Computational Engineering of Small
Molecules to Treat Infectious Diseases
by
Raja R. Srinivas
B.S. Biomedical Engineering, Applied Mathematics & Statistics Johns Hopkins University (2011)
Submitted to the Department of Biological Engineering in partial fulfillment of the requirements for the degree of:
Doctorate of Philosophy in Biological Engineering at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY February 2017 MASSACHUSETTS INSTITUTE OF TECHNOLOGY
MAY
E
2017
LIBRARIES
ARCHMWS
0 Massachusetts Institute of Technology 2017. All rights reserved.
Signature redacted
Signature of A uthor...Department of Biological Engineering December 5 th 2016
C ertified by...
Sic
Sianature redacted
...Bruce Tidor Professor of Biological Engineering and Computer Science
7 / Thesis Supervisor
nature red actedi
This thesis has been examined by a committee of the Department of Biological Engineering as follows:
K. Dane Wittrup
C. P. Dubbs Professor of Chemical Engineering and Biological Engineering
Chairman of Thesis Committee
Bruce Tidor
Professor of Biological Engineering and Computer Science Thesis Supervisor
Amy Keating
Professor of Biology
Computational Engineering of Small
Molecules to Treat Infectious Diseases
by Raja Srinivas
Submitted to the Department of Biological Engineering on December 8, 2016, in partial fulfillment of the
requirements for the degree of
Doctor of Philosophy in Biological Engineering Abstract
Rational drug design of small molecules has led to the development of robust therapeutics that are currently used in the clinic. However, key challenges remain in designing drugs against infectious disease targets that are susceptible to mutation. To achieve full clinical efficacy against rapidly-mutating targets, new methods must be developed for designing drugs. In this thesis, we utilize a three pronged approach for rational ligand design by developing methods, analyzing existing
experimental data, and designing novel therapeutics.
On-target mutations in infectious diseases often render inhibitors ineffective and are one of the key clinical failures of current therapies. We use HIV protease as a model system to understand mutation resistance. HIV protease substrates are unaffected or only moderately affected by resistance mutations that greatly decrease inhibitor binding. This idea has led to the design of broadly binding inhibitors using substrate mimicry. This is achieved by constraining inhibitors to bind within the consensus substrate volume, which we term the "substrate envelope". However, while the substrate envelope has been relatively successful, some inhibitors that are designed based on this model are sensitive to mutants. We performed a detailed biophysical binding energy decomposition of a flat and susceptible binder pair and found that the susceptible inhibitor forms stronger interactions with key residues. These residues are entirely characterized by examining known resistant mutants to approved HIV protease inhibitors. To generalize our findings, we cross-validate on a set of ten HIV protease inhibitors with previously measured sensitivity. We find that interaction energy successfully classifies susceptible and flat inhibitors. Based on these results, we extend the current design paradigm. We develop a methodology to minimize extraneous contacts with the active site and express it as an appropriate cost function, which is then minimized. We then implement this design scheme for HIV protease, yielding both flat and susceptible binders.
Next, we apply rational drug design principles to other infectious disease targets. We first focus on the tuberculosis specific CIpP1P2 peptidase to optimize the antibiotic, acyl depsipeptides (ADEPs). We use component analysis to understand the biophysics of ADEP binding to the active
treatment of Candida albicans. We use molecular docking to predict a binding mode for the lead compound and then account for receptor plasticity by performing molecular dynamics simulations. We use this improved receptor model to design novel analogs that are predicted to bind better than the parent compound. Lastly, we focus on disease diagnosis by developing a novel paradigm for MRI contrast agent design. We first integrate the governing thermodynamics and relevant parameters that influence imaging efficacy to develop an integrated workflow for contrast agent design. We then apply our methodology to the DOTA system and successfully explain differential activity of designed analogs. Put together, we demonstrate the power of rational design in various relevant biological contexts.
Overall, this thesis presents new techniques, analysis, and applications of rational design to address unmet clinical problems. Work from this thesis accelerates the field of computational drug design, which has implications in many uncured diseases and diagnostics.
Thesis Supervisor: Bruce Tidor
Acknowledgements
First and foremost, I want to thank Bruce Tidor for being an incredible mentor throughout my entire PhD journey. He has been extremely supportive and generous with his time and has taught
me the quantitative skills required for all the work presented in this thesis. Beyond that, he has been an invaluable mentor for my career. I also want to thank my committee, Dane Wittrup and Amy Keating, for meeting with me about this work and giving constructive feedback that helped
improve the science. Much of the improvements in the HIV protease chapters are a result of my committee meetings and for that I am grateful.
