HAL Id: tel-02865503
https://tel.archives-ouvertes.fr/tel-02865503
Submitted on 11 Jun 2020
HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Fonctionnalisation C-H dirigée d’hétérocycles azotés
Fares Roudesly
To cite this version:
Fares Roudesly. Fonctionnalisation C-H dirigée d’hétérocycles azotés. Chimie organique. Sorbonne Université, 2018. Français. �NNT : 2018SORUS354�. �tel-02865503�
Thèse de doctorat de
Sorbonne Université
Spécialité : Chimie organique
Ecole doctorale de Chimie Moléculaire de Paris Centre - ED406
Institut Parisien de Chimie Moléculaire / Equipe ROCS
Fonctionnalisation C-H dirigée d’hétérocycles azotés
Présentée par
Fares ROUDESLY
Pour obtenir le grade de
Docteur de Sorbonne Université
Soutenance publique prévue le 19 octobre 2018 Devant un jury composé du :
Pr Christophe HOARAU INSA de Rouen Rapporteur
Pr Jacques LEBRETON Université de Nantes Rapporteur
Dr Laurence GRIMAUD Ecole Normale Supérieure Examinateur
Dr Joanna WENCEL-DELORD Université de Strasbourg Examinateur
Dr Julie OBLE Sorbonne Université Examinateur
Thèse de doctorat de
Sorbonne Université
Spécialité : Chimie organique
Ecole doctorale de Chimie Moléculaire de Paris Centre - ED406
Institut Parisien de Chimie Moléculaire / Equipe ROCS
Fonctionnalisation C-H dirigée d’hétérocycles azotés
Présentée par
Fares ROUDESLY
Pour obtenir le grade de
Docteur de Sorbonne Université
Soutenance publique prévue le 19 octobre 2018 Devant un jury composé de :
Pr Christophe HOARAU INSA de Rouen Rapporteur
Pr Jacques LEBRETON Université de Nantes Rapporteur
Dr Laurence GRIMAUD Ecole Normale Supérieure Examinateur
Dr Joanna WENCEL-DELORD Université de Strasbourg Examinateur
Dr Julie OBLE Sorbonne Université Examinateur
1
Remerciements
Ces travaux de thèse ont été réalisés au sein de l’Institut Parisien de Chimie Moléculaire dirigé par le Professeur Louis Fensterbank.
Je tiens à remercier le Dr. Laurence Grimaud, le Dr. Joanna Wencel-Delord, le Pr. Christophe Hoarau et le Pr. Jacques Lebreton d’avoir accepté de juger mon travail de thèse.
Je remercie Giovanni Poli pour avoir accepté de m’accueillir au sein de son équipe durant ces trois années et demi. Merci de m’avoir fait confiance, de m’avoir donné la chance de travailler sur ce(s) projet(s) et de m’avoir toujours soutenu. Un grand merci à mon encadrante de thèse Julie Oble. Je te remercie pour ta présence quotidienne, pour tes conseils, pour ta bonne humeur contagieuse. Vous m’avez permis de devenir un meilleur chimiste et une meilleure personne et je vous en serai toujours reconnaissant.
Je tiens aussi à remercier Anne Lise Dhimane pour m’avoir permis de partir en échange à Montréal, pour tous ses conseils et encouragements.
Je tiens également à remercier Fabrice Chemla, Franck Ferreira, Alexandre Pradal,. Alejandro Pérez-Luna et Olivier Jackowski pour leurs conseils et les discussions qu’on a pu avoir lors des réunions d’équipe. Un grand merci pour leur bonne humeur, surtout pendant les repas annuels. Un grand merci à Yang pour avoir partagé le labo avec moi pendant ces trois année et qui a dû supporter mes commentaires incessants. La relève est plus que garantie avec l’arrivée de Karen dans l’équipe. Son acharnement au travail est formidable et exemplaire.
Je remonte quelques années en arrière maintenant et j’aimerai remercier tous les stagiaires, doctorants et post-docs qui sont passés par le labo pendant ma thèse. Une pensée pour Mélanie qui était là pour voir mes débuts au labo, Kévin qui aimait m’embêter avec ses imitations et ses blagues interminables. Un grand merci à Valentin qui a réussi à me supporter pendant 2 ans et qui aimait me rappeler à quel point je râlais. J’ai eu aussi le plaisir de travailler avec plusieurs étudiants italiens : Daria, Tea, Cristofer et Gabriele, des personnes très sympathiques qui se sont adaptées rapidement à la vie du labo et avec qui j’ai partagé des bons moments au labo et bon nombre de sorties. Je remercie aussi Kajetan et ses passages à répétition au labo, le genre de personne qu’on ne peut pas détester et ses petits gadgets rigolos. Je tiens aussi à remercier Valérie-Anne et Floriane pour avoir réussi à survivre à mes règles (plus ou moins strictes) au labo.
J’ai aussi le plaisir de côtoyer les gens des autres équipes dans les différents couloirs. J’aimerai remercier les anciens qui m’ont accueilli à mes débuts et qui m’ont très vite adopté, je pense particulièrement à Simon, Brendan, Ludwig, Laura, Julien, Sha (ma chinoise préférée) et Juan. Je remercie aussi les personnes avec qui j’ai commencé cette aventure et avec qui j’ai eu le plaisir de partager beaucoup de bons moments comme Jorge, Anthony et Amina (courage, c’est presque fini). Il y a aussi les petits nouveaux comme j’aime les appeler qui nous ont rejoints les années suivantes : Lorièn (pour toutes nos discussions et tes encouragements), Sawsen (comment on peut être aussi gentille que toi ?!), Thibault, Valérie-Anne, Gaetan, Antoine, Léonid, Floriane…) je tiens à les remercier et leur souhaiter beaucoup de courage pour la fin.
2
Je remercie également les autres permanents du labo que j’ai eu le plaisir de connaître pour leurs conseils et les discussions diverses et variées qu’on a pu avoir.
Un grand merci aux gestionnaires du labo avec une pensée spéciale pour Sylvie L. La vie au labo est beaucoup plus facile grâce à ces personnes qui s’occupent de nos commandes au quotidien.
J’aimerai aussi remercier les personnes qui s’occupent des plateformes d’analyses, particulièrement Aurélie et Claire pour la RMN. Je sais que j’ai beaucoup râlé mais vous faites un travail formidable et je vous souhaite beaucoup de courage pour la suite. Je remercie aussi Omar (pour toutes nos discussions sur le foot) et Cédric pour toutes les analyses HRMS.
Depuis mon arrivée à Paris, j’ai eu le plaisir de connaître beaucoup de personnes formidables qui m’ont beaucoup aidé. Je remercie tous les membres de l’association « les internationaux de l’UPMC ». Je remercie aussi toutes les personnes qui ont été présentes depuis le début et encore plus pendant ces 3 années de thèse (Max, Antoine, Diola, Baptiste, Adrien, Manfreed, Laurène, Tristan, Mathilde, Clément, Thelma, Kim). Promis, la thèse, on en parle plus.
Je remercie aussi les membres de ma famille et mes amis de Tunis pour avoir été présents toutes ces années et pour leurs encouragements incessants.
Je remercie aussi mes parents pour m’avoir poussé et permis d’aller le plus loin possible dans mes études, pour tous vos encouragements, pour tous vos conseils et les valeurs que vous m’avez transmises. J’espère que vous êtes fiers de moi. Je remercie mes deux frères, Houssem et Wael, pour leur soutien continu, leurs encouragements et tous les moments qu’on a pu partager ensemble. Je tiens également à remercier ma belle-sœur Ariane, tu t’es rapidement intégrée dans la famille avec ton caractère bien trempé que j’aime tant, je te remercie aussi pour tous ces conseils que tu as pu me donner.
Je remercie ma petite chérie Adèle qui était présente depuis quasiment le début de cette thèse et qui était là pour me remonter le moral dans les mauvais moments et me féliciter dans les bons. C’est ton tour de jouer maintenant. Je remercie également les membres de sa famille que j’ai appris à connaître ces dernières années et qui m’ont rapidement adopté. J’ai la chance de dire que j’ai une deuxième famille formidable.
