Conformation moléculaire,structure cristalline, spectroscopie vibrationnelle du dibromomésitylène
191
0
0
Texte intégral
(2)
(3) E DEMOCRATIQUE ET POPULAIRE MI
(4) ISTERE DE L’E
(5) SEIG
(6) EME
(7) T SUPERIEUR ET DE LA RECHERCHE SCIE
(8) TIFIQUE U
(9) IVERSITE ME
(10) TOURI-CO
(11) STA
(12) TI
(13) E FACULTÉ DES SCIE
(14) CES EXACTES DEPARTEME
(15) T DE PHYSIQUE
(16) ° d’ordre Série THESE Présentée pour obtenir le titre de DOCTEUR E
(17) SCIE
(18) CES E
(19) PHYSIQUE Option Cristallographie Intitulée. Conformation moléculaire, structure cristalline, spectroscopie vibrationnelle du dibromomésitylène. Conséquences des interactions intermoléculaires dans les halogénomésitylènes cristallisés PAR. Soria ZEROUAL Soutenue le : 15 / 12 / 2013 Devant le jury : Président:. Miloud SEBAIS. Prof.. Université Constantine 1. Rapporteur:. Ali BOUDJADA. Prof.. Université Constantine 1. Co-Rapporteur:. Jean MEI
(20)
(21) EL. Prof.. Université Rennes 1 France. Examinateurs:. Touhami LA
(22) EZ. Prof.. Université d’El-Oued. Omar KHALFALLAH. Prof.. Université Constantine 1. Azzeddine CHELLOUCHE M.C.A.. Université A. Mira-Bejaia.
(23)
(24) Remerciements Les travaux de recherches qui font l’objet de cette thèse ont été réalisés dans le cadre d’une collaboration entre le laboratoire de Cristallographie de la faculté des Sciences de l’université de Constantine et l’UMR Sciences Chimiques de Rennes.. J’exprime ma gratitude à Monsieur le Professeur Boudjada Ali, Ali directeur de thèse, pour avoir diriger cette thèse, et pour l’aide précieuse et le temps qu’il a bien voulu me consacrer. Je tiens a adresser mes vifs remerciements à Monsieur le Professeur Jean Meinnel, Meinnel co-directeur de ma thèse, il m’a transmis la passion de la recherche physique et n’a eu de cesse de m’encourager et de me soutenir durant mon séjour à Rennes. J’adresse mes remerciements les plus respectueux à Monsieur le professeur Sebais Miloud d’avoir accepté de présider mon jury. Je remercie très sincèrement Messieurs les professeurs Kalfallah Omar, Omar Lanez Touhami et le docteur Chell Chellouche Azzedine d’avoir accepté d’examiner le travail de ma thèse. J’ai effectué ma thèse au sein du groupe MaCSE au Laboratoire des Sciences Chimiques de Rennes 1, j’aimerais remercier son directeur Monsieur le professeur Marc Fourmigué pour son accueil. Je tiens a adresser mes plus chaleureux remerciements au Pr. Abdou Boucekkine, Boucekkine pour la réalisation des calculs DFT , pour sa gentillesse et sa disponibilité. Je souhaite remercier Mme.Marie le Flock pour les mesures RMN, Mr. Alain Moréac pour les spectres Raman et Mr.Yann Le Gal pour les spectres IR..
(25) Je remercie le ministère de l’enseignement supérieur et de la recherche scientifique Algérien qui en m’accordant une bourse BAF, m’a permis de finaliser ma thèse.. Enfin, je souhaite remercier les membres de ma famille pour leur soutien constant. Cette thèse, aboutissement de longues années d’études, je la dois beaucoup a mes défunts parents et à mes sœurs. Il m’est très difficile de trouver des mots pour dire a quel point je suis fier d’eux, et a quel point je les aime. Pour conclure, je souhaite bien évidemment remercier mon époux Sadek , merci de m’avoir soutenu toutes ces années..
(26) Dédicace. Je dédie cette thèse A la mémoire de mes parents A mon mari Sadek A mes filles Kinda et Minna A mes Sœurs et à mes Frères A mes beaux frères A mes Nièces et à mes Neveux A mes amies : Saliha, Nadjet, Samia, Assia, Nawel… A toutes mes collègues. Et à la mémoire de Miloud.
(27) Sommaire.
(28) Sommaire. Introduction générale ...........................................................................................................1 Chapitre I : Etude bibliographique : Structure cristalline des benzènes polysubstitués et effet de la substitution sur la symétrie moléculaire. .........7 Introduction .....................................................................................................................................8 I.1 Benzène monosubstitué..............................................................................................................9 I.2 Benzène disubstitué..................................................................................................................10 I.3 Benzènes trisubstitués ..............................................................................................................12 I.4 Benzènes tétrasubstitués...........................................................................................................12 I.5 Benzènes pentasubstitués .........................................................................................................13 I.6 Benzènes hexasubstitués ..........................................................................................................13 I .6.1 Benzènes hexasubstitués avec un même substituant........................................................13 I .6.2 Benzènes hexasubstitués avec différents substituants......................................................15 Chapitre II : Aperçu sur les concepts théoriques et les techniques expérimentales utilisées.....................................................................................................20 II.1 La diffusion neutronique.........................................................................................................21 II.1.1 Les propriétés du neutron.................................................................................................21 II.1.2 Concepts théoriques .........................................................................................................21 II.1.2.1 Définition des sections efficaces de diffusion .........................................................21 II.1.2.2 Diffusion inélastique des neutrons...........................................................................22 II.1.2.3 Diffusion quasi élastique..........................................................................................25 II.1.3 Modèle de la particule isolée ...........................................................................................26 II.2 Théorie des fonctionnelles de la densité (DFT)......................................................................27 II.2.1 Principe des calculs DFT .................................................................................................27 II.2.2 Méthode de Kohn-Sham ..................................................................................................28 II.2.3 Les principales méthodes de la DFT................................................................................29 II.3 Spectroscopie de résonance magnétique nucléaire .................................................................31 II.3.1 Principe de la RMN. ........................................................................................................31 II.3.1.1 Introduction...............................................................................................................31.
