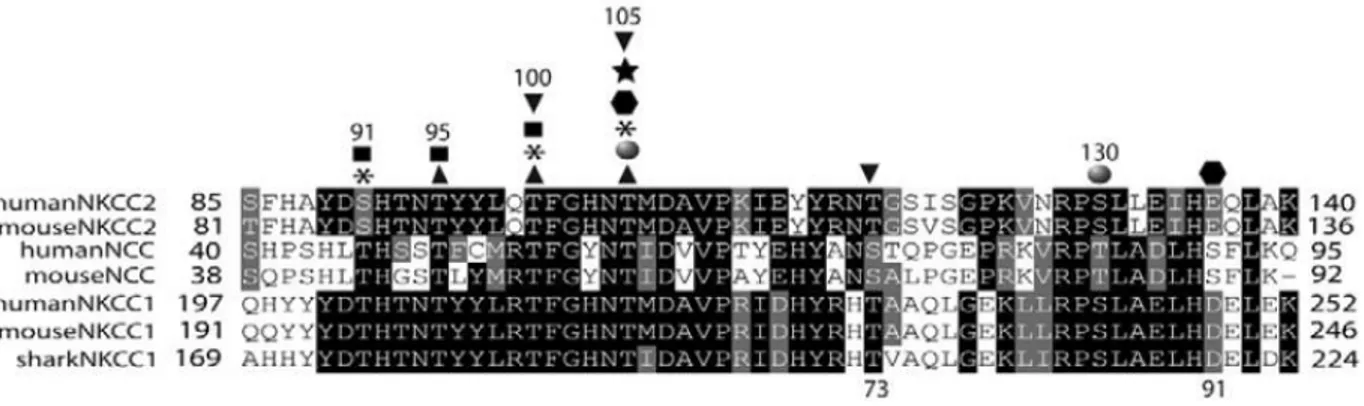
Deux promoteurs sont actifs dans le locus WNK1 (Delaloy et al., 2003; Vidal-Petiot et al., 2012). Le promoteur proximal (pP), situé en amont de l’exon 1, donne naissance à l’isoforme longue L-WNK1 (Long-WNK1), qui est exprimée de façon ubiquitaire. Le deuxième promoteur, rP (renal Promoter), est localisé dans l’intron 4. Il donne naissance à une isoforme dépourvue de la majorité du domaine kinase et exprimée uniquement dans le rein, d’où son nom, KS-WNK1 pour Kidney-Specific WNK1 (Delaloy et al., 2008). L-WNK1 et KS-WNK1 possèdent donc en commun le domaine auto-inhibiteur, les deux domaines coiled-coil (CC1 et CC2) et la portion distale du domaine kinase (Figure 8). Dans le rein, KS-WNK1 est exprimé très fortement dans le DCT et plus faiblement dans la branche large ascendante de l'anse de Henlé, le tubule connecteur et le canal collecteur cortical. L-WNK1 est détecté à faible niveau dans l’ensemble du néphron. Dans le DCT, l’expression de KS-WNK1 est 80 fois plus forte que celle de L-WNK1 (Vidal-Petiot et al., 2012). L'équipe de R. Brown a suggéré que les niveaux d’expression de L-WNK1 dans le néphron correspondraient à du bruit de fond (O’Reilly et al., β006).
Figure 8. Structure du gène WNK1 et de ses isoformes KS- et L-WNK1.
WNK1 possède deux promoteurs pP et rP qui donnent naissance à deux types d’isoformes : L-WNK1, l’isoforme longue qui possède l’intégralité du domaine kinase (rectangle rose) et qui s’exprime de manière ubiquitaire (résultat northern Blot), et l’isoforme KS-WNK1dépouvue du domaine kinase et qui est exprimée spécifiquement au niveau du rein (résultat northern Blot). Adapté de Hadchouel et al., 2006.
En plus des isoformes L-WNK1 et KS-WNK1 dont l'expression est contrôlée par des promoteurs alternatifs, WNK1 est soumis à des phénomènes d’épissage alternatif. 8 exons de WNK1, les exons 8b, HSN2, 9, 11, 12, 26, 26a et 26b, sont concernés. Les variants issus de ces épissages sont exprimés de manière ubiquitaire ou préférentiellement dans certains tissus (Figure 9).
L’étude de la neuropathie héréditaire sensitive et autosomique de type 2 (HSN2), une insensibilité congénitale à la douleur de transmission autosomique récessive, a permis la découverte des exon 8b et HSN2, localisés entre les exons 8 et 9 de WNK1 (Shekarabi et al., 2008). Cette maladie est causée par une mutation de l’exon HSNβ. Le variant contenant ces deux exons est exprimé de manière prépondérante dans le système nerveux périphérique, plus précisément dans la moelle épinière et les ganglions spinaux (Vidal-Petiot et al., 2012). Une faible expression est également détectée dans l'aorte, le rein et le système nerveux central (Vidal-Petiot et al., 2012).