T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Cancérologie
JURY
Bernard SALLES PU, IPBS/CNRS-UMR5089, Toulouse Président Patricia KANNOUCHE CR1, Institut Gustave Roussy, Villejuif Rapporteur Jean ROSENBAUM DR, INSERM-U889, Bordeaux Rapporteur Agnès TISSIER CR1, UPR3081 CNRS, Marseille Examinatrice Christophe CAZAUX PU, IPBS /CNRS-UMR5089, Toulouse Directeur de thèse
Ecole doctorale : Biologie Santé Biotechnologies Unité de recherche : IPBS/CNRS (UMR5089) Directeur(s) de Thèse : Christophe CAZAUX Rapporteurs : Patricia Kannouche (CR1-CNRS)
Jean Rosenbaum (DR-INSERM)
Présentée et soutenue par Fanny LEMEE Le 12 juin 2009
Titre : Dérégulation de l’expression des ADN polymérases translésionnelles, instabilité
Je souhaite tout d’abord remercier le Pr Salles, le Dr Kannouche, le Dr Rosenbaum et le Dr Tissier pour l’honneur qu’ils m’ont fait d’avoir accepté de faire partie de mon jury de thèse et de juger mon travail.
Merci à Christophe Cazaux et Jean-Sébastien Hoffman, co-directeurs de l’équipe IGC, qui m’ont accueillie dans leur équipe et m’ont accordé leur confiance, notamment entre le MR2 et la thèse en s’impliquant activement dans la recherche de financements nécessaires à la réalisation de cette thèse.
Je souhaite remercier plus particulièrement Christophe, mon directeur de thèse. J’ai été honorée de la grande confiance que tu m’as accordée durant ces quatre années. Tu m’as laissé la liberté de mes choix dans le développement de ce projet en sachant que l’autonomie et le développement du libre-arbitre sont au final particulièrement formateurs. Merci de m’avoir confié un projet
ambitieux et de m’avoir permis de m’impliquer dans toutes les étapes de son développement, de sa conception théorique à sa réalisation pratique, en passant par les mises au point d’outils qui se sont révélées particulièrement fastidieuses, mais qui m’ont conduite à apprécier davantage l’obtention d’un résultat positif ! Merci de m’avoir ainsi donné l’opportunité de travailler dans un contexte pluridisciplinaire riche, en relation étroite avec ce qui m’avait à l’origine conduite à Toulouse et qui reste mon cheval de bataille: la cancérologie. Je te remercie également de m’avoir permis de graver dans ma mémoire la Chine et sa Grande Muraille, St Petersburg et les sensations fortes d’un atterrissage d’urgence ainsi que la Californie et la danse des baleines…
Un grand merci à toi, Clarisse. C’est avec toi que tout a débuté. J’ai énormément apprécié travailler à tes cotés sur le projet « promoteurs » qui nous a toutes deux tenu à cœur et que tu m’as si
généreusement transmis.
Mille mercis Marie pour ton énergie si positive et si communicative qui m’a redonné la pêche plus d’une fois! Merci de m’avoir transmis avec patience et bonne humeur tout ton savoir sur le
combing et d’avoir ainsi pu partager en connaissance de cause toutes mes peines, mais aussi mes joies (car il y en a parfois !) relatifs à cette partie du projet. Je te remercie pour tous les bons moments partagés et j’espère que tu viendras vérifier par toi-même si tes prévisions se sont réalisées.
Merci Valérie, pour avoir allié tes forces aux miennes sur le projet PolQ, apportant l’aspect
clinique au projet. J’ai aimé le partage de nos idées, nos discussions pour essayer de faire avancer le « chmilblick » et ta manière si juste d’analyser les choses. Merci pour ton solide soutien ces
derniers mois.
Claire et Elena, comment vous remercier pour l’énorme contribution que vous avez apporté au projet PolQ ? Merci pour le temps et l’énergie que vous avez investis sans compter dans ce projet. Travailler avec vous a été pour moi d’un grand réconfort, car on réfléchit bien mieux à deux que tout seul, et nos discussions, nos « brainstormings » du soir ont permis peu à peu de faire mûrir ce projet. J’espère ne pas avoir été trop exigeante comme encadrante, mais vue l’amitié qui nous lie désormais, j’en conclue que vous ne m’en tenez pas trop rigueur. Je vous souhaite bonne chance à
as su te plonger rapidement dans l’univers de ce projet et y apporter ton savoir-faire et ton efficacité. Merci pour ta gentillesse et ta bonne humeur qui ensoleille les journées au labo. Merci Anne, pour m’avoir transmis ton savoir sur la PCR en temps réel et sur 1001 petites choses du labo. Merci surtout pour ta douceur et ta gentillesse qui m’ont apaisée bien des fois.
Merci aux membres de l’équipe IGC, passés et présents, pour vos conseils avisés et les discussions enrichissantes que nous avons pu avoir. François, merci de m’avoir initiée avec patience à la biochimie des protéines et merci pour ton aide précieuse sur cette partie du projet qui n’a
malheureusement pas eu de suite. Merci Nadine pour ton aide dans la stratégie de clonage de PolQ ainsi que pour tes nombreux conseils qui m’ont aidée dans mes laborieuses mises au point. Dennis, merci pour tous tes trucs et astuces qui font qu’un protocole fonctionne du premier coup! Yvan, merci pour tes questions déstabilisantes qui m’ont aidée à mieux maitriser mes projets ;) et pour nos discussions, scientifiques ou non. Merci, Anne, de prendre la suite du projet PolQ. Bonne chance avec cette protéine un peu capricieuse, mais si passionnante. Merci Remy pour nos discussions scientifico-politico-bibliographiques, pour m’avoir fait découvrir et aimer le trip-hop et pour toutes les fois où tu m’as fait rire, bien souvent malgré toi. Laurie, ma petite Laurie, je suis heureuse d’avoir pu partager avec toi les joies et peines du labo et surtout durant cette dernière longue ligne droite qui nous a rapprochées.
Merci Marlène d’avoir été ma co-équipière de choc dans l’aventure SILAC. Merci de m’avoir ouvert l’esprit à la protéomique avec patience et humour. Le sabotage n’aura pas eu raison de nous ! Merci à mes amis, précieuse soupape de décompression, qui m’ont entourée et chouchoutée.
Oriane, merci de toujours avoir été là pour moi et d’avoir supporté avec courage mes craquages migrainogènes du vendredi soir. Marine, merci pour ta joie de vivre et tes pâtisseries salvatrices. Claire, merci pour ces loulous-meetings d’anthologie et ton humour qui a musclé mes
zygomatiques. Camille, merci pour nos délires euphorisant. Eva, Anthony, Diane, Corentin, Clémentine et Paul, merci pour tous ces week-ends ressourçant. Soazig et Mélanie, merci pour votre amitié solide et inaltérable par-delà les années et les kilomètres…
Merci à ma mère, pour son amour et son soutien inconditionnel et irremplaçable. Merci à mon père, qui est ma leçon de courage et en qui je puise ma force.
