Le gène suppresseur de métastases Growth
Arrest-Specific 1 (Gas1) est réprimé épigénétiquement
chez le mélanome métastatique
Mémoire
Jimmy Savard-Lalancette
Maîtrise en Biologie Cellulaire et Moléculaire
Maître ès sciences (M. Sc.)
Québec, Canada
iii
Résumé
Nous avons récemment découvert que le gène Gas1 agit comme un suppresseur de métastases chez le mélanome. Ce gène pourrait être utilisé pour d’éventuelles thérapies contre la métastase. Afin de découvrir le mécanisme réprimant l’expression de Gas1 chez les cellules métastatiques, nous avons utilisé des ChIP, des inhibiteurs chimiques et des petits ARN interférents contre des molécules susceptibles de réprimer Gas1. Chez le mélanome métastatique murin B16-F10, nous avons observé plusieurs groupements méthyles sur l’îlot CpG du promoteur de Gas1, ainsi que la marque de répression H3K9me3
et l’absence de la polymérase II. Chez les cellules humaines C8161 et Lox-IMVI, nous avons découvert qu’il y avait une forte présence d’acétylation au niveau de l’histone 3 et de polymérase II. Par ailleurs, nous avons prouvé que c-MYC et BRD4 étaient impliqués dans la régulation de GAS1. Finalement, ces résultats pourront être utilisés pour développer d’éventuelles thérapies contre la métastase.
v
Table des matières
Résumé ... iii
Table des matières ... v
Liste des tableaux ... ix
Liste des figures ... xi
Liste des abréviations ... xiii
Remerciements ... xv
1. Introduction ...1
1.1. Le cancer ...1
1.2. Les métastases ...1
1.3. Les gènes suppresseurs de métastases ...2
1.4. Le gène Gas1 ...4
1.4.1. Gas1 et le cycle cellulaire ... 4
1.4.2. Les caractéristiques de Gas1 ... 5
1.4.3. Gas1 et les voies de signalisation GDNF et SHH ... 7
1.4.4. Gas1 et l’apoptose ... 8
1.4.5. Gas1, un gène suppresseur de métastases ... 11
1.4.6. Gas1, un gène à plusieurs rôles ... 12
1.4.7. La régulation de Gas1 ... 13
1.5. La régulation épigénétique ...14
1.5.1. Le code des histones ... 15
1.5.1.1. Les marques activatrices ...15
1.5.1.2. Les marques répressives ...16
1.5.1.3. Les régulateurs épigénétiques ... 17
1.5.2. La pause transcriptionnelle ... 18
1.5.3. La poly(ADP-ribosyl)ation ... 19
1.5.4. Les microARNs ... 20
1.6. L’oncogène c-Myc ...20
1.6.1. Le rôle de répresseur génique de c-Myc ... 21
1.7. Problématique et implications scientifiques de l’étude du gène Gas1 ...25
2. Méthodologie ... 27
2.1. Culture cellulaire ...27
2.2. Les vecteurs d’expression ...27
2.3. Traitements chimiques cellulaires ...28
2.4. Extraction d’ARN ...28
2.5. Dosage d’ARN ...28
2.6. Réaction de transcription inverse ...28
2.7. Réaction en chaîne par polymérase quantitative ...29
2.8. Immunoprécipitation de la chromatine ...29
2.8.1. Fixation et récolte cellulaire ... 29
2.8.2. Lyse cellulaire ... 30
2.8.3. Sonication ... 30
2.8.4. Immunoprécipitation ... 30
2.8.5. Préparation de l’ADN ... 31
2.8.6. Isolation de l’ADN ... 31
vi
2.8.8. Vérification de la sonication ... 32
2.9. Clonage moléculaire ... 32
2.9.1. Réaction en chaîne par polymérase ... 32
2.9.2. Gel d’agarose ... 33 2.9.3. Purification d’ADN ... 34 2.9.4. Digestion enzymatique ... 34 2.9.5. Déphosphorylation de vecteurs ... 34 2.9.6. Ligation ... 34 2.9.7. Transformation bactérienne ... 35
2.9.8. Extraction plasmidique de petites quantités (Miniprep) ... 35
2.9.9. Séquençage ... 35
2.9.10. Extraction plasmidique de moyennes quantités (Midiprep) ... 35
2.10. Extraction d’ADN génomique ... 36
2.11. Conversion d’ADN génomique au bisulfite de sodium ... 36
2.12. Répression d’ARNm par petits ARN interférents ... 37
2.12.1. Production des plasmides lentiviraux pLKO.1 ... 37
2.12.2. Production des lentivirus pLKO.1 ... 37
2.12.3. Transduction des lentivirus pLKO.1 ... 37
2.13. Essai luciférase ... 37
3. Résultats ... 39
3.1. Le mélanome murin ... 39
3.1.1. Gas1 est réprimé chez les cellules de mélanome murin B16-F10 ... 39
3.1.2. L'expression de Gas1 est modulable chimiquement par la TSA et le 5-Aza chez le mélanome métastatique murin B16-F10 ... 41
3.1.3. Cbx1 est impliqué dans la répression épigénétique du gène Gas1 chez les cellules de mélanome métastatique murin B16-F10 ... 42
3.2. Le mélanome humain ... 43
3.2.1. L'expression de GAS1 est réprimé chez les cellules de mélanome métastatique humain C8161 et Lox-IMVI ... 43
3.2.2. L'expression de GAS1 n'est pas modulable par la TSA et le 5-Aza chez les cellules C8161 et Lox-IMVI ... 44
3.2.3. L’expression de GAS1 n'est pas contrôlée par microARNs ... 46
3.2.4. L’expression de GAS1 est contrôlée par les gènes BRD4 et c-MYC chez les cellules de mélanome humain C81-61, C8161 et Lox-IMVI ... 47
3.2.5. La répression de GAS1 n'implique pas les complexes c-MYC/MAX/MIZ1 ou c-MYC/TFAP2C/KDM5B ... 50
3.2.6. La régulation de GAS1 implique d'autres protéines comme les PARPs ou BMI-1 52 3.3. Le cancer du sein murin ... 53
3.3.1. Gas1 est réprimé chez les cellules de cancer du sein murin 4T1 ... 53
4. Discussion ... 55
4.1. Les lignées cellulaires murines ... 55
4.1.1. Gas1 est réprimé chez les lignées cellulaires métastatiques murines B16-F10 55 4.1.2. L'expression de Gas1 est modulable chimiquement par la TSA et le 5-Aza chez la lignée cellulaire métastatique murine B16-F10 ... 56
vii 4.1.3. Cbx1 est impliqué dans la répression du gène Gas1 chez la lignée cellulaire
métastatique murine B16-F10 ... 59
4.1.4. Gas1 est réprimé chez les lignées cellulaires murines 4T1 ... 61
4.1.5. Conclusion ... 62
4.2. Les lignées cellulaires humaines ...62
4.2.1. GAS1 est réprimé chez les cellules métastatiques de mélanome humain C8161 et Lox-IMVI ... 62
4.2.2. GAS1 n'est pas régulé par les HDACs ou les DNMTS chez les cellules métastatiques C8161 et Lox-IMVI ... 63
4.2.3. L’expression du GAS1 est contrôlée par les gènes BRD4 et c-MYC chez les cellules métastatiques de mélanome humain C8161 et Lox-IMVI ... 67
