Développement d’une thérapie génique pour l’Ataxie de
Friedreich en induisant l’expression du gène de la frataxine
avec les TALEs-FT
Mémoire
Khadija Cherif
Maîtrise en biologie cellulaire et moléculaire
Maître ès sciences (M.Sc.)
Québec, Canada
© Khadija Cherif, 2017
Développement d’une thérapie génique pour l’Ataxie de
Friedreich en induisant l’expression du gène de la frataxine
avec les TALEs-FT
Mémoire
Khadija Cherif
Sous la direction de :
iii
Résumé
L’ataxie de Friedreich (FRDA) est la plus fréquente des ataxies héréditaires autosomiques récessives. FRDA est due à une mutation du gène de la frataxine (FXN) situé sur le chromosome 9, q13. Cette mutation est une augmentation du nombre de répétitions du trinucléotide GAA au niveau du 1er intron du gène de la frataxine (FXN). Le nombre de trinucléotides augmente de moins 30 chez les sujets normaux jusqu’à 1300 chez les patients. Cela a pour conséquence de diminuer l’expression de la protéine frataxine, une protéine qui joue un rôle important dans le métabolisme du fer dans la mitochondrie. Mon projet traite de l’utilisation des protéines TALE-platinum (plTALE) fusionnées avec des systèmes permettant une stimulation de la transcription (FT), tel que VP64 ou P300. Ces plTALEs ciblent spécifiquement la région régulatrice du gène FXN pour augmenter sa transcription et ainsi induire une augmentation de l’expression de la protéine frataxine. Les plTALEs contiennent des variations au niveau des acides aminés 4 et 30 pour chaque RVD (Repeat Variable Di-residues) en plus des variations des acides aminés 12 et 13. Ces variations permettent d’augmenter la spécificité des plTALEs pour les séquences nucléotidiques ciblées. L’assemblage des RVD de plTALEs a été fait par 4-modules RVD ce qui permet d’obtenir un rendement d’assemblage de 100%.
Nous avons produit 34 effecteurs plTALE-FT qui ciblent 14 séquences du gène FXN afin, de sélectionner 3 plTALE-VP64 et 2 plTALE-SunTag10X qui peuvent augmenter la transcription et l’expression du gène FXN de 2 fois jusqu’à 19 fois dans les différentes cellules modèles FRDA. Nous avons quantifié le taux de synthèse des ARNm et de la protéine FXN après le traitement in vitro avec ces plTALEs-FT par qRT-PCR et westerns. Les résultats montrent que ces plTALES-FT sélectionnés induisent l’activité transcriptionnelle du gène endogène FXN, ainsi que l’expression de la protéine frataxine in vitro. L’augmentation de la frataxine augmente l’activité d’aconitase qui est modulée réversiblement par le niveau de la frataxine dans la mitochondrie. Nous avons utilisé un virus AAV9 pour livrer plTALE-FT dans les souris modèles de FRDA dans le but de valider l’efficacité de ces effecteurs in vivo avant de passer aux tests précliniques. Les résultats de ces traitements in vivo ne sont pas encore disponibles.
iv
Abstract
Friedreich's ataxia (FRDA) is the most frequent autosomal recessive hereditary ataxia. FRDA is due to a mutation of the frataxin gene (FXN) located on chromosome 9, q13. This mutation is an increase in the number of repetitions of the trinucleotide GAA in the 1st intron of the frataxine gene (FXN). The number of trinucleotides increases from less than 30 in normal subjects up to 1300 in patients. This decreases the expression of the protein frataxin, a protein which plays an important role in the metabolism of iron in the mitochondria.
My project deals with the use of TALE-platinum (plTALE) proteins fused with transcription-enhancing systems (FT), such as VP64 or P300. These plTALEs specifically target the regulatory region of the FXN gene to increase its transcription and thus induce an increase in the expression of the frataxin protein.
The plTALEs contain variations in amino acids 4 and 30 for each Repeat Variable Diresidues in addition to the variations of amino acids 12 and 13. These variations make it possible to increase the specificity of the plTALEs for the targeted nucleotide sequences. The assembly of the RVDs of plTALEs was done using modules containing 4 RVDs, which makes it possible to obtain an assembly efficiency of 100%.
We produced 34 plTALE-FT effectors that target 14 sequences of the FXN gene to select 3 plTALE-VP64 and 2 plTALE-SunTag10X, which increased FXN gene transcription and expression by up to 19-folds in different FRDA model cells. We quantified the synthesis of mRNAs and of FXN protein after in vitro treatment with these plTALEs-FT by qRT-PCR and westerns. The results show that these selected PlTAL-FT induced the transcription of the endogenous FXN gene as well as the expression of the frataxin protein in vitro. The increase in frataxin increased the aconitase activity, which is modulated reversibly by the level of frataxin in the mitochondria. We used an AAV9 virus to deliver plTALE-FT in FRDA model mice to validate the efficacy of these effectors in vivo before proceeding to preclinical testing. The results of these in vivo treatments are not yet available.
v
Table des matières
Résumé ... iii
Abstract... iv
Table des matières ...v
Liste des tableaux ... xi
Liste des figures ... xii
Liste des abréviations ... xv
Remerciements ... xviii
Chapitre 1 : La maladie de Friedreich ... 1
1.1 L’ataxie de Friedreich (FRDA)... 1
1.1.1 Introduction générale ... 1
1.1.2. Historique ... 1
1.1.3 Incidence ... 2
1.1.4 Cause... 3
1.2 FRDA et le stress oxydatif ... 3
1.3 Symptômes cliniques de FRDA... 4
1.3.1 Symptômes neurologiques ... 4
1.3.2 Symptômes cardiaques ... 5
1.3.3 Autres symptômes cliniques ... 5
1.4 Anatomopathologie ... 6
1.4.1 Neuropathologie (Fig. 4) ... 6
1.4.2 Hypertrophique cardiaque ... 8
1.5 Frataxine ... 8
1.5.1 Gène de la frataxine (FXN) ... 8
1.5.2 La protéine de la frataxine ... 9
1.5.3 La fonction de la frataxine ... 10
1.5.4 L’absence de la frataxine ... 11
1.6 Déficience moléculaire de la FRDA ... 11
1.6.1 Formation d’ADN collant (Sticky DNA) ... 12
1.6.2 Formation d’hybride RNA-DNA ... 12
1.6.3 Changement d’épigénétique ... 13
1.7 Diagnostic de FRDA ... 13
vi
1.7.2 Triplet-repeat (TP-PCR) ... 14
1.7.3 RT-PCR quantitatif en temps réel (qRT-PCR) ... 15
1.7.4 Southern Blot ... 15
1.8 Thérapie ... 15
1.8.1 Principe et le but des traitements de FRDA ... 15
1.8.2 Exemple des thérapies ... 16
1.8.2.1 Idébénone ... 16
1.8.2.2 Inhibiteurs des histones désacétylases (HDACi) ... 17
1.8.2.3 La protéine frataxine exogène... 17
1.8.2.4 Thérapie génique ... 18