I have been surrounded by incredible scientists and people in the Tidor lab that has made coming to work a pleasure. First, I want to thank Brian Bonk who came into the program with me in Biological Engineering and has been my desk mate and great friend throughout this journey. Ishan Patel was instrumental in recruiting me to this lab and made lab memorable for the four years we overlapped. James Weis joined the lab in 2014 and has been an incredible friend and provided me with a new perspective on finance through his experiences. I have also enjoyed my conversations with all of my lab mates including David Flowers, Kevin Shi, Andrew Horning, David Hagen, Natasha Seelam, David Bloore, and Ishan Arora. Finally, I want am indebted to Yang Shen, who was a post-doc in our lab when I first joined and taught me the drug design code which really accelerated my research.
I want to thank Madhavi Gavini who has been my best friend for almost a decade. We have been through a lot of new experiences together and I am excited to see where the future goes. I was fortunate to start a company with her, with two incredible products that could reach the clinic soon. I also want to thank Joey Orofino who I have known since junior year of college. He has been a huge support to get through all those lousy New York Knicks seasons. Finally, I want to thank the boys, without whom graduate school would not have been as fun. We have had some crazy shared experiences and I am excited to see what the futures holds.
Finally, I want to thank my family for all their support. I was fortunate to be able to live with my sister for my first three years of graduate school and really got to know her in that time. I am lucky that she is a co-founder of the company. To my parents, I obviously would not be here without all your support and guidance. From bringing me food to giving me advice on my thesis, all your hard work is what made this thesis possible.
Table of Contents
I
Chapter 1: Introduction... 121.1 Ligand Design: Overview ... 12
1.2 Ligand design: a three prong approach ... 16
1.3 Sum m ary of thesis... 17
2 Chapter 2: Computational Analysis of HIV Protease Inhibitors with Differential Sensitivity To Resistant M utants ... 20
2.1 Introduction ... 21
2.2 Results ... 25
2.2.1 Calculations can capture sensitivity and insensitivity of KC-08 and KB-83 respectively ... 25
2.2.2 M olecular Understanding of Differential Binding... 30
2.2.3 Generalizing sources of differences for susceptible and flat inhibitors... 36
2.2.4 Testing set inhibitor selection ... 39
2.2.5 Binding Affinity Theory ... 43
2.2.6 Improvem ent to design ... 46
2.2.7 Future W ork ... 47
2.3 M ethods... 49
2.3.1 Receptor Structure Preparation ... 49
2.3.2 Scaffold Preparation/Ligand Optim ization ... 49
2.3.3 Ensem ble Calculations... 51
2.3.4 Energy decom position ... 52
3 Chapter 3: Development of Framework for Robust Inhibitors: Applied to HIV Protease.... 54
3.1 Introduction ... 55
3.2 M ethods: Fram ework Developm ent... 58
3.2.1 Fram ework Developm ent Overview ... 58
3.2.2 Criterion for Framework ... 58
3.2.3 Fram ework version 1 ... 59
3.2.4 Fram ework 2... 64
3.2.5 Fram ework version 3 ... 70
3.2.6 Selecting Fram ework for HIV protease ... 71
3.3.1 W hy is an approxim ation needed?... . . .. . . .. . . .. .. . .. . . 71
3.3.2 M onte Carlo Approxim ation... 72
3.3.3 Overall Implementation ... 75
3.4 M ethods: Operational... 75
3.4.1 Scaffold Preparation/Ligand Optim ization... 76
3.4.2 M C Approxim ation... 77
3.4.3 Energy decomposition ... 78
3.4.4 Emin Calculation ... 79
3.4.5 Param eter Calculation for each inhibitor ... 79
3.4.6 Inhibitor Analysis... 79
3.5 Results ... 80
3.5.1 Initial Design... 80
3.5.2 M C Approxim ation Results ... 81
3.5.3 0 Calculations have a high spread... 83
3.5.4 Additional parameters: O1...N, Interaction Energy, Residues of Interest ... 85
3.5.5 Compound Classification... 86
3.5.6 Compound Selection... 87
3.5.7 Interaction Energy Analysis... 92
3.5.8 Overall Summary of Compounds and Future Directions ... 94
4 Chapter 4: Design and Optimization of ADEPs to the Tuberculosis ClpP Peptidase... 97
4.1 Introduction ... 98
4.2 Results ... 100
4.2.1 Minimum energy conformation of the parent compound ... 100
4.2.2 Energetic decomposition of la... 102
4.2.3 Rational Design at R1 ... 103
4.2.4 M olecular dynamics sim ulation of RI designs ... 104
4.2.5 Experim ental Testing ... 107
4.2.6 Rational Design at R2... 108
4.2.7 Rational Design at R3 ... 110
4.2.11 l a-d do not bind to ClpP1 com pared to ClpP2 ... 118
4.2.12 Further Design at P1 Site... 120
4.2.13 Future W ork... 124
4.3 M ethods... 126
4.3.1 Structure Preparation ... 126
4.3.2 Scaffold Preparation and Enum eration... 127
4.3.3 Functional Group Library Preparation... 127
4.3.4 Inverse Design ... 128
4.3.5 Energetic Decom position... 129
4.3.6 M olecular Dynam ics... 130
5 Chapter 5: Methods and Design of Novel MRI Contrast Agents... 131
5.1 Introduction ... 132
5.2 M ethods: Fram ework Developm ent...134
5.2.1 Design Strategy... 134
5.3 M ethods... 142
5.3.1 Structure Preparation ... 142
5.3.2 Rotam er Library Preparation ... 143
5.3.3 W ater Rotam er Library Preparation... 143
