HAL Id: tel-01692360
https://tel.archives-ouvertes.fr/tel-01692360
Submitted on 25 Jan 2018HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
dans la BPCO à travers les effets de l’inflammation
associée à l’hypoxie sur un modèle murin d’hypertrophie
musculaire
Clovis Chabert
To cite this version:
Clovis Chabert. Compréhension de la non-réponse au réentraînement dans la BPCO à travers les effets de l’inflammation associée à l’hypoxie sur un modèle murin d’hypertrophie musculaire. Médecine humaine et pathologie. Université Grenoble Alpes, 2016. Français. �NNT : 2016GREAS042�. �tel-01692360�
THÈSE
Pour obtenir le grade de
DOCTEUR DE LA COMMUNAUTE UNIVERSITE
GRENOBLE ALPES
Spécialité : Biotechnologie, instrumentation, signal Arrêté ministériel : 7 août 2006
Présentée par
Clovis CHABERT
Thèse dirigée par M. le professeur Christophe PISON et codirigée par M. Hervé DUBOUCHAUD
préparée au sein du Laboratoire de Bioénergétique Fondamentale et Appliquée.
dans l'École Doctorale d’Ingénierie pour la Santé, la Cognition et l’Environnement.
Compréhension de la non-réponse
au réentrainement dans la BPCO à
travers les effets
de l’inflammation
associée à l’hypoxie sur un modèle
murin d’hypertrophie musculaire
Thèse soutenue publiquement le « 4 octobre 2016 », devant le jury composé de :
Mme. Esther BARREIRO
Professeur de l’université Pompeu Fabra, Barcelone (Présidente) M. Frédéric COSTES
Professeur du CHU de Clermont Ferrand (Rapporteur) M. Pascal POMIES
Docteur de l’Université de Montpellier (Rapporteur) M. Patrick MUCCI
Professeur de l’université de Lille (Membre) Mme. Sophie ROUSSEAUX
Ce manuscrit de thèse est la somme du travail effectué par de nombreuses personnes au cours de ces dernières années.
Pour cela, je tiens à remercier la fondation Agir pour les Maladies Chroniques qui m’a financé durant les trois premières années de doctorat, ainsi que le Centre Hospitalier Universitaire qui m’a permis de rédiger ce manuscrit dans les meilleures conditions.
Toute ma gratitude va à mon directeur de thèse Pr. Christophe Pison sans qui je n’aurai jamais pu réaliser ce doctorat. C’est grâce à son soutien indéfectible, de la recherche de financement de thèse à la rédaction du manuscrit, que ce travail a pu aboutir.
Merci également au Dr. Valérie Chaté et Dr. Hervé Dubouchaud qui se sont relayés dans l’encadrement de ce travail. Merci pour votre confiance et pour le temps que vous avez accordé à ce travail. Merci Valérie pour le plaisir que m’a procuré l’enseignement de la physiologie de l’exercice en ta compagnie.
Mes remerciements s’adressent aussi au directeur du laboratoire Pr. Uwe Schlattner qui m’a accepté au sein du Laboratoire de Bioénergétique Fondamentale et Appliquée, et a participé à la publication des travaux réalisés durant ce travail.
Je tiens également à remercier les membres du Jury d’avoir accepté d’évaluer le fruit de ces presque 4 années de recherche :
Aux Dr. Pascal Pomiès et au Pr. Frédéric Costes qui me font l’honneur d’être les rapporteurs de ce manuscrit malgré le travail que cela implique.
Aux Pr. Esther Barreiro qui a accepté d’être la présidente de ce jury de thèse malgré un emploi du temps déjà bien rempli.
Au Pr. Patrick Mucci et au Dr. Sophie Rousseaux pour l’intérêt qu’ils ont porté à cette étude en acceptant d’être les examinateurs de ce travail.
Je suis très honoré par votre présence et je vous prie de trouver ici l’expression de ma profonde gratitude et de mon respect le plus sincère.
Il m’est impossible de ne pas remercier toute l’équipe du CRSSA qui m’a fait confiance du Master 2 au début de la thèse. Merci donc à Hervé Sanchez, Alexandra Malgoyre, Adélie Meunier, Rachel Chapot, Bernard Serrurier, Nadine Simmler et Nathalie Koulmann, qui m’ont donné l’autonomie, les compétences et le soutien sans lesquels ce travail n’aurait pas abouti. Merci au Pr. Saadi Khochbin et au Dr. Sophie Rousseaux de l’IAB, d’avoir pris de le temps de partager leur expertise dans le domaine de l’épigénétique.
Merci bien sûr à tous les membres du LBFA pour leur accueil et leur disponibilité. Ces remerciements s’adressent tout particulièrement à Gérard, Sarah et Cindy pour leur support.
Un grand merci à l’ancienne (Evy, Marie, Sarah) et à la nouvelle génération de doctorants que j’ai eu le plaisir de côtoyer (Adeline, Arthur, Camille, Céline, Charline, Martin, Morgane, Rafaela). De la Grèce à l’Espagne en passant par le « Hasard » merci pour tous ces moments passés ensembles. Difficile de parler du Hasard sans remercier également Julien, Boris et Yannick pour leur présence constante et immuable. Merci aussi à tous ceux qui m’ont permis de continuer à voir le soleil pendant les week-end, mais aussi un peu entre midi et deux. Je pense à Baptiste et à mon frère Lucas, pour les sessions parapentes, montagnes, escalades, skis, buts météo, buts réveils, buts…. Impossible de ne pas remercier Pierre ici, sans avoir de réflexions douteuses de la part des personnes qui nous connaissent. Donc voici Pierre, mes remerciements pour, ton temps, ton travail, à peu près tous les sports possibles et imaginables que l’on peut faire dans la région (et ils sont nombreux), ta disponibilité… Grace à toi j’ai enfin pu collaborer avec un laboratoire d’astrophysique !
Impossible maintenant de ne pas aussi remercier Camille, ma compagne, pour m’avoir supporté pendant ces années plutôt intenses du doctorat. Merci pour ta patience, ton soutien, pour tous les bons moments que nous avons passé et que nous passerons ensemble !
Pour finir, merci à toute ma famille qui m’a soutenu, poussé et encouragé tout au long de ma scolarité ! Merci aussi à Mathilde, Lucas, Héloïse, et Samuel pour tous les bons moments passés avec vous durant les 30 dernières années…et oui déjà ! Je n’oublie pas non plus Emilie pour son aide précieuse dans la relecture du manuscrit.