1.9 Modèles animaux et cellulaires de FRDA ... 19
1.9.1 Modèle Animaux ... 20
1.9.1.1 Souris MCK et souris NSE ... 20
1.9.1.1.1 Souris MCK ... 20
1.9.1.1.2 Souris NSE ... 20
1.9.1.2 Souris knock-in ... 21
1.9.1.2.1 Souris KIKI et souris KIKO ... 21
1.9.1.2.2 Souris YG8R et souris YG22R ... 22
1.9.2 Modèle des cellules FRDA : fibroblastes, lymphoblastes et les
cellules iPSC dérivées de patients FRDA ... 23
Chapitre 2 : Thérapie génique... 25
2.1 Définition ... 25
2.2 Différents types de thérapie génique ... 25
2.2.1 Thérapie génique somatique ... 25
2.2.1.1 In vivo ... 25
2.2.1.2 Ex vivo ... 25
2.2.2 Thérapie génique germinale ... 26
2.3 Moyens de livraison du gène d’intérêt... 26
2.3.1 Les vecteurs... 26
2.3.2 Les vecteurs viraux ... 27
2.3.2.1 Adénovirus ... 27
2.3.2.2 Virus adéno-associé ... 28
2.3.2.3 Retroviridae ... 30
2.3.2.3.1 Gamma-rétroviral vecteurs ... 31
2.3.2.3.2 Les lentivirus ... 31
2.3.3 Les vecteurs non-viraux ... 32
2.3.3.1 Les vecteurs chimiques ... 32
vii
Chapitre 3 : Transcription effecteur activateur-like (TALE) ... 33
3.1 TALE/TALEN ... 33
3.2 La limite majore des effecteurs TALENs/TALEs ... 35
3.3 Construction des TALEs ... 35
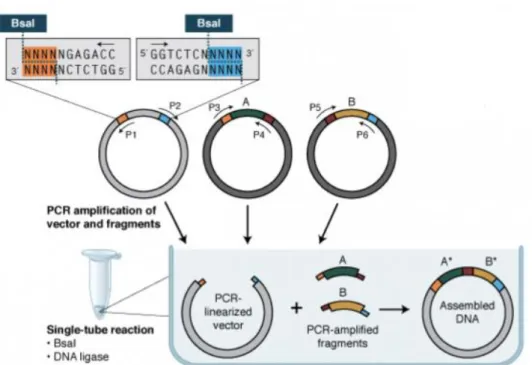
3.3.1 Réaction de Golden Gate ... 35
3.4 Platinum-TALE (plTALE) ... 37
3.5 Les facteurs de la transcription ... 38
3.4.1 VP64 ... 39
3.4.2 p300 ... 39
Chapitre 4 : Hypothèse et l’objectif ... 41
4.1 Hypothèse... 41
4.2 Objectif ... 42
Chapitre 5 : Matériels et méthodes ... 43
5.1 Platinum TALE-VP64 (plTALE-VP64) ... 43
5.1.1 Introduction ... 43
5.1.2 La construction plTALEs-VP64 ... 43
5.1.2.1 Première étape de la construction des plTALENs (pFUS2) . 44
5.1.2.2 La deuxième étape de la construction des plTALENs ... 45
5.1.2.3 La construction des plTALEs-VP64 ... 46
5.1.2.3.1 Le clonage In-fusion ... 47
5.1.2.3.2 La construction finale de plTALEs-VP64 ... 47
5.1.3 Test plTALEs-VP64 in vitro ... 48
5.1.3.1 Les cellules fibroblaste des patients FRDA ... 48
5.1.3.2 Nucléofection ... 48
5.1.3.3 qRT-PCR... 49
5.1.3.4 Western blot ... 49
5.2 Les vecteurs lentivirus ... 50
5.2.1 Introduction ... 50
5.2.2 La construction des pLenti-plTALEs-VP64... 51
5.2.3 La production purification et titrage des Lentivirus dans 293T .. 51
5.2.4 Traitement des cellules FRDA par les lentivirus exprimant
plTALEs-VP64 ... 52
5.3 La mutagenèse des RVD ... 53
5.3.1 Introduction ... 53
5.3.2 Mutagénèse des modules RVD ... 53
viii
5.3.4 Test nplTALEs-VP64 in vitro... 54
5.4 plTALEs-p300 ... 54
5.4.1 Introduction ... 54
5.4.2 La construction plTALEs-p300 ... 54
5.4.3 Test plTALEs-p300 in vitro... 55
5.5 plTALEs-SunTag (plTALE-ST) ... 55
5.5.1 Introduction ... 55
5.5.2 La construction plTALEs-ST10X et plTALEs-ST24X ... 56
5.5.2.1 La construction de plTALE-ST10X et de
pCR3.1-plTALEs-ST24X ... 56
5.5.2.2 La construction de scFV- SpGFP-Vp64-GB1-NLS (scFV) . 57
5.5.3 Test plTALEs-ST in vitro ... 57
5.6 pAAV-plTALEs ... 58
5.6.1 La construction des pAAV-plTALEs-FT ... 58
5.6.1.1 La construction pAAV-plTALEs-VP64 ... 59
5.6.1.2 La construction pAAV-plTALEs-ST10X ... 59
5.7 Test d’aconitase ... 60
Chapitre 6 : Résultats ... 61
6.1 L’induction du gène FXN in vitro dans les fibroblastes FRDA
avec plTALEs-VP64 ... 61
6.1.1 La détermination des séquences ciblées ... 61
6.1.2 La détermination des séquences des RVD ... 62
6.1.3 Les plTALEs-VP64 ... 63
6.1.3.1 La première étape de la construction des plTALENs ... 63
6.1.3.1.1 pFUS2-RVD (pour les plTALENs à 13 RVD) ... 63
6.1.3.1.2 pFUS2-RVD (pour les plTALENs à 15 RVD) ... 63
6.1.3.2 La deuxième étape des plTALENs ... 64
6.1.3.3 Les constructions pCR3.1-plTALEs-VP64 ... 65
6.1.3.3.1 Les construction ptCMV-plTALEN-VP64-XbaI ... 65
6.1.3.3.2 Les constructions pCR3.1-plTALEs-VP64 ... 66
6.1.4 L’induction du gène FXN in vitro par plTALEs-VP64 ... 68
6.1.4.1 La sélection des meilleurs plTALEs-VP64 ... 68
6.1.4.2 L’effet synergique des plTALEs-VP64 sur l’induction du
gène FXN des cellules FRDA4078... 71
6.1.4.3 L’induction de l’expression de la protéine frataxine par les
effecteurs plTALEs-VP64 ... 71
6.1.4.4 Traitement des cellules FRDA provenant de 4 patients avec
des plTALEs-Vp64 sélectionnés. ... 72
ix
6.1.4.5 L’induction de l’expression de la protéine frataxine par les
effecteurs plTALEs-VP64 dans des cellules provenant de 2 patients
FRDA ... 73
6.2 Les vecteurs lentiviraux comme outils pour l’expression
génétique des plTALEs-VP64 dans les cellules FRDA. ... 74
6.2.1 Les vecteurs pLenti-plTALE-VP64 ... 74
6.2.2 L’infection des cellules FRDA4078 avec les lentivirus qui
expriment plTALEs-VP64 ... 76
6.3 L’amélioration de la spécificité et de l’affinité des plTALEs avec
de nouvelles séquences nRVD (nplTALEs) ... 78
6.3.1 Les nplTALEs-VP64 ... 78
6.3.2 Des nplTALE-VP64 qui ciblent le promoteur du gène FXN pour
induire la transcription du gène ... 80
6.4 Le changement épigénomique du gène FXN avec
l’acétyltransférase p300 fusionné avec des plTALEs ... 81
6.4.1 Les plTALEs-p300 ... 81
6.4.2 L’induction du gène FXN par plTALE-p300 ... 82
6.5 L’induction du gène FXN avec plTALEs-SunTag qui recrute 10
ou 24 copies de VP64 ... 85
6.5.1 La construction plTALEs-SunTag10X et plTALE-SunTag24X 85
6.5.2 Traitement in vitro des cellules FRDA4078 par le système de
plTALE-ST ... 88
6.5.3 L’effet synergique des plTALEs-ST10X et plTALEs-ST24X sur
l’augmentation de l’activité du gène FXN ... 90
6.5.4 Activation du gène FXN endogène dans des fibroblastes de 4
patients FRDA avec plTALEs-ST (Fig. 70 et 71) ... 91
6.5.5 L’induction de l’expression de la protéine frataxine avec les
effecteurs plTALEs-VP64 dans les cellules provenant de 4 patients
FRDA (Fig. 72) ... 94
6.6 Les pAAV-plTALEs-VP64 et ST ... 95
6.6.1 Les pAAV-plTALEs-VP64 ... 95
6.6.2 Les pAAV-plTALEs-ST10X ... 96
6.6.3 Traitement des cellules FRDA4078 par pAAV-plTALEs-ST10X
... 96
6.7 L’augmentation de l’activité d’aconitase dans les cellules traitées
par les effecteurs plTALE-ST10X ... 99
x
Chapitre 8 : Conclusion et perspective ... 106
Bibliographie ... 107
xi
Liste des tableaux
Tableau 1: Historique de la découverte de la maladie de l'ataxie de Friedreich. ... 2 Tableau 2: les principaux signes de la maladie de FRDA. ... 4 Tableau 3: Liste des noms des plTALEs-VP64 avec la séquence des RVD qui sont
complémentaires aux nucléotides ciblés. ... 62
Tableau 4: Les codons des nouveaux et des anciens RVD avec leurs nucléotides cibles.
