FACULTE DES SCIENCES Rabat N° d’ordre : 2788
THESE DE DOCTORAT
Présentée par
Halima ZAARI
Discipline:Physique informatique.
Spécialité: Matière condensée et modélisation des systèmes
Etude ab initio des propriétés optiques des
matériaux :
Cas de ZnTe, CdFe
2O
4et MgB
2Soutenue le 22-07-2015
Devant le jury :
Président :
Abdelilah BENYOUSSEF
PES, Faculté des Sciences, Rabat
Examinateurs
:
Abdelmajid AINANE
PES, Faculté des Sciences, Meknès
Lahoucine BAHMAD
PES, Faculté des Sciences, Rabat
Mohammed BENAISSA
PES, Faculté des Sciences, Rabat
Youssef EL AMRAOUI
PES, Faculté des Sciences, Rabat
Abdallah EL KENZ
PES, Faculté des Sciences, Rabat
Hamid EZ-ZAHRAOUY
PES, Faculté des Sciences, Rabat
Mohammed LOULIDI
PES, Faculté des Sciences, Rabat
Faculté des sciences, 4 Avenue Ibn Battouta B.P. 1014 RP, Rabat-Maroc
Remerciement
Cette thèse a été réalisée au sein du Laboratoire du Magnétisme et Physique des Hautes Energies (LMPHE), de la Faculté des Sciences de l’Université Mohammed V-Rabat sous la direction de Monsieur le professeur Abdelilah Benyoussef.
Je tiens à remercier Monsieur Abdelilah Benyoussef , Professeur d’enseignement supérieur de la Faculté des Sciences Rabat, mon directeur de thèse et président du jury de cette thèse qui fut pour moi un directeur attentif , généreux et disponible malgré ses nombreuses charges. Il avait des solutions à tous les problèmes rencontrés, une vision claire sur ma recherche et n’hésitait pas à présenter ses conseils et ses orientations qui témoignent de sa compétence et de sa rigueur scientifiques. Il est pour moi un privilège d'être son étudiante.
Je tiens à exprimer mes sincères remerciements au Professeur Abdallah El Kenz, Professeur d’enseignement supérieur de la Faculté des Sciences Rabat, pour avoir accepté de participer à ce jury en tant que rapporteur et examinateur.
Je tiens à exprimer mes sincères remerciements au professeur Mohammed Louilidi, Professeur d’enseignement supérieur de la Faculté des Sciences Rabat, d’avoir accepté de participer à ce jury en tant qu’examinateur.
Je tiens à exprimer mes sincères remerciements au Professeur Mohammed Benaissa, Professeur d’enseignement supérieur de la Faculté des Sciences Rabat, d’avoir accepté de participer à ce jury en tant qu’examinateur, aussi bien je le remercie pour nos discussions sur les propriétés optiques soit de point de vue théorique ou expérimentale.
Je tiens à exprimer mes sincères remerciements à Youssef El Amraoui, Professeur d’enseignement supérieur de la Faculté des Sciences Rabat, d’avoir accepté de participer à ce jury en tant qu’examinateur.
Je tiens à exprimer mes sincères remerciements à Hamid. Ez-Zahraouy, Professeur d’enseignement supérieur de la Faculté des Sciences Rabat, d’avoir accepté de participer à ce jury en tant qu’examinateur.
Je tiens à exprimer mes sincères remerciements à Lahoucine BAHMAD, Professeur d’enseignement supérieur de la Faculté des Sciences Rabat, d’avoir accepté de participer à ce jury en tant qu’examinateur.
Je tiens à exprimer mes sincères remerciements au Professeur Abdelmajid AINANE, Professeur d’enseignement supérieur de la Faculté des Sciences Meknès, pour avoir accepté de participer à ce jury en tant que rapporteur et examinateur.
Un remerciement particulier à deux amies très chères à moi, Errafig aicha et Nihal Ferhane pour leurs soutien et disposition.
S.Naji et B.Khalil, et ceux qui sont encore là Lakouari Noureddine, Abdel-Ghafor, Chaimae, Bhihi, Maryem El-khatabi, Abbassi, Marwan,Mourad, Khalid, Rachid, Souad et Siham … Un grand merci à l’équipe du code Wien2k prof. P. Blaha, prof. K. Schwartz and WIEN2K group, ils étaient tout le temps disponibles pour répondre à nos questions au forum sur le code, les méthodes de calculs. En particulier Prof A.Gavin , R.Oleg
Je remercie aussi les membres du forum “PWSCF” et “Yambo “ d’avoir accepté ma participation à la formation qui a eu lieu à Lausanne, merci pour les moment que j’ai passé là-bas, avec un équipe trop serviable et gentil.
Je voulais remercier tous les amis qui m’ont soutenue depuis ma Première année à la faculté des sciences, mes anciens professeurs du lycée Abou bakr Essaddek, spécialement Madame Elmounadi Kalthoum, mes anciens professeurs de l’école normale supérieur Takkadoum. Je remercie mes collègues du lycée Ennassim-Temara El-Bousaidi, Kenza, Fatima, Saadia, Adnani…
Enfin j’ai une pensée particulière pour mes parents et toute la famille qui m’ont toujours encouragée et soutenue, et qui m’ont appris la valeur du travail et du savoir, ce dont je leur en suis chaleureusement reconnaissant.
Lors des dernières décennies, l'accroissement de la puissance informatique disponible ainsi que le développement d'algorithmes de plus en plus performants ont contribué à l'évolution des techniques de modélisation des matériaux à l'échelle atomique. Il est actuellement possible de caractériser fidèlement les propriétés de nombreux matériaux en appliquant des méthodes basées sur les lois fondamentales de la mécanique quantique. Même si l'étude pratique des systèmes complexes nécessite quelques approximations, les résultats ne dépendent d'aucun paramètre empirique ajustable. C'est la raison pour laquelle ces techniques sont communément appelées calculs ab-initio. Depuis 1990, ces méthodes ont été largement appliquées à l'étude des matériaux et ont contribué à améliorer notre compréhension de l'origine microscopique de leurs propriétés.
Le présent travail peut être vu comme un premier pas dans l'étude ab-initio des propriétés optiques et magnéto-optiques des matériaux semi-conducteurs et l’étude de la dynamique du réseau des structure cubiques et hexagonales, qui ont suscité un intérêt croissant au sein de la communauté scientifique vu qu'ils sont potentiellement intéressants pour de nombreuses applications. Nous avons choisi certains types de matériaux, à savoir : ZnTe, CdFe2O4, MgB2 et BaF2, vu leurs applications énormes en optoélectronique, photovoltaïque ou détecteurs de rayonnement.
Les calculs réalisés tout au long de ce travail s'inscrivent dans le cadre de la théorie de la fonctionnelle de la densité (DFT) que nous décrirons dans un premier temps, et puis on présentera les équations de base pour la description des propriétés optiques et magnéto-optiques.
Nous verrons que les calculs relatifs à l’optique peuvent se faire à différents niveaux d'approximation et que les fonctionnelles élémentaires de la DFT ne traitent pas toujours correctement les matériaux caractérisés par un certain ordre magnétique, aussi bien pour des structure en mode de vibration. On propose des méthodes correctives pour surmonter le problème lié au premier cas, et on introduit la théorie de perturbation TDDFT pour le 2eme cas.
Ainsi cette thèse donnera une vision générale sur l’application des méthodes ab initio et puis une constatation générale sur la validité des approches utilisées.
Mots clé
: étude ab initio, propriétés optiques, propriétés magnéto-optiques, phonon, dynamique du
réseau, supraconducteurIn recent decades, the increasing of the availability of computing power has contributed to the development of modeling techniques of materials at the atomic scale. Since 1990, the ab initio methods have been widely applied to the study of materials and contributed to improving our understanding of the microscopic origin of their properties.
This work can be seen as a first step in the study ab initio optical and magneto-optical properties of semiconductor materials and dynamics study of the cubic and hexagonal structure and have attracted growing interest in the scientific community as they are potentially interesting for many applications.
