Ecole doctorale de Chimie Physique et Chimie analytique de Paris Centre (ED388)
Laboratoire d’Electrochimie Moléculaire
Approche électrochimique multiéchelle de l’activation
réductrice de O2 par un complexe de fer bioinspiré.
Par Célia ACHAIBOU
Thèse de doctorat d’Electrochimie
Dirigée par Elodie ANXOLABEHERE-MALLART Présentée et soutenue publiquement le 14 novembre 2019
Devant un jury composé de :
M. Nicolas LE POUL CR Université de Bretagne Occidentale, Brest Rapporteur
M. Mathieu ETIENNE DR Université de Lorraine, Nancy Rapporteur
Mme Petra HELLWIG PR Université de Strasbourg Examinatrice
Mme Carole DUBOC DR Université de Grenoble Alpes Examinatrice
Mme Elodie ANXOLABEHERE-MALLART
DR Université Paris Diderot Directrice
3
dirigé par Mme Elodie Anxolabéhère-Mallart. Je la remercie de m’avoir accueillie entre ses murs et d’avoir encadré ma thèse avec beaucoup d’enthousiasme, de disponibilité et de bienveillance. Je tiens à lui exprimer dans ces quelques mots ma plus profonde gratitude.
Je tiens à remercier les rapporteurs, M. Nicolas Le Poul et M. Mathieu Etienne qui ont accepté de consacrer de leur temps à l’examen de ce manuscrit ainsi que Mme Petra Hellwig et Mme Carole Duboc qui ont accepté de participé à mon jury de thèse.
Je remercie très vivement M. Frédéric Kanoufi qui a gardé un œil attentif et bienveillant sur l’ensemble des travaux de cette thèse.
Mes remerciements les plus sincères à M. Jean-Marc Noël. C’est d’ailleurs sa bonne humeur, son dynamisme et son éternel optimisme envers ces travaux qui m’ont permis de continuer à avancer ! J’ai pris beaucoup de plaisir à travailler dans l’équipe TERS. Je remercie Mme Catherine Combellas de m’avoir accueillie dans cette équipe et de s’être toujours montrée disponible et bienveillante lors de nos différents échanges. Je remercie tous les membres de cette équipe formidable, M. Jean Pinson, M. Jérôme Médard et M. Jean-François Lemineur.
Mes remerciements vont également à Mme Claire Fave pour avoir contribué au bon déroulement de ma thèse, pour nos échanges constructifs, enrichissants et toujours très agréables.
Je tiens à remercier l’équipe REACTE au LEM dirigée par M. Marc Robert, grâce à qui tous les moyens, aussi bien matériels qu’immatériels, ont toujours été mis pour que nos recherches soient réussies.
Je vais remercier l’ensemble des membres du laboratoire, notamment Julien et son humour, Sihem notre tata préférée, François, Mathieu, Niklas, Orestes, Rabia… vous avez su égayer mon quotidien entre les murs verts de Lavoisier.
Ensuite je vais remercier tous les thésards et post-docs que j’ai côtoyé durant ces années de thèse au LEM en commençant par la « dream-team » Etienne, Charlie, Jérémy, Claudio, Khalil et Martin, par Lydia et Justine mes acolytes de pause post-diurétiques puis Hussein, Min, Bing et Telmo avec qui nous avons partagés bien plus qu’un bureau. Je remercie également Nikos mon petit « partner in crime », Thomas, Tamal, Mickaël, Benji, Daniela, Marie, Martin et Dorian. Je remercie également les thésards croisés au 6ème (Vitor, Alice D, Mathias, Alice F) et ailleurs dans le bâtiment (Brandon, Thomas…). Si j’en ai oublié, je m’en excuse.
Je remercie du fond du cœur mes parents qui m’ont toujours soutenue et ma famille sans qui rien n’aurait été possible.
Enfin, je remercie mon mari Massine qui a été d’un soutien infaillible durant cette longue aventure et bien d’autres...
4
synthèse chimique respectueux de l'environnement, peu coûteux et faciles à mettre en œuvre.
Pour le développement des procédés d’oxydation économiquement et écologiquement viables, l’utilisation de O2, abondant et peu coûteux, apparait comme un choix idéal. Pour cela, certaines
metalloenzymes, telles que les cytochromes P450 capables de réaliser des réactions d’oxydation efficacement et sélectivement dans des conditions douces, constituent une source d’inspiration. Cependant, une telle réactivité nécessite l’activation de O2. Ce processus, appelé activation réductrice,
implique la coordination de O2 sur un ion Fe du site actif et une succession d’étapes de réduction et
protonation conduisant à la formation d’intermédiaires réactionnels tels que FeIII
OO•, Fe IIIOO-, FeIIIOOH et FeVO après la coupure de la liaison O–O, espèce hautement réactive et capable d’oxyder un substrat organique.
L'enjeu de ma thèse est de comprendre les mécanismes et déterminer les paramètres permettant de contrôler cette activation. Nous avons alors choisi un complexe porphyrine modèle [FeII(F20TPP)Cl] et
une approche électrochimique multiéchelle, basée sur l’utilisation de la voltamétrie cyclique (CV), la microscopie électrochimique à balayage (SECM) et la spéctroéléctrochimie UV-Vis (SEC). Dans un premier temps, l’étude du système [O2 + e
+ H+] en absence de Fe, complétée par une étude en présence d’ion Li+
et Na+ constitue une point de départ et une mise au point expérimentale. Par la suite, l’étude par SECM du système [FeII
+ O2 + e
-] a mis en évidence la détection de l’oxydation à un
électron de FeIIIOO. En présence de protons, l’étude du mécanisme réactionnel du système [FeII + O2 +
e- + H+] a montré la formation de l’intermédiaire FeIVO par SEC et sa détection par SECM. Enfin, des études en réactivité en présence de Cl- sont présentées comme résultats prometteurs de ce travail.
Abstract
Economic and ecological issues call for the development of new environmentally friendly, inexpensive and easy to implement chemical synthesis processes. To turn towards economically and environmentally viable oxidation processes, the use of molecular O2 which is abundant and inexpensive
is an ideal strategy. Some metallo-enzymes such as cytochromes p450 whose active site contains a Fe ion, are able to use O2 to perform efficient and selective oxidation reactions in mild conditions.
However, O2 must be first be activated to achieve these reactions. This process called reductive
activation involves several steps of different reduction and protonation leading to the formation of reactive intermediate species such as FeIIIOO•, FeIIIOO-, FeIIIOOH and FeVO after the cleavage of the O-O that is able to oxidize an organic substrates.
The challenge of my phD thesis is to understand the mechanisms and to determine the parameters to control this activation. Firstly, the study of the [O2 + e
+ H+] system in the absence of Fe, completed by a study in the presence of Li+ and Na+ ion is a starting point. Subsequently, the SECM study of the [FeII + O2 + e
-] system demonstrated the detection of FeIIIOO- oxidation at one electron. In the presence of protons, the study of the mechanism of the [FeII + O2 + e
-
+ H+] system is completed and the formation of the FeIVO intermediate is evidenced by SEC. Finally, reactivity studies in the presence of Cl– are presented as promising results of this work.
5
Table des matières
Table des matières ... 5 Liste des figures ... 8 Introduction générale ... 17 I.
Chapitre I : Utilisation du dioxygène comme oxydant, contexte, état de l’art et stratégie I.
novatrice. ... 21 L’oxydation dans l’industrie chimique... 22 1.
La réaction d’oxydation dans l’industrie ... 22 1.1.
Intérêt de la chimie verte ... 23 1.2.
L’utilisation de O2 dans l’industrie ... 23
1.3.
Les systèmes naturels d’activation de O2 ... 25
2.
Métalloenzymes et activation de O2... 25
2.1.
Cytochrome c oxydase: réduction de O2 à 4 électrons ... 26
2.2.
Cytochrome P450 : activation de O2 à 2 électrons... 29
2.3.
Les systèmes bio-inspirés ... 33 3.
Les métalloporphyrines, modèles des cyt P450 ... 34 3.1.
Espèces intermédiaires du cycle catalytique du cyt P450 ... 36 3.2.
Conclusion ... 41 3.3.
Activation de O2 par voie électrochimique ... 42
4.
Etat de l’art de l’activation de O2 par voie électrochimique ... 42
4.1.
Résultats antérieurs ... 43 4.2.
Méthodologie ... 47 4.3.
Conclusion et présentation du sujet ... 54 5.
6
Chapitre II : Etude électrochimique de la réduction du dioxygène en présence de protons II.
et d’acide de Lewis en milieu aprotique. ... 55 Réaction de réduction de O2 ... 56
1.
LA ORR en milieu organique ... 57 2.
Réduction de O2 en absence de protons ... 57
2.1.
Réduction de O2 en présence de protons ... 60
2.2.
Etude mécanistique par SECM ... 64 2.3.
ORR en présence d’acide de Lewis ... 68 3.
Introduction ... 68 3.1.
ORR en présence de Li+ et Na+ : formation de la SEI ... 69 3.2.
Etude des mécanismes de dissolution de la SEI par SECM ... 72 3.3.
Conclusion ... 76 4.
Chapitre III : Etude SECM de l’activation réductrice de O2 par le complexe
III.
[FeIII(F20TPP)Cl]. ... 77
Etude du complexe [FeIII(F20TPP)Cl] sous argon en mode SG-TC. ... 78
1.
[FeIII(F20TPP)Cl] sous O2 en mode SG-TC ... 80
2.
Suivi de la formation de FeIIIOO- à l’UME. ... 81 3.
Analyse quantitative des données expérimentales. ... 84 4.
4.1. Simulation en configuration SG-TC, potentiel fixe au substrat ... 84 4.2. Simulation en configuration SG-TC, potentiel fixe à l’UME ... 89 Conclusion ... 91 5.
Chapitre IV : Rôle des protons sur l’activation réductrice de O2 par [Fe(F20TPP)Cl].