... 79
Tableau 5: La liste des nplTALE-VP64 avec leurs séquences des nRVD et leurs
séquences cibles ... 79
Tableau 6: La liste des plTALE-p300 avec leurs séquences RVD et leurs séquences
cibles. ... 82
Tableau 7: La liste des plTALEs-ST10X. ... 87 Tableau 8: La liste des plTALEs-ST24X. ... 87
xii
Liste des figures
Figure 1: Des signes classiques de FRDA:... 6
Figure 2: La coupe transversale de moelle épinière d’un patient FRDA atteint au niveau du cordon postérieur et le faisceau pyramidal (cordon latéral). ... 7
Figure 3: L’aspect des noyaux dentelés (DN) dans un patient FRDA. ... 7
Figure 4: L’anatomopathologie des cerveaux FRDA. ... 8
Figure 5: L’aspect brut du cœur dans les patients FRDA. ... 8
Figure 6: La représentation moléculaire du gène FXN (entre chaque 2 exons il y a un intron). ... 9
Figure 7: Les principaux sites régulateurs du gène FXN. ... 9
Figure 8: La localisation de la frataxine dans la mitochondrie et dans le cycle de la production d’énergie. ... 10
Figure 9: La dérégulation de la chaine respiratoire et le cycle de la production d’énergie en absence et en diminution de l’expression de la frataxine dans la mitochondrie. ... 11
Figure 10: Une présentation schématique de la forme d’ADN anormal dans le cas de FRDA. En haut l’ADN triplex et en bas l’ADN collant. ... 12
Figure 11: La formation d’hybride RNA-DNA dans le cas FRDA. ... 13
Figure 12: Schéma représentatif des différents produits d’amplification avec 3 amorce dans Triplet-repeat (TP-PCR). ... 15
Figure 13: Présentation de la structure d’euchromatine (gène actif) et de la forme l’hétérochromatine (gène silencieux). ... 17
Figure 14: Les domaines de Tat-FXN protéine de fusion. ... 18
Figure 15: Présentation de la structure moléculaire de complexe TALE-FT ... 19
Figure 16: Présentation schématique de la génération des souris MCK et des souris NSE. ... 20
Figure 17: Représentation schématique de la génération des souris KIKI et KIKO. .... 21
Figure 18: Représentation schématique de la génération des souris YG8R et les souris YG22R. ... 23
Figure 19: Présentation des différences majeures entre thérapie génique in vivo et ex vivo ... 26
Figure 20: La structure et l'organisation du génome du virus adéno-associé (AAV). ... 29
Figure 21: Cycle de vie de l’AAV. ... 29
Figure 22: Cycle de réplication des rétrovirus. ... 31
Figure 23: Les différentes parties des TALEs. ... 33
Figure 24: Les différents domaines liés après le N-ter des TALEs. ... 34
Figure 25: Deux paires des TALENs ciblent deux séquences spécifiques d’ADN. ... 34
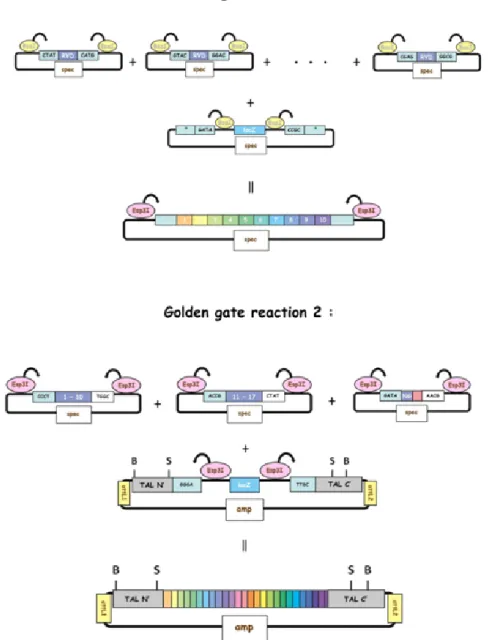
Figure 26: Les deux grandes étapes de l’assemblage des RVDs avec la réaction Golden Gate ... 36
Figure 27: L’assemblage plusieurs inserts dans un seul vecteur selon la réaction Golden Gate de NEB. ... 37
Figure 28: L’assemblage des RVDs Platinum-TALE (plTALE) en deux grande étapes. ... 38
Figure 29: Activation de la transcription après le recrutement du complexe transcriptionnel par le domaine d’activation fusionné avec TALE. ... 39
xiii
Figure 30: Le VP64 regroupe 4 séquences de VP16 (image du bas), pour activer la
transcription après le recrutement du complexe transcriptionel (image du haut)... 39
Figure 31: Le rôle de p300 dans l’activation du gène après l’acétylation et le
remodelage de la chromatine. ... 40
Figure 32: La présentation schématique pour les grandes étapes de la construction des
plTALEs-VP64 ... 44
Figure 33: Le protocole de la réaction d’assemblage des 4 modules des RVD d’étape 1
... 45
Figure 34: Le protocole de la réaction d’assemblage finale de l’ensemble des RVD dans
un vecteur ptCMV afin de construire plTALENs... 46
Figure 35: Le schéma de test in vitro des plTALEs-VP64 dans des cellules FRDA. .... 49 Figure 36: pAAV_TALE-TF(VP64) -BB_V3 ... 58 Figure 37: La localisation schématique des régions ciblées du promoteur et de l’intron 1
du gène FXN ... 61
Figure 38: Position des séquences ciblées. ... 62 Figure 39: PCR pour sélectionner les bonnes constructions de l’assemblage de 4 RVD
pour plTALENs contenant 13 RVD. ... 63
Figure 40: PCR pour sélectionner l’assemblage de 4 et 2 RVD pour plTALENs à 15
RVD. ... 64
Figure 41: PCR pour sélectionner l’assemblage final des plTALENs 13 et 15 RVD. .. 64 Figure 42: L’image du vecteur et de l’insert utilisés dans la réaction de clonage
In-fusion pour faire ptCMV-plTALENs-VP64-XbaI :... 65
Figure 43: Le produit de PCR de la sélection des clones positifs
plTALENs-VP64-XbaI. ... 66
Figure 44: La présentation de l’insert plTALE-VP64 et du vecteur pCR3.1 digéré par
MluI et XbaI pour faire la construction pCR3.1-plTALE-VP64... 66
Figure 45: Le produit de la digestion pour sélectionner les constructions positives ayant
pCR3.1-plTALE-VP64. ... 67
Figure 46: Les séquences des acides aminés de N_ter-15 RVD-C_ter-NLS-VP64 de la
construction plTALE-6-15 qui a été séquencée avec 4 amorces. ... 68
Figure 47: L’image représentative des fibroblastes FRDA4078 en culture, 24 h après la
nucéofection avec un plasmide contenant le gène GFP. ... 70
Figure 48: L’induction du gène FXN dans les cellules FRDA4078 par des
plTALEs-VP64. ... 70
Figure 49: L’induction de l’expression de la protéine frataxine dans les cellules
FRDA4078 ... 72
Figure 50: L’induction du gène FXN dans 4 différents types de cellules FRDA avec les
plTALEs-PV64-6-15, 8-15 et F4-15 par rapport aux témoins. ... 73
Figure 51: L’induction de l’expression de la protéine frataxine dans deux autre cellules
FRDA traitées avec les plTALEs-VP64 par rapport aux contrôles négatifs. ... 74
Figure 52: Les produites de digestion purifiée des vecteur pCR3.1 et des inserts digérés
par BstI/XbaI pour faire les constructions pLenti-plTALEs-VP64. ... 75
Figure 53: La sélection des clones positives pLenti-plTALEs-VP64 par une double
digestion (BstI/XbaI)... 75
xiv
Figure 55: L’activité transcriptionnelle du gène FXN dans les cellules FRDA4078
traitées avec les lentivirus qui expriment plTALEs-VP64 par rapport aux témoins négatifs. ... 77
Figure 56: L’analyse de l’expression de la protéine frataxine dans les cellules
FRDA4074 traitées avec les lentivirus qui expriment plTALEs-VP64 par rapport aux témoins négatifs ... 78
Figure 57: Les produits de purification du vecteur pCR3.1 et des nplTALEs-VP64-6-15
et 11-15 digérés par MluI et XbaI pour faire les constructions pCR3.1-nplTALEs-VP64. ... 79
Figure 58: Le résultat du séquençage de nplTALE-8-15-VP64 avec 4 amorces. ... 80 Figure 59: L’activité de gène FXN dans les cellules FRDA4078 traitées par
nplTALEs-VP64 et dans les cellules témoins. ... 81
Figure 60: La structure moléculaire de plTALE-P300 dans le pCR3.1 contient le
promoteur CMV, le N-ter, le RVD, le C-ter, le NLS-VP64 et le p300. ... 82
Figure 61: La présentation des fragments du vecteur linéaire et de l’insert utilisés dans
le clonage In-fusion pour construire les plTALEs-p300. ... 82
Figure 62: La transcription du gène FXN dans les cellules traitées par les
plTALEs-p300 et les cellules témoins. ... 83
Figure 63: L’activité transcriptionnelle du gène FXN avec l’effet synergique des
effecteurs VP64 + p300 et plusieurs p300 ensemble qui ciblent séquences 6, 8 et F4. .. 84
Figure 64: L’analyse de l’expression de la protéine frataxine dans les cellules traitées
avec plTALE-p300 et avec plusieurs effecteurs VP64 et p300 et dans les cellules
témoins. ... 85
Figure 65: La structure moléculaire de plTALE-ST10X/24X... 86 Figure 66: Le résultat de séquençage pCR3.1-plTALE-ST10X-4-13 avec 4 amorces. . 88 Figure 67: L’induction de l’activité du gène FXN des cellules FRDA4087 par les
plTALEs-ST10X et plTALEs-ST24X normalisée par GAPDH et HPRT et par rapport aux témoins. ... 89
Figure 68: Le taux d’expression de la frataxine dans les cellules traitées par
plTALEs-ST10X et plTALEs-ST24X normalisé par GAPDH et par rapport au contrôle. ... 90
Figure 69: L’effet synergique des plTALEs-ST10X sur l’activation de la transcription
du gène FXN dans les cellules FRDA4078. ... 91
Figure 70: Le nombre de copies d’ARNm FXN après une induction du gène endogène
FXN avec 2 meilleurs plTALEs-ST10X-4-15 et plTALEs-ST10X-6-15 dans 4 cellules FRDA par rapport aux fibroblastes normaux et témoins négatifs. ... 93
Figure 71: L’augmentation de l’activité du gène endogène FXN normalisé par les gènes
GAPDH et HPRT dans les 4 FRDA traitées par les plTALEs-ST10X-ST10X par