5.3.4 Attachm ent to Scaffold... 143
5.3.5 Pre-screening... 144
5.3.6 Lead Candidate Identification... 144
5.3.7 M olecular D ynam ics Sim ulations... 145
5.4 Results... 146
5.4.1 C Term inus Results... 146
5.4.2 N-Term inus Design... 150
5.4.3 N-Term inus Analysis ... 151
5.4.4 Future Directions ... 154
6 Chapter 6: Design of Novel Inhibitors Combining Docking, Molecular Dynamics, and Inverse Design ... 156
6.1 Introduction ... 157
6.2 Results... 159
6.2.3 M odel Refinement ... 166
6.2.4 Receptor Plasticity... 168
6.2.5 New Ligand Identification ... 171
6.3 M ethods... 176
6.3.1 Structure Preparation ... 176
6.3.2 Glide Docking... 177
6.3.3 Inverse Design ... 177
6.3.4 M olecular Dynam ics and highres calculations... 178
7 Chapter 7: Conclusion ... 179
7.1 Conclusion... 180
8... 183
Chapter 1: Introduction
1.1 Ligand Design: Overview
The field of rational drug design, and broadly all-atom modeling, has matured remarkably in the past few decades. In 2013, three founders of the field, Martin Karplus, Michael Levitt and Arieh Warshel, were awarded the Nobel Prize in chemistry due to their joint contribution of molecular dynamics simulation for biomolecules. Initially, most molecular dynamics modeling simulation focused on native proteins and their interactions. However, as the state of the art for technology increased, the capability of biomolecular simulation progressed likewise and it became more feasible to study and understand detailed molecular interaction from both a scientific and engineering perspective. This was further aided by substantial leaps in experimental crystallography - as more proteins were discovered and structurally resolved, the dataset for molecular modeling expanded exponentially.
In the late 1980s-early 1990s, an alarming epidemic of human immunodeficiency virus (HIV) and acquired immune deficiency syndrome (AIDS) plagued the United States along with other nations. Interestingly, the field of rational drug design was one of the biggest contributors in this collective battle. The Cambridge, MA based company, Vertex, had built its reputation on the ability to crystalize protein targets and subsequently rationally engineer drugs. As a pioneer, Vertex was the first company to use rational engineering could be an alternative to the then-current paradigm of high-throughput-screening of chemical libraries.
Vertex worked on drugging HIV targets, and specifically the viral protease required for maturation of the virus. Through extensive rational design from the crystal structure, along with collaboration with leading experts, one of the first HIV protease inhibitors was developed'. The heralded story of Vertex and their successes is well documented in the book "The Billion Dollar Molecule". While this HIV protease inhibitor would later be optimized to yield drugs with better pharmacokinetics and toxicity properties, the barrier to generating an initial therapy would have been substantially higher without the rational drug design field. Since that early success, the field has blossomed and key clinical challenges can be solved using rational drug design principles such as drugging infectious disease targets.
Acquired drug resistance remains a key challenge in the effective treatment of various diseases such as infectious pathogens2-7. One common failure mode for therapies is target-based
mechanisms, which decreases inhibitor function without a significant reduction in substrate recognition. Key residues in the binding site mutate and inhibitors lose affinity and become clinically invalid. However, substrate functionality is retained. Drug resistance results in massive burden on our healthcare system, which costs billions of dollars when approved therapeutics are rendered non-viable due to mutation of their target8'9. Therefore, there is a huge economic and
patient need to develop drugs that are robust against various mutation resistance. Rational drug design can be used to specifically engineer therapeutics that are not prone to mutation resistance.
There are various computational approaches that are currently implemented to design small molecule drugs such as high-throughput screening or fragment based design 0.12. For the former,
is optimized rationally. Other methods include fragment-based design in which the binding areas within the active site are initially considered independently 3. Small chemical moieties are
designed for each site and then the various fragments are then joined together by a scaffold. Such methods have been used in a vast number of biological systems, ranging from metabolic targets such as butyrylcholinesterase to infectious disease targets such as HIV reverse transcriptase14-6 In these examples, a co-crystal structure of the lead bound to the target is optimized to improve binding affinity via additional energetic interactions, for example, by introducing a fluorine (increased bulk for better vdW packing) or altering a methyl group to a hydroxyl (to form a key hydrogen bond) 17,18. Computational ligand design can be used to not just develop high-affinity
inhibitors but to also address key biological challenges such as drug resistance.