4.2.4. Conclusion ... 70
5. Conclusion générale et perspectives ... 73
6. Références ... 77
7. Annexes ... 97
7.1. Annexe un - Candidats criblés par petits ARN interférents ... 97
7.2. Annexe deux - Séquence des constructions 3'UTRhGAS1-A.1 et B.1 ... 99
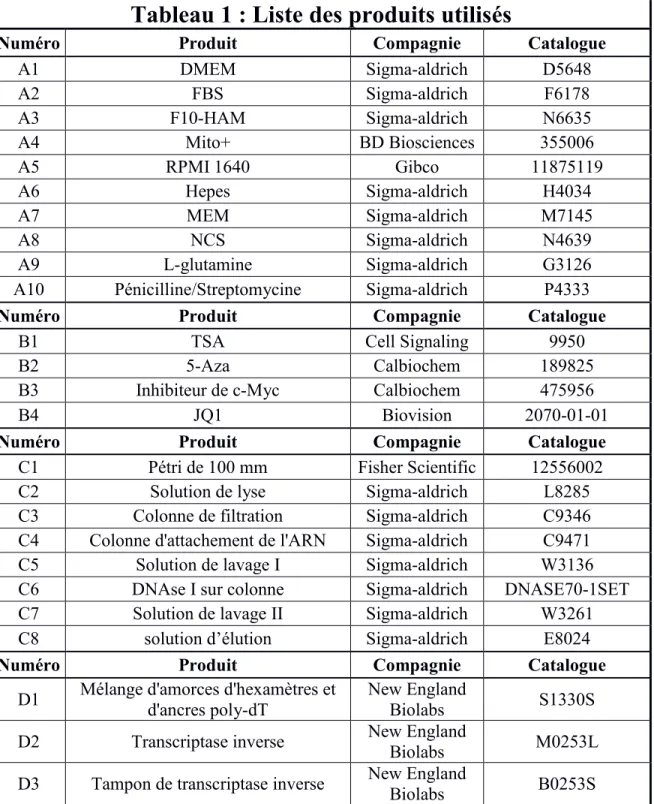
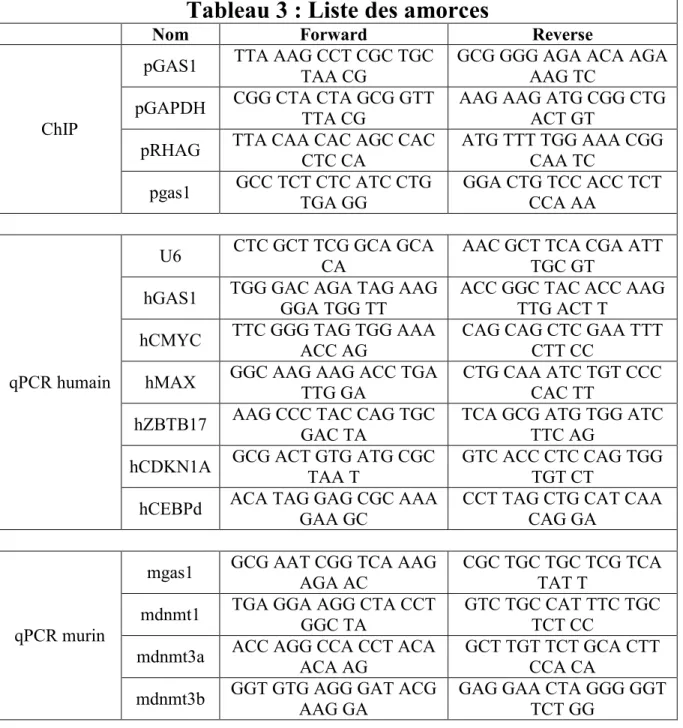
7.3. Annexe trois - Liste des produits, des anticorps et des amorces utilisés ... 101
ix
Liste des tableaux
Tableau 1 : Liste des produits utilisés...101 Tableau 2 : Liste des anticorps...104 Tableau 3 : Liste des amorces...105
xi
Liste des figures
Figure 1 : Mécanisme d'apoptose induite par les récepteurs Gas1 et Ret...9
Figure 2 : Métastases in vivo issues d'une injection de cellules B16-F10...12
Figure 3 : Mécanisme de répression de la protéine Dnmt1 par PARylation...20
Figure 4 : Différents mécanismes de répression employés par l'oncoprotéine c-Myc...23
Figure 5 : Mécanisme de pause transcriptionnelle induite par c-Myc...24
Figure 6 : Gas1 est réprimé chez les cellules B16-F10...40
Figure 7 : Gas1 est modulable chimiquement chez les cellules B16-F10 par la TSA et le 5-Aza...42
Figure 8 : Le gène Cbx1 est impliqué dans la répression de Gas1...43
Figure 9 : GAS1 n'est pas modulable chimiquement par la TSA ou le 5-Aza chez les lignées métastatiques C8161 et Lox-IMVI...44
Figure 10 : Le promoteur de GAS1 sont fortement acétylées au niveau des histones et est caractérisé par la présence de polymérase...45
Figure 11 : GAS1 n’est pas contrôler par microARNs...46
Figure 12 : BRD4 régule directement le gène GAS1 chez la lignée faiblement métastatique C81-61...48
Figure 13 : c-MYC est impliqué dans la répression du gène GAS1 chez les cellules métastatiques C8161 et Lox-IMVI...49
Figure 14 : c-MYC réprime directement le gène GAS1 chez les lignées métastatiques C8161 et Lox-IMVI...50
Figure 15 : Le complexe c-MYC/MAX/MIZ1 n’est pas impliqué dans la répression du gène GAS1 chez les C8161...51
Figure 16 : La répression de GAS1 n’implique pas les facteurs de transcription KDM5B, PARG ou NELF...52
Figure 17 : Gas1 est réprimé chez les cellules métastatiques murines du cancer du sein 4T1...54
Figure 18 : Schéma récapitulatif des mécanismes de répression du gène GAS1...74
Figure 19 : Candidats transduits par petits ARN interférents chez les cellules C8161...97
Figure 20 : Clonage moléculaire de la région 3’UTR du gène GAS1...99
Figure 21 : Vecteur pGEM-T Easy...107
Figure 22 : Vecteur pLKO.1...108
xiii
Liste des abréviations
5-Aza 5-Aza-2'-deoxycytidine
Ac Acétyl
Acta2 Alpha-actin-2
ADN Acide Désoxyribonucléique
ADNc ADN complémentaire
ARN Acide Ribonucléique
ARNm ARN messager
BRD4 Bromodomain-containing Protein 4
C/EBP-delta CCAAT/Enhancer Binding Protein-delta
Cggbp1 CGG Triplet Repeat-binding Protein 1
CTCF CCCTC-binding Factor
Ctso Cathepsin O
DEPC Diethylpyrocarbonate
Dmp1 Dentin Matrix Acidic Phosphoprotein 1
Dnmt DNA methyltransferase
DSIF DRB Sensitivity Inducing Factor
ERK Extracellular Signal-regulated Kinases
FBS Fetal Bovine Serum
Gas1 Growth Arrest-Specific 1
GDNF Glial Cell Line-derived Neurotrophic Factor
GFR GDNF Family Receptor Gly Glycine GPI Glycophosphatidylinositol H3K4 Histone 3 Lysine 4 H3K9 Histone 3 Lysine 9 H3K14 Histone 3 Lysine 14 H3K27 Histone 3 Lysine 27 H3K36 Histone 3 Lysine 36 H4K20 Histone 4 Lysine 20
HAT Histone acétyltransférase
Hdac Histone déacétylase
HP1 Heterochromatin Protein 1
HSP70 Heat Shock Protein 70
IgG Immunoglobulin G
kDa Kilo Dalton
KMT Histone Demethylase
LSD1 Lysine (K)-specific demethylase 1A
xiv
MKK4 Mitogen-activated Protein Kinase Kinase 4
NELF Negative Elongation Factor
NF-kb Nuclear Factor-Kappa B
NMDA N-methyl-D-Aspartic Acid
ORF Open Reading Frame
PARG Poly ADP Ribose Glycohydrolase
PARP Poly ADP Ribose Polymerase
PCR Polymerase Chain Reaction
PEI Polyethylenimine
PEITC Phenethylisothiocyanate
RET Rearranged During Transfection
PI3-K Phosphoinositide 3-Kinase
Pol II Polymérase II
PRC Polycomb Repressive Complex
Rkip Raf Kinase Inhibitor Protein
Ser Sérine
SHH Sonic Hedgehog
shRNA Small Hairpin RNA
SIRT Sirtuin
TGF-beta Transforming Growth Factor Beta
TSA Trichostatin A
xv
Remerciements
Je voudrais souhaiter tous mes remerciements à l’équipe du Dr. Stéphane Gobeil. Au travers de deux années de maîtrise, vous avez tous su m’aider et apporter votre grain de sel dans une excellente équipe de travail. Je vous en suis très reconnaissant.
Pour le Dr. Gobeil, je suis très heureux d’avoir été le premier étudiant à la maîtrise au sein de votre laboratoire. Avec une belle complicité, nous avons réussi à développer une méthodologie de travail très intéressante et stimulante. De plus, je voudrais vous remercier pour toutes les connaissances et les techniques scientifiques que vous m’avez transmises. Vous avez su tirer le meilleur de moi-même afin de toujours repousser mes limites et atteindre mes objectifs. Merci beaucoup!
Pour tous les étudiants ou professionnels de recherche avec qui j’ai travaillé, vous êtes parvenu à enrichir mes semaines au laboratoire. Louis-Jean a toujours été à disposition pour m’enseigner les voies les plus rapides pour résoudre mes problèmes, en plus d’être un excellent partenaire de laboratoire. Merci aussi à Yann, Rosette et Josie pour votre bonne humeur quotidienne. Un gros merci à Coraline et Olivier pour votre aide dans mon projet de maîtrise, mais aussi pour votre agréable compagnie.
Également un gros merci pour tous les stagiaires avec lesquels j’ai travaillé, ce fût un plaisir. Finalement, j’aimerais souhaiter merci à ma conjointe qui a toujours été là pour m’encourager et me soutenir, tout comme notre famille. J’ai été ravi de travail au sein de l’équipe du Dr. Gobeil et je vous souhaite un très bel avenir!
1
1.