rapport aux contrôles négatifs... 94
Figure 72: L’induction de l’expression de la protéine frataxine dans 4 cellules FRDA
traitées avec les plTALEs-ST10X-ST10X par rapport aux contrôles négatifs. ... 95
Figure 73: Les noyaux des cellules FRDA4078 traitées par pAAV-plTALEs-ST10X. 97 Figure 74: Les cellules FRDA4078 traitées par plTALE-ST10X. ... 97 Figure 75: Les résultats d’induction du gène endogène FXN avec les effecteurs
plTALEs-ST10X exprimés par les vecteurs pCR3.1 et pAAV. ... 99
Figure 76: L’augmentation de l’activité d’aconitase dans les cellules FRDA4078
xv
Liste des abréviations
AAV Adeno-Associated virus
AAV9 Adeno-Associated Virus Sérotype 9
ADN Acide Désoxyribonucléique
ADNc Acide Désoxyribonucléique Complémentaire
Amp Ampicilline
AAC Adénine-Adénine-Cytosine
ARN Acide Ribonucléique
ARNm Acide Ribonucléique Messager
ATP Adénosine Triphosphate
Bax Protéine Bcl-2–Associated X
BGH Bovine Growth Hormone
BSA Bovine serum albumin
CAG CMV Early Enhancer/Chicken β Actin
CMV Cytomégalovirus
CHU Centre Hospitalier Universitaire
C-ter Extrémité Carboxy-Terminale
CI Cystéine- Isoleucine
CMH Recepteur Protéine
Cas9 CRISPR Associated Protein 9
Cre Cycling Recombination
CpG Cytosine–Phosphate–Guanine
Ca2+ Calcium Ions
CRISPR Clustered Regularly Interspaced Short Palindromic Repeats
ChIP Chromatin ImmunoPrecipitation
DRG Ganglions de la Racine Dorsale
DTT Dithiothreitol
DNase I DNA Plymerase I
DMEM Dulbecco's Modified Eagle Medium
EF1 Elongation factor 1 alpha
EN Acide glytamique- Asparagine
EIAV Virus de l'Anémie Infectieuse Equine
ELISA Enzyme-Linked Immunosorbent Assay
EGR3 Early Growth Response Gene
FBS Fetal Bovin Serum
FXN Frataxine
FT Facteur Transcription
FRDA Friedreich Ataxia
Fe-S Fer-Soufre
FAST-1 FXNAntisense Transcript - 1
FoKI Flavobacterium Okeanokoites
FR Facteur de Répression
GAG Guanine-Adénine- Guanine
GFP Green Fluorescent Protein
GAA Guanine-Adénine-Adénine
GAPDH Glycéraldéhyde-3-Phosphate Déshydrogénase
GABA Acide Gamma-Aminobutirique
xvi
H2O2 Peroxyde d'Hydrogène
HCl Acide Chlorhydrique
HBSS Hanks Balanced Salt Solution
HPRT Hypoxanthine phosphoribosyltransferase
HG Histidine- Glycine
HD Histidine-Acide aspartique
HIV Human Immunodeficiency virus
His6 Hexa-Histidine Tag
hFxn Human Frataxin
H3K27me3 Histone3 Lysine27 Trimethylation
H3K9me3 Histone3 Lysine9 Trimethylation
HDACi Inhibiteurs des Histones Désacétylases
H3 Histone 3
HRP Horseradish Peroxidase
iPSC Induced Pluripotent Stem Cells
IRM Image par Résonance Magnétique
KCl chlorure de potassium
KH2PO4 Potassium dihydrogen phosphate
kDa KiloDalton
Klf4 Kruppel-Like Factor 4
Kb Kilobase
KO Knock-Out
LB Luria-Bertan
LTR Longue Répétition Terminale
LoxP Locus of X-over P1
MCK Muscle Creatine Kinase
ml Millilitre
mg Milligramme
mm Millimètre
mM Milli molaire
MoMLV Virus de le Leucémie Murine de Moloney
MyoD Myogenic Differentiation
NSE Neuron Specific Enolase
NaCl Chlorure de Sodium
Na2HPO4 Sodium phosphate dibasic
nm Nanonètre
NHEJ Non Homologous End Joining
NLS Signal de localisation Nucléaire
NI Asparagine-Isoleucine
NG Asparagine-Glycine
NN Asparagine-Asparagine
N-ter Terminaison Amine
OH Groupe Hydroxyle
O2- Superoxyde
Oct4 Octamer-Binding Transcription Factor 4
PBS Phosphate-Buffered Saline
P300 Co-activating Proteins
PCR Polymerase Chain Reaction
PUMA Upregulated Modulator of Apoptosis
xvii
P65 Transcription Factor
pb Paire de Bases
qPCR Quantitative Polymerase Chain Reaction
qRT-PCR Quantitative Real-time Polymerase Chain Reaction
RVD Repeat Variable Diresidues
RT-PCR Reverse Transcription Polymerase Chain Reaction
ROS Reactive Oxygen Species
Rev Regulator of virion protein
rAAV Adeno-Associated Virus Recombinante
Sox2/SRY Sex Determining Region Y
SsRNA Single-Stranded Ribonucleic Acid
SRF Serum Response Factor
SP1 Specificity Protein 1
SDS Sodium Dodécyl Sulfate
SDS-PAGE sodium dodecyl sulfate polyacrylamide gel electrophoresis
SAGA Serum Response Factor
sfGFP Superfolder Green Fluorescent Protein
TGC Thymine-Guanine- Cytosine
Tris-HCl Tris (hydroxymethyl) aminomethane hydrochloride
TP-PCR Triplet-Repeat PCR
TSS Transcription Start Site
TFAP2 Transcription Factor AP-2 Alpha
TTC Thymine- Thymine-Cytosine
TAT Peptide Trans-Activateurs
TBP TATA-Binding Protéine
TFIIB Transcription Factor II B
TFAP2α transcription factor Activator Protein 2 alpha TALE Transcription Activator-like Effector
TALEN Transcription Activator-like Effector Nucleases
VP64 4 X VP16
VP16 Herpes Simplex Virus Protein vmw65
VIF Virus de l'immunodéficience Féline
YAC Yeast Artificial Chromosomes
µg Microgramme
xviii
Remerciements
Tout d’abord, je tiens à exprimer mes sincères remerciements à mon directeur de recherche Dr Jacques-P. Tremblay pour m’avoir accepté au sein de son équipe, de m’avoir formé puis m’encadrer durant ma Maitrise, ainsi que de m’avoir fait confiance et cru en moi pour réaliser ce projet, mais également pour sa patience, sa disponibilité, ses nombreux conseils scientifiques et ses suggestions pertinentes.
Mes sincères remerciements aussi à Joël Rousseau pour sa collaboration essentielle à la réussite de mon projet, sa disposition, et le temps consacré pour me faire profiter de sa grande expérience. C’est principalement, grâce à son aide que j’ai pu avancer ce projet. Je remercie Pierre Chapdelaine, qui m’as apporté son expertise de la technologie des TALEs et pour les expériences réalisées dans les cultures de fibroblaste. De m’avoir aidé à faire les premiers pas dans ce projet, de m’avoir inculqué la patience ainsi que la fascination pour la recherche et enfin, je le remercie pour sa présence à mon séminaire.
Je remercier Dominique Ouellet pour son aide et ses conseils, ses réflexions qui mènent souvent à des solutions et des suggestions très importantes, ainsi que de m’avoir aidé à apprendre la production des Lentivirus.
Je remercier Catherine Gérard la collaboratrice principale de ce projet qui est responsable de la partie in vivo, je lui souhaite une bonne continuation avec de bons résultats in vivo merci pour le cadeau de 13-01-2017.
Je remercie mes chers collègues pour nos conversations scientifiques et intellectuelles et pour l’environnement au sein du laboratoire, particulièrement Benjamin Duchene pour sa bonne humeur et ses débats que j’ai très souvent apprécié. Jean-Paul Iyombe-Engembe pour son encouragement et son aide moralement et scientifiquement et surtout au niveau de la langue française. Je n’oublierai jamais mon ami, mon camarade de classe et mon collègue du laboratoire, Arnaud Perrin, qui as un bon cœur, aimable et très serviable, avec qui j’ai partagé des moments de pause, j’ai apprécié nos discussions, sans oublié ses aides depuis mes premiers jours au laboratoire jusqu’à aujourd’hui et qui a était ma référence de la langue française, ses conseils qui m’ont aidée d’amélioré mon niveau de la langue française. Arnaud, n’oublie pas qu’il te reste que 2 chances!
xix
Mes remerciements aussi à toutes les personnes rencontrées dans le laboratoire et qui ont contribué à nos travaux, Véronique Dorval, Daniel Skuk et sa pertinente expertise dans les greffes de myoblastes, merci de m’avoir rappelé des bons souvenirs de mon pays natal, Chantale Maltais et sa petite princesse, William-Edouard Gravel et son animation positive dans le laboratoire, Daniel Agudelo avec qui nous n’avions pas les mêmes préférences de température ambiante, mais nous partagions le même amour pour la thérapie génique, Anteneh Argaw et ses discussions riches et Antoine Guyon pour son sérieux au laboratoire.
Je remercie tous les membres du centre génomique du CHU de Québec (Plate-forme de l'expression génétique, CHU de Québec, Québec) particulièrement Nathalie Paquet pour les tests de qRT-PCR réalisés tout au long de ce projet et pour sa collaboration professionnelle.
Je remercie la société Amorchem qui nous a fait confiance en finançant ce projet
Je remercie la directrice de programme Dr Josée N Lavoie, ainsi que Madame Chantal Joubert pour leurs orientations et leurs conseils pendant ma maitrise.
Je remercie les professeurs Dr Sébastien Hébert et Dr Bruno Gaillet d’avoir accepté de corriger ce travail
Aux différentes personnes des services administratifs uLaval.
Pour terminer, un grand Merci à ma mère, à mes frères et mes sœurs, à mes amis pour leurs aides, leurs amours, leurs pensées, leurs soutiens et leurs encouragements.