The calculations carried out throughout this work within the framework of the density functional theory (DFT) are to be described at first, and then we will present the basic equations for describing the optical and magneto optical properties.
We are going to see that the calculations for optics can be done at different levels of approximation and basic functional DFT do not always properly handle materials characterized by a magnetic order, both for vibration mode structure. Firstly, we are going to propose corrective methods to overcome this problem, and secondly we will introduce TDDFT perturbation theory. Examples of materials are the subject of detailed studies to illustrate each case separately.
Key-word: ab initio study, optical, magneto-optical properties, phonon dynamics of the network, superconducting
Remerciement ... i
Résumé ... iv
Abstract ... iv
Introduction ... 1
I. Contexte général et Champ d’application : ... 1
II. Les Objectifs de la thèse ... 2
III. Plan générale ... 4
Partie I : Généralités ... 6
Chapitre I : Théorie de la fonctionnelle de la densité ... 7
I. Historique ... 8
1. Introduction ... 8
2. L’approximation de Born-Oppenheimer ... 8
3. L’approximation à un électron ... 10
4. Approximation de Hartree ... 10
II. Théorie de la fonctionnelle de la densité : DFT ... 11
1. Principe de la théorie de la fonctionnelle de la densité ... 11
2. Théorème de Hohensberg et Kohn ... 12
3. Approche Kohn et Sham. ... 13
4. Fonctionnelle d’échange-corrélation ... 14
III. Résolution des équations de Kohn-Sham ... 17
1. Introduction ... 17
2. La méthode FP-LAPW ... 18
3. La méthode des pseudo-potentiels (ondes planes) ... 19
IV. Approches Correctifs ... 22
1. Introduction ... 22
2. Origines et formalisme de la méthode LDA+U ... 23
3. L’approche GW ... 25
4. Potentiel de Becke et Johnson modifié mBJ ... 26
V. Protocole numérique ... 28
VI. Conclusion du chapitre ... 29
Chapitre II : Rappel théorique sur les propriétés optiques et XMCD ... 30
I. Propriétés optiques ... 31
3. Propriétés Optiques ... 32
4. Les excitons dans les semi-conducteurs massifs ... 33
II. Etude théorique des propriétés optiques ... 34
1. Calcul de la réponse d’un système à une excitation électrique ... 34
III. Le dichroïsme magnétique circulaire (XMCD) ... 37
1. Introduction ... 37
2. Théorie de XMCD: ... 38
IV. Mesures expérimentales des propriétés électroniques ... 40
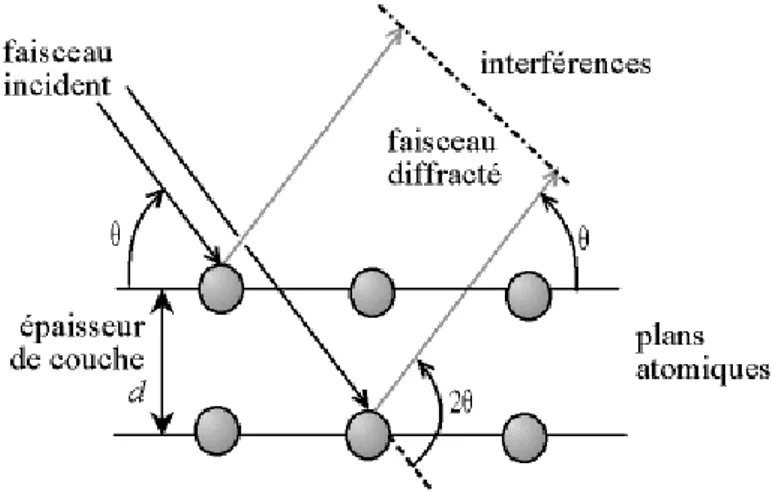
1. Interaction des rayons X avec la maille cristalline ... 40
2. Caractérisation du matériau ... 41
V. Mesure de propriétés optiques ... 42
1. Généralités ... 42
2. La photoluminescence ... 43
4. Diagramme de Jablonski ... 44
5. La Spectroscopie des pertes d'énergie ... 45
6. La spectrométrie photo-électronique X ... 46
7. La spectroscopie angulaire d’électrons photo-émis (ARPES) ... 46
Partie II : Résultats ... 50
Chapitre I : Propriétés optiques des semi-conducteurs type zinc blende ... 51
I. Introduction ... 52
II. Propriétés optiques des semi-conducteurs type zinc blende ... 52
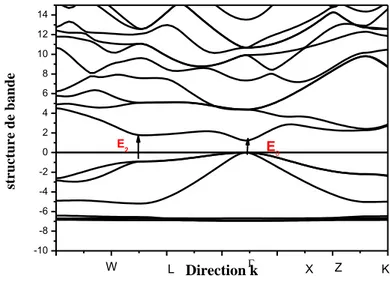
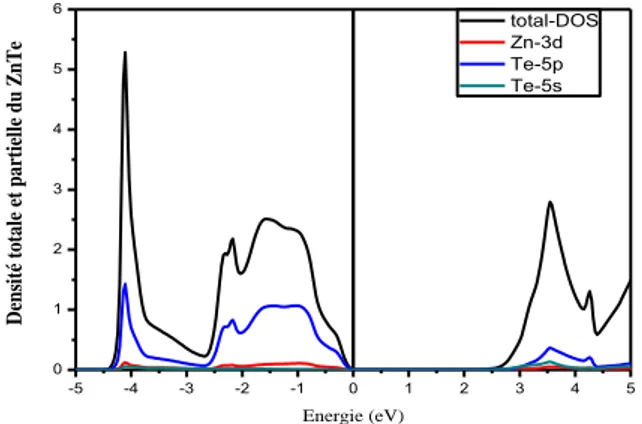
1. Semi-conducteur ZnTe ... 52
2. Propriétés électroniques... 53
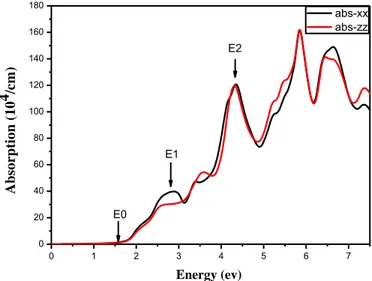
3. Propriétés optiques ZnTe ... 54
III. Correction TB-mBJ ... 56
1. Cas du ZnTe massif ... 56
2. Cas du ZnTe dopé ... 57
3. Conclusion ... 57
IV. Propriétés magnétiques du ZnTe dopé par TM ... 57
1. Propriétés électroniques et structurales: ... 58
2. Propriétés Magnéto-optiques ... 58
V. Dopage ZnTe avec Nd (terres rares) ... 60
3. Effet spin-orbite (XMCD) ... 62
4. Conclusion ... 64
Chapitre II : Etude comparative des approches correctives ... 65
I. Spinnel ferrites CdFe2O4 ... 66
1. Introduction ... 66
2. La structure de type spinelle AB2O4 ... 67
3. Propriétés électroniques CdFe2O4 ... 68
II. Propriétés magnétiques du CdFe2O4 ... 70
1. L’exploitation du spectre XMCD ... 70
Partie III : Dynamique du réseau ... 72
Chapitre I : Rappel théorique sur la dynamique du réseau ... 72
I. Introduction ... 73
II. Théorie de la dynamique du réseau... 73
1. Dynamique du réseau ... 73
2. Notion de phonon ... 74
III. Etude théorique ... 74
1. Cas d’une chaine unidimensionnelle ... 74
2. Généralisation à 3D ... 75
3. Cas d’une chaine diatomique... 76
IV. Classement des modes de vibration du réseau Phonons ... 78
1. Les phonons acoustiques-optiques ... 78
2. Cas des matériaux polaires ... 78
3. LO-TO Splitting ... 79
V. Mesures expérimentales des modes de vibrations ... 79
1. Spectroscopie infra-rouge ... 79
VI. Méthodologie ... 80
Chapitre II : MgB2 phénomène de la supraconductivité ... 82
I. Introduction sur la supraconductivité ... 83
1. Aperçu historiques ... 83
2. La nature physique de la supraconductivité : ... 84
II. La théorie BCS de la supraconductivité: ... 84
1. Définition... 84
IV. Application : Etude des propriétés physiques du supraconducteur : MgB2 ... 87
1. Structure cristalline et propriétés électroniques ... 87
2. Propriétés électronique MgB2 en couches ... 88
3. Propriétés optiques et vibration du réseau : cas massif ... 90
4. Propriétés optiques et vibration du réseau : cas 2D ... 91
Chapitre III : Etude du réseau dynamique cas d’un isolant BaF2 ... 93
I. Eude de propriétés physiques du BaF2 ... 94
1. Motivation de l’étude ... 94
2. Propriétés électroniques et structurales du BaF2 ... 94
II. Eude de réseau dynamique de BaF2 ... 95
1. Propriétés optiques du BaF2 ... 95
2. Effet de pression sur la stabilité du réseau ... 96
I. Conclusion ... 98
Conclusion ... 101
Références : ... 103
Listes de publications: ... 109
10
1
Introduction
I.