Introduction
1.1. Le cancer
Le cancer est défini comme un désordre génétique causant une multiplication cellulaire anarchique. Tout débute par quelques mutations au niveau de l’ADN. Ces dernières peuvent être spontanées ou héréditaires. Dans la plupart des cas, les mutations spontanées sont induites par des expositions aux composés mutagènes. Suite à l’accumulation de mutations, la cellule peut perdre le contrôle de sa croissance et devenir tumorale197.
Le cancer est donc défini par dix caractéristiques importantes pour sa progression, soit l’immortalité, l’évitement du système immunitaire, l’évasion des suppresseurs de la croissance, le maintient de signaux de survie, la dérégulation énergétique, la résistance à la mort cellulaire, l’instabilité du génome, l’angiogénèse, la promotion inflammatoire et l’activité métastatique110. Le cancer touche donc plusieurs processus cellulaires et est d’autant plus très complexe à étudier.
Malheureusement, le cancer est devenu la première cause de décès en Amérique du Nord, dépassant les maladies cardiovasculaires202. Il est donc vital d’accroître les connaissances sur le cancer pour contribuer à la mise en place de nouvelles thérapies pour le vaincre.
1.2. Les métastases
Responsables de 90% des décès issus d’une tumeur primaire, les métastases sont l’étape avancée du cancer69. Fréquemment retrouvées aux os, au foie, aux poumons ou au cerveau, les cellules métastatiques devront traverser une série d’étape pour y parvenir, appelée la cascade métastatique69-201. Par conséquent, les cellules doivent envahir l’environnement autour de la tumeur primaire, procéder à l’intravasation dans le système de circulation, être transportées jusqu’au site secondaire, effectuer l’extravasation dans le tissu et finalement former la métastase201. En d’autres mots, les cellules métastatiques devront accumulées des modifications qui accroîtront leur pouvoir métastatique afin de parvenir à franchir toutes les étapes de la cascade métastatique. Parmi ces modifications, la perte de gènes suppresseurs de métastases a été démontrée comme très importante pour faciliter le passage vers les sites secondaires. Par exemple, la répression de l’E-Cadherin permet de réduire l’attachement
2
aux cellules entourant la tumeur primaire pour augmenter l’invasion et la migration vers le second site113. Au final, étant donné la mortalité associée aux métastases, il apparait impératif d’étudier en détail les mécanismes l’entourant afin de développer de nouveaux outils thérapeutiques contre la métastase.
1.3. Les gènes suppresseurs de métastases
Les métastases sont la principale cause de décès chez les patients atteints du cancer. Par exemple, chez le cancer du sein, ces tumeurs secondaires causeront la plupart des décès69. Les patients ayant été opérés pour la tumeur primaire devront être suivis jusqu’à dix ans afin de déceler rapidement l’apparition de métastases. Cependant, plusieurs ne survivront pas aux colonies secondaires111, d’où l’importance d’amener de nouveaux outils contre les métastases. C’est dans ce contexte que les gènes suppresseurs de métastases sont de plus en plus étudiés.
Tout d’abord, il faut souligner le potentiel important pour combattre les colonies secondaires grâce aux gènes suppresseurs de métastases. Par exemple, notre laboratoire a démontré au cours d’essais in vitro et in vivo qu’il était possible de réduire le nombre de métastases pulmonaires, causées par des cellules de mélanome64. Il est important de mentionner qu’afin d’obtenir le titre de gène suppresseur de métastases, le gène doit réduire la capacité de formation de colonies secondaires in vivo, sans toutefois affecter la capacité tumorigène de la cellule111-112. Ceci démontre bien la possibilité d’utiliser ces gènes comme outil thérapeutique contre la métastase.
Par ailleurs, plusieurs publications s’accordent à décrire les gènes suppresseurs de métastases comme de bons biomarqueurs de la progression métastatique111. En effet, des études ont démontré l’utilité des gènes NM23207 et E-Cadherin208 dans le cadre d’un diagnostique de la tumeur primaire. Ainsi, lorsque ces deux gènes y sont absents, le diagnostique est plutôt mauvais111. Il est donc intéressant d’accroître les connaissances à propos de ces gènes suppresseurs de métastases, autant dans un contexte thérapeutique que diagnostique.
Au final, bien que la liste des gènes suppresseurs de métastases reste restreinte, plusieurs nouveaux gènes comme Gas1, Acta2 ou Ctso64 s’ajoutent aux premiers gènes découverts
3 comme NM23, KAI1, KISS1 ou MKK4111-112. Il est maintenant possible de compter au dessus d’une quarantaine de ces suppresseurs de métastases au sein de la littérature. Afin de bien illustrer leur rôle important en oncologie, voici une description des principaux gènes suppresseurs de métastases :
NM23 : Découvert en 1988, il est le premier gène suppresseur de métastases. Il a été démontré que la perte de ce gène aide à promouvoir la survie, la mobilité et bien d’autres processus cellulaires lors de la cascade métastatique111-112-113.
KAI1 : Dans les premiers gènes identifiés, il a illustré sa capacité à supprimer la formation métastatique chez le cancer de la prostate. Il a été associé à l’accroissement de la capacité d’invasion autour de la tumeur primaire111-112.
RKIP : Également capable d’empêcher la formation métastatique chez le cancer de la prostate, le gène RKIP a été associé à des dérégulations signalétiques au sein des cellules, menant à une augmentation de la croissance, de l’invasion et du pouvoir métastatique112-114.
KISS1 : Découvert chez le mélanome, KISS1 a été démontré comme empêchant la colonisation métastatique en y maintenant les cellules en dormance112.
GAS1 : Découvert par notre laboratoire comme gène suppresseur de métastases, il a été publié comme un frein du cycle cellulaire, en plus d’induire l’apoptose64.
NDRG1 : Ce gène a été montré comme absent chez plusieurs lignées cellulaires métastatiques du cancer du colon. De plus, il a été impliqué dans la capacité d’invasion cellulaire113.
E-Cadherin : L’absence d’expression de ce gène a été prouvée chez le cancer du colon. Il a été démontré que la perte de ce récepteur permettait de diminuer les interactions cellules-cellules afin d’accroitre le potentiel métastatique des cellules tumorales113.
4
1.4. Le gène Gas1
1.4.1. Gas1 et le cycle cellulaire
Le cancer est défini comme une prolifération cellulaire anormale au sein d’un tissu. Au cours des années, plusieurs scientifiques se sont donc penchés sur la question et ont étudié le cycle cellulaire. Il était connu que plusieurs gènes promouvaient le cycle cellulaire. Schneider (Schneider, 1988) avait ainsi émit l’hypothèse que d’autres gènes freinaient le cycle cellulaire. C’est donc en 1988 qu’il découvrit six gènes ayant les capacités d’induire l’arrêt du cycle cellulaire en G0. Il les appela les gènes Growth Arrest-Specific (Gas).60
Parmi ceux-ci, Gas1 a été démontré comme ayant le meilleur rôle de frein du cycle cellulaire.
Poursuivant plus profondément ses études, Schneider (Schneider, 1988) a décrit Gas1 comme modulateur négatif du cycle cellulaire chez les cellules de fibroblastes murines NIH 3T3. Ainsi, il a illustré qu'en situation de sevrage nutritionnel, l'expression de Gas1 s'accroît après trois heures. Il a déterminé que les cellules sont ensuite maintenues en arrêt lors de la phase G0, bloquant l'accès vers les phases G1/S. Par la suite, en stimulant les
fibroblastes avec du sérum nutritionnel, il a établi que les cellules redémarrent le cycle cellulaire et que les niveaux d'expression de Gas1 s’amenuisent. Ensuite, d'autres laboratoires ont confirmé le rôle de frein cellulaire de Gas1 chez les fibroblastes2-16-17-45, les cellules musculaires54 ou les adipocytes10. Également, Evdokiou (Evdokiou, 1998) a utilisé un vecteur inductible au dexamethasone pour obtenir des niveaux de Gas1 plus physiologique chez les 3T3. Il a illustré qu'avec ces conditions de surexpression, la croissance cellulaire était grandement ralentie et qu'il y avait un changement morphologie des cellules. En outre, il a prouvé qu'il était possible de rétablir la croissance cellulaire par un ARN anti-sense contre Gas116. Fait intéressant, un autre scientifique a trouvé de l'ARNm de Gas1 chez des cellules murines de foie en G0, mais également en G1/S, suite à
une hépatotectomie, signe qu’il est possible d’observer une hétérogénéité au sein d’une population des niveaux d'expression de Gas156. En plus, Schneider (Del Sal et Schneider, 1992) a montré que ce gène pouvait être impliqué également lors de l'inhibition de
5 croissance de contact entre les cellules1. En bref, ces expériences ont bien imagé l'implication de Gas1 comme frein du cycle cellulaire chez les cellules de fibroblastes.