There is considerable work in the area of rational drug design to overcome mutation resistance. One strategy is to rationally engineer drugs that target common drug resistance mutants; this technique has been used in the development of inhibitors for cancer, infectious diseases, and anti-microbials. For instance, despite its high efficacy for cancer patients, the BCR-ABL inhibitor imatinib results in clinical resistance'9 2 1. Crystal structures of imatainib bound to both WT
BCR-ABL as well as clinical mutants led to the rational design of nilotinib, which is extremely effective against the common imatanib-resistant variant22,23. This strategy requires knowledge of the
mutations -the drawback being that other mutants can induce resistance. In fact, nioltonib, while active against key imatinib drug resistant mutants, is still susceptible to other mutations. Other strategies for rational design around mutations include designing inhibitors that target multiple epitopes.
An alternate to the mutation-targeted design is using substrates as templates for drug design, or substrate mimicry 24. For a successful substrate mimic, any mutation that knocks out inhibitor
function would also diminish substrate activity, rendering the enzyme inactive. Previous work from our group has implemented this principle for HIV protease by designing drugs to stay within the consensus volume of HIV protease substrates, which is termed the "substrate envelope"2426 Inhibitors designed to stay in the substrate envelope exhibited a broader resistance profile compared to therapeutics that violate the envelope26
To implement the substrate envelope constraint, our group has developed a new method of drug design, which we refer to in this thesis as "inverse-design",2 . Unlike top-down approaches, which
dock fully formed molecules into the active site and then score the results, the inverse approach creates a library of potential small molecules that are combinatorially designed from a commonly shared scaffold. Scaffold positions are enumerated in the active site and functional groups are attached by finding optimal rotamers for each position. In this context, the inverse drug design framework is analogous to the protein design problem of finding the global minimum energy conformation for a particular protein mutation2 7. Framed this way, the tree-pruning and search
algorithms, dead-end elimination and A*, can be used to generate the global minimum solution along with a gapless list of the low-energy conformations28
While the techniques and principles discussed above have had remarkable success, there are many unaddressed aspects in drug design. In this thesis, we use and improve rational approaches to solve various challenges in drugging infectious disease targets. One theme in this thesis is developing
new approaches and novel designs for drug resistance, and much of our work uses the inverse-design framework discussed above.
1.2 Ligand design: a three prong approach
There are three critical components to rational drug design that all feed into one another: methods development, analysis/understanding, and novel design. This is somewhat analogous to the biological engineering paradigm of "make, model, manipulate" which encompasses the field in a simple three block diagram.
Rational Drug Design
Methods2 Anlysis& DevelopmentUndrtnding HIV Protease DOTAla Cytochrome B %,- Novel Design *r Applications
HIV Protease HIV Protease
BCR-ABL DOTAla
CIpP Peptidase
Cytochrome 8
Figure 1-1:The three prongs of rational drug design, with the projects we worked on below
Methods development focuses on designing new techniques and computational frameworks that can aid in molecular modeling. This can be either hardware or software centric. For example, the development of the Anton chip from D.E Shaw research, which enables faster molecular dynamics non-bond calculation, is an example of hardware development for MD simulations29. Examples of
and providing new scientific insights. One early example from our group was in the role of salt bridges in protein stability. After analyzing many protein structures, Hendsch et al. found that salt bridges actually destabilize many protein complexes30
. This important study shifted paradigm on protein design and ligand design by providing new context for evaluating electrostatic contacts. Finally, novel design uses existing methods and insights to engineer new ligands for biologically relevant targets.
These three overarching aspects are all required to further the field of ligand design. In this thesis, the five chapters (and one project that is not presented) encompass all three phases of ligand design.
1.3 Summary of thesis
The first two chapters in the thesis describe our work on HIV protease, which we use as a model system to study rapidly mutating targets. Our group has used HIV protease to develop a framework for inhibitors that can evade mutation resistance. We leverage the known data on this system to augment this design scheme.
In chapter 2, we perform detailed molecular computation on two inhibitors with different sensitivity profiles against mutant HIV- 1 protease to understand the sources of difference. We find that the robust inhibitor forms less interaction in the active site compared to the susceptible binder. We cross-validate this on a set of ten inhibitors with known resistance profiles and are able to correctly classify each inhibitor. These results provide a concrete basis of understanding for molecular drivers of susceptibility.