Pour finir, merci à mon homme, Mathieu, qui chaque jour, avec amour, écarte mes doutes, soigne mes blessures et partage mes joies… merci pour la promesse de ces longues années à venir…
Sommaire ...1
Abréviations ...7
Résumé...11
Abstract...12
INTRODUCTION...13
CHAPITRE I – Les ADN polymérases et le maintien de la stabilité du génome...14
1. Les ADN polymérases réplicatives et la réplication génomique... 14
1.1 La réplication de l’ADN chez les eucaryotes supérieurs ... 14
1.1.1 L’initiation et Polα ... 14
a) L’activation des origines... 16
b) L’initiation de la réplication ... 16
c) L’ADN polymérase alpha (Polα) ... 17
d) La régulation de l’activation des origines... 17
1.1.2 L’élongation et les ADN polymérases Polδ et Polε ... 20
1.1.3 La terminaison... 24
1.2 La fidélité de la réplication ... 26
1.2.1 La sélection des nucléotides... 27
1.2.2 L’activité 3’ Æ 5’ exonucléase... 28
1.2.3 La réparation des mésappariements par le MMR ... 29
2. Les ADN polymérases translésionnelles (TLS) et la gestion des lésions de l’ADN .. 31
2.1 Les barrières de fourches de réplication ... 31
2.1.1 L’ADN structuré ... 31
a) Les lésions endogènes... 35
b) Les lésions exogènes ... 36
2.2 Rôle des ADN polymérases TLS dans la tolérance des lésions... 39
2.2.1 Mono et poly-ubiquitinylation de PCNA ... 40
2.2.2 Le contournement de la lésion ... 43
2.2.3 La synthèse translésionnelle ... 44
a) Contrôle de la synthèse translésionnelle ... 45
b) Le choix des ADN polymérases translésionnelles. ... 48
2.3 Rôles des ADN polymérases TLS dans la réparation des lésions ... 50
2.3.1 Rôles dans la réparation par excision de base et dans la réparation des cassures simple-brin de l’ADN ... 50
2.3.2 Rôles dans la réparation par excision de nucléotides ... 53
2.3.3 Rôle dans la réparation des cassures double-brin de l’ADN ... 53
2.4 Caractéristiques et fonctions des ADN polymérases TLS et plus particulièrement de Polη, Polκ et Polθ... 57
2.4.1 Fidélité des ADN polymérases translésionnelles... 57
2.4.2 L’ADN polymérase η... 58
2.4.3 L’ADN polymérase κ... 60
2.4.4 L’ADN polymérase θ... 62
3. Régulation de l’expression des ADN polymérases... 66
3.1 Régulation de l’expression des ADN polymérases réplicatives... 66
3.2 Régulation de l’expression des ADN polymérases translésionnelles... 68
CHAPITRE II- La perturbation de la réplication : Réponses cellulaires et conséquences sur la stabilité du génome...71
1. Le point de contrôle de phase S... 71
1.1.2 La signalisation issue de l’activation d’ATR... 75
a) Activation et rôles de Chk1 ... 75
b) Les autres substrats d’ATR ... 76
1.2 La voie dépendante d’ATM... 77
1.2.1 Les mécanismes d’activation d’ATM... 78
1.2.2 L’amplification du signal... 79
1.2.3 La propagation du signal par Chk2... 81
1.3 Inter-conversion de ces deux voies de signalisation. ... 81
2. Rôle des protéines du point de contrôle de phase S lors d’une phase S normale .... 83
2.1 Régulation de l’initiation et de la progression des fourches de réplication ... 83
2.2 Le cas particulier des sites fragiles communs... 84
3. Stress réplicatif et cancer ... 85
Chapitre III- La dérégulation de l’expression des protéines de la réplication comme acteur du développement des cancers et facteur pronostique des cancers colorectaux et mammaires ...89
1. L’instabilité génétique dans le développement tumoral ... 89
2. Mutations et dérégulations de l’expression des ADN polymérases dans les cancers92 2.1 Mutations et dérégulations d’expression des ADN polymérases réplicatives .... 92
2.2 Mutations et dérégulation de l’expression des ADN polymérases translésionnelles. ... 93
3. La réplication de l’ADN comme source de nouveaux marqueurs pronostiques des cancers colorectaux et mammaires... 99
3.1 Les cancers colorectaux et mammaires ... 99
3.1.1 Le cancer colorectal ... 99
3.1.2 Le cancer du sein ... 101
CHAPITRE IV- Présentation des travaux de thèse...108
RESULTATS...110
CHAPITRE I- Régulation de l’expression des ADN polymérases TLS Polκ et Polη. Recherche des causes possibles de la dérégulation de Polκ dans les cancers colorectaux. ...111
1. Introduction... 111
2. Analyse de l’expression de Polκ et Polη dans les cancers colorectaux ... 112
3. Caractérisation d’éléments régulateurs du promoteur de POLΚ impliqués dans la sous-expression de cette polymérase dans les cancers colo-rectaux. ... 114
3.1 Publication... 114
3.2 Résultats complémentaires : Effet de p53 sur la transcription de POLK ... 123
4. Etude de la régulation transcriptionnelle du gène POLH... 124
4.1 Caractérisation du promoteur de POLH ... 124
4.2 Identification de séquences régulatrices de l’expression de POLH ... 125
4.3 Identification de facteurs de transcription régulant l’expression de POLH .... 127
5. Conclusions et discussion... 128
CHAPITRE II- La surexpression de l’ADN polymérase théta (Polθ) dans les cancers du sein : acteur du développement tumoral et facteur de mauvais pronostic. ...133
1. Introduction... 133
2. Résultats... 134
2.1 Publication... 134
2.2 Résultats complémentaires ... 172
2.2.1 Validation de la surexpression de Polθ dans les clones cellulaires... 172
2.2.2 L’expression ectopique de Polθ affecte la prolifération cellulaire et augmente l’apoptose... 173
175
2.2.5 La surexpression de Polθ induit de l’instabilité chromosomique ... 177
3. Conclusion et discussion... 179
Conclusions et discussion ...189
Annexe 1 : La classification TNM ...197
Annexe 2 : Matériels et méthodes...199
1. Lignées cellulaires ... 199
2. Clonages... 199
2.1 Clonage des promoteurs tronqués de POLK et POLH ... 199
2.2 Clonage du cDNA de Polθ ... 201
3. Mutagenèse dirigée des promoteurs de POLK et POLH... 203
4. Transfections transitoires et tests luciférase ... 204
5. Transfections stables ... 205
6. Mesure de l’expression des transcrits par PCR en temps réel :... 206
6.1 Extraction d’ARN : ... 206
6.2 Rétro-transcription : ... 206
6.3 PCR quantitative en temps réel :... 206
6.4 Analyse des résultats : ... 207
7. Préparation d’extraits nucléaires ... 207
8. Retard sur gel... 207
9. Extraits protéiques... 209
10. Western Blot... 209
11. Calcul du temps de doublement ... 209
12. Etude de l’apoptose... 210
13. Analyse du cycle cellulaire par cytométrie en flux ... 210
17. Peignage moléculaire de l’ADN... 213
17.1 Marquage de l’ADN en cours de réplication et préparation des blocs d’agarose ... 213
17.2 Peignage... 214
17.3 Immunodétection... 214
ADN: Acide-Desoxyribo-Nucleique ADNsb: ADN simple-brin
AF: Anémie de Fanconi
AP: apurinique, ou apyrimidinique ARN: Acide Ribo-Nucleique
AT: Ataxia Telangiectasia
ATM: ataxia-telangiectasia mutated
ATR: ataxia-telangiectasia and RAD3-related ATRIP: ATR Interacting Protein
B[a]P: Benzo[a]pyrène
BPDE: 7,8-diol-9,10,-epoxide BrdU: Bromo-désoxy-uridine CDB : Cassure Double Brin CDK: Cycline-dependant kinase
CGH: Comparative Genomic Hybridization Chk1: checkpoint kinase-1
Chk2: checkpoint kinase-2 CIN: Chromosomal Instability Cis-Pt: cisplatine
CPD: Cyclobutane Pyrimidine Dimer CRE: cAMP response element
DDK: Dbf4-dependant kinase Fen1: Flap endonuclease 1
HLTF: Helicase Like Transcription Factor HMS: hypermutation somatique
HU: Hydroxyurée Lig1: DNA ligase 1
MCM: minichromosome maintenance
MDC1: mediator of DNA damage checkpoint-1 MEF: Mouse Embryo fibroblastes
MIN: microsatellite instability miARN: microARN MMC: Mitomycine C MMP: Matrix Metalloproteinases MMR: Mismatch Repair MMS: Methyl-méthane sulfonate MNNG: N-methyl-N'-nitro-N-nitrosoguanidine MNU: N-methyl-N-nitrosurea
MRE11: meiotic recombination protein-11 MRN: MRE11/ Rad50/ NBS1
NBS1: Nijmegen breakage syndrome protein-1 NHEJ: Non Homologous End Joining
ORC: Origin Recognition Complex pb: paires de bases
PIM: Point Mutation Instability PIP: PCNA interacting Peptide PI3K: phosphoinositide-3-kinase PKA: Protein Kinase A
PKC: Protein Kinase C Plk1: Polo-like-kinase 1 Pol: polymérase
PréIC: complexe de pré-initiation PréRC: complexe pré-réplicatif RPA: Replication Protein A RFC: Replication Factor C
RAT: Recombinaison Associée à la Transcription REB: Réparation par Excision de Base
REN: Réparation par Excision de Nucléotides RH: Recombinaison Homologue
RI: Radiations Ionisantes ROS: Reactive Oxygen Species SCE: sister chromatin exchange SFC: Sites Fragiles Communs
SHPRH: SNF2 Histone Linker PHD RING Helicase SIT: Site d’Initiation de la Transcription
TLS (Polymérase): Polymérase translésionelle TOPBP1: Topoisomerase-II Binding Protein 1 UBM: ubiquitin-binding motif
UBZ: ubiquitin-binding zinc finger UV: Ultra-Violet
XRE: Xenobiotic Responsive Element XP-V: Xeroderma pigmentosum variant UTR: untranslated region
53BP1: p53-binding protein-1
Dérégulation de l’expression des ADN polymérases translésionnelles
(TLS), instabilité génétique et progression des cancers colorectaux et
du sein.
L’instabilité génétique est un facteur clé dans le développement de la plupart des cancers solides. En plus des ADN polymérases « réplicatives » impliquées dans la réplication conventionnelle du génome, un nombre croissant d’ADN polymérases alternatives, appelées ADN polymérases « translésionnelles » (TLS), ont été récemment identifiées et jouent un rôle majeur dans la gestion des lésions de l’ADN. Ces enzymes interviennent en effet dans les mécanismes de réplication, de réparation et de recombinaison (les « 3R »), jouant un rôle primordial dans le maintien de la stabilité du génome. Notre hypothèse de travail est qu’un déséquilibre entre l’expression des ADN polymérases réplicatives et TLS peut entrainer un stress réplicatif, générant alors de l’instabilité génétique.
Dans ces travaux, nous montrons que Polκ et Polη, ADN polymérases TLS de la famille Y, sont sous-exprimées dans les cancers colorectaux. Dans le but de comprendre les mécanismes moléculaires responsables de ce défaut d’expression, nous avons cartographié leurs promoteurs et montré qu’ils contenaient des régions répressives et activatrices. Nous avons identifié CREB et Sp1 comme des facteurs activateurs du promoteur de POLK, et observé que la sous-expression de ces facteurs de transcription dans les tumeurs colorectales était corrélée à la sous-expression de Polκ. Ces observations suggèrent que l’inhibition de CREB et Sp1 puisse contribuer à l’expression réduite de Polκ dans les cancers colorectaux. Dans une deuxième partie, nous montrons que Polθ est la seule ADN polymérase TLS surexprimée dans les tumeurs du sein et que sa surexpression corrèle avec un mauvais pronostic. D’autre part, dans des cellules MRC5-SV, l’excès de Polθ engendre un défaut de progression des fourches de réplication, une induction de cassures double-brin et une activation du point de contrôle de réponse aux dommages d’ADN, résultant en une induction d’anomalies chromosomiques. Nous proposons que Polθ en excès engendre un stress réplicatif,
Misregulation of TLS DNA polymerases expression, genetic instability
and progression of colorectal and breast cancers.