Afin de mieux comprendre le rôle de Gas1 au sein du cycle cellulaire, Schneider (Del Sal et Schneider, 1992) a utilisé un vecteur exprimant Gas1 et l’a inséré par microinjection chez les cellules de fibroblastes NIH 3T3. En accord avec son hypothèse, les cellules exprimant le vecteur de Gas1 ont inhibé la croissance cellulaire, en plus de la synthèse d'ADN. Pour valider ces recherches en oncologie, Schneider a introduit son vecteur de Gas1 chez des cellules transformées par un oncogène, soit v-fos, v-myc, v-src, v-ras ou SV40. Ainsi, il a montré que l'effet de Gas1 sur l’arrêt du cycle cellulaire était similaire chez toutes ces cellules transformées, exception faite pour SV401-2 ou par un adénovirus9. Par contre, il a été prouvé qu'une insertion d'un vecteur v-src diminue l'expression de Gas1 chez ces cellules NIH 3T3 non transformées, permettant le passage des cellules de G0 vers la phase S, impliquant donc potentiellement la voie de régulation Src pour ce gène18. Par conséquent, il est intéressant de noter que l’action du gène Gas1 n’est pas seulement visible chez les cellules saines, mais également chez les cellules transformées et potentiellement tumorigéniques.
Compte tenu de cet effet de frein du cycle cellulaire, Del Sal (Del Sal, 1995) a étudié plus en profondeur les raisons derrières ce phénomène, pour finalement conclure que l'arrêt cellulaire chez les 3T3 induite par Gas1 dépendait de p53, un gène inactif chez les cellules transformées par SV40 ou par un adénovirus9. De plus, ce chercheur a démontré que le domaine transactivateur de p53 n'était pas nécessaire pour observer l’effet de frein du cycle cellulaire9, résultat appuyé par un autre groupe qui en plus a montré qu'il pouvait y avoir compétition entre la signalisation de Gas1 et 53BP213. Fait intéressant, un autre groupe a démontré qu'il n'y avait pas d'expression de Gas1 chez les cellules NIH 3T3 transformées par v-mos en situation de privation nutritionnelle. Selon eux, cet effet serait causé par un arrêt des cellules en phase de G1 au lieu de G05. Ces travaux ont permis de démontrer plus en détail le rôle de Gas1 lors du cycle cellulaire.
1.4.2. Les caractéristiques de Gas1
L’existence de Gas1 est connue depuis plusieurs années maintenant. Suite à de nombreux travaux, il a été possible de comprendre plus en détail les caractéristiques entourant la
6
structure de cette protéine, souvent associée à la famille GDNF23-35. Malgré cela, il reste encore beaucoup de recherches à effectuer pour mieux comprendre les utilités des différents domaines présents au cœur de la protéine Gas1.
Pour ce faire, Schneider (Del Sal et Schneider, 1992) a débuté l'étude de Gas1 au niveau de l'ARNm lui-même. Par analyse d'extension d'amorces, il a démontré que l’ARNm de Gas1 murin commence à 14 nucléotides en amont d'un site de restriction BamHI. Son ARNm comporte un 5’UTR de 425 nucléotides ayant trois ATG devant trois sites ORF interrompus par trois codons d’arrêt. De plus, il a déterminé que le nucléotide 425 code pour l’ATG de départ, terminant au nucléotide 1579, pour un total de 384 acides aminés. Par ailleurs, Schneider a trouvé que le 3’UTR était de 1386 nucléotides de long. Or, il a démontré que ce 3'UTR contient deux sites consensus AUUUA, impliqués dans la déstabilisation d’ARNm. Pour sa part, ce scientifique a décrit que la protéine contient une région membranaire entre les acides aminés 38-74. Ensuite, il a indiqué que la région entre les acides aminés 75 et 384 semblait être une partie extracellulaire, car elle contient une séquence semblable à des récepteurs à intégrine, deux sites N-glycosylation et des résidus Ser-Gly, tous caractérisant de manière similaires les parties de domaines extracellulaires1. En bref, Schneider a mis en lumière la similitude de Gas1 avec d’autres récepteurs extramembranaires.
En complément, d'autres caractéristiques de Gas1 ont été publiées au fil des années. En 1993, un laboratoire a démontré que Gas1 était présent sur le chromosome 9 au niveau de la région 9q21.3-q223-6, une région absente chez le syndrome de Gorlin. De plus, d'autres scientifiques ont analysé que le gène GAS1 humain comportait 85 % d'homologie avec le gène murin4. Au niveau de la protéine, Gas1 a été démontré comme ayant un signal peptide et s'illustrant comme étant un ancre-GPI, situé à la membrane22, dans des radeaux lipidiques35, et codant une protéine d’un total de 45 kDa30. En contrepartie, un autre chercheur a prouvé que l'ancre-GPI de Gas1 n'était pas essentiel pour que ce gène soit effecteur. En effet, avec l'aide d'une protéine de Gas1 soluble, l'effet de frein cellulaire de Gas1 restait le même chez les cellules NIH 3T323, mais également chez des cellules mesangiales58. En outre, il est justifié d’affirmer maintenant que le gène Gas1 code une protéine similaire aux récepteurs de la famille GDNF, possédant eux-mêmes un ancre-GPI209.
7
1.4.3. Gas1 et les voies de signalisation GDNF et SHH
Découlant des faits que Gas1 pouvait freiner le cycle cellulaire et induire l’apoptose, des chercheurs se sont penchés sur l’option d’utiliser ce gène comme cible thérapeutique pour le traitement du cancer. Plusieurs laboratoires ont donc cherché à découvrir les partenaires d’interaction de Gas1 pour comprendre les voies de signalisation derrière ces complexes, avec pour objectif d’induire son expression. C’est ainsi que certains laboratoires ont illustré l’implication de la famille GDNF avec le gène Gas1.
Par exemple, Ruaro (Ruaro, 2000) a émis l'hypothèse que Gas1 pouvait interagir au sein de la signalisation GDNF grâce aux ligands GDNF, perséphine, artémine ou neurturine, au récepteur Ret23-35 ou aux co-récepteurs GFR-α ou GFR-β23-27-30-34-37. De même, un chercheur a démontré que Gas1 pouvait réduire la phosphorylation de la tyrosine 1062 du récepteur Ret et en diminuer sa signalisation43. Dans un même ordre d'idée, il a été dévoilé que la structure de Gas1 était similaire aux récepteurs GFR-α, en plus de pouvoir induire la mort cellulaire lorsqu’il y avait une liaison à un ligand GDNF en condition de sevrage nutritionnel35. L'hypothèse que Gas1 pourrait freiner AKT et induire ERK a donc été émise35 et prouvé pour AKT chez le neuroblastome SH-SY5Y43. D'un autre côté, un chercheur a montré l'association de Gas1 avec la voie de signalisation Sonic Hedgehog (Shh) par une co-immunoprécipitation des deux protéines26-41-42-49. Ce résultat coïncide avec celui d'autres chercheurs qui ont démontré que la signalisation de la voie Shh était fortement moins induite chez des souris comportant une ablation de Gas140, en présence d’un ARN anti-sens contre Gas155 ou chez un mutant N115K, limitant l'interaction avec Gas1, à la surface de Shh46. Également, il a été illustré que Gas1 agissait avec Patched1 pour faire la liaison avec Shh lors du développement murin38, avec Cdo39 ou même Boc52. Bien évidemment, tous ces chercheurs ont réussi à prouver que Gas1 semble agir en partenariat pour induire sa signalisation par les voies GDNF ou Shh. Plus précisément, il a même été souligné que Gas1 pouvait joue un rôle au sein des voies de signalisation d’AKT et de ERK, des joueurs importants en oncologie.
8
1.4.4. Gas1 et l’apoptose
Il est maintenant bien connu que Gas1 possède un rôle important lors du mécanisme apoptotique. En effet, dès les années 90, les scientifiques avaient remarqué qu’une surexpression de Gas1 entraînait une mort cellulaire67. Plusieurs ont par la suite tenté de déceler les mécanismes entourant l’apoptose enclenché par Gas1.
Le premier exemple impliquant Gas1 avec l’apoptose a été démontré chez une lignée cellulaire de gliome. En surexprimant la protéine Gas1, les chercheurs ont constaté qu’il y avait une augmentation de l’activité de la caspase-3, un joueur important de l’apoptose, in vitro67. Par la suite, ce même groupe a démontré qu’il était possible avec l’aide d’un rétrovirus exprimant Gas1, d’induire in vivo l’apoptose66. En plus, un autre groupe a publié qu’une version soluble de Gas1 était également capable d’induire l’apoptose61. Également, il a été validé que ce gène cause l'apoptose chez les cellules mammaires11. Il était maintenant clair que Gas1 possédait un rôle lors de la mort cellulaire, lorsque ce dernier est exprimé fortement à l’aide d’un vecteur.