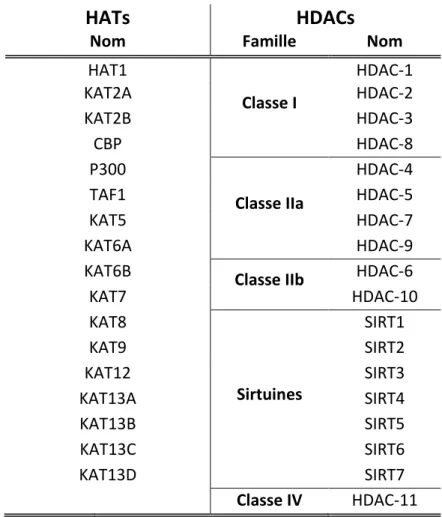
Liste des abréviations
µARN les micros-ARN 3-MH la 3-Methyl Histidine
4E-BP1 Eukaryotic translation initiation factor 4E-binding protein 1 AA Acides Aminés
Ac-CoA Acétyl Coenzyme A ADNc ADN complémentaire ADP Adénosine Di-Phosphate ALD Affection de Longue Durée AMP Adénosine Mono-Phosphate AMPk AMP kinase
APS Activité Physique Spontanée ARNm Acide RiboNucléique messager ATP Adénosine Tri Phosphate BNIP3 BCL2/adenovirus E1B
19kd-Interacting Protein BPCO Broncho-Pneumopathie
Chronique Obstructive Brd Bromodomaines BSA Bovine Serum Albumin
CaMKII Calmodulin-dependent protein kinase II CK Créatine Kinase CPT1 Carnitine Palmitoyltransférase 1 CPT2 Carnitine Palmitoyltransférase 2 CRP C-Reactive Protein CS Citrate Synthase
CSF Colony Stimulating Factors CVF Capacité Vitale Forcée DNMTs DNA Methyl Transferases
dP/dt max vitesse de mise en tension des fibres cardiaques
dP/dt min vitesse de relâchement des fibres
DTNB acide dinitrothiobenzoïque DTT Dithiothréitol
EGTA acide éthylènediamintétra-acétique
eIF2B eukaryotic initiation Factor 2B EPO Erythropoïétine
ERK Extracellular signal-Regulated Kinase
ET-1 Endothelin-1 FBXO40 F-Boxes Other 40 Fi.O2 Fraction inspirée d’O2 FoxO Forkhead Box O
GAPDH Glycéraldéhyde-3-phosphate Déshydrogénase
GLUT Transporteurs du Glucose GOLD Global initiative for chronic
Obstructive Lung Disease H3K27 ysine 37 de l’histone H3 HA Hypoxie Aigue
HAT Histone AcetyTransférase HC Hypoxie Chronique HDAC Histone Déacétylase HEPES acide
4-(2-hydroxyéthyl)-1-pipérazine éthane sulfonique HIF-1 Hypoxia Inducible Factor 1
HIRA protéine Replication-Independante Histone Chaperone
HK Hexokinase
HRE Hypoxia Response Element HTAP Hypertension Artérielle
Pulmonaire
HTP Hypertension Pulmonaire I-BET inhibiteur des Bromodmaines et
des domaines Extra-Terminaux IBP Institut de Biologie et de
Pathologie
IGF-1 Insuline Growth Factor 1 IL Interleukines
IL1-b Interleukine 1-b
IMC Indice de Masse Corporelle INVS Institut National de Veille
Sanitaire
IP Inflammation Pulmonaire LC3 protéines 1 light chains 3 LDH Lactate Dehydrogénase LPS Lipopolysaccharides MAFBx atrogine-1
MAPK Mitogen-Activated Protein Kinase
Mef Myocite enhanced factor MHC chaines lourdes de myosine MIP-1 Macrophage Inflammatory
Protein-1
MITR Mef2-Interacting Transcription Repressor
mTOR mammalian Target Of
Rapamycin C1 MuRF-1 Muscle Ring Factor-1 MyoG Myogénine
N Normoxie
NFAT Nuclear Factor of Activated T-cells
NF-kB Nuclear Factor kappa B
NIRS Spectroscopie Proche Infrarouge NO Oxyde Nitrique
OMS Organisation Mondiale de la Santé
PaO2 Pression artérielle en Oxygène PCr Phospho-Créatine PDH Pyruvate Déshydrogénase PDK-1 Pyruvate Dehydrogenase Kinase-1 PFK Phospho Fructokinase PGC-1α PPAR coactivator-1α PGK Phosphoglycerate Kinase pH potentiel Hydrogène PHDs Proly-4-HyDroxylases PI3K Phosphoinositide 3-kinase PMSF Fluorure de
Phénylméthylsulfonyle Pol II Polymérase II
PPAR-γ Protein Proliferator-Activated Receptor-γ
pVHL proteine Von Hippel Lindau Rack-1 Receptor for Activated C
Kinase-1
RT Reverse Transcription
chaine en temps réel SaOS Syndrome de l’Apnée du
Sommeil
Six/Eya sine oculis homeobox 1/eyes absent 1
Smad2/3 small body size mothers against decapentaplegic 2 et 3
SmO2 Saturation de l’oxymyoglobine SQSTM1 Sequestrosome 1
SUMO Small Ubiquitin-like Modifier SUP Système Ubiquitine/Protéase–
dépendant
TA Température Ambiante TCA Acide Trichloracétique
TEMED Tetramethyl-Ethylenediamine TGF Facteurs de croissance et
transformation
TLR Toll-Like Receptors TNF Tumor Necrosis Factor TNFR1 TNF Récepteur 1
TNFR2 TNF Récepteur 2
TSC1-2 Tuberous Sclerosis Complex 1-2 Va/Q Ventilation alvéolaire sur
Perfusion
VD Ventricule cardiaque Droit VEF Volume Expiratoire Forcé VEGF Vascular endothelial Growth
Factor
VEGFR Vascular Endothelial Growth Factor Receptor
VEMS Volume d’Expiration Maximal par Seconde
INTRODUCTION ... 1
I LA BPCO : une pathologie systémique d’origine pulmonaire ... 2
I.A La Bronchopneumopathie Chronique Obstructive ... 2
I.B Diagnostic et classification ... 5
I.C Une évolution en lien avec le tissu musculaire ... 7
II Le muscle strié : du sujet sain au BPCO ...12
II.A La composition du tissu musculaire ... 12
II.A.1 La composition structurelle musculaire du sujet sain ... 13
II.A.2 La composition musculaire du sujet BPCO ... 15
II.B Les propriétés métaboliques du tissu musculaire ... 17
II.B.1 Le muscle du sujet sain ... 17
II.B.2 Le muscle du patient BPCO ... 19
II.C Les facteurs impliqués dans le contrôle de la masse musculaire ... 23
II.C.1 Les voies anaboliques ... 23
II.C.2 Les voies cataboliques ... 28
II.D La régulation de la masse musculaire dans la BPCO ... 30
II.D.1 Le statut anabolique des patients BPCO ... 31
II.D.2 Le rôle du catabolisme dans l’altération musculaire du BPCO ... 31
II.D.3 Les effets du réentrainement physique dans la BPCO. ... 32
III Quel rôle de l’inflammation dans la BPCO ? ...36
III.B Le paradoxe de l’inflammation chronique ... 40
III.C L’inflammation dans la BPCO ... 42
IV L’Hypoxie Chronique dans la BPCO ...48
IV.A Les mécanismes de régulation de HIF-1 ... 48
IV.B Répercussions de l’hypoxie sur la masse musculaire ... 50
IV.C L’hypoxémie et stress oxydant dans la BPCO ... 55
V Epigénétique, du microenvironnement à l’atteinte musculaire...60
V.A Définition de l’épigénétique ... 61
V.B Les mécanismes de l’acétylation ... 62
V.C Conséquences des mécanismes de l’acétylation sur le muscle ... 65
V.D L’acétylation dans la BPCO ... 67
V.E Les mécanismes autres que l’acétylation ... 68
OBJECTIFS SCIENTIFIQUES ... 73
MATERIEL ET METHODES ... 77
I Le conditionnement animal ...77
I.A Animaux ... 77
I.B Le développement du modèle de non réponse musculaire ... 77
I.C L’étude des effets de l’I-BET sur le muscle soléaire ... 79
I.D Le modèle de surcharge musculaire par chirurgie ... 80
I.E Les conditionnements hypoxiques ... 83
I.E.2 L’hypoxie aigüe ... 83
I.F L’instillation de Lipopolysaccharides ... 85
I.G Le traitement à l’inhibiteur des bromodomaines et domaines extra-terminaux ... 85
II Les procédures d’euthanasie et de prélèvements ...86
III La mesure du taux d’hématocrite ...86
IV La mesure de l’activité locomotrice spontanée des animaux ...87
V Les mesures des pressions intracardiaques in situ par microsonde Millar ...87
VI La quantification des niveaux protéiques par Western-blot ...90
VI.A L’extraction des protéines ... 90
VI.B L’électrophorèse et l’éléctrotransfert des protéines ... 91
VI.C L’immunorévélation des protéines ... 92
VI.D La déshybridation des anticorps ... 95
VII Les mesures du niveau d’acétylation global des histones ...95
VIII La mesure de la typologie musculaire ...96
IX Mesure de l’activité de la citrate synthase ...97
X L’expression des ARN messagers ...98
XI Les analyses statistiques ... 103
RESULTATS ... 105
I Etude 1 : Développement d’un système d’acquisition vidéo de l’activité physique spontanée. ... 105
I.A Introduction ... 105
I.B Principaux résultats ... 107
I.C Conclusion ... 109
I.D ”A low-cost system to easily measure spontaneous physical activity in rodents”. ... 109
II Etude 2 : Influence de la typologie dans la sensibilité spécifique de deux muscles différents à l’hypoxie chronique et/ou l’inflammation pulmonaire. ... 117
II.A Introduction ... 117
II.B Principaux résultats ... 119
II.C Conclusion ... 122
II.D “ Influence of typology on heterogeneous response of two different muscles to hypoxia and pulmonary inflammation.” ... 122
III Etude 3: Hypertrophie musculaire en situation hypoxique et inflammatoire: contrôle par les bromodomaines et les domaines extra-terminaux des protéines. ... 154
III.A Introduction ... 154
III.B Principaux résultats ... 157
III.C Conclusion ... 160
III.D “Muscle hypertrophy in hypoxia with inflammation is controlled by bromodomain and extra-terminal domain proteins.” ... 160
IV Etude 4: Rôle des bromodomaines et des domaines extra-terminaux dans le développement de l’hypertension artérielle pulmonaire et de ses conséquences sur l’hypertrophie du ventricule droit. ... 187
IV.A Introduction ... 187
IV.B Principaux résultats ... 189
IV.C Conclusion ... 191
IV.D “Inhibition of Bromodomain and Extra-terminal domain proteins reduces right ventricle hypertrophy resulting from combined hypoxia and pulmonary inflammation.” ... 191
DISCUSSION GENERALE ... 211
I L’influence de l’activité locomotrice spontanée des animaux dans l’hypertrophie musculaire ... 211
II L’hypoxie chronique et l’inflammation pulmonaire ont des effets spécifiques sur les capacités d’hypertrophie du soléaire et du plantaire. ... 213
III L’altération de l’hypertrophie du soléaire par l’inflammation pulmonaire semble impliquer les mécanismes de l’acétylation des histones. ... 219
CONCLUSION ET PERSPECTIVES ... 227
I Conclusion... 227
II Perspectives ... 228
II.A Comprendre les mécanismes impliqués dans la sensibilité spécifique du plantaire et du soléaire à l’hypoxie chronique et à l’inflammation pulmonaire. ... 228
II.B Déterminer le rôle de l’inflammation pulmonaire et de l’hypoxie chronique dans une situation d’atrophie musculaire. ... 229
II.C Comprendre les mécanismes épigénétiques à l’origine des altérations des capacités de réponse du soléaire présence d’une inflammation pulmonaire. ... 230
II.D Les perspectives cliniques des I-BET dans la BPCO. ... 231
BIBLIOGRAPHIE ... 232
ANNEXES ... 256
I Etude 1 : Développement d’un système d’acquisition vidéo de l’activité physique spontanée. ... 256
II Etude 2 : Influence de la typologie dans la sensibilité spécifique de deux muscles différents à l’hypoxie chronique et/ou l’inflammation pulmonaire. ... 257
III Etude 3: Muscle hypertrophy in hypoxia with inflammation is controlled by Bromodomain and Extra-terminal domain proteins. ... 260
I.A Supplemental data ... 260
I.B Résultats supplémentaires ... 264
INTRODUCTION
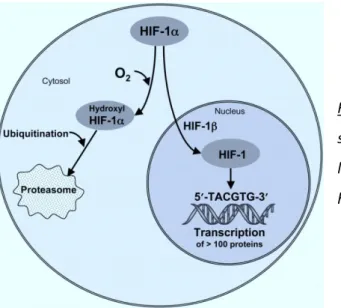
Selon le dernier rapport du groupe de travail Global Initiative for Chronic Obstructive Lung Disease (GOLD), la Broncho-Pneumopathie Chronique Obstructive (BPCO) est une pathologie commune évitable et traitable, qui se caractérise par une limitation persistante et habituellement progressive du flux d'air. Cette pathologie est associée à une réponse inflammatoire chronique accrue des voies respiratoires et des poumons aux particules ou aux gaz nocifs. Les exacerbations et les comorbidités contribuent à la sévérité globale individuelle des patients. A l’heure actuelle, les mécanismes impliqués dans l’évolution d’une atteinte pulmonaire à systémique ne sont que très partiellement compris. Toutefois l’importante prévalence d’un signal hypoxique associé à une inflammation pulmonaire chronique, semble placer ces 2 stimuli comme des acteurs centraux de cette affection.