The faithful maintenance of the genome is essential for the prevention of most of human solid cancers. Beside “replicative” DNA polymerases, a growing number of alternative mammalian DNA polymerases, called translesional (TLS), have been recently identified. These enzymes play specialized roles at the interplay of DNA replication, repair and recombination (the “3Rs”) transactions, critical pathways to preserve the genome integrity. Our working hypothesis is that an unbalanced expression of replicative/TLS DNA polymerases can modify the replication program (the so-called “replicative stress”), and can therefore trigger genetic instability.
Here, we show that the specialized Y-family TLS DNA polymerases Polκ and Polη are downregulated in colorectal tumor biopsies compared with adjacent normal tissues. With the aim to investigate the molecular bases underlying such defective expression, we carried out a genetic cartography of the promoters and found that they contain repressive as well as activating regions. We identified CREB and Sp1 as transcriptional activators of the POLK promoter, and we observed that a decreased level of these factors in colorectal biopsies correlated with Polκ downregulation. These observations suggest that inhibition of CREB and Sp1 proteins could contribute to the reduced level of Polκ in colorectal tumours.
In a second part we report that the recently identified Polθ was the only specialized/TLS DNA polymerase significantly regulated in breast tumours. More importantly, Polθ up-regulation significantly correlated with poor clinical outcome. Aiming to decipher the molecular consequences of Polθ up-regulation, we generated human cellular models which stably overexpressed this polymerase. Our data show that high level of Polθ expression is associated with defective DNA replication fork progression, double-strand breaks induction and activation of the DNA damage checkpoint, resulting in chromosomal aberrations. We propose that excess Polθ results in a replicative stress, which in turn generates genetic instability, allowing variant tumour cells to survive and proliferate.
CHAPITRE I – Les ADN polymérases et le maintien
de la stabilité du génome.
1. Les ADN polymérases réplicatives et la réplication
génomique
1.1
La réplication de l’ADN chez les eucaryotes supérieurs
Comme le disait François Jacob, « le rêve d’une cellule est de devenir deux cellules » contenant une copie identique du génome d’origine. La duplication fidèle de l’information génétique est réalisée grâce à un processus cellulaire appelé réplication. La réplication de l’ADN est un processus complexe, extrêmement régulé et hautement organisé qui se déroule en trois étapes : l’initiation, l’élongation et la terminaison. Les ADN polymérases réplicatives jouent un rôle majeur dans ce processus. Polα, Polδ et Polε assurent la réplication du génome nucléaire « non endommagé », Polγ étant affectée à celle du génome mitochondrial.
1.1.1
L’initiation et Polα
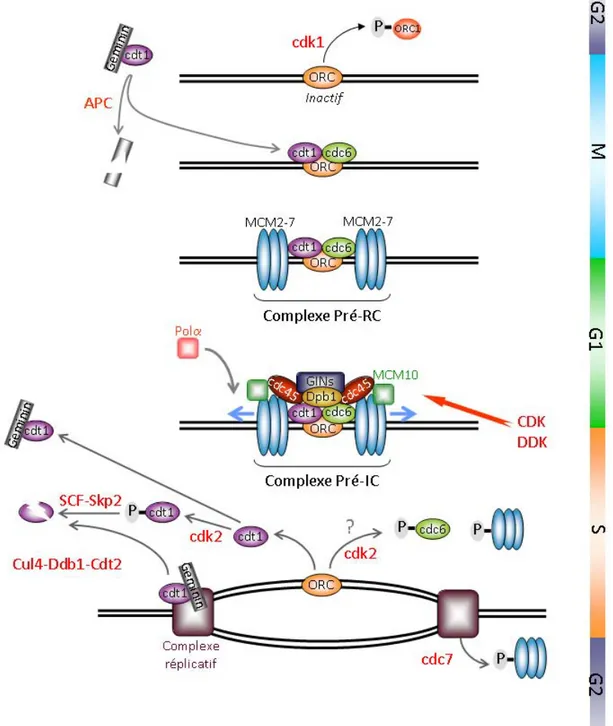
La réplication du génome débute au niveau des origines de réplication. C’est à partir de ces séquences d’ADN, estimées à environ 50 000 dans le génome humain, que la double-hélice d’ADN s’ouvre pour former les fourches de réplication qui progressent de manière bidirectionnelle afin d’assurer la réplication complète du génome avant la ségrégation des chromosomes. Les hélicases ont évidemment un rôle important à jouer au cours de ce processus, mais un grand nombre d’autres protéines sont aussi importantes à la bonne coordination des événements (Figure 1).
FIGURE 1. Activation et inactivation de l’initiation de la réplication chez les mammifères. Les Pré-RC sont assemblés à la transition M-G1 par l’addition séquentielle de Cdc6, Cdt1 et des hélicases MCM2-7 sur les complexes ORC1-6 fixés aux origines. Les Pré-RC inactifs sont activés en Pré-IC à la transition G1-S par le recrutement de Dpb1, Cdc45, le complexe GINS et MCM10. L’initiation de la réplication de l’ADN durant la phase S nécessite la phosphorylation de composants du Pré-RC par les CDK et les DDK. Au cours de la phase S, Cdt1 est phosphorylée par Cdk2 et dégradée par le protéasome. Cdt1 liée au complexe réplicatif est également dégradée par le protéasome. La Géminine se lie à Cdt1 et l’inhibe. Les MCM sont phosphorylées par Cdc7-Dbf4 en S et G2 et libérées de la chromatine. En début de phase M, la sous-unité ORC1 est phosphorylée et libérée de la chromatine rendant le complexe ORC inactif. En fin de M, la Géminine est dégradée sous l’action d’APC et Cdt1 redevenu actif peut se lier aux complexes ORC1-6 pour une nouvelle initiation. Adapté de DePamphilis, 2006, Aladjem, 2007 et Arias et Walter, 2007 (Aladjem, 2007; Arias and
a) L’activation des origines
Le complexe ORC (Origin Recognition Complex), composé des protéines ORC1-6, reconnaît les origines de réplication et s’y fixe de manière constitutive tout au long du cycle cellulaire, à l’exception de la sous-unité ORC1 qui est dégradée durant la phase S (Li and DePamphilis, 2002). A la transition M-G1, le complexe ORC lié à la chromatine recrute Cdc6 et Cdt1, qui vont alors faciliter le chargement des hélicases MCM (Minichromosome maintenance) 2-7 sur la chromatine. Cette étape qui voit la formation du complexe pré-réplicatif (PréRC) est appelée « licensing » ou « permis de répliquer » et rend l’ADN compétent pour la réplication. Aucune nouvelle ouverture de l’ADN n’est détectée jusqu’à la phase S, indiquant que les hélicases MCM2-7 chargées au niveau des origines durant la phase G1 restent inactives.
b) L’initiation de la réplication
L’événement clé de l’initiation de la réplication est sans doute l’activation du complexe hélicase MCM2-7, qui requière l’assemblage du complexe MCM2-7-Cdc45-GINS (composé des protéines Sld1 et Psf1-3). A la transition G1-S, les CDK (cyclin-dependent kinase) et les DDK (Dbf4 et Drf1-dependent kinase) coopèrent avec de nombreux facteurs, dont MCM10 et Dpb11, afin de recruter Cdc45 et GINS au niveau du complexe MCM2-7. L’assemblage de ces facteurs au niveau des origines active le complexe PréRC qui devient alors un complexe de pré-initiation (PréIC). Il semble que la formation du complexe PréIC soit rapidement suivie par l’activation finale des origines, appelée « firing », permettant l’ouverture de la double hélice d’ADN et son déroulement. L’ADN simple-brin formé est alors protégé et stabilisé par la protéine RPA (Replication Protein A). L’ADN polymérase α/primase est à son tour recrutée par Cdc45 (Mimura and Takisawa, 1998) et MCM10 (Zhu et al., 2007). La réplication chez les eucaryotes est bidirectionnelle, alors que la synthèse par les ADN polymérases s’effectue exclusivement dans le sens 5’ vers 3’ du brin néo-synthétisé. Le fonctionnement de la machinerie réplicative s’est adapté selon qu’elle se trouve sur le brin continu ou discontinu. La réplication de brin continu nécessite un seul événement d’initiation, alors que la synthèse de plusieurs amorces est nécessaire à la
réplication du brin discontinu. Ainsi, on estime à 4 .104, soit environ le nombre d’origines dans une cellule de mammifère, le nombre d’événements d’initiation sur le brin à synthèse continue, et à 2.107 le nombre d’initiations sur le brin à synthèse discontinue dans les cellules mammifères (Hubscher et al., 2002).
c) L’ADN polymérase alpha (Polα)
Polα, appelée aussi primase, joue un rôle essentiel dans l’initiation de la réplication. Elle appartient à la famille B des ADN polymérases et est composée de quatre sous-unités, dont la sous-unité primase de 49 kDa et la sous-unité catalytique de 180 kDa codée par le gène
POLA1 en copie unique sur le chromosome X. Le rôle des deux autres sous-unités n’est à
ce jour pas clairement défini. Polα a tout d’abord été décrite comme étant la principale réplicase, avant la découverte de Polδ et Polε. Son implication dans la réplication de l’ADN a été étayée par sa forte activité dans les cellules se divisant rapidement, sa localisation nucléaire et son induction lorsque les cellules quiescentes entrent en prolifération. Son rôle de primase est désormais admis. En effet, la primase initie la réplication en synthétisant une amorce ARN d’une dizaine de ribonucléotides environ, puis son activité catalytique étend la synthèse sur 20 désoxynucléotides supplémentaires. Son recrutement n’est possible que si l’ADN a été déroulé et si le simple-brin est recouvert par RPA. Après synthèse de l’amorce ARN/ADN, le complexe RFC (Replication Factor C) charge le facteur de processivité PCNA (Proliferating Cell Nuclear Antigen) autour de l’ADN pour permettre l’élongation des amorces.
d) La régulation de l’activation des origines
Il est essentiel que les origines de réplication ne s’activent qu’une seule et unique fois au cours du cycle cellulaire. En effet, la ré-activation d’une seule origine au cours du même cycle cellulaire peut causer une forte instabilité génétique. Les cellules ont donc mis en place plusieurs mécanismes empêchant la re-formation du complexe PréRC.
de la réplication dans deux phases du cycle qui s’excluent mutuellement (respectivement G1 et S). En effet, les hélicases MCM2-7 restent inactives tout au long de la phase G1, sans doute parce que leur activation nécessite l’activité des CDK (Figure 2) qui est très faible durant cette phase. Une fois que les complexes MCM2-7 sont activés et qu’ils initient la réplication, aucun autre complexe MCM2-7 ne peut être recruté au niveau des origines, ceci jusqu’au cycle cellulaire suivant.