3
Sommaire
Introduction générale ... 7
Chapitre I : Activation de liaison C-H ... 11
I. Introduction – Historique ... 13
II. Mécanismes et exemples pionniers ... 15
1. Activation - fonctionnalisation de liaison C-H : quelques définitions ... 15
2. Mécanisme par sphère externe ... 16
3. Mécanisme par sphère interne ... 17
III. Sélectivité dans l’activation C-H ... 22
1. Couplage croisé déshydrogénant ... 22
2. Activation dirigée de liaison C(sp2)-H aromatique ... 26
3. Activation de liaison C(sp3)-H ... 29
4. Application de l’activation C-H en synthèse totale ... 32
5. Conclusion ... 34
Chapitre II : Oléfination et allylation de la pyridineN-oxyde par activation C-H ... 35
I. Introduction ... 37
II. Réactivité générale de la pyridine et de la pyridine N-oxyde ... 39
1. Réactivité classique de la pyridine ... 39
2. Fonctionnalisation de la pyridine N-oxyde ... 43
III. Fonctionnalisation par activation C-H de la pyridine ... 46
IV. Fonctionnalisation par activation C-H de la pyridine N-oxyde ... 53
V. Réaction d’allylation par activation C-H... 62
1. Allylation catalysée par du palladium ... 63
2. Allylation catalysée par du ruthénium ... 64
3. Allylation catalysée par du rhodium ... 66
4. Allylation catalysée par du cobalt ... 69
VI. Oléfination et allylation palladocatalysée d’azines N-oxyde ... 70
1. Objectif ... 70
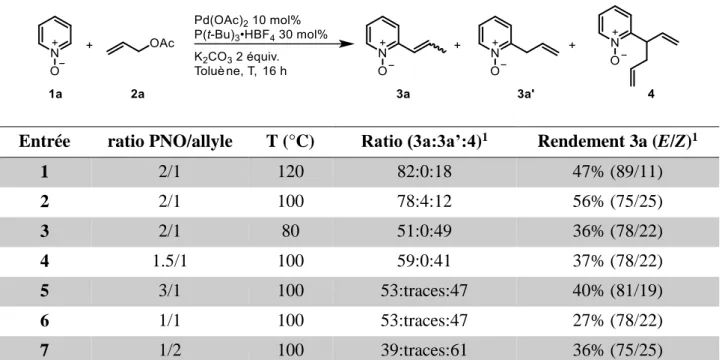
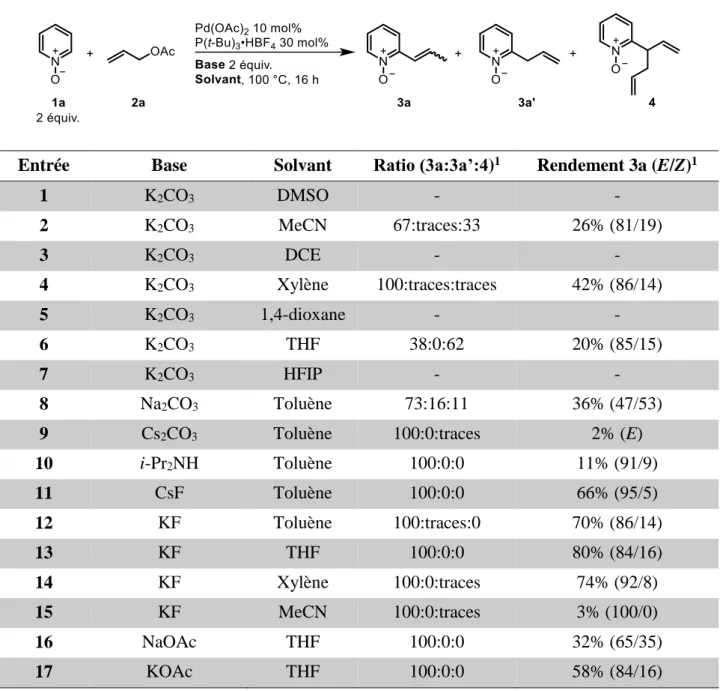
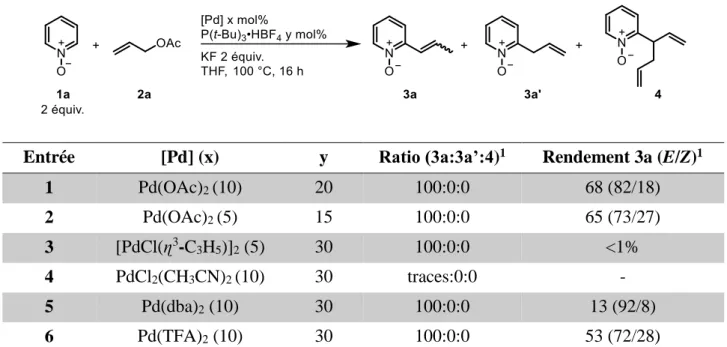
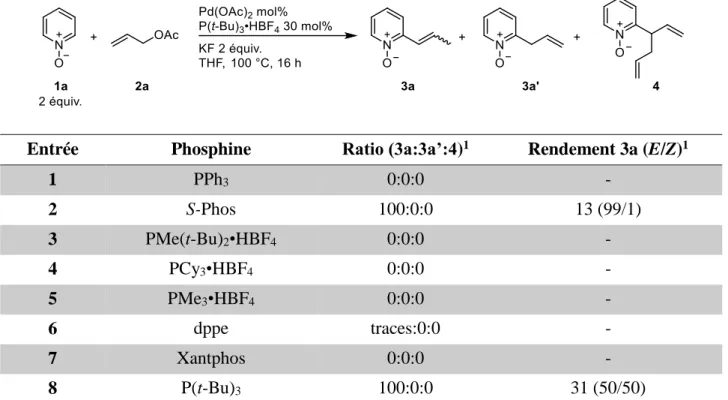
2. Choix de la réaction modèle et optimisation des conditions de réaction ... 71
3. Synthèse des substrats ... 75
4. Champ d’application des conditions d’oléfination ... 77
5. Post-fonctionnalisation des produits d’oléfination ... 80
6. Etudes mécanistiques ... 81
4
Chapitre III : Réaction de Murai carbonylative sur les pyrroles ... 93
I. Introduction ... 95
1. Fonctionnalisation du pyrrole ... 96
2. Fonctionnalisation par activation catalytique de liaison C-H ... 98
II. Réaction de Murai-Historique ... 107
1. Premiers travaux ... 108
2. Etudes mécanistiques ... 111
3. Autres applications de la réaction de Murai ... 115
4. Réaction de Murai carbonylative ... 118
III. Résultats et discussion ... 122
1. Précédents travaux de l’équipe et objectif du projet ... 122
2. Synthèse des substrats ... 123
3. Choix de la réaction modèle et optimisation des conditions de réaction ... 125
4. Champ d’application des conditions optimisées ... 130
5. Mécanisme proposé ... 132
IV. Conclusion et perspectives ... 134
Conclusion générale ... 137
Partie expérimentale ... 141
I. General remarks ... 143
II. Pd-Catalyzed Direct C-H Alkenylation and Allylation of Azine N-Oxides with Allyl Acetates ... 144
1. Mechanistic studies ... 144
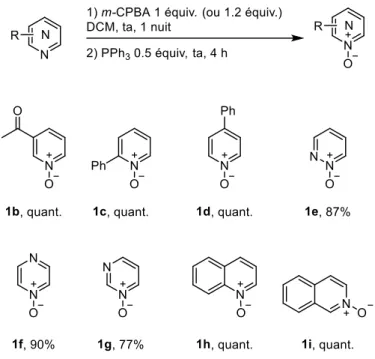
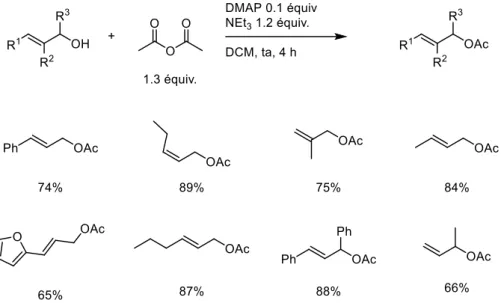
2. Preparation and characterization of starting materials ... 147
3. Preparation and characterization of C2-alkenylated and allylated N-Oxides ... 150
4. Further derivatizations of C2-alkenylated N-Oxides ... 159
III. Carbonylative Murai reaction on Pyrrole derivatives ... 162
1. Preparation and characterization of starting materials ... 162
5
Abréviations
Ac-Phe-OH N-acétylphénylalanine
Ac2O anhydride acétique
AcOH acide acétique
Ad adamantyle
AL acide de Lewis
APTS acide paratoluènesulfonique
Bn benzyle Boc tert-butoxycarbonyle BQ benzoquinone chromat. chromatographie Cp cyclopentadiényle Cp* perméthylcyclopentadiényle Cyp cyclopentyle dba dibenzylidèneacétone DCE 1,2-dichloroéthane DCM dichlorométhane DMA diméthylacétamide DMF diméthylformamide dmpe 1,2-bis(diméthylphosphino)éthane DMSO diméthylsulfoxyde dppe 1,2-bis(diphénylphosphino)éthane dppf 1,1’-bis(diphénylphosphino)ferrocène d. r. rapport diastéréomérique ee excès énantiomérique équiv. équivalent Et2O éther diéthylique ETOH éthanol HFiP hexafluoropropan-2-ol k constante de vitesse MeCN Acétonitrile MeOH Méthanol M Métal
6 MT métal de transition OAc acétate OBz benzoate OPiv pivalate OTf triflate S-Phos 2-Dicyclohexylphosphino-2′,6′-diméthoxybiphényle Ta température ambiante
TBAF fluorure de tetrabutylammonium
TBDMS tert-butyle-diméthylsilyle TFA trifluoroacétate THF tétrahydrofurane TIPS tri-iso-propylsilyle Ts tosyle TSE (triméthylsilyl)éthyle Xantphos 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthène µW micro-ondes
7
9
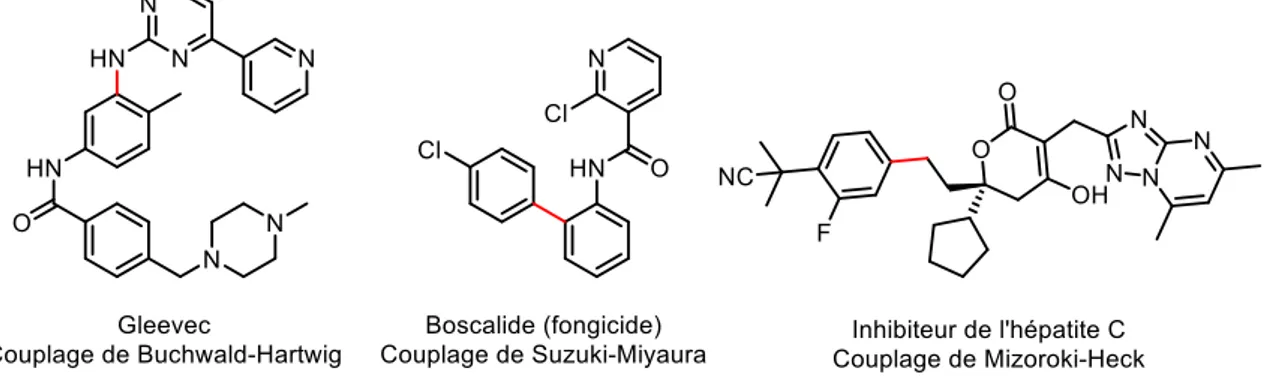
L’utilisation des métaux de transition en catalyse est certainement une des méthodes les plus répandues en synthèse organique que ce soit en milieu académique ou dans l’industrie.1 Leur efficacité et leur polyvalence pour certaines réactions font qu’ils sont devenus des outils incontournables. Par exemple, les réactions de couplage croisé catalysées au palladium représentent de nos jours une des méthodes les plus utilisées pour la synthèse des principes actifs de médicaments et des pesticides (Figure 1).2 Le large champ d’application et l’efficacité prouvée de cette chimie a valu le prix Nobel de chimie en 2010 au trio Heck, Negishi et Suzuki.