IV.
Proposition de mécanisme ... 93 Etude électrochimique de l’adduit FeII
OO en présence de protons... 94 1.
Etude par voltamétrie cyclique ... 94 1.1.
Etude par spectroélectrochimie ... 98 1.2.
Etude par SECM ... 106 1.3.
7
Conclusion et mécanisme global ... 110 2.
Chapitre V : Conclusion et perspectives ... 111 V.
Approche multiéchelle. ... 112 Etude de la réactivité en présence de Cl- ... 113 3.
Optimisation des conditions expérimentales pour une catalyse d’oxydation efficace. . 115 4.
Source d’acide ... 115 Vers une catalyse hétérogène... 117 Conclusion ... 117 5. Conclusion générale ... 119 Partie expérimentale ... 123 VI. Produits chimiques ... 124 1. Gaz ... 124 2. Solutions ... 124 3. Techniques analytiques ... 125 4. La voltammétrie cyclique... 125 4.1.
La microscopie electrochimique à balayage ... 126 4.2.
Spéctroélectrochimie ... 127 4.3.
Annexes ... 129 VII.
Préparation et caractérisation de FeIIOH. ... 130 1.
Préparation et caractérisation de FeIVO. ... 132 2.
Caractérisation électrochimique de [FeIII(F20TPP)DMF]. ... 133
3.
Bibliographie ... 135 VIII.
8
Liste des figures
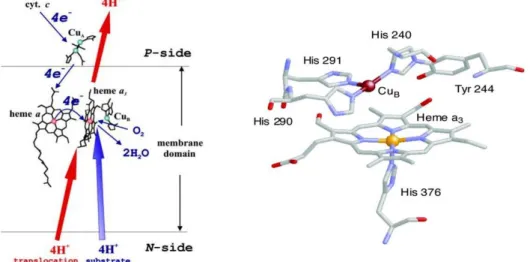
Figure I-1 : Représentation schématique de la chaîne respiratoire mitochondriale. ... 26
Figure I-2 : A gauche, centres oxydoréducteurs de la CcO.17 L’acheminement des réactifs (électrons, protons et dioxygène) est indiqué par les flèches bleues. Les flèches rouges indiquent le pompage transmembranaire de protons couplé à cette réaction du « N-side » (chargé négativement) vers le « P side » (chargé positivement) de la membrane. A droite, structure du site actif binucléaire de CcO.18 ... 27
Figure I-3 : Mécanisme de réduction de O2 par la cytochrome c oxydase. 20 ... 28
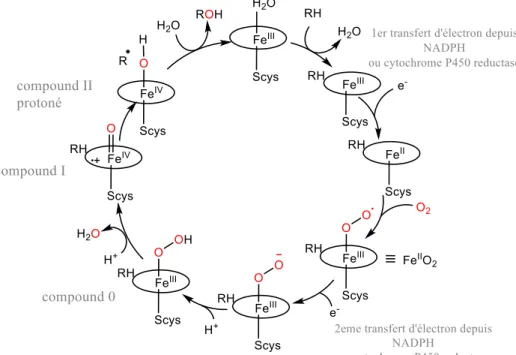
Figure I-4 : Structure du site actif du cytochrome P450 ... 30
Figure I-5 : Cycle catalytique du cytochrome P450.34 ... 30
Figure I-6 : Mécanisme d'hydroxylation d'un alcane R-H. 38 ... 31
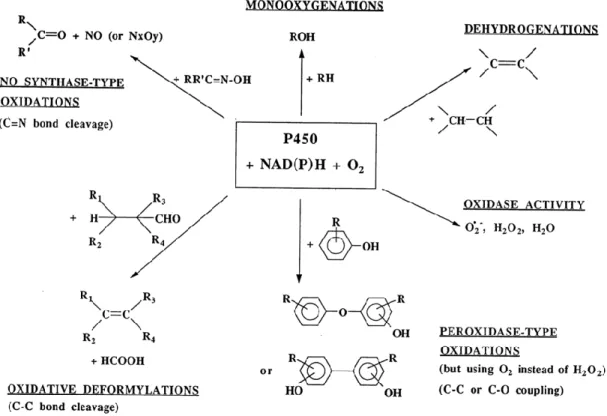
Figure I-7 : Réactions d’oxydation catalysées par les cytochromes P450 d’après Mansuy.42 ... 32
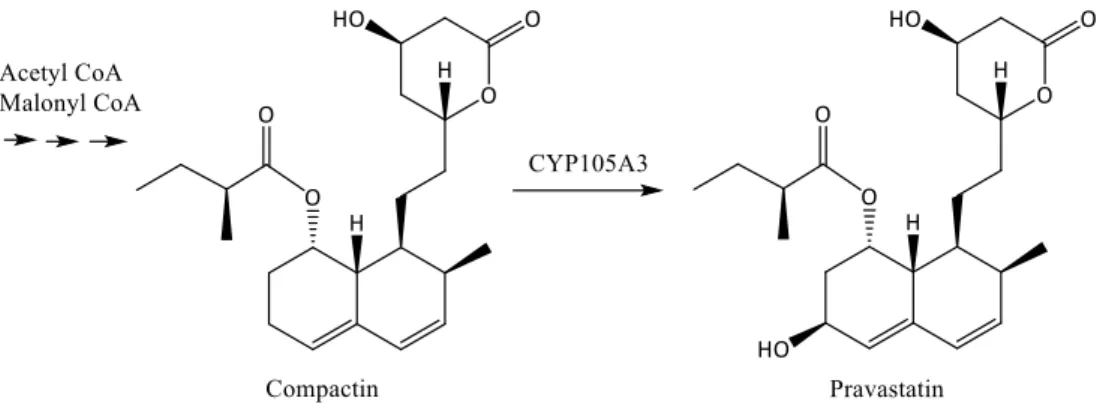
Figure I-8 : Biotransformation de la compactine en pravastine par CYP105A3 dans l’organisme de S.carbophilus. ... 33
Figure I-9 : Porphyrines de Fer majoritairement utilisées comme modèle moléculaire des CytP450.(a) Structure de la protoporphyrin IX avec R1 CH=CH2 , R2 CH3 et R3 (CH2)2-COOH. (b) Structure de 5,10,15,20-tétraphénylporphyrine de Fer ,Fe(TPP) ; avec Ar groupement aryl. ... 34
Figure I-10 : Structure de la porphyrine pyrroheme-N-[3-(1-imidozolyl)propyl]-amide utilisée par Chang et Taylor. X= pas de substituant, H2O, O2 ou CO le Fer est alors au degré d’oxydation +II ou Cl avec le Fer au degré d’oxydation +III. ... 35
Figure I-11 : Formation de l’intermédiaire FeIII-peroxo par voie chimique.59 Le premier équivalent de O2•- sert à réduire l’ion FeIII en FeII puis le second équivalent de O2•- se coordine au FeII. ... 37
Figure I-12 : Structure end-onou η1-peroxo et side on ou η2-peroxo du Fe-peroxo. ... 37
Figure I-13 : Spectre UV-Vis de FeIII-peroxo, formé à partir de [FeIII(F20TPP)Cl] et KO2 dans CH3CN.63 ... 38
Figure I-14 : Epoxydation d’une oléfine pauvre en électrons (2-methyl-1,4-naphtaquinone) par un FeIII-peroxo. ... 38
9
Figure I-15 : Formation de l’intermédiaire FeIII-hydroperoxo par voie chimique selon Tajima.66
... 39
Figure I-16 : Formation de l’intermédiaire FeIII-hydroperoxo.67 ... 40
Figure I-17: Formation de l'intermédiaire FeV=Opar voie chimique.68 ... 41
Figure I-18 : Structure du complexe [FeIII(F20TPP)Cl] ... 44
Figure I-19 : A gauche, CV dans DMF + 0.1 M TBAPF6 de [Fe(F20TPP)Cl] à 1 mM sous argon (noir), solution saturée en air ([O2] = 1 mM) (rouge), solution saturée en air seule (rouge pointillés). A droite, fenêtre de potentiel plus petite. Vitesse de balayage à 0.1 V.s-1, électrode de travail en carbone vitreux (0.07 cm2), T=293K. ... 44
Figure I-20 : Spectres UV-Vis (bande de Soret (A) et bandes Q (B)) obtenus au cours des expériences de SEC d’une solution de DMF +0.2 M TBAPF6 contenant [Fe(F20TPP)C]l à 0.025 mM. Fe sous argon avant électrolyse (tracé noir pointillé),T = 243K. Electrolyse à Eapp = -0.25 v vs ECS (tracé noir) sous argon, et en présence de O2(tracé bleu). Electrolyse à Eapp = -0.6 v vs ECS (tracé rouge) sous O2 ... 46
Figure I-21 : CV de [FeIII(F20TPP)Cl] à 0.5 mM dans DMF + 0.2 M TBAPF6, vb =0.1 V.s-1 : sous argon, à T = 293 K (tracé noir en pointillés), après électrolyse à Eapp = -0.25 V vs SCE sous O2 à T = 243K (tracé bleu), après électrolyse à Eapp = -0.6 V vs SCE sous O2 à T = 243K (tracé rouge). ... 47
Figure I-22 : Représentation du montage SECM à quatre électrodes utilisé. ... 48
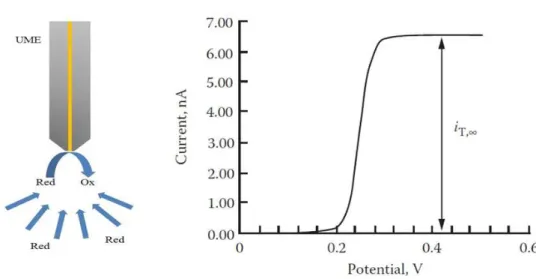
Figure I-23 : A gauche, représentation de la diffusion hémisphérique d’une espèce redox à l’UME. A droite : réponse en voltamétrie cyclique typique à l’UME. ... 49
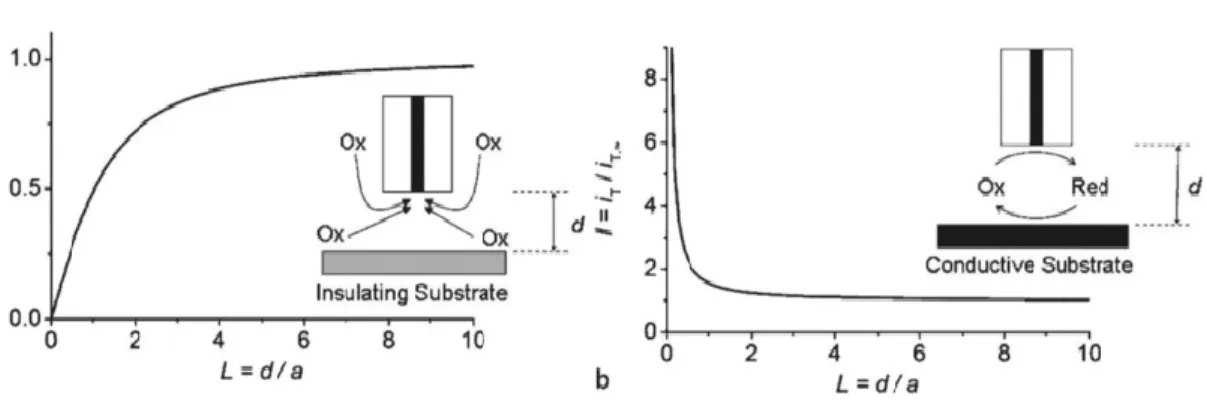
Figure I-24 : A gauche, feedback négatif : mesure du courant lorsque l’UME est approchée d’un substrat isolant. A droite, feedback positif : mesure du courant lorsque l’UME est approchée d’un substrat conducteur.81 ... 50
Figure I-25 : Représentation du mode G/C en présence d’une espèce redox... 50
Figure I-26 : Représentation du mode compétition en présence d’une espèce rédox. ... 51
Figure I-27 : Schéma de la cellule de spectroélectrochimie ... 53 Figure II-1 : CV d’une solution de DMF contenant 0.1 M de TBAPF6 et saturée en air ([O2]=1
10
mV.s-1. A droite, CV obtenu à une ultramicroélectrode d’or de 25 µm de diamètre, vb = 50 mV.s-1. Les CV sont obtenus dans la cellule SECM. ... 58 Figure II-2: A gauche, principe du SECM en mode substrat générateur- pointe collectrice pour sonder la ORR à une électrode génératrice de carbone vitreux de 3 mm et à une UME d’or de 25µm de diamètre polarisée à Eume = 0.5 V vs Ag.AgAgCl et placée à 10µm du substrat. A
droite, CV obtenus par SECM en mode SG-TC dans une solution de DMF contenant 0.1 M de TBAPF6 saturée en air ([O2]= 1 mM). En bleu, CV obtenu au substrat. En rouge, réponse à la
pointe. vb = 50 mV.s-1. ... 60
Figure II-3 : CV d’une solution de DMF contenant 0.1 M de TBAPF6. En haut, obtenu à une
électrode de carbone vitreux de 3 mm de diamètre, en noir 1 mM de O2, en rouge 1 mM O2 et
2 mM HClO4, en vert 1 mM O2 et 10 mM HClO4,vb = 100 mV.s-1. En bas, obtenu à l’UME
d’or de 25 µm de diamètre, en noir 1 mM O2 , en bleu 2 mM H+, en rouge 1 mM O2 et 2 mM
HClO4, vb = 50 mV.s-1 ... 61
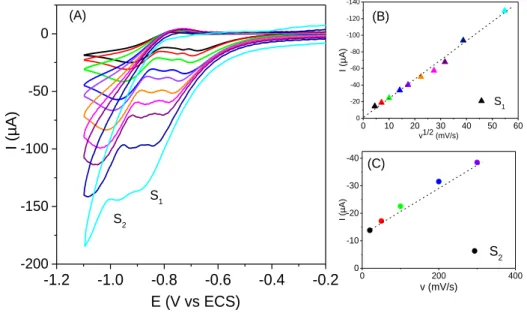
Figure II-4 : A) CV à l’électrode de carbone vitreux à différentes vitesses de balayage (20 mV à 3000 mV) d’une solution de DMF + 0.1 M TBAPF6 saturée en O2 en présence de 2 mM
HClO4. (B) Evolution du courant du pic S1 en fonction de la racine carré de la vitesse de