FIGURE 2. Formation des complexes cycline/CDK au cours du cycle cellulaire.
(D’après R. Boutros, 2007)
Le facteur majeur et limitant responsable de l’initiation de la réplication est Cdt1. L’inactivation de ce facteur dès le début de la phase S, essentielle à la prévention contre une éventuelle re-réplication, peut être le fruit de diverses voies indépendantes les unes des autres.
La Géminine a été identifiée comme étant un inhibiteur du « licensing » régulant l’activité de Cdt1. La Géminine, par sa liaison avec Cdt1, empêche le recrutement de MCM2-7 sur la chromatine et donc la formation du complexe PréRC (Wohlschlegel et al., 2000). Cet inhibiteur est exprimé au cours des phases S, G2 et M et déstabilisé dans les cellules juste avant la phase G1. En effet, à la transition métaphase-anaphase, la Géminine est ubiquitinylée par APC et dégradée par le protéasome permettant la formation du complexe PréRC durant la phase G1 (McGarry and Kirschner, 1998).
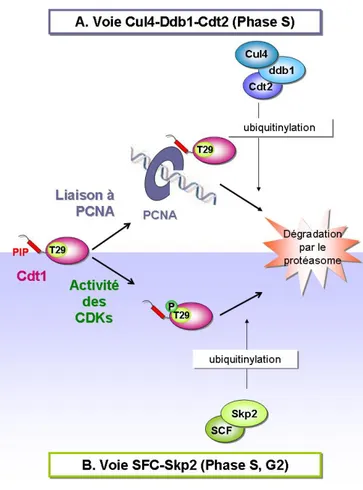
La dégradation de Cdt1 durant les phases S et G2 est également un moyen de contrôler l’initiation de la réplication. Deux voies peuvent conduire à cette dégradation (Nishitani et al., 2006) (Figure 3).
FIGURE 3. Les deux voies majeures de protéolyse de Cdt1. La boîte PIP de Cdt1 est représentée en rouge. (A) La dégradation de Cdt1 dépendante de Cul4-Ddb1-Cdt2 se passe en deux étapes. Tout d’abord Cdt1 se fixe à PCNA par l’intermédiaire de son domaine PIP au niveau des fourches de réplication. Ensuite, Cdt2 interagit avec le complexe Cdt1-PCNA et ubiquitinyle Cdt1. (B) La dégradation dépendante de SCF-Skp2 se produit également en deux étapes. Cdk2 phosphoryle Cdt1 sur la Thr29 puis SCF-Skp2 se lie à Cdt1 phosphorylé est l’ubiquitinyle. Adapté d’E. Arias et J. Walter, 2007 (Arias and Walter, 2007)
La première à avoir été identifiée est déclenchée par la kinase Cdk2 qui phosphoryle Cdt1 sur la thréonine 29. Cette forme phosphorylée de Cdt1 est alors reconnue par le complexe E3-ubiquitine ligase SCF-Skp2 et adressée au protéasome pour être dégradée. Cdt1 peut également être protéolysé par une voie dépendante de PCNA. En effet, Cdt1 peut se lier par le biais de son domaine PIP (PCNA Interacting Peptide), à PCNA au niveau des fourches de réplication. Cdt1 lié à PCNA est reconnu par la complexe E3-ubiquitine ligase Cul4-Ddb1-Cdt2 conduisant de même à sa dégradation par le protéasome. Cette dernière voie semble être prépondérante en phase S, alors que la voie SCF-Cdk2 prédominerait en phase G2.
initiation au cours des phases G2 et M. En effet, l’inactivation conditionnelle de Cdk1 induit de la re-réplication dans les cellules cyclantes (Itzhaki et al., 1997). Il a été proposé qu’au cours de la mitose, Cdk1 activé phosphorylerait la sous-unité ORC1, empêchant ainsi sa liaison à la chromatine et donc la formation de nouveaux complexes PréRC (Li and DePamphilis, 2002). Cependant le devenir d’ORC1 au cours des phases S et G2 reste incertain. Certaines études indiquent qu’ORC1 est partiellement détruit par protéolyse en phase S et qu’il n’est pas détecté au niveau de certaines origines de réplication (Li and DePamphilis, 2002; Mendez et al., 2002), alors que d’autres études indiquent que le niveau d’expression d’ORC1 ne varie pas au cours de la phase S (Okuno et al., 2001).
Le rôle de la régulation de Cdc6 dans la prévention de la re-réplication reste assez controversé. En effet, des études ont initialement montré que des formes de Cdc6 transfectées dans les cellules étaient exportées du noyau sous contrôle de l’activité des CDKs (Petersen et al., 1999). Cependant des travaux ultérieurs ont montré que la protéine Cdc6 endogène reste liée à la chromatine en phase S et G2 et que seulement la fraction soluble de Cdc6 était exportée du noyau (Alexandrow and Hamlin, 2004).
1.1.2
L’élongation et les ADN polymérases Polδ et Polε
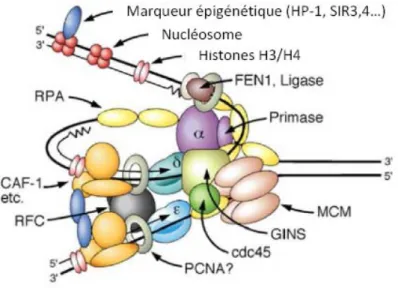
Une fois l’initiation de la réplication de l’ADN mise en place, un large complexe multi-protéique appelé réplisome est requis afin de propager les fourches de réplication, de réguler leur avancement ainsi que la coordination de leur activité avec d’autres machineries métaboliques de l’ADN. Il y a quinze ans, la reconstitution du processus complet de réplication d’un plasmide d’ADN contenant une origine virale SV40 a permis d’identifier Polα/Primase, Polδ, RPA, RFC, PCNA, Flap endonuclease 1 (Fen1), RNase H1 et la DNA ligase 1 (Lig1) comme étant des composants essentiels du réplisome eucaryote (Waga and Stillman, 1994). Depuis, plusieurs autres facteurs ont été identifiés comme faisant partie du réplisome et ayant un rôle important à jouer dans la propagation des fourches de réplication. La double hélice d’ADN ayant ses deux brins enroulés dans des sens opposés, et les ADN polymérases ne pouvant étendre les amorces uniquement à partir de l’extrémité 3’, la réplication des 2 brins va se faire de façon différente. Un des brins sera
répliqué de manière continue, alors que le brin opposé sera copié par la succession de petits fragments d’Okazaki d’environ 200 paires de bases. (Figure 4).
FIGURE 4. Modèle de fourche de réplication eucaryote. La fourche de réplication est initiée par le déroulement de la double-hélice d’ADN grâce à l’activité hélicase du complexe Cdc45-MCM-GINS (CMG), le recouvrement de l’ADN simple-brin par RPA et la synthèse d’amorces ARN/ADN par Polα/Primase. RFC, qui reconnaît cette amorce hybride, charge PCNA auquel se lient les ADN polymérases réplicatives afin de gagner en processivité. Polε réplique le brin continu et Polδ le brin discontinu sur lequel FEN1 et la ligase 1 assurent la maturation des fragments d’Okazaki. D’après B. Stillman, 2008 (Stillman, 2008).
A l’avant de la fourche, l’hélicase de la réplication ouvre la voie en décomplexant les deux brins d’ADN. Bien que ces dernières années ont vu croître nos connaissances sur les mécanismes moléculaires de la réplication eucaryote, l’identification de l’hélicase de l’élongation est restée pendant longtemps en suspens. Un certain nombre de données indirectes semblaient accorder ce rôle au complexe MCM2-7, déjà identifié comme l’hélicase de l’initiation. Curieusement, ce complexe MCM2-7 n’a pas montré d’activité hélicase in vitro, à l’inverse du sous-complexe MCM4/6/7. (Ishimi, 1997). Des études de biochimie ont également mis en évidence que l’activité de ce sous-complexe était inhibée par MCM2, MCM3 ou MCM5 (Ishimi et al., 1998; Lee and Hurwitz, 2000). De plus, le complexe MCM2-7 est distribué largement sur la chromatine et n’est pas retrouvé concentré au niveau des foyers de réplication (Madine et al., 1995). La découverte de cofacteurs pouvant réguler l’activité de MCM2-7 à permis d’appuyer un peu plus sa
stimule son activité hélicase (Masuda et al., 2003) (Pacek and Walter, 2004). De plus, un complexe de haut poids moléculaire contenant Cdc45, MCM2-7 et le complexe GINS a été isolé à partir d’extraits d’embryons de drosophile et présente une activité hélicase in vitro
(Moyer et al., 2006). Ce complexe alors nommé CMG (Cdc45/ MCM2-7/ GINS) s’affiche désormais comme le meilleur candidat pour l’hélicase de la réplication. Seul un faible pourcentage de protéines MCM ferait partie de ce complexe CMG et serait donc localisé au niveau des foyers de réplication. Enfin, il a été suggéré que MCM8 pouvait jouer également un rôle dans le déroulement de la double-hélice au cours de l’élongation. En effet, MCM8 présente une activité hélicase in vitro, se localise au niveau des foyers de réplication et son absence empêche la liaison de la sous-unité RPA34 et de Polα à la chromatine (Maiorano et al., 2005). Cependant, son absence dans les extraits d’œufs de Xénopes n’inhibant pas complètement la synthèse d’ADN, MCM8 partage donc sans doute son rôle dans l’élongation avec le complexe CMG.