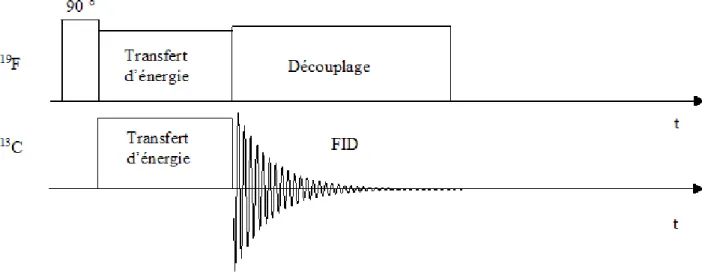
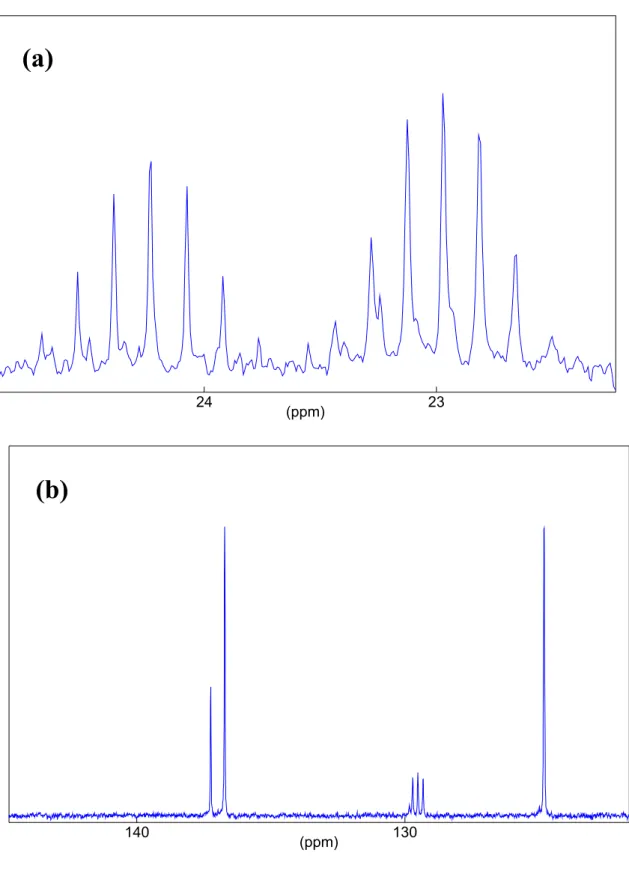
(29) II.3.1.2 Principe .....................................................................................................................31 II.3.1.3 Interaction dipolaire ..................................................................................................33 II.3.1.4 Phénomène de relaxation ..........................................................................................33 II.3.1.5 Blindage et déblindage des protons ..........................................................................35 II.3.1.6 Déplacements chimiques ..........................................................................................35 II.3.1.7 L'interaction dipolaire ...............................................................................................37 II.3.1.8 L'interaction quadrupolaire .......................................................................................38 II.3.2 Techniques expérimentales..............................................................................................38 II.3.2.1 Séquence simple........................................................................................................39 II.3.2.2 Séquence CP-MAS ...................................................................................................39 II.3.2.3 Séquence de saturation – récupération......................................................................41 II.4 Spectroscopie Raman et infrarouge (IR).................................................................................42 II. 4.1 La spectroscopie Raman .................................................................................................42 II.4.1.1 Principe .....................................................................................................................42 II.4.1.2 La polarisabilité ........................................................................................................44 II.4.1.3 Dispositif expérimental.............................................................................................45 II.4.2 La spectroscopie Infrarouge.............................................................................................47 II.4.2.1 Principe .....................................................................................................................47 II.4.2.2 Dispositif expérimental.............................................................................................47 Chapitre III : Synthèse, spectroscopie de masse et spectroscopie de résonance magnétique nucléaire du DBM(10H) et DBM(10D) .........................49 III.1 Synthèse du dibromo-mésitylène (DBM) deutéré .................................................................50 III.2 Spectroscopie de masse du DBM deutéré et hydrogéné........................................................50 III.3 Spectroscopie RMN liquide et solide du DBM (10H) et DBM(10D) : résultats expérimentaux ...............................................................................................................................53 III.3.1 Spectres RMN liquide 1H et 13C du DBM (10H) ...........................................................53 III.3.2 Spectres RMN 2H et 13C du liquide du DBM (10D) ......................................................55 III.3.3 Spectres RMN CP/MAS 1H et 13C du DBM ..................................................................57 III.3.4 Attribution spectrale .......................................................................................................59 III.3.5 Interprétation et discussion des résultats ........................................................................61 III.4 Conclusion .............................................................................................................................62.
(30) Chapitre IV : Calorimétrie différentielle à balayage (DSC), structure cristalline, calcul DFT et calcul des interactions intermoléculaires du DBM.............................................................................................................................................63 IV. 1 Calorimétrie différentielle à balayage (DSC) du DBM........................................................63 IV.2 Structure cristalline du dibromo-mésitylène (DBM) hydrogéné (étude antérieure) ............67 IV.3 Conformation moléculaire calculée par la théorie de la fonctionnelle de la densité (DFT) du DBM hydrogéné et deutéré ...........................................................................................................71 IV.4 Calcul du potentiel intermoléculaire dans la phase monoclinique du DBM........................74 IV.4.1 Introduction ....................................................................................................................74 IV.4.2 Description de la position relative des proches voisins autour d'une molécule témoin .77 IV.4. 3 Choix des coefficients d'interaction interatomiques en (exp-6) pour le calcul du potentiel de VdW du DBM........................................................................................................82 IV.4.4 L'énergie de VdW due à l'interaction de la molécule témoin 0M0 avec chacun de ses voisins........................................................................................................................................84 IV.4.5 L’énergie d'empilement dipolaire de la paire (0M0 + bm0) ou (0M0+ am0), énergie d'interaction entre des paires voisines. ......................................................................................87 IV.4.6 Calcul de la part du potentiel gênant la rotation du Me2, créé par son environnement moléculaire. ...............................................................................................................................88 IV.5 Conclusion.............................................................................................................................91 Chapitre V : Spectroscopie vibrationnelle et tunneling du dibromomésitylène hydrogéné et deutéré. ................................................................92 V.1 Spectroscopie vibrationnelle du DBM hydrogéné et deutéré .................................................93 Introduction ...............................................................................................................................93 V.1.1 Spectroscopie vibrationnelle du DBM hydrogéné...........................................................96 V.1.1.1 Spectres IR, Raman et INS du DBM hydrogéné ......................................................96 V.1.1.2 Modes internes du DBM (10H) calculés à partir de la DFT et attribution expérimentale.......................................................................................................................101 V.1.2 Effet isotopique dans le DBM (10H) et DBM (10D) : Spectres Raman et IR du DBM deutéré, calcul de DFT.............................................................................................................119 V.2 Eclatement tunnel des groupes méthyle ...............................................................................125.
(31) V.2.1 Eclatement tunnel des groupements méthyle dans les dihalogéno-mésitylènes hydrogénés...............................................................................................................................127 V.2.2 Tunneling dans le dibromo-mésitylène hydrogéné .......................................................128 V.2.3 Etude de l’éclatement tunnel du CD3 dans le dibromomésitylène deutéré....................133 V.2.4 Effet de la température sur le tunneling.........................................................................136 V.2.5 Tunneling dans le dichloro-mésitylène hydrogéné........................................................138 V.3 Conclusion ............................................................................................................................142 Chapitre VI : Interactions intermoléculaires dans les trihalogénomésitylènes : tribromomésitylène (TBM), trichloromésitylène (TCM) et triiodomésitylène (TIM)……………………………………………………144 Introduction .................................................................................................................................145 VI .1 Calcul de la conformation moléculaire moyenne...............................................................145 VI .2 Calcul du potentiel d’interaction .......................................................................................156 VI .2.1 Définition de l’espace d’interaction ...........................................................................156 VI .2.2 Paramètres utilisés dans le calcul du potentiel de Van der Waals ..............................157 VI .2.3 Résultats et discussions ..............................................................................................159 VI .2.4 Potentiel rotationnel moléculaire ................................................................................165 VI .3 Conclusion .........................................................................................................................166 Conclusion générale...........................................................................................................167 Références……...……………………………………………………………………………172.
(32) Introduction générale.
(33) Introduction générale. Il est communément admis que les transitions de phase structurales dans les cristaux moléculaires sont intimement corrélées à la liberté d'orientation des molécules ou de leurs groupes moléculaires (par exemple NH3, CH3 . . .). C'est la raison pour laquelle l'étude du comportement des molécules ou des rotateurs en fonction de la température et de l'environnement moléculaire dans l'état solide présente un grand intérêt. Les particules légères se déplaçant dans un potentiel avec une dégénérescence topologique manifeste leur nature quantique par effet tunnel (tunneling en anglais). Dans la matière condensée, l'observation du tunneling, jette la lumière sur les problèmes fondamentaux de la mécanique quantique dans des environnements complexes. Pour un groupe isolé en rotation autour de son axe d'inertie supposé être fixe, l'amplitude de l’éclatement tunnel dépend de la masse des particules et de la forme du potentiel. La limite supérieure de l’éclatement tunnel est la constante de rotation B = h 2 2 I (I est le moment d’inertie du rotateur). En particulier, à basse température, la rotation d'un groupe tel que le méthyle ne se fait plus par des sauts thermiquement assistés, mais obéit à un mécanisme quantique : l'effet tunnel du proton à travers une barrière de potentiel [1]. Ce phénomène, dépendant des faibles changements de l'environnement moléculaire, constitue une sonde sensible aux forces interatomiques. Cependant, l'observation des transitions tunnel pour les analogues deutérés est gênée par la section efficace beaucoup plus faible des atomes D et par la baisse spectaculaire de l’éclatement après deutération, donc la deutération a des conséquences importantes sur les fréquences tunnel [2]. Les rotateurs quantiques libres sont des systèmes intéressants parce que, en plus du traitement des problèmes d'intérêt fondamental, ils sont sensibles au désordre [3] et permettent l'observation des effets de couplage entre les rotateurs ou les modes rotationnels et vibrationnels avec une précision particulière [4]. Cependant, on ne connaît que seulement quelques solides dans lesquels les dynamiques de rotation approchent les limites des rotations quantiques libres, donc la comparaison entre les prédictions théoriques et les résultats expérimentaux est limitée à seulement quelques exemples [4,5].. 2.