balayage. (C) Evolution du courant de pic S2 en fonction de la vitesse de balayage. ... 63
Figure II-5 : CV obtenus par SECM en mode SG-TC dans une solution de DMF contenant 0.1 M de TBAPF6 En bleu, CV obtenu au substrat. En rouge, réponse à la pointe pour la solution
saturée en O2 (1 mM).En pointillés en présence de 2 mM d’HClO4. Eume = 0.2 V vs Ag/AgCl,
distance pointe substrat 10µm, vb = 20 mV.s-1 ... 64
Figure II-6 : Principe du mode SECM compétition. CV obtenus par SECM en mode compétition dans une solution de DMF contenant 0.1 M de TBAPF6, 1 mM O2 et 2 mM
HClO4. Distance pointe substrat 10µm, vb = 10 mV.s-1 En haut : CV au substrat (bleu), réponse
à la pointe (rouge) Eume = -1V vs Ag/AgCl. En bas : dérivée du courant de pointe. ... 67
Figure II-7 : Schéma représentant les réactions au cours de la charge et décharge d’une batterie Li-air dans une solution électrolytique aprotique.105 ... 69 Figure II-8 : CV à l’électrode de carbone vitreux de 3 mm de diamètre d’une solution de DMF + 0.1 M TBAPF6 saturée en O2. En rouge, en présence de 4 mM LiPF6. En bleu, en présence de
11
Figure II-9 : Principe du SECM en mode SG/TC. SEI générée au substrat puis oxydée à celui-ci. UME placée à 10 µm du substrat et polarisée à E =2.5 V vs Li/Li+. ... 72 Figure II-10 : CV obtenus par SECM en mode SG-TC dans une solution de DMF contenant 0.1 M de TBAPF6 et 1 mM O2, UME placée à 10 µm du substrat et polarisée à E =2.5 V vs
Li/Li+. A gauche, en présence de 4 mM LiPF6, vb = 2 V.s-1. A droite, en présence de 2 mM
NaBF4 ,vb = 2 V.s-1 72
Figure II-11: CV obtenus par SECM en mode SG-TC dans une solution de DMF contenant 0.1 M de TBAPF6 et 1 mM O2, en présence de 4 mM LiPF6, UME placée à 10 µm du substrat et
polarisée à E =2.5 V vs Li/Li+ et vb = 2 V.s-1.En bleu, CV au substrat lors du balayage en
oxydation normalisé par le courant du pic S2 et multiplié par 2. En rouge, dérivée du courant à
l’UME normalisé par le courant du pic P2 et ramené à la même échelle de courant... 73
Figure II-12 : CV obtenus par SECM en mode SG-TC dans une solution de DMF contenant 0.1 M de TBAPF6 et 1 mM O2, en présence de 2 mM NaBF4 , UME placée à 10 µm du substrat
et polarisée à E =2.5 V vs Li/Li+ et vb = 2 V.s-1.En bleu, CV au substrat lors du balayage en
oxydation normalisé par le courant du pic S2 et multiplié par 2. En rouge, dérivée du courant à
l’UME normalisé par le courant du pic P2 et ramené à la même échelle de courant... 75
Figure III-1 : A gauche, schéma du SECM en mode génération collection, balayage en potentiel au substrat et potentiel fixe à l’ume. A droite, CV obtenus par SECM en mode SG-TC dans une solution de DMF contenant 0.1 M de TBAPF6 et 0.25 mM de [Fe(F20TPP)Cl]
sous argon. En noir, CV obtenu à l’électrode génératrice de carbone vitreux de 3 mm de diamètre. En rouge, réponse à la pointe polarisée à Eume = 0.5 V vs Ag/AgClet placée à 10µm
du substrat. vb = 50 mV.s-1. ... 79
Figure III-2 : SECM en mode SG-TC. Balayage au substrat et potentiel fixe à l’UME placée à 10µm du substrat. ... 80 Figure III-3 : CVs obtenus par SECM en mode SG-TC dans une solution de DMF contenant 0.1 M de TBAPF6 et 0.25 mM de [Fe(F20TPP)Cl]. En noir, sous argon. En rouge, sous O2. A
gauche Eume = 0.2 V vs Ag/AgClet placée à 10µm du substrat. vb = 50 mV.s-1.A droite Eume =
0.5 V vs Ag/AgClet placée à 10µm du substrat. vb = 50 mV.s-1. ... 80
Figure III-4 : SECM en mode SG-TC lorsque le potentiel est fixe au substrat (E = -0.6 V vs Ag/AgCl) et balayage en potentiel en oxydation effectuée à l’UME placée à distance fixe (10µm). ... 82
12
Figure III-5 : CV obtenus par SECM en mode SG-TC dans une solution de DMF contenant 0.1 M de TBAPF6 et 0.25 mM de [FeIII(F20TPP)Cl]. Le substrat est placé à potentiel fixe Esubstrat =
- 0.6 V vs A.AgCl et un balayge en oxidation est réalisé à l’UME placée à 10µm du substrat. vb = 20 mV/s. ... 82
Figure III-6 : A gauche :courbes simulées obtenues en configuration SG / TC obtenues à une UME de 25 µm de diamètre placée à 8 µm du substrat avec 0,25 mM [Fe (F20TPP) Cl] en
utilisant les paramètres définis dans la table 3-1. Le potentiel est balayé à l'UME de -0.3 V à 0,8 V à une vitesse de balayage de 20 mV.s-1 alors que le potentiel est fixé au substrat Esub = -0.6 V vs Ag / AgCl. Les valeurs de la constante de dismutation kd Fe(III)OO- / Fe(III) ont été prises comme suit: 0 s-1 (noir), 0,1 s-1 (rouge), 0,5 s-1 (vert), 1 s-1 (bleu ), 2 s-1 (rose), 5 s-1 (jaune), 10 s-1 (orange), 100 s-1 (marron). A droite : courbe expérimentale (rouge) et courbe simulée (noir) obtenue utilisant les paramètres définis dans la table III-1 avec kd Fe(III)OO-/Fe(III) =
1.5 s-1 ... 86 Figure III-7: CV obtenu au substrat, électrode de travail en carbone vitreux (0.07 cm2), d’une solution de DMF + 0.1 M TBAPF6 et de [FeIII(F20TPP)Cl] à 0.25 mM saturée en air (O2 à 1
mM ) Vitesse de balayage à 1 V.s-1. ... 87 Figure III-8 : CA de 60 s puis CV vers les potentiels anodiques à l’électrode de carbone vitreux (0.07 cm²) d’une solution de DMF + 0.1 M TBAPF6 et de [FeIII(F20TPP)Cl] à 0.25 mM saturée
en air (O2 à 1 mM) obtenus après chronoampérométrie pendant 60s. Vitesse de balayage à 0.1
V.s-1. En vert, chronoampérométrie à E = -0.2 V vs ECS. En rouge, chronoampérométrie à E = -0.7 V vs ECS. En noir, CV de 0.25 mM de [FeIII(F20TPP)Cl] en présence d’O2. ... 88
Figure III-9 : Courant à l’UME en mode SG-TC d’une solution de DMF + 0.1 M TBAPF6
contenant 0.25 mM de [FeIII(F20TPP)Cl] et saturée en air (O2 à 1 mM ). En rouge, courbe
expérimentale, en noire courbe simulée lorsque le potentiel à l’UME est fixé à Eume = 0.5 V vs
Ag/AgCl et qu’elle est placée à 8 µm du substrat alors que le substrat est balayé en potentiel de 0.2 à -0.7 V vs Ag/AgCl à vb = 20 mV.s-1. La courbe simulée est obtenue en utilisant les
paramètres de la table III-1 et kd Fe(III)OO- / Fe(III) = 1.5 s-1. ... 89
Figure III-10 : CV simulé au substrat, électrode de travail en carbone vitreux (0.07 cm2), d’une solution de DMF + 0.1 M TBAPF6 et de [FeIII(F20TPP)Cl] à 0.25 mM saturée en air (O2 à 1
mM ) La courbe simulée est obtenue en utilisant les paramètres de la table III-1 et kd Fe(III)OO- / Fe(III) = 1.5 s-1. Vitesse de balayage à 20 mV.s-1. ... 90
13
Figure III-11 : Courant à l’UME en mode SG-TC d’une solution de DMF + 0.1 M TBAPF6
contenant 0.25 mM de [FeIII(F20TPP)Cl] et saturée en air (O2 à 1 mM ). En rouge, courbe
expérimentale, en noire courbe simulée lorsque le potentiel à l’UME est fixé à Eume = 0.2 V vs
Ag/AgCl et qu’elle est placée à 8 µm du substrat alors que le substrat est balayé en potentiel de 0.2 à -0.7 V vs Ag/AgCl à vb = 20 mV.s-1. La courbe simulée est obtenue en utilisant les
paramètres de la table III-1 et kd Fe(III)OO- / Fe(III) = 1.5 s-1. ... 91