Juste après l’initiation, le remplacement de la Polα/Primase par une ADN polymérase réplicative nécessite le chargement de PCNA par RFC au niveau de l’amorce ARN/ADN, que ce soit sur le brin continu ou au niveau de chaque fragment d’Okazaki sur le brin discontinu. En effet, l’amorce ARN/ADN synthétisée par Polα est reconnue par le facteur RFC. Dans une réaction dépendante de l’ATP, RFC charge PCNA à la jonction matrice/extrémité 3’ de l’amorce puis se dissocie permettant à PCNA de fonctionner avec les ADN polymérases réplicatives. PCNA est un homotrimère prenant une structure en anneau capable d’encercler l’ADN et de glisser librement dans les deux directions. Son nom vient de l’observation de son abondance dans les cellules qui prolifèrent. L’anneau PCNA recrute les ADN polymérases réplicatives et leur permet une association forte avec l’ADN, augmentant ainsi largement leur processivité. Ce trimère est capable d’interagir avec diverses autres protéines par le biais de leur motif PIP (PCNA Interacting Peptide), jouant ainsi un rôle de plateforme impliquée dans de nombreux mécanismes (réplication, réparation, synthèse translésionnelle…).
Le rôle de la synthèse d’ADN lors de l’élongation revient à deux ADN polymérases réplicatives : Polδ et Polε.
Polδ et Polε humaines appartiennent à la famille B, sont toutes deux composées de quatre sous-unités et possèdent une activité 3’-5’ exonucléase appelée également « correctrice d’erreur ». Cette capacité les rend particulièrement fidèles lorsqu’elles répliquent l’ADN. Sur la fourche de réplication, Polδ s’associe à PCNA et cette interaction augmente sa processivité. En effet, des premières études in vitro avaient montré qu’en présence de PCNA la processivité des ADN polymérases passait d’une dizaine à plusieurs milliers de nucléotides. Cependant, à part en condition de forte force ionique, PCNA ne semble pas nécessaire à l’activité de Polε. En effet, Polε semble être recrutée à la fourche de réplication avant PCNA et ces deux protéines ne colocalisent en immunofluorescence qu’en fin de phase S (Fuss and Linn, 2002).
Pendant plusieurs décennies, la fonction exacte de ces deux ADN polymérases et leur organisation sur la fourche de réplication est restée très vague. Les toutes premières études développées grâce au système de réplication in vitro SV40 avaient fait émerger un modèle très simple, où Polα/primase initiait la réplication par la synthèse d’amorces ARN/ADN et où Polδ, sur le brin continu, prolongeait cette synthèse d’ADN jusqu’à ce que tout l’ADN soit répliqué et, sur le brin discontinu, remplaçait Polα sur chaque fragment d’Okasaki afin de les allonger. Ce système compact et efficace semblait tout à fait pouvoir se passer d’une troisième ADN polymérase. Néanmoins, en 1990, Polε a été identifiée chez la levure comme étant, tout comme Polδ, une protéine essentielle à la viabilité cellulaire ainsi qu’à la réplication (Morrison et al., 1990). De plus, des expériences d’immuno-déplétion réalisées dans des œufs de Xénopes ont montré une réduction à peu près équivalente de la quantité d’ADN synthétisé dans des extraits déplétés en Polδ ou Polε (Fukui et al., 2004). Enfin, sa capacité de correction d’erreur a achevé de légitimer son rôle en tant qu’ADN polymérase réplicative. Ce n’est que très récemment que des arguments forts ont permis d’éclaircir l’affectation des deux ADN polymérases sur la fourche de réplication. Ces études menées chez la levure ont tiré partie de formes mutées de Polδ et de Polε sur un acide aminé particulier très conservé. Cette mutation produit des ADN polymérases dont le taux de synthèse n’est pas perturbé, mais dont le taux d’erreurs au cours de cette
rapporteur dans une orientation ou une autre, d’un côté ou de l’autre d’une origine de réplication dans le génome de la levure, le laboratoire de Thomas Kunkel a pu mettre en lumière que Polδ est majoritairement la polymérase du brin discontinu et que Polε est principalement celle du brin continu (Nick McElhinny et al., 2008; Pursell et al., 2007). Cette répartition des rôles restent encore à être démontrée chez les eucaryotes supérieurs.
1.1.3
La terminaison
La terminaison de la réplication a lieu lorsque deux fourches de réplication se rencontrent. Les connaissances concernant cette étape chez les eucaryotes sont relativement limitées. Contrairement aux procaryotes, aucune séquence spécifique d’ADN ne semble requise pour cette étape. L’ADN ligase I catalyse la formation de la liaison phosphodiester entre les deux brins d’ADN néo-synthétisés. Les contraintes topologiques engendrées par la réplication sont annulées par l’action de l’ADN topoisomérase I tout au long de la réplication et les molécules d’ADN filles sont finalement déconcaténées par la topoisomérase II.
Cependant la terminaison de la réplication aux extrémités des chromosomes (télomères) est un tout autre problème. Dès 1972, James Watson avait prédit avec justesse que le brin discontinu d’un chromosome linéaire recopié par une synthèse semi-conservative ne pouvait pas être complet. L’incapacité des ADN polymérases réplicatives à répliquer totalement des molécules d’ADN linéaires est appelé « le problème de fin de réplication ». Chez l’homme, un des modèles de fin de réplication propose que la synthèse du brin discontinu s’arrête une fois que le brin continu a terminé la réplication du brin parental matrice. Si l’on estime que le retard de synthèse entre les deux brins correspond au temps nécessaire à la synthèse d’un fragment d’Okazaki (environ 200 nucléotides chez l’homme), on s’attend alors à ce que le brin discontinu soit plus court d’environ 200 nucléotides par rapport au brin parental lui servant de matrice. D’autres étapes de maturation des extrémités peuvent avoir lieu, notamment des étapes de résection, entrainant alors la perte de plusieurs centaines de nucléotides au final (Figure 5).
FIGURE 5. Fin de la réplication au niveau des télomères et maturation chez l’homme. La réplication semi-conservative des télomères génère une extrémité franche sur le brin continu et une extrémité 3’ sortante sur le brin à synthèse discontinu. Cette extrémité sortante d’environ 200 nucléotides est due à une inhibition de la synthèse au niveau du dernier fragment d’Okazaki. Une digestion nucléolytique par des facteurs de résection dans le sens 5’ vers 3’ génère alors des extrémités 3’ sortantes aux deux extrémités du chromosome, permettant la formation d’une structure télomérique fonctionnelle (non illustrée ici). D’après E. Gilson et V. Geli, 2007 (Gilson and Geli, 2007).
1.2
La fidélité de la réplication
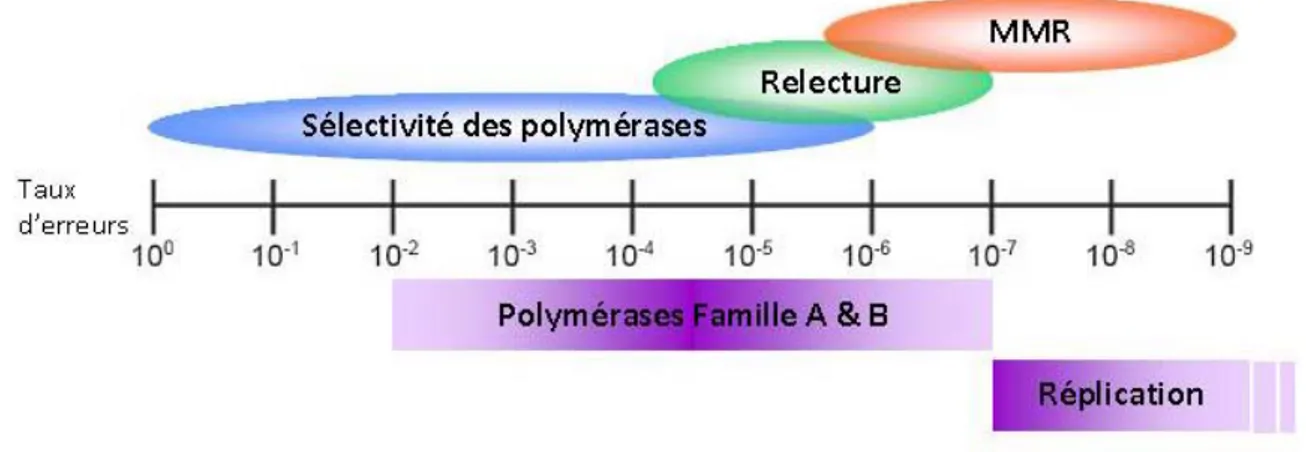
Un des grands challenges d’une cellule est de dupliquer six milliards de nucléotides dans le cas d’une cellule humaine avec la fidélité et la précision nécessaires au maintien de la stabilité du génome à travers les générations. Cette grande fidélité de la réplication est assurée par la combinaison de plusieurs mécanismes. En effet, la majorité des ADN polymérases réplicatives sont capables de sélectionner avec précision le nucléotide approprié à la base qu’elles vont répliquer. De plus, la plupart de ces ADN polymérases réplicatives possèdent une activité 3’Æ 5’ exonucléase qui leur permet de corriger l’incorporation d’une base non appropriée sur la chaîne d’ADN en cours d’élongation. Dans l’éventualité où une erreur de copie aurait échappé à l’activité correctrice des ADN polymérases, un mécanisme post-réplicatif de reconnaissance et de réparation des mésappariements, le MMR (Mismatch Repair), rétablit la séquence d’ADN correcte. L’ensemble de ces trois mécanismes résulte en un taux d’erreurs in vivo estimé à moins de 1x10-9 (Figure 6).