Parmi les répercussions systémiques de la BPCO, une atteinte des paramètres fonctionnels et quantitatifs du tissu musculaire des membres inférieurs est présente, en lien avec la sévérité de l’atteinte [1]. Or, le lien étroit entre le tissu musculaire et la probabilité de survie des patients BPCO [2-4], a placé l’homéostasie du tissu musculaire au cœur de la prise en charge de cette pathologie [5, 6]. Basé sur ce constat, le réentrainement s’est progressivement imposé comme un outil incontournable avec un niveau de preuve élevé [7-10]. Malheureusement, l’efficacité de cette prise en charge est hétérogène puisque 30 à 40 % des patients ne répondent pas à un protocole de réentrainement musculaire [2, 11-13]. Pour expliquer cette non-réponse, de nombreuses hypothèses ont été avancées, impliquant le statut nutritionnel [14], les voies de protéolyse/synthèse [15], ou encore la composante nerveuse [16]. Toutefois, aucune de ces hypothèses n’est encore suffisamment aboutie, pour totalement expliquer les altérations fonctionnelles et quantitatives des muscles du patient BPCO.
Récemment, des chercheurs se sont intéressés au rôle de l’épigénétique dans cette atteinte musculaire. Les premiers résultats mettent déjà en évidence une perturbation du motif global épigénétique de ces patients, potentiellement en lien avec les altérations de la réponse musculaire à un protocole de réentrainement [13]. Or l’Hypoxie Chronique (HC) et l’Inflammation Pulmonaire (IP) sont deux stimuli connus pour induire des perturbations du microenvironnement cellulaire qui induisent des perturbations des facteurs de régulations
épigénétiques [17-20]. Ainsi, HC et IP seraient les initiateurs du remodelage de l’épigénome des cellules musculaires du BPCO [21], conduisant à l’altération des capacités de réponse du tissu musculaire à un protocole de réentrainement.
Afin de comprendre les mécanismes impliqués dans la non-réponse au réentrainement, le travail présenté dans ce manuscrit s’est intéressé au rôle de l’HC et l’IP sur la régulation du tissu musculaire. Pour cela, nous avons développé un modèle murin combinant une HC environnementale avec des instillations intra-pulmonaires répétées de lippopolysaccharides, sur un modèle d’hypertrophie musculaire chirurgical. L’étude de 2 muscles locomoteurs ayant une typologie diamétralement opposée nous a permis d’étudier l’influence de la typologie musculaire sur la sensibilité des muscles à l’HC ou l’IP. Ainsi le muscle soléaire, composé à plus de 90 % de fibres oxydatives, et le muscle plantaris, composé à plus de 90 % de fibres glycolytiques, nous ont permis d’essayer de comprendre les mécanismes impliqués dans l’hétérogénéité de l’altération musculaire observée dans la BPCO.
I LA BPCO : une pathologie systémique d’origine pulmonaire I.A La Bronchopneumopathie Chronique Obstructive
La BPCO est actuellement la 4ème cause de mortalité dans le monde avec la projection
qu’elle devienne la 3ème cause de décès en 2030 selon l’Organisation Mondiale de la Santé
[22]. En 2007, une étude a montré que 10,1 % de la population mondiale de plus de 40 ans présentait une BPCO modérée à très sévère.
En 2010, on pouvait déplorer 2,9 millions de décès liés à la BPCO dans le monde [23-25]. Sur la population d’âge supérieur à 40 ans, la prévalence de la BPCO, de modérée à très sévère, est estimée à plus de 10 % avec de grandes différences selon le sexe et le pays [26]. Selon l’Institut National de Veille Sanitaire (INVS), la BPCO concernait 7,5 % de la population adulte en France en 2010, soit 3,5 millions de patients avec 200 000 patients en Affection de Longue Durée (ALD), dont 93 000 avaient recours à une oxygénothérapie de longue durée [27].
La BPCO est une maladie respiratoire chronique évolutive, définie par une obstruction permanente et progressive des voies aériennes [27-29]. Cette obstruction est causée par
l’association d’une diminution du calibre des bronchioles par remodelage et d’une distension permanente associée à une destruction des parois des alvéoles pulmonaires, appelée emphysème. Cette pathologie touche principalement les populations ayant un faible niveau d’activité physique, âgées de 45 ans et plus, qui ont été exposées de manière prolongée (de 15 à 20 ans) aux fumées de cigarette autant de manière active que passive (environ 80 % des BPCO sont ou ont été des fumeurs) [30, 31].
L’évolution de la BPCO est rythmée par l’alternance d’états dits « stables » au cours desquels la sévérité de la pathologie n’évolue que très lentement, avec des phases de décompensations aiguës, appelées exacerbations. Cette évolution se traduit par un déclin accéléré de la fonction respiratoire qui augmente la fréquence d’épisodes de décompensations pouvant mettre en jeu le pronostic vital et réduire l’activité physique quotidienne. L’origine d’un épisode d’exacerbation est mal connue et très probablement multifactorielle. Cependant, les exacerbations ont comme dénominateur commun un épisode inflammatoire aigu qui déclenche une réponse disproportionnée des voies de l’inflammation, comme cela est le cas lors d’une infection pulmonaire, même bégnine [32]. Les conséquences des exacerbations à court terme (2,5-25 % décèdent durant l’hospitalisation), à moyen terme (25-50 % de décès au cours de l’année qui suit l’hospitalisation [33]), et à long terme avec une réduction de 55 % de la survie à 5 ans pour les patients ayant 3 ou plus exacerbations sévères par an [34], placent ces épisodes de décompensation au cœur de la BPCO. Ce lien entre les exacerbations et l’évolution de la pathologie est encore renforcé par la récupération souvent partielle de la qualité de vie, de l’autonomie et des paramètres physiologiques antérieurs à l’exacerbation [35]. Une étude prospective a montré que 6 mois après une exacerbation, seulement 27 % des patients estiment avoir une qualité de vie bonne à excellente alors que 49 % la qualifient de moyenne à faible [36]. La fonction pulmonaire est également impactée avec une diminution plus rapide du Volume d’Expiration Maximal par Seconde (VEMS) chez les patients ayant des exacerbations récurrentes (> 2,92 exacerbations.an-1 [37]). Le poids de
ces épisodes de décompensations pour le patient BPCO est bien illustré par les courbes de survie abaissée des patients lorsqu’ils sont classés en fonction de la fréquence de leurs exacerbations et de leurs sévérités (Figure 1).
