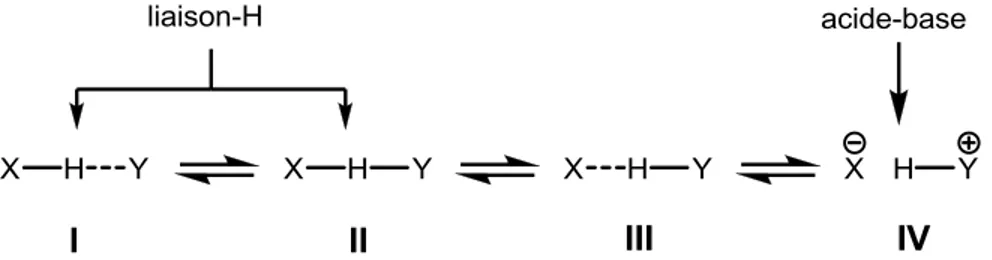Activation de liens C–F à l’aide de liaisons hydrogène
Thèse
Pier Alexandre Champagne
Doctorat en chimie
Philosophae Doctor (Ph.D.)
Québec, Canada
Résumé
L’activation de liens C–F a été un sujet de recherche prolifique des dix dernières années, considérant l’utilité de telles études pour mieux comprendre et utiliser le comportement des composés fluorés. Toutes les méthodes qui existent pour réaliser la substitution nucléophile de fluorures aliphatiques tirent profit d’interactions faibles en conditions acides ou basiques, ou de métaux de transition. Cependant, une interaction comme la liaison hydrogène avec un lien C–F, certainement faible en énergie, n’a toujours pas été utilisée pour réaliser l’activation en conditions neutres.
Après une introduction sur l’activation C–F et leur capacité d’accepteur de liaisons hydrogène, divers systèmes utilisant cette faible interaction pour la substitution nucléophile de fluorures aliphatiques, qui ont été développés dans cette thèse, seront présentés. Plusieurs types de donneurs de liaisons hydrogène (DLH) ont été utilisés au courant de la thèse, notamment l’eau, les alcools polyfluorés, ainsi qu’une large gamme d’alcools, diols, triols et autres groupements donneurs. En présence de nucléophiles variés, il a été démontré que les liaisons hydrogène sont suffisamment puissantes pour permettre la transformation de composés organofluorés par l’activation et la substitution de leur lien C–F.
L’analyse des mécanismes réactionnels a aussi démontré qu’en plus de nécessiter des activateurs pour pouvoir le substituer, le lien C–F conserve un certain caractère spécial dans nos conditions avec les DLHs. Pour nos systèmes de substitution nucléophile, un mécanisme SN2 semble correct, tandis que dans les réactions de Friedel-Crafts, des paires
d’ions fortement retenues par les liaisons hydrogène semblent expliquer la réactivité observée mieux qu’un mécanisme ionisant SN1 classique ne pourrait le faire.
Abstract
C–F activation has been a subject of intensive research over the last decade, considering the usefulness of such studies to understand, predict and profit from the unusual behaviour of fluorinated organic compounds. All methods that are known to enable the nucleophilic substitution of aliphatic fluorines use weak interactions in acidic or basic conditions, or require transition metals. However, hydrogen bonds with fluorine, energetically-weak interactions, have not yet been harnessed to activate C–F bonds in neutral conditions.
After an introduction on C–F activation and on the possibility of these bonds to accept hydrogen bonds, various systems that exploit this weak interaction for the nucleophilic substitution of aliphatic fluorines and that were developed during the thesis, will be presented. Multiple types of hydrogen-bond donors (HBD) were used to affect this transformation, notably water, polyfluorinated alcohols and a wide variety of alcohols, diols, triols, and other donating groups. In the presence of varied nucleophiles, it will be shown that hydrogen bonds are strong enough to permit the transformation of organofluorine compounds through activation and substitution of their C–F bond.
Thorough analysis of the reaction mechanisms demonstrated that, in addition to needing activating agents to be able to substitute it, the C–F bond keeps its unusual behaviour in our conditions with HBDs. In our systems of nucleophilic substitution, a SN2 mechanism seems
to operate, while for the Friedel-Crafts reactions, ion pairs strongly kept together by hydrogen bonds are a better explanation for the observed reactivity than the traditional SN1
Table des matières
Résumé ... iii
Abstract ... v
Table des matières ... vii
Liste des figures ... xi
Liste des schémas ... xiii
Liste des tableaux ... xv
Liste des abréviations... xvii
Remerciements ... xxi
Avant-propos ... xxv
Chapitre 1 : Introduction ... 1
1.1 Généralités sur le fluor et le lien C–F ... 1
1.1.1 Le fluor... 1
1.1.2 Le lien C–F ... 1
1.1.3 Les composés organofluorés... 2
1.2 Activation de liens C–F ... 3
1.2.1 Préambule ... 3
1.2.2 Méthodes d’activation C–F de composés aliphatiques ... 6
1.3 Le fluor comme accepteur de liaisons hydrogène ... 25
1.3.1 Définition d’une liaison hydrogène ... 25
1.3.2 Liaisons hydrogène avec le fluor : un historique ... 27
1.3.3 Récents exemples de liaisons hydrogène avec un lien C–F comme accepteur ... 30
1.4 Objectifs de la thèse ... 33
Chapitre 2 : Permettre les réactions de substitution nucléophile des fluorures d’alkyles activés par la donation de liaisons hydrogène / Enabling Nucleophilic Substitution Reactions of Activated Alkyl Fluorides through Hydrogen Bonding ... 35
2.1 Résumé ... 36
2.2 Abstract ... 36
2.3 Introduction ... 36
2.4 Results and discussion ... 38
2.5 Acknowledgment... 47
2.6 Annexe 1 : activation séquentielle d’un chlorure puis d’un fluorure benzylique ... 47
2.8 Supporting Information Available ... 55
2.8.1 General information ... 55
2.8.2 Materials and methods ... 56
2.9 Partie expérimentale des résultats non publiés (section 2.6) ... 69
Chapitre 3 : Activation de liens C–F promue par les triols : Amination de fluorures benzyliques en conditions hautement concentrées mediée par le 1,1,1-tris(hydroxyméthyl)propane / Triol-promoted activation of C–F bonds: Amination of benzylic fluorides under highly concentrated conditions mediated by 1,1,1-tris(hydroxymethyl)propane ... 71
3.1 Résumé ... 72
3.2 Abstract ... 72
3.3 Introduction ... 73
3.4 Results and discussion ... 75
3.5 Conclusion ... 80
3.6 Acknowledgments ... 81
3.7 Annexe : Discussion ... 81
3.8 Supporting Information ... 83
3.8.1 General information ... 83
3.8.2 Materials and methods ... 84
Chapitre 4 : Explication mécanistique révisée pour l’amination des fluorures benzyliques promue par des alcools dans des conditions hautements concentrées : Preuves computationnelles et expérimentales sur un substrat modèle / Revised Mechanistic Explanation for the Alcohol-Promoted Amination of Benzylic Fluorides under Highly Concentrated Conditions: Computational and Experimental Evidence on a Model Substrate ... 93
4.1 Résumé ... 94
4.2 Abstract ... 94
4.3 Introduction ... 95
4.4 Results and discussion ... 96
4.5 Annexe 1 : exploration de catalyseurs variés ... 109
4.6 Experimental ... 118
4.6.1 General information and materials ... 118
4.6.2 General procedure for the reaction of 4.1 promoted by alcohols ... 118
4.7 Acknowledgments ... 119
4.8 Partie expérimentale des résultats non-publiés (section 4.5) ... 119
Chapitre 5 : Réaction de Friedel–Crafts des fluorures benzyliques : Activation sélective de liens C–F permise par la donation de liaisons hydrogène / Friedel–Crafts Reaction of Benzyl Fluorides: Selective Activation of C– F Bonds as Enabled by Hydrogen Bonding ... 121
5.1 Résumé ... 122
5.2 Abstract ... 122
5.3 Introduction ... 122
5.4 Results and discussion ... 125
5.5 Conclusion ... 134
5.6 Acknowledgements ... 135
5.7 Annexe 1 : Développement de systèmes connexes pour la réaction de Friedel-Crafts ... 135
5.7.1 Activation in situ d’alcools benzyliques par le XtalFluor-E pour la formation de diaryl- et de triarylméthanes ... 135
5.7.2 Réaction de Friedel-Crafts de fluorures benzyliques activés par l’acide trifluoroacétique ... 141