FIGURE 6. La fidélité de la réplication. La contribution relative des trois principales composantes de la fidélité de la réplication est représentée au-dessus de l’échelle, estimée à partir du taux de mutations de systèmes déficients en une ou plusieurs de ces composantes. Les barres horizontales sous l’échelle représentent l’intervalle du taux d’erreurs estimé in vitro des ADN polymérases des familles A et B et du taux d’erreurs estimé in vivo du mécanisme global de réplication. La barre en pointillé à droite indique que ce taux peut être plus faible que celui indiqué. D’après S. D. McCulloch et T. A. Kunkel, 2008 (McCulloch and Kunkel, 2008)
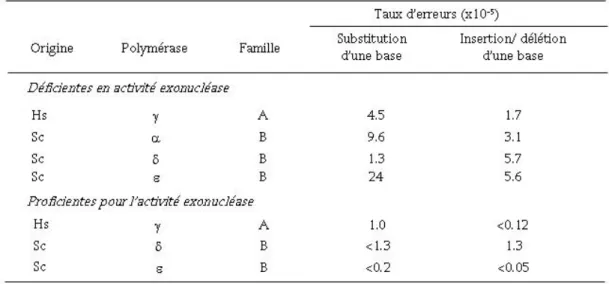
TABLE 1. Taux d’erreurs des ADN polymérases réplicatives
Hs = Homo sapiens ; SC = Saccharomyces cerevisiae
Les valeurs représentent la moyenne des taux d’erreurs obtenus en utilisant le système de synthèse d’ADN M13mp2
D’après S. D. McCulloch et T. A. Kunkel, 2008 (McCulloch and Kunkel, 2008)
1.2.1
La sélection des nucléotides
L’incorporation initiale d’un nucléotide par la grande majorité des ADN polymérases réplicatives est quasiment toujours correcte. Cette capacité à sélectionner avec précision le bon nucléotide est sans doute la plus simple, mais également la plus efficace contribution à la fidélité de la réplication. En effet, Polα, qui n’a naturellement pas d’activité correctrice d’erreurs et des mutants de Polε et Polδ ayant leur activité 3’Æ 5’ exonucléase inactivée ont tout de même de faibles taux de substitutions, insertions ou délétions (Table 1), illustrant leur haute sélectivité du nucléotide à incorporer.
Des études structurales sur les ADN polymérases ont permis d’expliquer en partie cette propriété. Des complexes ternaires formés entre ADN polymérase, matrice et dNTP ont mis en évidence les contraintes stériques expliquant que seul un dNTP respectant l’appariement de Watson-Crick peut se positionner de manière stable dans le site actif de l’enzyme afin d’y subir la réaction chimique lui permettant d’être incorporé. La présence
diminuer l’affinité de liaison du dNTP incorrecte au site actif de l’enzyme, d’affecter les changements conformationnels nécessaires à la réaction de polymérisation et enfin de réduire le taux de formation de liaisons phosphodiesters nécessaires à l’incorporation du nucléotide.
1.2.2
L’activité 3’ Æ 5’ exonucléase
Les ADN polymérases réplicatives, mise à part Polα, présentent également une activité exonucléase orientée dans le sens inverse de polymérisation, c’est-à-dire dans le sens 3’ Æ 5’. Cette activité exonucléase est localisée à proximité du site catalytique. Au cours de la synthèse d’ADN, la polymérase oscille constamment entre son activité polymérase et exonucléase. La balance entre ces deux activités dépend de l’extrémité 3’ OH à étendre. Lorsqu’un dNTP incorrect est incorporé dans l’ADN, l’extension du mésappariement est environ mille à un million de fois plus lente qu’une extrémité correcte (Perrino and Loeb, 1989). Ce délai dans l’extension permet à l’exonucléase de cliver le nucléotide incorrect. Après clivage, l’activité polymérase peut reprendre normalement à partir de bases correctement appariées.
La contribution de ce mécanisme dans le contrôle de la fidélité de réplication est illustrée par le faible taux de substitutions des ADN polymérases Polδ et Polε, comparées à leurs versions déficientes en activité 3’ Æ5’ exonucléase (Table 1). De manière plus large, cette activité de relecture joue un rôle critique dans le maintien de la stabilité du génome. En effet, des souris déficientes en activité exonucléase de Polδ voient leur durée de vie raccourcie et leur prédisposition à différents types de cancers augmentée (Goldsby et al., 2001; Goldsby et al., 2002).
Polα, qui est dépourvue de cette activité exonucléase, synthétise 10 nucléotides sur chaque fragment d’Okazaki du brin discontinu, ce qui représente à peu près 2% du génome. Son taux d’erreur étant d’environ 10-4 (Table 1) sa contribution à la réplication du génome devrait générer 12 000 mésappariements, ce qui n’est concrètement pas le cas. Des études chez la levure ont soulevé et appuyé l’hypothèse d’une correction des erreurs
générées par Polα lors de l’initiation des fragments d’Okazaki, par l’activité exonucléase de Polδ (Pavlov et al., 2006a).
1.2.3
La réparation des mésappariements par le MMR
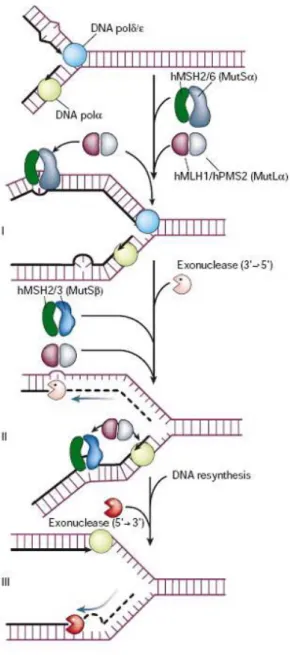
Le système de réparation des mésappariements, le MMR est un mécanisme de réparation de l’ADN conservé des bactéries aux êtres humains. Il cible les mésappariements qui peuvent être dus à des substitutions, des insertions ou des délétions résultant d’erreurs de réplication ayant échappé à l’activité de relecture des ADN polymérases. De cette façon, le MMR améliore la fidélité de la réplication d’un facteur 50 à 1000 (Figure7). Le mésappariement est reconnu par l’hétérodimère Msh2/Msh6, alors que les insertions ou délétions sont détectées par le dimère Msh2/Msh3. L’ATP permet un changement de conformation de ce complexe qui agit alors comme un anneau glissant de manière latérale le long de l’ADN, permettant au complexe d’orienter sa réparation en identifiant le brin parental du brin néo-synthétisé à corriger. L’identification du brin à corriger est en effet orientée par la recherche de brèches simple-brin dans l’ADN entre les fragments d’Okazaki sur le brin discontinu ou d’extrémités 3’OH libres sur le brin continu. Msh2/Msh6 se lie à l’hétérodimère Mlh1/Pms2 et cet ensemble déplace PCNA et l’ADN polymérase du brin en cours de synthèse au niveau du mésappariement et recrute l’exonucléase I (Exo I). Cette dernière dégrade le brin néo-synthétisé jusqu’à l’élimination du mésappariement (Genschel et al., 2002) (Figure 7). Cette exonucléase est stimulée par la présence de PCNA et RFC (Dzantiev et al., 2004). RPA stabilise alors la partie d’ADN simple-brin découverte et la resynthèse se fait grâce à l’action de Polδ.
FIGURE 7. Modèle de réparation des mésappariements par le MMR.
Les hétérodimères Msh2/6 se focalisent sur les mésappariements, alors que les dimères Msh2/3 reconnaissent les insertion ou délétions (étape I). Le dimère Mlh1/Pms2 interagit avec Msh2/6 et les facteurs de réplication. Plusieurs protéines sont impliquées dans l’excision du nouveau brin en aval du mésappariement et dans l’étape de resynthèse, dont Polδ/ε, RPA, PCNA, RFC et l’exonucléase 1 (étapes II et III)
2. Les ADN polymérases translésionnelles (TLS) et la
gestion des lésions de l’ADN
La réplication de l’ADN a été décrite jusqu’ici dans un contexte théorique simplifié, afin de mettre en évidence les rôles respectifs des protéines impliquées dans un but simple et unique : répliquer dans chaque cellule les trois milliards de paires de bases constituant le génome, dans leur intégralité et sans erreur. En réalité, la réplication s’effectue dans un contexte bien plus hostile, puisque l’ADN est continuellement endommagé par des processus endogènes et des facteurs mutagènes environnementaux, générant des obstacles à la progression des fourches de réplication, le site actif contraignant des ADN polymérases réplicatives ne leur permettant pas de répliquer les bases endommagées. Cependant, un nombre croissant d’ADN polymérases alternatives, appelées ADN polymérases « translésionnelles » (TLS), ont été récemment identifiées et jouent un rôle majeur dans la gestion des lésions de l’ADN. Ces enzymes interviennent en effet dans les mécanismes de réplication, de réparation et de recombinaison (les « 3R »), qui jouent un rôle primordial dans le maintien de la stabilité du génome.