Figure 1 : Courbes de survie de patients BPCO en fonction de la fréquence des exacerbations (A) et de leur sévérité (B) [34]. A : patients non-exacerbateurs ; B : Patients avec 1-2 hospitalisations pour exacerbation par an ; C : Patients avec plus de 3 exacerbations par an. 1 : patients non-exacerbateurs ; 2 : patients avec exacerbations ne nécessitant pas d’hospitalisation ; 3 : patients avec exacerbations nécessitant 1 hospitalisation ; 4 : patients avec réhospitalisation.
Le déclin des patients BPCO est accéléré par l’augmentation de la fréquence des épisodes d’exacerbation en lien avec la sévérité de la maladie. En effet, une moyenne de 2,68 exacerbations par an a été relevée par Donaldson et coll. dans une population de patients modérés, contre 3,43 pour les patients sévères [37].Cependant d’autres travaux ont montré qu’une différence de la fréquence des exacerbations existe entre les patients dès les premiers stades de la BPCO [38]. Il est donc possible que le lien entre la sévérité et la fréquence des exacerbations ne soit seulement dû au fait que les patients les plus sévèrement atteints soient principalement des patients exacerbateurs fréquents.
Figure 2 : Nature et fréquence comorbidités recensées dans la population des différentes BPCO [39].
De nombreux patients BPCO présentent également une ou plusieurs comorbidité(s), associant ainsi l’altération du système respiratoire à des troubles métaboliques, osseux, musculaires et neurologiques [40]. Selon Vanfleteren et coll. [39], 98 % des patients BPCO sont diagnostiqués pour au moins une comorbidité et 53 % en présentent 4 et plus (Figure 2). Cependant, il est compliqué de déterminer si les comorbidités observées sont dues aux répercussions directes de la BPCO, ou aux facteurs de risques communs entre elles (tabagisme, sédentarité,…). Or, leur incidence dans la mortalité de ces patients est non négligeable puisque seuls 36 % des décès sont attribuables à une défaillance respiratoire, les 76 % restant étant associés aux comorbidités [41].
I.B Diagnostic et classification
L’hétérogénéité de la BPCO, son caractère systémique et progressif, et les nombreuses comorbidités rendent difficile l’évaluation des patients. Afin de pallier à cette difficulté, plusieurs indices ont été créés dont le GOLD standard [42]. Depuis sa création en 1997, cette classification de la sévérité d’atteinte des patients à déjà été révisée à plusieurs reprises [43]. Actuellement, cet indice de classification se base sur la sévérité de l’atteinte fonctionnelle de voies aériennes évalué à partir du VEMS selon l’ancien GOLD standard (GOLD I-IV), le nombre d’exacerbations au cours de l’année écoulée, la sévérité de la dyspnée évaluée par l’échelle modifiée «Medical Research Council Dyspnea Scale» (mMRC), et les symptômes ressentis par le patient quantifiés au moyen du COPD Assessment Test (CAT). Cette classification qui catégorise les patients en 4 groupes référencés sous l’appellation GOLD A à D (Figure 3B),
place les exacerbations au centre de la BPCO et permet de prendre en compte la différence des symptômes perçus entre les patients pour une même atteinte fonctionnelle. Toutefois, cette nouvelle classification reste discutée car elle possède une moins bonne valeur pronostic que l’ancienne classification GOLD I-IV [44]. Cela est due au fait que la nouvelle classification GOLD a été conçue dans une optique de stratification du risque associé à la BPCO et non pour prédire la mortalité. Ainsi ces 2 systèmes de classifications peuvent cohabiter en fonction de l’information que l’on souhaite obtenir.
Figure 3 : Classification des patients BPCO selon l’indice de GOLD (A) et le New GOLD (B) GOLD: Global initiative for chronic Obstructive Lung Disease. VEMS: Volume d’Expiration Maximal par Seconde, CVF: Capacité Vitale Forcée, CAT: COPD Assessment Test, mMRC: Medical Research Council Dyspnea Scale.
Le suivi longitudinal de la mortalité de 15 632 patients issus de 22 cohortes de BPCO différentes a cependant montré que ni le GOLD 2007, ni le GOLD 2011 n’étaient suffisamment
aboutis pour prédire la mortalité des patients BPC0 à 3 ans [45]. Cette étude montre également que la nouvelle classification GOLD 2011 ne permet pas d’améliorer la prise en charge des patients puisque leur espérance de survie reste inchangée. Ainsi, il semble nécessaire de trouver de nouveaux critères prédictifs pour améliorer la pertinence de la classification des patients, et donc de leur prise en charge. La multiplication des données intrinsèques à chaque patient devrait conduire vers une médecine plus prédictive et personnalisée, qui semble être la clef de la prise en charges de pathologies hétérogènes telle que la BPCO.
I.C Une évolution en lien avec le tissu musculaire
Il a été montré par de nombreux auteurs dont Marquis et coll. (Figure 3) que l’espérance de survie des patients était très fortement corrélé à leur masse musculaire [3, 46, 47].
Figure 3 : courbes de survies basées sur le modèle de Cox pour 4 sous-groupes de patients [46]. FEV1 :
Volume d’expiration forcée en 1 seconde ; MTCSA : Surface de section musculaire à mi-cuisse.
En effet, la comparaison de 2 populations de patients ayant la même limitation pulmonaire (VEMS ≥ 50 % prédit ou VEMS < 50 % prédit) montre que les patients ayant une surface de section musculaire de la cuisse préservée (MTCSA ≥ 70 cm2) ont une probabilité de
survie supérieure. Il est intéressant de noter que la diminution de la probabilité de survie est accentuée dans les groupes ayant l’atteinte pulmonaire la plus sévère (VEMS < 50 % prédit, en rouge sur la figure). En effet la probabilité de survie à 2000 jours des patients ayant un MTCSA ≤ 70 cm2 est 4 fois plus faible dans les groupes avec un VEMS < 50 % prédit (en rouge)
par rapport aux groupes avec un VEMS > 50 % prédit (en bleu sur la Figure 3).
Figure 4 : Relation entre la mortalité sur une période de 4 ans et la distance parcourue lors d’un test de marche de 6 min initial [48]. 6MWD : 6 Minutes Walking Distance. Pour le groupe <100m, n=19 ; 101-200m, n=61 ; 201-300m, n=57 ; 301-400m, n=46 ; >400m, n=15.
Outre la quantité de masse musculaire, la fonctionnalité de celle-ci est également un facteur prédictif fiable de l’espérance de survie des patients BPCO. Le suivi sur 4 ans d’une cohorte de 198 patients BPCO, présentant un VEMS/CVF et un VEMS < 70 % de la valeur prédite, montre bien la relation entre la distance initiale parcourue durant un test de marche de 6min, et la mortalité des patients à l’issue de l’étude [48]. En effet, sur la Figure 4 nous pouvons voir que les patients ayant parcouru moins de 100 m au test initial, ont une mortalité 5,6 x plus élevée que ceux ayant parcouru une distance de marche supérieure à 400 m.
De manière intéressante, il a été montré que les patients BPCO avaient un volume et une intensité d’activité physique spontanée (APS) journalière, bien plus faible que des sujets sains du même âge (respectivement 57 % et 75 % des valeurs des sujets contrôle), dès les premiers stades [49]. Cette réduction progressive de l’activité physique est associée à la sévérité de l’atteinte des patients, de leur fonction pulmonaire, et à la déplétion de la masse musculaire [50]. En effet l’absence de sollicitation régulière des muscles locomoteurs diminue leur masse [51], leurs capacités oxydatives et leur force [52].
Figure 5 : La spirale du déconditionnement. Concept présenté A. Young et coll. [53].
Pour le patient BPCO, l’augmentation de la sédentarité augmente progressivement la difficulté respiratoire, dans un premier temps au cours de l’effort puis à des intensités de plus en plus faibles. De plus, la survenue d’une dyspnée précoce lors d’un effort physique, diminue les possibilités de sollicitation de la masse musculaire du patient, accélérant encore l’évolution de la dyspnée. Le patient entre donc dans une spirale du déconditionnement dont le concept a été présenté par A. Young et coll. (Figure 5), qui diminue leur autonomie et de leur qualité de vie. Ainsi, que ce soit une cause ou une conséquence, l’inactivité physique du patient BPCO joue un rôle central dans l’évolution de la maladie. Toutefois, le fait que la diminution de l’APS soit présente dès les premiers stades de la BPCO, avant même l’apparition de dyspnée que l’on retrouve dans les stades avancés [54], suggère que les altérations du tissu musculaire du patient BPCO ne sont pas seulement dues à l’inactivité mais également à la présence d’une régulation parallèle. Les effets bien connus de l’HC et de l’IP sur l’équilibre protéolyse/protéosynthèse en font des candidats idéaux que nous décrirons dans les sections à venir respectivement (respectivement section III.C page 42 et section IV.C, page 55).