1
Chapitre 1 : La maladie de Friedreich
1.1 L’ataxie de Friedreich (FRDA) 1.1.1 Introduction générale
L’ataxie de Friedreich (FRDA) est la plus fréquente des ataxies héréditaires autosomiques récessives [1] [2]. Cette maladie est la conséquence de la diminution du niveau de la protéine frataxine dans les cellules jusqu’à 5-35 % par rapport aux taux normaux [3]. Les porteurs asymptomatiques produisent environ 50% de la protéine frataxine [4]. Ce déséquilibre est le résultat d’une mutation au niveau de 1er intron du gène FXN situé dans
le chromosome 9q13. Cette mutation se traduit par une hyper expansion du nombre de répétitions du triplet GAA jusqu’à 1300 [3], par rapport aux personnes normales dont le nombre des répétitions est compris entre 5 à 70 [5]. 98% des patients sont homozygotes pour cette mutation, les autres cas ont une répétition du triplet GAA sur un allèle et une mutation ponctuelle sur l’autre allèle [6] [7] [8]. Cette expansion du triplet GAA est due à un déplacement du brin qui contient la répétition GAA, lors de la réplication de l’ADN, pour former une structure secondaire qui contient un nombre élevé de répétitions GAA. Cette anomalie génétique empêche le glissement de l'ARN polymérase, ce qui bloque la transcription. Il y a aussi formation d’hétérochromatine ce qui rend silencieux le promoteur du gène FXN, ce qui diminue l’expression de la protéine frataxine. La frataxine est une protéine mitochondriale importante dans la régulation du fer et le métabolisme cellulaire. La sévérité de cette anomalie est corrélée avec l’allèle qui a moins de répétitions [3]. La maladie FRDA cause des impacts profonds sur la santé humaine, présentés sous forme des différents symptômes qui apparaissent à partir de l'âge 10-15 ans et dure environ 25 ans conduisant à l'utilisation du fauteuil roulant [3]. Les symptômes les plus marquants de FRDA sont des troubles de la coordination des mouvements, la neuro-dégénérescence, la cardiomyopathie, la dérégulation de la glycémie avec diabète sucré, l'atrophie optique, la perte auditive et l'apnée du sommeil [1] [2] [9] [6] [7]. Ces symptômes sont dus au stress oxydatif (un évènement secondaire de la progression de FRDA, qui cause le dommage des cellules des tissus ciblés) [1].
1.1.2. Historique
La maladie a été découverte par un médecin allemand, Nikolaus Friedreichentre 1863-1877, comme une maladie héréditaire. C’est lui qui a identifié les premières caractéristiques et décrit certains symptômes de FRDA. Quelques années plus tard, Mott
2
[10], a étudié la lésion du noyau dentelé chez un seul cas de FRDA, afin d’expliquer des détails neuropathologiques de cette maladie. À partir des années 80, les études montrent les liens entre FRDA, le déficit en frataxine ainsi que l’excès de fer. À ce jour, les rôles précis de la frataxine dans la biogenèse de complexe fer-soufre (Fe-S), chaperonne de fer et l’accumulation de fer au niveau des mitochondries des cellules restent inconnus [8].
Année
1863–1877 Friedreich publie des descriptions détaillées de la maladie FRDA [11]. 1907 Mott [10] décrit des symptômes neuropathologiques d'un seul cas de
FRDA, y compris la lésion du noyau dentelé.
1957 Urich et al. [8] ont montré l'existence de lésions "suprasegmental" dans FRDA.
1980 Lamarche et al. [12] ont découvert des granules contenant du fer dans les cardiomyocytes des patients atteints de FRDA.
1996 Campuzano et al. [13] ont identifié la mutation commune chez des patients atteints de FRDA, par la suite ils ont identifié la protéine frataxine et son rôle dans le métabolisme du fer.
1997 Rötig et al. [14] ont découvert le complexe "fer-soufre" et la carence des protéines (complexes I, II et III de la chaine de transport mitochondrial d'électrons) et l’aconitase, dans les biopsies de l’endocarde des patients FRDA.
2002 Mühlenhoff et al. [15] ont reconnu l'importance de la frataxine dans la biogenèse du complexe fer-soufre (Fe-S). La déficience du complexe Fer-S est un facteur critique dans la pathogenèse de FRDA.
Tableau 1: Historique de la découverte de la maladie de l'ataxie de Friedreich [8].
1.1.3 Incidence
La fréquence de la maladie FRDA est déterminée avec le développement de la biologie moléculaire pour confirmer la mutation car souvent les patients ont un mauvais diagnostique. Les études actuelles estiment qu’une personne sur 29 000 est atteinte de cette maladie. Un homme et une femme sur 85 sont porteurs de la mutation. Ceci est une augmentation par rapport aux anciennes statistiques indiquant qu’une personne sur 50 000 était atteinte par la maladie de FRDA et qu’une personne sur 110 était porteuse de la mutation [16] [17].
3
La maladie de FRDA est fréquente en Europe, en Amérique, en Afrique du Nord, au Moyen-Orient, et aux Indes. La maladie est particulièrement absente dans les populations sahariennes et d'Extrême-Orient. Le nombre de personnes avec FRDA aux États-Unis est estimé à 9000 [8].
1.1.4 Cause
La maladie FRDA est une maladie héréditaire autosomique récessive due à la transmission de la mutation du gène FXN situé sur le chromosome 9 locus 13 (9q13), issus de deux parents porteurs de la mutation. Les porteurs hétérozygotes pour le gène muté n’ont pas de symptômes. La maladie s’exprime exceptionnellement sur deux générations successives [2] [7].
1.2 FRDA et le stress oxydatif
Le stress oxydatif est le principal symptôme des conséquences cellulaires de la diminution d’expression de la frataxine dans les cellules [9]. En général, c’est le résultat du déséquilibre de la production des dérivés réactifs de l’oxygène (ROS, Reactive Oxygen Species) qui dépasse le niveau de leurs défenses anti-oxydantes utilisées par la cellule pour neutraliser et empêcher la production des ROS. Les ROS sont des produits incomplets de la réduction d’oxygène avec l’eau pendant la respiration mitochondriale par le métabolisme en aérobie cellulaire. Ils peuvent être l’anion superoxyde (O2-) et le
radical hydroxyle (OH) avec un électron des radicaux libres, ainsi que le peroxyde d’hydrogène (H2O2). L’effet négatif des molécules ROS est l’oxydation des différents
composants cellulaires tels que les protéines, les lipides et l’ADN, pour favoriser des maladies ou un vieillissement accéléré [2] [9] [18].
Dans le cas de FRDA, la diminution de l’expression de la frataxine provoque le dysfonctionnement des complexes fer-soufre qui sont nécessaires pour la chaine respiratoire mitochondriale, et l’augmentation de fer libre dans la matrice mitochondriale. Ces anomalies favorisent la catalyse de la dismutation du superoxyde en dioxygène et peroxyde d’hydrogène par la superoxyde dismutase, ce qui provoque les dommages de la chaine respiratoire et la production de ROS. Donc la carence en frataxine pourrait rendre la cellule plus susceptible aux dommages induits par le stress oxydatif. Ces dommages induisent l’apparition des symptômes typiques de la maladie FRDA [2] [9] [19].
4 1.3 Symptômes cliniques de FRDA
Les patients touchés sont normaux à la naissance et pendant une période, jusqu'à l’âge d'apparition des premiers symptômes (10-15ans) [3]. Les patients hétérozygotes peuvent avoir des caractéristiques cliniques atypiques, avec une tendance à avoir moins de dysarthrie que les patients homozygotes, mais ont une atrophie optique avec une plus grande fréquence [8].
La réduction de l'expression de la frataxine dans les cellules cause plusieurs symptômes (Tableau 2), qui touchent les organes les plus sensibles au stress oxydatif, comme le système nerveux pour la dégénérescence et le cœur pour l’hypertrophie cardiaque. L’absence totale de la biosynthèse de la frataxine provoque une létalité embryonnaire chez les modèles murins de FRDA [2] [8].
Tableau 2: les principaux signes de la maladie de FRDA[20].
Les symptômes les plus souvent associés à la progression de la maladie FRDA sont des symptômes neurologiques et des symptômes cardiaques.
1.3.1 Symptômes neurologiques
En général, le nombre de tri-nucléotides GAA dans l’intron 1 du gène FRDA est inversement corrélé avec l’âge d’apparition de la maladie et directement corrélé avec la sévérité des symptômes neurologiques [20] (des troubles de coordination des mouvements, la perte des réflexes, et des troubles de l’équilibre) [9] [17]. Ces signes commencent par une faiblesse des membres inférieurs chez la majorité des patients
5
FRDA, qui cause l’instabilité avec des chutes lorsqu‘ils sont debout ou lorsqu’ils exécutent des mouvements. Par la suite les symptômes progressent aux membres supérieurs et jusqu’à la tête [17].
La dégénérescence spinocérébelleuse cause la dysarthrie, la perturbation des réflexes posturaux et l’hypotonie musculaire. L’atteinte corticospinale est responsable de l’abolition des réflexes ostéotendineux et de l’apparition du signe de Babinski. D’autre part, l’atteinte des cordons postérieurs permet la réduction de la sensation tactile et cause des problèmes de proprioception [20].
D’autres symptômes neurologiques liés à la maladie de FRDA sont des douleurs, des spasmes musculaires, l'atrophie optique et / ou une déficience visuelle chez 5 à 25% des FRDA et la neuropathie auditive avec une déficience auditive [17].