Figure IV-1 : Voltamétrie cyclique d’une solution de DMF contenant 0.1 M TBAPF6 et 1 mM
de [Fe(F20TPP)Cl]. A gauche : sous argon (noir), saturé en air, [O2] = 1 mM (rouge) et en
présence de 10 équivalents de HClO4 (en vert). A droite : balayage de la vague FeII/FeIII en
ajoutant successivement une quantité croissante de HClO4. vb = 0.1 V.s-. ... 94
Figure IV-2 : schéma carré représentant l’équilibre entre les espèces complexées par le DMF et les ions Cl-. KCl-constante de dissociation. ... 95
Figure IV-3 : Mécanisme proposé pour l’activation réductriced’O2 par [Fe(F20TPP)Cl] à E = -
0.2 V vs ECS selon un chemin PET (vert) et à E = -0.6 V vs ECS selon un chemin EPT (bleu) ... 96 Figure IV-4 : Mécanisme du transfert de protons et d’électrons à E = -0.2 V vs ECS. ... 97 Figure IV-5: Spectres UV visible d’une solution de DMF + 0.2 M TBAPF6 et [FeIII(F20TPP)Cl]
à 0.05 mM enregistrés lors d’une expérience de SEC à Tamb. En noir, sous argon pas de
potentiel appliqué. En orange, sous argon Eapp = -0.2 V vs ECS. En rouge, solution saturée en
air ([O2]= 1 mM) Eapp = -0.2 V vs ECS. ... 98
Figure IV-6 : Spectre UV Visible d’une solution de DMF + 0.2 M TBAPF6 et [FeIII(F20TPP)Cl]
à 0.05 mM enregistré lors d’une expérience de SEC à T = 258K. En noir, sous argon pas de potentiel appliqué. En rouge, sous O2 avec Eapp = -0.25 V vs ECS. En bleu, solution saturée en
O2 (1 mM) et 0.05 mM d’HClO4 (70%) avec Eapp = -0.2 V vs ECS. ... 99
Figure IV-7 : Spectre UV visible d’une solution de DMF + 0.2 M TBAPF6 et [FeIII(F20TPP)Cl]
à 0.05 mM enregistré lors d’une expérience de SEC à T = 258K. En noir pointillé, sous argon pas de potentiel appliqué. En bleu, solution saturée en O2 (1 mM) et 0.05 mM d’HClO4 (70%)
Eapp = -0.25 V vs ECS. En magenta, solution saturée en O2 (1 mM) et 0.5 mM d’HClO4 (10
14
Figure IV-8 : Evolution du Spectre UV visible de la solution précédente de DMF + 0.2 M TBAPF6, [FeIII(F20TPP)Cl] à 0.05 mM et 0.5 mM d’HClO4 (10 équivalents) enregistré lors
d’une expérience de SEC à T = 258K. avec Eapp = -0.25 V vs ECS. ... 101
Figure IV-9 Evolution du Spectre UV visible de la solution précédente de DMF + 0.2 M TBAPF6, [FeIII(F20TPP)Cl] à 0.05 mM et 0.5 mM d’HClO4 (10 équivalents) enregistré lors
d’une expérience de SEC à T = 258K. avec Eapp = -0.25 V vs ECS. Ajout de 0.5 mM d’HClO4
(20 équivalents au total). ... 102 Figure IV-10 : mécanisme proposé pour l’activation réductriced’O2 par [Fe(F20TPP)Cl] à E = -
0.2 V vs ECS. ... 103 Figure IV-11 : Génération de l’intermédiaire FeIvO par voie chimique par réaction de [FeIII(F20TPP)CL] et de 8 équivalents de m-CPBA sous argon à T = 263 K. ... 103
Figure IV-12 : Spectre UV visible d’une solution de DMF + 0.1 M TBAPF6 et [FeIII(F20TPP)Cl]
à 0.1 mM enregistré lors d’une expérience de SEC à T = 263K. En noir, sous argon. En magenta, 8 équivalents de m-CPBA ajoutés à la solution, sous argon. ... 104 Figure IV-13 : CV d’une solution de DMF + 0.1 M TBAPF6 et [FeIII(F20TPP)Cl] à 0.1 mM à T
= 263K. En noir, Fe sous argon. En magenta, 8 équivalents de m-CPBA ajoutés à la solution, sous argon. vb = 0.1 V/s ... 105
Figure IV-14 SECM en mode génération collection d’une solution de DMF + 0.1 M TBAPF6
et 0.25 mM [Fe(F20TPP)Cl], 0.5 mM d’O2 et 0.25 mM H+. Balayage en potentiel cathodique
réalisé au substrat et pointe fixée à 10 µm du substrat avec variation du potentiel de pointe de Eume = 0.4 V vs Ag/AgCl (noir) , Eume = 0.6 V vs Ag/AgCl (rouge) , Eume = 0.8 V vs Ag/AgCl
(vert) et Eume = 1 V vs Ag/AgCl (bleu). En gris pointillés, vague du FeII sous argon. En haut,
courant à la pointe. En bas, courant au substrat. ... 107 Figure IV-15 : CV obtenus en mode SG-TC dans une solution de DMF contenant 0.1 M de TBAPF6 et 0.5 mM de [FeIII(F20TPP)Cl]. En noir, solution saturée en air ([O2] = 1mM), en
rouge ajout de traces d’eau (1µL) à la solution Le substrat est placé à potentiel fixe Esubstrat = -
0.6 V vs A/AgCl et un balayage en oxydation est réalisé à l’UME placée à 10µm du substrat. vb = 50 mV.s-1... 109
Figure V-1: Montage de spectroélectrochimie en intégrant une UME au plus proche de la grille de Pt. ... 112
15
Figure V-2 : Schéma carré représentant l’équilibre entre les espèces complexées par le DMF et les ions Cl-. KCl-constante de dissociation. ... 113
Figure V-3 : Voltamétrie cyclique d’une solution de DMF contenant 0.1 M TBAPF6 et 1 mM
de [Fe(F20TPP)Cl]. En gris, solution saturée en air ([O2] = 1mM). En noir, solution saturée en
air et en présence de 10 mM TBACl. En bleu pointillés, en présence d’O2 et 5 mM HClO4, en
bleu trait plein, ajout de 10 mM TBACl. En rose pointillés, en présence d’O2 et 20 mM HClO4,
en rose trait plein, ajout de 10 mM TBACl. ... 113 Figure V-4 : Réaction de la chloration du thymol par une chloroperoxidase (CPO).134 ... 115 Figure V-5 : Différentes voies réactionnelles de l’intermédiaire P•+FeIVO. ... 115 Figure V-6 : Mécanisme proposé pour la formation de FeIVO par activation réductrice d’O2 via
un précurseur Sc3+-peroxo-Fe.136 ... 116
Figure PE- 1 : Montage des débitmètres pour diluer O2 par Ar. ... 124
Figure PE- 2: Cellule électrochimique expérimentale. CE, contre-électrode, E.T électrode de travail et Réf électrode de référence. ... 125 Figure PE- 3 : Montage SECM utilisé pendant les expériences... 126 Figure PE- 4 : Courbe d’approche à 1 µm.s-1 dans une solution de DMF + 0.1 M TBAPF6
contenant 0.25 mM de [FeIII(F20TPP)Cl] sous argon. L’UME est placée à potentiel de Eume = -
0.4 V vs Ag.AgCl réduisant le FeIII en FeII et le potentiel au substrat est de E = + 0.5V vs Ag.AgCl oxydant le FeII en FeIII. ... 127 Figure PE- 5 : Cellule de spéctroélectrochimie UV-Visible ... 128
Figure A- 1 : Evolution du Spectre UV visible de la solution de DMF + 0.2 M TBAPF6,
[FeIII(F20TPP)Cl] à 0.05 mM et excès de LiOH enregistré lors d’une expérience de SEC à T =
258K. Eapp = -0.2 V vs ECS. ... 130
Figure A-2 : Evolution du Spectre UV visible de la solution précédente de DMF + 0.2 M TBAPF6, [FeIII(F20TPP)Cl] à 0.05 mM et excès de LiOH enregistré lors d’une expérience de
SEC à T = 258K. Eapp = +0.5 V vs ECS. ... 131
Figure A-3 : CV de Fe(F20TPP) à 0.1 mM dans DMF + 0.2 M TBAPF6 à T = 263 K sous
16
Figure A-4 : CV Fe(F20TPP) à 0.5 mM dans DMF + 0.1 M TBAPF6 à l’électrode de carbone
vitreux ( d= 3 mm) à T = 293 K sous argon (en noir) et après ajout d’une quantité croissante, 0, 1, 2, 3, 4 et 5 mM de AgClO4 sous argon (en rouge) .vb =0.1 V.s1... 133
Figure A- 5 : CV Fe(F20TPP) à 0.5 mM dans DMF + 0.1 M TBAPF6 à l’électrode de carbone
vitreux ( d= 3 mm) à T = 293 K sous argon (en noir),après ajout de 5 mM de TBACl (en bleu) et après ajout de 5 mM de AgClO4 sous argon (en rouge) .vb =0.1 V.s1 ... 134
Liste des tables
Table I-1 : Produits de base de chimie organique synthétisés à partir de procédés d'oxydation.10 ... 24 Table II-1 : Produits de réduction de O2 en présence de Li+ et Na+ lors de la décharge dans les