L’évolution de la BPCO est rythmée par des phases de décompensations qui accélèrent la fonte musculaire en associant l’inactivité [55] à une réduction de la prise alimentaire, une augmentation de la dépense énergétique [56], une majoration de l’inflammation [57], et à la
présence d’un stimulus hypoxique [58]. Tous ces facteurs sont connus pour leur implication dans la fonte musculaire [59]. Cette perturbation du microenvironnement cellulaire perturbe l’équilibre protéolyse/synthèse en faveur de cette première. De plus, le stress provoqué par ces différents stimuli peut modifier la signature épigénétique de la chromatine, perturbant ainsi l’expression des gènes impliqués dans la régulation de la masse musculaire du BPCO [60].
Ces résultats, associés à ceux obtenus avec le cancer, le diabète, l’obésité, les cardiopathies, etc. [61, 62] placent le maintien de la masse musculaire comme une priorité dans la prise en charge des maladies chroniques [63]. Les effets de l’entrainement physique sur la quantité et la fonctionnalité de la masse musculaire, ont progressivement imposé cette intervention dans la prise en charge de la BPCO [5, 28, 64]. Malheureusement, 30 à 40 % des patients ayant suivi un protocole de réentrainement ne retirent aucun des bénéfices attendus, que ce soit en terme de qualité de vie, d’amélioration de la tolérance à l’effort, et/ou de la composition corporelle [2, 11-13].
En résumé :
La BPCO représente un enjeu économique, social et humain majeur, puisqu’elle est en passe de devenir la 3ème cause de mortalité dans le monde en 2030. Cette pathologie fréquente,
est caractérisée par une limitation du débit d'air, habituellement progressive, associée à une réponse inflammatoire accrue dans les voies respiratoires et les poumons lors d’une exposition à des particules ou gaz. Les exacerbations et les comorbidités contribuent à la gravité individuelle globale des patients. En effet, l’évolution de la BPCO se fait principalement lors des phases de décompensations aiguës des patients, appelées exacerbations, qui alternent avec des phases dites stables moins inflammatoires.
La pluralité des atteintes dans la BPCO ainsi que les comorbidités qui lui sont associées, rendent impossible l’utilisation de score unique de sévérité ou pronostique. L’indice de GOLD ABCD, basé sur la fonction respiratoire, la fréquence des exacerbations, la dyspnée et le ressenti du patient est actuellement la classification multifactorielle la plus aboutie. Toutefois celle-ci ne permet pas de prédire l’évolution de la pathologie à long terme. Il est donc nécessaire d’améliorer notre compréhension de la BPCO afin d’identifier les facteurs clefs impliqués dans son évolution.
Le faible niveau d’activité physique associé à l’hypoxémie et à l’inflammation pourrait être à l’origine de l’altération de la masse musculaire des patients, particulièrement importante lors des exacerbations. Or la masse musculaire est étroitement corrélée avec la survie des patients. C’est pourquoi, des protocoles de réentrainement physique sont utilisés afin de développer les paramètres fonctionnels et quantitatifs de leur tissu musculaire. Bien que bénéfique pour la majorité des patients, cette approche reste inefficace pour 30-40 % d’entre eux, qui ne retirent aucun bénéfice de cette prise en charge. Les mécanismes impliqués dans la non-réponse au réentrainement de plus d’un tiers des patients BPCO restent à élucider.
II Le muscle strié : du sujet sain au BPCO II.A La composition du tissu musculaire
Chez l’adulte, le muscle strié compose environ 40 % du poids corporel. Sa consommation énergétique correspond à 25 % du métabolisme de base au repos, et peut être multipliée par 50 lors d’un effort physique intense. C’est le tissu le plus actif métaboliquement. Il joue un rôle central dans l’homéostasie énergétique de l’organisme.
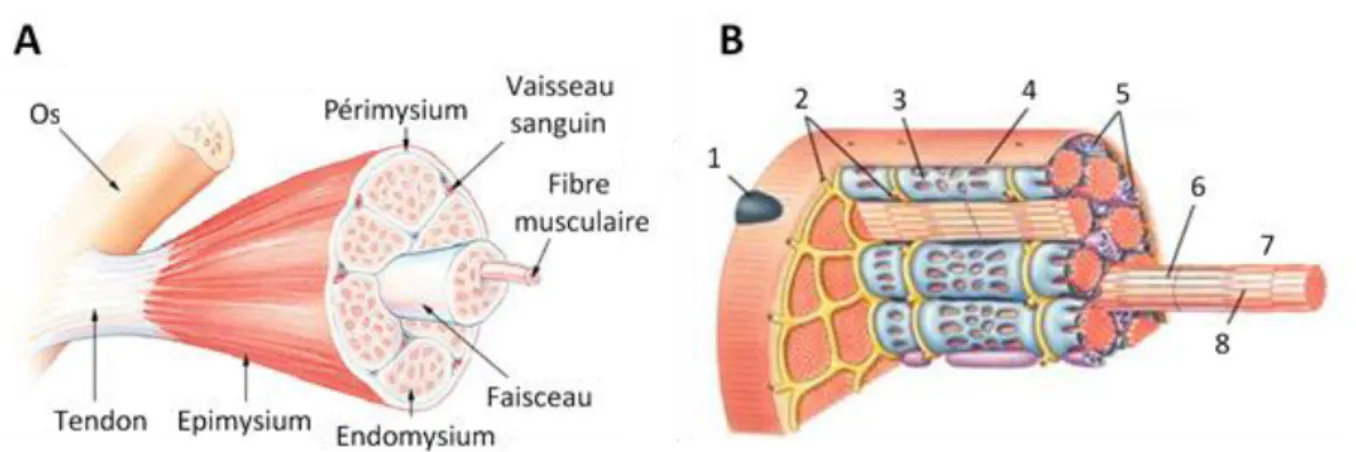
D’un point de vu structurel, le muscle squelettique est composé de nombreuses cellules musculaires appelées myocytes, qui sont entourés par une membrane de tissu conjonctif appelé endomysium, rassemblés en faisceau par le périmysium puis par l’épimysium (Figure 6).
Figure 6 : Représentation structurelle d’un muscle squelettique.1 : noyau ; 2 : tubules T ; 3 : réticulum sarcoplasmique ; 4 : sarcolemme ; 5 : mitochondries ; 6 : actine ; 7 : myofibrille ; 8 : myosine.
Le cytoplasme de la cellule musculaire (le sarcoplasme), a la particularité de posséder de nombreux noyaux (jusqu'à 500 par fibre), de renfermer principalement des protéines contractiles (l’actine et la myosine) entourées de canaux calciques (réticulum sarcoplasmique). L’orientation énergétique des myocytes est directement dépendante de la fréquence de stimulation du motoneurone efférent [65-67]. Les fibres musculaires innervées par les axones avec une basse fréquence de stimulation produisent des protéines contractiles oxydatives (fibres de type I), caractérisées par une grande endurance, une faible vitesse et
puissance de contraction, ainsi qu’un recrutement à l’effort pour de faibles intensités. Alors que les grandes fréquences de stimulation forment des protéines contractiles glycolytiques (Fibres de type IIa, IIx, IIb), possédant une grande puissance et vitesse de contraction, une endurance faible, et un recrutement spécifique lors des efforts de hautes intensités [68]. Ces variations structurelles se répercutent sur la densité mitochondriale, le diamètre des fibres musculaires, la densité capillaire, et le développement spécifique des voies de resynthèse de l’Adénosine TriPhosphate [69].
Tableau 1: Caractéristiques fonctionnelles et morphologiques des fibres musculaires en fonction de leur typologie adapté des travaux de Bacou et Vigneron, 1988 [70]
II.A.1 La composition structurelle musculaire du sujet sain
Suivant la nature de leur fonction, les muscles ont une typologie très différente pouvant être oxydative pour les muscles posturaux comme le muscle soléaire (87,7±9,1 % de fibres I chez le jeune adulte), ou inversement, plus glycolytique pour les muscles sollicités dans les efforts dynamiques et intenses comme l’extenseur long des doigts (47,3±5,2 % de fibres I chez le jeune adulte) [71]. La correspondance entre les typologies d’un même muscle sur
Physiologie Type l Type IIa Type IIb-IIx
Vitesse de contraction lente rapide rapide
Durée de contraction longue courte courte
Résistance à la fatigue +++ ++ +
Recrutement à l'effort tôt intermédiaire tard
Morphologie
Couleur rouge rouge blanche
Aire des fibres + +++ +++
Densité capillaire +++ ++ +
Mitochondries +++ +++ +
différentes espèces de mammifères est compliquée, car bien que le profil oxydatif ou glycolytique global reste le même, les proportions de fibres varient différemment d’un muscle à un autre. Par exemple, chez le rat mâle adulte, le soléaire a une composition similaire à l’homme (89±5,0 % de fibres I chez le rat), mais l’extenseur long des doigts est beaucoup plus glycolytique (3,9±1,6 % de fibres I chez le rat) [72]. Ainsi, le transfert des résultats obtenus d’une espèce à une autre nécessite la connaissance de la typologie musculaire intrinsèque à chaque espèce.