Contexte général et Champ d’application:
La science des matériaux repose sur la relation entre les propriétés, la morphologie structurale et la mise en œuvre des matériaux qui constituent les objets qui nous entourent (métaux, polymères, semi-conducteurs, céramiques, composites, etc.). Elle est au cœur de beaucoup des grandes révolutions techniques. Particulièrement depuis un siècle : électronique (ordinateurs, lecteurs de CD et DVD…), aéronautique, énergies renouvelables (panneaux solaires…), nanosciences, nanotechnologies, etc.
La connaissance et la maîtrise des phénomènes microscopiques (diffusion, arrangement des atomes, recristallisation, apparition de phases, etc.) confèrent aux scientifiques et aux industriels la possibilité d'élaborer de nouveaux matériaux aux propriétés et aux performances voulues pour différentes applications.
La classification des matériaux dépend de l’application souhaitée. En particulier pour une application optoélectronique ou photovoltaïque on s’intéresse aux semi-conducteurs qui ont suscités un vif intérêt tant dans leur analyse expérimentale que dans leur développement théorique. Les cristaux monoatomiques dont le chef de file et premier représentant est le silicium, sont placés au premier rang de ces semi-conducteurs. En effet, le silicium représentait le candidat par excellence pour différentes applications, il est synthétisé avec une très haute pureté, puis élargie plus tard à des composés binaires de la même lignée du type GaAs, à structure dite zinc blende.
Désormais la course à l’intégration en microélectronique à base de silicium, poussée par des raisons économiques, doit atteindre ses limites. Depuis quelques années, des solutions alternatives à la microélectronique silicium sont apparues. Différents composés semi-conducteurs suscitent un grand intérêt, commençant par les semi-semi-conducteurs à large bande interdite (GaN, AlN, SiC, ZnO, Diamant, etc.) qui font l’objet d’études depuis plusieurs années, dans un contexte technologique très compétitif.
Au-delà du progrès énorme enregistré dans la technologie des semi-conducteurs, la communauté s’est trouvée contrainte à consacrer de nombreux travaux à la conception de nouveaux matériaux, capables d’améliorer les propriétés structurales et électroniques de ces dispositifs et ainsi élargir leur champ d’application. Les applications potentielles sont nombreuses : sources lasers dans le bleu ou l’Ultraviolet (UV), source de lumière blanche,
1 détecteurs UV, électronique de puissance, des dispositifs optoélectroniques pour les télécommunications et des cellules photovoltaïques, les oxydes transparents, phénomène de « up-conversion » ou « down-conversion ».
Ces applications sont basées sur des éléments communs qui sont :
une source de lumière : considérée comme un émetteur et peut être représentée par : une diode électroluminescente (D.E.L) pour les liaisons à faible distance. une diode laser pour les liaisons à grande distance.
une fibre optique pour la transmission de la lumière.
un détecteur de rayonnement optique placé en bout de chaîne, pour la conversion du signal lumineux en signal électrique en utilisant le principe de l'effet photoélectrique
Un détecteur de scintillation : qui permet la conversion d’un rayonnement UV ou RX à un rayonnement visible ou infrarouge.
Dans tous les cas le processus d'échange d'énergie se fait par transitions radiatives. Il existe principalement 3 types de transitions radiatives :
Absorption d'un photon par un électron qui le fait passer de la bande de valence dans la bande de conduction. (Photo-détecteurs).
Emission spontanée d'un photon par un électron effectuant une transition de la bande de conduction à la bande de valence (Diodes Electro Luminescentes).
Emission stimulée d'un photon en phase avec un photon incident (Diodes LASER).
La conversion ascendante de photon est le processus par lequel un atome absorbe consécutivement deux photons (ou plus) et en émet un seul, de longueur d'onde inférieure. Les matériaux capables d'une telle conversion ascendante contiennent souvent des ions appartenant au bloc f et au bloc d de la classification périodique.
La longueur d'onde du rayonnement émis dépend essentiellement de la largeur du Gap du matériau utilisé. Dans le cas des diodes électroluminescentes du visible, la largeur du gap doit être comprise entre 1,8 eV et 2,6 eV. Ce type de largeur de gap ne peut être obtenu qu'avec des alliages semi-conducteurs tels que l'alliage GaAs1-yPy constitué d'Arséniure de Gallium et de Phosphore ayant une proportion y introduite dans le réseau, ou une autre technique permettant le réglage du gap nommée : le dopage soit de type p ou n. A titre d’information, le dopage permet aussi le contrôle du gap suivant les besoins, par exemple : le développement de couches
2 conductrices transparentes (TCOs) à base de ZnO dopées avec un ou plusieurs ions de terres rares (Tb, Yb, Nd…) afin d’obtenir les propriétés de conversion recherchées.
Vu ces applications énormes, nous avons choisi un exemple de semi-conducteurs ZnTe, un candidat compétitif au CdTe, et qui sera exploité dans une cellule solaire pour une application photovoltaïque, et puis notre étude va s’étendre à d’autres exemples de matériaux tel CdFe2O4 , MgB2 et BaF2 dans le but de mieux s’approfondir sur les différentes interactions que peut avoir la matière avec le champ électromagnétique (rayon lumineux).
II. Les Objectifs de la thèse
La réponse d’un matériau à un champ de rayonnement hγ ou Rayon X, peut prendre plusieurs formes, il s’agit d’abord d’interpréter cette réponse, ensuite étudier les propriétés optiques d’un tel système utilisant les méthodes ab initio.
En se basant sur les travaux antérieurs sur La simulation numérique, nous avons commencé d’abord les problématiques reliés à la fois aux méthodes de simulation et le système étudié lui-même. De ce fait il y a avait plusieurs questions ont été posées.
Posant la première question :
Serait-il correcte de se baser sur des approches théoriques pour révéler les propriétés physiques, optiques en particulier, d’un tel matériau ?
La réponse sera donnée au chapitre I qui traite la théorie de la fonctionnelle de la densité depuis les premières tentatives jusqu’à l’état actuel en donnant quelques détails sur les codes que nous avons utilisé dans nos calculs.
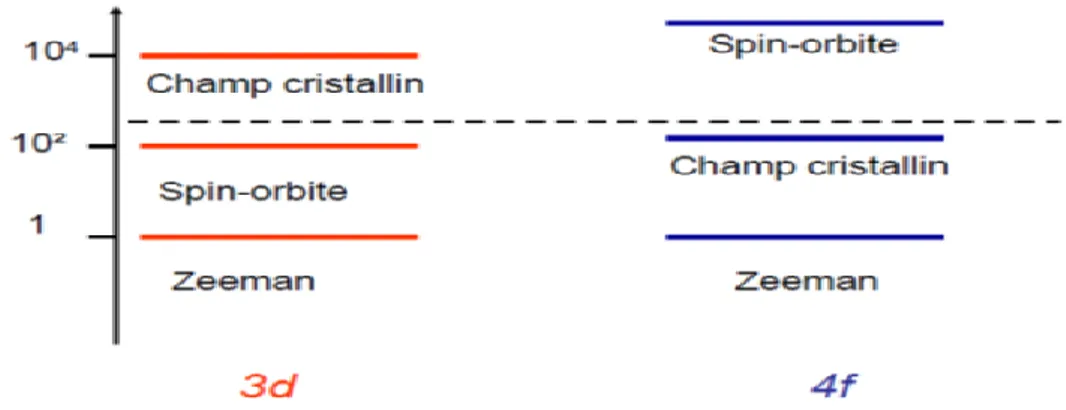
En principe, pour calculer les propriétés optiques d’un tel système, nous avons pris plusieurs considérations pour faire le calcul, partant du ZnTe par exemple, nous avons supposé que le système est non magnétique et par conséquent, la polarisation en spin n’est pas prise en compte dans le calcul, or le dopage de ZnTe par des éléments de transition (3d) apporte généralement du magnétisme, même remarque pour les terres rares (4f). Cette étude nous a menés à une 2ème question :
Quelle est la relation entre propriétés magnétiques et optiques ? Y a-t-il un effet du couplage spin-orbite et champ cristallin sur les phénomènes optiques ?