batteries Li-air et Na-air.106 ... 70 Table II-2 : Rapport du nombre d’électron sur le nombre de O2 obtenu à chaque pic... 74
Table II-3 : Rapport du nombre d’électron sur le nombre de O2 obtenu à chaque pic... 75
Table III-1 : Paramètres utilisés pour les simulations SECM à l’aide du programme COMSOL. ... 86
17
Introduction générale
18
Les procédés de catalyse d’oxydation sont indispensables dans l’industrie chimique pour la production d’une grande quantité de produits d’intérêt (chimie fine, industrie pharmaceutique…). En effet, 30% de la production mondiale de l’industrie chimique utilise un procédé d’oxydation, ce qui en fait l’une des réactions chimiques les plus importantes.
Énergivores et polluants, les procédés d’oxydation actuels utilisent des conditions drastiques de température (T) et de pression (P), des catalyseurs onéreux et des oxydants chimiques nocifs. A titre d’exemple, la synthèse du cyclohexanol, précurseur du nylon, est issue de l’oxydation du cyclohexane en présence d’un catalyseur de cobalt en phase aqueuse à 140-180°C et à 8-20 bar.
Compte tenu des enjeux économiques et environnementaux actuels, la recherche d’alternatives plus respectueuses de l’environnement et économiquement viable est devenu un enjeu majeur. Dans la nature, la catalyse d’oxydation de molécules organiques est effectuée en condition douce de façon très sélective et efficace par des métalloenzymes en utilisant le dioxygène comme oxydant. A l’état fondamental, O2 est inerte de part son état de spin triplet. Il est donc
nécessaire de « l’activer » afin de franchir la barrière cinétique l’empêchant de réagir avec des espèces diamagnétiques. Dans la nature, cette activation est en particulier réalisée par des métalloenzymes à Fer, capable de révéler le caractère oxydant de O2 en présence d’une source
d’électrons.
Par une approche bio-inspirée, en utilisant un modèle moléculaire proche du site actif des métalloenzymes à fer-hémique, nous nous intéressons à comprendre et à reproduire cette remarquable activité catalytique. L’électrochimie, pour fournir les électrons nécessaire à l’activation réductrice du dioxygène et pour détecter les différents intermédiaires réactionnels, est une technique de choix pour mener notre étude.
Nous avons choisi de présenter nos résultats comme suit :
Le chapitre I décrit les systèmes enzymatiques mononucléaires capables d’oxyder un substrat organique. En particulier, la structure et le cycle catalytique des cytochromes
19
P450 sont décrits. Par la suite, nous présentons des systèmes bio-inspirés synthétiques et décrivons plus particulièrement les voies d’obtention des différents intermédiaires rencontrés au cours du cycle catalytique.
Nous détaillerons également notre approche électrochimique avec la présentation des différentes techniques complémentaires utilisées ainsi que l’étude antérieure réalisée au LEM sur l’activation réductrice de O2 à l’aide d’une porphyrine de fer, point de départ
des travaux présentés dans ce manuscrit.
Le chapitre II décrit la réduction de O2 en milieu organique aprotique (DMF) par
voltamétrie cyclique (CV) et par microscopie électrochimique à balayage (SECM). Cette étude, en absence de porphyrine de fer, constitue une première étape importante. La réduction de O2 est étudiée en présence de protons puis en présence d’acides de
Lewis Li+ et Na+.
Le chapitre III détaille l’étude par SECM de l’activation réductrice de O2 par le
complexe [Fe(F20TPP)Cl] en milieu DMF et la mise en évidence de la formation de
l’espèce Fe-peroxo.
Par la suite, le chapitre IV est consacré à l’évolution du système décrit dans le chapitre III en présence de protons. L’analyse des résultats obtenus par SECM et spectroéléctrochimie (SEC) permet de proposer un mécanisme d’activation de O2 par la
porphyrine [Fe(F20TPP)Cl] en milieu organique.
Enfin, le chapitre V complète l’étude en présentant les résultats préliminaires obtenus en réactivité sur des substrats organiques. Ces résultats prometteurs permettent de présenter les perspectives de ce travail de thèse.
21
Chapitre I : Utilisation du dioxygène
comme
I.
oxydant, contexte, état de l’art et stratégie
novatrice.
22
L’oxydation dans l’industrie chimique
1.
La réaction d’oxydation dans l’industrie
1.1.
La réaction d’oxydation est une réaction majeure en synthèse organique et permet l’insertion d’un ou plusieurs atomes d’oxygène. Elle joue un rôle majeur dans la production d’une très grande quantité d’intermédiaires et de monomères à forte valeur ajoutée, par exemple, pour la fabrication de polymères. En effet, près d’un quart des produits chimiques sont synthétisés à partir de procédés d’oxydation catalytique (hétérogène ou homogène) en phase liquide ou gaz.1
Pour réaliser ces réactions, l’industrie chimique utilise, dans la plupart des cas, des catalyseurs souvent coûteux à base de métaux nobles tels que le platine et le palladium, des oxydants nocifs ou toxiques (CrO42-, MnO4–, S2O82-…) utilisés en quantité stœchiométrique et des
conditions drastiques de température et de pression.2
Typiquement, l’oxydation de molécules inertes telles que les alcanes nécessite une activation à très haute température (>200°C) pour franchir la barrière énergétique nécessaire pour casser les liaisons C-H fortes. Les rendements de ces réactions demeurent cependant faibles et le manque de sélectivité est une limitation majeure à leur développement.3
Prenons l’exemple de la synthèse de l’oxyde d’éthylène, deuxième produit chimique issu d’un procédé d’oxydation en termes de quantité (30x106
t/a en 2016), impliqué dans de nombreuses applications, comme la synthèse d’éthylène-glycol, de détergents, de polymères… Sa production industrielle se fait à partir d’un mélange de dioxygène et d'éthylène qui réagissent entre 200 °C et 300 °C et à des pressions comprises entre 10 et 20 bar sur un catalyseur d'argent dispersé sur alumine, selon la réaction chimique 1.1 et avec un rendement de 70%.4
Cette réaction ne produit pas uniquement de l’oxyde d’éthylène mais également une quantité indésirable de dioxyde de carbone (réaction 1.2).5
De nombreuses études sont menées afin d’améliorer le procédé de cette synthèse, notamment en réduisant la consommation énergétique et en améliorant la sélectivité, la durée de vie et l’activité du catalyseur. Des voies de synthèse électrochimiques6
ou enzymatiques7 sont proposées mais ces études sont encore loin d’une possible application industrielle.
23
Les procédés d’oxydation industriels ont donc des inconvénients majeurs tels que l’utilisation d’oxydants dangereux parfois même explosifs engendrant ainsi des risques pour l’homme, la production de déchets polluants, un fort coût énergétique et un manque de sélectivité.