(34) Introduction générale. Un des problèmes fondamentaux dans les cristaux moléculaires concerne la dynamique des protons, l'influence des rotateurs tels que le groupe méthyle (CH3) dans les transitions de phase structurales, les couplages entre rotateurs et enfin l’existence des transitions par effet tunnel de ces derniers. Les techniques les plus couramment utilisées pour étudier le comportement des molécules ou des rotateurs sont la diffraction des rayons X et des neutrons, la diffusion inélastique des neutrons (INS), la résonance magnétique nucléaire (RM
(35) ), les spectroscopies infrarouge (IR) et Raman et la calorimétrie. Bien que ces techniques aient leurs avantages et inconvénients, elles fournissent des données complémentaires. Cette accumulation d’excitations d’origine diverses pose le problème de leur attribution ainsi que celui de leur couplage éventuel, en particulier entre les rotateurs méthyles et les vibrations moléculaires. Afin d'élucider les propriétés structurales et les comportements conformationnels d'une molécule, les calculs de mécanique quantique sont très importants. Parce que l’interprétation des résultats expérimentaux repose sur la stabilité conformationnelle [6]; ils se sont avéré un outil essentiel pour l'interprétation des résultats expérimentaux et la prévision des spectres de vibration [7,8]. En raison de l'augmentation de la fiabilité avec laquelle les champs de force peuvent être calculés avec les méthodes de chimie quantique, basées sur la théorie de la fonctionnelle de la densité (DFT), la compréhension des spectres de vibration des molécules organiques de petite et grande taille a fait des progrès considérables ces dernières années. La précision de la méthode de calcul est établie par la capacité de reproduire les spectres expérimentaux. Ces calculs, fournissent dans la plupart des cas un accord presque parfait avec l'expérience, de sorte que l’attribution des modes normaux fondamentaux n'est plus soumise à des incertitudes associées aux champs de force empiriques. Mais, l’accord peut être moins satisfaisant, comme par exemple pour les mouvements impliquant des liaisons hydrogène, qui continuent de présenter un intérêt en ce qui concerne la perfection des méthodes de mécanique quantique. Les interactions de Van der Waals et, par conséquent, les caractéristiques spectrales comme les modes externes à basses fréquences ne sont pas nécessairement bien reproduits [9].. 3.
(36) Introduction générale. En outre, pour un rotateur Me peu gêné, les protons du Me sont largement réparties autour de l'atome de carbone, en conséquence, la validité et l'exactitude des calculs théoriques qui sont fondés sur l’approximation. harmoniques sont soumis à prudence pour touts les. mouvements impliquant des groupes Me. En plus, à l’état solide, les changements de la conformation du Me peuvent se produire à cause du champ cristallin, ce qui modifie le spectre de torsion. En conséquence, les fréquences des modes de torsion seraient indicatives de la conformation du Me et, simultanément, de ses potentiels gêneurs. Dans le cas de molécules avec des groupes méthyle entouré par un atome substituant lourd (Cl, Br, ou I), une difficulté apparaît dans l’attribution des fréquences de torsion du Me, car ils apparaissent dans la même gamme de fréquence que les vibrations du réseau et quelques modes internes. Dans le présent travail on est particulièrement intéressé par l’étude des benzènes polysubstitués, en particulier les halogénomésitylènes où la substitution est la même sur les deux cotés d’un groupement méthyle ; Ces molécules se distinguent à la fois par leurs haute symétrie et par l’existence d’un éclatement tunnel important indiquant que les groupements méthyle sont peu gênés dans leurs réorientation. Par conséquent, nous avons entrepris une étude détaillée par RMN,. spectroscopie. IR,. Raman. et. INS. du. 1,3-dibromo-2,4,6. triméthylbenzène. (dibromomésitylène : DBM) hydrogéné et entièrement deutéré, en plus d’une étude théorique par DFT. Dans ce composé, le groupement méthyle entouré par deux atomes de brome se comporte comme un rotateur quantique quasi-libre. En revanche, la rotation des deux autres groupements méthyle situés entre un atome de brome et un atome d’hydrogène est fortement gênée. Cette étude nous permettra de détecter d’autres conséquences de la présence de deux types de groupes méthyle dans la même molécule. D’un autre coté, l'objectif commun de plusieurs études est principalement de déterminer la nature du potentiel cristallin (amplitude et symétrie) et de décrire le mouvement des molécules ou des rotateurs en fonction de la température. Dans les matériaux organiques, les liaisons chimiques fortes assurent la structure chimique des molécules. L’organisation cristalline reste possible à l’état solide mais par arrangement, cette fois-ci, des molécules qui interagissent par l’intermédiaire essentiellement de. 4.
(37) Introduction générale. liaisons de Van der Waals, qui sont des interactions faibles vis-à-vis des liaisons chimiques. L’état fondu est également possible par changement de conformation et d’orientation relative des molécules [10]. Les interactions de Van der Waals agissent entre atomes non directement liés car n’appartenant pas à la même molécule ou parce qu’ils sont trop éloignés sur la même molécule pour être impliqués dans une liaison covalente. Le modèle le plus classique est le modèle de Lennard-Jones avec une variante plus réaliste proposée par Buckingham [10]. Elles sont à la base d’un grand nombre de phénomènes plus ou moins complexes, présents aussi bien en chimie, qu’en biologie et qu’en physique du solide. En conséquence, elles reçoivent une attention toute particulière tant au niveau expérimental que théorique. Expérimentalement, elles sont, par exemple, observées et désignées comme responsables de la cohésion de nombreux cristaux liquides et moléculaires, mais aussi présentes dans les phénomènes de polymérisation [11], et assurent la cohésion de matériaux graphitiques [12] et d’autres systèmes lamellaires. Il a d’ailleurs été montré récemment toute leur importance dans les phénomènes d’adhésion [13] et de physisorption. De plus, il est bon de rappeler le rôle joué par ces interactions dans la biologie, et de manière plus générale dans la science du vivant : la spécificité de site de l’ADN ou encore sa réplication sont autant d’exemples frappants pour lesquels une compréhension parfaite des mécanismes est absolument nécessaire. Citons enfin le rôle central de ces forces dans les processus "hôte-récepteur" lors de réactions impliquant des protéines [14] et plus simplement encore lors de la formation de complexe au cours d’une réaction chimique. Enfin, l’autre intérêt de l’étude de ces forces est le problème qu’elles posent théoriquement quant à leur inclusion dans les méthodes de chimie quantique ab initio mais aussi dans le cadre spécifique de la théorie de la fonctionnelle de la densité (DFT) [15]. Pour examiner l’emballage des molécules et comprendre l’effet de l’environnement, nous avons calculé les interactions intermoléculaires dans les trihalogénomésitylènes (tribromo, trichloro et triiodomésitylène : TBM, TCM, TIM). L'INS sur ces monocristaux [16,17] a été utilisée pour établir que les trois groupes méthyle dans chaque molécule ont un éclatement tunnel différent, trois excitations tunnel sont ainsi observées dans chaque composé, allant de 4 à 13µeV pour le TCM, de 14 à 49 µeV pour le TBM, et 14 à 88 µeV pour le TIM ; ce qui est inattendu pour une molécule de symétrie C3. Et contrairement à la tendance attendue compte tenu de la molécule isolée, la hauteur de la barrière gênant la rotation du groupe méthyle diminue avec l'augmentation de la taille de l’atome de l'halogène. Tous ces résultats. 5. indiquent que la.