1.3.2 Symptômes cardiaques
La cardiomyopathie hypertrophique est le symptôme cardiaque le plus commun. C’est le second symptôme clinique important le plus commun dans deux tiers des patients [9] [17]. Cette cardiomyopathie hypertrophique est la cause principale de décès précoces chez 83,3% des FRDA. En général ce symptôme est moins fréquent chez les FRDA avec le nombre de répétitions de triplet GAA très petit, et au début de la maladie [8] [17]. La cardiomyopathie chez les patients FRDA est caractérisée par l’augmentation de l’épaisseur de la paroi du ventricule gauche (due à l'hypertrophie des myocytes), qui cause une insuffisance cardiaque et la mort. Cette hypertrophie est corrélée avec le nombre de répétitions GAA dans l’allèle qui contient le moins de répétitions [3]. La détection de la cardiomyopathie hypertrophique se fait par l’électrocardiographie et l’échocardiographie [17], plus récemment par l'IRM et la spectroscopie 31P [8].
1.3.3 Autres symptômes cliniques
Un diabète sucré, présent chez 8-32% des FRDA [avec leur besoin en insuline], qui requièrent de l’insuline, l’apparition du diabète est corrélée avec le nombre de répétitions de GAA [8]. De plus, 60 à 80 % des patients de FRDA ont des complications orthopédiques et 80% ont des malformations au niveau des jambes avec pied creux bilatéral (Fig. 1) [17]. Plusieurs patients FRDA souffrent aussi d’une scoliose grave et d’un mal de dos, en raison d'une combinaison de spasmes musculaires para-spinaux, d’une déformation de la colonne, de malformations du squelette et d’une mauvaise assise qui nuit au confort du patient même dans un fauteuil roulant (Fig. 1) [9] [17]. Le temps
6
moyen avant la nécessité d’utiliser un fauteuil roulant dépend de l'apparition des symptômes (10 à 15 ans). L'espérance de vie moyenne des patients FRDA est de 25 à 36,5 ans [17].
Figure 1: Des signes classiques de FRDA:
Des pieds creux bilatéraux, des déformations de la colonne vertébrale et des malformations du squelette. © ELSEVIER.INC.-NETTERIMAGES.com
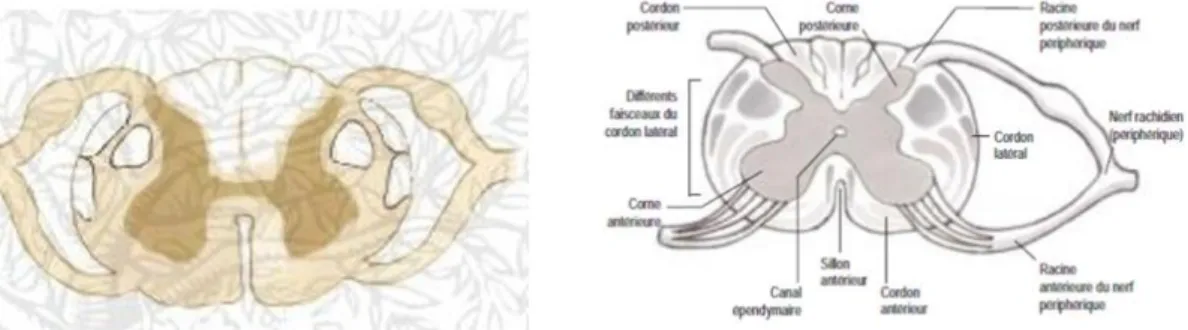
1.4 Anatomopathologie 1.4.1 Neuropathologie (Fig. 4)
La neuropathologie de l’ataxie de Friedreich est caractérisée par une sévère atrophie des ganglions de la racine dorsale (DRG), ce qui les rendent difficiles à reconnaitre lors de la dissection. Cette lésion est causée par une destruction progressive des neurones les plus grands et des axones myélinisés plus épais. La dégénération réduit principalement le diamètre de la moelle épinière (Fig. 2), ou les voies spinocérébelleuses. Les cordons postérieurs et les voies cortico-spinales sont atteints de façon constante [8] [9] [20]. Au niveau thoracique, la lésion implique les faisceaux graciles et cunéiformes dans la même mesure, mais dans les segments du col utérin. L'apparence gélatineuse n'est pas toujours en forme de coin, mais semble parfois comme un bouchon sur les colonnes dorsales. La perte de fibres dans les domaines antérolatérales correspondant aux
spino-7
cérébelleuse et cortico-spinales peuvent également être visibles à l'œil nu (Fig. 2) [8] [9] [20].
La dégénérescence cérébelleuse tardive est présente plus au niveau du noyau dentelé que dans le vermis du cortex cérébelleux, cette lésion est variable d’un individu à l’autre (Fig.
3) [8] [9] [20].
Figure 2: La coupe transversale de moelle épinière d’un patient FRDA atteint au niveau du cordon postérieur et le faisceau pyramidal (cordon latéral).
(http://www.afaf.asso.fr/).
Figure 3: L’aspect des noyaux dentelés (DN) dans un patient FRDA [8].
(a) FRDA, (b) le contrôle normal. Les lignes interrompues indiquent l'emplacement approximatif de la matière grise des noyaux dentelés. La petite taille du noyau dans le patient FRDA est particulièrement visible par la taille réduite de la zone marquée pour le fer (a). Le noyau dentelé normal montre le ruban typique de méandres gris (b). Le produit de la réaction pour le fer ne se localise pas avec la matière grise, mais se prolonge dans la substance blanche.
Les lésions de dégénérescence spinale diminuent la concentration de glutamate et de glycine dans la substance grise de la moelle épinière (Fig. 2 et 3), et les lésions de dégénérescence cérébelleuse causent la diminution des concentrations de l’acide gamma-aminobutirique (GABA) et de glutamate dans les régions vermiennes et hémisphériques du cervelet ainsi que la diminution des récepteurs des benzodiazépines au niveau du noyau dentelé et de l’une des régions afférentes principales du cervelet. Il y a une corrélation entre, la sévérité des symptômes, le dysfonctionnement métabolique et
8
la diminution de la quantité de glucose utilisée. Également, il y a une diminution de la concentration du cytochrome oxydase, l’enzyme mitochondrial impliqué dans la phosphorylation oxydative dans cervelet [20].
Figure 4: L’anatomopathologie des cerveaux FRDA[21].
a) l’absence d’atrophie cérébelleuse après une année, b) une légère atrophie cérébelleuse après 10 ans, c) une atrophie cérébelleuse marquée après 20 ans.
1.4.2 Hypertrophique cardiaque
Les études montrent la présence d’une hypertrophie cardiaque par l’augmentation de l’épaisseur de la paroi de ventricule gauche (due à l'hypertrophie des myocytes) (Fig. 5). La cardiomyopathie dans les patients FRDA peut être concentrique, hypertrophique et dilatée. Elle est caractérisée par des thrombus muraux dans le ventricule gauche et par l’accumulation des granules riches en fer [8].
Figure 5: L’aspect brut du cœur dans les patients FRDA [8].
(a) L'hypertrophie cardiaque accompagnée de la décoloration du myocarde induit le rétrécissement des ventricules. (b) L'hypertrophie cardiaque affectant seulement la paroi ventriculaire gauche et le septum interventriculaire induit une augmentation du ventricule droit.
1.5 Frataxine
1.5.1 Gène de la frataxine (FXN)
Le gène de la frataxine (FXN) situé sur le chromosome 9q13, contient cinq exons et 4 introns, avec 3 dinucléotides CpG (sites de méthylation) [2] [22]. Le premier intron contient la répétition du triplet GAA jusqu’à 70 pour les personnes normales (Fig. 6) [22]
9
[23], et de plus de 100 jusqu'à 1300 répétitions pour les cas de FRDA [3]. Cette répétition bloque la transcription et cause une modification épigénétique qui réduit l’activité du promoteur du gène FXN [3] [22].
Figure 6: La représentation moléculaire du gène FXN (entre chaque 2 exons il y a un intron)
[22].
Le promoteur du gène FXN contient des sites de liaisons pour 4 facteurs de transcription majeurs (TSS1 (Transcription Start Site 1), TSS2 (Transcription Start Site 2), TFAP2 (transcription factor Activator Protein 2) et EGR3 (Early Growth Response Gene)). Les 2 premiers sont situés en amont du site d’initiation de la traduction à une distance de 221 pb et de 60 pb de la région du promoteur intro\PDE. Le 3eme facteur TFAP2 est situé entre
les sites d’initiation de la transcription, TSS1 et TSS2, et le 4eme facteur EGR3 se localise
au niveau de l’intron 1. Il existe également une séquence E-box qui régule l’expression des gènes dans les neurones et dans les muscles (Fig. 7) [22].
Figure 7: Les principaux sites régulateurs du gène FXN [22].
1.5.2 La protéine de la frataxine
La frataxine est une protéine hautement conservée de la bactérie à l'homme. Elle est le résultat d'un transcrit de 1.3 Kb. Elle est destinée à la mitochondrie sous forme d'un précurseur de 23 kDa (forme intermédiaire de 210 AA). Après un clivage enzymatique, elle se localise à l’intérieur de la mitochondrie sous une forme active de 14 kDa [2] [8].