In chapter 3, we leverage our results from chapter 2 and develop a framework for the design of inhibitors that will not fail via mutation resistance. We go through multiple evolutions in our design as we conceptualize failure modes and use the data from chapter 2 to provide guidance and context. We then apply these principles to design new inhibitors to HIV protease. As a validation, we recover all binders studied in the previous chapter with the correct sensitivity predicted. We then suggest a subset of 12 for synthesis to our collaborators.
In chapter 4, we use current ligand design techniques to optimize an antibiotic (ADEP) for the tuberculosis ClpP peptidase system. We first understand the biophysics of ADEP binding via component analysis and then design a series of analogs that functionalize various parts of the molecule. We are able to achieve a 2-fold improvement in binding affinity along with enhanced peptidase activity. To address this phenomena, we conceptualize different models that explain data and perform design calculations to validate our models.
In chapter 5, we shift our focus and develop methods to design novel MRI contrast agents. In contrast to other work provided in this thesis, the design objective function is quite different, as we design analogs to control the rotation of the paramagnetic water via a hydrogen bond. We develop a framework for design by considering the various factors that govern efficacy of contrast agents and incorporate thermodynamic models to achieve the objective. This framework is then validated on a set of existing MRI contrast agents, and then applied to design new leads.
In chapter 6, we develop a new method for to optimize a novel inhibitor for Candida albicans cytochrome bcl. For this optimization, we start with a crystal structure bound to a non-specific
inhibitor and develop methods to predict the bound structure conformation for our lead using molecular docking. We then account for receptor plasticity via molecular dynamics simulations and tested these conformations in silico. By combining these three tools, we generate a scheme for rational design with imperfect experimental data.
Chapter 2: Computational Analysis of
HIV Protease Inhibitors with
Differential Sensitivity to Resistant
Mutants
Abstract: HIV protease substrates are unaffected or only moderately affected by resistance
mutations that greatly decrease inhibitor binding. This idea has led to the successful design of broadly binding inhibitors using substrate mimicry in an idea termed the "substrate envelope hypothesis". However, not all binders that respect the envelope are insensitive to HIV protease mutants. In this chapter, we select one susceptible and one flat inhibitor and decompose the binding energy to wild-type and the sensitive mutant. We find that the susceptible binder forms tighter overall interaction energy and stronger contacts with certain residues compared to the flat binder. Encouraged by these results, we cross-validate this finding on ten HIV-protease
inhibitors with known sensitivity profiles. We successfully classify the flat and susceptible inhibitors based on interaction energy. Finally, we develop theory to reconcile binding affinity and interaction energy. Taken together, this body of work provides mechanistic evidence for drivers of susceptibility.
2.1 Introduction
Acquired drug resistance is a major clinical problem in the treatment of various diseases including infectious diseases and cancer2-7. There are various mechanisms for drug resistance some of which do and others of which do not operate primarily through mutation of the target 31-35. Examples of
non-target induced mutations include efflux pumps and drug modifying enzymes. Target-based mechanisms must somehow decrease inhibitor function without greatly reducing substrate recognition. Despite the number of resistance mechanisms, a substantial number of drugs lose efficacy via on-target mutations. One strategy to combat this problem uses substrate recognition as a principle to design drugs to structurally and chemically resemble the substrate36-39. For a
successful mimic, mutations that decrease inhibitor function will also greatly reduce native substrate processing. For targets that are essential for the maturation of a bacteria or virus, the loss-of function mutations will never be favored.
Drug resistance has been extensively studied in the rapidly mutating human immunodeficiency virus (HIV), particularly for the aspartyl protease enzyme from HIV40-48. HIV protease is a well-studied and characterized enzyme with extensive structural, inhibitor, and mutation data. Existing crystal structures of HIV protease include wild-type enzyme with clinical inhibitors, wild-type enzyme with inactivated substrates (chemically modified substrates that do not react upon binding), and mutants in complex with clinical inhibitors45,4 95051. Furthermore, the mutation profile for HIV protease has been broadly studied in both a clinical context (characterizing the mutations
Efforts to design inhibitors for HIV protease using substrate mimicry require a priori knowledge of the mechanism for substrate recognition. The Schiffer group crystallized five unique enzyme-substrate complexes5 2. The structural data suggests that the enzyme recognizes substrates that
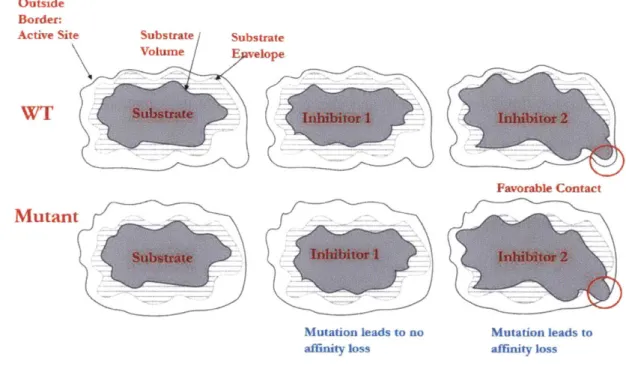
adopt an asymmetric shape. Therefore, a substrate mimic should occupy the consensus volume required for recognition, which we refer to as the "substrate envelope" (Figure 2-1, Figure
2-2)24,25,53.