(38) Introduction générale. contribution dominante pour la barrière de rotation est due à l'interaction des molécules voisines avec les groupes méthyles situés dans des sites inéquivalents dans un cristal triclinique, plutôt que de l'interaction intramoléculaire. L'influence des interactions intra et intermoléculaires sur les propriétés du tunneling des groupes méthyle dans le cas du dibromomésitylène DBM (totalement hydrogéné ou deutéré) est encore plus spectaculaire. Pour comprendre pourquoi l'environnement du Me2 dans le DBM n’ajoute que seulement une faible contribution à la barrière empêchant sa rotation, il était nécessaire d'examiner en détail la structure du cristal, et ensuite évaluer quantitativement les différentes contributions aux forces de cohésion, nous avons évalué séparément les interactions de Van der Waals et les interactions électrostatiques. Le travail de cette thèse est présenté dans six chapitres : Le premier chapitre, est une synthèse bibliographique de la structure cristalline des benzènes polysubstitués et l’effet de la substitution sur la symétrie moléculaire. Dans le deuxième chapitre, nous abordons quelques éléments théoriques indispensables à la compréhension de ce travail, et les techniques expérimentales utilisées dans cette étude. Dans le troisième chapitre, nous présentons la synthèse, la spectroscopie de masse et la RMN en solution et du solide, du dibromomésitylène hydrogéné et deutéré (DBM(H) et DBM(D)). La première partie du quatrième chapitre est consacrée à la DSC du DBM(H) et DBM(D), la structure cristalline et la conformation moléculaire en utilisant les calculs DFT, tandis que dans la deuxième partie on étudie les interactions intermoléculaires dans le DBM. La spectroscopie de vibration et l’étude du tunneling font l’objectif du cinquième chapitre Le dernier chapitre, est consacré à la présentation des résultats du calcul des interactions intermoléculaires dans les trihalogénomésitylènes (TCM, TBM et TIM). Nous terminons par une conclusion générale qui résume les différents résultats obtenus.. 6.
(39) Chapitre I. Etude bibliographique : Structure cristalline des benzènes polysubstitués et effet de la substitution sur la symétrie moléculaire.. 7.
(40) Chap. I. Etude bibliographique. Introduction Le benzène a été découvert en 1825 par le scientifique britannique Michael Faraday qui l'isola du pétrole et le baptisa bicarburet of hydrogen. Sa structure n’a été résolue qu’en 1932, puis affinée en 1958 par Cox [18]. Il cristallise dans le groupe d’espace Pbca avec quatre molécules par maille. La molécule présente une symétrie D6h parfaite avec des angles C-C-C et C-C-H de 120°. En 1964 Bacon [19] a repris l’affinement des longueurs des liaisons C-C et le calcul des positions des atomes d’hydrogènes par diffraction neutronique à 218K et138K ; il a trouvé la valeur 1.393Å pour la longueur de la liaison C-C, 1.086Å pour la liaison C-H et 120° pour les angles C-C-C et C-C-H. Pour mieux voir l’effet de la substitution sur le cycle benzénique, nous avons rassemblé ici les études faites sur un nombre de composés benzéniques substitués. La résolution de la structure d’un grand nombre de composés substitués du benzène solide à température ambiante dans les années soixante a montré que la substitution par des groupements donneurs ou accepteurs provoque une modification de la géométrie hexagonale du noyau benzénique (Fig.1.1), en agissant particulièrement sur les angles autour du point de substitution, avec une légère variation des longueurs des liaisons géminales. Dominicano et al [20] et après une étude détaillée sur la géométrie moléculaire de plusieurs dérivés du benzène ont montré que si le substituant est un groupe fonctionnel attractif, la déformation globale a pour effet d’abaisser la symétrie du noyau de D6H à C2V et peut se décrire comme : •. Un accroissement de quelques degrés au dessus de 120° pour l’angle α du cycle en face du substituant X.. •. Une décroissance mineure des deux angles adjacents endocycliques ß.. •. Un raccourcissement de 10-2 à 2. 10-2 Å des liaisons a par rapport aux liaisons adjacentes b.. Les angles restants du cycle γ et δ semblent très peu affectés.. 8.
(41) Chap. I. Etude bibliographique. a δ. α γ. X. β. Fig.1.1. Schéma représentatif de la notation des angles du benzène substitué. I.1 Benzène monosubstitué Les composés benzéniques mono ou disubstitués sont généralement liquides à la température ambiante (293K), ce qui fait que relativement peu de structures ont été faites à des températures suffisamment éloignées de la température de fusion afin d’avoir un nombre de réflexions important en évitant des corrections de longueurs de liaison tenant compte des agitations thermiques. * Cas du fluorobenzène Nyggard (1968) a étudié la structure du fluorobenzéne [21], il a trouvé une valeur de 123°4 pour l’angle α en face du fluor et 117.°9 pour les angles β et des liaisons géminales raccourcies à 1.383 Å, ce qui est en bon accord avec les observations de Dominicano [20]. •. Cas du chlorobenzène. André et al (1971) [22] ont montré l’influence significative de la substitution d’un hydrogène par un atome de chlore, qui apparait dans l’augmentation de l’angle α du cycle de 2°4 et le raccourcissement de la liaison C-C à 1.372 Å. •. Cas de l’iodobenzène. R.Ugolini et P.Diehl (1989) [23] ont donné la conformation moléculaire de l’iodobenzène en utilisant la spectroscopie RMN. Ils ont trouvé la valeur 121°13(13) pour l’angle intracyclique face à l’iode, et en comparant ce composé avec le chlorobenzène, le fluorobenzène et le benzène, ils ont conclu que cet angle varie linéairement avec l’électronégativité du substituant (Fig.1.2).. 9.
(42) Etude bibliographique. Angle alpha. Chap. I. Électronégativité Fig.1.2.Variation de l’angle face au substituant en fonction de son électronégativité [23]. I.2 Benzène disubstitués Notre intérêt porte sur du benzène substitué par des halogènes et des méthyles et nous avons limité notre recherche aux dihalogéno-mésitylènes dont la formule brute est C6H4X2 ( X= Cl, Br, I). Les halogènes sont substitués soit en position ortho, méta ou para. * Méta-chlorobenzène La structure moléculaire du méta-chlorobenzène (Fig.1.3) a été déterminée par Anders et al [24] par diffraction neutronique. Elle montre une déformation de la géométrie du cycle due à la substitution par le chlore. Les distances C-C adjacentes au Cl sont plus courtes que les autres et les angles intracycliques concernant les atomes de carbones liés au chlore sont plus grands que 120°.. Fig.1.3. Structure moléculaire du m-dichloro-mésitylène [24]. 10.