Un autre joueur important impliqué lors de l’apoptose est la protéine p53. En effet, cette molécule est une pierre angulaire de la mort cellulaire, à un point où les cellules tumorales s’en débarrassent régulièrement pour survivre109. Un groupe a donc mis au point un système bicistronique d’expression de Gas1 et p53 grâce à un adénovirus. Ainsi, en infectant des cellules de gliome, ils ont réussi à induire l’apoptose chez elles. Cependant, ils ont constaté que Gas1 seul était suffisant pour obtenir la mort cellulaire65. Ils ont donc démontré que l’apoptose stimulée par Gas1 était indépendant de p53.
D’un autre point de vue, d’autres scientifiques ont démontré une implication de l’apoptose causée par Gas1 via la voie de signalisation de Ret. En effet, ils ont constaté qu’une expression de Gas1 menait à une diminution de l’autophosphorylation du récepteur Ret. En plus, ils ont observé qu’il y avait une déphosphorylation par AKT de la protéine Bad, impliqué dans la répression de l’apoptose via l’inhibition du relâchement du cytochrome C, une molécule essentielle à l’apoptose. Ils ont d’ailleurs enregistré une augmentation du niveau du cytochrome C lors de la surexpression de Gas1, en plus d’une augmentation de
9 l’activité de la caspase-9 et de la caspase-3. Par la suite, ils ont remarqué qu’il n’y avait pas plus d’activité de la caspase-8 (Figure 1). Finalement, ils ont exposé qu’une privation nutritionnelle entraîne une augmentation de Gas1 et cause l’apoptose, mais que s’il y a en plus une inhibition par petits ARN interférents de Gas1, les cellules ne mourraient plus61. Aussi, ce même groupe avait également illustré l’implication des GDNF avec Gas1. En effet, ils ont découvert que si Gas1 est présent, les GDNF ne peuvent plus activer le récepteur Ret43. Par ces expériences, il apparait maintenant plus clair le mécanisme par lequel Gas1 induit l’apoptose chez les cellules gliales.
Par la suite, un autre laboratoire a affirmé l’implication de Gas1 au sein de l’apoptose, mais chez le cancer gastrique. Ils ont illustré qu’une augmentation de Gas1 diminue l’activation de Bad, augmente la phosphorylation de Bax et de la caspase-3 et induit ainsi l’apoptose. Dans une autre expérience, ils ont inhibé l’expression de Gas1, diminuant alors l’activation
10
de la caspase-3 et augmentant la survie cellulaire57. Ce groupe a une fois de plus confirmé le rôle de Gas1 lors de l’apoptose, mais chez un nouveau type de cancer.
Par ailleurs, d’autres chercheurs ont vérifié la possibilité d’un mécanisme d’activation de Gas1 via la voie de signalisation Sonic Hedgehog (Shh). Ils ont prouvé que chez des cellules de glioblastomes et astrocytomes, la surexpression de Gas1 cause l’apoptose sans toutefois passer par la voie de Shh63. Finalement, notre laboratoire a également démontré pour la première fois que Gas1 est impliqué dans l’apoptose par l’activation de la caspase-3, mais chez le mélanome métastatique murin B16-F1064.
En contrepartie, il est important de souligner que certaines études attribuent le titre de gène suppresseur de tumeurs à Gas1, pour ses caractéristiques apoptotiques. En effet, un chercheur a testé des injections de cellules d'adénocarcinomes du poumon A549 ou de fibrosarcomes HT1080 comportant un vecteur inductible de Gas1 chez des souris. Il a observé moins de tumeurs et de plus petit volume lorsque Gas1 est exprimé. Il a donc émis l'hypothèse que Gas1 était un gène suppresseur de tumeurs, faute de tumeurs viables en présence de Gas115.
Au final, Gas1 a été identifié lors d'un criblage pour déceler les gènes impliqués durant la mort neuronale induite par le NMDA. Pour valider ce résultat, le chercheur a exprimé Gas1 par un vecteur chez des cellules de neuroblastome BM69 et a observé une forte diminution de la viabilité cellulaire. Par contre, ce phénomène pouvait être restreint si le vecteur de Gas1 était exprimé en même temps qu'un codant pour la protéine Bcl-2 ou OpIAP2, sans toutefois co-immunoprécipiter ensemble28. Ces résultats impliquent potentiellement Gas1 durant une mort cellulaire différente des autres études publiées jusqu’à ce jour.
L’apoptose est un processus biologique contre le cancer très intéressant dans l’optique qu’elle permet d’induire la mort des cellules, éliminant la tumeur. Il est donc très captivant de poursuivre les recherches entre Gas1 et l’apoptose avec pour cible un traitement par l’expression de ce gène. Les études présentées ci-haut illustrent bien le potentiel et l’utilité de Gas1 pour réaliser cet objectif.
11
1.4.5. Gas1, un gène suppresseur de métastases
L’étude fondamentale entourant les mécanismes métastatiques du cancer prend de l’ampleur chaque jour. Les puces à ADN et l’utilisation des petits ARN interférents sont des exemples de nouvelles technologies utilisées pour l’étude des métastases. C’est en utilisant un criblage pangénomique par petits ARN interférents que le Dr. Gobeil a décelé un nouveau rôle de gène suppresseur de métastases de Gas1 chez le mélanome64. Il s’agit d’une découverte importante pour l’avancement de la recherche contre la métastase.
Pour atteindre un site secondaire, les cellules de la tumeur primaire devront parvenir à franchir les étapes de la cascade métastatique69. Pour ce faire, elles vont accumuler des modifications qui vont augmenter leur potentiel métastatique. Ces modifications vont amener l’activation de gènes pro-métastatiques et la perte d’expression de gènes dont le rôle réduit directement ou indirectement la capacité métastatique des cellules68. Dans ce cas, il s’agit de gènes suppresseurs de métastases. Il est donc fréquent de ne pas retrouver leur expression chez des cellules métastatiques. Ainsi, il a été démontré que l’expression de Gas1 est diminuée ou absente chez certains mélanomes64, cancers de la prostate21-31, cancers de la thyroïde33, cancers colorectaux50 ou gastriques57. Pour cette raison, il est important de souligner que Gas1 peut servir de biomarqueur de la progression tumorale44. Par conséquent, il est possible de croire en l’importance de la perte de Gas1 chez plusieurs types de cancer pour le développement des métastases.
Jusqu’à ce jour, le Dr. Gobeil a démontré que Gas1 agit comme gène suppresseur de métastases chez le mélanome. Tout d’abord, il a illustré qu’il y avait moins de Gas1 chez les cellules métastatiques de mélanome B16-F10 en comparaison avec les lignées primaires B16-F0. De plus, il a démontré que la perte d’expression de Gas1 par petits ARN interférents chez les B16-F0 augmente leur potentiel métastatique. Par la suite, le Dr. Gobeil a surexprimé Gas1 chez le mélanome métastatique B16-F10, ce qui a provoqué une diminution de la capacité de formation de métastases. Ces résultats ont également été confirmés in vivo64 (Figure 2). Ces expériences ont bien démontré le rôle de Gas1 comme gène suppresseur de métastases chez le mélanome. En outre, bien que l’étude de Gas1
12
comme gène suppresseur de métastases soit jeune, il n’y a aucun doute de son fort potentiel contre ce stade avancé du cancer.
1.4.6. Gas1, un gène à plusieurs rôles
En dernier lieu, il faut souligner que le gène Gas1 est également étudié pour la résistance aux agents thérapeutiques, en reproduction, mais aussi chez d’autres pathologies comme l’Alzheimer.
Un groupe de chercheurs a souligné un effet de résistance au médicament epirubicine chez le cancer gastrique lors d'une diminution de Gas1. Ils ont émis l'hypothèse que le gène réduit l'apoptose en dérégulant le ratio des protéines Bcl-2 et Bax47. Il s’agit d’une première concernant un rôle de résistance accrue en l’absence de Gas1.
Une autre implication pour le gène de Gas1 concerne son rôle lors du développement. En effet, ce gène a été associé à l'organogénèse chez les fœtus de rat lors des stages préliminaires du développement20. Ensuite, Gas1 a été démontré comme étant exprimé au sein de plusieurs régions lors de l'embryogénèse murine24, en fonction du temps25. Plus précisément, Gas1 a pleinement été impliqué lors de la mort cellulaire interdigitale pendant le développement25. Ces études ont permis d’accroître encore plus les connaissances entourant le gène Gas1.
13 En plus d'être impliqué pleinement au sein des recherches portant sur l’oncologie, Gas1 fait l'objet d'études chez la pathologie de l'Alzheimer. En effet, un chercheur a prouvé qu'il y avait une augmentation du taux de production d'amyloïde-β lorsque Gas1 n'est pas exprimé51. Cette recherche illustre bien le rôle important que joue le gène Gas1 contre la pathologie de l’Alzheimer.