De plus, l’importante plasticité du muscle en fait un tissu dont la masse et la composition varient tout au long de l’existence. En effet, chez les hommes le développement du tissu musculaire se stabilise aux alentours de 20-25 ans et tend à s’inverser dès l’âge de 50 ans [73]. La perte musculaire accélère progressivement en lien direct avec l’âge des sujets (Figure 7). Ainsi, la masse musculaire d’une population de sujets âgés de 80 ans est significativement inférieure d’environ 20 à 30 % à celle d’une population âgée de 20 ans [74].
Figure 7 : Evolution de la masse musculaire squelettique totale de l’homme et de la femme au cours de la vie [75]
Outre la diminution quantitative, le tissu musculaire subit également des altérations qualitatives telle qu’une perte des MHC de type II [76], une réduction de l’activité enzymatique impliquées dans les voies aérobies [77] et anaérobies [78] et une altération du fonctionnement mitochondrial à l’origine d’un stress oxydatif marquée au-delà de 65 ans [79]. Toutes ces altérations diminuent la fonctionnalité du muscle squelettique, dont les capacités d’endurance et de force décroissent au cours du temps [80]. De manière intéressante, cette
étude montre que lorsque les résultats obtenus sont normalisés par la masse musculaire des sujets, la différence entre le groupe 45 ans et celui de 65 ans est minorée. Cela laisse donc supposer que pour la majorité des intensités de contraction, la fonctionnalité propre de chaque fibre musculaire est préservée chez les personnes âgées saines. Les atteintes fonctionnelles observées sur la population vieillissante seraient uniquement dues à la diminution de la masse musculaire.
II.A.2 La composition musculaire du sujet BPCO
Le patient BPCO présente une altération des qualités de forces, de la masse et de l’endurance de son tissu musculaire très proche de celle observée au cours du vieillissement. De plus il est extrêmement difficile de différencier l’atteinte musculaire observée due à la pathologie elle-même de celle liée au vieillissement normal puisque la moyenne d’âge du diagnostic de la maladie est supérieure à 40 ans. Cependant, le déclin de la fonction musculaire du patient BPCO est beaucoup plus rapide que celle du sujet âgé sain. En effet, de nombreuses études ont montré que les patients BPCO avaient une diminution de la force de 20-30 % plus importante que celle de sujets contrôles [81, 82]. Ce résultat est vrai dans différents groupes musculaires tels que le quadriceps, le grand pectoral et le grand dorsal (Figure 8A).
Figure 8 : Répercussions de la BPCO sur les qualités de force (A) et d’endurance (B) du muscle quadriceps (figure issu de Kim et coll., 2008 ; [83] et Swallow et coll., 2007 ; [84]).BPCO : Bronchopneumopathie Chronique Obstructive. * : BPCO diff. de contrôle (p<0,05).
Parallèlement à l’atteinte de la force, le patient BPCO présente une altération encore plus marquée de ses capacités d’endurance. Une étude publié par Swallow et coll. [84] a utilisé des stimulations magnétiques répétées du nerf fémoral afin d’évaluer l’endurance du muscle quadriceps lors d’une succession de contractions. Celle-ci a montré une diminution plus précoce et plus importante de la contraction musculaire du quadriceps des sujets BPCO, comparées à des sujets sains (Figure 8B). La stimulation magnétique du nerf fémoral permet de s’affranchir de la composante nerveuse, ainsi, la diminution de la force de contraction du groupe BPCO est due uniquement à la composante musculaire, et non à une fatigue précoce du système nerveux central comme cela est le cas lors d’une exposition à l’hypoxie [85]. La plus faible distance parcourue par des patients BPCO comparés à des sujets sains au test de marche de 6 min, traduit bien la diminution des qualités d’endurance de ces patients. De plus ces auteurs ont également montré un lien entre la distance parcourue et la sévérité de la BPCO [48]. Ces répercussions fonctionnelles sont partiellement dues à la réduction significative de la proportion de fibres oxydatives dans le vaste latéral du muscle quadriceps [86-88]. Une méta-analyse de 2007 [89] a montré qu’une proportion de fibres I inférieure à 27 % associée à une proportion de fibres IIx supérieure à 29 % devait être considérée comme pathologique dans le vaste latéral du quadriceps des patients BPCO. De plus, cette même étude a confirmé l’étroite corrélation qu’il existe entre la sévérité de la BPCO et les répercussions typologiques et fonctionnelles du vaste latéral.
Toutefois, la disparition des fibres I au profit des fibres II ne peut expliquer à elle seule, toute l’atteinte musculaire du patient BPCO. En effet, contrairement aux observations réalisées avec des sujets sains vieillissants les patients BPCO présentent une diminution de la force normalisée par la section du muscle. Cette atteinte qualitative, pousse même certains auteurs à parler de myopathie du BPCO [90]. Ces études ont montré que, malgré un indice de masse maigre identique, la force du quadriceps des patients BPCO était inférieure aux contrôles [91]. Toutefois, ce résultat est soumis à controverse puisque d’autres travaux rejettent l’hypothèse d’une atteinte fonctionnelle intrinsèque des fibres musculaires, attribuant la faiblesse du BPCO uniquement à la diminution de la surface de section du muscle [92, 93]. L’explication la plus probable impliquerait une hétérogénéité d’atteinte muscle spécifique en fonction de la typologie, de la localisation et de la sollicitation du muscle par le patient BPCO [94, 95]. Ainsi, les différences de zone où la biopsie est réalisée et la proportion
différente des fibres la composant, expliqueraient ces résultats obtenus divergents.
La diminution de la vascularisation des fibres musculaires, due à une moindre expression du Vascular endothelial Growth Factor (VEGF) [90, 96] et de la capillarisation [97] [98] des muscles du membre inférieur des patients avec BPCO, participe à la perturbation du microenvironnement cellulaire. La diminution de la surface d’interaction entre le compartiment cellulaire et sanguin diminue la diffusion de l’oxygène dans le tissu musculaire ce qui pourrait être une hypothèse séduisante pour expliquer l’atteinte des capacités oxydatives et l’augmentation du stress oxydant observé dans cette pathologie.
II.B Les propriétés métaboliques du tissu musculaire
II.B.1 Le muscle du sujet sain
Les fibres musculaires sollicitent des voies différentes de resynthèse de l’Adénosine Tri Phosphate (ATP) afin de répondre au mieux à leurs besoins énergétiques. Ces différences sont conditionnées par le type de fibre, et donc par la nature de la tâche qui leur est attribuée. 3 grandes voies de production énergétique existent : l’aérobie, l’anaérobie lactique et l’anaérobie alactique.
Figure 9 : Représentation simplifiée des 3 grandes voies de libération d’énergie participant à la resynthèse de l’Adénosine Triphosphate. CK : Créatine Kinase ; Cr : Créatine ; P : Phosphate
L’anaérobie alactique participe à la resynthèse de l’ATP en libérant l’énergie contenue dans la liaison de la Phospho-Créatine sous l’action de la Créatine Kinase (CK).
L’anaérobie lactique libère de l’énergie en dégradant les molécules de glucose dans le cytosol. Cette lyse du glucose est réalisée par plusieurs enzymes dont les principales sont l’hexokinase et la phosphofructokinase. En anaérobie, le pyruvate ainsi formé est converti en acide lactique par la lactate dehydrogénase (LDH) en vue d’une oxydation future.
L’oxydation du glucose est commune avec la glycolyse anaérobie jusqu’à l’étape de synthèse du pyruvate. A cette étape, le pyruvate entre dans la mitochondrie sous l’action de la pyruvate déshydrogénase (PDH) pour être complètement dégradé en H20, CO2 et énergie
par le cycle de Krebs. Le cycle de Krebs, sous l’action d’une série d’enzymes, sépare les électrons des molécules, pour alimenter la chaine respiratoire, fournissant ainsi à la pompe ATPase, l’énergie nécessaire pour re-synthétiser de l’ATP à partir d’Adénosine Di-Phosphate (ADP) [99]. Le métabolisme aérobie est également capable d’oxyder les substrats lipidiques
pour fournir de l’énergie à l’organisme. Pour cela, les acides gras libres entrent passivement dans la mitochondrie, si ce sont des acides gras à chaines courtes et moyennes, ou sous l’action des transporteurs Carnitine Palmitoyltransférase 1 et 2 (CPT1 et 2) si ce sont des acides gras à chaines longues. Une fois dans la matrice mitochondriale, une étape intermédiaire de dégradation des molécules par la -oxydation, leur permet de rejoindre le cycle de Krebs [100].