(43) Chap. I. Etude bibliographique. * Para-dibromobenzène (PDBB) Le para-dibromobenzène cristallise dans le système monoclinique, groupe d’espace P21/a avec deux molécules par maille [25]. Sa structure moléculaire par diffraction des électrons en phase gazeuse a été étudiée en utilisant différents jeux de données expérimentales. L’angle du cycle en face du substituant est monté à 121°8(2). * Para-diiodobenzène (PDIB) La structure du para-diiodobenzène a été étudiée par Lyan et al [26], il cristallise dans le système Pbac avec quatre molécules par maille et les paramètres sont : a=17.008 (2) Å, b=7.321 (2) Å et c=5.949(2) Å. C. A. Meriles et al [27] ont étudié la structure cristalline du p-chlorobromobenzène et pchloroiodobenzène. Les deux composés cristallisent dans le système monoclinique P21/c avec les paramètres suivants : 1-Cl-4-Br-benzène (PCBB): a=4.0238 (6) Å, b=5.8034 (10) Å, c=15.083 (2) Å et. β =112.23(1) ° 1-Cl-4-I-benzène (PCIB): a=4.1762 (4) Å, b=5.8814(9) Å, c=15.79(2) Å et β =113.42(1) ° Ces structures présentent un désordre réorientationnel, chaque molécule peut se pointer dans deux directions possibles par une rotation de 180° (Fig.1.4). La molécule perd la symétrie hexagonale du benzène et un accroissement de l’angle face aux substituants apparait : α =121.6 (4)° dans le cas du PCBB et 121.5(5)° pour le PCIB.. (b). (a). Fig.1.4. Schéma représentatif des ellipsoïdes de l’agitation thermique à une probabilité de 30% [27] de : (a) La molécule du PCBB. (b) La molécule du PCIB.. 11.
(44) Chap. I. Etude bibliographique. I.3 Benzènes trisubstitués Dans le même but : Pour mieux voir l’influence du substituant sur la symétrie du cycle benzénique, des dérivés 1-3-5 (des halogènes et des méthyles) du benzène ont été étudiés par diffraction électronique [28]. Dans le cas du trinitrobenzène, une diminution de la longueur de la liaison C-C à 1.388 Å a été observée avec un accroissement de l’angle α à 123°2. Ce qui est l’opposé du cas du triméthylbenzène, où l’angle α a diminué à 118.2°, toutefois, la distance C-C ne diffère pas de celle du benzène. Les effets observés sont régis par le caractère accepteur-donneur caractéristique des substituants. Pour le tribromobenzène, une étude similaire a été faite en phase gazeuse pour déterminer les paramètres structuraux de la molécule ; la valeur trouvée pour la longueur de la liaison C-C est 1.395Å et. l’angle α =120.4°. Ces valeurs sont intermédiaires entre les valeurs des deux. premiers composés, ce qui est dû à l’atome du brome dont l’électronégativité est intermédiaire entre celles des groupes NO2 et CH3.. I.4 Benzènes tétrasubstitués Prenant comme exemple le 1, 2, 4, 5 tétrabromobenzène étudié par Gafner et al [29,30]. Ce composé possède deux phases : la phase β en dessous du point de transition à 319.5 K et la phase γ au dessus de cette température et avant le point de fusion. Les deux phases cristallisent dans le système monoclinique P21/c avec les paramètres de maille suivant : Pour la phase β [12]: a= 10.323Å, b=10.705 Å, c=4.018 Å et β =102°22. Pour la phase γ [13]: a= 10.00Å, b=11.18Å, c=4.07 Å et β =103°48. La molécule possède une symétrie D2h , elle est représentée par la figure 1.5.. Fig.1.5. Symétrie de la molécule du tétrabromobenzène [29]. 12.
(45) Chap. I. Etude bibliographique. I.5 Benzènes pentasubstitués Parmi les benzènes pentasubstitués étudiés, nous examinons le dibromomésitylène, que nous allons étudier en détail dans ce travail. Il subit un changement de phase à 19°C. La phase basse température a été étudiée par Hernandez et al [31]. Ce composé cristallise dans le système monoclinique P21/n avec quatre molécules par maille. Les paramètres de maille trouvés à 14 K sont : a=7.691 (13) Å, b=14.41 (2) Å, c=8.909 (11) Å et β=113.13(2) °.. I.6 Benzènes hexasubstitués. I .6.1 Benzènes hexasubstitués avec un même substituant. Les résultats obtenus pour les benzènes hexasubstitués avec un même substituant donnent bien l’image d’un noyau aromatique hexagonal, mais ne permettent pas d’obtenir toute la précision souhaitée par suite de la forte agitation thermique à laquelle sont soumises ces molécules qui sont sujettes à des mouvements rapides de réorientation à 300K (et parfois à des températures bien inférieures) [32] ce qui donne une allure elliptique aux cartes de densité électronique des substituants. Dans le cas de molécules asymétriquement substitués, les mouvements moléculaires sont de beaucoup plus faible amplitude (cas des produits que nous étudierons dans cette thèse : benzène pentasubstitué). Les. benzènes. hexasubstitués. par. des. halogènes. (l’hexaclorobenzène :. HClB,. l’hexabromobenzène :HBrB et l’hexaiodobenzène : HIB) ont tous la même structure à la température ambiante : monoclinique ( P21 /C) avec deux molécules par maille chacune placée sur un centre de symétrie. Nous prenons comme exemple l’HClB. •. Hexachlorobenzène. L’HClB est le prototype des composés hexahalogénés. Tulinsky et White [33] ont confirmé la structure trouvée en 1931 par Lonsdale [34]. Ils ont établi qu’il cristallisait dans le système monoclinique P21/c avec deux molécules par maille et avec les paramètres : a= 8.08(2) Å, b=3.87(1) Å, c=16.65(3) Å et β =117°0(2).. 13.
(46) Chap. I. Etude bibliographique. La molécule est approximativement hexagonale avec des distances C-C=1.39(2) Å et C-Cl=1.70(2) Å. Les plans moléculaires sont inclinés de 20° par rapport à l’axe b, et comme on l’a déjà mentionné pour tous les produits hexasubstitués par un même substituant, la molécule de l’HClB a une agitation thermique importante qui est compatible avec celle d’une molécule rigide exécutant de fortes rotations autour de l’axe sénaire perpendiculaire à son plan. •. Hexaméthylbenzène (HMB). A cause de sa haute symétrie, l’HMB est un produit de référence après le benzène. Son étude se heurte aux difficultés rencontrées pour la localisation des hydrogènes des groupes méthyles. La première étude de sa structure a été faite en 1929 par Lonsdale [35], il proposait une −. structure triclinique P 1 avec les paramètres : a=9.01 Å. b=8.926 Å. c=5.344 Å. α =44°27’. β =116°43’. γ =119°34’. Cette étude montre que les atomes de carbone du cycle benzénique sont arrangés suivant un anneau hexagonal (ou pseudohexagonal) de diamètre 2.96Å alors que les liaisons Ca-Cm=1.48Å. En 1939, Brockway et al [36] reprenaient la détermination de la structure de l’HMB et proposaient pour la longueur de la liaison Ca-Ca la valeur 1.39 Å et 1.53 Å pour le Ca-Cm . Une vingtaine d’années après, Tulinsky [33] a confirmé aussi cette structure avec : a=8.92(2) Å. b=8.86(2) Å. c=5.30(1) Å. α =44°5’. β =111°7’. γ =119°6’. Il confirmait aussi l’anisotropie de l’agitation thermique de l’ensemble considéré comme corps rigide. Afin de traiter le problème des hydrogènes des groupes méthyles, Hamilton et al [37] ont repris l’étude structurale aux neutrons à deux températures 298K et 130K. L’analyse des données à 130K par une synthèse différentielle de Fourier a permis de bien localiser les atomes d’hydrogène et proposer la valeur 1.10(2) Å pour la liaison C-H et 1.506(1) Å pour le Car-Cm et 111°7(2) pour l’angle Car-Cm-H. Le noyau benzénique est rigoureusement plan alors que les groupes méthyles adoptaient une configuration cahotante, alternativement à 0.05 Å au dessus, puis en dessous du plan aromatique.. 14.