Figure 1. Structures de quelques principes actifs synthétisés en utilisant une étape de couplage croisé palladocatalysé (en rouge la liaison formée lors du couplage).
En outre, avec l’émergence des notions de chimie verte et d’économie d’atomes, les réactions d’activation de liaison C-H catalysées par des métaux de transition ont gagné en importance. Les liaisons C-H étant fortes et peu polarisées, leur activation représente un défi d’autant plus important pour le chimiste. Cette technique permet de diminuer le nombre d’étapes généralement nécessaires pour l’introduction et la suppression des groupes fonctionnels, et de former ainsi des liaisons C-C ou C-X par fonctionnalisation directe de liaison C-H avec une économie évidente d’étapes et d’atomes. On peut donc considérer que cette stratégie constitue une suite logique aux travaux réalisés sur les couplages croisés classiques.
Durant les trois dernières décennies, les réactions impliquant une activation catalytique de liaison C-H sont devenues un sujet de recherche des plus importants menant à l’attribution du prestigieux Prix Wolf en 2017 à Robert Bergman pour la « découverte de l’activation de liaison C-H d’hydrocarbures par des complexes de métaux de transition ». Cette thématique est aussi considérée comme un sujet pouvant être récompensé par un prix Nobel.3 Comme on peut le voir sur la figure 2,
1 (a) Beller, M.; Bolm, C. Transition Metals for Organic Synthesis, 2nd ed., Wiley-VCH, Weinheim, 2004; (b) Torborg,
C.; Beller, M. Adv. Synth. Catal. 2009, 351, 3027-3043.
2 (a) Johansson Seechurn, C. C. C.; Kitching, M. O.; Colacot, T. J.; Snieckus, V. Angew. Chem. Int. Ed. 2012, 51,
5062-5085; (b) Colacot, T. New Trends in Cross-Coupling: Theory and Applications, RSC press, Cambridge, 2015.
10
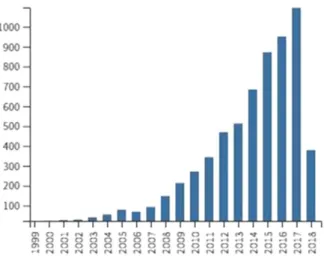
le nombre de publications qui portent sur les thématiques d’activation C-H et de fonctionnalisation C-H, a augmenté de manière significative.4
Figure 2. Nombre de publications par an sur les thématiques « C-H activation » « C-H functionalization ».
Ce manuscrit est construit en trois parties. Tout d’abord, un chapitre au cours duquel nous présenterons un bref historique de l’activation de liaison C-H et les différents mécanismes qui peuvent entrer en jeu. Les deux chapitres suivants porteront sur la fonctionnalisation de liaison C(sp2)-H de deux hétérocycles azotés. D’une part, nous présenterons la mise au point d’une nouvelle méthode d’oléfination et d’allylation d’azines N-oxydes ; des hétérocycles appauvris en électrons. D’autre part, nous décrirons une nouvelle méthode d’acylation en position C3 du 2-pyrrole carboxaldéhyde, un hétérocycle riche en électrons, en utilisant un groupement directeur facilement détachable.
11
Chapitre I
13
I.
Introduction – Historique
5La recherche de méthodes qui permettent l’exploitation des ressources les plus abondantes afin de synthétiser des molécules complexes est un objectif de longue date pour les chimistes. Ces dernières décennies, en accord avec les enjeux sociétaux et environnementaux, la notion d’activation de liaison C-H a pris une place plus grande dans ce domaine de recherche. Plus particulièrement, l’utilisation des métaux de transition (MT) en catalyse homogène a permis le développement de plusieurs stratégies efficaces.6,7,8
En plus de permettre la diminution du nombre d’étapes de synthèse et de purification, réduisant ainsi la quantité de déchets, cette technique présente l’avantage de pouvoir utiliser directement les matières premières. En effet, la transformation directe d’alcanes en alcools, en acides ou en d’autres dérivés mono-oxydés représente un des défis les plus stimulants et encore difficiles à atteindre. Dans ce contexte, le méthane est une source considérable pour la production d’énergie et son oxydation en méthanol faciliterait énormément son transport.
Pour ce qui concerne les substrats à chaine plus longue que le méthane, le plus gros défi est le contrôle de la sélectivité du site. En effet, une liaison C-H dans une position non activée est très peu polarisée et faiblement acide. En présence de plusieurs liaisons C-H non activées, la sélectivité peut alors être difficile à réaliser. En plus, l’activation directe d’une liaison C-H peut s’avérer très compliquée en présence d’autres liaisons plus facilement activables, ce qui présente un problème de chimiosélectivité.
En conséquence, ce type de réactivité entraîne donc un changement dans le raisonnement synthétique. Au lieu de concevoir des chemins se basant sur des groupes fonctionnels (GFs) classiques, la nouvelle approche est de choisir une méthode permettant la fonctionnalisation contrôlée d’une liaison C-H
5 Ce chapitre est basé sur une revue tutorielle écrite récemment par l’équipe : Roudesly, F.; Oble, J.; Poli, G. J. Mol. Cata. A Chemical 2017, 426, 275-296.
6 Pour une sélection de livres, voir : (a) Yu, J.-Q. Catalytic Transformations via C-H Activation, vols 1 and 2, Science of
Synthesis, Thieme, Stuttgart, 2016; (b) Goldberg, K. I.; Goldman, A. S. Activation and Functionalization of C-H Bond, ACS Symposium Series 885, American Chemical Society, Washington DC, 2004.
7 Pour une sélection d’issues spéciales, voir : (a) Chem. Rev. 2017, 117, 8481-9520; (b) Beilstein J. Org. Chem. 2012, 8,
1552; (c) Acc. Chem. Res. 2012, 45, issue 6, 777-958; (d) Chem. Soc. Rev. 2011, 40, issue 4, 1845-2040; (e) Chem. Rev. 2010, 110, issue 2, 575-1211.
8 Pour une sélection de revues récentes, voir : (a) Gensch, T.; Hopkinson, M. N.; Glorius, F.; Wencel-Delord, J. Chem. Soc. Rev. 2016, 45, 2900-2936; (b) Théveau, L.; Schneider, C.; Fruit, C.; Hoarau, C. ChemCatChem 2016, 8, 3183-3194;
(c) Hartwig, J. F. J. Am. Chem. Soc. 2016, 138, 2-24; (d) Davies, H. M. L.; Morton, D. J. Org. Chem. 2016, 81, 343-350; (e) Ackermann, L. Acc. Chem. Res. 2014, 47, 281-295; (f) Franzoni, I. ; Mazet, C. Org. Biomol. Chem. 2014, 12, 233-241; (g) Li, B.-J.; Shi, Z.-J. Chem. Soc. Rev. 2012, 41, 5588-5598; (h) Chen, X.; Engle, K. M.; Wang, D. H.; Yu, J.-Q.
14
donnée. Cette approche fait que la liaison C-H peut alors être considérée comme un groupe fonctionnel pour une synthèse directe et efficace (Schéma 1).
Schéma 1. Schéma général de la fonctionnalisation de liaison C-H.
La faible réactivité de la liaison C-H est une conséquence directe de sa haute énergie de dissociation de liaison (BDE) et une valeur de pKa très élevée. Comme nous pouvons le voir dans le Tableau 1, la réactivité de la liaison C-H dépend du type d’hybridation de l’atome de carbone. En effet, en allant d’un carbone C(sp) vers un carbone C(sp2) puis C(sp3), la BDE diminue alors que dans le même temps le pKa augmente.5,9
Type de
liaison C-H C(sp) C(sp
2)
arom C(sp2)vinyl C(sp3)1° C(sp3)2° C(sp3)3° C(sp3)allylique
Structure BDE
(kJ/mol) 552.2 473.0 460.2 410.8 397.9 389.9 361.1
pKa ~25 43 44 ~50 ~50 ~50 43
Tableau 1. pKa et BDE de certaines liaisons C-H.