5.8 Annexe 2 : Discussion ... 147
5.9 Supporting Information ... 150
5.9.1 General information ... 150
5.9.2 Materials and methods ... 151
5.9.3 Activation using XtalFluor-E... 189
Chapitre 6 : Détermination des mécanismes réactionnels des réactions d’activation C–F ... 191
6.1 Mise en contexte ... 191
6.2 Substitution énantiospécifique de fluorures d’alkyle secondaires chiraux... 194
6.2.1 Synthèse et réaction d’α-fluorocétones énantioenrichies ... 194
6.2.2 Synthèse et réactivité de fluorures benzyliques secondaires énantioenrichis ... 197
6.3 Synthèse et réactivité de fluorures benzyliques deutérés, primaires et chiraux ... 201
6.3.1 Origines synthétiques et caractérisations des substrats ... 202
6.3.2 Réactions d’activation C–F et résultats obtenus ... 205
6.3.3 Explications mécanistiques... 219
6.4 Effets cinétiques isotopiques du deutérium lors de la substitution nucléophile de fluorures benzyliques primaires par les amines ... 221
6.4.1 Introduction sur les effets isotopiques ... 221
6.4.2 Effet secondaire du deutérium α : sonde mécanistique ... 228
6.4.3 Développement de la méthode ... 231
6.4.4 Résultats et discussion ... 235
6.4.5 Perspectives avec cette méthode analytique ... 237
6.5 Conclusion partielle ... 238
6.6 Partie expérimentale ... 240
6.6.2 Méthodes expérimentales ... 241
Chapitre 7 : Conclusion et travaux futurs... 261
7.1 Retour sur les objectifs ... 261
7.1.1 Activation de fluorures d’alkyles activés par l’eau ... 261
7.1.2 Activation à l’aide de triols, diols, alcools et autres DLHs ... 262
7.1.3 Activation par le HFIP ... 263
7.2 Perspectives... 265
7.2.1 Substitution nucléophile aromatique de fluorures d’aryles ... 265
7.2.2 Étude de DLHs puissants pour l’activation C–F ... 267
7.2.3 Étendue de la réaction d’activation C–F par le HFIP ... 268
Liste des figures
Figure 1.1. Différentes hybridations possibles d’un carbone lié à un fluor. ... 4
Figure 1.2. Spectre des états possibles lors du transfert d’un proton de X à Y. ... 27
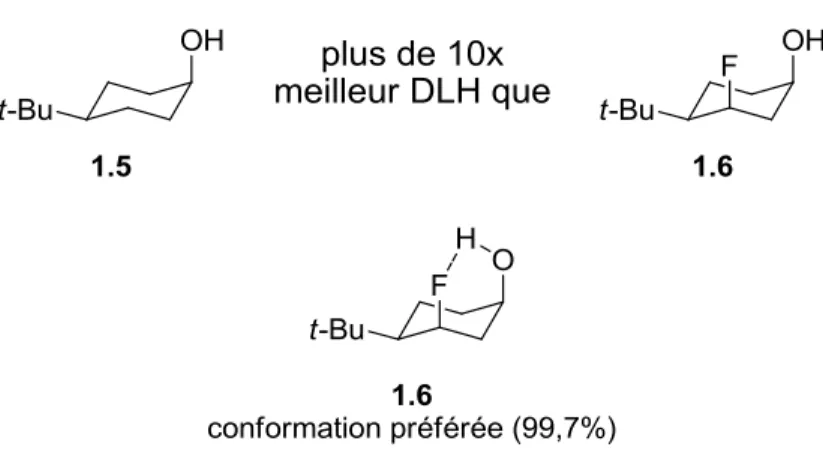
Figure 1.3. Différence de capacité de DLH entre deux alcools et conformation majoritaire du composé 1.6, calculée au niveau de théorie MPWB1K/6-31+G(d,p). ... 32
Figure 1.4. Composé « cage » synthétisé par le groupe de Lectka. ... 33
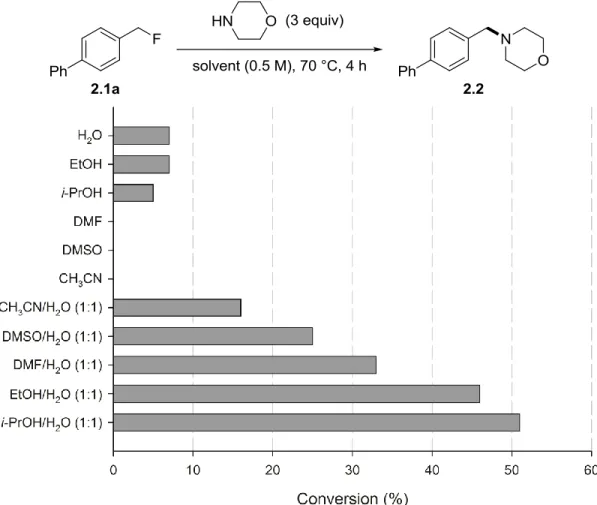
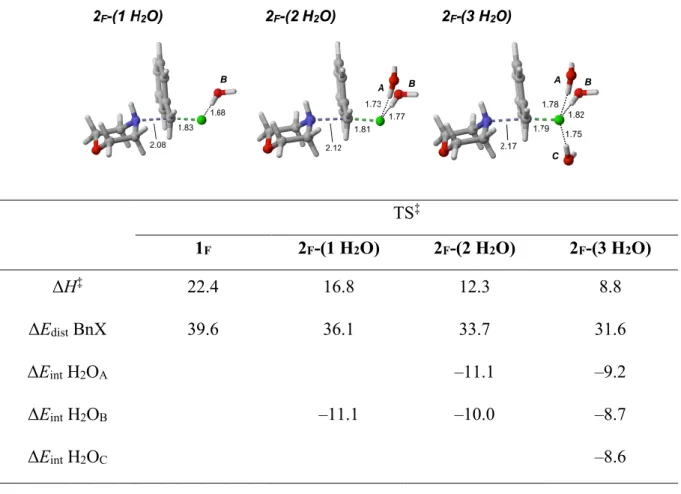
Figure 2.1. Evaluation of the effect of the solvent on the SN2 reaction of benzyl fluoride 2.1a with morpholine. Conversions of 2.1a to 2.2 were estimated by 1H NMR of the crude mixture. ... 40
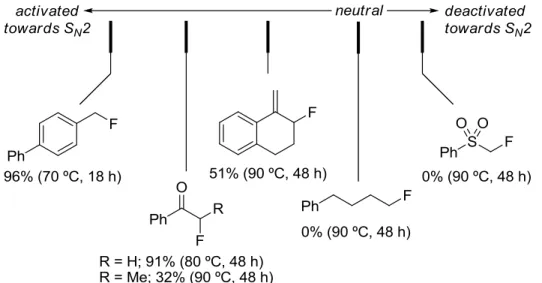
Figure 2.2. Establishing the reactivity of various alkyl fluorides. ... 46
Figure 2.3. Structures des produits secondaires obtenus dans la réaction du substrat bifonctionnel. ... 49
Figure 2.4. Différence énergétique schématisée entre les deux chemins réactionnels pour la réaction d’un fluorure benzylique. ... 54
Figure 3.1. SN2 reaction of activated alkyl fluorides and calculated transition state for the reaction of morpholine with benzyl fluoride with three molecules of water. ... 74
Figure 3.2. Proposed activation of C–F bonds mediated by a triol. ... 74
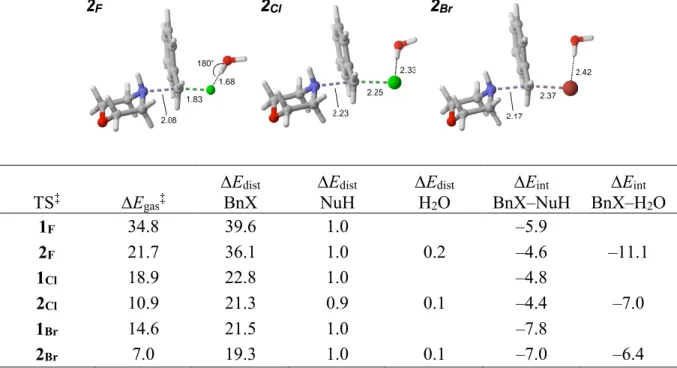
Figure 4.1. Representative TSs of coordination modes of the triol. ... 98
Figure 4.2. M06-2X optimized structure of the fluoride anion-triol adduct. ... 99
Figure 4.3. Improved proposed mechanism for the substitution of 4.1 promoted by 1,1,1-tris(hydroxymethyl)propane. R–O–H represents an alcohol from another triol molecule. ... 106
Figure 4.4. Structures des catalyseurs envoyés par le groupe de B. Bibal. ... 111
Figure 5.1. Conversion RMN 1H en fonction du temps de réaction, pour les systèmes d’activation au HFIP ou au TFA. ... 146
Figure 6.1. Exemples de fluorures benzyliques primaires achiraux ou chiraux. ... 201
Figure 6.2. Spectres RMN 2H en matrice chirale γ-PBLG : CHCl3. (a) (R)-6.11a synthétisé à partir de (S)-6.10a sans TMS-morpholine. (b) (R)-6.11a. (c) (S)-6.11b. Figure reproduite de la référence 266 et légèrement modifiée; les structures ont été ajoutées pour faciliter la compréhension. ... 204
Figure 6.3. Spectres RMN 2H (76,7 MHz) en matrice de γ-PBLG (13% dans CHCl3) des deux échantillons du composé 6.13 obtenus à partir de la réaction de Friedel-Crafts de (a) (R)-6.11a et (b) (S)-6.11b. ... 210
Figure 6.4. Structure confirmée du produit secondaire de la réduction. ... 215
Figure 6.5. Structure de 6.23, déterminée par diffraction des rayons X. ... 217
Figure 6.6. Spectre RMN 2H {1H} du composé (S)-6.13 dans une matrice chirale de γ-PBLG (13% dans CHCl3). ... 218
Liste des schémas
Schéma 1.1. Mécanismes classiques de substitution nucléophile aromatique (SNAr) ou de substitution
nucléophile vinylique (SNV), où les molécules fluorées sont de bons électrophiles. ... 5
Schéma 1.2. Exemples d’activation C–F à l’aide d’acides de Lewis classiques.- ... 8
Schéma 1.3. Exemples représentatifs de réactions d’activation C–F catalysées par des acides de Brønsted., 11 Schéma 1.4. Activation de liens C–F à l’aide de cations silylium. A : Principe général de la génération catalytique de cations silylium; B-D : Exemples tirés de la littérature.- ... 13
Schéma 1.5. Bris d’une liaison C(sp3)–F par l’action d’un cation aromatique formé à partir d’un aryle diazonium. ... 15
Schéma 1.6. Activation C–F par transfert monoélectronique d’un sel de samarium. ... 16
Schéma 1.7. Substitution d’un seul fluor par une combinaison bimétallique. ... 17
Schéma 1.8. Exemples représentatifs d’activation de liens C–F aliphatiques par des complexes de métaux de transition.- ... 18
Schéma 1.9. Exemples récents d’activation de composés aliphatiques fluorés en conditions basiques.- ... 20
Schéma 1.10. Ordre de basicité des amines synthétisées par Johnson et Lectka, ainsi que l’explication pour la basicité supérieure de 1.3. ... 31
Schéma 2.1. Réaction envisagée d’un composé bifonctionnel. ... 47
Schéma 2.2. Synthèse du substrat bifonctionnel. ... 48
Schéma 2.3. Premier essai pour la réaction du substrat bifonctionnel. ... 49
Schéma 2.4. Second essai de la réaction séquentielle du substrat bifonctionnel. ... 50
Schéma 2.5. Dernier essai de la réaction du substrat bifonctionnel. ... 50
Scheme 4.1. C–F bond activation by water and calculated transition state (A); Activation promoted by a triol and its proposed coordination to the fluorine atom (B). ... 96
Scheme 5.1. A previous study, an unexpected result, and the goal of this study... 124
Scheme 5.2. Benzylation of 5.3 with various arenes. The yields given are for the isolated product. The ratio of regioisomers was determined by 1H NMR spectroscopic analysis and is given in parentheses with the major isomer shown. [a] The solvent used was HFIP/benzene (1:9). [b] The reaction was carried out with 10 equivalents of the arene. [c] Reaction conditions: HFIP/DCE (1:9), N-methylpyrrole (10 equiv), 60 °C. [d] The solvent used was HFIP/fluorobenzene (1:9). [e] Ratio of the para:ortho:meta isomers. ... 126
Scheme 5.3. Benzylation of various benzyl fluorides with p-xylene. The yields given are for the isolated product. [a] The solvent used was HFIP/CH2Cl2 (1:9). [b] Reaction conditions: HFIP/DCE (1:1), 60 °C. [c] The solvent used was HFIP/CH2Cl2 (1:1). [d] Decomposition was observed. [e] No reaction was observed. [f] The solvent used was HFIP/CH2Cl2 (9:1). [g] The reaction was carried out with 10 equivalents of p-xylene. [h] The solvent used was HFIP (100%). [i] Reaction conditions: p-xylene (20 equiv), HFIP/CH2Cl2 (1:9, 0.1 M). ... 128