2.1
Les barrières de fourches de réplication
2.1.1
L’ADN structuré
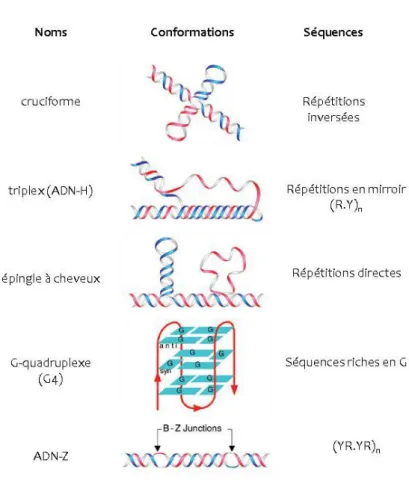
La majorité de la molécule d’ADN adopte une conformation en hélice droite nommée ADN-B. Cependant, au moins dix types différents de conformations non-B ont été identifiés, dont les principaux sont présentés sur la Figure 8.
FIGURE 8. Conformations non-B de l’ADN. Le nom, la conformation et les séquences de cinq formes d’ADN non-B majeures sont présentés. D’après R. Wells, 2007 (Wells, 2007).
Ces séquences d’ADN capables d’adopter naturellement des conformations non-B constituent des sources d’interférence avec la machinerie de réplication. Les séquences répétées qui représentent plus de 50% du génome, contribuent majoritairement à la formation de structures non-B de l’ADN et l’expansion des séquences répétées trinucléotidiques sont associées à plusieurs syndromes neurologiques hériditaires (X fragile ou maladie de Huntington) (Bowater and Wells, 2001). Des difficultés de progression des fourches de réplication surviennent également au niveau de séquences riches en G capables d’adopter une structure d’ADN en G-quadruplex (G4) grâce à des liaisons hydrogènes entre les résidus guanine. Ces régions d’ADN sont capables d’influencer l’expression des gènes et la stabilité du génome (Maizels, 2006) et le nombre de sites
distincts capables de former des G4 est estimé à plus de 300 000 dans le génome humain (Eddy and Maizels, 2006). Ces séquences G4 sont très difficiles à répliquer et leur duplication nécessite l’intervention d’hélicases spécialisées dont le défaut conduit également à des désordres génétiques majeurs et à des maladies héréditaires. Les structures d’ADN non-B sont également associées à une variété d’autres types d’instabilité génétique
in vivo. Certaines régions génomiques appelées « sites fragiles » sont ainsi
préférentiellement sujettes aux cassures double brin de l’ADN et aux translocations, délétions ou amplifications chromosomiques (Popescu, 2003). A l’heure actuelle, plus de cent sites fragiles ont été identifiés. Ils se subdivisent en deux catégories : les sites fragiles rares et les sites fragiles communs. Les sites fragiles rares sont présents chez moins de 5% des individus et se ségrégent de façon mendélienne. L’augmentation des CDBs à leur niveau est le plus souvent causée par des expansions de répétitions de nucléotides. Les sites fragiles communs (SFC) sont eux présents chez chaque individu et représentent la classe de sites fragiles la plus importante. Ces sites fragiles communs comportent des séquences d’ADN structuré et constituent des régions du génome hypersensibles aux stress de la réplication provoqués par l’aphidicoline, un inhibiteur des ADN polymérases réplicatives (Durkin and Glover, 2007). De plus, ces SFC sont répliqués tardivement en phase S, voire pendant la phase G2, ce qui pourrait rendre compte de la difficulté à les répliquer (Durkin and Glover, 2007). Enfin, leur implication dans le développement tumoral est de plus en plus reconnue. En effet, de nombreux réarrangement au niveau de ces SFC ont été mis en évidence comme étant une caractéristique des stades précoces de développement des cancers (Bartkova et al., 2005; Gorgoulis et al., 2005).
2.1.2
Les collisions avec la machinerie de transcription
Continuellement, les complexes protéiques de transcription permettent l’expression des gènes requis pour l’ensemble des fonctions cellulaires. Il est alors possible que la machinerie de transcription puisse interférer avec le processus de réplication et que le réplisome puisse entrer en collision avec les protéines de la transcription. Afin d’éviter cela, la cellule a mis en place des mécanismes de « pause » de la réplication en amont des
gènes hautement transcrits, comme par exemple au niveau de l’ADNr codant pour les ARN ribosomiques chez la levure. Au niveau des gènes transcrits par l’ARN polymérase III (produisant les ARNm), il apparaît que la collision entre réplication et transcription soit une source naturelle de cassure de l’ADN et soit à la base du mécanisme de Recombinaison Associée à la Transcription (RAT) (Prado and Aguilera, 2005). Afin d’éviter au maximum les collisions entre les deux machineries, sur plus d’un quart du génome, les gènes fortement transcrits sont orientés dans la même direction que le sens d’élongation de la réplication (Huvet et al., 2007).
2.1.3
Les lésions de l’ADN
Le génome des cellules est sans cesse la cible d’un large spectre de lésions d’origine endogène ou causées par une exposition à des agents génotoxiques environnementaux
(Figure 9). De plus, tous les composants de l’ADN peuvent être sujets à ces dommages (les
bases, les sucres et les liaisons phosphodiesters).
FIGURE 9. Les lésions de l’ADN. Les agents génotoxiques les plus communs sont représentés en haut et les dommages de l’ADN qu’ils engendrent sont représentés en bas. D’après J. H. J Hoeijmakers, 2001 (Hoeijmakers, 2001)
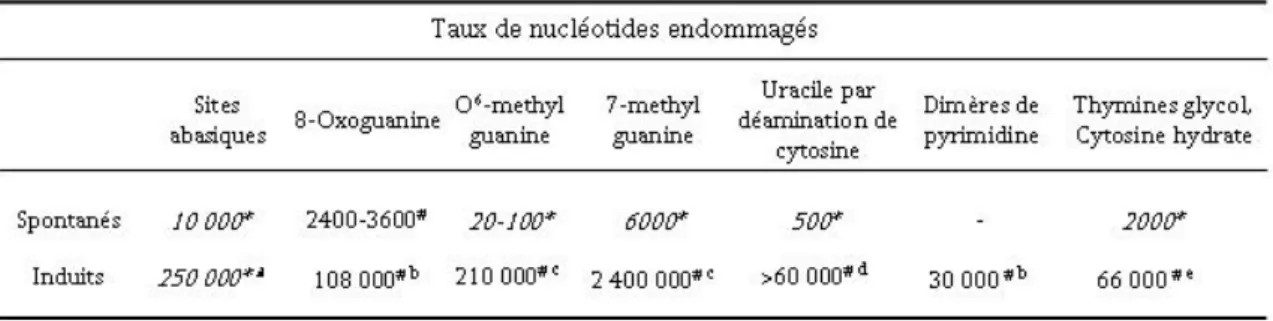
TABLE 2. Types et quantités de nucléotides endommagés retrouvés communément dans l’ADN.
* Taux de lésions exprimé en nombre de lésions par cellules par jour, considérant six millions de nucléotides par cellule diploïde.
# Taux de lésions exprimés pour six millions de bases d’ADN
a Traitement de la peau par 200 nM d’œstradiol-3,4-quinone
b Induit par la dose d’UVA, qui est estimée correspondre à une heure d’exposition au soleil en été à midi à
45° de latitude (200kJm-2 par heure)
c Taux de lésions dans de l’ADN isolé de tissu hépatique de rat 1-2 heures après adminitration d’une simple
dose (60 mg/kg) de méthyl nitrosourée (MNU)
d Induit par une surproduction de AID. C’est une estimation minimale basée sur le taux de mutations CÆ T. e Basé sur le taux de formation de thymine glycol par Gy de γ-irradiation d’une solution de thymus de veau.
D’après Y. I. Pavlov, P. V. Shcherbakova, I. B. Rogozin (Pavlov et al., 2006b)
a) Les lésions endogènes
Nous savons désormais que la structure primaire de l’ADN peut être largement altérée en réagissant avec des molécules présentes dans l’environnement cellulaire normal. Plus de 10 000 résidus purines ou pyrimidines sont perdus chaque jour dans une cellule par hydrolyse de la liaison reliant la base et le squelette phosphate de l’ADN (Table 2)
(Lindahl and Nyberg, 1972; Nakamura et al., 1998). Cette réaction produit des sites abasiques, ou AP (apurinique, ou apyrimidinique). Une autre source de lésion endogène de l’ADN est la transformation de résidus cytosine en résidus uracile par désamination. Cela se produit environ 500 fois par jour dans chaque cellule de mammifère (Frederico et al., 1990; Shen et al., 1994) (Table 2). Les produits de désamination peuvent donner naissance à des mutations au cours de la synthèse d’ADN, car la perte du groupe amine change les propriétés d’appariement de la base. Enfin, le stress oxydatif est une conséquence inévitable de la vie en atmosphère riche en oxygène et les ROS (Reactive oxygen species) sont générés de manière continue comme produits annexes du métabolisme aérobique.
Cependant, ces ROS sont extrêmement réactifs et endommagent l’ADN. En effet, l’attaque du résidu sucré conduit à une fragmentation, une perte de base et des cassures de l’ADN essentiellement simple-brin et l’attaque au niveau des bases peut conduire par exemple au thymine-glycol ou à la 7,8-dihydro-8-oxoguanine, plus connue sous l’abréviation 8-oxoG
(Figure 10).
FIGURE 10. Exemples de modifications des bases de l’ADN par le stress oxydatif.
b) Les lésions exogènes
Des agents physiques naturels venant de sources externes peuvent également porter atteinte à l’intégrité du matériel génétique. Les rayonnements ultra-violets (UV) du soleil sont en effet à l’origine de lésions toxiques de l’ADN et représentent la source majeure de développement de cancers de la peau. La plupart des effets mutagènes des rayonnements UV proviennent des UV-B et UV-C qui génèrent des photoproduits de l’ADN, majoritairement des Dimères de Pyrimidines Cyclobutane (CPD) (Figure 11A) et des adduits (6-4) pyrimidine-pyrimidone [(6-4)PP] (Figure 11B) qui distordent la structure de l’ADN (Table 2). Les CPD sont les photoproduits les plus délétères pour la réplication car ils induisent un ralentissement de la synthèse globale d’ADN au cours de la phase S.