En faisant une comparaison entre un spectre d’absorption théorique et un autre expérimental, pour identifier les différentes transitions, dans certains cas on n’arrive pas à
3 donner une interprétation de quelques transitions qui apparaissent. Cela dit qu’on a négligé un terme de spin-orbite ou du champ cristallin dans le calcul qu’il ne fallait pas négliger.
La 2ème question sera abordée dans la 2ème partie du manuscrit avec plus de détails. Les méthodes ab initio en général traitent les systèmes à T=0 K, dans le cadre d’une approximation mono-électronique, et une approximation adiabatique ou de Born-Oppenheimer cela dit que les mouvements nucléaires et électroniques sont séparés, il n’y a donc pas de couplage électron-phonon, et les transitions indirectes sont interdites en supposant que la structure étudiée est stable.
Physiquement parlant, il faut faire l’étude des deux cas :
une structure supposée stable
une structure dynamiquement instable,
Dans le 2ème cas, nous parlons de vibration du réseau, phénomène observé généralement pour les semi-conducteur à gap indirect où les transitions sont accompagnées par une vibration de la structure et apparition d’une quasi-particule appelée phonon.
Un phonon peut être couplé avec un photon, ce qui peut affecter les propriétés optiques d’un tel matériau. Vu que les vibrations de la structure ne doivent pas être négligées :
Quels sont les contributions des phonons sur les propriétés optiques et les propriétés thermodynamiques ?
Les méthodes ab initio permettent-elles toujours de reproduire cet effet ?
Ces questions seront discutées en détails dans le dernier chapitre, ou nous avons traité un exemple d’un système isolant BaF2 qui présente énormément d’applications par exemple un scintillateur, qui permet la conversion de l’énergie reçue sous forme de radiations ionisantes RX ou rayon γ en lumière visible ou proche visible, cette propriété nous a poussé à donner plus de détails sur ce système en terme de propriétés électroniques et optiques ,nous avons aussi mis le point sur l’effet de pression sur le changement de la structure de la phase cubique, à la phase orthorhombique et puis la phase hexagonale.
Jusqu’à maintenant nous avons parlé de systèmes autant qu’un réseau 3D (massif), alors que les études s’orientent de plus en plus aux systèmes 2D (les couches minces) ou 1D (nanoparticules), nous avons pris l’exemple du MgB2 pour étudier le phénomène de la supraconductivité, le couplage électron-phonon, et les modes de vibration dans le cas du massif, d’une monocouche et deux couches de MgB2
Dans le cas du MgB2, les modes de vibrations optiques et acoustiques contribuent aux propriétés optiques et thermodynamiques, dans la dernière partie nous allons discuter la
4 variation de ces propriétés entre le massif et une couche. Plusieurs modèles théoriques ont été proposés pour expliquer la supraconductivité autant que phénomènes physique, nous avons pris juste un exemple de systèmes supraconducteurs, pour voir l’interaction de la lumière avec ce type de matériaux.
Ainsi on pourra généraliser cette étude sur l’ensemble de réponse que peut avoir un matériau suite à une excitation par un rayonnement.
Cette thèse ne prêtant pas l’exhaustivité, en effet plusieurs questions n’ont pas été traitées ; à fin de préserver l’unité du thème de notre recherche de façon à concentrer l’intérêt et les efforts sur l’objectif de cette thèse. Ainsi cette recherche se prolongera pour aborder ces questions restées en suspend dans cette recherche. Elles ouvriront d’autres perspectives et répondront à d’autres ambitions.
III. Plan générale
Ce travail de thèse contribue à l’étude des propriétés électroniques, optiques, magnétiques et la dynamique du réseau d’un ensemble de matériaux. La totalité du travail est numérique et utilise les méthodes de calcul du premier principe implémentées dans les deux codes Wien2k et le programme d’accès public : PWscf (Quantum Espresso).
Ce travail sera divisé en 3 parties. La première, incluant les chapitres 1 et 2, donne le cadre théorique dans lequel ce travail a été effectué avec les principes fondateurs de la théorie de la fonctionnelle de la densité
La compréhension du rôle des interactions est certainement l’un des phénomènes les plus difficiles et les plus importants à résoudre dans la physique de la matière condensée. Dans l’étape suivante, nous présentons les deux approches permettant l’application de cette théorie ; à savoir l’approximation du pseudo-potentiel (PW) et la méthode "Full Potential Linearized Augmented Plane Wave (FP LAPW)", développée par l’équipe de Schwarz, basée sur la résolution auto-cohérente des équations de Kohn-Sham dans deux régions arbitrairement définies de la maille élémentaire.
Pour contribuer à une meilleure compréhension des propriétés des semi-conducteurs à base de Zinc, nous avons choisi d’étudier dans le deuxième chapitre quelques généralités sur les propriétés des semi-conducteurs particulièrement les semi-conducteurs II-VI et leurs caractéristiques selon leur domaine d’application, ensuite un rappel théorique et la mise en équations des propriétés optiques et la technique XMCD.
5 La deuxième partie de ce travail, sujet de 5 publications est subdivisé en 2 chapitres. La première section décrit l’ensemble des travaux consacrés au traitement des semi-conducteurs de structure cubique, à savoir le ZnTe, ZnTe dopé ou nous appliquons les différentes méthodes d’approximation tel l’Approximation du Gradient Généralisé (GGA) puis la correction mBJ et la correction GGA+U. Au cours de cette section nous allons confronter les prédictions aux résultats déjà acquis expérimentalement ainsi qu’aux travaux théoriques consacrés à cet égard.
Dans la deuxième section qui sera le chapitre 2 intitulé étude comparatives des approches utilisés, nous allons voir les limites d’utilisation de chaque approches et leurs validité.
La vibration des atomes dans un réseau cristallin est accompagnée généralement par une dissipation de l’énergie, phénomène qui peut influencer sur les propriétés optiques et thermodynamique, deux exemples de systèmes seront étudiés dans la dernière partie, après un rappel théorique à propos de la dynamique du réseau sur le chapitre 1 et puis l’étude des propriétés optiques du MgB2 et BaF2 l’objet du chapitre II et III respectivement.
6
7
Chapitre I : Théorie de la fonctionnelle de la
8
I. Historique
1. Introduction
La description du comportement d’un tel matériau a connu plusieurs tentatives d’explication. Les premières tentatives ont commencé avec le modèle de Drude [1]. Ce dernier proposa une vision classique de l’électron dont le point de départ est la théorie cinétique des gaz appliquée à l’électron [2]. Cette approche est très instructive et importante. Pauli a apporté une amélioration au modèle de Drude en introduisant la notion de fermions dans son modèle. Puis, Fermi a développé les conséquences du principe d’exclusion de Pauli vers une formule générale de la statistique de particules indépendantes [3].