(47) Chap. I. Etude bibliographique. En calculant la barrière de potentiel s’opposant à la rotation des CH3, Hamilton a trouvé une valeur de 1.90±0.3KJ/mole. Deux modes de vibration internes (hors du plan) ont de faibles basses fréquences : 139cm-1 et 196cm-1 et de grandes amplitudes ont été trouvées pour l’HMB, ces mouvements justifient l’interprétation faite par RX et neutrons concernant la position des CH3.. I .6.2 Benzènes hexasubstitués avec différents substituants. Le groupe de recherche « effet tunnel » de Rennes s’est intéressé ça fait plusieurs années au benzène hexasubstitué. par des méthyles et des halogènes, afin de mieux connaitre le. comportement des méthyles dans différents champs cristallins. Les trihalogénomésitylènes (1, 3, 5-trimethyle- 2, 4, 6-trihalogénobenzéne) ont été choisis pour être étudier à cause de la haute symétrie de la molécule (symétrie trois quasi parfaite) et d’autre part les CH3 d’une même molécule sont isolés les uns des autres par des halogènes et certainement moins couplés que ceux de l’hexaméthylbenzène. •. 1, 3, 5-trichloro-2, 4, 6-triméthylebenzène ou trichloromésitylène (TCM) Tazi [32] a étudié, sur monocristal, la structure cristalline à 297K et 148K (phase III) et les. propriétés spectroscopiques du TCM. À ces deux températures, le TCM cristallise dans le −. système triclinique P 1 aves deux molécules par maille, les paramètres de maille sont : a= 7.738(6) Å b=8.842(4) Å α=59.74(3)°. c=8.880(3) Å. β=66.51(5)° γ=73.06(4)°. A priori une étude à partir de la diffraction des rayons X (à la température ambiante) a montré que la structure cristalline ne manifeste pas de désordre moléculaire, contrairement à la plus part des autres benzènes hexasubstitués et l’agitation thermique est très importante, notamment pour les hydrogènes. Mais une étude en résonance magnétique protonique a révélé une réorientation moléculaire par sauts de 120° dans le plan du noyau benzénique pour toutes les températures supérieures à 160K, et un changement de phase vers 320K. L’étude en diffusion incohérente inélastique des neutrons a permis de mettre en évidence les excitations attribuables aux torseurs CH3. Trois excitations ont été décelées à 4, 9 et 13µeV, ce. 15.
(48) Chap. I. Etude bibliographique. qui indique que les trois méthyles ne sont pas soumis au même potentiel, ceci pourrait être dû à la symétrie triclinique du cristal. Hernandez et al [38] ont repris la détermination de la structure des deux autres phases: II et IV, sur poudre, soit complètement deutérée soit hydrogénée (qui ont quasiment les mêmes paramètres), en utilisant la diffractométrie à haute résolution (synchrotron ou neutron). La phase II (au dessus de TIII. II. =314K) est désordonnée, elle possède une symétrie. monoclinique et les molécules se réorientent par un saut de 60° dans leur plan. La phase basse température (phase IV) en dessous de 160K est ordonnée avec une symétrie triclinique et présente la même topologie structurale que la phase à température ambiante du TBM ou TIM. L’empilement est significativement plus dense que celui dans la phase III. La transition à TIII. IV. induit des rotations des molécules de 16° le long d’un axe perpendiculaire au. plan moléculaire en passant par l’atome C1, associé à des déplacements de l’ordre de 1.2 Å (Fig.1.6).. Fig.1.6. Superposition de la molécule du TCM dans la phase III à 298K et la phase VI à 2K [38]. La structure à température ambiante a été aussi réaffinée par Hernandez et al [38], en utilisant une poudre de TCM deutéré, ils ont confirmé les résultats de Tazi [32] avec les paramètres : a=7.76357(6) Å b=8.86462(6) Å. c=8.90417(6) Å. 16.
(49) Chap. I. Etude bibliographique. α=59.6418(4)°. β=66.4330(6)° γ=72.8861(8)°. V=481 Å3. Ce qui indique que la deutération du TCM a très peu d’influence sur sa structure. •. 1, 3, 5-tribromo- 2, 4, 6-triméthylebenzène ou tribromomésitylène (TBM) −. Comme le TCM, le TBM cristallise dans le système triclinique P 1 avec deux molécules par maille, les paramètres cristallographiques obtenus par Mani [39] sont : a=7.81(1) Å b=9.12(1) Å α=59.78(3)°. β=67.95(3)°. c=9.15(1) Å. Z=2. γ=73.21(3)°. V=517.5 Å3. Pour le TBM deutéré, des résultats similaires ont été obtenus [40] à 295K avec les paramètres de maille suivants : a=7.810(4) Å b=9.131(6) Å. c=9.153(6) Å. Z=2. α=59.75(5)°. γ=73.22(5)°. V=497.7 Å3. β=67.99(6)°. Les molécules forment un arrangement pseudo hexagonal dans le plan bc, ces feuillets s’empilent ensuite suivant l’axe « a » ; Ce dernier est oblique par rapport au plan bc, il correspond à la direction de croissance la plus rapide. F. Boudjada [40] a aussi résolu la structure du TBM hydrogéné et deutéré à basse température (14K). La structure du TBM (9D) est analogue à celle du TBM (9H) (Fig.1.7). À cette température les trois groupes CD3 reliés aux carbones C7, C8 et C9 sont quasi-équivalents par symétrie.. (a). (b). Fig.1.7. Structure cristalline du tribromo 1, 3, 5-triméthyle 2, 4, 6- benzène à 14K [40]: (a) TBM (9D). (b) TBM (9H).. 17.
(50) Chap. I. Etude bibliographique. Dans les deux produits, les cartes de différence de Fourier obtenues, montrent clairement trois maximas de densité de présence des protons pour chaque méthyle ; ce qui prouve la conservation de la symétrie trois dans la disposition des protons, contrairement à ce qui semblait se produire à 293K où les méthyles ne sont pas représentés comme un ensemble à symétrie ternaire mais plutôt comme un groupe de 4 particules (Fig.1.8).. (2). (1). Fig.1.8. Densité nucléaire localisée des protons des groupes méthyles [40] : (a) CH3 relié au carbone C7, (b) CH3 relié au carbone C8, (c) CH3 relié au carbone C9. (1) à 295K. (2) à 14K.. 18.
(51) Chap. I. •. Etude bibliographique. 1, 3, 5 -Triiodo-2,4,6-triméthylebenzène ou triiodomésitylène (TIM). La structure cristalline du TIM a été déterminée à 293K et 100K par diffraction des rayons X et à −. 60 et 15K par diffraction neutronique par Boudjada [41]. Elle est triclinique (P 1 et Z=2), aucun désordre n’a été décelé dans la gamme de température 0-300K. La conformation moléculaire du TIM à été ainsi calculée par les méthodes de DFT avec les fonctionnelles de la densité B3LYP et MPW1PW91. Tenant compte des atomes d’hydrogènes des groupes méthyles, deux conformations ont quasiment la même énergie de formation, une a la symétrie C3h, l’autre CS. Expérimentalement la structure observée est beaucoup plus proche de CS que C3h. Les paramètres de maille trouvés expérimentalement ( à 100K) sont : a=7.90446 (1) Å b=9.5564 (1) Å α=60.2429 (6)°. β=66.6597(7)°. c=9.5600 (1) Å γ=85.6158(7)°. V=569.05(2)Å3. Concernant la géométrie moléculaire, elle est caractérisée par une distorsion significative des angles endocycliques du cycle benzénique ; En moyenne, l’angle est de 123.6(1)° pour les atomes faces à l’iode et de 116.4(1)° pour les atomes faces au groupes méthyles. À ±0.1°, ces angles correspondent à ce qui est prédit par les calculs de DFT avec la fonctionnelle MPW1PW91 (Fig.1.9). Meinnel et al [42] ont attribué les modes de vibration interne du TIM en comparant ses vibrations avec celles du 1,3,5 triméthylbenzène.. (a). (b). Fig.1.9. Conformation de la molécule du TIM [41] : (a) Expérimentale à 15K (D9, ILL, Grenoble, France). (b) Calculée par DFT (avec une symétrie CS). 19.