Le travail de Chatt, en 1965, est considéré comme le premier exemple d’activation C-H mettant en jeu un complexe de métal de transition. En effet, en présence de deux équivalents d’un anion naphtylique, le complexe [RuCl2(dmpe)] est préalablement réduit en un complexe de Ru(0). Ce dernier peut alors s’insérer par addition oxydante dans une liaison C-H du naphtalène conduisant à un complexe naphtyl-Ru(II)-H (Schéma 2).10
9 Blanksby, S. J.; Ellison, G. B. Acc. Chem. Res. 2003, 36, 255-263. 10 Chatt, J.; Davidson, J. M. J. Chem. Soc. A 1965, 843-855.
15
Schéma 2. Activation C-H du naphtalène.
Dans la suite de ce chapitre, nous allons nous focaliser sur les différents mécanismes qui peuvent être mis en jeu dans ce type de réactivité. Nous présenterons ensuite quelques travaux pionniers et pertinents dans ce domaine.
II. Mécanismes et exemples pionniers
1. Activation - fonctionnalisation de liaison C-H : quelques définitions
Dans le domaine de l’activation/fonctionnarisation C-H promue ou catalysée par un métal (M), Shilov et Shul’pin définissent « une véritable activation» comme étant la formation d’une liaison σ(M-C), suite à l’entrée de la liaison C-H dans la sphère de coordination du métal (Schéma 3).11
Schéma 3. Activation C-H selon Shilov et Shul’pin.
Crabtree, quant à lui, décrit deux types d’approche. « L’approche organométallique », où l’activation C-H est définie aussi comme étant le résultat d’une interaction directe entre le métal et la liaison C-H, suivie de la rupture de cette dernière, avec la formation d’une liaison M-C. Puis « l’approche de chimie de coordination » qui mime l’oxydation des alcanes réalisée par les enzymes. Dans ce dernier cas, la liaison C-H interagit directement avec un des ligands du complexe.12
Ces définitions ont été reprises ensuite par Sanford, qui utilise le terme « sphère interne » pour désigner « l’approche organométallique » et le terme « sphère externe » pour désigner « l’approche de chimie de coordination ».13
11 Shilov, A. E.; Shul'pin, G. B. Chem. Rev. 1997, 97, 2879-2932. 12 Crabtree, R. H. J. Chem. Soc., Dalton Trans. 2001, 2437-2450. 13 Dick, A. R.; Sanford, M. S. Tetrahedron, 2006, 62, 2439-2463.
16
De nos jours, les termes « activation C-H » et « fonctionnalisation C-H » sont utilisés de manière interchangeable. Toutefois, l’usage du terme « activation C-H » se limite souvent à un mécanisme de type sphère interne, alors que le terme « fonctionnalisation C-H » est utilisé pour décrire un mécanisme à sphère externe.
2. Mécanisme par sphère externe
Comme nous l’avons mentionné plus haut, ce type de mécanisme est basé sur l’action de certaines enzymes d’oxydation, comme le cytochrome P450 ou la méthane mono-oxygénase, qui permettent l’hydroxylation de liaisons C-H. Ces réactions passent généralement par un complexe métallique de type oxo, nitrène ou carbène, pour former respectivement des liaisons O, N ou C-C. Notons toutefois, que la Nature utilise cette chimie seulement pour le premier cas de figure. La première étape consiste en la génération d’un complexe métallique à haut degré d’oxydation et comportant un ligand hautement réactif. La deuxième étape peut être une étape métalloradicalaire suivant un mécanisme de type « rebond » radicalaire. C’est le cas du complexe oxo, qui va capter un atome d’hydrogène par abstraction homolytique, suivie de la réaction de l’espèce radicalaire ainsi formée sur le complexe hydroxyle résultant pour donner l’alcool correspondant (Schéma 4, haut). Dans le cas d’un carbène ou d’un nitrène, cette deuxième étape est une insertion directe dans la liaison C-H en parallèle de la dissociation du ligand du complexe métallique (Schéma 4, bas).
Schéma 4. Activation C-H par sphère externe : cas des complexes de type oxo (haut) et de type nitrène ou carbène (bas)
Un exemple qui illustre ce type de réactivité a été décrit par l’équipe de Doyle.14 L’utilisation d’un catalyseur chiral à base d’un dimère de rhodium [Rh2(4R-MPPIM)4] a permis la synthèse de lactones avec de bons rendements et de bons excès énantiomériques (Schéma 5). La génération du carbène se fait in situ par réaction entre le diazo-carbonyle et le complexe de rhodium. Cette méthode a été utilisée pour la synthèse de quelques produits naturels tels que la (+)-enterolactone.
17
Schéma 5. Synthèse de lactone à partir de composé diazo.
3. Mécanisme par sphère interne
Historiquement, trois grandes classes de mécanismes ont été différentiées en se basant sur des études mécanistiques : l’addition oxydante (AO), la métathèse de liaison σ (σ-BM) et l’activation électrophile (AE). Cependant, ces dernières années, d’autres types de mécanismes ont été suggérés en se basant sur différentes observations expérimentales et études computationnelles.15
Addition oxydante
Ce mécanisme est généralement admis pour un complexe comportant un MT nucléophile, à valence faible, situé en deuxième ou troisième rang.
Parmi les premiers groupes à découvrir ce type de réactivité, on peut citer ceux de Green,16 Bergman,17 Graham18 et Jones.19 En effet, dès 1970, Green a montré qu’un complexe Cp2*W était capable de s’insérer dans une liaison C-H du benzène. Plus tard, Bergman a décrit l’utilisation d’un complexe dihydrido iridium qui, sous irradiation, pouvait subir une élimination réductrice de H2, suivie d’une addition oxydante en présence du benzène, du cyclohexane ou du néopentane comme solvant (Schéma 6).
15 Vastine, B. A.; Hall, M. B. J. Am. Chem. Soc. 2007, 129, 12068-12069.
16 (a) Green, M. L. H.; Knowles, P. J. J. Chem. Soc. Chem. Comm. 1970, 1677; (b) Giannotti, C.; Green, M. L. H. J. Chem. Soc. Chem. Commun. 1972, 1114-1115.
17 Janowicz, A. H.; Bergman, R. G. J. Am. Chem. Soc. 1982, 104, 352-354. 18 Hoyano, J. K.; Graham, W. A. G. J. Am. Chem. Soc. 1982, 104, 3723-3725. 19 Jones, W. D.; Feher, F. J. Organometallics 1983, 2, 562-563.
18
Schéma 6. Activation C-H du benzène par addition oxydante.
Métathèse de liaison σ (σ-BM : σ-Bond Metathesis)
Pour les métaux de transition de configuration électronique d0 à haut degré d’oxydation, l’addition oxydante est impossible. Dans ce cas, l’activation C-H passe par un mécanisme de type
σ-BM.
Le premier exemple décrivant une telle réactivité a été décrit par Watson en 1983.20 Elle a montré qu’un complexe de type Cp2*Lu(CH3) peut réagir avec du méthane en échangeant le ligand CH3 par un 13CH3 (Schéma 7). La même réaction est possible avec des complexes du même type basés sur l’Yttrium.
Schéma 7. Activation du méthane par métathèse de liaison σ.
Ces résultats ont été ensuite exploités par d’autres équipes qui ont contribué de manière remarquable à ce domaine.21
Par ailleurs, les différentes études réalisées par Hartwig sur la borylation directe des alcènes, des arènes et des alcanes l’ont amené à considérer un autre type de mécanisme qui serait entre une addition oxydante et une σ-BM.22 Ce type de réactivité a été observé en présence de complexe de type CpM(CO)n(BOR2) (où M = W, Fe, Co). Il a appelé ce type de mécanisme « transfert d’hydrogène par
20 Watson, P. L. J. Am. Chem. Soc. 1983, 105, 6491-6493.
21 (a) Thompson, M. E.; Baxter, S. M.; Bulls, A. R.; Burger, B. J.; Nolan, M. C.; Santarsiero, B. D.; Schaefer, W. P.;
Bercaw, J. E. J. Am. Chem. Soc. 1987, 109, 203-219; (b) Jordan, R. F.; Taylor, D. F. J. Am. Chem. Soc. 1989, 111, 778-779,
22 (a) Waltz, K. M.; He, X.; Muhoro, C.; Hartwig, J. F. J. Am. Chem. Soc. 1995, 117, 11357-11358; (b) Waltz, K. M.;
Hartwig, J. F. Science 1997, 277, 211-213; (c) Waltz, K. M.; Muhoro, C.; Hartwig, J. F. Organometallics 1999, 18, 3383-3393; (d) Webster, C. E.; Fan, Y.; Hall, M. B.; Kunz, D.; Hartwig, J. F. J. Am. Chem. Soc. 2003, 125, 858-859; (e) Hartwig, J. F. Chem. Rev. 2010, 110, 890-931.
19
l’intermédiaire d’un métal aidé par le bore », qui est un cas particulier de la métathèse assistée par un complexe σ (σ-CAM).
D’autres types de mécanismes peuvent être retrouvés en littérature : état de transition additionné oxydativement (OATS) et la migration oxydative d’hydrogène (OHM).
Activation électrophile
Par ailleurs, dans le cas d’un complexe comportant un MT pauvre en électrons, à haut degré d’oxydation tels que PdII, PtII, RhIII, le mécanisme le plus susceptible d’intervenir est de type activation électrophile.