Partant d’un système mono-électronique à un système multiélectronique, les équations d’états deviennent plus en plus compliqués et difficile à résoudre basant sur des théories cités, et par conséquent il fallait chercher d’autres formalismes qui essaient de décrire mieux les propriétés d’un système, d’où la naissance de la théorie de la densité fonctionnelle (DFT) (density fonctionnel theroy) vers les années 1967.[4]
La théorie de la densité fonctionnelle « DFT » et les méthodes ab initio qui en découlent sont maintenant devenues incontournables pour l’étude des propriétés des matériaux. Les progrès considérables dans les dix dernières années, à la fois dans le domaine de la théorie, des implémentations de cette théorie et des outils et méthodes informatiques, font que la modélisation de composés réels (à structures éventuellement complexes) devient parfaitement envisageable avec des moyens et un temps de calcul plus ou moins rapide.
2. L’approximation de Born-Oppenheimer
L’étude à l’échelle atomique des propriétés structurales, électroniques et optiques d’un cristal périodique est un des sujets traditionnels de la physique du solide [5]. Leurs objectifs étaient l’interprétation des mesures expérimentales, prévoir des effets nouveaux ou de concevoir de nouveaux matériaux. Plusieurs modèles théoriques ont été proposés, du côté microscopique le problème peut être établi d’une manière simple. Ceci consiste à résoudre l’équation de Schrödinger décrivant un système cristallin périodique, composé de noyaux atomiques (n) et d’électrons de spin σi.
Vu la complexité de l’équation stationnaire, sa résolution reste une tache très difficile lors de son application à des systèmes réels incluant plusieurs atomes et électrons.
La recherche des solutions de ce système macroscopique (énergie et fonctions d’onde) est appelée problème à plusieurs corps (ou à N-corps).
9 2 2 2 2 2 2 2 , 0 0 0 1 1 1 ˆ 2 2 4 8 8 Ri ri i i i i i i i i j i j i j i j i j i j e Z e Z e Z h h H M m R r r r R R
(I.1) 2 2 2 Ri i i h M
: L’opérateur d’énergie cinétique des noyaux de masse Mi et cordonnés Ri 2 2 2 ri i i h m
: L’opérateur d’énergie cinétique des électrons de masse mi et de cordonnés ri 2 , 0 1 4 i i j i j e Z R r
: L’interaction Coulombienne entre électronset noyaux2 0 1 8 i i j i j e Z r r
: L’interaction Coulombienne entre électrons-électrons 2 0 1 8 i i j i j e Z R R
: L’interaction Coulombienne entre noyaux-noyaux
Ou encore on peut écrire : H=HN+Hel
L’Hamiltonien électronique Hel dépend des coordonnées nucléaires, c'est pourquoi la partie de l'Hamiltonien correspondant à l'énergie cinétique des noyaux ne commute pas avec l'Hamiltonien électronique. Ainsi, nous ne pouvons pas, en toute rigueur, écrire la fonction d'onde totale tot( , )R r comme étant le produit d'une fonction d'onde pour les noyaux par une fonction d'onde pour les électrons. Cependant les noyaux étant beaucoup plus lourds que les électrons, nous pouvons les considérer comme étant fixes lors du mouvement des électrons. Nous pouvons donc effectuer une séparation adiabatique et écrire ce que nous appelons l'approximation de Born-Oppenheimer[6] c'est-à-dire une séparation de la fonction d'onde entre une partie pour les noyaux (fonction de R) et une autre pour les électrons lorsque les noyaux sont dans une position R0 (fonction de ri connaissant R0)
( , ) ( ) ( )
tot R r n R ri R
(I.2)
L’équation de Schrödinger relative aux électrons s’écrit sous la forme:
e e e e H E Ou 0 ( , ) ( , ) ( ) e e r R r R e r (I.3)
Représente la fonction d’onde des électrons, et Ee leur énergie. Ψe et Ee ne dépendent que des coordonnées des noyaux R0
10 Grâce à cette approximation qui permet de séparer le mouvement des électrons à ceux des noyaux, le problème de la résolution de l'équation de Schrödinger se réduit à celui du comportement des électrons, mais il reste encore très complexe à cause des interactions électrons-électrons. Ces simplifications ne suffisent pas donc pour résoudre le problème, donc on recourt à d’autres approximations complémentaires.
3. L’approximation à un électron
Dans le cadre de l’approximation adiabatique, l’approximation à un électron consiste à globaliser les interactions individuelles électron-électron et à écrire que chaque électron évolue dans un potentiel moyen (ou champ moyen) résultant de la présence de l’ensemble des autres électrons. Cette approximation donc ramène le problème de plusieurs électrons en interaction à celui d’un seul électron. Le modèle est qualifié de particules indépendantes car l’interaction entre deux particules n’existe plus. Toutes les méthodes qui adoptent l’approximation à un électron s’appuient sur le modèle de particules indépendantes à fin d’écrire l’équation de Schrödinger à un seul électron (mono-électronique) :
2 2 ( ) ( ) ( ) 2 i eff i i i i i i h V r r r m
(I.4)Où Veff( )ri est un potentiel effectif qui tient compte de l’interaction de l’ième électron avec les
noyaux et de l’interaction moyenne avec les autres électrons et i( )ri
est la fonction d’ondemono-électronique [7]:
4. Approximation de Hartree
En 1928, Hartree [8]a proposé une méthode, dans celle-ci, la fonction d’onde à N électrons
1 2
( ,r r ,....,rN)
est représentée comme le produit des fonctions d’ondes à un électron
(mono-électroniques) : 1 2 1 1 2 2 1 ( , ,...., ) ( ) ( )... ( ) ( ) N N N N i i i r r r r r r r (I.5)
Avec cette approximation, l’énergie totale du système ee en E H T V V (I.5.a) 2 2 2 , 1 1 ( ) ( ) ( ) ( ) ( ) ( ') ( ) ( ') 2 2 ' H i i i i ext i i j i j i i i j Eext E h ke E r r r V r r r r r m r r
(I.5.b)11
H ee
E V L’énergie coulombienne entre les électrons (énergie de Hartree).
ext en
E V L’énergie d’interaction coulombienne des électrons avec les noyaux.
Avec : 2 , ( )i en ext i K K i i ZK V ke V r R r
Bien que la fonction d’onde proposée par Hartree soit une solution de l’équation (I.3), elle ne tient pas en compte la nature des électrons comme des fermions, et elle ne respecte pas aussi le principe d’exclusion de Pauli, parce qu'elle n'est pas antisymétrique par rapport à l'échange de deux particules quelconques En dépit de ces manques, l’approximation de Hartree peut être très utile, par exemple quand appliqué aux atomes à plusieurs électrons. Elle est aussi utile pour avoir une compréhension brute de la validité du modèle des électrons quasi-libres de métaux.
Finalement, c'est plus facile de comprendre la méthode de Hartree-Fock étant donné qu’elle est considérée comme la première étape dans l’évolution théorique de la DFT.
II. Théorie de la fonctionnelle de la densité : DFT
1. Principe de la théorie de la fonctionnelle de la densité
La solution d’un tel problème ainsi que sa représentation analytique s’annonce une tâche difficile compte tenu de la mémoire limitée des outils informatiques. Cependant, il est possible de reformuler le problème en employant les théorèmes et les approximations adaptés. La théorie de la densité fonctionnelle aurait suscité peu de curiosité de nos jours, si ce n’est dans le cadre du théorème établi par Kohn et Sham [9], ce qui l’a rendue utile à travers des approximations sur les fonctionnelles de l’état fondamental, et ce, afin de décrire les systèmes réels à plusieurs électrons. L’idée originale de cette théorie a vu le jour dans les travaux de
Thomas [10] et Fermi [11] en 1927. Bien que leurs approximations ne soient pas
suffisamment appropriées pour des calculs de structure électronique, cette approche élucide la manière de fonctionnement de la DFT. Dans leur premiers travaux, Thomas et Fermi ont écarté les interactions entre les électrons, considérant ainsi le système comme un gaz homogène et son énergie cinétique comme fonctionnelle de la densité (locale). Les deux auteurs ont négligé les effets d’échange-corrélation qui surgissent entre les électrons, cependant ce défaut fut corrigé par Dirac [12] en 1930, qui a introduit l’approximation d’échange locale.
12 La DFT s’est donnée pour le but de déterminer, à l’aide de la seule connaissance de la densité électronique ( )r les propriétés de l’état fondamental d’un système composé d’un nombre fixé d’électrons, en interaction avec les noyaux ponctuels.