Outside
Border:
Active Site Substrate Substrate
volume Epe WT Sbtrtehibitor Mutant Inhbitor I Mutation leads to no affinity loss Inhibitor 2 Favorable Contact Irdlibitor 2 Mutation leads to affinity loss
Figure 2-1: Cartoon representation of the substrate envelope. Inhibitor one respects the envelope and is not prone to mutation resistance. However, inhibitor 2 violates the envelope and mutation leads to susceptibility.
rbstrate E Q
HIV Piotea4.,e
Figure 2-2: The substrate envelope as defined for HIV protease. Our group has used the envelope successfully to design robust inhibitors against HIV protease.
Our group has previously implemented a computational inverse design method to design compounds that stay within the substrate envelope25. Through two rounds of design, we designed
36 compounds with nM to pM activity which were then assayed against a panel of clinical HIV protease mutants2 4. The sensitivity of an inhibitor is defined as the fold inhibition lost compared to wild-type. A subset of the compounds that obeyed the envelope exhibited relatively flat binding profiles (fold loss < 20) while other compounds were sensitive to mutants (fold loss >100). Strikingly, inhibitors designed with no constraint exhibited a susceptible resistance profile54.
Shen et al. further provided validation of the substrate envelope by designing compounds that gradually violated the envelope26. Interestingly, sensitivity to mutants was directly correlated to
In this work, we use computational analysis to better understand drivers of mutation resistance. We first choose two compounds, one flat and one susceptible binder. We compute and subsequently decompose the binding of both compounds to WT and a mutant. The binding energy includes terms for configurational entropy, interaction energy between the inhibitor and target, and self-energy. We find that the susceptible inhibitor forms stronger interactions in the binding site compared to the flat binder. Furthermore, the susceptible binder makes more contacts with a certain subset of residues, which can be entirely categorized by examining known resistance mutants.
We then use a set of HIV protease inhibitors with known sensitivity to cross-validate our hypothesis. We find that the interaction energy completely separates susceptible and flat inhibitors, and furthermore this difference in interaction energy is mostly due to this subset of residues. These results suggest a design scheme for inhibitors that avoid making excessive interaction energy with the active site.
2.2 Results
2.2.1 Calculations can capture sensitivity and insensitivity of KC-08
and KB-83 respectively
We first sought to understand the differential interaction profiles for one flat (KB-83) and one susceptible (KC-08) binder. To capture the increased sensitivity of the susceptible binder, free energy of binding was computed for the pair to both wild-type (WT) and M1 mutant. Given the relatively small free energy gap between the inhibitors, a single conformation-based free energy cannot accurately resolve the free energy difference; in contrast, enumerating a configurational ensemble more accurately captures the differential binding (Figure 2-3) 5. The procedure is
detailed in the methods section, but in short, each inhibitor was divided into a common scaffold and five functional groups. The scaffold degrees of freedom were sampled via a Monte Carlo search, and the functional group torsions explored using the tree-pruning dead-end elimination
Capture Appropriate Experimental Results for Inhibitors: Governing Thermodynamics: AGsI=Gsw- Gs- Ggn (1) AGF1"T =Gr~ - GF- GuT (2) AGS5 = - Gs - Gat (3) AGFM=G - GE - G. (4)
Individual inhibitor free energy
Want sensitivity of susceptible over flat to mutant vs WT
AAG7 AAGM AAAG = [(1) -(2)] - [(3) -(4)] (Gswr - GF-r ) - (sm - GF-1nt) Sampling of Inhibitor - Conformational Enumeration L 7- 7 Accurat compute binding energy Compute: 1. AG/H 2. AAG/H 3. AAAG/H Fumd Receptor
Required erent ligand
for nformations
AAAG/H
s
Bound state enumerated
Conformational Space
Required for AG/H 2nd AAG/H
(DEE) and A* search algorithm to generate a gap-free list of low energy conformations. These conformations were then Boltzmann weighted to compute free energy of binding.