Ainsi, les fibres de type I sollicitent principalement le métabolisme aérobie par oxydation des substrats lipidiques ou glucidiques dans la mitochondrie. Cette voie de resynthèse de l’ATP permet aux cellules de maintenir un apport énergétique sur de longues périodes, mais cette filière possède une puissance assez faible. Inversement, le métabolisme anaérobie sollicité par les fibres de type II permet un apport en énergie important, mais extrêmement limité dans le temps. En effet, la dégradation du glucose par les enzymes de la glycolyse, ou de la Phospho-Créatine (PCr) par la créatine kinase, libère une puissance 2 à 4 fois plus importante que l’aérobie, mais seulement sur une période maximum de quelques minutes [101].
II.B.2 Le muscle du patient BPCO
Comme cela a été présenté dans la section II.A.2, la BPCO se traduit par une disparition progressive des fibres oxydatives de type I au profit des fibres de type II, dites fibres glycolytiques [102]. Ce remodelage typologique réoriente le métabolisme musculaire des patients, vers un profil favorisant la production énergétique par la voie métabolique anaérobie [103]. Cependant, la même augmentation de l’anaérobie a été obtenu en comparant des cerveaux de sujets sains avec des patients alors qu’aucune modification de la composition cellulaire n’était présente [104]. Ce résultat suggère que la réorientation métabolique dans le muscle du BPCO n’est pas une conséquence de l’augmentation de la proportion de fibres de type II, mais un mécanisme parallèle à ce remodelage.
Des études de la fonction mitochondriale des muscles locomoteurs des patients BPCO ont également mis en évidence une détérioration du couplage et de l’activité des complexes de la chaine respiratoire [105, 106]. Cette dysfonction mitochondriale semble particulièrement marquée dans le vaste latéral de patients BPCO ayant une IMC inférieur à 20
[107]. Toutefois, un travail publié par Picard et coll. en 2008 [108] conclut que ces perturbations de l’activité mitochondriale pourraient être dues à l’augmentation de la proportion de fibres II, qui produisent intrinsèquement plus de radicaux libres que les fibres I. Cette reorientation de la typologie pourrait également expliquer la diminution de la 3-Hydroxyacyl-CoA déshydrogénase, une enzyme clef de la -oxydation, chez les patients BPCO comparés à des sujets sains [109]. Le rôle de la diminution de l’efficacité mitochondriale dans l’atteinte musculaire est développé de manière plus approfondi par l’intermédiaire de la production musculaire de radicaux libre (Cf. section IV.C, page 55).
Les patients BPCO montrent également une altération des acides aminés plasmatiques à chaine branchés [110], et du glutamate associé à une réduction du glutathion intramusculaire des patients emphysémateux [111]. Cette perturbation systématique du profil des acides aminés est étroitement liée au degré du catabolisme musculaire des patients. La diminution de la concentration plasmatique en glutamate semble être un témoin de la capacité de diffusion de l’O2 réduite par l’emphysème des patients. Lorsque la réduction du
glutamate est associée à une diminution du glutathion, la sollicitation de la glycolyse augmente, comme en témoigne l’augmentation accélérée de la lactatémie lors d’un exercice physique [110, 112]. Les fortes perturbations de la concentration en glutamate ainsi que la diminution du glutathion des muscles du membre inférieur alors que celles des muscles respiratoires tel que le diaphragme sont épargnés [113], montrent encore une fois une atteinte très hétérogène ente les groupes musculaires. La diminution significative des acides aminés après exercice dans le quadriceps de patients BPCO, couplée à leur augmentation plasmatique [114], semble indiquer une augmentation du turnover des acides aminés [115]. Cela pourrait être une conséquence de l’inflammation chronique de ces patients [116], qui traduit bien une perturbation du métabolisme musculaire. Cependant, ces données sont à prendre avec prudence et ne sont pas la preuve formelle d’une modification de l’équilibre de la masse musculaire du patient BPCO.
Dans la population BPCO une forte prévalence du diabète existe, sans que les mécanismes ne soient clairement identifiés. Quelques études ont cependant montré que l’inactivité, la corticothérapie, ainsi que des altérations mitochondriales pourraient être impliquées dans les mécanismes initiateurs de ces troubles métaboliques [112]. Or, l’atteinte métabolique et mitochondriale observée chez les patients uniquement diabétiques, est très
proche de celle observée dans la BPCO (avec ou sans diabète) [117]. Ces altérations communes concernent la réduction des capacités d’oxydation du pyruvate et des acides gras [118, 119], la diminution de la densité mitochondriale [120], ainsi que de la forme protéique de la protein proliferator-activated receptor-γ (PPAR-γ) [121] et de la transcription du facteur de transcription PPAR coactivator-1α (PGC-1α) [122]. La diminution de ces 2 facteurs de transcriptions responsables de l’initiation du développement du métabolisme oxydatif [123], concorde avec les altérations musculaires observées dans la BPCO.
En résumé :
Chaque type de fibre musculaire possède des caractéristiques structurelles et fonctionnelles qui leurs permettent de répondre au mieux à la nature du travail mécanique demandé. Ainsi les fibres de type IIx et IIb sollicitent principalement le métabolisme anaérobie qui permet la libération d’une grande quantité d’énergie sur une courte période de temps. Les fibres IIa ont un profil métabolique mixte et les fibres de type I sont quasiment exclusivement oxydatives, ce qui leur permet de maintenir une faible intensité de contractions sur de longues périodes. Cette orientation fonctionnelle se répercute sur leur phénotype. Les fibres de type IIb et IIx (dites glycolytiques), ont un diamètre important associé à une faible vascularisation alors que les fibres I (dites oxydatives) ont un faible diamètre avec un réseau capillaire dense. Les fibres IIa présentent encore un phénotype intermédiaire avec un diamètre important associé à une vascularisation développée. Le phénotype propre à chaque type de fibre est à l’origine de variations de la surface d’échange entre le compartiment sanguin et cellulaire qui pourrait expliquer l’hétérogénéité de l’atteinte musculaire observée chez les patients. En effet, chaque groupe musculaire des patients présente une diminution de la proportion de fibres I plus ou moins marquée. Cette disparition des fibres I au profit des fibres II est la symétrie parfaite de l’évolution du muscle du sujet sain âgé, qui perd ses fibres II au profit des fibres I. L’altération des fibres I du patient BPCO peut être en partie expliquée par la diminution de la vascularisation des muscles, induite par une diminution de VEGF, qui ne permet plus à ces cellules de subvenir à leur besoin en O2. Cela pourrait également expliquer la transition du
métabolisme aérobie vers l’anaérobie lactique qui est observée chez les patients BPCO. Cette transition se retrouve également dans le tissu cérébral, ce qui semble indiquer qu’elle n’est pas seulement due à l’augmentation de la proportion des fibres II.
Au niveau cellulaire, la BPCO est à l’origine d’une diminution de la densité mitochondriale, d’une réduction des protéines responsables de l’oxydation, et d’une augmentation de l’activité des enzymes de la glycolyse. Ces modifications augmentent le stress oxydant et la production de lactate intramusculaire des muscles du membre inférieur. Cependant la moyenne d’âge des patients BPCO étant supérieure à 45 ans, il est difficile de différencier les atteintes musculaires liées au vieillissement de celles induites par la pathologie elle-même.
Toutefois, le déclin plus rapide des qualités de force et d’endurance des muscles, ainsi que la disparition progressive des fibres de type I, sont des signatures caractéristiques de l’évolution de cette pathologie, qui poussent certains auteurs à parler de myopathie du BPCO.
II.C Les facteurs impliqués dans le contrôle de la masse musculaire
Le tissu musculaire est régulé par un équilibre dynamique entre les facteurs anaboliques qui initient la protéosynthèse, et cataboliques à l’origine de la protéolyse. Cette balance est régulée à différents niveaux cellulaires par de nombreux mécanismes qui orientent l’évolution de la masse musculaire et ses caractéristiques fonctionnelles.
II.C.1 Les voies anaboliques
Toute sollicitation mécanique de la masse musculaire, perturbe l’homéostasie cellulaire. L’hypertrophie musculaire est l’une des principales conséquences des nombreuses adaptations tissulaires initiées par la contraction musculaire. Cette croissance musculaire est le reflet direct d’une augmentation de la synthèse protéique par les cellules, dépendante de 2 étapes majeures, la transcription et la traduction des Acides RiboNucléiques messagers (ARNm).
Chez l’adulte, la croissance musculaire induite par une surcharge mécanique est principalement régulée par une augmentation du contenu protéique des cellules existantes. Il est maintenant bien reconnu que la nature des contractions, ainsi que la durée de l’effort réalisé, n’active pas les mêmes voies de transcription (Figure 10). Cette spécificité d’activation des voies de transcription des ARNm permet au muscle de s’adapter spécifiquement au stress auquel il est soumis. Lors d’un protocole d’entrainement en force, les fibres de type II s’hypertrophient car la surface de section d’un muscle conditionne sa capacité à produire de la force [124], alors que les fibres I développent plutôt des aspects qualitatifs à l’issue d’un entrainement en endurance. En effet, les contractions de haute intensité favorisent le développement des fibres glycolytiques en augmentant la transcription des protéines impliquées dans les filières anaérobies et dans l’hypertrophie des fibres II, alors que les efforts d’endurance augmentent l’expression de gènes en lien avec le métabolisme oxydatif [125].