2. Théorème de Hohensberg et Kohn
L’approche de Hohenberg et Kohn vise à faire de la DFT une théorie exacte pour les systèmes à plusieurs corps, basée sur 2 théorèmes :
Théorème 1 : « pour un système d’électrons en interaction, le potentiel externe Vext(r) est déterminé d’une façon unique, à une constante près, par la densité électronique de l’état fondamental ( )r .
Ce théorème met en évidence une correspondance unique entre le potentiel extérieur et la densité électronique. Une conséquence immédiate de ce théorème est que la densité électronique détermine de façon unique l’opérateur Hamiltonien et à travers ce dernier, les propriétés du système peuvent être calculées.
Mathématiquement parlant: la valeur attendue de l’état fondamental de toute observableOˆ, est une fonctionnelle unique de la densité électronique exacte à l’état fondamental :
ˆ
O O
Théorème 2 : Le minimum de la fonctionnelle d’énergie totale E () du système correspond à la densité exacte de l’état fondamental0( )r , par conséquent, la densité de l’état
fondamental peut être obtenue à partir du principe variationnel :
0 ( 0) min ( )
E E E
(I.6)
Pour trouver 0correspondant à l’état fondamental, on doit minimiser E( ) avec la condition
3 ( )r d r N
3
( ) ( ) 0 E r d r
(I.6.a)En principe le problème se résume à minimiser l’énergie totale du système en respectant les variations de la densité régie par la contrainte sur le nombre de particules 3
( )
n r d rNe
à cestade la DFT permet de reformuler le problème mais pas de le résoudre compte tenu de la méconnaissance de la forme de la fonctionnelle EHK n avec :
13
3int ( ) ( )
HK ext nn
E n T n E n
d rV r E R (I.6.b)En résumé Hohenberg et Kohn énoncèrent les théorèmes suivant:
THÉORÈME 1 La densité électronique ρ(r) détermine le potentiel extérieur Vext. Quand on connaît la densité, on connaît le potentiel extérieur.
THÉORÈME 2 L'énergie de l'état fondamental est obtenue à partir de la densité électronique exacte. Cela établit un principe variationnel pour l'énergie.
Cependant ces théorèmes ne permettent pas de construire l'application ρ(r)→Ψ[ρ] qui permet de déterminer FHK[ρ].
3. Approche Kohn et Sham.
La théorie de la fonctionnelle de la densité demeure la méthode la plus utilisé dans les calculs de la sturcture électronique, elle doit succès à l’approche proposée par Kohn et Sham en 1965.[13] cette approche a pour but de determiner les propriétés exactes d’un système à plusieurs particules en utilisant des méthodes à particules independantes, en pratique cette révolution en la matière a permis d’effectuer certaines approximations qui se sont révèlées très satisfaisantes. L’approche de Kohn et Sham remplace le système à particules interagissant entre elles par un systeme moins complexes facilement résolvables, ceci implique des équations de particules independants obtenues en regroupant tous les termes compliqués et difficile à évaluer dans une fonctionnelle d’échange-correlation Exc[n] :
3
3( ) ( )
KS ext s xc ext
E E n
d rV r T n E n
d rV r (I.7)Ts est l’énergie cinétique d’un système de particules indépendantes noyées dans un potentiel effectif qui n’est autre que celui du système réel.
2 1 1 2 Ne s e i i i T n T
EH l’énergie de Hartree ou l’énergie d’interaction de coulomb associé à l’auto interaction de la densité électronique définie par :
1 3 3 ( ) ( ') ' 2 ' Hartree n r n r E n d rd r r r
2 1 ( ) Ne i( ) i n r
rLa solution du système auxiliaire de Kohn et Sham pour l’état fondamental peut être vu tel un problème de minimisation tout en respectant la densité n(r) .L’exception de Ts qui est fonctionnelle des orbitales, tous les autres termes dépendent de la densité, par conséquent il est possible de faire varier les fonctions d’onde en ainsi l’équation variationnelle :
14 * * * ( ) 0 ( ) ( ) ( ) ( ) ( ) ( ) KS s ext Hartree xc i i i E T E E E n r r r n r n r n r r (I.8)
Avec la contrainte d’ortho-normalisation i j ij.Ceci donne la forme de Kohn-Sham pour les équations de Schrodinger : (HKS i) ( )i r 0(I.9)
i
représente les valeurs propres et HKS est l’Hamiltonian effectif
2 1 ( ) 2 ( ) ( ) ( ) ( ) KS KS Hartree xc KS ext H V r E E H r V r n r n r
Les équations (I.7)-(I.8) sont connues sous le nom des équations de Kohn et Sham, avec la densité n(r) et l’énergie totale EKS résultante. Ces équations sont indépendantes de toute
approximation sur la fonctionnelle Exc[n], leur résolution permet d’obtenir les valeurs exactes de la densité et l’énergie de l’état fondamental du système interagissant, à condition qu’Exc[n] exacte soit connue. Cette dernière peut être décrite en fonction de la fonctionnelle de Hohenberg Kohn
(
) xc HK Hartree E n E n Ts n E n (I.10) Ou plus explicitement
xc s Hartree E n T T n E n (I.11)Cette énergie est associée au potentiel d’échange-corrélation :
( ) xc xc E V n r 4. Fonctionnelle d’échange-corrélation
Comme il est décrit au-dessus, la DFT est au stade des équations de Khon-Sham, une théorie parfaitement exacte dans la mesure où la densité électronique qui minimise l'énergie totale est exactement la densité du système de N électrons en interaction. La seule ambiguïté dans l’approche de Kohn et Sham (KS) est le terme d’échange-corrélation qui est inconnu. La complexité formelle de ce dernier rend la résolution des équations de KS difficile, néanmoins cette fonctionnelle peut être soumise à des approximations de l’ordre local ou proche local de la densité, ceci dit l’énergie Exc peut être écrite sous la forme :
3 ( ) ( , ) xc xc E n
n r n r d r (I.12)
( , ) xcE n r est l’énergie d’échange-corrélation par électron au point r, elle dépend de n(r) au
15 Ces approximations ont suscité l’intérêt de plusieurs scientifiques et enregistré d’énormes progrès en la matière. Nous allons apporter quelques définitions des plus populaires d’entre elles à savoir qu’il s’agit de deux types d'approximations [14]
l'approximation de la densité locale (LDA)
l'approximation du gradient généralisé (GGA) ainsi que les méthodes dérivées qui se fondent sur une approche non locale.
a. Approximation de la densité locale (LDA)
Dans leur article original, [9] Kohn et Sham ont souligné le fait que l’on peut considérer les solides très proches d’un gaz d’électrons homogène. Dans cette limite, il est soutenu que les effets d’échange-corrélation ont un caractère local. Les deux auteurs ont proposé l’utilisation de l’approximation de la densité locale (LDA).
Cette approximation n’est autre qu’une intégrale sur tout l’espace, en supposant que
εxc[n(r)], est l’énergie d’échange-corrélation par particule d’un gaz d’électrons homogène de
densité n
hom
3
hom
hom
3( ) ( ) ( ) ( ) ( )
LDA
xc xc x c
E n
n r n r d r
n r n r n r d r(I.13)Le terme d’échange hom
x
peut être exprimé analytiquement, tandis que le terme de corrélation est calculé avec précision, utilisant la technique de Monte Carlo, par Ceperley et Alder (CA) [15] et ensuite paramétré en différentes formes. Hormis la nature locale du terme d’échange-corrélation, L’approximation LDA suppose que la distribution de la densité ne donne pas une variation rapide. En dépit de sa simplicité cette approximation a fait ses preuves notamment dans le cas traitant les systèmes non homogènes.