Les travaux de Shilov sur l’oxydation du méthane sont parmi les plus marquants dans ce domaine.23 Les auteurs décrivent un système combinant du PtII catalytique et du PtIV stœchiométrique pour obtenir sélectivement du méthanol. Cette réaction, en dépit du fait qu’elle soit stœchiométrique en Pt, représente une grande avancée dans la fonctionnalisation sélective d’alcanes. Après la complexation de l’eau, puis du méthane, un ion chlorure déprotone le méthane coordonné. Ensuite, le complexe de Pt(II) est oxydé en Pt(IV) en présence de K2PtCl6. Enfin, une molécule d’eau se décoordonne et attaque le groupement CH3 pour donner le méthanol. Il est à noter que certaines études décrivent que l’étape d’activation C-H passe par un mécanisme de type addition oxydante (Schéma 8).24
23 Goldshleger, N. F.; Eskova, V. V.; Shilov, A. E.; Shteinman, A. A. Russ. J. PhysChem. 1972, 46, 785-786. 24 Stahl, S. S.; Labinger, J. A.; Bercaw, J. E. Angew. Chem. Int. Ed. 1998, 37, 2180-2192.
20
Schéma 8. Mécanisme de la réaction de Shilov pour l’oxydation du méthane.
Plusieurs années plus tard, Periana25 et son équipe de l’entreprise Catalytica ont rapporté une amélioration du système de Shilov en utilisant un ligand de type bipyrimidine (bpmd) pour éviter la dégradation du catalyseur de platine. En présence d’acide sulfurique, ils ont réussi à obtenir avec 81% de sélectivité l’acide méthylsulfonique (Schéma 9).
Schéma 9. Oxydation du méthane via le process Catalytica.
Dans le cas de l’activation de liaison C(sp2)-H aromatique, si l’arène est électron-riche, cette étape suit un mécanisme de type substitution électrophile aromatique (SEAr). En revanche, si l’arène est électron-appauvri, le mécanisme implique normalement une déprotonation intramoléculaire par un ligand du métal (halogénure, alkoxy, acyloxy ou carbonate) comme étape lente. Ce dernier mécanisme est appelé « activation ambiphilique ligand-métal » (AMLA), « métalation-déprotonation concertée » (CMD) ou encore « substitution électrophile interne » (IES). Maseras et Echavarren26 ont décrit une telle approche dans leur étude sur l’arylation intramoléculaire pallado-catalysée. De même,
25 Periana, R. A.; Taube, D. J.; Gamble, S.; Taube, H.; Satoh, T.; Fujii, H. Science 1998, 280, 560-564.
21
lors de leur étude du mécanisme de l’arylation directe de différentes arènes et hétéro-arènes, Gorlesky et Fagnou27 ont proposé un état de transition similaire pour l’étape d’activation C-H (Schéma 10).
Schéma 10. Etats de transition proposés pour l’activation C-H aromatique.
Par ailleurs, dans le cas de certains noyaux hétéroaromatiques, un mécanisme de type métalation-déprotonation non concertée (nCMD) est proposé qui peut être par sphère interne ou par sphère externe. Dans leur étude sur la réaction d’arylation de 4-carboxylate oxazole et thiazole, le groupe de Hoarau a montré que la maîtrise des conditions de réaction permettait de contrôler la compétition entre une CMD et une nCMD.8b,2829 En effet, le choix de la base, du ligand et de l’halogénure d’aryle permet d’avoir un régio-contrôle de la réaction d’arylation (Schéma 11).
Schéma 11. Différences entre CMD et nCMD.
Addition 1,2
En 1988, Bergman30 et Wolczniacki31 ont rapporté, dans la même issue du journal JACS, l’activation de liaison C-H d’alcanes et du benzène par addition 1,2 en utilisant des complexes de zirconium de type amido alkyle. Ce complexe subit une première élimination du méthane pour former
27 (a) Lafrance, M.; Rowley, C. N.; Woo, T. K.; Fagnou, K. J. Am. Chem. Soc. 2006, 128, 8754-8756; (b) Gorelsky, S. I.;
Lapointe, D.; Fagnou, K. J. Am. Chem. Soc. 2008, 130, 10848-10849.
28 (a) Verrier, C.; Martin, T.; Hoarau, C.; Marsais, F. J. Org. Chem. 2008, 73, 7383-7386; (b) Martin, T.; Verrier, C.;
Hoarau, C.; Marsais, F. Org. Lett. 2008, 10, 2909-2912; (c) Théveau, L.; Verrier, C.; Lassalas, P.; Martin, T.; Dupas, G.; Querolle, O.; Van Hijfte, L.; Marsais, F.; Hoarau, C. Chem. Eur. J. 2011, 17, 14450-14463; (d) Théveau, L.; Querolle, O.; Dupas, G.; Hoarau, C. Tetrahedron 2013, 69, 4375-4380.
29 Bellina, F.; Cauteruccio, S.; Rossi, R. Curr. Org. Chem. 2008, 12, 774-790.
30 Walsh, P. J.; Hollander, F. J.; Bergman, R. G. J. Am. Chem. Soc. 1988, 110, 8729-8731. 31 Cummins, C. C.; Baxter, S. M.; Wolczanski, P. T. J. Am. Chem. Soc. 1988, 110, 8731-8733.
22
l’intermédiaire imido. Ce dernier s’additionne alors sur une liaison C-H d’un alcane ou du benzène (Schéma 12).
Schéma 12. Activation C-H du benzène par addition 1,2.
III. Sélectivité dans l’activation C-H
Comme nous l’avons mentionné dans l’introduction de ce chapitre, un des défis les plus importants de l’activation de liaison C-H est la sélectivité. Pour relever ce défi, plusieurs techniques ont été développées au fil des ans. Nous pouvons notamment citer d’une part la stratégie qui implique l’intervention d’un groupement directeur (sélectivité dirigée) et d’autre part celle qui fait appel à une situation électronique intrinsèquement biaisée du substrat (sélectivité non-dirigée). Dans le premier cas, une pré-complexation du métal, soit par un hétéroatome, soit par une insaturation, permet de rentrer le substrat dans la sphère de coordination du métal, facilitant ainsi l’interaction du centre métallique avec une liaison C-H du substrat. On obtient alors un métallacycle dont la taille adéquate favorise la régiosélectivité de l’activation C-H. D’autre part, la sélectivité peut aussi être contrôlée par la richesse électronique du substrat, en particulier dans le cas des noyaux aromatiques.
Dans la suite de ce chapitre, nous allons présenter une sélection d’exemples pertinents. 1. Couplage croisé déshydrogénant
L’activation de liaison C-H d’arènes et d’hétéroarènes est généralement contrôlée par des effets électroniques. En effet, dans la plupart des cas, c’est la liaison C-H la plus riche en électrons qui réagit. Toutefois, dans d’autres cas, la réaction se fait au niveau de la liaison C-H la plus acide.
23
Couplage alcène/arène : réaction de Fujiwara-Moritani
Dès 1967, Fujiwara et Moritani32 ont décrit leur étude sur l’oléfination d’arènes en présence d’un sel de Pd(II) par réaction entre un arène, présent en grand excès, et un alcène. Cette réaction est sans doute une très grande découverte dans le domaine de l’activation C-H. Deux années plus tard, la même équipe rapporte la version catalytique aérobique de cette réaction en utilisant un système Cu(OAc)2/O2 pour réoxyder le Pd(0) et régénérer à la fin du cycle catalytique le complexe de Pd(II) (Schéma 13).
Schéma 13. Synthèse palladocatalysée du E-stylbène.
Le mécanisme proposé pour ce type de réaction débute par une activation C-H du benzène formant un intermédiaire aryl-Pd(OAc). Il est suggéré que cette étape passe par un mécanisme de type AMLA/CMD. Après coordination du complexe aryl-Pd(OAc) de la part de l’alcène, une insertion migratoire suivie d’une déshydropalladation donnent le stylbène et l’hydrure de palladium HPd(OAc). Ce dernier subit une élimination réductrice conduisant à un complexe de Pd(0). Enfin, en présence de Cu(OAc)2 et de dioxygène, l’espèce de Pd(II) catalytique est régénérée (Schéma 14).
32 (a) Moritani, I.; Fujiwara, Y. Tetrahedron Lett. 1967, 8, 1119-1122; (b) Fujiwara, Y.; Moritani, I.; Danno, S.; Asano,
24
Schéma 14. Mécanisme de la réaction de Fujiwara-Moritani.
Couplage arène/arène
Les biaryles sont des motifs très importants dans l’industrie. On les retrouve dans la structure de plusieurs molécules d’intérêt biologique ainsi que celle de certains principes actifs. La stratégie la plus utilisée pour la synthèse de tels composés est le couplage croisé, généralement catalysé par du cuivre ou du palladium.33 En particulier, le couplage de Suzuki-Miyaura est un outil très efficace pour atteindre ce but.
Les premiers exemples de couplage croisés d’arènes non activés ont été décrits en même temps par Fagnou34 et DeBoef.35 Durant son étude, l’équipe de Fagnou a rapporté le développement de conditions pour la réaction entre des dérivés d’indole et le benzène (Schéma 15, a). DeBoef, quant à lui, a rapporté le couplage entre le benzofurane et le benzène en utilisant un système hétéropolymolybdovanadique HMPV/O2 comme oxydant (Schéma 15, b).
33 Hassan, J.; Sévignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Chem. Rev. 2002, 102, 1359-1470. 34 Stuart, D. R.; Fagnou, K. Science 2007, 316, 1172-1175.