Dans ce contexte environnemental et financier, l’industrie chimique a alors tout intérêt à trouver des alternatives plus respectueuses pour l’environnement et moins couteuses pour réaliser les réactions d’oxydation.
Intérêt de la chimie verte
1.2.
Un pan de la chimie s’est développé à partir des années 1990 pour répondre à ces problématiques écologiques et économiques, il s’agit de la chimie dite «Verte » .8 Celle-ci répond à des enjeux capitaux, d’une part le développement de voies de synthèses propres, économes (économie d’atomes) et sécuritaire et d’autre part la mise en œuvre de matières premières renouvelables lors de l’élaboration de produits finis. Pour la synthèse d’intermédiaires organiques clés de l’industrie chimique, dont la demande est en constante augmentation (tels que les acides acryliques comme précurseurs de monomères), des efforts ont été réalisés afin d’améliorer leur procédés de synthèse9
tel que le développement de nouveaux catalyseurs moins coûteux avec un impact environnemental plus faible.
Ainsi, les procédés de catalyse d’oxydation, compte tenu de leurs rôles clés dans l’industrie chimique, doivent évoluer et s’adapter pour répondre aux besoins de notre société moderne tout en préservant les ressources de la planète.
En ce sens, la communauté scientifique tente d’une part d’implémenter de plus en plus l’utilisation du dioxygène comme oxydant « vert » et d’autre part de développer une approche bio-inspirée voir bio-mimétique, basée sur la réactivité des systèmes naturels. Cette deuxième voie nécessite une compréhension fine de la réactivité des sites actifs des métalloenzymes. Ce point sera décrit dans la partie 2 de ce chapitre.
L’utilisation de O2 dans l’industrie
1.3.
Afin d’illustrer mon propos sur l’utilisation de O2 dans l’industrie, quelques exemples de
24
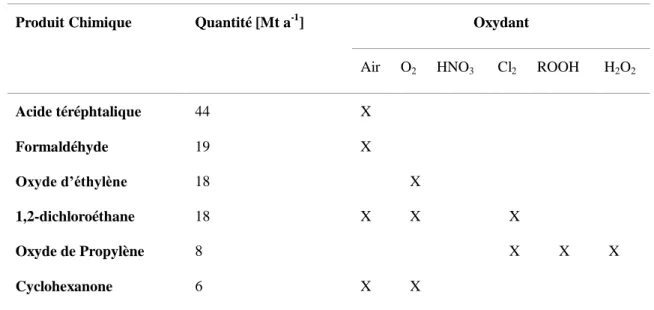
Produit Chimique Quantité [Mt a-1] Oxydant
Air O2 HNO3 Cl2 ROOH H2O2
Acide téréphtalique Formaldéhyde Oxyde d’éthylène 1,2-dichloroéthane Oxyde de Propylène Cyclohexanone 44 19 18 18 8 6 X X X X X X X X X X X
Table I-1 : Produits de base de chimie organique synthétisés à partir de procédés d'oxydation.10
Actuellement, O2 est utilisé comme oxydant dans la grande majorité des procédés d’oxydation
qui se veulent ainsi plus « durables » en faveur d’autres oxydants chimiques tels que Cl2 ou
H2O2.
L’avantage de l’oxydation utilisant O2 est notamment la possibilité de réaliser des oxydations
contrôlées à 2 ou 4 électrons avec la formation de H2O comme sous-produit. Cependant son
utilisation nécessite la présence de catalyseurs et la mise en place de procédés beaucoup plus sophistiqués pour atteindre des rendements satisfaisants comme l’emploi d’oxydants plus forts tels que HNO3.
En effet, comme précisé dans l’introduction générale, le dioxygène paramagnétique est à l’état de spin triplet, ce qui l’empêche de réagir, selon les règles d’interdiction de spins, avec des molécules organiques diamagnétiques à l’état de spin singulet. Il faut donc un catalyseur pour surmonter cette barrière cinétique. En catalyse hétérogène, la surface de différents métaux nobles (Pd, Ni, Pt…) est capable d’abaisser les barrières cinétiques en adsorbant O2 à leur
surface créant ainsi des intermédiaires oxydants.11 En catalyse homogène, des complexes de métaux de transition (Fe, Mn, Cr), par leurs états de spins, sont capables d’activer le dioxygène. De plus, en catalyse homogène aérobie, les risques liés à la combustion imposent des contraintes drastiques sur les conditions de synthèse telles que les concentrations en réactifs, les températures et les solvants utilisés.12
L’utilisation de O2 comme oxydant est donc une voie à grand potentiel dans l’industrie
25
efficacement l’oxydation de molécules organiques en condition douce est le grand défi à relever.
Pour repenser ces procédés d’oxydation, créer des systèmes artificiels biomimétiques économiquement et écologiquement viables semble avoir un bel avenir.
Les systèmes naturels d’activation de O
22.
Parallèlement aux travaux sur la mise au point de ces procédés d’inspiration bio-mimétiques, les chercheurs s’intéressent également à l’étude de systèmes naturels et plus particulièrement à la compréhension des mécanismes d’action et à la relation structure-réactivité à l’origine de leur unique efficacité catalytique.
Métalloenzymes et activation de O2
2.1.
Le dioxygène joue un rôle majeur dans de nombreux processus biologiques tels la métabolisation de substances xénobiotiques et la production d’énergie dans la cellule. Ces réactions sont catalysées par des métalloenzymes dont la réactivité spécifique est liée à la nature du site actif.
Présentes tant chez les bactéries que chez l’homme et les plantes, certaines de ces métalloenzymes sont capables de réaliser l’insertion d’un ou de plusieurs atomes d’oxygène dans les substrats organiques. Il s’agit des oxygénases, découvertes en 1955 par Hayaishi au Japon et Mason aux Etats-Unis et longuement étudiées depuis pour comprendre leurs propriétés chimiques et physiques.13,14 Essentiellement, ces oxygénases utilisent O2 afin de
réaliser des réactions d’oxygénation, avec un apport d’électrons et de protons via des cofacteurs (NAD, FAD, FMN…).
Nous avons fait le choix de restreindre cette présentation au cas des enzymes à fer même si des enzymes à Cu et à Mn existent dans les systèmes naturels. Plus précisément, nous nous intéresserons dans ce chapitre aux systèmes à site actif contenant un hème tel que le cytochrome P450 et la cytochrome c oxydase.
Le cytochrome P450 est capable de réaliser une réaction d’oxygénation en insérant un atome d’oxygène dans un substrat organique selon la réaction (1.3) et donc de réaliser la réduction contrôlée de O2 à deux électrons.
26
La cytochrome c oxydase quant à elle est capable de réaliser la réduction de O2 à quatre
électrons en H2O selon la réaction (1.4)
Cytochrome c oxydase: réduction de O2 à 4 électrons
2.2.
La cytochrome c oxydase (CcO) est une enzyme membranaire au cœur de la chaine respiratoire (figure 1-1), présente chez les organismes eucaryotes comme chez les procaryotes et qui catalyse la réduction de O2 en H2O selon la réaction (1.4). Cette réaction nécessite
l’apport de quatre électrons et quatre protons. Les électrons sont fournis par le cytochrome c, petite protéine et les protons sont pompés au travers la membrane.
Figure I-1 : Représentation schématique de la chaîne respiratoire mitochondriale.
Dans cette partie, nous nous intéresserons uniquement à la CcO de la chaine respiratoire.
Structure de la cytochrome c oxydase
La structure tridimensionnelle de la cytochrome c oxydase de la bactérie Paracoccus
denitrificans a été obtenue dans les années 90 par diffraction des rayons X, ce qui a permis des
avancées significatives dans la compréhension du fonctionnement de cette enzyme.15
La CcO est une protéine transmembranaire de plusieurs sous-unités (8 à 13 sous-unités) se présentant comme un dimère dans la membrane. Les sous unités I et II contiennent quatre
27
centres rédox, deux hèmes à fer (hème a et a3) et deux centres à Cu (CuA et CuB). La structure
du site actif de la CcO, représentée en figure I-2 est donc une structure bi-nucléaire avec un hème à fer ainsi d’un centre à cation cuivre proximal.16
Cette structure élaborée permet de contrôler très finement l’apport des électrons et des protons nécessaires à la réaction et d’empêcher la production d’espèces réduites de O2 (ROS).
Figure I-2 : A gauche, centres oxydoréducteurs de la CcO.17 L’acheminement des réactifs (électrons, protons et dioxygène) est indiqué par les flèches bleues. Les flèches rouges indiquent le pompage transmembranaire de protons couplé à cette réaction du « N-side » (chargé négativement) vers le « P side » (chargé positivement) de la membrane. A droite, structure du site actif binucléaire de CcO.18
Mécanisme catalytique
De nombreux groupes travaillent encore sur la compréhension du mécanisme catalytique de la CcO bien que les travaux de Wikström et Babcock par des techniques spectroscopiques ont permis des avancées majeures.19 Le cycle catalytique de réduction du dioxygène en eau est composé de réductions et de protonations successives.
28
Figure I-3 : Mécanisme de réduction de O2 par la cytochrome c oxydase. 20
Le complexe oxydé [Fe(III)-OH Cu(II)] subit dans un premier temps deux réductions à un électron consécutives depuis le cytochrome c pour prendre une forme [Fe(II) Cu(I)]. Au cours de ce processus, un proton est transféré et une molécule d’eau est libérée. Le dioxygène peut alors se lier au Fe(II) pour former le composé oxy, puis par réarrangement électronique, le complexe P oxyferryle [Fe(IV)=O2- HO—Cu(II)] est obtenu. Une troisième réduction à partir du cytochrome c associé à l’acquisition de deux protons donne le composé F [Fe(IV)=O
2-Cu(II)] avec libération d’une seconde molécule d’eau.Un dernier transfert d’électron associé à un transfert de proton donne le composé oxydé [Fe(III)-OH Cu(II)] et referme le cycle catalytique. La réaction de réduction de O2 en eau est présentée par le bilan suivant :
Pour réaliser cette réaction, 4 protons sont pompés au travers de la membrane de l’intérieur vers l’extérieur (figure I-2). Bien que ce mécanisme de pompage soit encore mal connu, il contribue au gradient de protons qui assure la synthèse de l’ATP.