En conclusion, il est crucial de souligner que les études effectuées permettent d’accroître le savoir entourant le gène Gas1. Ainsi, les recherches ont contribué à connaître mieux le mécanisme d’opération de Gas1 lors du cycle cellulaire, de l’apoptose et du développement. Les futures publications pourront donc permettre de mieux comprendre les procédés derrières la répression de Gas1 chez les cellules les plus agressives et d’obtenir de meilleurs résultats pour les thérapies contre les métastases.
1.4.7. La régulation de Gas1
Au fil des ans, plusieurs scientifiques se sont intéressés au gène Gas1 et les mécanismes entourant sa régulation. Parmi eux, c’est la répression de Gas1 par l’oncogène c-Myc qui a été la mieux détaillée. Par contre, il est important d’ajouter que d’autres recherches ont réussi à bien illustrer l’implication directe de protéines dans la régulation du gène Gas1 comme le facteur de transcription Cggbp1, par exemple53.
Dans un premier temps, des chercheurs ont confirmé l'implication potentielle de certains gènes comme Tgf-β ou c-Myc dans la régulation de Gas1. En effet, lorsque les niveaux d'expression de c-Myc augmente, les niveaux d'expression de Gas1 diminue, et vice-versa
1-2-29. L'expérience a été réalisée également chez les cellules Rat-1, mais avec plus de
profondeur. En effet, ce groupe a inséré un vecteur luciférase de Gas1 et c-Myc, pour illustrer la répression de Gas1 par c-Myc et aussi avec un vecteur MycER inductible. Dans les deux cas, lorsque c-Myc est présent, l’expression de Gas1 est réduite14. Plus précisément, un autre laboratoire a montré avec l'aide d'un vecteur MycS, dont la protéine est tronquée pour inactiver la fonction transactivatrice, qu'il était encore possible de réprimer un vecteur exprimant Gas1, selon une expérience de luciférase19. Il en va de même pour Tgf- β. En sa présence, le niveau d’expression de Gas1 est diminué chez les 3T37. De plus, d'autres protéines ont été associées à Gas1. Par exemple, un groupe a dévoilé qu'une liaison de l'intégrine α5-β1 et de la fibronectine amenait une réduction du niveau
14
d'expression du gène Gas18. Finalement, un autre laboratoire a démontré chez des souris ayant une ablation du gène Dmp1 qu'il y avait une augmentation de l'expression de Gas1, indiquant un nouveau répresseur potentiel48. Ces gènes pourraient donc être tous des répresseurs directs ou indirects de Gas1.
Par la suite, d'autres se sont penchés sur les activateurs potentiels de Gas1. Ainsi, O'Rourke (O’Rourke, 1997) a stipulé que C/ebpδ pourrait être un activateur direct de Gas1, puisqu'il est exprimé lors de l'arrêt du cycle cellulaire chez les fibroblastes murins12. D'autre part, la protéine Cggbp1 a été confirmée dans la régulation de Gas1 chez la lignée de gliomes humains U-2987 Mg. En effet, sa réduction par ARN interférent stimule l'expression de Gas1 et p21, tout en diminuant les marques de répression H3K9me353. Par ailleurs, un autre
groupe a démontré que Gas1 pourrait être réprimé par la protéine Menin chez les cellules MEF59. Des voies de signalisation ont également été scrutées pour induire l'expression de Gas1. En effet, un laboratoire a prouvé que la VE-cadhérine pouvait induire l'expression de Gas1 chez les cellules HEK293. En plus, avec l'aide d'une protéine VE-cadhérine tronquée, il a observé l'implication de la protéine PI3-K pour induire Gas1. Au final, il a également émis l'hypothèse que Gas1 pourrait être activé par NFkB32. De surcroît, un groupe a montré que l'expression de Gas1 peut être induite par un stress engendré par un sevrage en méthionine, une méthode utilisée pour combattre les cancers de la peau36.
Par conséquent, il est important de comprendre la portée de ces études pour accroître les outils face aux pathologies comme le cancer. Ainsi, en démontrant que Gas1 peut être modulé par des répresseurs comme l’oncogène c-MYC, il est possible de croire en l’utilisation du gène Gas1 comme mécanisme de frein du cycle cellulaire et pour amener les cellules tumorales en apoptose. Ces recherches des protéines responsables de la régulation de Gas1 sont donc particulièrement intéressantes.
1.5. La régulation épigénétique
Bien que les mutations génétiques comme les délétions soient essentielles pour l’évolution des espèces, la régulation épigénétique tient également un rôle vital au sein de la biologie cellulaire. Avec l’afflux de signaux externes, une cellule du système immunitaire peut s’adapter en permettant l’expression de gènes pour la mobilité pour se déplacer vers un site
15 d’une blessure, par exemple197. Bref, l’épigénétique permet une réactivité cellulaire importante pour répondre aux différents signaux environnementaux.
Au niveau du cancer, il a été montré que la régulation épigénétique était souvent déstabilisée afin de permettre l’activation d’oncogènes comme c-MYC185 et d’empêcher la transcription de gènes anti-tumoraux comme p21104. Par conséquent, il a été publié fréquemment des régulations aberrantes de protéines impliquées dans l’épigénétique comme les HDACs ou les KMTs chez les cellules cancéreuses122. En somme, la régulation épigénétique est cruciale pour la survie des cellules tumorales.
1.5.1. Le code des histones
Initialement, les chercheurs attribuaient l’acétylation des histones à des gènes ouverts, permettant leur expression, et les marques de méthylation à des gènes fermés115. Rapidement, ces derniers ont compris que l’épigénétique était beaucoup plus subtile et complexe qu’à prime abord. En effet, il a été démontré au fil des ans que l’acétylation et la méthylation dépendaient plutôt de la position de ces marques sur les histones. Par exemple, la méthylation au niveau H3K4 ou H3K36 a été associée à l’activation115. De plus, une
balance entre l’acétylation et la méthylation a été décrite régulièrement, impliquant la présence des deux groupements au même promoteur. De ce fait, la transcription est active ou non dépendamment de la proportion d’un groupement en comparaison à l’autre56. Il apparait évident que beaucoup de recherches reste à effectuer pour déceler toute la finesse auquel le code des histones est soumis.
1.5.1.1. Les marques activatrices
Il existe plusieurs marques d’histones présentes au promoteur des gènes en transcription. Cependant, H3K4 et H3K36 sont les deux marques les mieux connues et étudiées
présentement pour mieux comprendre les mécanismes entourant leur régulation chez les cellules tumorales.
H3K4 : La marque H3K4 a été associée à l’activation de gènes lorsqu’elle était di- ou
tri-méthylée. Par conséquent, il a été démontré que les cellules tumorales pouvaient utiliser des histones déméthylases comme LSD1 ou JARID1 pour éliminer des groupements méthyles
16
et restreindre l’activation du gène118. Dans de telle situation, il a été montré qu’il est fréquent de retrouver de la méthylation à l’ADN apporté par la famille DNMT afin d’y maintenir le gène fermé196. De plus, la marque d’activation H3K4me3 a été rapportée pour
être important pour la phase d’initiation de la transcription avec les marques H3K9ac et
H3K14ac198. En outre, la marque H3K4 est l’une des principales menant à l’activation
génique afin d’entamer la transcription des gènes.
H3K36 : Autre marque d’importance, H3K36 a été associé avec l’élongation lors du processus
de transcription. Par conséquent, il est fréquent d’y retrouver un groupement tri-méthyle lorsque le gène est activé et produit de l’ARNm198. De plus, il a été démontré que des histones méthylases comme KDM5B pouvaient éliminer des groupements méthyles sur H3K36 afin d’induire la polymérase en pause transcriptionnelle107. Par conséquent, il s’agit
d’une marque d’activation importante pour permettre l’élongation de la polymérase.
1.5.1.2. Les marques répressives
Dans le cadre de la biologie cellulaire, les marques de répression au niveau des histones sont très importantes. En effet, elles permettent d’atténuer l’expression de gènes néfastes pour la cellule comme ceux impliqués dans les voies apoptotiques, mais importants lorsque survient une nécessité, ce qui ne serait plus possible en présence d’une délétion197. Par contre, la répression épigénétique permet aux cellules tumorales d’éteindre des gènes qui seraient susceptibles de les restreindre. Or, ces études sont très importantes pour mieux cerner les processus cellulaires comme la répression épigénétique dans l’optique d’apporter de nouvelles thérapies anti-tumorales.
H3K9 : La marque de répression H3K9me3 a été associée à l’hétérochromatine, dévoilant
bien son rôle lors de la répression115-116-117. La famille de protéine HP1 a également été prouvée comme élément stabilisateur de cette marque de répression et de l’état d’hétérochromatine115. En effet, cette liaison a été montrée comme possible par le chromodomaine de la protéine HP1 se liant à la marque H3K9me3115. D’une part, les
protéines SETDB1, SUV39H1, SUV39H2, EHMT1 et EHMT2 ont été publiées comme des actrices lors de la modification de l’histone 3 en H3K9me3116-117.