(52) Chapitre II. Aperçu sur les concepts théoriques et les techniques expérimentales utilisées. 20.
(53) Chap. II. Concepts théoriques et techniques expérimentales utilisées. II.1 La diffusion neutronique II.1.1 Les propriétés du neutron L'utilisation du neutron dans l'étude de la matière condensée provient de ses propriétés : •. La longueur d'onde des neutrons thermique (à 300K) est du même ordre de grandeur que les distances interatomiques dans les solides ou liquides, ce qui fait du neutron un outil très puissant pour l'étude microscopique des propriétés tant dynamiques que géométriques de la matière.. •. Les effets interférentiels renseigneront sur la structure du système diffuseur d’où la possibilité d’étudier les structures cristallines de matériaux organiques ou biologique où il y a des hydrogènes avec grande précision : c’est la diffraction.. •. Le neutron, particule neutre, possède un grand pouvoir de pénétration dans la matière et l’interaction neutron-matière est nucléaire.. •. L'énergie des neutrons thermiques est du même ordre que beaucoup d'excitations dans la matière condensée. Ainsi, quand un neutron est diffusé inélastiquement, la mesure de son énergie fournit des informations précises sur les énergies d'excitation et par conséquent sur les forces interatomiques dans l'échantillon.. •. L'interaction entre son moment magnétique et celui des atomes (dû aux électrons non appariés) permet de connaître l'arrangement des moments magnétiques de ces atomes dans les cristaux ainsi que les énergies des excitations magnétiques.. II.1.2 Concepts théoriques II.1.2.1 Définition des sections efficaces de diffusion La résultante entre l'interaction d'un faisceau de neutrons incidents d’énergie E et un solide cristallin peut s'exprimer en termes de sections efficaces. Ce sont ces quantités que l’on mesure dans une expérience de diffusion de neutrons. Le nombre de neutrons diffusés dans r r r r r l'angle solide dΩ , avec un transfert de moment q = k − k ' ( k et k ' étant les vecteurs d'onde des. (. ). neutrons incidents et diffusés respectivement) et un transfert d'énergie hω = E − E ' = h 2 k 2 − k '2 2m. (m étant la masse du neutron) s'exprime mathématiquement dans le cadre de l'approximation de Born par la somme de deux termes :. 21.
(54) Chap. II. Concepts théoriques et techniques expérimentales utilisées. d 2σ d 2σ d 2σ = + ………………………………………………………..(2.1) dΩdE ' dΩdE ' coh dΩdE ' incoh d 2σ 1 k ' σ coh Où = ' dΩdE coh 2πh k 4π. +∞. ∑∫. j ' j −∞. {. } {. }. r r r r exp − iq.R j ' (0) exp iq.R j (t ) × exp(− iωt )dt ……...…..(2.2). représente la diffusion cohérente des neutrons avec σ coh = 4π (b ). 2. la section efficace cohérente. ( b : la longueur de diffusion elle traduit l’amplitude de diffusion du neutron par un noyau). Et. d 2σ 1 k ' σ incoh = ' dΩdE incoh 2πh k 4π. +∞. ∑∫. j ' j −∞. {. } {. }. r r r r exp − iq.R j ' (0) exp iq.R j (t ) × exp(− iωt )dt …...… (2.3). {. représente la diffusion incohérente des neutrons avec σ inc = 4π b 2 − (b ). 2. }. la section efficace. incohérente. - La diffusion cohérente dépend de la corrélation entre les positions d'un même noyau au cours du temps. Elle dépend également de la corrélation entre les positions des différents noyaux au cours du temps. Il en résulte des phénomènes interférentiels dus à la diffusion par un système vu par les neutrons comme un système `moyen’, elle pourra être définie comme la moyenne de la longueur de diffusion de tous les noyaux de l’échantillon. - La diffusion incohérente dépend uniquement de la corrélation entre les positions du même noyau au cours du temps. Il n'y a pas d'effets interférentiels. La diffusion incohérente est proportionnelle à l'écart à la moyenne des longueurs de diffusion. Elle est le terme que l'on doit ajouter pour tenir compte de la vraie diffusion du système. L'intensité diffusée dépend par ailleurs de deux paramètres : r r r • Le transfert de moment q = k − k ' : l'information est spatiale.. (. • Le transfert d'énergie hω = E − E ' = h 2 2m k 2 − k '2. ). : l'information concerne les excitations. dans la matière. Le cas où la mesure se fait avec conservation de l'énergie initiale du neutron lors de sa diffusion. hω = 0 est la diffusion élastique des neutrons ou diffraction.. II.1.2.2 Diffusion inélastique des neutrons r Dans le cadre de l'étude du tunneling, l'analyse se fait de façon spatiale ( q ) et en. énergie hω ≠ 0 . Le formalisme qui se prête le mieux à l'analyse théorique des spectres mesurés par diffusion inélastique des neutrons est celui introduit par Van Hove [43] : il relie les sections. 22.
(55) Chap. II. Concepts théoriques et techniques expérimentales utilisées. efficaces doubles-différentielles aux fonctions de corrélation. D'après l'équation générale 2.1, la diffusion inélastique des neutrons s'écrit :. σ r r d 2σ k ' σ coh = $S (q , ω ) + inc $S inc (q , ω ) ……………………………………………. (2.4) ' k 4π 4π dΩdE Le premier terme représente la diffusion cohérente et le second la diffusion incohérente r définissant ainsi la fonction de diffusion cohérente S (q , ω ) et la fonction de diffusion r incohérente S inc (q , ω ) . * Fonction de diffusion cohérente La fonction de diffusion cohérente s’explicite par : r 1 S (q , ω ) = 2π. +∞. rr r r iq r − iω t e e ∫ ∫ .G ( r , t ) d r dt ................................................................................ (2.5). −∞. r Où G (r , t ) est la fonction de corrélation de van Hove qui donne la probabilité de trouver une r particule j en r à l'instant t, sachant qu'une particule j', différente de j, était en O, origine. arbitraire du repère à l'instant 0. La fonction de diffusion cohérente est par conséquent connectée aux propriétés collectives du système. * Fonction de diffusion incohérente La diffusion inélastique incohérente de neutrons est une technique de spectroscopie vibrationnelle complémentaire à l’absorption infra rouge ou à la diffusion Raman. Grace au spin du neutron, elle permet d’induire des transitions entre niveaux de différents états de spin, ce qui permet, entre autre, de mesurer directement l’éclatement tunnel de l’état fondamental. La gamme d’énergie accessible et la résolution varient selon l’instrument. La diffusion incohérente s'exprime par : +∞. rr r r r 1 iq r − i ωt S inc (q , ω ) = e e .Ginc (r , t )dr dt ................................................................................ (2.6) ∫ ∫ 2π −∞ r Où Ginc (r , t ) est la fonction d'auto-corrélation de van Hove qui donne la probabilité de trouver r r une particule en r à l'instant t, sachant que cette même particule était en r0 à l'instant 0.. 23.