Ce système enzymatique, dont la structure est très élaborée, possède une activité catalytique remarquable suscitant l’intérêt de nombreux groupes qui tentent de mimer cette réactivité par des modèles moléculaires. Cela représente un intérêt majeur pour des applications énergétiques comme dans les piles à combustibles et les batteries métal air où la réaction de réduction de O2
29
Cytochrome P450 : activation de O2 à 2 électrons
2.3.
Le cytochrome P450 est la monooxygénase la plus étudiée, capable d’insérer un atome d’oxygène dans un substrat organique.
Cette enzyme, fortement oxydante, peut réaliser diverses réactions telles que l’hydroxylation de liaisons C-H, l’époxydation de liaisons C=C, l’oxydation d’aromatiques. Une molécule de O2 est utilisée pour insérer un atome d’oxygène à un substrat organique et le second oxygène
est réduit pour produire une molécule d’eau (réaction 1.3). Les électrons nécessaires à la réactivité du cytochrome P450 sont apportés par un cofacteur NADPH.
En 1958, Garfinkel et Klingenberg ont reporté pour la première fois l’apparition d’un pigment jaune orangé dans les fractions microsomales hépatiques de rat et de cochon en présence de monoxyde de carbone.25,26 Par la suite, Omura et Sato élucidèrent en 1962 la structure biochimique de ces pigments, montrant qu’il s’agissait d’hémoprotéines appelées ensuite cytochrome P450 en raison d’un maximum d’absorption à 450 nm en présence de CO.27,28
Des études au début des années 1960 ont permis de démontrer l’implication de cette enzyme dans la transformation des stéroïdes par Estabrook, Cooper et al en 1963 et dans l’oxydation de composés exogènes par Cooper et al quelques années après.29,30 A la fin des années 1960, le premier cytochrome P450 a été purifié à partir de la bactérie Pseudomonas putida qui catalyse l’hydroxylation du camphre en 5-exo-hydroxycamphre.31
Structure du cytochrome P450
Des études spectroscopiques et structurales menées sur les cytochromes P450 ont permis de résoudre la structure de cette enzyme. Après l’obtention de la première structure 3D du cytochrome P450 du camphre par Poulos et al en 1986, la structure de cette enzyme et sa fonction ont été de mieux en mieux compris.32
Le site actif de l’enzyme comporte un ion FeIII
coordiné à quatre atomes d’azote d’une protoporphyrine IX et relié à une chaîne polypeptidique par un ligand axial thiolate cystéine. Ce ligand thiolate est responsable de l’absorption de Soret maximum à 450 nm caractéristique pour l’adduit FeII
-CO. Une sixième position de coordination du Fe, vacante ou occupée par une molécule d’eau, est susceptible de fixer une molécule de O2.
30
Figure I-4 : Structure du site actif du cytochrome P450 Cycle catalytique du cytochrome P450
De nombreuses études ont permis d’élucider le mécanisme du cycle catalytique de cette enzyme.33 Le cycle communément admis est présenté sur la figure I-5 et illustré dans le cas de réactivité avec un substrat RH.
Figure I-5 : Cycle catalytique du cytochrome P450.34
Le fer initialement dans un état d’oxydation +III est coordiné par une molécule d’eau en position axiale. Lorsque le substrat arrive à proximité du site actif, la molécule d’eau se décoordine facilitant ainsi la réduction de l’ion fer par une réductase à un état d’oxydation +II. Une molécule de O2 peut alors se coordiner au FeII pour former l’adduit FerIII-superoxo (FeIII
31
FeIII-hydroperoxo (FeIII-OOH), un intermédiaire à très courte durée de vie. Après une seconde protonation, la liaison O-O est coupée de façon hétérolytique libèrant une molécule d’eau et formant l’espèce FeIV
=oxo radical cation (P+•FeIV=O) avec le radical cation délocalisé sur la porphyrine. Cette espèce est également notée FeV=O en particulier dans le cas des monooxygénases non hémiques. Dans les systèmes porphyriniques, cet intermédiaire, appelé compound I, est l’espèce oxydante capable de réaliser l’insertion d’atome d’oxygène sur un substrat organique. L’espèce FeIII
-H2O est alors régénérée après libération du produit de la
réaction terminant le cycle catalytique.
Différents intermédiaires réactionnels impliqués dans le cycle catalytique du cytochrome P450 ont pu être observés par des techniques spectroscopiques. En effet, l’adduit FeIII
-OO•- a été étudié par spectroscopie Raman.35 Les espèces FeIII-OO- et FeIII-OOH ont été caractérisées par spectroscopie Raman et RPE lors de la réduction de l’adduit FeIII
-OO•- à basse température (T= 77 K). L’espèce P+•FeIV=O a été caractérisée par spectroscopie Mössbauer, RPE et UV-Visible en 2010 par Green et al.36
Dès l’étape d’addition du deuxième électron dans le cycle catalytique, la réaction d’hydroxylation a lieu. Le mécanisme d’oxydation ou « Oxygen Rebound Mechanism » proposé par Groves et al dans les années 1970, est centré sur l’ion Fe qui reçoit l’atome d’oxygène et le cède au substrat organique.37
Il a été montré que le substrat RH passe par un intermédiaire à l’état radicalaire R• dû à la cassure homolytique de la liaison C-H engendrée par l’espèce à haute valence P+•FeIV
=O, communément appelée FeV=O.38,39 Un intermédiaire [FeIVOH │ R• ] est alors formé avant d’obtenir la formation de la liaison C-O et retour à l’état Fe(III) du CytP450.
Figure I-6 : Mécanisme d'hydroxylation d'un alcane R-H. 38
Les cytochromes P450 peuvent également réaliser de façon efficace et stéréoséléctive, l’époxydation de différentes oléfines.40
Dans ce cas, il a été montré que l’intermédiaire FeIII -hydroperoxo est l’espèce oxydante impliquée dans une telle réactivité mais les mécanismes restent encore à élucider.41
32
Ainsi, cette variété de réactivité est due au fait qu’il existe une multitude de cyt P450 bien spécifique à une réaction. Ces cyt P450 présentent des différences structurales, notamment au sein de leur seconde sphère de coordination, à l’origine leur spécificité.
Réactions catalysées par le cyt P450
Comme indiqué précédemment, les cyt P450 sont capables de réaliser des réactions très variées. Il ne s’agit pas ici d’en faire une liste exhaustive, cependant le schéma ci-dessous rassemble quelques exemples de ces réactions répertoriées.
Figure I-7 : Réactions d’oxydation catalysées par les cytochromes P450 d’après Mansuy.42
Par ailleurs, des réactions de déshydratation, des isomérisations ou encore des réductions ont également été observées, montrant ainsi cette grande versatilité de réactivité dont les chercheurs peuvent s’inspirer pour développer de nouvelles voies de synthèse.43
Application des cytochromes P450 dans l’industrie
Il est difficile d’isoler et de produire des cyt P450 pour une utilisation commerciale vu la complexité de ces voies enzymatiques, de ces différents partenaires protéiques et des différents cofacteurs nécessaires à sa fonction. Leur faible stabilité et leur nombre de cycles catalytiques peu élevés sont également des inconvénients majeurs. Cependant, leur exceptionnelle réactivité
33
a conduit les chercheurs à fournir des efforts considérables pour isoler et manipuler ces enzymes pour en faire des catalyseurs à application commerciale notamment dans l’industrie pharmaceutique. Même si on trouve peu d’exemples dans la littérature, il est cependant possible de citer le cas de l’utilisation de cette enzyme dans la chaine de production de la pravastatine, médicament utilisé mondialement et produit à échelle industrielle pour traiter l’hyperlipidémie, soit la présence de taux élevés de lipides dans le sang. En effet, la pravastatine est produit via la biotranformation de la compactine, un précurseur dans l’organisme de S. carbophilus où le gène d’une enzyme P450 (CYP105A3) est hyper-exprimé.
4445
Figure I-8 : Biotransformation de la compactine en pravastine par CYP105A3 dans l’organisme de S.carbophilus.
L’une des approches utilisées en biotechnologie pour contrer les limitations de l’utilisation de cette enzyme est la mutagénèse pour créer de nouvelles enzymes P450 plus efficaces et robustes et de trouver des coenzymes redox pour assurer le fonctionnement de ces enzymes. Cependant cette approche est peu rationnelle, très coûteuse et chronophage. L’intérêt de développer des modèles moléculaires à ces enzymes est alors d’autant plus justifié.
Les systèmes bio-inspirés
3.
Dans cette partie, nous présenterons l’approche historique de l’activation réductrice de O2 par
des modèles moléculaires synthétiques du cytochrome P450. Nous ne donnerons pas de modèles de la cytochrome c oxydase dont la structure décrite en partie 2.2 montre l’extrême complexité. De nombreux travaux, utilisant des modèles moléculaires pour mimer cette enzyme, ont été développés ces dernières années mais ne seront pas décrits dans ce manuscrit, dans la mesure où les travaux de cette thèse sont centrés sur l’activité d’oxydation des cytP450 et non sur la catalyse de la réaction de réduction de O2.
34
Les métalloporphyrines, modèles des cyt P450
3.1.
Après la découverte des cyt P450 et de leur structure dans les années 50, les premiers modèles synthétiques sont apparus à la fin des années 70. L’objectif est alors de synthétiser des complexes modèles des sites actifs afin d’étudier la réactivité et produire des connaissances sur les mécanismes réactionnels de ces enzymes. Les complexes porphyriques de fer sont utilisés pour tenter de (i) reproduire les intermédiaires connus du cycle catalytique, (ii) reproduire l’activité d’oxydation de substrat organique des cyt P450.