25
Schéma 15. Formation de biaryles par couplage croisé déshydrogénant.
DeBoef et Gorelsky ont ensuite mené des études permettant de mieux comprendre le mécanisme qui régit cette réaction.36 D’abord, ils ont étudié le mécanisme de la palladation de l’indole. Deux voies possibles ont été proposées : une palladation électrophile via SEAr ou un processus CMD. La combinaison de l'étude de deutération et des résultats des expériences de compétition a permis aux auteurs de conclure qu'un mécanisme CMD est le plus envisageable dans ce cas. Ensuite, une série d'expériences comprenant une étude de l'effet isotopique cinétique a montré que la palladation de l'arène se produit également via le mécanisme CMD. Cette observation est en accord avec le travail de Gorelsky et Fagnou réalisé pour étudier le mécanisme de l'arylation directe d’(hétéro)arènes.27b,37
En se basant sur ces études, un mécanisme général pour le couplage croisé palladocatalysé d’arènes impliquant une double activation C-H a été proposé. Une première activation de liaison C-H sur un Ar1-H en procédant par un processus AMLA/CMD a lieu, qui est suivie d'une seconde activation C-H sur un Ar2-H se produisant par le même mécanisme. Enfin, l'élimination réductrice permet d'obtenir les produits biaryles et une espèce de Pd(0) qui sera réoxydée en Pd (II) par le système oxydant (Schéma 16).
Schéma 16. Schéma général du couplage croisé déshydrogénant arène/arène.
36 Potavathri, S.; Pereira, K. C.; Gorelsky, S. I.; Pike, A.; LeBris, A. P.; DeBoef, B. J. Am. Chem. Soc. 2010, 132,
14676-14681.
26
2. Activation dirigée de liaison C(sp2)-H aromatique
Dans ce contexte d’activation de liaison C-H, la fonctionnalisation sélective des noyaux aromatiques est, depuis plusieurs années, sujette à un très grand nombre de progrès. En se basant sur l’introduction d’un groupe directeur (GD), différentes techniques ont été développées.
2.1. Activation C-H dirigée en position ortho
C’est la méthode la plus étudiée pour l’activation C-H des arènes et à laquelle plusieurs groupes se sont intéressés. Cet intérêt a donné lieu au développement de plusieurs groupements de type carboxylate, cétone, pyridine, amide ou quinoléine… On peut classer ce type d’activation en deux grandes catégories : activation C-H nucléophile ou électrophile.
Au début des années 1990, Kakiuchi et Murai38 ont décrit l’utilisation de catalyseur de Ru pour l’insertion d’oléfine en positon ortho sur des dérivés d’acétophénone. Ces travaux sont, en partie, à l’origine des différentes études sur l’activation C-H dirigée (Schéma 17). L’étape d’activation C-H passe dans ce cas par un mécanisme de type addition oxydante par le complexe de Ru(0) formé in
situ. Ce mécanisme sera détaillé dans le chapitre III.
Schéma 17. Ortho-alkylation d’acétophénone catalysée par un complexe de ruthénium.
D’autre part, les groupes de Miura39 et de Vries40 ont décrit une version dirigée en ortho de la réaction de Fujiwara-Moritani. Tandis que les premiers ont utilisé des sulfonamides comme groupement directeur (Schéma 18, haut), les seconds ont introduit un groupe amide pour diriger l’activation C-H (Schéma 18, bas).
38 Murai, S.; Kakiuchi, F.; Sekine, S.; Tanaka, Y.; Kamatani, A.; Sonoda, M.; Chatani, N. Nature 1993, 366, 529-531. 39 Miura, M.; Tsuda, T.; Satoh, T.; Pivsa-Art, S.; Nomura, M. J. Org. Chem. 1998, 63, 5211-5215.
40 Boele, M. D. K.; van Strijdonck, G. P. F.; de Vries, A. H. M.; Kamer, P. C. J.; de Vries, J. G.; van Leeuwen, P. W. N.
27
Schéma 18. Réaction de Fujiwara-Moritani dirigée en ortho.
Plus récemment, les groupes de Yoshikai41 et Nakamura42 ont décrit la fonctionnalisation ortho-sélective d’arènes catalysée par des complexes de cobalt. D’une part, la réaction d’hydroarylation d’alcynes a été rapportée par Yoshikai en utilisant la pyridine pour diriger l’activation C-H (Schéma 19, haut). D’autre part, Nakamura a décrit l’alkylation de benzamide en présence de chlorure d’alkyle et en utilisant le groupement amide comme groupement directeur (Schéma 19, bas). Nous remarquons que dans ces deux méthodes, les auteurs utilisent un réactif de Grignard, qui joue le rôle de réducteur du Co(II) en Co(0). Ce dernier s’insère ensuite dans la liaison C-H en position ortho par addition oxydante.
Schéma 19. Exemples d’activation C-H ortho-dirigée catalysée par du cobalt.
41 Gao, K.; Lee, P.-S.; Fujita, T.; Yoshikai, N. J. Am. Chem. Soc. 2010, 132, 12249-12251. 42 Chen, Q.; Ilies, L.; Nakamura, E. J. Am. Chem. Soc. 2011, 133, 428-429.
28
2.2. Activation C-H dirigée en position meta
C’est en 2012 que le groupe de Yu43 décrit l’activation C-H dirigée en position méta par rapport au carbone portant le groupe directeur. Pour ce faire, les auteurs ont introduit une nouvelle classe de groupement directeur basé sur un nitrile qui permet de passer outre la réactivité intrinsèque des substrats due aux effets électroniques et stériques. Dans cet exemple impliquant une réaction de Fujiwara-Moritani dirigée en meta, les auteurs proposent une approche de type AMLA/CMD pour l’étape d’activation C-H. En général, un tel mécanisme passe par un état de transition à six ou sept chainons (pour une activation ortho), alors que dans ce cas c’est un macrocycle à douze chainons qui est proposé (Schéma 20).
Schéma 20. Activation C-H dirigée en postion meta.
Cet exemple fut le point de départ pour le développement d’un grand nombre de fonctionnalisation C-H en position meta par la synthèse de différents groupements directeurs possédant un motif nitrile relié par un bras espaceur à l’arène, comme en témoignent les travaux des équipes de Yu,44 Maiti,45 Kuninobu et Kana.46
2.3. Activation C-H dirigée en position para
Récemment, Maiti47 a décrit un exemple dans lequel un groupe directeur en « forme de D » a été conçu en vue d’une activation sélective en position para.
Dans cette étude de conception, le défi majeur était la définition de la longueur de chaîne du modèle qui régit la fonctionnalisation sélective ; ce qui permet la formation d’un état de transition
43 Leow, D.; Li, G.; Mei, T.; Yu, J.-Q. Nature 2012, 486, 518-522.
44 Shen, P.-Q.; Wang, X.-C.; Wang, P.; Zhu, R.-Y.; Yu, J.-Q. J. Am. Chem. Soc. 2015, 137, 11574-11577. 45 Bera, M.; Modak, A.; Patra, T.; Maji, A.; Maiti, D. Org. Lett. 2014, 16, 5760-5763.
46 Pour un exemple de borylation, voir : Kuninobu, Y.; Ida, H.; Nishi, M.; Kanai, M. Nat. Chem. 2015, 7, 712-717. 47 Bag, S.; Patra, T.; Modak, A.; Deb, A.; Maity, S.; Dutta, U.; Dey, A.; Kancherla, R.; Maji, A.; Hazra, A.; Bera, M.;
29
macrocylique de grande taille donc moins tendu et le réglage de la proximité de l’hétéroatome coordonnant du GD vers la liaison C-H ciblée.
Le GD ainsi conçu et synthétisé leur a permis d’obtenir le produit de la réaction d’oléfination d’arènes avec de très bons rendements et une bonne sélectivité. Dans cet exemple, l’état de transition formant le métallacyle au cours de l’étape d’activation C-H est un macrocycle à 17 chainons (Schéma 21). Cette réaction a ensuite été étendue à des dérivés du phénol.48
Schéma 21. Activation C-H dirigée en postion para.
3. Activation de liaison C(sp3)-H
Le premier exemple décrivant l’activation catalytique de liaison C(sp3)-H a été rapporté par Dyker en 1992.49 Il a montré que l’o-iodoanisole pouvait subir une trimérisation par une séquence domino en présence de Pd(OAc)2. La formation du dibenzopyrane est le résultat d’une séquence d’addition oxydante et de trois activation C-H successives intramoléculaires. La première est celle du groupement méthoxy par le complexe aryl-Pd-X formé après addition oxydante de l’o-iodoanisole sur le Pd(0) (Schéma 22, a). Cette même réactivité a été appliquée pour la dimérisation du
2-iodo-tert-butylbenzène.50
48 Patra, T.; Bag, S.; Kancherla, R.; Mondal, A.; Dey, A.; Pimparkar, S.; Agasti, S.; Modak, A.; Maiti, D. Angew. Chem. Int. Ed. 2016, 55, 7751-7755.
49 Dyker, G. Angew. Chem. Int. Ed. Engl. 1992, 31, 1023-1025. 50 Dyker, G. Angew. Chem. Int. Ed. Engl. 1994, 33, 103-105.
30
Schéma 22. Activation de liaison C(sp3)-H palladocatalysée.