The binding free energy (AG) and enthalpy (AH) for the inhibitors KB-83 and KC-08 binding to the wild-type (WT) and MI mutant of HIV- 1 protease was calculated and is compared with the corresponding experimental measurements (Table 2-1). Calculations and experimental data agree that KC-08 is more susceptible than KB-83 with diminished binding to the Ml mutant relative to wild-type (AAAGexp=-1.61 kcal/mol; AAAGcaic = -2.77 kcal/mol). Interestingly, the susceptible inhibitor KC-08 binds more tightly to both wild-type (AAGexpwT = -3.25 kcal/mol; AAGcaicw =
-3.25 kcal/mol) and the Ml mutant ( AAGxpMI = -1.18 kcal/mol; AAGcaicMi = -0.48 kcal/mol) but the difference is significantly smaller in the mutant. The individual AG and AH values are not comparable between experiment and calculation because of reference states and other differences that cancel out when differences are taken.
Experimental (Free Energy) Computed Free Energy Computed Enthalpy KC-08 WT -119.50 -109.45 KB-43_Wi -109.70 -99.62 KC-O8_Ml -101.33 -91.66 KB-83_Ml -94.30 -84.41 KC-O8 Unbound -49.90 -29.90 KB-83 Unbound -43.35 -23.30 AKC-OS WT -16.43 -69.60 -79.55 AKB-83 WI -13.64 -66.35 -76.32 AKC-08_Ml -13.55 -51.43 -61.76 AKB-93 Ml -12.37 -50.95 -61.11 AAWt -2.79 -3.25 -3.23 A&Ml -1.18 -0.48 -0.65 AA -1.61 -2.77 -2.58
Table 2-1: Computed free energy and enthalpy agrees with experimental measurements. Individual states cannot be measured experimentally, and therefore those fields are left blank
Because they are readily decomposable into additive pairwise contributions, the AH values are used in the analysis that follows. As shown above, the computed AAH and AAAH values agree extremely well with the corresponding AAG and AAAG values. Specifically, the overall AAAG (AAAH) = -2.77 (-2.58) kcal/mol and the corresponding AAG's and AAH's also agree well. (AAGcaicwT (AAHcaCwT) =-3.25 (-3.23) kcal/mol; AAGcacMI (AAHcac"l) =-0.48 (-0.65) kcal/mol ).
Together these results show that the calculations capture the experimental differences that we aim to understand, and that the computed enthalpies are sufficiently close to the free energies that their decomposition can be the basis for the molecular decomposition.
Cutoff: [ H_(KC-08 wt) or H_(K-83 wt) I > 1.5 kcal/mol Definitions H_(KC-08_wt) H_(KB-83_wt) AAH(KC-08 wt-KB-83 wt) A23 -1.47 -0.99 -0.48 A25 -6.60 -6.20 -0.40 A27 -2.92 -3.05 0.13 A28 -2.27 -3.75 1.48 A29 -3.25 -5.09 1.84 A30 -2.49 -2.71 0.22 A32 -0.95 -0.76 -0.18 A47 -1.67 -1.11 -0.56 A48 -2.91 -0.97 -1.94 A49 -3.51 -2.82 -0.69 A50 -5.86 -5.58 -0.28 ASl -0.54 -0.54 0.00 A81 -2.22 -1.27 -0.95 A82 -2.21 -0.94 -1.27 A84 -2.62 -2.65 0.03 A87 -0.67 0.16 -0.83 B8 -1.74 -0.97 -0.77 B25 -5.85 -5.52 -0.33 B27 -2.40 -2.21 -0.19 B28 -3.18 -3.29 0.11 B29 -2.39 -2.66 0.27 B30 -2.99 -2.90 -0.09 B32 -1.17 -1.28 0.11 B47 -2.01 -2.08 0.07 B48 -1.54 -1.26 -0.28 B49 -1.43 -1.63 0.20 B50 -3.92 -3.10 -0.82 B81 -1.27 -1.28 0.01 B82 -2.21 -2.23 0.02 B84 -2.91 -2.97 0.06 W102 -2.53 -2.30 -0.23 Total -79.69 -73.96 -5.73 Sum (AAH >= 10.51) -26.90 -22.17 -4.74 Interaction Total -86.99 -81.64 -5.35
Remaining Interaction Energy -7.30 -7.68 0.38
Table 2-2: Any residue with an interaction energy value greater than 0.5 kcal/mol. The highlighted residues have a difference greater than 0.5 kcal/mol, implying that they are important for understanding the sources of differences
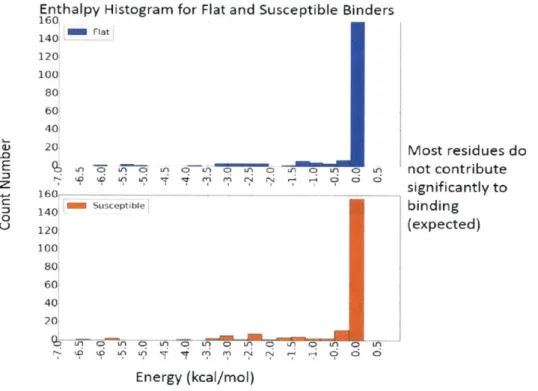
The ensemble-interaction energy (receptor-ligand interaction) for the susceptible binder KC-08 and the flat binder KB-83 were decomposed into pairwise additive contributions from each residue (Table 2-2). The susceptible inhibitor makes stronger interaction (-86.99 kcal/mol) compared to the flat inhibitor (-81.39 kcal/mol) to the wild-type HIV-1 protease. Residues that form greater than 10.51 kcal/mol interaction were considered for further structural analysis. This subset cumulatively accounts for -76.69 kcal/mol and -73.71 kcal/mol for KC-08 and KB-83 respectively. All other residues contribute -- 7.5 kcal/mol for both binders, which suggests that this threshold sufficiently captures the key differences between the inhibitors, as evidenced by the histogram (Figure 2-4). In total, 10 residues have a AAH > 10.51 kcal/mol between the susceptible binder KC-08 and the flat binder KB-83.