Les premières porphyrines pentacoordinées comportant un ligand axial thiolate ont été obtenues en 1975 par Collman et Holm.46,47 Les caractéristiques spectrales (UV-Vis, RPE) obtenues avec ces complexes de type [Fe(P)(SR)] (SR groupement thiolate) sont très proches de celles enregistrées pour l’état Fe(III)SH pentacoordiné du cycle catalytique du cyt P450 (figure I-5) et constituent de bons modèles du site actif de l’enzyme.48
Figure I-9 : Porphyrines de Fer majoritairement utilisées comme modèle moléculaire des CytP450.(a) Structure de la protoporphyrin IX avec R1 CH=CH2 , R2 CH3 et R3 (CH2)2-COOH. (b) Structure de
5,10,15,20-tétraphénylporphyrine de Fer ,Fe(TPP) ; avec Ar groupement aryl.
Les premières études de réactivité des porphyrines de fer vis-à-vis de O2 sont réalisées dans le
contexte de l’étude de transporteurs de O2, modèle de l’enzyme myoglobine. Il est alors montré
que les porphyrines de Fe (II) peuvent réagir de façon réversible avec O2 (réaction 1.6)
où L est un ligand axial (H2O, Cl…)
Citons l’exemple de la porphyrine à « bras imidazole » publiée en 1973 par l’équipe de Taylor (figure I-10).49
35
Figure I-10 : Structure de la porphyrine pyrroheme-N-[3-(1-imidozolyl)propyl]-amide utilisée par Chang et Taylor. X= pas de substituant, H2O, O2 ou CO le Fer est alors au degré d’oxydation +II ou Cl
avec le Fer au degré d’oxydation +III.
L’étude par spectroscopie UV-Vis montre que la porphyrine de Fe(II) peut complexer de façon réversible O2 en présentant un comportement similaire à la myoglobine. Cependant, la réaction
de O2 avec les complexes porphyriniques Fe(II) peut conduire à la formation de dimères µ-oxo
selon la réaction 1.7.50
Les chercheurs ont alors développé des stratégies afin de minimiser cette réaction de dimérisation en choisissant par exemple de travailler à basse température afin de ralentir la réaction de dimérisation, très lente par rapport à celle des intermédiaires d’intérêt.51 Une autre approche fut de fonctionnaliser la porphyrine de façon à l’encombrer stériquement et prévenir ainsi les réactions de dimérisation même à température ambiante. On pourra citer l’exemple emblématique de la « picket-fence » porphyrine de Collman52 mais également les modèles développés par le groupe de Momenteau.53 Enfin, adsorber le catalyseur sur une surface de façon à ce que deux atomes de Fer ne puissent pas interagir constitue également une stratégie utilisée.54
Cette courte présentation de quelques exemples de porphyrines de fer utilisées comme modèles des cytochromes P 450 a pour ambition d’introduire la famille des complexes de cette étude. Le présent travail de thèse, comme nous le verrons plus loin en section 4.1, est réalisé sur un modèle unique et commercial. Les revues de D. Mansuy publiées en 2007 et de J. Groves en 2018 offre aux lecteurs le désirant une présentation détaillée de la contribution des métalloporphyrines à l’étude de la réactivité du cyt P450.
Dans le paragraphe suivant, nous présenterons les intermédiaires successifs proposés pour le cycle catalytique du cyt P450.
36
Espèces intermédiaires du cycle catalytique du cyt P450
3.2.
Au cours de ces vingt dernières années, différents intermédiaires réactionnels de type Fe-oxygène ont été identifiés dans le cycle catalytique du cyt P450, soit par des études spectroscopiques sur l’enzyme elle-même (section 2.3), soit à l’aide de composés modèles. Dans ce dernier cas, les espèces intermédiaires Fe-oxygène sont obtenues par voie chimique, c’est-à-dire à partir d’un précurseur FeIII
ou FeII et d’un réactif chimique source de donneur d’atome d’oxygène. Ce réactif peut être l’ion superoxyde O2•-, H2O2, des peracides (m-CPBA)
ou encore PhIO (iodosylbenzene). Dans certains cas, O2 lui-même est utilisé comme donneur
d’atome d’oxygène en présence alors d’un réducteur chimique tel que NaBH4. Nous donnons
ci-dessous une brève présentation des intermédiaires réactionnels modèles et leur réactivité associée.
L’adduit Fe(II)-O2
Dans les systèmes naturels, la première étape de l’activation réductrice de O2 par les systèmes
hémiques est la complexation de O2 après réduction du FeIII en FeII (figure I-5). Le FeIII ne
présentant aucune affinité vis-à-vis de O2, celui-ci va se lier au FeII et va s’accompagner d’un
transfert de charge formant ainsi un adduit Fe-superoxo FeIII-OO•-.
Collman a caractérisé par cristallographie une telle espèce FeIII-OO•- dans le cas de l’étude de l’affinité Fe/O2 du complexe hémique « picket fence porphyrin » modèle de la myoglobine.55
Ce modèle moléculaire présente un bras imidazole en position axiale qui prévient la formation de dimère µ- oxo mais qui permet également d’améliorer l’affinité de O2 au fer. Traylor et al
montrent que l’affinité de O2 pour des porphyrines présentant un ligand imidazole est plus
importante car l’imidazole fortement π conjugué stabilise la liaison Fe-O faible.56
L’espèce FeIII
-superoxo est souvent considérée comme une espèce de passage vers des intermédiaires de haute valence plus réactives. Dans la nature, la réactivité de cet intermédiaire existe pour des métalloenzymes non hémiques et des modèles synthétiques ont permis de confirmer un caractère oxydant de FeIII-superoxo.57,58 Cependant il n’existe à notre connaissance aucun exemple de réactivité d’un adduit FeIII-superoxo hémique dans la littérature.
Au stade FeIIIOO•- du cycle catalytique du cyt P450, O2 est réduit partiellement à un électron et
37
Fe(III)-Peroxo
C’est la réduction de l’adduit Fe-superoxo qui conduit à la formation de l’intermédiaire FeIII
-peroxo (figure I-5).
Valentine et al ont préparés pour la première fois cet intermédiaire dans un solvant aprotique à partir de FerIII(TPP)Cl par ajout de 2 équivalents de KO2 ou à partir de FeII(TPP) et d’un
équivalent de KO2 (figure I-11).59 Le premier équivalent de O2•- sert à réduire l’ion FeIII en FeII
puis le second équivalent de O2•- se coordine au FeII.
Figure I-11 : Formation de l’intermédiaire FeIII-peroxo par voie chimique.59 Le premier équivalent de O2•- sert à réduire l’ion Fe
III
en FeII puis le second équivalent de O2•- se coordine au Fe II
.
L’intermédiaire FeIII
-peroxo peut donc être obtenu par voie chimique par ajout d’équivalents réduits de dioxygène O2•- sur un modèle moléculaire hémique. L’intermédiaire FeIII-peroxo
peut alors prendre une configuration dite « end-on » (η1-peroxo) ou « side-on » ( η2-peroxo) après réarrangement électronique.60
Figure I-12 : Structure end-onou η1-peroxo et side on ou η2-peroxo du Fe-peroxo.
La structure η1-peroxo présente une seule liaison Fe-O alors que dans la structure η2-peroxo, les deux atomes d’oxygène sont liés au fer rendant cette structure plus stable. Ces deux structures peuvent être discriminées par des techniques de spectroscopie vibrationnelle IR ou Raman.61 Bien que le FeIII-peroxo est plus favorablement en configuration « side-on », l’intermédiaire peroxo « end-on » a pu être observé par Jin-Gang-Liu et al en 2010.62
Parmi les techniques de caractérisation des intermédiaires du cycle catalytique, la spectroscopie UV-Vis a été largement utilisée pour déterminer la formation de l’intermédiaire FeIII-peroxo.
Typiquement, le spectre UV-Vis de la porphyrine [FeIII(F20TPP)Cl] avec F20TPP le ligand
38
une bande de Soret à λ = 410 nm et des bandes Q à λ = 528 et 568 nm. Après génération par voie chimique du FeIII-peroxo, un spectre présentant une bande de Soret à λ = 431 nm et deux bandes Q à λ = 538 et 589 nm est obtenu, caractéristique de la formation de cet intermédiaire FeIII-peroxo.63
Figure I-13 : Spectre UV-Vis de FeIII-peroxo, formé à partir de [FeIII(F20TPP)Cl] et KO2 dans
CH3CN. 63
La réactivité catalytique de Fe-peroxo vis-à-vis de substrats organiques a également été étudiée. Dans la littérature, la réactivité du peroxo dans le cas des cytochromes P450 n’a jamais été mis en évidence. Valentine et al ont montré qu’un η2-FeIII-peroxo pouvait réaliser l’époxydation d’oléfines déficientes en électrons selon le mécanisme en figure I-14.64
Figure I-14 : Epoxydation d’une oléfine pauvre en électrons (2-methyl-1,4-naphtaquinone) par un FeIII-peroxo.
L’utilisation de substrats riches en électrons comme le styrène n’a pas donné de résultats concluants, démontrant ainsi le caractère plutôt nucléophile de l’intermédiaire FeIII
-peroxo. Cette nucléophilie résulte des caractéristiques électroniques du macrocycle porphyrinique, structures riches en électrons, et par la présence d’un ligand axial.

![Figure I-13 : Spectre UV-Vis de Fe III -peroxo, formé à partir de [Fe III (F 20 TPP)Cl] et KO 2 dans CH 3 CN](https://thumb-eu.123doks.com/thumbv2/123doknet/14495548.718229/38.893.253.646.237.538/figure-spectre-vis-iii-peroxo-formé-partir-iii.webp)