(56) Chap. II. Concepts théoriques et techniques expérimentales utilisées. La fonction de diffusion incohérente est donc liée aux propriétés individuelles du système et par conséquent elle donne des informations sur la densité d’états des excitations. Donc les fonctions de diffusion exprimées dans les équations 2.5 et 2.6 contiennent à elles seules toute l'information concernant l'interaction entre les atomes de l'échantillon étudié et les neutrons. Expérimentalement, l'intensité diffusée dépendra donc des déplacements atomiques et des sections efficaces σ coh et σ inc des atomes présents dans le matériau [44]. L'élément hydrogène est présent en très grande concentration dans les composés que nous étudions. Il possède une section efficace de diffusion incohérente 40 fois supérieure à celle des autres atomes (C, N, O). La contribution incohérente de l'hydrogène domine donc largement les contributions cohérentes et incohérentes des autres atomes (Tableau 2.1) [45]. Il peut donc être facile d'étudier la diffusion incohérente des atomes d'hydrogène dans de tels composés :. k ' σ inc r d 2σ ≈ $S inc (q , ω ) ………………………………………………………….. (2.7) ' k 4π dΩdE H ∈CH 3 La diffusion inélastique des neutrons paraît, par conséquent, comme la technique par excellence la plus adaptée pour l'étude de la dynamique du groupe méthyle. Tab. 2.1 Spin et sections efficaces de diffusion des atomes présents dans les halogénomésitylènes (1barn=10-24cm2) Atomes. Spin. σ coh (barn). σ inc (barn). H. 1/2. 1.8. 79.9. D. 1. 5.6. 2. C. 0. 5.6. 0. Cl35. 3/2. 17.1. 4.5. Cl37. 3/2. 1.19. 0.0001. Br79. 3/2. 5.81. 0.04. Br81. 3/2. 5.78. 0.03. I53. 5/2. 3.5. 0. 24.
(57) Chap. II. Concepts théoriques et techniques expérimentales utilisées. II.1.2.3 Diffusion quasi élastique Des processus de relaxation pure ou de réorientation incohérente ne donnent pas lieu à des pics inélastiques dans le spectre, mais une distribution d’excitations qui mène à un élargissement du pic élastique. Ils sont caractérisés par un taux d’orientation, qui est déterminé r par la mesure de la largeur spectrale du pic élastique, et dont la dépendance en q est souvent significative. Il s’agit par exemple de mouvements de diffusion, ou de rotations moléculaires de grande amplitude. La « Fixed window scan » est une technique dans laquelle l’intensité du pic élastique est mesurée en fonction de la température. Cette technique consiste à mesurer, en fonction de la température, l’intensité collectée dans une fenêtre fixe en énergie centrée sur le pic élastique. La perte d’intensité dans le pic élastique audelà d’une certaine température, au profit d’une composante quasi-élastique, est signe de l’excitation thermique de l’échantillon. Dans cette technique on ne mesure pas la largeur de la composante quasi-élastique, mais seulement la perte d’intensité du pic élastique en fonction de la température. Plazanet [46] a étudié la variation de l’intensité élastique diffusée du dibromo-mésitylène en fonction de la température (Fig.2.1). La décroissance rapide autour de 100K est attribuée à la l’excitation du mouvement classique de réorientation des deux groupement méthyles fortement gênés situés entre les bromes et l’hydrogène du cycle (les réorientations peuvent se produire lorsque la particule isolée a suffisamment d’énergie thermique pour affranchir la barrière de potentiel).. Fig. 2.1. Dépendance en température de l’intensité élastique diffusée par une poudre de DBM hydrogéné mesuré sur IN10 [46]. 25.
(58) Chap. II. Concepts théoriques et techniques expérimentales utilisées. II.1.3 Modèle de la particule isolée. Les niveaux d’énergie E et les fonctions d’onde ψ du rotateur quantique d’une molécule avec une constante rotationnelle B = h 2 2 I ( I est le moment d’inertie), dans l’approximation de la particule isolée sont donnés par les solutions de l'équation de Schrödinger : Hψ = Eψ avec l’hamiltonien H = B ∇ 2 + V (ω ) . Dans le cas de la rotation à une dimension, on a un seul angle ∂2 ∂2 caractéristique : ω = θ , ∇ = − 2 et H = − B 2 + V (θ ) ………………………………(2.8) ∂θ ∂θ 2. Pour la molécule isolée, la symétrie du potentiel rotationnel ne pourra pas être plus faible que celle du rotateur. Ainsi, dans le cas des groupements méthyle et en raison de la symétrie ponctuelle 3m des trois protons du méthyle, le potentiel V (θ ) auquel est soumis l’ensemble des trois protons du groupe CH3, s'écrit sous la forme d'un développement en série de Fourier respectant cette symétrie : V (θ ) = ∑ (V3n 2)[1 + cos(3nθ − φ3n )] ……………………………………..…………………. (2.9) n. V3n représente la hauteur de la barrière de potentiel et φ3n un terme de phase généralement pris égal à 0 ou π . Par commodité dans le calcul, ce développement est généralement tronqué au premier ou deuxième ordre. Dans le cas du groupement méthyle : I : le moment d'inertie du groupement méthyle autour de son axe ternaire. ICH3=1/2 ICD3=10,6 10-47kg.m2 BCH3=2BCD3=0.655meV. θ l’angle de rotation du rotateur (indépendant du temps). L'Hamiltonien que nous venons de définir est seulement applicable dans un régime de basse température où le groupe méthyle est considéré isolé de son environnement thermique : c'est le modèle de la particule isolée [47,48,49]. Dans le cas d'un rotateur présentant un potentiel de symétrie 3 pure (Fig.2.2), les niveaux d'énergie peuvent être décrits comme une séquence de niveaux de libration (de l'ordre du meV) éclatés chacun en deux niveaux de symétrie A (singulets avec un spin nucléaire 3/2) et E (dégénérés de spin ½) (écart de l'ordre du µeV pour le niveau fondamental).. 26.
(59) Chap. II. Concepts théoriques et techniques expérimentales utilisées. Effet tunnel ̴ µeV Libration ̴ meV. E A. Fig.2.2. Exemple de potentiel V3 et des niveaux d'énergie associés.. On peut distinguer deux types d’excitations (Fig.2.2) : a-Transitions torsionnelles ou librationnelles, avec des énergies dans le domaine des meV, qui décrivent l’oscillation de la molécule dans un minimum du potentiel. b- Transition tunnel, avec des énergies dans le domaine des µeV , chaque état de torsion n est éclaté en deux états tunnel (An, En) en raison de la superposition des fonctions d’onde dans les puits adjacent du potentiel [48]. Dans une compilation, Prager et Heidemann [49] ont cité toutes les références jusqu’en 1997.. II.2 Théorie des fonctionnelles de la densité (DFT) II.2.1 Principe des calculs DFT. Une fonction f d’une variable x associe un scalaire y = f(x) à tout scalaire x. Une fonctionnelle associe un scalaire x = F[f] à toute fonction f. Une intégrale définie de f(x), par exemple, est une fonctionnelle de f.. 27.
Figure



+7





Documents relatifs
Dans ce chapitre nous rappellerons des travaux déjà faits sur des molécules polycycliques, organoséléniés,similaires de nos produits,
Deux raisons principales expliquent pourquoi nous avons sélectionné ces molécules : (i) la caractérisation expérimentale de la liaison halogène est plus facilement mise
جقو ماق باتكك ةعػبصسلا ايشم تافلؤم ةجع فيلأتب " ملأا " لآاو هحيملات ىمع هلامأو خخ هخرتخا ماقو ب غم وعسج ايرخش انأ ويمع تجستعا جقو( ةعػبصسلا وبتك
Chem ins mouillés, verglacés, en n eigés; routes accidentées, cols escarpés et tortueux, rien ne rebute la VW.. |ole et satisfaction
(Les produits pV sont pratiquement égaux sous la pression de 10 5 Pa.) Son volume final est plus grand dans le cas du gaz de Van der Waals que dans celui du gaz parfait.
Vers l’équation d’état de Van der Waals A.. Caractère non ponctuel des molécules
De telles cellules d’´ epaisseurs microm´ etriques sont connues et ´ etudi´ ees dans l’´ equipe de- puis quelques ann´ ees pour des exp´ eriences de spectroscopie sub-Doppler `
Le fluide qui permet ce stockage est un fluide diphasique, constitué par un mélange fluide d’eau et d’hydrates d’un sel que l’on nommera TBAB pour les besoins de la cause.. Un