Scheme 5.4. Probing the reaction mechanism. ... 130
Scheme 5.5. Mechanistic hypothesis. ... 131
Scheme 5.6. Selective activation of a C–F bond in the presence of a similar C–Cl bond. ... 134
Schéma 5.7. (a) Voie actuelle de synthèse des diarylméthanes par isolation de fluorures benzyliques, puis réaction de Friedel-Crafts. (b) Voie proposée impliquant la génération in situ de fluorures benzyliques par déoxofluoration d’alcools correspondants. ... 136
Schéma 5.8. Synthèse de 1,1,1-triarylméthanes.a,b ... 139
Schéma 6.1. Représentation de la réaction hypothétique du substrat benzylique chiral 6.1 avec un nucléophile NuH par les mécanismes SN1 et SN2, incluant l’intermédiaire et l’état de transition correspondants. Les DLHs
activateurs sont omis. ... 192
Schéma 6.2. Synthèse de l’α-fluorocétone énantioenrichie 6.5. ... 195
Schéma 6.3. Synthèse de la propiophénone fluorée racémique. ... 196
Schéma 6.4. Substitution des cétones fluorées par la pipéridine. ... 196
Schéma 6.5. Synthèse des fluorures benzyliques secondaires racémique ou énantioenrichi. ... 198
Schéma 6.6. Réaction tentée du fluorure benzylique secondaire énantioenrichi. ... 200
Schéma 6.7. Synthèse de fluorures benzyliques deutérés énantiopurs, rapportée par le groupe de O’Hagan. ... 202
Schéma 6.8. Réactions des fluorures benzyliques énantiopurs par divers modes d’activation. (a)(b) Substitution par la morpholine activée par l’eau; (c)(d) Substitution par la morpholine activée par un triol; (e)(f) Réaction de Friedel-Crafts promue par le HFIP. ... 206
Schéma 6.9. Synthèse des produits 6.16 à 6.19. ... 209
Schéma 6.10. Voie de synthèse envisagée vers le composé racémique (rac)-6.13. ... 211
Schéma 6.11. Substitution énantiospécifique par un énolate de lithium d’un alcool benzhydrylique chiral activé sous la forme d’un tosylate. ... 212
Schéma 6.12. Conditions optimisées pour la réduction de l’alcool benzhydrylique. ... 215
Schéma 6.13. Voie de synthèse énantiosélective du diarylméthane deutéré 6.13. ... 216
Schéma 6.14. Réactions de Friedel-Crafts partiellement énantiospécifiques des fluorures benzyliques deutérés chiraux 6.11. ... 219
Schéma 6.15. Ionisation d’un fluorure benzylique en plusieurs étapes, et conséquences de l’attaque du nucléophile sur chacun des intermédiaires... 220
Schéma 6.16. Réaction étudiée et effets isotopiques potentiellement utilisés. ... 224
Schéma 6.17. (a) Réaction de compétition entre deux fluorures benzyliques isotopologues. (b) Spectre RMN 19F (126 MHz) du mélange des isotopologues, démontrant une bonne séparation des déplacements chimiques en fonction de l’isotope adjacent. ... 228
Schéma 6.18. Synthèse du fluorure benzylique mono-deutéré. ... 232
Schéma 7.1. Mécanisme SNAr typique pour la réaction de substitution nucléophile aromatique d’un fluorure d’aryle. ... 266
Schéma 7.2. Réaction de Friedel-Crafts potentielle de fluorures d’alkyle activés. ... 269
Schéma 7.3. Réaction d’un fluorure benzylique avec l’allytriméthylsilane. ... 269
Liste des tableaux
Tableau 1.1. Enthalpies de dissociation (énergies) de liaisons entre le fluor et les atomes avec lequel
l’activation C–F se produit, en ordre croissant de numéro atomique., ... 25
Table 2.1. DFT Analysis of the SN2 Reaction of Benzyl Halides with Morpholine: Strain Model Analysis of the SN2 Reaction Transition Structures Including Explicit Water Molecules ... 43
Table 2.2. Effect of Water Molecules on the Stabilization of the Transition Structure of SN2 Reaction with BnF ... 45
Table 2.3. SN2 reaction of 4-phenylbenzyl fluoride (2.1a) with N-, O-, S- and C-nucleophiles. ... 62
Table 3.1. Initial Screening. ... 76
Table 3.2. Fine-tuning. ... 78
Table 3.3. Substrate scope. ... 79
Table 4.1. Relative energies, hydrogen-bond distances and angles for the calculated transition state structures of Figure 4.1. ... 99
Table 4.2. Substitution of 4.1 using different OH donors. ... 100
Table 4.3. Substitution of 4.1 using various triols and carbohydrate derivatives. ... 102
Table 4.4. Substitution of 4.1 using various diols and dimethoxyethane. ... 104
Table 4.5. Substitution of 4.1 using various primary, secondary and tertiary alcohols. ... 105
Table 4.6. Initial results for catalytic triol activation of 4.1. ... 108
Tableau 4.7. Résultats obtenus à l’aide des catalyseurs du groupe de B. Bibal. ... 113
Table 5.1. Comparison of different leaving groups. ... 133
Tableau 5.2. Sélection de résultats d’optimisation. ... 138
Tableau 5.3. Effet de la catalyse acide sur la réactivité de 5.1 avec le p-xylène... 142
Tableau 5.4. Quelques exemples de réaction de Friedel-Crafts des fluorures benzyliques avec le p-xylène, activée par le TFA. ... 144
Table 5.5. Selected optimization data for the Friedel-Crafts benzylation of 5.3 with p-xylene. ... 168
Table 5.6. Results of selectivity experiments. ... 188
Tableau 6.1. Réaction de (rac)-6.8 avec la morpholine, en fonction des conditions. ... 199
Tableau 6.2. Tentatives de réduction de l’alcool benzhydrylique ... 214
Tableau 6.3. Quelques exemples de masses réduites de liaisons chimiques. ... 223
Tableau 6.4. Effets isotopiques secondaires du deutérium-α lors de réactions de substitution nucléophile d’halogénures d’alkyle. ... 229
Tableau 6.5. Temps de relaxation T1 en RMN 19Fdes trois espèces fluorées. ... 234
Tableau 6.6. Effets isotopiques (kH/kD) secondaires α de la substitution nucléophile de fluorures benzyliques par la pyrrolidine, promue par l’eau. ... 236
Liste des abréviations
Ac acétyle Ar aromatique Boc tert-butyloxycarbonyle Bn benzyle BSA N,O-bis(triméthylsilyl)acétamide COD cyclooctadièneCSA acide camphresulfonique
Cp cyclopentadiényle
DAST trifluorure de diéthylaminosoufre DBU 1,8-diazabicyclo[5.4.0]undéc-7-ène DCC dicyclohexylcarbodiimide
DCE 1,2-dichloroéthane
DFT théorie de la fonctionnelle de densité
DHP dihydropyrane
DLH donneur de liaisons hydrogène DMAP N,N-diméthyl-4-aminopyridine DMF N,N-diméthylformamide DMSO diméthylsufoxyde DMTFAc N,N-diméthyl-1,1,1-trifluoroacétamide DPEN 1,2-diphényléthylènediamine DPEPhos (oxydi-2,1-phénylène)bis(diphénylphosphine) dppf diphénylphosphinoferrocène EDCI 1-éthyl-3-(3-diméthylaminopropyl)carbodiimide ee excès énantiomérique équiv équivalent er ratio énantiomérique es énantiospécificité
ET (TS) état de transition (transition state)
GEA (EWD) groupe électroattracteur (electron-withdrawing group) GED (EDG) groupe électrodonneur (electron-donating group)
HBD hydrogen-bond donor
HFIP 1,1,1,3,3,3-hexafluoropropan-2-ol HMDS hexaméthyldisilazane
HPLC chromatographe en phase liquide à haute pression NMR nuclear magnetic resonance
Nu nucléophile
Ph phényle
pin pincacolato
RMN résonance magnétique nucléaire RT room temperature
Tf trifluorométhanesulfonyle
TFA acide trifluoroacétique TFE 2,2,2-trifluoroéthanol THF tétrahydrofurane THP tétrahydropyranyle T. M. tamis moléculaire TMS triméthylsilyle Ts p-toluènesulfonyle
Remerciements
Considérant que la présente thèse est l’accumulation de longues années de travail en équipe, je n’ai pas l’intention d’être bref dans mes remerciements mais plutôt d’encenser tous ceux et celles qui ont aidé à la réalisation de cette œuvre.