17 H3K27me3 : Cette marque de répression a été prouvée comme étant ajoutée par le complexe
PRC2116-117. Récemment, deux autres protéines, AEBP2 et JARID2, ont été illustrées comme pouvant jouer un rôle lors de la modification de cette marque de répression116. Au final, il faut ajouter qu’il existe d’autres marques de répression permettant la régulation épigénétique comme H4K20me3129, une marque pouvant jouer des rôles d’activation, de
répression ou même de réparation de l’ADN selon les circonstances. Ces marques sont de plus en plus étudiées pour apporter de nouvelles perspectives contre le cancer.
1.5.1.3. Les régulateurs épigénétiques
La régulation épigénétique est un processus biologique très complexe impliquant plusieurs joueurs comme les facteurs de transcription ou des protéines modifiant la configuration de la chromatine pour permettre la transcription ou non. Au fil du temps, les chercheurs ont su démontrer toute l’importance des protéines responsables de la modulation de la chromatine, soit les DNA méthyltransférases, les histones méthyltransférases, les histones déacétylases, les histones acétylases ou les histones deméthylases118-122-130. De plus, il a été prouvé que ces acteurs pouvaient être dérégulés afin de transcrire des gènes favorables au cancer et d’empêcher la transcription d’autres gènes nuisibles au développement des cellules tumorales130.
Tout d’abord, la famille HDAC a été illustrée pour enlever les groupements acétyle des histones130. Elle se divise en quatre classes composées des HDAC1 à HDAC11 et des SIRT1 à SIRT7203. Par ailleurs, il a été publié fréquemment que les HDACs pouvaient être dérégulés chez plusieurs types de cancer comme celui du colon ou du sein122. Par exemple, les gènes p15139, p21140-141 et p27141 ont été dévoilés comme pouvant être réprimés par les HDACs. Il existe donc des inhibiteurs chimiques contre les HDACs en essais cliniques afin d’être utiliser pour les empêcher de réprimer des gènes anti-tumoraux132. Cette famille est donc pleinement impliquée dans la régulation de la transcription génique chez le cancer. Ensuite, la famille HAT a été associée pour sa part avec l’acétylation des histones, permettant par le fait même la transcription des gènes130. Cependant, il a été montré que les cellules tumorales pouvaient diminuer l’expression des HATs pour atténuer la transcription de gènes anti-tumoraux ou d’en augmenter l’expression pour stimuler des gènes
pro-18
cancer204-205. En d’autres termes, il a été prouvé que la hausse ou la réduction des HATs pouvaient influencer la régulation de plusieurs gènes chez les cellules tumorales.
Par la suite, une autre famille a été dévoilée pour jouer un rôle important lors de la régulation épigénétique, soit les KMTs. Ces protéines ont été démontrées comme responsables de la déméthylation de lysine sur les histones118-119. Cette famille a été associée à plusieurs protéines, soit les gènes KDM1 à KDM6118. Régulièrement, les KMTs sont dérégulés chez les cellules tumorales. Par conséquent, les protéines KDM1, KDM2, KDM4 et KDM5 ont été découvertes comme déméthylant les marques de transcription H3K4 et H3K36 pour empêcher la transcription de gènes anti-tumoraux alors que KDM3,
KDM4 et KDM6 déméthylent les marques de répression H3K9 et H3K27 pour permettre la
transcription d’oncogènes206. En outre, il est facile de comprendre l’implication cruciale des KMTs au sein des cellules cancéreuses.
Au final, la famille DNA méthyltransférase a été publiée pour jouer un rôle lors de la répression des gènes. En effet, cette famille comportant les gènes DNMT1, DNMT3A, DNMT3B et DNMT3L a été prouvée comme responsable de la méthylation de l’ADN lors de la fermeture de gènes au niveau d'îlots CpGs130. Par ailleurs, les DNMTs ont été montrés comme pouvant réprimer les gènes anti-tumoraux comme p14142, p21 ou p16143. En somme, cette famille permet de maintenir fermés les gènes agissant contre les cellules cancéreuses.
1.5.2. La pause transcriptionnelle
La pause transcriptionnelle est un mécanisme qui a été mis en lumière grâce à des études sur les protéines Hsp70 au cours des années 90. Il a été démontré que la polymérase était présente au promoteur du gène Hsp70 et parée pour la transcription. Pour ce faire, deux protéines ont été illustrées comme rétentrices de la transcription, soit NELF et DSIF. En présence d’un stimulus de choc thermique, le gène peut être rapidement activé pour permettre aux cellules de résister à la chaleur199. L’étude des Hsp a permis de mieux comprendre le mécanisme entourant le maintient de la polymérase dans un état de pause transcriptionnelle.
19 Dans un contexte oncologique, il arrive régulièrement que les cellules tumorales aient recours à ce type de mécanisme pour restreindre la transcription de certains gènes pouvant être utiles dans d’autres circonstances. C’est notamment le cas des gènes c-Myc et c-Fos200. Il a été prouvé qu’en premier lieu, les protéines NELF et DSIF étaient responsables d’un maintient de la polymérase en pause transcriptionnelle en empêchant sa phosphorylation. Pour sa part, P-TEFb était responsable de la phosphorylation du domaine c-terminal de la polymérase, inhibant la répression de NELF et DSIF, et permettant la transcription167. Ce type de mécanisme est donc très important afin de moduler rapidement l’expression de certains gènes en toute circonstance.
1.5.3. La poly(ADP-ribosyl)ation
Bien qu’ayant un rôle très important lors de la réparation de l’ADN, la poly (ADP-ribosyl)ation est un mécanisme épigénétique également utilisé par les processus biologiques pour l’activation et la répression génique178. Par exemple, il a été illustré
qu’afin de résister au stress inflammatoire, la PARP-1 pouvait activer p65NF-κB lors de la tumorigénèse179. De plus, il a été démontré que certains facteurs de transcription comme KDM5B pouvaient dépendre d’une PARylation par PARP-1 pour être inactivés, les empêchant d’effectuer une répression180. D’ailleurs, il a été publié que la combinaison PARP-1/CTCF pouvait inhiber la protéine DNMT1 (Figure 3), permettant donc à la chromatine de rester hypométhylée et donc de permettre la transcription181. Par contre, il a été montré que la PARP-2 pouvait agir comme répresseur en recrutant des HDACs183, tout comme les isolateurs CTCF avec la cohésine184. D’un autre côté, le rôle inverse de la PARG a été dévoilé comme une balance à la PARP. Ainsi, la PARG pouvait réduire la PARylation de DNMT1 et permettre la méthylation de l’ADN et la répression génique182. En outre, il est facile de voir l’équilibre derrière les mécanismes épigénétiques impliquant la poly(ADP-ribosyl)ation et le rôle important pour les PARPs, la PARG et les isolateurs CTCF au sein du cancer.
20
1.5.4. Les microARNs
Au cours des dernières années, les scientifiques ont porté leur attention sur les mécanismes entourant la régulation des gènes par les microARNs. Leur présence permet donc d’ajuster finement les niveaux d’ARNm de gènes comme p21194. Pour ce faire, certaines régions génomiques contenant les microARNs sont transcrites, libérant des pri-microARNs. Ces derniers sont catalysés en pré-microARNs par le complexe DGCR8/Drosha. Ensuite, les pré-microARNs sont clivés en duplex microARN/microARN par un autre complexe contenant la protéine DICER. Par la suite, le complexe RISC assemble l'ARNm, le microARN et la protéine Ago, responsable de la dégradation de l'ARNm212. Ainsi, par des mécanismes de dérégulation génétique ou épigénétique, il a été démontré que les microARNs sont régulièrement impliqués dans la répression génique chez les cellules tumorales195. Par conséquent, il est bien d’ajouter que les cellules cancéreuses peuvent utiliser ce mécanisme pour réduire l’expression des gènes anti-tumoraux.
1.6. L’oncogène c-Myc
Les oncogènes peuvent mener au développement néoplasique, seuls ou en présence d’autres facteurs185. Ras, raf, src et myc sont des exemples de ces gènes186. Fréquemment, les
21 oncogènes seront exprimés constitutivement. Ces gènes vont permettre la prolifération cellulaire aberrante en dérégulant des voies de signalisation comme AKT ou ERK186. Par exemple, b-Raf est souvent activé constitutivement chez les mélanomes et les cancers colorectaux, ovariens187, thyroïdiens188-189 et pulmonaires190-191. De tous ces oncogènes, c-Myc est le plus étudié pour ces multiples rôles lors de la régulation cellulaire.
Parmi les facteurs nécessaires pour favoriser l’apparition tumorale, plusieurs types de cancer possèdent une expression aberrante de proto-oncogènes185. Le gène c-Myc fait partie de cette catégorie. Chez de nombreuses tumeurs, c-Myc est surexprimé, causant un dérèglement du cycle cellulaire. C-Myc est un facteur de transcription ciblant des gènes impliqués dans la promotion du cycle cellulaire. De plus, il est connu pour réprimer plusieurs candidats freinant le cycle cellulaire192. Finalement, il possède également un rôle de protection contre l’apoptose193. Il est possible de concevoir la présence particulièrement grande de c-Myc au sein des processus biologiques.