L'emplacement de la frataxine peut être proche ou dans la membrane mitochondriale interne, suggérant qu’elle peut jouer plusieurs rôles dans le métabolisme et la production d’énergie cellulaire. Elle pourrait donc être impliquée dans le transport du fer à travers la
10
membrane mitochondriale ou dans le transport d'électrons par des complexes fer-soufre [2] [7] [8].
1.5.3 La fonction de la frataxine
La fonction principale de la frataxine est encore controversée, bien que les différentes recherches montrent que cette protéine peut intervenir dans plusieurs fonctions cellulaires (Fig. 8) [1] [2]:
• Régulation du fer dans les mitochondries
• Régulation du métabolisme cellulaire dans les mitochondries • Homéostasie du fer intracellulaire
• Biogénèse de l’hème • Catalyse enzymatique • Régulation des gènes
• Empêche l’accumulation de fer • Désintoxication
• Activité chaperon de fer
• Assemblage des complexes fer-soufre dans la mitochondrie et dans le
compartiment extra-mitochondrial • Empêche la génération de stress oxydatif
Figure 8: La localisation de la frataxine dans la mitochondrie et dans le cycle de la production d’énergie.
(Adapté de www.grand-est.inserm.fr).
La protéine frataxine peut intervenir dans plusieurs fonctions cellulaires au niveau de la mitochondrie, telle que : la régulation du métabolisme cellulaire, la biogenèse de l’hème, et la catalyse enzymatique.
11 1.5.4 L’absence de la frataxine
La diminution de la production de la frataxine dans les cas de FRDA peut causer différents problèmes dans la cellule [2] (Fig. 9) :
• Déficiences enzymatiques mitochondriales telles que l’aconitase et les complexes I, II et III des mitochondries,
• Empêche la biosynthèse du complexe fer-soufre • Problème dans la respiration mitochondriale • Problème de la production d’ATP
• Fer libre et l’accumulation de fer mitochondrial
• Augmentation de la production des ROS dans la mitochondrie
• Dommage oxydatif qui inactive les enzymes mitochondriaux et induit la production d’autres radicaux libres
Figure 9: La dérégulation de la chaine respiratoire et le cycle de la production d’énergie en absence et en diminution de l’expression de la frataxine dans la mitochondrie.
(Adapté de www.grand-est.inserm.fr).
L’absence ou la diminution de la protéine frataxine dans la mitochondrie peut induire différents problèmes au niveau de ce dernier, tel que; le stress oxydatif, l’accumulation de Fer dans la mitochondrie et la dérégulation de la chaine respiratoire.
1.6 Déficience moléculaire de la FRDA
L’hyper-expansion de la répétition GAA au niveau de l'intron 1 provoque la mise sous silence de la transcription par la formation des structures anormales d'ADN (triplex ou ADN collant), la formation d'un hybride ADN-ARN, ou la formation d'hétérochromatine. Le triplex (ADN collant) formé par la séquence de longue répétition GAA provoque aussi des instabilités génétiques et des recombinaisons génétiques [7] [24].
12 1.6.1 Formation d’ADN collant (Sticky DNA)
La répétition GAA peut provoquer la formation d’une structure d’ADN de triple hélice (ADN triplexes ou Sticky DNA) due à l’association de deux brins ayant la répétition GAA et d’un brin ayant la répétition TTC (Fig. 10). La structure de l’ADN triplex inhibe la transcription en piégeant l’ARN polymérase hors de la transcription des séquences répétées triplet GAA [7] [24].
Figure 10: Une présentation schématique de la forme d’ADN anormal dans le cas de FRDA. En haut l’ADN triplex et en bas l’ADN collant [24].
1.6.2 Formation d’hybride RNA-DNA
Lors de la transcription des séquences répétées GAA, un hybride ARN-ADN est formé au cours de déplacement de l'ARN-polymérase et avec le sur-enroulement négatif de l'ADN derrière l'ARN-polymérase. Le brin non-transcrit se replie alors en arrière pour former un triplex d'ADN. Le nouvel ARN naissant s’hybride au brin matrice formant un hybride ARN-ADN (Fig. 11) [7] [24].
13
Figure 11: La formation d’hybride RNA-DNA dans le cas FRDA [24].
A) La transcription au niveau de la répétition GAA, B) formation un triplex d'ADN par le brin non-transcrit et le double brin d’ADN au niveau de la répétition GAA, C) et D) formation de l’hybride ADN-ARN.
1.6.3 Changement d’épigénétique
L’expression du gène FXN chez les patients FRDA, peut être affectée par les différents changements épigénétiques au niveau de l’ADN, tel que la méthylation de l’ADN et des modifications des histones, à cause de la répétition du triplet GAA au niveau de l’intron 1 du gène FXN. Dans certaines cellules des FRDA, il y a plus de méthylation des résidus CpG en amont de la répétition et moins de méthylation en aval de la répétition par rapport aux cellules saines. Des études sur l’analyse des modifications des histones par ChIP et qPCR ont montré que dans cette anomalie de FRDA il y a aussi une diminution du niveau de l’acétylation des lysines 9 et 14 de l’histone H3 et une augmentation de triméthylation de H3K9. Des chercheurs ont également prouvé que le niveau de formation d'hétérochromatine est augmenté beaucoup plus avec un nouveau transcrit antisens (FAST-1) du gène FXN, en tant que médiateur de la formation de l’hétérochromatine [3] [7] [25].
1.7 Diagnostic de FRDA
Les maladies génétiques sont généralement difficiles à diagnostiquer en utilisant seulement leurs symptômes, car de nombreuses maladies peuvent partager des symptômes similaires. Par exemple, 3 maladies, l’Ataxie avec déficience en vitamine E, A-lipoproteinaemia et la maladie de Refsum, ont des symptômes semblables à FRDA. Afin de diagnostiquer plus précisément la maladie FRDA, il existe des tests de diagnostic
14
moléculaire qui sont utilisés pour confirmer et détecter la présence de la mutation de gène FXN qui est différente des autres maladies génétiques ayant des symptômes similaires. Les techniques de diagnostic qui peuvent être utiles, dans le cas de la maladie FRDA, comprennent des procédés et des outils qui sont basés sur l'analyse de l'ADN, ainsi que l'analyse des niveaux d'ARNm et de protéine frataxine [7]. Ces techniques permettent aussi de détecter des répétions du triplet GAA dans le premier intron du gène FXN, ces techniques sont basées sur des réactions PCR, RT-PCR ou Southern/Norton blot [7].
1.7.1 PCR
La PCR utilise des amorces qui flanquent la région d'intérêt afin de l’amplifier. Les produits de PCR peuvent ensuite être analysés en utilisant une électrophorèse sur gel d'agarose ou d'autres techniques. Cependant, la limite majeure de cette technique et la taille du fragment à amplifier (3-4 kb). Par conséquent, il peut être difficile d'amplifier un allèle contenant une très grande répétition de GAA par une PCR classique. D'autres techniques basées sur la PCR sont explorées afin de trouver des méthodes efficaces pour le diagnostic moléculaire de FRDA [7].
1.7.2 Triplet-repeat (TP-PCR)
Le Triplet-repeat PCR (TP-PCR) est évalué comme méthode de dépistage pour FRDA. Cette technique a été développée par J P Warner et al, 1996 [26], afin d'identifier l'expansion de répétition CAG de la dystrophie myotonique de Steinert. Elle utilise un ensemble de trois amorces pour amplifier la région contenant la répétition, afin de déterminer leur taille exacte (Fig. 12) [7].
15
Figure 12: Schéma représentatif des différents produits d’amplification avec 3 amorce dans Triplet-repeat (TP-PCR) [7].
1.7.3 RT-PCR quantitatif en temps réel (qRT-PCR)
La RT-PCR est une méthode alternative de diagnostic de FRDA, elle permet de quantifier des niveaux d'ARNm du gène FXN, en utilisant une PCR quantitative en temps réel spécifique, qui mesure les produits de PCR amplifiés générés au cours de chaque cycle du processus de PCR sur l’ADNc, produit par transcription inverse de l'ARNm du gène FXN. Le niveau d’ARNm chez les patients FRDA est inférieur au niveau d’ARNm des individus sains. Avec une forte corrélation inverse entre les niveaux d’ARNm du gène FXN et le nombre de répétitions GAA [3] [7] [27].
1.7.4 Southern Blot
La technique Southern Blot permet la détection de molécules spécifiques d’ADN dans un mélange séparé par un gel. Lors de cette technique, l'ADN extrait, puis purifié, est digéré par des enzymes de restriction, les fragments obtenus sont séparés par une électrophorèse sur gel d’agarose, puis transférer sur une membrane de nitrocellulose ou de nylon, les séquences des fragments d’ADN d’intérêt sont détectées dans la membrane par hybridation moléculaire avec des sondes marquées d'acides nucléiques (Département de biochimie, Université d’Oxford, 2006) [7].
Le Southern Blot a été utilisé comme test moléculaire, de dépistage des patients FRDA, pour détecter l’expansion du triplet GAA du gène FXN[7].
1.8 Thérapie
1.8.1 Principe et le but des traitements de FRDA
Actuellement il n’existe aucun traitement (thérapie) curatif connu pour FRDA. Par conséquent, la plupart des traitements potentiels sont encore en phase d’essais cliniques
16
en France, aux États-Unis et au Canada. Ils visent la réduction et la gestion des symptômes pour ralentir la progression de la maladie en :
• Aidant les mitochondries à mieux fonctionner
• Réduisant les contraintes imposées à la mitochondrie
• Vérifiant si les fonctions normalement effectuées par la frataxine peuvent être remplacées par d’autres protéines.