J’aimerais tout d’abord remercier chaleureusement mon directeur de thèse, Jean-François Paquin. Derrière le stagiaire désordonné et arrogant que j’étais en 2008, tu as su reconnaître mon potentiel et tu as tout fait pour me permettre de l’atteindre. Aujourd’hui, si je suis un scientifique plus créatif, plus curieux, plus critique et plus rigoureux, c’est grâce aux longues et passionnées discussions scientifiques que nous avons souvent eues; elles me manqueront cruellement. Merci surtout de ta confiance et de ton aide constantes, qui auront contribué à faire de moi le chercheur que je suis maintenant. Cette incroyable générosité qui t’anime m’a inspiré à devenir moi-même une meilleure personne et j’espère pouvoir rendre à d’autres le support que j’ai reçu de ta part.
Un énorme merci à mon mentor et ami, Grégory. À tous ceux qui ont voulu l’entendre, j’ai répété que tu es la personne qui m’a tout appris, et je le crois sincèrement. Tout au long de mes études, tu m’as guidé et je t’ai toujours considéré comme le meilleur maître duquel il soit possible d’apprendre. Tu es une source inépuisable de connaissances, de conseils et de bonne humeur et je suis très chanceux d’avoir pu te suivre pendant si longtemps.
Merci à ma bonne amie Marie-France pour ton amitié pendant toutes nos années ensemble aux études graduées. Ton attitude fonceuse et travaillante a été d’une grande inspiration pour moi. J’aspire encore aujourd’hui à atteindre ton niveau de détermination, mais je me suis grandement amélioré grâce à toi!
J’aimerais aussi remercier Yasmine, qui a été la personne la plus gentille, la plus attentionnée, la plus altruiste et la plus rayonnante qu’il m’a été donné de rencontrer au cours de ma vie. Dès ton arrivée, tu as su inspirer confiance à tous par ton sourire et ton écoute et tu as semé de la bonne humeur partout sur ton passage. C’est incroyable tout ce qu’on a vécu et traversé ensemble! Je suis content de te compter parmi mes amis!
Merci à tous ceux avec qui j’ai collaboré étroitement au laboratoire et spécialement aux stagiaires qui ont travaillé sur les multiples projets auxquels j’ai contribué. Parfois les résultats ont été au rendez-vous et parfois non, mais vous avez toujours su rester positifs et persévérants. Marie-Ève, Julien, Yasmine, Myriam, Alexandre, Clément, Mélina, Samuel et Olivier, vous êtes tous des personnes exceptionnelles et des scientifiques intelligents. Je suis choyé d’avoir pu travailler en si bonne compagnie et je n’ai nul doute que vous prouverez tous vos qualités individuelles dans de grandes réalisations personnelles et professionnelles au fil des années.
Merci à tous mes collègues au sein du groupe Paquin, présents ou passés. À tous vous côtoyer, j’ai pris conscience que l’ambiance de travail au sein d’une équipe est primordiale, mais aussi qu’elle ne se crée pas toute seule. Vous avez tous contribué à rendre cette ambiance agréable et je vous souhaite encore de beaux moments de groupe après mon départ. Merci pour toutes les activités parascolaires qui nous ont rapprochés et pour les bruyantes périodes d’hilarité au bureau ou au laboratoire. Ces moments ont rendu le travail amusant et m’ont motivé à y venir tous les matins (de la semaine) depuis de nombreuses années. J’appréciais particulièrement l’ambiance de famille qui règne dans le groupe et je suis très heureux d’avoir pu être votre doyen pendant quelques temps! Merci à l’équipe revue (JD, Justine et Mathilde) pour avoir réussi à ne pas s’arracher mutuellement la tête malgré les demandes des évaluateurs! Un merci tout spécial à Myriam et Elsa, dont l’amour
du café m’a convaincu à adhérer à leur pratique et a clairement sauvé ma dernière année quasi-entièrement consacrée à la rédaction!
Merci à tous les employés du département, qui ont toujours été des plus courtois et aidants. Parmi ceux avec qui j’ai interagi le plus et qui méritent une mention, notons Pierre Audet, Jean Laferrière, Christian Côté, Mélanie Tremblay, Denyse Michaud et Marie Tremblay.
Merci à tous mes amis, qu’ils soient au sein du département ou à l’extérieur de l’Université. Pour ceux à l’interne, merci pour les discussions scientifiques et les bons moments lorsqu’on s’est retrouvés ensemble sur différents comités. Pour les autres, merci de m’avoir encouragé à poursuivre mes études, même si ça faisait déjà bien longtemps que j’étais sur les bancs d’école! Une mention spéciale aux gars de l’ADF (Charles, David et Benjamin) s’impose, eux qui ont fait de moi la personne que je suis aujourd’hui. Jamais je n’aurai la chance d’être dans un groupe de gens aussi brillants, aussi hilarants et aussi supportifs que vous. Les trois années que l’on a passées ensemble resteront pour toujours parmi les plus belles de ma vie!
Merci à mes parents pour leur support inconditionnel tout au long de mes études. Bien avant que j’en prenne conscience, vous aviez réalisé mon potentiel et vous m’avez toujours poussé au-delà de mes limites pour que je m’améliore. Vous m’avez encouragé dans tous les projets un peu débiles que j’ai entrepris, que ce soit un voyage en sac à dos à l’autre bout du monde, ou un doctorat en chimie. Je crois qu’on peut maintenant dire que vos efforts et vos sacrifices ont porté fruit et je vous en serai éternellement reconnaissant. Je vous aime.
Finalement, j’aimerais remercier la plus importante de toutes, Laurence. Jamais je ne me suis senti aussi prêt à affronter les épreuves qui m’attendent à la sortie de l’Université, que
maintenant que je suis avec toi. Ton amour sans bornes et ton support inconditionnel sont des certitudes sur lesquelles j’ai pu prendre appui pour progresser, tant au niveau personnel que professionnel. J’espère pouvoir être à la hauteur de ton amour et te le rendre pour encore de longues années. Je t’aime et merci pour tout!
Avant-propos
La présente thèse est divisée en 7 chapitres, dont cinq principaux décrivent de la recherche originale. Pour tous les chapitres tirés d’articles scientifiques, le texte et les figures ont été recopiées sans modifications, sauf bien sûr pour adapter la numérotation des schémas, tableaux, figures et molécules.
Le chapitre 2 est basé sur un article scientifique paru dans la revue Organic Letters et officiellement publié en ligne le 24 avril 2013. Ce travail a été réalisé avec différents étudiants stagiaires au sein du groupe, et en collaboration avec le groupe d’un autre chercheur. Julien Pomarole était un stagiaire de l’Université de Nantes, Marie-Ève Thérien une stagiaire de l’Université Laval et Yasmine Benhassine une étudiante en stage pré-maîtrise. Samuel Beaulieu était stagiaire au sein du groupe du Pr. Claude Y. Legault, à l’Université de Sherbrooke. Les trois premiers ont aidé à la synthèse des substrats et à la réalisation des expériences. Les deux derniers ont effectué les calculs DFT et écrit la section du texte s’y rapportant. Quant à moi, j’ai réalisé la majorité des expériences et j’ai participé à l’écriture en tant qu’auteur partiel, avec mon directeur Jean-François Paquin. J’ai aussi préparé le document d’informations supplémentaires. Les annexes du chapitre décrivent des travaux que j’ai réalisés moi-même.