Près d’une décennie plus tard, Baudoin a rapporté la synthèse de bicycle par arylation intramoléculaire de liaison C-H (Schéma 22, b).51 L’utilisation d’un ligand phosphine volumineux permet d’éviter l’oligomérisation observée par Dyker.
Malgré le développement de différentes méthodes pour l’activation et la fonctionnalisation de liaison C-H, la conception de conditions permettant l’activation catalytique de liaisons C(sp3)-H inactives n’était pas encore bien connue. Dans ce contexte, les travaux de l’équipe de McDonald et Shaw52 représentent le premier exemple d’une activation de liaison C(sp3)-H dirigée en position β du groupement directeur. Les auteurs ont décrit la palladation d’oxime en présence de quantité stœchiométrique de NaPdCl4. Ils ont, en outre, réussi à isoler et caractériser le palladacyle qui s’est avéré être un complexe dimérique (Schéma 23).
Schéma 23. Activation stœchimétrique de liaison C-H dirigée en positon β.
Au fil des ans plusieurs exemples de métallation de ce type ont été décrits. Mais il a fallu attendre le début des années 2000 pour voir le premier exemple catalytique d’une telle réactivité.
Le groupe de Sanford53 a rapporté la réaction d’acétoxylation de liaison C(sp3)-H en utilisant du Pd(OAc)2 comme catalyseur et PhI(OAc)2 comme oxydant. Cette réaction est possible grâce à l’utilisation d’un GD de type O-méthyle oxime ou pyridine (Schéma 24). Après l’étape de
51 Baudoin, O.; Herrbach A.; Guéritte, F. Angew. Chem. Int. Ed. 2003, 42, 5736-5740; (b) Hitce, J.; Retailleau P.; Baudoin,
O. Chem. Eur. J. 2007, 13, 792-799; (c) Chaumontet, M.; Piccardi, R.; Audic, N.; Hitce, J.; Peglion, J.-L.; Clot E.; Baudoin, O. J. Am. Chem. Soc. 2008, 130, 15157-15166.
52 Constablew, A. G.; McDonald, A.; Sawkins, L. C; Shaw, B. J. Chem. Soc., Chem. Commun. 1978, 1061-1062. 53 Desai, L. V.; Hull, K. L.; Sanford, M. S. J. Am. Chem. Soc. 2004, 126, 9542-9543.
31
cyclopalladation dirigée suivant un mécanisme de type AMLA/CMD, PhI(OAc)2 oxyde le complexe de Pd(II) en Pd(IV). Enfin, une étape d’élimination réductrice permet de former la liaison C-O et régénérer le catalyseur de Pd(II).
Schéma 24. Activation catalytique de liaison C-H dirigée en positon β.
Juste après, Daugulis a décrit l’arylation sélective de la 2-éthylpyridine sur la liaison C(sp3)-H terminale.54 Les mêmes auteurs ont ensuite décrit la même réactivité en utilisant une approche légèrement différente. En effet, l’introduction d’un GD bidente (pyridine/amide) a permis l’activation de liaison C(sp3)-H en position β et γ. Cette réaction est possible par l’utilisation de Pd(OAc)
2 comme catalyseur en présence d’acétate d’argent (Schéma 25). Le mécanisme proposé pour cette réaction fait intervenir une étape d’activation C-H par une approche de type AMLA/CMD. Le complexe de Pd(II) ainsi formé subit une addition oxydante par le p-iodoanisole pour donner un complexe de Pd(IV). Ce dernier subit une élimination réductrice pour donner le produit d’arylation et le complexe I-Pd-OAc. La présence du sel d’argent permet d’extraire l’anion iodure et régénérer l’espèce catalytique par échange de ligands.
Schéma 25. Activation catalytique de liaison C-H dirigée par un groupement directeur bidentate.
D’autre part, le groupe de Yu a contribué largement à ce domaine depuis 2005 et ses travaux sur l’iodation et l’acétoxylation diastéréosélective d’oxazoline.55 Les auteurs se sont intéressés à la
54 (a) Shabashov, D.; Daugulis, O. Org. Lett. 2005, 7, 3657-3659; (b) Zaitsev, V. G.; Shabashov, D.; Daugulis, O. J. Am. Chem. Soc. 2005, 127, 13154-13155.
55 (a) Giri, R.; Chen, X.; Yu, Q. Angew. Chem. Int. Ed. 2005, 44, 2112-2115; (b) Giri, R.; Liang, J.; Lei, G.; Li,
32
réaction de couplage croisé impliquant une liaison C(sp3)-H. Ils ont réussi à développer une méthode permettant l’alkylation de liaison C(sp3)-H dirigée par un noyau pyridine en combinant une étape d’activation C-H avec une étape de transmétallation avec des organoboranes (Schéma 26).56 Le mécanisme de cette réaction commence donc par une activation de liaison C-H après complexation du palladium à la pyridine. Ensuite, une séqence de transmétallation avec le partenaire boronique et une élimination réductrice donne le produit de couplage et le Pd(0). Ce dernier est réoxydé en Pd(II) en présence de sel d’argent et de la benzoquinone.
Schéma 26. Alkylation palladocatalysée en présence d’organoboranes.
4. Application de l’activation C-H en synthèse totale
Comme mentionné au début de ce chapitre, un des objectifs majeurs du développement de l’activation de liaison C-H est de pouvoir faciliter l’accès à des molécules complexes à partir de molécules simples et abondantes. En parallèle de ces découvertes, plusieurs équipes se sont intéressées à l’application et à la mise en valeur de ce type de réactivité. En effet, plusieurs synthèses de produits naturels ont été décrites dont une des étapes invoque une activation C-H. Certains de ces produits ont des structures simples, l’application d’une stratégie basée sur l’activation C-H ayant pour objectif de démontrer son utilité. D’autres part, même si l’utilisation de l’activation C-H n’a pas permis d’améliorer, de manière significative, certaines synthèses traditionnelles, son application et son efficacité ont été prouvées pour la synthèse de différentes molécules complexes.57
Par exemple, l’équipe de Du Bois58 a décrit la synthèse de la tétrodotoxine en utilisant deux étapes faisant intervenir une activation C-H. La première étape se base sur la formation d’une liaison C-C d’une diazocétone par catalyse au rhodium, tandis que la deuxième étape fait intervenir la formation d’une liaison N-C par l’intermédiaire d’un complexe Rh-nitrène. Cette stratégie a permis de réduire le nombre d’étapes de 67 à 32 (Schéma 27).
56 Chen, X.; Goodhue, C. E.; Yu, J.-Q. J. Am. Chem. Soc. 2006, 128, 12634-12635.
57 Yamaguchi, J.; Yamaguchi, A. D.; Itami, K. Angew. Chem. Int. Ed. 2012, 51, 8960-9009. 58 Hinman, A.; Du Bois, J. J. Am. Chem. Soc. 2003, 125, 11510-11511.
33
Schéma 27. Synthèse de la (-)-tétrodotoxine.
En 2008, l’équipe de Gaunt a rapporté la synthèse de la rhazinicine.59 Les auteurs ont mis au point une stratégie reposant sur une borylation par activation C-H catalysée par un complexe d’iridium(I), puis une réaction intramoléculaire de type Fujiwara-Moritani palladocatalysée (Schéma 29).
Schéma 28. Synthèse de la rhazinicine.
Plus récemment, l’équipe de Baran a décrit la synthèse des pipérarbornines B et D, deux molécules à base d’un noyau de cylcobutane.60 La méthode utilisée par les auteurs repose sur deux étapes d’arylation par activation C-H palladocatalysée (Schéma 28).
59 Beck, E. M.; Hatley, R.; Gaunt, M. J. Angew. Chem. Int. Ed. 2008, 47, 3004-3007. 60 Gutekunst, W. R.; Baran, P. S. J. Am. Chem. Soc. 2011, 133, 19076-19079.
34
Schéma 29. Synthèse de la pipérarborenine B.
IV. Conclusion
Comme nous venons de le voir, plusieurs méthodes existent pour le contrôle de la sélectivité lors de l’étape d’activation C-H. En effet, les différents exemples que nous avons présentés ne sont qu’une petite sélection de ce que les chimistes ont réussi à faire dans ce domaine. Cependant, les réactions possédant une large tolérance pour les groupes fonctionnels restent très limitées. La plupart des méthodes développées nécessitent l’introduction d’un groupement directeur ou d’un groupement protecteur et sont généralement dépendantes des substrats.
Dans notre équipe, une des thématiques de recherche sur lesquelles nous travaillons est la découverte de nouvelles méthodes de fonctionnalisation par activation de liaison C-H. La suite de ce manuscrit est divisée en deux parties : une première portant sur les travaux réalisés sur l’allylation et l’oléfination des azines N-oxyde ; dans la deuxième partie, nous allons présenter nos travaux sur l’application de la réaction de Murai sur le 2-pyrrole carboxaldéhyde. Dans chaque chapitre, nous commencerons par décrire l’état de l’art, les exemples les plus pertinents sur la fonctionnalisation de ce type d’hétérocycles et la réactivité exploitée, avant de présenter les résultats obtenus.
35
Chapitre II :
Oléfination et allylation de la pyridine
N-oxyde par activation C-H
37
I. Introduction
Figure 3. Structures de la pyridine et de la pyridine N-oxyde.