Globalement, on peut réecrire l’epxression (1.13)
𝜀
𝑥𝑐(𝑛) = 𝜀
𝑥(𝑛) + 𝜀
𝑐(𝑛) (1.13a)
La contribution provenant de l'échange électronique dans l'approximation de la densité locale est connue et provient de la fonctionnelle d'énergie d'échange formulée par Dirac [12]
16 L'approximation LDA peut être formulée de manière plus générale prenant en compte le spin de l'électron dans l'expression de la fonctionnelle, on parle alors d'approximation LSDA (pour
local spin density approximation). Cette approche fut initialement proposée par John C. Slater
(1900-1976) [13] et permet de résoudre certains problèmes liés à une approche LDA, notamment le traitement de systèmes soumis à des champs magnétiques et les systèmes où les effets relativistes deviennent importants. En prenant en compte l'approximation LSDA, la fonctionnelle d'échange est exprimée comme :
1/3 1/3 3 3 4/3 4/3 3 2 . ( ) ( ) 4 n r n r d r
Où α et β expriment les spins up et down.
La réussite de cette approximation dans le traitement des systèmes différents, l’a rendue très réputée et a donné naissance à de nouvelles idées pour l’améliorer. Bien que la détermination de la structure électronique des solides soit possible, notons qu’avec cette méthode, les énergies de cohésion sont systématiquement surestimées, et l’erreur augmente au fur et à mesure que la taille ou la dimensionnalité du système diminue. Cette méthode sous-estime également les gaps dans un matériau isolant, les longueurs de liaison à l’équilibre, tandis que les fréquences de vibration des petits systèmes sont généralement surestimées. Ces erreurs proviennent du modèle de gaz d’électrons homogène, car l’approximation n’est correcte que dans la limite d’une distribution de densité variant infiniment lentement. Une amélioration a ensuite été apportée à la LDA et cette nouvelle approximation prend en compte non seulement la densité électronique locale mais également un gradient local dans cette densité (qui rend ainsi compte de l’hétérogénéité de la densité). Il s’agit de l’approximation du gradient généralisé (GGA, Generalized Gradient Approximation).en particulier, les formalismes PBE (Perdew-Burke-Ernzerhof), [16] WC (Wu-Cohen)[17] et EV (Engel Vosko) [18] ont été utilisés dans cette étude. Ces méthodes non-empiriques sont simples et précise. Elles permettent de garder le meilleur de la méthode LDA tout en l’améliorant.
b. L’approximation du gradient généralisé
Cette approximation revient à considérer le terme d’échange-corrélation non plus comme une fonction uniquement de la densité, mais de manière plus générale comme une fonction de la densité n et de sa variation locale| n |. Une première approche ; l’approximation du Gradient d’Expansion (GEA) a été introduite par Kohn et Sham, ensuite elle sera utilisée par d’autres
17 auteurs notamment dans les travaux d’Herman et al. [19]. Cependant, cette approximation n’a pu apporter les améliorations escomptées à la LDA, aboutissant à de faux résultats. La notion d’approximation du gradient généralisé (GGA) réside dans le choix des fonctions, permettant une meilleure adaptation aux larges variations de telle sorte à préserver les propriétés désirées. L’expression du terme exchange-corrélation s’écrit dans sa forme générale [16]:
3
, ( ) , ,
GGA
xc xc
E n n
n r n n n nd r (I.14.a) Et le potentiel d’échange corrélation est calculé à partir de :) ( ) ( ) , ( ) , ( ) ( ) ( r r r E x V XC XC XC LDA XC (I.14.b) Où hom ( ) x n
est l’énergie d’échange d’un système non polarisé de densité n(r). Il existe plusieurs versions de la GGA, les plus utilisées sont celles de Perdew et Wang et Perdew .Meta-GGA introduite par Tao al en 2003 et GGA-WC introduite par Wu-Cohen en 2006.
c. L’approximation EV-GGA (Engel- Vosko):
Dans les deux approximations LDA et GGA, une lacune majeure apparait dans l’estimation d’énergie du gap qui est essentiellement due au terme de corrélation, et qui est jugé trop simple. Alors pour corriger cette lacune Engel et Vosko ont montré que la GGA ne s’améliore pas sur l’expansion du second ordre du gradient généralisé due généralement à l’annulation des erreurs locales ; d’où la correction apportée aux termes d’échange et corrélation, En mixant le second ordre avec le terme d’échange et corrélation de Hartre-Fock ; cette nouvelle forme (EV-GGA) améliore le calcul du gap.
III. Résolution des équations de Kohn-Sham
1. Introduction
Diverses méthodes peuvent être utilisées pour résoudre les équations de Kohn-Sham. On différencie ces méthodes selon :
le potentiel d’interaction électron-noyau (Vext).
le potentiel d’échange-corrélation.
la base d’onde sur laquelle sont développées les fonctions d’onde. Considérant l’équation (I.15)
18 2 2 ( ) ( ) ( ) ( ) ( ) 2 Ne Hartree xc i b c d a h V r V r V r i r i r m (I.15)
(a) Energie cinétique déterminée par un calcul relativiste ou non. (b) Potentiel d’interaction électron-noyau VNe.
Il existe deux grandes classes de potentiels :
Les pseudo-potentiels
Les potentiels tous électrons : type Muffin-tin ou Full Potential (c) Potentiel d’échange-corrélation :
LDA
GGA
(d) Base sur laquelle est développée la fonction d’onde:
Base numérique de type ondes planes.
Base optimisée : Orbitales linéarisées Muffin Tin (LMTO),
Ondes planes augmentées (LAPW).
Quelle que soit l’approche, les états de cœur et de valence sont traités séparément. L’équation de Schrödinger est appliquée aux électrons de valence, les électrons de cœur sont soit traités par un calcul atomique séparé, soit leur contribution est introduite dans un potentiel effectif.
2. La méthode FP-LAPW
La méthode « Full Potential Linearized Augmented Plane Wave (FP-LAPW) » développée par l’équipe de Schwarz, [20- 21] est basée sur la résolution auto-cohérente des équations de Kohn-Sham dans deux régions arbitrairement définies de la maille élémentaire.
La région I correspond à des sphères atomiques ne se recouvrant pas, de rayon Rmt (mt = muffin tin), ou on utilise une série de combinaisons linéaires de fonctions radiales et angulaires.
La région II est la région interstitielle entre les sphères. Elle est décrite par une expansion d’ondes planes. Les deux types de région, représentées schématiquement sur la Figure (I-1). La convergence de cette base est contrôlée par un paramètre de « cut-off » Rmt *Kmax qui est le produit du rayon de la plus petite sphère de muffin-tin (Rmt) par l’énergie de coupure de la base d’ondes planes. Cette méthode permet la considération d’un potentiel réaliste (FP = Full Potential) qui ne se restreint pas à la composante sphérique. Contrairement aux méthodes utilisant des pseudo-potentiels, les électrons de cœur sont intégrés dans le calcul. On obtient
19 ainsi une description correcte des fonctions d’onde près du noyau. C’est la méthode la plus précise, mais elle est lourde en temps de calcul.
Cette méthode est implémentée dans le code WIEN2k, développé par Blaha et Schwarz depuis 1990 à l’Institut de Chimie des Matériaux de Vienne.
Figure I.1 : Partition de la maille unitaire en sphères atomiques I et en région interstitielle II[22]
3. La méthode des pseudo-potentiels (ondes planes)
a. Pseudo-potentiels
En théorie de pseudo-potentiel, on part du principe que la liaison chimique dans les molécules et les solides est dominée par les électrons de valence de chacun des atomes. Un pseudo-potentiel est une approximation du pseudo-potentiel réel. En effet, cette approximation vise à substituer le potentiel d'interaction coulombien du noyau et les effets des électrons dits de cœur, considérés comme fortement liées, par un potentiel effectif interagissant uniquement avec les électrons de valence. Ainsi, on ne traite le problème des potentiels qu'avec les électrons qui jouent un rôle dans les liaisons chimiques. Cette approximation apporte un gain en ressources informatiques. L'approche du pseudo-potentiel est basée sur le fait qu'une grande majorité des propriétés physiques et chimiques des matériaux ne dépend que du comportement des électrons de valence. En effet, dans un atome, seuls les électrons périphériques (minoritaires) contribuent à la formation des liaisons chimiques alors que les électrons de cœur (majoritaire) sont ceux fortement liés au noyau atomique et donc peu sensibles à l'environnement proche des électrons de valence. On considère que les orbitales de cœur sont gelées d'où l'expression en anglais « frozen-core approximation ». Ceci consiste à
20 résoudre des équations plus réduites puisque seuls les électrons de valence sont pris en compte. [23]
Une base d’ondes planes nécessite un très grand nombre d’ondes pour décrire au mieux le système. Un moyen de réduire la base est de supprimer les ondes dont l’énergie cinétique est supérieure en valeur absolue à une certaine énergie nommée Ecut-off. On supprime alors les ondes de faible participation au développement de la base. Cette base, bien que réduite, n’est toutefois pas bien adaptée car il faut toujours un très grand nombre d’ondes planes pour décrire correctement les orbitales électroniques fortement liées. Les éléments qui possèdent peu d’électrons nécessiteront peu d’ondes planes, tandis que les éléments lourds ou les métaux de transition, vont nécessiter des moyens de calcul extrêmement puissants.