Le chapitre 3 est basé sur une publication parue dans la revue Beilstein Journal of Organic
Chemistry qui a été publiée le 13 novembre 2013. Ce travail a été entièrement réalisé au
sein du groupe de recherche. Alexandre Saint-Martin était un étudiant stagiaire de l’Université de Poitiers qui a réalisé une partie des expériences préliminaires. Mélina Drouin était stagiaire de l’Université Laval et a aidé à évaluer l’étendue de la réaction. Quant à moi, j’ai planifié et réalisé les expériences. Avec les directives de mon directeur, j’ai aussi écrit l’article à titre d’auteur principal et colligé tout le document d’informations supplémentaires. Les annexes sont des résultats que nous avons obtenus en collaboration
avec le groupe de la Pr. Brigitte Bibal, de l’Université de Bordeaux. Celui-ci nous a fourni les composés mais j’ai réalisé les expériences moi-même.
Le chapitre 4 provient d’un article soumis au Journal of Fluorine Chemistry et officiellement publié le 6 septembre 2014. La contribution du Pr. Claude Legault a été nécessaire, car il a effectué les calculs DFT sur le système et composé le texte s’y attachant. Mélina Drouin, encore stagiaire, a réalisé une majorité des expériences présentées, tandis que Clément Audubert, stagiaire de l’Université de Reims, signe quelques résultats utilisant les sucres comme activateurs. De mon côté, j’ai principalement dirigé les travaux, et participé au laboratoire en effectuant une partie des expériences. À toute fin pratique, j’ai complètement écrit le texte de la publication et sa partie expérimentale, sous la supervision de mon directeur.
Le chapitre 5, quant à lui, est une publication parue dans la revue Angewandte Chemie
International Edition et publié en ligne le 10 octobre 2014. Il représente des travaux
réalisés par trois étudiants gradués au sein du groupe. Yasmine Benhassine, étudiante à la maîtrise, a réalisé la plupart des expériences ayant trait à l’étendue de la réaction (substrats et nucléophiles). Justine Desroches, étudiante au doctorat, a contribué ponctuellement à quelques réactions. De mon côté, j’ai réalisé la majorité des expériences, dont la totalité de celles pour élucider le mécanisme de la réaction. En collaboration avec mon directeur, j’ai écrit le texte et le document d’informations supplémentaires de l’article. Les annexes discutent de résultats obtenus dans d’autres projets, où j’ai aidé Justine Desroches ou Rémy Hemelaere, un stagiaire postdoctoral.
Finalement, le chapitre 6 est une addition de différentes explorations que j’ai réalisées seul la plupart du temps et parfois en collaboration avec des groupes de recherche à l’étranger. Bien qu’ils n’aient pas encore été officiellement publiés, les résultats qu’il contient sont très pertinents pour la thèse.
En fonction de ce qui a été dit ci-haut et des multiples scientifiques qui ont contribué à mes travaux, ma thèse est écrite à la première personne du pluriel par respect pour tous ceux avec qui j’ai travaillé : la recherche qui y est présentée a toujours été un travail d’équipe, même lorsque j’ai réalisé les expériences seul.
Chapitre 1
:
Introduction
1.1 Généralités sur le fluor et le lien C–F
1.1.1 Le fluor
Le fluor est le plus léger de la série des halogènes, un atome ne contenant que neuf protons. Le fluor naturel n’est composé que d’un seul isotope, le 19F et celui-ci est retrouvé dans
différents minerais dans la croûte terrestre, à la hauteur de 0,027% m/m.1 Selon les
estimations de Bondi, le rayon de Van der Waals du fluor est de 1,47 Å, ce qui le place au deuxième rang du plus petit atome derrière l’hydrogène, à 1,20 Å.2 Il est l’atome le plus
électronégatif du tableau de classification périodique (3,98 sur l’échelle de Pauling, vs l’oxygène à 3,44), et tout ceci fait en sorte qu’il est aussi le moins polarisable (0,557 Å3 vs
l’hydrogène à 0,667). Le fluor est donc un atome aux propriétés extrêmes.
1.1.2 Le lien C–F
Lorsqu’il forme une liaison covalente avec un atome de carbone, le fluor conserve de ses propriétés exceptionnelles.2 Le lien C–F est la deuxième plus courte liaison après le lien C–
H (1,38 vs 1,09 Å), mais est la liaison simple la plus forte que peut faire le carbone avec n’importe quel atome, avec une énergie de liaison de 486 kJ/mol (CH3–F). Le lien C–F est
donc encore plus fort que le lien C–H (414 kJ/mol) qui est considéré comme relativement inerte, mais surtout beaucoup plus fort que les autres liens carbone–halogène, qui ont des énergies entre 339 et 213 kJ/mol, du chlore vers l’iode.
1 Kirsch, P. Introduction dans Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications;
Wiley-VCH: Weinheim, Germany, 2013, p. 3.
2 Hiyama, T. Organofluorine Compounds; Chemistry and Applications; Yamamoto, H. (Éd.): Springer, 2000,
Cette force de liaison est certainement expliquée par la faible distance entre les atomes et de leur grande différence d’électronégativité qui lui confère un important caractère ionique. La combinaison de toutes ces propriétés fait du lien C–F une liaison fortement polarisée (moment dipolaire de 1,41 D), mais étonnamment très peu polarisable. Le lien C–F peut donc réaliser de bonnes interactions électrostatiques lorsque la situation se présente.
1.1.3 Les composés organofluorés
Les composés organofluorés sont ceux qui contiennent au moins une liaison C–F. Malgré l’abondance du fluorure dans la croûte terrestre, on ne retrouve que 18 composés naturels qui comportent une liaison C–F, alors qu’il en existe des milliers qui soient répertoriés avec un autre halogène, qui sont pourtant tous moins abondants.3 Les principales hypothèses
pour expliquer un tel phénomène sont que les fluorures sont peu disponibles puisqu’ils sont extrêmement stables dans les minerais naturels, ou alors que le F- n’est pas très nucléophile
en milieu aqueux dû à la forte énergie de solvatation.
Ironiquement peut-être, le fluor induit de telles propriétés aux composés organiques qu’il est maintenant primordial dans de nombreux domaines, tels les matériaux comme les cristaux liquides, les polymères fluorés de type Teflon ou les composés pharmaceutiques et agrochimiques.4 À titre d’exemple, on estime que 20 à 25% des molécules en
développement dans les compagnies pharmaceutiques contiennent un atome de fluor, et que ce pourcentage est plus élevé encore dans le domaine agrochimique.5 Dû au grand intérêt
apporté aux composés organofluorés et à l’absence de source naturelle de tels composés, il est clair que la seule solution offerte aux chimistes est de synthétiser eux-mêmes les composés organofluorés désirés. L’approche de construction de la liaison C–F a été
3 Deng, H.; O’Hagan, D.; Schaffrath, C. Nat. Prod. Rep. 2004, 773. 4 Tressaud, A. Angew. Chem. Int. Ed. 2006, 45, 6792.
5 (a) Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320; (b) Müller, K.;
grandement explorée dans les soixante dernières années et maintenant les méthodes pour introduire un atome de fluor dans une molécule organique sont multiples et efficaces.6
Comme les composés organofluorés font partie intégrante de l’environnement chimique, que ce soit dans des composés bioactifs ou non, il est important de bien comprendre comment ces composés se comportent. L’étude de leur réactivité chimique, tout comme l’étude de leurs interactions avec les biomolécules du vivant, ont intéressé plusieurs groupes de chercheurs dans les dernières années. Au courant de la présente thèse, nous ne nous intéresserons qu’aux transformations sur les composés organiques fluorés.
1.2 Activation de liens C–F
1.2.1 Préambule
Dues aux propriétés uniques du lien C–F, dont sa forte énergie de liaison et sa faible polarisabilité, celui-ci est plutôt difficile à briser. C’est pourquoi l’un des domaines qui a intéressé les scientifiques dans les dernières années est celui de l’activation C–F.7 Ce
domaine comprend toutes les transformations qui permettent de faire réagir un fluor organique, que ce soit par une réaction d’élimination ou de substitution nucléophile, qui sont les deux transformations majoritaires qui se font sur ce motif. Bien que l’on ne parle pas d’activation lorsque l’on fait réagir des groupes sortants traditionnels pour ce genre de réactions (p. ex. Cl, Br, I, OTs, OMs, etc.), le statut du fluor comme d’un atome électronégatif non-nucléofuge implique qu’il faut l’assister pour déclencher sa réactivité, d’où le nom du domaine.
6 Pour quelques revues récentes, voir (a) Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem. Int. Ed. 2013,
52, 8214; (b) Ma, J.-A.; Cahard, D. Chem. Rev. 2008, 108, PR1; (c) Furuya, T.; Kamlet, A. S.; Ritter, T. Nature 2011, 473, 470; (d) Yang, X.; Wu, T.; Phipps, R. J.; Toste, F. D. Chem. Rev. 2015, 115, 826; (e)
Campbell, M. G.; Ritter, T. Chem. Rev. 2015, 115, 612; (f) Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073.