1.6.1. Le rôle de répresseur génique de c-Myc
Il n’y a plus de doute possible, le facteur de transcription c-Myc possède un rôle d’activateur, mais également de répresseur génique. En effet, les évidences se sont accumulées au cours des années, portant sur différents complexes répresseurs comme c-Myc/Miz1/Max82-89 ou c-Myc/KDM5B/TFAP2C105. Par conséquent, il est facile de visualiser toute la finesse cellulaire derrière les mécanismes de répression impliquant c-Myc et son importance pour mieux s’outiller face au cancer.
En premier lieu, il incombe de porter l’attention sur l’un des premiers complexes répressifs publiés au fil des ans, soit l’interaction c-Myc/Miz1/Max82-89-92-97 (Figure 4A). En effet, plusieurs chercheurs ont démontré l’étendu d’un tel partenariat pour la répression génique au sein des cellules tumorales. Par exemple, des chercheurs ont prouvé que ce complexe était présent au promoteur du gène C/EBPδ et le réprimait, chez des cellules épithéliales mammaires en prolifération82-89. De plus, ce complexe a été découvert au promoteur du gène GADD153, par immunoprécipitation de la chromatine chez les cellules Rat-192. Le complexe c-Myc/Max/Miz1 s’ait aussi révélé être répresseur du gène p15 chez des cellules MEFs97. Dans le même sens, un autre laboratoire a utilisé un criblage pour découvrir 67 gènes inhibés par le complexe c-Myc/Miz1. En utilisant un mutant (MycV394D)
22
empêchant le lien entre c-Myc et Miz1, il a trouvé qu’il y avait une forte diminution de l’expression de ces 67 gènes seulement avec un vecteur exprimant c-Myc en comparaison au vecteur mutant88. Par la suite, il a été démontré que Miz1 et c-Myc répriment plusieurs gènes de la famille Hox100, p21104, des membres DNRG102-103 ou un membre de la voie de signalisation Wnt, WIF-1101. Il est intéressant d’ajouter que le complexe de répression entourant c-Myc peut varier. En effet, il a été confirmé que c-Myc/Miz1/Gfi-1 pouvait réprimer le gène p2190, mais également le gène p1591. Ces expériences ont donc permis d’approfondir les connaissances entourant le dynamisme derrière la répression par c-Myc, passant par le partenaire Miz1.
Ensuite, bien que certaines études aient stipulé une répression de c-Myc/Miz1/Max, d’autres laboratoires ont illustré différents mécanismes de répression entre c-Myc et Max indépendant de Miz1. Une étude a même mis en lumière lors d’un criblage la dépendance de c-Myc à Max pour réprimer plusieurs gènes93. De plus, en 2004, c-Myc/Max ont été directement trouvés au promoteur des gènes GADD45a et GADD153 chez les cellules Rat-192. Également, ces deux protéines ont été associées à la répression du gène p2796. Il est
facile de deviner toute la complexité en arrière des processus de répression par c-Myc et l’étendu des recherches qu’il reste à faire pour mieux les comprendre.
Il est à souligner également que plusieurs publications ont établi qu’il y a un équilibre entre les complexes de facteurs de transcription, c-Myc/Max, Max/Max et Max/Mad, pour réguler les gènes cibles95-99 comme ODC94 ou hTERT98. Cet équilibre est particulièrement pratique pour établir des ajustements fins lors des processus biologiques chez les cellules saines, mais également très complexe à étudier lors de pathologies comme le cancer.
23 Par la suite, il faut mentionner qu’un autre complexe a été impliqué lors de la répression de plusieurs gènes. Il s’agit du complexe c-Myc/Sp1 (Figure 4B). En fait, Gartel (Gartel, 2001) a illustré le rôle de ce complexe pour la répression du gène p21 chez les cellules d’adénocarcinome du colon Caco-278-81. Également, un autre scientifique a montré qu’un complexe Sp1/Smad2-3/c-Myc pouvait interagir ensemble pour causer une répression du gène p15Ink4B chez la lignée cellulaire HaCaT79-81 (Figure 4C). Lors de l’utilisation d’un nouvel inhibiteur d’histones déacétylases, le Phenethylisothiocyanate (PEITC), Wang (Wang, 2008) a démontré une nouvelle fois l’action du complexe répressif c-Myc/Sp1 sur le gène p21, mais chez le cancer de la prostate. Ainsi, suite au traitement avec le PEITC, le promoteur du gène p21 avait perdu la protéine c-Myc, en plus de devenir hypométhylé et actif84. De surcroît, la présence d’un complexe répresseur c-Myc/Sp1/Max au promoteur du gène BRD7 a également été publiée, bien que seul c-Myc semblait le réguler négativement85. Une expérience similaire a montré que Sp1/c-Myc pouvait réprimer le gène BAG2 chez les cellules TRE29387. Par ailleurs, le complexe c-Myc/Sp1 a même été publié chez des cellules T du système immunitaire de patients infectés par le HIV en latence. En fait, la présence des trois protéines c-Myc/Sp1/Hdac1 a été remarquée au niveau du promoteur HIV latent83. Ces résultats dévoilent une nouvelle fois la variabilité entre les différents mécanismes de répression cellulaire engendrés par la protéine c-Myc.
24
Au cours des dernières années, plusieurs laboratoires ont découvert d’autres partenaires ou processus permettant une répression par c-Myc. Par exemple, il a bien été illustré qu’une interaction entre c-Myc/KDM5B/TFAP2C (Figure 5) menait à la répression du gène p21 chez les cellules MCF-7105, confirmant l’implication potentiel de membres de la famille d’histone 3 lysine 4 demethylases JARID1 avec c-Myc106. De plus, il a été prouvé que la répression par c-Myc pouvait impliquer des histones déacétylases comme HDAC183 et HDAC3108 ou des DNA methyltransférases comme DNMT3a et DNMT3b109, chez plusieurs gènes. Cependant, c-Myc a été démontré comme pouvant réprimer non seulement des gènes, mais aussi des microARNs. Ainsi, il a été publié que c-Myc était responsable de la répression du miR-26a, un microARN ayant des vertus d’atténuation de la prolifération cellulaire, chez des cellules d’un lymphome murin107.
En conclusion, il apparait évident que c-Myc joue un rôle majeur lors de la répression de plusieurs gènes engagés dans une grande variété d’activité cellulaire comme p2178-81 ou Gas129. Avec les années, beaucoup études ont démontré l’implication de plusieurs partenaires potentiels pour exercer le rôle de répresseur attribué à c-Myc. Bien que Sp1 ou Miz1 semble faire partie de ce complexe empêchant la transcription de gènes, certains scientifiques se posent la question à savoir si ces deux partenaires sont bien nécessaires pour que c-Myc parvienne à exécuter sa répression86. D’autres publications futures pourront permettre de mieux cerner les différents rôles intermoléculaires joués par cet ensemble très complexe de facteurs de transcription.
25
1.7. Problématique et implications scientifiques de l’étude du gène
Gas1
L’étude des gènes suppresseurs de métastases comme Gas1 est très important pour la recherche contre le cancer. En effet, cette pathologie est devenue la première cause de décès en Amérique du Nord. De surcroît, les métastases sont responsables d’un très grand nombre de cette mortalité. L’acquisition de connaissances entourant les mécanismes biologiques menant à la formation de colonies secondaires est donc cruciale dans cette lutte contre le cancer. Nous nous sommes donc penché à découvrir comment le gène Gas1 est régulé chez les lignées métastatiques étudiées. À la lumière de la littérature illustrant l’implication de c-Myc lors du processus de répression du gène Gas1, nous avons émis l’hypothèse que cet oncogène était responsable de l’arrêt de la transcription de Gas1 par le recrutement de complexes répresseurs.
Pour en découvrir la véracité, nous avons d’abord fixé comme objectifs de trouver un modèle cellulaire de lignées primaires et métastatiques pour en vérifier les niveaux d’expression du gène Gas1. Par la suite, nous voulions déterminer la composition moléculaire au niveau du promoteur de ce gène, soit en étudiant les groupements d’acétylation et de méthylation sur l’ADN et les histones. En plus, nous voulions démontrer l’implication de complexes par l’utilisation d’inhibiteurs chimiques empêchant la répression de Gas1. Ensuite, nous désirions établir les facteurs de transcription responsables d’empêcher la transcription de ce gène par des immunoprécipitations de la chromatine et l’utilisation de petits ARN interférents. Finalement, notre objectif principal était de détourner la répression du gène Gas1 dans l’optique d’une utilisation thérapeutique contre la métastase.