La pyridine est un motif hétérocyclique d’importance majeure en chimie organique, et plus particulièrement dans la chimie du vivant ainsi qu’en chimie médicinale (Figure 3). Plus spécifiquement, on retrouve ce motif dans plusieurs produits naturels, cofacteurs, ainsi que dans un grand nombre de principes actifs (Figure 4).61 Ce n’est que logiquement que les chimistes se sont intéressés à cette molécule et au développement de nouvelles méthodes de sa fonctionnalisation, à la fois plus efficaces et plus éco-compatibles pour la construction de structures plus complexes.
Figure 4. Structures de certaines molécules contenant le motif pyridine.
D’un point de vue structurel, la pyridine peut être considérée comme l’équivalent du benzène où un groupement « CH » est remplacé par un atome d’azote. Les principales différences entre ces deux cycles aromatiques sont : 1) la présence d’un doublet non liant dans le plan du cycle, à la place d’un
61 (a) Pelttari, E.; Matikainen, J.; Elo, H. Z. Naturforsch. 2002, 57c, 548-552; (b) Altaf, A. A.; Shahzad, A.; Gul, Z.;
38
atome d’hydrogène, qui n’est pas impliqué dans l’aromaticité de la molécule ; 2) la diminution de la longueur des liaisons C=N ; 3) la présence d’un dipôle permanent ainsi que l’appauvrissement électronique du système π du fait de l’électronégativité de l’azote en comparaison au carbone (Figure 5).62
La présence de l’atome d’azote induit une polarisation qui stabilise les structures limites dans lesquels l’atome d’azote est chargé négativement, ce qui implique à son tour la présence de charges positives partielles sur les atomes de carbone en positions 2, 4 et 6. De ce fait, la pyridine fait partie des hétérocycles aromatiques à noyau électron-appauvri ou π–déficient. De plus, les structures limites, montrent que les positions 2, 4 et 6 sont des sites d’attaques nucléophiles, alors que les positions 3 et 5 sont des sites à caractère faiblement électrophile.
Figure 5. Strucutres limites significatives de la pyridine.
D’autre part, la pyridine N-oxyde (PNO) représente aussi une molécule de grand intérêt. On retrouve ce motif dans la structure de plusieurs principes actifs (Figure 4). L’oxydation de la pyridine en PNO augmente le moment dipolaire de 2.22 D à 4.25 D. En plus, la présence du motif N-oxyde a un impact important sur la réactivité de la molécule, car, en fonction des conditions réactionnelles, les positions 2, 4 et 6 peuvent être des sites électrophiles ou nucléophiles (Figure 6).
Figure 6. Structures limites significatives de la PNO.
Il est à noter que la fonctionnalisation de la pyridine ne donne pas seulement accès à des dérivés de pyridine, mais aussi à des dihydro- et tetrahydropyridines, ainsi que des pipéridines.63 Dans la suite de ce chapitre, nous allons nous intéresser dans un premier temps à la réactivité classique de la pyridine et de la pyridine N-oxyde. Ensuite, nous présenterons certains exemples portant sur la fonctionnalisation catalytique de la pyridine ou de la pyridine N-oxyde faisant intervenir une étape d’activation C-H.
62 Joule, J. A.; Mills, K. Heterocyclic Chemistry, Wiley, 5th edition, 2010.
39
II. Réactivité générale de la pyridine et de la pyridine N-oxyde
En comparaison au benzène, la substitution électrophile sur la pyridine (positions 3 et 5) est beaucoup plus difficile, alors que la substitution nucléophile (positions 2, 4 et 6) est plus favorable. En plus, la réaction d’un électrophile sur l’atome d’azote est assez facile.
1. Réactivité classique de la pyridine
1.1. Réaction sur l’azote
Le doublet libre de l’azote n’étant pas impliqué dans l’aromaticité de la pyridine, il peut se comporter de manière similaire à celui des amines tertiaires en termes de basicité et de nucléophilie. En 1965, l’équipe d’Olah décrit la nitration et la nitrosation de la pyridine en la faisant réagir respectivement avec du tetrafluoroborate de nitronium et de nitrosium (Schéma 30).64 Le N-nitropyridinium peut être à son tour utilisé pour le transfert du groupement nitro vers le benzène ou le toluène.65
Schéma 30. N-nitration et nitrosation de la pyridine.
La pyridine peut aussi réagir avec des halogénures d’acyle ou de sulfonyle.66 Les sels d’acyl- ou de sulfonyl-pyridinium ainsi formés peuvent être isolés, ou bien réagir in situ pour la conversion d’alcools en esters d’acides carboxyliques ou sulfoniques, et d’amines en carbox- et sulfonamides. D’autres réactions peuvent avoir lieu sur l’atome d’azote telles que l’amination, la sulfonation, l’halogénation et l’alkylation.62
1.2. Réactivité des atomes de carbone
Comme nous l’avons mentionné, la nitration de la pyridine est très difficile. Par exemple, le traitement avec KNO3/H2SO4 à 300 °C ne donne que 6% de 3-nitropyridine.62 D’autre part,
64 Olah, G. A.; Olah, J. A.; Overchuk, N. A. J. Org. Chem. 1965, 30, 3373-3376.
65 Olah, G. A.; Narang, S. C.; Olah, J. A.; Pearson, R. L.; Cupas, C. A. J. Am. Chem. Soc. 1980, 102, 3507-3510. 66 King, J. A.; Bryant, G. L. J. Org. Chem. 1992, 57, 5136-5139.
40
l’utilisation de pyridine 2,6-dihalosubstituées en présence de tetrafluoroborate de nitronium, inhibe la nitration sur l’azote et permet d’obtenir la 3-nitropyridine avec un bon rendement (Schéma 31).67
Schéma 31. C-nitration de la pyridine.
D’autre part, la réaction d’halogénation est plus facile à réaliser. Ainsi, la réaction entre la pyridine et le dibrome dans l’oléum permet d’obtenir la 3-bromopyridine avec un très bon rendement.62 La 3-chloropyridine peut être préparée directement en présence du dichlore à 200 °C ou en présence d’AlCl3 à 100 °C (Schéma 32).68
Schéma 32. Halogénation de la pyridine.
Les substitutions nucléophiles aromatiques sont possibles sur la pyridine, en particulier dans le cas des halopyridines. Comme pour toutes les SNAr, la facilité de ces réactions dépend de l’électronégativité de l’atome d’halogène et non pas de l’aptitude nucléofuge de l’halogénure.69 En général, les 4-halopyridines sont plus réactives que les 2-halopyridines, tandis que les 3-halopyridines sont très peu réactives.
Parmi les réactions de substitution nucléophile sur les pyridines non halo-substituées, la réaction de Chichibabin est sans doute la plus connue. Cette réaction, qui consiste dans le déplacement d’un anion hydrure en position 2 par un anion amidure, est accomplie par traitement de la pyridine avec de l’amidure de sodium. C’est un mécanisme d’addition-élimination qui est généralement proposé pour cette réaction. L’anion amidure attaque la pyridine en position 2 pour former l’intermédiaire de type Meisenheimer. Ensuite, l’hydrure est déplacé et l’aromaticité est régénérée après déprotonation de l’amine (Schéma 33).70
67 Duffy, J. L.; Laali,, K. K. J. Org. Chem. 1991, 56, 3006-3009.
68 Pearson, D. E.; Hargrove, J. W. W.; Chow, K. T.; Suthers, B. R. J. Org. Chem. 1961, 26, 789-792. 69 Fier, P. S.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136, 10139-10147.
41
Schéma 33. Réaction de Chichibabin.
De manière analogue, en présence d’organo-lithiens, dans la plupart des cas, la substitution nucléophile intervient en position 2. Giam et Stout71 ont montré que cette réaction passe par un mécanisme de type addition-élimination : l’étape d’addition donne le sel de lithium de la dihydropyridine puis l’élimination de l’hydrure de lithium donne le produit d’alkylation ou d’arylation en partant respectivement d’un alkyle ou d’un aryle de lithium (Schéma 34).
Schéma 34. Substition nucléophile en présence d’organolithiens.
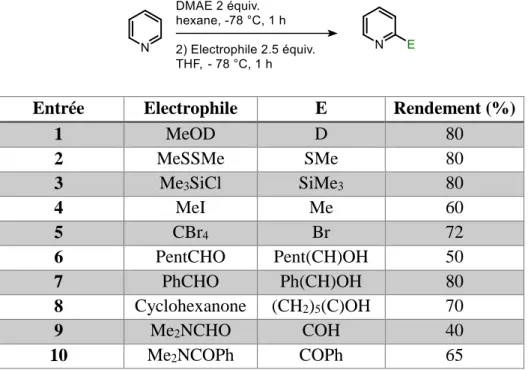
Par ailleurs, la métallation directe des pyridines en position 2 est possible en présence d’un mélange de base. En effet, l’équipe de Caubère et Fort72 a montré que le couple n-BuLi/Me2N(CH2)2OLi (LiDMAE), formé in situ entre du n-BuLi et du diméthylaminoéthanol (DMAE), permet de synthétiser des pyridines substituées en position 2 avec de bons rendements. Plusieurs électrophiles tels que l’iodure de méthyle, le benzaldéhyde et la cyclohexanone ont été utilisés pour piéger l’espèce lithiée (Tableau 2).
71 Giam, C. S.; Stout, J. L. Chem. Commun. 1969, 142.
72 (a) Gros, P.; Fort, Y.; Caubère, P. J. Chem. Soc. Perkin Trans. 1 1997, 3597-3600; (b) Gros, P.; Fort, Y. Eur. J. Org. Chem. 2002, 3375-3383.