Il existe plusieurs types de carbones auxquels peut être attaché un atome de fluor (Figure 1.1). Avec un carbone hybridé sp, on obtient un fluoroalcyne, un motif souvent instable et difficile à synthétiser, dont la réactivité est limitée à des réactions de cycloadditions.6f Avec
un carbone sp2, on peut obtenir deux motifs distincts, soit les aromatiques fluorés ou les fluoroalcènes. Ceux-ci sont très répandus en chimie organique et les systèmes nécessaires pour en faire l’activation C–F ont été parmi les premiers à être découverts.7 Finalement,
lorsqu’un fluor est lié à un carbone sp3, le motif obtenu est un fluor aliphatique, dont les propriétés sont bien différentes des autres motifs. Dues à ces différences entre les motifs, il est important de comprendre que les catalyseurs/activateurs utilisés pour faire réagir le lien C–F seront différents en fonction du motif, et qu’ils seront aussi différents en fonction du nombre de fluors présents sur un même carbone (p. ex. un groupement CH2F vs CF3).
Figure 1.1. Différentes hybridations possibles d’un carbone lié à un fluor.
Très tôt dans un cours classique de chimie organique, on enseigne aux étudiants que le fluor, contrairement à ses collègues halogènes, n’est pas un bon nucléofuge.8 Ceci n’est pas
tout à fait vrai, car les aromatiques fluorés et les difluoroalcènes sont des électrophiles compétents dans des réactions d’addition-élimination nucléophiles (Schéma 1.1).7,9 Dans
ces deux situations, l’étape déterminante de la vitesse n’est pas le bris de la liaison C–F, mais bien l’addition du nucléophile sur le motif insaturé, facilitée par l’effet électronégatif du fluor qui rend l’insaturation plus électrophile. Dans le cas des alcènes fluorés, seuls les motifs difluorés sont suffisamment réactifs et pour les deux types de substrats, les réactions sont considérablement lentes et le fluor est un meilleur groupement sortant que les autres nucléofuges traditionnels.
8 Solomons, T. W. G.; Fryhle, C. B. Organic chemistry; 10ème éd.; Wiley: Hoboken, NJ, 2011. 9 Paquin, J. F.; Landelle, G.; Bergeron, M.; Turcotte-Savard, M.-O. Chem. Soc. Rev. 2011, 40, 2867.
Schéma 1.1. Mécanismes classiques de substitution nucléophile aromatique (SNAr) ou de substitution nucléophile vinylique (SNV), où les molécules fluorées sont de bons
électrophiles.
Comme les motifs C(sp2)–F peuvent déjà réagir dans des réactions de substitution C–F lorsqu’ils sont placés dans des conditions favorables, il n’est pas surprenant que ces deux motifs aient été les premiers sur lesquels l’activation C–F par d’autres méthodes a été tentée en premier.7 Sans entrer dans les détails, nous mentionnerons ici que ces méthodes
incluent l’addition oxydante avec des métaux de transition et les transferts électroniques à l’aide de métaux alcalins ou alcalino-terreux pour les fluoroarènes, tandis qu’il s’agit davantage de réactions d’addition-élimination ou de couplages catalysés aux métaux de transition pour les difluoroalcènes.
Pour les motifs aliphatiques cependant, la simple substitution nucléophile a longtemps été reconnue comme impossible à réaliser dans les conditions réactionnelles qui étaient pourtant utilisées avec les autres halogènes ou pseudo-halogènes.10 Avec ce type de
substrats, les mécanismes de substitution nucléophile sont principalement la SN1 et la SN2,
10 Dörwald, F. Z. dans Side Reactions in Organic Synthesis: A Guide to Successful Synthesis Design;
deux mécanismes qui impliqueraient le bris de la liaison C–F lors de l’étape déterminante de la vitesse, et qui ne sont donc pas favorisés due à la force du lien C–F et à sa faible polarisabilité. Bien sûr, il n’est pas impossible de réaliser de telles réactions, comme nous le démontrerons dans la prochaine section et dans la présente thèse. Cependant, des conditions gagnantes doivent être réunies pour permettre une telle réactivité de la liaison C–F.
1.2.2 Méthodes d’activation C–F de composés aliphatiques
Les travaux réalisés dans le cadre de la thèse se sont concentrés sur la réactivité des composés fluorés aliphatiques monofluorés. C’est pourquoi nous présenterons ci-bas les principales méthodes pour activer de tels composés. Certaines méthodes, réalisées sur des substrats difluorés (R2CF2) ou trifluorés (RCF3) seront aussi présentées dans la prochaine
revue de la littérature. Comme les liaisons C–F dans de tels motifs sont plus fortes que dans un composé monofluoré,2 ces méthodes devraient être aussi applicables aux substrats qui
comportent moins de fluors géminaux. Le contraire n’est pas nécessairement vrai et donc certaines méthodes d’activation de composés monofluorés ne permettent pas l’activation de CF2 ou de CF3. Dépendamment du mécanisme d’activation, la présence de groupements sur
la molécule peut aussi influencer la réactivité. Par exemple, les fluorures benzyliques, allyliques ou tertiaires sont plus réactifs que les fluorures aliphatiques simples.11
1.2.2.1 Activation en conditions acides
La principale façon de déclencher une réaction de substitution nucléophile de fluorures d’alkyle est de les placer dans des conditions acides (de Lewis ou de Brønsted).12 En effet,
le fluor dans un lien C–F est une base de Lewis compétente, mais un accepteur de liaisons
11 Smith, M. B.; March, J. Aliphatic Substitution: Nucleophilic and Organometallic. March’s Advanced
Organic Chemistry: Reactions, Mechanisms and Structure, 6th ed.; Wiley-Interscience: Hoboken, NJ, 2007;
pp. 425-656.
hydrogène (voir section 1.3) et une base de Brønsted plutôt faible.13 Malgré tout, si des
interactions favorables entre l’atome de fluor (et surtout le fluorure éjecté lors de la réaction) et l’acide de Lewis sont possibles, les énergies associées à ces interactions sont suffisantes pour briser la liaison carbone–fluor.
Après quelques exemples d’activation de fluorures de benzyle avec BF3 par le groupe de
Miller,14 Olah explora la généralité de l’activation par acides de Lewis à base de bore pour
faire la substitution sélective d’un lien C–F versus un lien C–Cl (Schéma 1.2A).15 Cette
activation produit un carbocation qui subit un réarrangement et est ensuite trappé par réaction de Friedel-Crafts. Une activation similaire de fluorures tertiaires en présence d’éthers d’énols silylés a permis au groupe d’Oshima d’obtenir des cétones α-fonctionnalisées.16 Plus récemment, Caputo et Stephan ont exploré la réactivité de B(C
6F5)3
pour l’activation de fluorures d’alkyle simples en présence d’un silane comme nucléophile, générant des alcanes saturés en de courts temps réactionnels (Schéma 1.2B).17
13 Le pK
a dans l’eau du super acide H2F+ n’a jamais été mesuré. Cependant, on peut l’approximer à environ
-12 en fonction de la constante d’autodissociation du HF pur (50 M). Le pKa de RFH+ devrait être d’un ordre
de grandeur similaire si R = aliphatique. Voir a) Toteva, M. M.; Richard, J. P. J. Am. Chem. Soc. 2002, 124, 9798; b) Gillespie, R. J.; Liang, J. J. Am. Chem. Soc. 1988, 110, 6053.
14 a) Bernstein, J.; Roth, J. S.; Miller, W. T. J. Am. Chem. Soc. 1948, 70, 2310; b) Miller, W. T.; Bernstein, J.
J. Am. Chem. Soc. 1948, 70, 3600.
15 a) Olah, G. A.; Kuhn, S. J. J. Org. Chem. 1964, 29, 2317; b) Olah, G. A.; Kobayashi, S.; Tashiro, M. J. Am.
Chem. Soc. 1972, 94, 7448.
16 Hirano, K.; Fujita, K.; Yorimitsu, H.; Shinokubo, H.; Oshima, K. Tetrahedron Lett. 2004, 45, 2555. 17 Caputo, C. B.; Stephan, D. W. Organometallics 2012, 31, 27.