• Utilisant desanti-oxydants
• Augmentant le niveau de la frataxine par l’utilisation des histones désacétylases (HDAC), l’interferon gamma (montré dans les deux modèles cellulaires et animaux de l'ataxie de Friedreich), l’érythropoïétine (EPO) (hormone produite dans le corps pour augmenter le nombre de globules rouges)
• Travaillant sur le développement de thérapies basées sur la livraison systémique des protéines ou des gènes exprimant la protéine frataxine capables de corriger la mutation du gène FRDA
1.8.2 Exemple des thérapies 1.8.2.1 Idébénone
L'idébénone est une molécule de type quinone permettant le transport des électrons dans la membrane interne de la mitochondrie. C’est un antioxydant qui protège contre les lésions oxydatives. Il est en phase d’essais en Europe. Son effet est variable selon les patients, ce médicament permet de protéger les constituants cellulaires contre l’action pro-oxydante du fer. In vivo, il peut réduire l’hypertrophie des parois du myocarde, la fatigabilité, la chélation de fer et l'amélioration des mouvements fins chez certains patients. Cependant, une amélioration significative de l’ataxie n’a pas été obtenue par ce traitement (http://www.afaf.asso.fr/).
Bien que le site d’Ataxie Canada, a annoncé en Juin 2014, que Santé Canada avait approuvé avec conditions le SNT-MC17/l’idébénone pour le traitement de l’ataxie de Friedreich, cette permission a depuis été retirée.
Aux États-Unis, l’essai clinique de phase III IONA (Idebenone effects On Neurological ICARS Assessments) est en cours sur plus 50 patients selon leur disponibilité. (Http://lacaf.org/fr/tag/idebenone/).
17
Ce traitement produit par le groupe Pharmaceutique Takeda (Japon), et commercialisé par le laboratoire Santhera dans l’Union Européenne, en Suisse, en Amérique du nord sous le nom de Catena, Raxone ou Sovrima (http://www.kjer-france.org/).
1.8.2.2 Inhibiteurs des histones désacétylases (HDACi)
Les inhibiteurs des histones désacétylases (HDACi) de la classe III activent le gène FXN dans plusieurs modèles de FRDA, par une réduction de H3K9me3 et H3K27me3. Ceci permet une accessibilité accrue de la DNase I, et l’induction d’histones acétylés de la chromatine (Fig. 13). Ce traitement peut augmenter de 67% de l’expression de la frataxine, en réactivant le contrôle épigénétique du gène FXN traité [1] [28] [29] [30].
Figure 13: Présentation de la structure d’euchromatine (gène actif) et de la forme l’hétérochromatine (gène silencieux) [28].
Selon la Figure 14, l’histone acétylée change l’organisation des nucléosomes, et active le gène FXN. Une longue répétition de GAA chez les patients FRDA conduit à la formation de l’hétérochromatine méthylée (gène non actif), avec la condensation des nucléosomes, qui va bloquer le recrutement du complexe transcriptionnel et réduire l’accessibilité de DNaseI (Fig. 14) [28] [29] [30].
1.8.2.3 La protéine frataxine exogène
L’administration de la protéine frataxine exogène, fusionnée au peptide trans-activateurs de la (TAT) pour transduction aux mitochondries des cellules de FRDA in vitro et in vivo. TAT-FXN (Fig. 14) injecté réduit la caspase-3, qui est activée à cause du stress oxydatif chez des cellules de FRDA. Chez les souris injectées, TAT-FXN (Fig. 14) augmente la vitesse de croissance et la durée de vie moyenne de 53% après la diminution des symptômes cardiaque, elles augmentent aussi l'activité de l'aconitase dans les mitochondries [31].
18
Figure 14: Les domaines de Tat-FXN protéine de fusion [31].
TAT de côté 5’ sous forme précurseur de la ADNc de hFXN, avec un promoteur T7 et His6 pour la purification par chromatographie d’affinité
1.8.2.4 Thérapie génique
Actuellement, il existe plusieurs stratégies de thérapie ‘moléculaire’ en phase de recherche pour traiter la maladie FRDA, on trouve :
-L’administration du gène de la hFXN in vivo, pour ré-exprimer le taux normal de la frataxine dans les cellules de FRDA à l’aide d’un vecteur viral, AAV sérotype 9 pour livrer hFXN humaine par la voie intrapéritonéale dans des souris modèles de la FRDA (NSE-Cre et MCK-Cre), différentes doses d’injection ont été testées pour optimiser celle qui permettra de doubler la durée de vie de souris FRDA traitées et permettant de diminuer les ou certains des symptômes de FRDA. Les résultats d’ELISA dosage d'immuno-absorption par enzyme liée montrent la présence de la frataxine dans les organes les plus touchés par la maladie FRDA, le cœur, le cerveau, les muscles, les reins et le foie [32].
-L’augmentation de l’expression de la frataxine à l’aide de AAV-TALE-FT. Le FT est un domaine d’activation de la transcription (ex. VP64) (Fig. 15), qui permet de recruter le complexe transcriptionnel et d'activer la transcription du gène, ce domaine d’activation a été fusionné avec TALE (Le nucléase effectrice de type activateur de transcription), une protéine de liaison à l’ADN ciblant la région régulatrice du gène de FXN. Les premiers résultats montrent que le domaine d’activation VP64 augmente l’expression de la protéine jusqu’à 2 fois dans des cellules de fibroblastes de patients de FRDA, in vivo le TALE-VP64 a été livré à l’aide du AAV9. Le AAV9-TALE-TALE-VP64 injecté par voie intrapéritonéale dans des souris YG8R (les souris modèles de FRDA), augmente l’expression de la protéine dans 3 organes (le cœur, les muscles et le foie), cette approche thérapeutique reste un traitement potentiel de FRDA qui nécessite d’être développée dans le futur [33] [34].
19
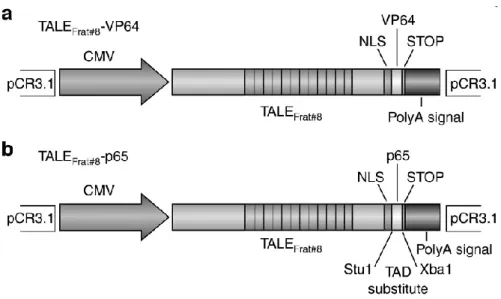
Figure 15: Présentation de la structure moléculaire de complexe TALE-FT
Le TALE fusionné avec le domaine d’activation de la transcription VP64 (a) et p65 (b) a été fait dans le plasmide pCR3.1 pour but d’induction le gène endogène FXN [33] [34].
Toutefois l’approche par l’édition du génome (Genome Editing) avec les technologies CRISPR/Cas9 et TALEN (transcription activator-like effector nuclease) pour corriger in vitro et in vivo les mutations au niveau du gène FXN, est une approche prometteuse pour ce type de maladie.
1.9 Modèles animaux et cellulaires de FRDA
Les chercheurs ont mis au point des modèles cellulaires et animaux, d’une part pour mieux comprendre l’aspect et les mécanismes moléculaires et biochimiques de la maladie de FRDA et, d’autre part, pour développer des stratégies thérapeutiques. Ces modèles sont des mammifères les plus proches physiologiquement et génétiquement des patients FRDA humains. Ils existent des modèles de FRDA favoris qui diffèrent par leurs avantages et leurs inconvénients [35].
Un bon modèle de FRDA devrait avoir un niveau très réduit de la frataxine, c’est pourquoi les cellules directement issues de patients FRDA sont les candidats idéals en tant que modèle cellulaire. Le modèle animal, quant à lui, devrait avoir des signes cliniques de la FRDA, à savoir des neuro-dégénérescences ainsi que la cardiomyopathie observée chez les patients FRDA.
![Figure 16: Présentation schématique de la génération des souris MCK et des souris NSE [35]](https://thumb-eu.123doks.com/thumbv2/123doknet/3642275.107325/39.892.293.590.280.610/figure-présentation-schématique-génération-souris-mck-souris-nse.webp)
![Figure 17: Représentation schématique de la génération des souris KIKI et KIKO [35].](https://thumb-eu.123doks.com/thumbv2/123doknet/3642275.107325/40.892.330.576.742.1064/figure-représentation-schématique-génération-souris-kiki-kiko.webp)
![Figure 18: Représentation schématique de la génération des souris YG8R et les souris YG22R [35]](https://thumb-eu.123doks.com/thumbv2/123doknet/3642275.107325/42.892.239.652.107.565/figure-représentation-schématique-génération-souris-yg-souris-yg.webp)
![Figure 20: La structure et l'organisation du génome du virus adéno-associé (AAV) [41].](https://thumb-eu.123doks.com/thumbv2/123doknet/3642275.107325/48.892.275.607.104.378/figure-structure-organisation-génome-virus-adéno-associé-aav.webp)
![Figure 25: Deux paires des TALENs ciblent deux séquences spécifiques d’ADN [45].](https://thumb-eu.123doks.com/thumbv2/123doknet/3642275.107325/53.892.167.732.406.521/figure-paires-talens-ciblent-séquences-spécifiques-adn.webp)