Lors d’un effort de haute intensité, une diminution de la PO2 intramusculaire augmente
la stabilisation de l’Hypoxia Inducible Factor 1HIF-1, et le relargage massif du calcium intramusculaire qui active la calmoduline (voir Figure 10). La stabilisation de HIF-1 est à l'origine d’une inhibition de la phosphorylation oxydative [126] parallèlement à une
augmentation d’enzymes clefs de la glycolyse dont la phosphofructokinase, et la lactate déshydrogénase [127]. La calmoduline active la calmodulin-dependent protein kinase II (CaMKII) et de la voie des Mitogen-Activated Protein Kinase (MAPK) qui inhibent la myogénine (MyoG) et le Myocite enhanced factor (Mef) (voir Figure 10). MyoG et Mef sont 2 protéines impliquées dans le développement des capacités oxydatives [128, 129]. Inversement CaMKII et les MAPK augmentent l’activité du complexe sine oculis homeobox 1/eyes absent 1 (Six/Eya), impliqué dans la réorientation du muscle vers un profil glycolytique [130].
L’adaptation aux efforts d’endurance est principalement médiée par le métabolisme et le statut énergétique cellulaire [131]. En effet le statut énergétique de la cellule, reflété par le rapport de la concentration cytosolique en Adénosine Mono-Phosphate (AMP) sur l’Adénosine Tri-Phosphate (ATP), joue un rôle majeur dans la plasticité musculaire. Ainsi, une forte consommation en ATP ou une perturbation de son turn-over est à l’origine d’une augmentation du rapport AMP/ATP, conséquence d’une activation de l’AMPkinase (AMPK). Dans un premier temps l’AMPK inhibe les fonctions cellulaires énergivores de la cellule tel que la synthèse protéique et lipidique [132]. Lorsque l’homéostasie énergétique est rétablie l’augmentation de la protéine PGC1 connue pour ses effets pro-oxydatifs [133], par l’AMPk participe activement au développement du métabolisme aérobie [134, 135]. L’AMPk est donc un senseur cellulaire du statut énergétique [136]. L’activation de PPAR par la présence d’acides gras libres [137] augmente également les capacités oxydatives musculaires sans perturbation de la typologie [138]. En présence d’une concentration modérée en ions calcium, la calmoduline favorise l’activité de la calcineurine [139] dont l’action augmente les capacités oxydatives du muscle par l’intermédiaire de Mef et du Nuclear Factor of Activated T-cells (NFAT) [140].
Figure 10 : Représentation schématique de l’état des connaissances actuelles de l’influence de la nature des contractions sur les voies de signalisations impliquées dans le couplage excitation-transcription [125]. AMP: Adenosine monophosphate; AMPK : AMP-activated kinase; CaMK : Ca2+/calmodulin-dependent protein kinase; cPKC: conventional protein kinase C; ERK: extracellular signal-regulated kinase; [Ca2+]: free intracellular calcium concentration; HPH: HIF-1α prolyl hydroxsylases; HDAC: histone deacetylase; PO2i: intracellular oxygen pressure; MAPK: mitogen-activated kinase; MEF2:
myocyte enhancer factor 2; MyoD: myogenic differentiation factor; nPKC: novel protein kinase C; NFAT : nuclear factor of activated T-cells; PPAR : peroxisome proliferator-activated receptor; PGC-1α: PPARγ coactivator-1α ; HIF-1α : hypoxia inducible factor 1α; PKD1:protein kinase D1; SIK1: salt inducible kinase 1; Six1/Eya1: sine oculis homeobox 1/eyes absent 1.
La traduction des ARNm en protéines est également sujette à de nombreuses régulations favorisant ou non la synthèse protéique. Au cours de ce processus la voie Akt/mTOR est la charnière centrale de transduction du signal anabolique [141]. Akt/mTOR est principalement activé par la phosphorylation de la protéine Phosphoinositide 3-kinase (PI3K), activée par l’insuline ou l’Insuline Growth Factor 1 (IGF-1) (Figure 11B). PI3K initie de multiples processus anaboliques tels que la prolifération, la croissance, la différenciation et la survie cellulaire [142]. Pour cela, PI3K phosphoryle la protéine kinase Akt (également connu sous l’appellation PKB) qui participe, d’une part à l’activation du facteur eukaryotic initiation Factor 2B (eIF2B) [143] et d’autre part, à l’activation de la mammalian Target Of Rapamycin (mTORC1) par l’intermédiaire d’une phosphorylation de la Tuberous Sclerosis Complex 1 et 2 (TSC1-2) [144]. La phosphorylation de mTORC1 active à son tour la protéine S6 Kinase-1 (S6K1) parallèlement à une inhibition de 4E-BP1 (Eukaryotic translation initiation factor 4E-binding protein 1). Ces 2 facteurs de transcriptions, situés à l’extrémité terminale de l’axe Akt/mTOR, régulent le niveau d’expression des gènes puisque S6K1 augmente la traduction des ARNm [145], alors que 4E-BP1 la réprime [146].
Figure 11 : représentation schématique de la voie des MAPK (A), de la voie Akt/mTOR (B), ainsi que de leurs principales interactions (d’après Mendoza et al, 2011 [147]). (--- : activation de la protéosynthèse ; --- : inhibition de la protéosynthèse). 4E-BP: Eukaryotic translation initiation factor 4E binding protein; AKT: AKT ou protein kinase B; ERK: extracellular signal-regulated kinase; GAP: GTPase-activating protein; GDP: Guanosine diphosphate; GF: Growth Factor; Grb2: Growth factor receptor-bound protein 2; GTP: Guanosine triphosphate; IRS: Insulin receptor substrate; MEK: MAPK/ERK kinase; mTOR: mammalian target of rapamycin; PDK1: Phosphoinositide-dependent kinase 1; PI3K: phosphatidylinositol 3-kinase; PIP3: phosphatidylinositol 3,4,5 trisphosphate; PKC: Protein kinase C; PTEN: Phosphatase and TENsin homolog; RHEB: Ras homolog enriched in brain gene; RSK: Ribosomal s6 kinase: S6K: ribosomal protein S6K; SOS: Son of sevenless; TSC: tuberous sclerosis complex.
Parallèlement à cette voie d’activation, mTORC1 est également activé par la voie des MAPK par l’intermédiaire de la protéine Extracellular signal-Regulated Kinase (ERK) (Figure 11A). La levée de l’inhibition de mTORC1 par TSC2 [148], ainsi que son activation directe par l’activité des MAPK permettent une transduction du signal anabolique indépendante d’Akt. La disponibilité en acide aminé intracellulaire [149], et la transduction des contraintes mécaniques induites par les contractions musculaires [150], sont d’autres mécanismes
![Figure 2 : Nature et fréquence comorbidités recensées dans la population des différentes BPCO [39]](https://thumb-eu.123doks.com/thumbv2/123doknet/14412505.512064/18.892.115.787.106.310/figure-nature-fréquence-comorbidités-recensées-population-bpco.webp)
![Figure 5 : La spirale du déconditionnement. Concept présenté A. Young et coll. [53].](https://thumb-eu.123doks.com/thumbv2/123doknet/14412505.512064/22.892.256.627.111.469/figure-spirale-déconditionnement-concept-présenté-young-coll.webp)
![Figure 8 : Répercussions de la BPCO sur les qualités de force (A) et d’endurance (B) du muscle quadriceps (figure issu de Kim et coll., 2008 ; [83] et Swallow et coll., 2007 ; [84]).BPCO : Bronchopneumopathie Chronique Obstructive](https://thumb-eu.123doks.com/thumbv2/123doknet/14412505.512064/28.892.116.769.766.1026/répercussions-qualités-endurance-quadriceps-swallow-bronchopneumopathie-chronique-obstructive.webp)
![Figure 11 : représentation schématique de la voie des MAPK (A), de la voie Akt/mTOR (B), ainsi que de leurs principales interactions (d’après Mendoza et al, 2011 [147])](https://thumb-eu.123doks.com/thumbv2/123doknet/14412505.512064/40.892.119.777.104.555/figure-représentation-schématique-voie-mapk-principales-interactions-mendoza.webp)
![Figure 22 : Cascade de régulation du peroxisome proliferator-activated receptor coactivator-1 [311]](https://thumb-eu.123doks.com/thumbv2/123doknet/14412505.512064/66.892.165.729.580.953/figure-cascade-régulation-peroxisome-proliferator-activated-receptor-coactivator.webp)