Les acides de Lewis à base d’aluminium ont aussi connu un grand succès pour l’activation C–F sélective, débutant avec les travaux de Maruoka sur les fluorures tertiaires.18 Les
alanes sont effectivement très utiles, car en plus de l’interaction favorable C–F···Al, les groupements sur l’aluminium agissent comme nucléophiles. Ainsi, peu importe les substrats et la quantité de fluor présents, l’utilisation d’alanes variés permet d’obtenir des produits de réactions variés (Schéma 1.2C).19 Les mécanismes de bris du lien C–F, dans ces
cas, sont probablement des ionisations de type SN1 avec formation de carbocation. Il est
cependant aussi possible d’utiliser un réactif à base d’aluminium pour faire la substitution SN2’ de 3,3-difluoropropènes fonctionnalisés (Schéma 1.2D).20
Plus récemment, le groupe de Hilmersson a publié des articles décrivant l’activation C–F de composés aliphatiques à l’aide de sels de lanthanides. Dans leurs systèmes, la réaction de substitution nucléophile semble se produire par un mécanisme SN2, car les substrats les
moins encombrés réagissent plus rapidement. Ainsi, en utilisant YbI3, on peut remplacer
des fluorures aliphatiques par des iodes (Schéma 1.2E),21 tandis qu’avec La[N(SiMe 3)2]3 il
est possible de substituer des fluorures primaires par des amines secondaires.22
L’activation C–F s’est aussi trouvé une niche importante pour les réactions de glycosylations, où les fluorures de glycosyle sont préférés aux autres halogénures correspondants puisqu’ils sont plus stables et donc plus faciles à manipuler.23 Depuis leur
première utilisation en synthèse par Mukaiyama en 1981 (Schéma 1.2F),24 une multitude de
systèmes activateurs ont été développés,25 incluant entre autres SnCl
2/AgClO4,24
18 Ooi, T.; Uraguchi, D.; Kagashima, N.; Maruoka, K. Tetrahedron Lett. 1997, 38, 5679.
19 a) Terao, J.; Begum, S. A.; Shinohara, Y.; Tomita, M.; Naitoh, Y.; Kambe, N. Chem. Commun. 2007, 855;
b) Ali, M.; Liu, L.-P.; Hammond, G. B.; Xu, B. Tetrahedron Lett. 2009, 50, 4078; c) Gu, W. X.; Haneline, M. R.; Douvris, C.; Ozerov, O. V. J. Am. Chem. Soc. 2009, 131, 11203; d) Terao, J.; Nakamura, M.; Kambe, N.
Chem. Commun. 2009, 6011.
20 Yanai, H.; Okada, H.; Sato, A.; Okada, M.; Taguchi, T. Tetrahedron Lett. 2011, 52, 2997. 21 Träff, A. M.; Janjetovic, M.; Ta, L.; Hilmersson, G. Angew. Chem. Int. Ed. 2013, 52, 12073. 22 Janjetovic, M.; Träff, A. M.; Hilmersson, G. Chem. Eur. J. 2015, 21, 3772.
23Carmona, A. T.; Moreno-Vargas, A. J.; Robina, I. Curr. Org. Synth. 2008, 5, 33.
24 Mukaiyama, T.; Murai, Y.; Shoda, S. Chem. Lett. 1981, 431. 25 Mukaiyama, T. Angew. Chem. Int. Ed. 2004, 43, 5590.
BF3•OEt2,26 Cp2MCl2/AgClO4 (M = Ti, Zr, Hf),27 Hf(OTf)4,28 et La(ClO4)3•nH2O.29 Même
des acides forts comme TfOH30 et différents carbocations ont été utilisés.31
L’effet des acides de Brønsted sur les composés fluorés aliphatiques a été remarqué il y a plus de 60 ans. En 1948, Miller a démontré que l’alcoolyse des fluorures benzyliques était grandement accélérée par l’ajout de HCl dans la solution, un effet qui n’est pas observé lors de la solvolyse des chlorures correspondants.32 En absence d’un solvant nucléophile, la
substitution du fluor par un chlorure est observée. Avec l’acide perchlorique, l’hydrolyse de ces composés benzyliques se produit aussi dans un mélange acétone/H2O.33 Le groupe de
Levy a enchaîné en s’intéressant à l’hydrolyse du fluorure de tert-pentyle dans un mélange EtOH/H2O, possible seulement en présence de HCl.34 Toutes ces réactions procédaient par
l’ionisation du lien C–F pour former un cation comme intermédiaire réactionnel. Comme les acides mentionnés ici ne sont pas assez forts pour formellement protoner le lien C–F, les auteurs des études citées ci-haut ont tous proposé que le rôle de l’acide activateur est de faire des liaisons hydrogène avec le fluor, engendrant un affaiblissement de la liaison C–F et une meilleure stabilisation du fluorure lorsqu’il serait éjecté. À cette époque cependant, il était difficile de prouver une telle hypothèse.
26 a) Nicolaou, K. C.; Chucholowski, A.; Dolle, R. E.; Randall, J. L. J. Chem. Soc. Chem. Commun. 1984,
1155; b) Kunz, H.; Waldmann, H. J. Chem. Soc. Chem. Commun. 1985, 638; c) Kunz, H.; Sager, W. Helv.
Chim. Acta 1985, 68, 283; d) Yamaguchi, M.; Horiguchi, A.; Fukuda, A.; Minami, T. J. Chem. Soc. Chem. Commun. 1990, 1079.
27 a) Matsumoto, T.; Maeta, H.; Suzuki, K.; Tsuchihashi, G. Tetrahedron Lett. 1988, 29, 3567, 3571, 3575; b)
Suzuki, K.; Maeta, H.; Matsumoto, T. Tetrahedron Lett. 1989, 30, 4853; c) Suzuki, K.; Matsumoto, T. J.
Synth. Org. Chem. Jpn. 1993, 51, 718.
28 Manabe, S.; Ito, Y. J. Org. Chem. 2013, 78, 4568.
29 Kim, W.-S.; Hosono, S.; Sasai, H.; Shibasaki, M. Tetrahedron Lett. 1995, 36, 4443.
30 a) Mukaiyama, T.; Takeuchi, K.; Jona, H.; Maeshima, H.; Saitoh, T. Helv. Chim. Acta 2000, 83, 1901; b)
Mukaiyama, T.; Jona, H.; Takeuchi, K. Chem. Lett. 2000, 696; c) Jona, H.; Takeuchi, K.; Mukaiyama, T.
Chem. Lett. 2000, 1278; d) Jona, H.; Mandai, H.; Mukaiyama, T. Chem. Lett. 2001, 426; e) Jona, H.; Mandai,
H.; Chavasiri, W.; Takeuchi, T.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2002, 75, 291.
31 Yanagisawa, M.; Mukaiyama, T. Chem. Lett. 2001, 224.
32 a) Bernstein, J.; Roth, J. S.; Miller, W. T. J. Am. Chem. Soc. 1948, 70, 2310; b) Miller, W. T.; Bernstein, J.
Am. Chem. Soc. 1948, 70, 3600.
33 Swain, C. G.; Spalding, R. E. T. J. Am. Chem. Soc. 1960, 82, 6104. 34 Chapman, N. B.; Levy, J. L. J. Chem. Soc. 1952, 1677.
D’un point de vue plus synthétique, quelques articles sur l’activation de fluorures aliphatiques par action d’acides sont parus plus récemment. Par exemple, Barrio a montré qu’on pouvait faire la substitution de fluorures d’alkyle simples par les autres halogénures en utilisant l’acide halogénohydrique correspondant (Schéma 1.3A), un protocole qui fonctionne sur une variété de composés monofluorés.35 De plus, deux groupes distincts ont
publié exactement la même réaction d’activation de groupement aryl–CF3 en cétones
diaromatiques par l’acide trifluorométhanesulfonique (Schéma 1.3B), et ce, à plusieurs années d’intervalle.36
Schéma 1.3. Exemples représentatifs de réactions d’activation C–F catalysées par des
acides de Brønsted.35,36
Le domaine d’activation C–F en conditions acides qui a le plus été exploré dans les dernières années est cependant l’activation par les cations silylium, une espèce cationique très électrophile, et dû à la forte affinité entre le silicium et le fluor,2 très fluorophile.
Contrairement aux acides de Lewis comme BF3 ou AlCl3, les cations silylium ne sont pas
des espèces facilement disponibles en quantité stœchiométrique. Il fallait donc trouver une façon de les générer à l’aide d’un catalyseur, une percée majeure qui a été réalisée par le groupe de Ozerov en 2005 et dans les années subséquentes.37
35 Namavari, M.; Satyamurthy, N.; Phelps, M. E.; Barrio, J. R. Tetrahedron Lett. 1990, 31, 4973.
36 a)Wang, F.; Hu, J. Chin. J. Chem. 2009, 27, 93; b) Kethe, A.; Tracy, A. F.; Klumpp, D. A. Org. Biomol.
Chem. 2011, 9, 4545.
37 a) Scott, V. J.; Çelenligil-Çetin, R.; Ozerov, O. V. J. Am. Chem. Soc. 2005, 127, 2852; b) Ozerov, O. V.;
Douvris, C. Science 2008, 321, 1188; c) Douvris, C.; Nagaraja, C. M.; Chen, C. H.; Foxman, B. M.; Ozerov, O. V. J. Am. Chem. Soc. 2010, 132, 4946.
L’idée est simple (Schéma 1.4A) : en utilisant un silane stœchiométrique et un cation en quantité catalytique, on peut obtenir un cycle catalytique où les cations silylium, l’espèce active qui s’attaque aux liens C–F, est regénérée au fil de la réaction, provoquant une activation complète des liens carbone-fluor aliphatiques. Avec un tel système, les chercheurs ont réussi l’hydrodéfluoration complète de perfluoroalcanes, au départ d’un cation trityle muni d’un contre-ion carborane non-nucléophile (Schéma 1.4B).37b La
présence de produits de réarrangements indique que des intermédiaires cationiques sont bel et bien en jeu, même pour les fluors sur des carbones primaires.