FIGURE 11. Lésions majoritaires induites par les radiations UV
Les radiations ionisantes sont une autre source de rayonnements subie par les être vivants. Ces rayonnements proviennent essentiellement des radiations cosmiques ou encore de la radioactivité naturelle des roches, mais aussi de sources artificielles telles que les rayons X ou les composés radioactifs utilisés en médecine nucléaire. La formation de molécules excitées ou ionisées par les radiations ionisantes induit des dommages à tous les composants cellulaires et provoque une grande variété de lésions sur l’ADN, notamment des cassures simple-brin (CSB) ou double-brin (CDB) ainsi qu’une multitude de dommages oxydatifs.
De nombreux composés chimiques peuvent réagir avec l’ADN et altérer sa chimie ainsi que sa structure. Les agents alkylant sont capables d’attacher de manière covalente un groupement alkyl aux bases de l’ADN, rendant ces bases difficiles à répliquer par les ADN polymérases et donc pouvant être à l’origine de mutations. Le methyl methanesulfonate (MMS), le methylnitrosourea (MNU) ou le temozolomide sont utilisés en laboratoire dans des études de réparation de l’ADN, et le temozolomide est également utilisé comme agent anticancéreux en chimiothérapie. Les agents bi-fonctionnels peuvent réagir avec deux centres nucléophiles différents sur l’ADN. Si ces deux sites sont sur le même brin d’ADN
opposés, il en résulte un pontage inter-brin (Figure 12). Ces derniers représentent des dommages chimiques important de l’ADN, car ils empêchent la séparation des deux brins d’ADN et constituent de ce fait un blocage complet des ADN polymérases réplicatives Polδ, ε et α (Hoffmann et al., 1995). C’est pour cette raison qu’un certain nombre d’agents de ce type, tels que le cisplatine (cis-Pt), la mitomycine C (MMC), les moutardes azotées ou certains psoralènes photo-activés ont été largement utilisés en chimiothérapie anticancéreuse.
FIGURE 12. Lésions générées par les agents bi-fonctionnels tels que le cisplatine.
Le benzo[a]pyrène est une molécule hautement cancérigène retrouvée dans le goudron, la fumée de cigarette, les gaz d’échappement des véhicules. Non modifié, ce composé n’est pas réactif. Cependant, lorsqu’il est inhalé, sa métabolisation par ce cytochrome P-450 dans les cellules le transforme en 7,8-diol-9,10,-epoxide (BPDE) qui se lie de manière prépondérante aux guanines (Figure 13). Cette lésion particulièrement encombrante entraîne le blocage d’une fourche de réplication du virus SV40 par la machinerie réplicative d’une cellule de mammifère (Rinaldy et al., 1982).
FIGURE 13. Formation d’un adduit BPDE sur l’ADN après métabolisation du Benzo[a]pyrene par le cytochrome P-450 des cellules.
2.2
Rôle des ADN polymérases TLS dans la tolérance des
lésions
Malgré l’ensemble de ces lésions, les cellules en division doivent à tout prix maintenir et achever la réplication de leur ADN. Cependant, les ADN polymérases réplicatives sont incapables de répliquer la plupart de ces bases endommagées, leur site actif étant trop étroit et pas assez flexible. Ces lésions peuvent alors se révéler très dangereuses, car elles sont capables de bloquer la machinerie de réplication. Si ce blocage perdure, il peut conduire à l’effondrement de la fourche de réplication et à la production de CDBs de l’ADN et de réarrangements chromosomiques pouvant aboutir à la mort cellulaire. Cependant, les cellules ont mis en place des mécanismes permettant de tolérer ces lésions, d’éviter un blocage de fourche prolongé et de permettre à la réplication de se poursuivre malgré les dommages de l’ADN. Ces lésions seront alors réparées plus tard dans le cycle, en phase G2, par divers mécanismes de réparation. Chez les eucaryotes, on distingue deux voix de tolérance des lésions. La première, connue sous le nom de « contournement de lésion », est fidèle et utilise la chromatide sœur non endommagée comme matrice. La seconde voie, appelée synthèse translésionnelle (STL) fait intervenir les ADN polymérases TLS pour répliquer l’ADN en regard des lésions de manière fidèle ou mutagène (Figure 14).
FIGURE 14. Mécanismes possibles de tolérance des lésions de l’ADN au niveau d’une fourche de réplication bloquée. Le triangle représente une lésion bloquant la machinerie de réplication. Les ADN polymérases TLS sont capables de réaliser de la synthèse translésionnelle, mécanisme qui peut être mutagène. Le contournement de la lésion semble être un mécanisme fidèle qui peut s’effectuer soit par une régression de fourche de réplication, soit par l’invasion de la chromatide sœur suivi d’une synthèse et d’une résolution des jonctions de Holliday. D’après P. L. Andersen (Andersen et al., 2008).
2.2.1
Mono et poly-ubiquitinylation de PCNA
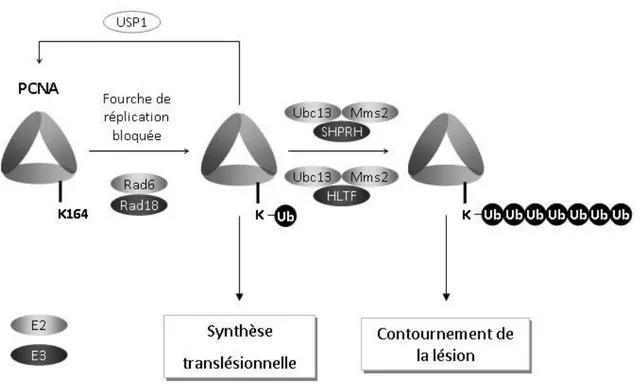
Le choix de tolérer les lésions par l’une ou l’autre de ces deux voies dépend des modifications post-traductionnelles de PCNA (Figure 15).
La première modification, commune à ces deux voies est la mono-ubiquitinylation de PCNA sur le résidu K164 de chacune de ses trois sous-unités réalisée grâce à l’action combinée de l’enzyme E2 Rad6 et de l’enzyme E3 ubiquitine-ligase Rad18. Lorsque la fourche de réplication est bloquée par une lésion, il se produit un découplage entre les hélicases qui précèdent la fourche et les ADN polymérases, ce qui conduit à une accumulation d’ADN simple-brin rapidement recouvert et stabilisé par la protéine RPA. Des études ont montré que l’ADN simple-brin généré par ce découplage était requis pour activer le complexe Rad6/Rad18 et induire la mono-ubiquitinylation de PCNA (Chang et
al., 2006). Récemment, l’importance de RPA dans l’ubiquitinylation de PCNA en réponse aux dommages à été mise en évidence. RPA interagit directement avec Rad18, que ce soit chez la levure ou les cellules de mammifères, et recrute Rad18 au niveau des régions simple-brin de l’ADN in vitro. Cette interaction entre RPA et Rad18 semble jouer un rôle important pour l’ubiquitinylation de PCNA in vivo (Davies et al., 2008). Même si le blocage de fourches de réplication et le découplage entre hélicases et ADN polymérases a également pour conséquence d’induire le point de contrôle médié par ATR, ces deux événements semblent indépendants, puisqu’un taux réduit d’ATR dans les cellules humaines n’a pas d’effet significatif sur la mono-ubiquitinylation de PCNA (Niimi et al., 2008). Cette mono-ubiquitinylation peut survenir en réponse à une grande diversité de lésions de l’ADN, telles que celles induites par les UV, le MMS et l’hydroxyurée (HU) qui agit en sevrant la cellule en désoxynucléotides, toutes ces lésions générant un découplage entre hélicases et polymérases. Cependant, les radiations ionisantes et la camptotécine, qui n’induisent pas de découplage mais des CDBs de l’ADN n’induisent pas la mono-ubiquitinylation de PCNA (Niimi et al., 2008). Ceci appuie l’importance de l’ADN simple-brin et de RPA dans ce processus d’activation de PCNA. L’hypersensibilité aux UV, au MMS ou encore au Cisplatine des cellules embryonnaires de souris dépourvues de Rad18 (Tateishi et al., 2003) démontre l’importance de la mono-ubiquitinylation de PCNA pour la résistance aux agents génotoxiques. De plus des mutants de PCNA sur la lysine 164 présentent également une hypersensibilité aux UV ainsi qu’au MMS (Niimi et al., 2008). L’action du complexe Rad6/Rad18 peut être contrebalancée par l’activité d’USP1. Cette désubiquitinase est capable d’exciser le résidu ubiquitine sur PCNA. En réponse aux UV, il a été montré qu’USP1 s’auto-dégrade permettant ainsi l’apparition de la forme mono-ubiquitinylée de PCNA (Huang et al., 2006). La mono-ubiquitinylation de PCNA favoriserait le recrutement de certaines ADN polymérases TLS au niveau de la fourche de réplication aiguillant le mécanisme de tolérance de la lésion vers la voie de synthèse translésionnelle.
![FIGURE 13. Formation d’un adduit BPDE sur l’ADN après métabolisation du Benzo[a]pyrene par le cytochrome P-450 des cellules](https://thumb-eu.123doks.com/thumbv2/123doknet/2259159.19535/42.892.166.736.122.320/figure-formation-adduit-metabolisation-benzo-pyrene-cytochrome-cellules.webp)