Les premiers pseudo-potentiels étaient empiriques mais une amélioration importante a été apportée par l’introduction de la condition de conservation de la norme[24] qui a permis de définir des pseudo-potentiels (PP) sans paramètres ajustables, et qui a eu une meilleure précision et une meilleure transférabilité.
Étant donné que les PP pour un système donné ne sont pas uniques, ils peuvent être crées pour un besoin spécifique et avec des propriétés spécifiques. Par exemple, un PP général pour un système donné conduit généralement à des pseudo-fonctions d’ondes qui ne sont pas orthonormées. Et puisque l’ortho-normalité est une propriété fortement appréciée pour les fonctions d’ondes, des PP à norme conservée NCPP sont développés. Les pseudo-fonctions d’ondes de ces derniers sont solutions normalisées d’un PP choisi pour reproduire les propriétés des électrons de valence dans un calcul tout-électron, ce qui les rend plus précises. Malgré les performances da la méthode PP, un problème numérique de taille persistait du fait de la "dureté" de ces pseudo-potentiels, qui nécessitait un très grand nombre d’ondes planes. L’approche "ab initio norm-conserving" a été étendue notamment par David Vanderbilt [25], avec la création des pseudopotentiels ultra-doux. En effet, les pseudo-potentiels associés à des énergies de coupure (Ecut-off) élevées sont appelés « hard », par comparaison aux pseudo- potentiels appelés «soft».
Vanderbilt a développé une base d’ondes plus réduite que ces pseudo-potentiels traditionnels,
avec des énergies de coupure encore plus basses. Ces pseudo-potentiels sont appelés «ultra-soft ». Ceci a permis de considérer des systèmes plus complexes et/ou de grande taille.
Une autre approche du cœur gelé qui évite les problèmes dus aux pseudo-potentiels « ultra-soft est le formalisme PAW (Projector Augmented-Wave), développé par Blöch [26]
implementé dans le code QUANTUM ESPRESSO. [27] Cette méthode permet de décrire, de
21 les fonctions d’ondes varient fortement. La taille de la base d’ondes planes est équivalente à celles des pseudo-potentiels ultra softs (taille minimale), mais nous avons de plus accès à la « vraie » densité électronique du système. La méthode PAW est donc a priori aussi précise qu’une méthode « tous électrons ».
b. La génération d’un pseudo-potentiel
La génération d’un pseudo-potentiel s’effectue en deux étapes : tout d’abord un calcul “tout électron” de l’atome seul dans une configuration atomique de référence, en général l’état fondamental, puis la pseudisation des fonctions d’ondes “tout-électron” obtenues, afin de générer les paramètres du pseudo-potentiel. Lors de ce dernier processus, un certain nombre de conditions sont vérifiées :
– les énergies des états propres “tout électron” et “pseudisés” sont égales ;
– les fonctions d’ondes “tout électron” et “pseudisés” sont identiques au-delà du rayon de coupure rc ;
– les dérivées logarithmiques des fonctions d’onde “tout électron” et “pseudisés” calculées en rc sont égales pour les énergies des états propres.
Figure III.2 : Diagramme illustrant le replacement d’une fonction d’onde et un potentiel de cœur par un pseudo-potentiel et une pseudo-fonction [30]
c. Procédure de culcul.
Le calcul “tout électron” est effectué dans le formalisme DFT, en utilisant une fonctionnelle d’échange-corrélation (LDA ou GGA), et de manière relativiste [28] ou non. Le calcul étant à symétrie centrale, la résolution se fait sur une grille radiale. Il faut donc spécifier pour cette partie plusieurs paramètres :
– le numéro atomique de l’atome.
22
– les effets relativistes sur les électrons de cœur peuvent être pris en compte, et c’est fait lors de la génération du pseudo-potentiel.
– la précision de la grille radiale, donne la précision du calcul, mais a peu d’impact sur la rapidité du pseudo-potentiel.
– la configuration électronique de référence, est en général l’état fondamental non polarisé. En résumé, cette méthode introduit comme seule approximation le fait que les électrons de cœur ne sont pas influencés par l’environnement des atomes. On parle de l’approximation de cœur gelé. Cette approximation peut provoquer des erreurs non négligeables dans le cas de calcul d’énergie avec extraction d’un électron de cœur. Même s’il est possible à partir du formalisme PAW de prendre en compte la relaxation des électrons de cœur, comme l’ont montré M. Marsman en 2006 [29].
IV. Approches Correctifs
1. Introduction
La théorie de la fonctionnelle de la denité a été reconnue pour son aptitude théorique à décrire les propriétés de l'état fondamental des molécules et des solides. Cette méthode est valable en principe pour des densités lentement variables. Son mérite tient essentiellement au fait qu'elle traite de manière égale l’énergie d'échange et l'énergie de corrélation, ce qui n'est pas réalisé dans la méthode de Hartree-Fock. Cependant, certaines prédictions de la LDA ne s'accordent pas avec l'expérience. Par exemple, l'énergie de cohésion de la plupart des solides et l'énergie de liaisons des molécules sont souvent surestimées, alors que leur distance à l'équilibre est sous-estimée. Autre exemple, l'approximation LSDA est fondée sur la théorie du gaz homogène d'électrons, il semble logique d'imputer le désaccord relevé pour ce type de matériaux à la présence d’électrons fortement localisés. Cette forte localisation donne lieu à une densité qui n'est plus lentement variable et engendre des inhomogénéités. L’énergie de corrélation évaluée par LSDA est alors sous-évaluée et ne corrige pas suffisamment le terme de répulsion coulombienne. Ce défaut apparaît essentiellement pour des systèmes comportant des électrons d ou f en couche ouverte. Pour réduire le désaccord survenant lors de l'application pratique de la théorie de Kohn-Sham, de nombreuses améliorations ont été proposées, elles tentent de corriger l’approximation elle-même. La question est donc de savoir si ce désaccord et notamment l'erreur sur la prédiction de la bande interdite dans les calculs DFT-GGA est imputable à l’utilisation de la GGA ou inhérente la DFT elle-même.
Pour les systèmes périodiques, l’équation de KS pour une fonction d’onde d’un seul électron s’écrit:
![Figure III.2 : Diagramme illustrant le replacement d’une fonction d’onde et un potentiel de cœur par un pseudo-potentiel et une pseudo-fonction [30]](https://thumb-eu.123doks.com/thumbv2/123doknet/2198829.12266/32.892.150.644.585.794/figure-diagramme-illustrant-replacement-fonction-potentiel-potentiel-fonction.webp)
![Figure I.3 représentation schématique de différentes méthodes basées sur la DFT[44]](https://thumb-eu.123doks.com/thumbv2/123doknet/2198829.12266/39.892.147.779.689.1052/figure-representation-schematique-methodes-basees-dft.webp)
![Figure I I.1 : Transitions inter bandes a) Gap directe b) Gap indirecte [46]](https://thumb-eu.123doks.com/thumbv2/123doknet/2198829.12266/43.892.209.682.394.613/figure-transitions-inter-bandes-gap-directe-gap-indirecte.webp)
![[PDF] Cours sur les bases des feuilles de style css | Formation css](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)