D’autres initiateurs cationiques ont aussi été utilisés par d’autres groupes de recherche. Par exemple, un cation disilylhydronium (Schéma 1.4C)38 ou fluorophosphonium39 permettent
d’initier le cycle catalytique directement par un évènement d’activation C–F. On peut aussi déclencher la réaction par une quantité catalytique d’un complexe cationique de ruthénium,40 à l’aide d’une surface composée de chlorofluorures d’aluminium qui ont de
bonnes affinités pour les silanes,41 ou avec des silylènes N-hétérocycliques sur des substrats
bien spécifiques.42
38 a) Panisch, R.; Bolte, M.; Müller, T. J. Am. Chem. Soc. 2006, 128, 9676; b) Lühmann, N.; Panisch, R.;
Müller, T. Appl. Organomet. Chem. 2010, 24, 533.
39 Caputo, C. B.; Hounjet, L. J.; Dobrovetsky, R.; Stephan, D. W. Science 2013, 341, 1374. 40 Stahl, T.; Klare, H. F. T.; Oestreich, M. J. Am. Chem. Soc. 2013, 135, 1248.
41 Ahrens, M.; Scholz, G.; Braun, T.; Kemnitz, E. Angew. Chem. Int. Ed. 2013, 52, 5328. 42 Azhakar, R.; Roesky, H. W.; Wolf, H.; Stalke, D. Chem Commun. 2013, 49, 1841.
Schéma 1.4. Activation de liens C–F à l’aide de cations silylium. A : Principe général de la
Finalement, le groupe de Shibata a récemment publié une étude intéressante sur le dédoublement cinétique de fluorures allyliques par trifluorométhylation énantiosélective (Schéma 1.4D).43 Selon les auteurs, la réactivité provient d’une interaction entre le réactif
de Ruppert-Prakash (CF3SiMe3) et l’atome de fluor, qui permet le bris C–F sélectif d’un
des énantiomères du substrat, générant du même coup un anion CF3- qui s’additionne par la
suite sur la molécule.
En plus d’être pour la plupart très actifs et capables d’activer n’importe quelles liaisons C(sp3)–F, les précédents systèmes d’activation par les cations silylium ne sont pas limités à la réaction d’hydrodéfluoration. En présence de composés aromatiques, il est souvent possible de réaliser les réactions de Friedel-Crafts à l’aide des cations générés par l’hétérolyse des liens C–F. Dans ces réactions, le cycle catalytique est modifié légèrement, mais est en essence identique : un cation silylium finit par être généré au terme du cycle et l’activation C–F se poursuit.
À l’exception des cations silylium, peu d’exemples exploitant d’autres types de cations ont été rapportés pour l’activation des liens C–F. Dans un exemple unique en son genre, Lectka a démontré que des carbocations aryles sont plus fluorophiles que les carbocations alkyles générés lors d’une hétérolyse aliphatique et que ceux-là peuvent servir à abstraire le fluorure d’un groupement CF3 (Schéma 1.5).44 Le cation RCF2+ ainsi obtenu peut terminer
sa course par une réaction de Friedel-Crafts intramoléculaire ou une simple hydrolyse, en fonction des conditions réactionnelles.
43 Nishimine, T.; Fukushi, K.; Shibata, N.; Taira, H.; Tokunaga, E.; Yamano, A.; Shiro, M.; Shibata, N.
Angew. Chem. Int. Ed. 2014, 53, 517.
Schéma 1.5. Bris d’une liaison C(sp3)–F par l’action d’un cation aromatique formé à partir d’un aryle diazonium.44
Les interactions entre les composés organofluorés et d’autres cations ont aussi été explorées, notamment par l’étude en phase gazeuse des collisions entre le fluorométhane et 46 monocations des 4ème, 5ème et 6ème rangées du tableau de classification périodique.45
Parmi toutes les réactions possibles, l’abstraction d’un fluorure est un processus qui n’est possible principalement qu’avec les cations des groupes II (Ca+, Sr+, Ba+), III (Sc+, Y+,
La+), IV (Ti+, Zr+, Hf+) et V (V+, Nb+, Ta+). Les métaux de la gauche semblent donc plus
fluorophiles, étant d’ailleurs moins polarisables. Les résultats expérimentaux à propos des alcalino-terreux46 et de toute la série des lanthanides47 ont aussi été comparés aux valeurs
obtenues par calculs ab initio et DFT.
En somme, différentes entités acides peuvent être utilisées pour l’activation C–F : des acides de Lewis classiques (BF3 et AlCl3), aux acides de Brønsted qui agissent
probablement comme donneurs de liaisons hydrogène, en passant par différents cations fluorophiles comme les silylium, carbénium ou autres. Dans ces conditions acides très ionisantes, la substitution nucléophile du lien C–F se produit principalement par un mécanisme de type SN1, avec formation de carbocations, même pour les substrats primaires
qui n’ont pourtant pas cette habitude. Les transformations réalisées incluent l’hydrodéfluoration et la réaction de Friedel-Crafts, deux procédés qui s’accommodent bien de la formation de cations.
45 Zhao, X.; Koyanagi, G. K.; Bohme, D. K. J. Phys. Chem. A 2006, 110, 10607.
46 a) Varela-Álvarez, A.; Rayón, V. M.; Redondo, P.; Barrientos, C.; Sordo, J. A. J. Chem. Phys. 2009, 131,
144309; b) Varela-Álvarez, A.; Sordo, J.; Redondo, P.; Largo, A.; Barrientos, C.; Rayón, V. Theor. Chem.
Acc. Theo. Comput. Mod. 2011, 128, 609.
1.2.2.2 Activation en conditions neutres
Substituer un lien C–F dans des conditions « neutres » ne se fait pas aussi aisément et globalement qu’en conditions acides. Le mode d’activation principal consiste à l’utilisation de conditions réductrices pour briser la forte liaison carbone-fluor en y injectant initialement un ou deux électrons, générant des intermédiaires radicalaires, des anions ou des complexes organométalliques.7
Pour ce qui est des réactions par transferts monoélectroniques, l’utilisation de lithium métallique et de complexes réducteurs à base de fer, zirconium ou titane a été rapportée, entre autres. Récemment, une réaction d’activation C–F impliquant un sel de samarium a été décrite par le groupe de Hilmersson (Schéma 1.6).48 Cette hydrodéfluoration, malgré
qu’elle nécessite une quantité surstœchiométrique de Sm(II), est expliquée par les auteurs selon un mécanisme radicalaire amorcé par un transfert électronique du métal vers la liaison C–F. Les résultats obtenus ont aussi permis aux auteurs de formuler l’hypothèse selon laquelle une interaction Sm2+···F–C est importante pour la réactivité, rappelant ce
qu’on connaît des interactions entre les cations des lanthanides et les liens carbone-fluor.
Schéma 1.6. Activation C–F par transfert monoélectronique d’un sel de samarium.48
Il est également possible d’engendrer la scission d’un lien C–F par le transfert successif ou simultané de deux électrons. À cet égard, l’utilisation de méthodes électrochimiques ou de métaux réducteurs comme le magnésium ou le zinc sont les plus populaires.7 Un système
contemporain basé sur de tels concepts a été publié en 2013 par le groupe de Katagiri et
implique un mélange bimétallique de Mg et de CuCl en présence de TMSCl pour réaliser la monosubstitution d’un groupement trifluorométhyle (Schéma 1.7).49
Schéma 1.7. Substitution d’un seul fluor par une combinaison bimétallique.49
Du point de vue des réactions organométalliques, de nombreux exemples ont été rapportés depuis que les méthodes pré-2009 ont été passées en revue.7 En 2009, Gouverneur et ses
collaborateurs ont démontré que les monofluorures allyliques sont de bons substrats pour une réaction de Tsuji-Trost catalysée au palladium (Schéma 1.8A).50 Dans leurs conditions,
les fluorures étaient même de meilleurs groupes sortants que les acétates, pourtant utilisés dans ce genre de réaction. En 2012, le même groupe a étendu sa méthodologie par l’utilisation d’un catalyseur de Pt(0), qui permet l’addition de nucléophiles variés, dont les amines aliphatiques (Schéma 1.8B).51 Le groupe de Paquin, quant à lui, a fait réagir les
gem-difluorures allyliques dans des conditions similaires dans le but d’obtenir des motifs
d’intérêt pharmaceutique, les β-amino-monofluoroalcènes. Tout d’abord réalisées à l’aide d’un catalyseur de Pd (Schéma 1.8C),52 ces transformations ont ensuite été effectuées avec
l’équivalent au Pt, sans toutefois permettre la même généralisation de nucléophiles que dans l’étude de Gouverneur.53 L’activation de composés difluorés est donc
substantiellement plus difficile que celle des composés monofluorés.
49 Utsumi, S.; Katagiri, T.; Uneyama, K. J. Fluorine Chem. 2013, 152, 84.
50 Hazari, A.; Gouverneur, V.; Brown, J. M. Angew. Chem. Int. Ed. 2009, 48, 1296.
51 Benedetto, E.; Keita, M.; Tredwell, M.; Hollingworth, C.; Brown, J. M.; Gouverneur, V. Organometallics
2012, 31, 1408.
52 Pigeon, X.; Bergeron, M.; Barabé, F.; Dubé, P.; Frost, H. N.; Paquin, J.-F. Angew. Chem. Int. Ed. 2010, 49,
1123.
Schéma 1.8. Exemples représentatifs d’activation de liens C–F aliphatiques par des