Enthalpy Histogram for Flat and Susceptible Binders
160 i Flatf 140 120 100 80 60 40[ 20! Ui iqU LA 160 141-susceptible 1201 100! 801 601 40! 20 Most residues do not contribute significantly to binding (expected) '0 LA 0? qA 03A0LA0L LF Ln 0 iA Energy (kcal/mol)
Figure 2-4: Histogram of the flat and susceptible binder interaction energy for all residues. Notably, most residues do not contribute to interaction enegy for both binders.
-0
E
z
0
U-binding to the wild-type were considered as a subset for analysis. Interestingly, the AAAH of these residues (-2.77 kcal/mol) can account for the overall AAAH (-2.54 kcal/mol) (Table 2-3). Furthermore, the AAAH > 10.51 kcal/mol (-2.74 kcal/mol) accounts for the overall AAAH. To gain a structural understanding of the differential interaction of both KC-08 and KB-83 in both wild-type and the M1 mutant, the interactions for each important residue (highlighted in Table 2-3) were examined in detail.
Cutoff: [ H_(KC-OS wt) or H_(KB-83_Wt) I >.5I kcal/mol
Definitions H_(KC-08 wt) H_(KB-83,wt) AAH(KC-08 wt-KB-83 wt) H_(KC-08_ml) H,(KB-83 m1) MH(KC-08 M1-KB-83_M1) fAiH
A29 -2.27 -3.75 1.48 -1.55 -3.66 2.11 -0.63 A29 -3.25 -5.09 1.84 -2.51 -3.01 0.50 1.34 M7 -1.67 -1.11 -0.56 -0.73 -1.22 0.49 -LO5 M -2.91 -0.97 -1.94 -3.87 -1.64 -2.23 0.29 M9 -3.51 -2.82 -0.69 -3.36 -2.52 -0.84 0.15 A81 -2.22 -1.27 -0.95 -1.32 -1.20 -0.12 -0.83 A22 -2.21 -0.94 -1.27 -1.31 -0.76 -0.55 -0.72 AB7 -0.67 0.16 -0.83 -0.43 0.15 -0.58 -0.25 88 -1.74 -0.97 -0.77 -0.87 -0.32 -0.55 -0.22 B50 -3.92 -3.10 -0.82 -3.20 -3.23 0.03 -0.85 Computed AMH -2.54
SUm (AMH For 11 Residues) -2.77
SUm(AM l=I0.5I) -2.74
Table 2-3: The residues that are the sources of difference for the WT explain the sensitivity difference between KC-08 and KB-83.
2.2.2
Molecular Understanding of Differential Binding
RI Group
HO Ns
N
o
,
fH NSH H OH N
KB-83 KC-08
Figure 2-5: The Ri group for both KB-83 and KC-08
this difference results in differential binding interactions to four residues in the wild-type: AlaA28
AspA2 9, fleA4 7, and fleB5 0.
The R1 group of KB-83 has a stronger interaction with the sidechain of AlaA28 (HA28-WTKB-8 3 3.75 kcal/mol, 3.6 A closest atom to CP to AlaA28) compared to the RI group of KC-08 (HA28-WTKC-08-2.27 kcal/mol, 4.0 A closest atom to CO to AlaA2 8) resulting in a
AAHA28 = 1.48 kCal/mol (Figure 2-6a-c). In the M1 mutant, the R1 sidechain of the susceptible binder KC-08 loses the favorable vdW interaction with the sidechain (HA28-MlKC-O8= -1.55 kcal/mol, 3.3
A
closest atom to CO toAlaA2 8), while there is no loss for the 3-hydroxyphenyl group at the RI position of KB-83 (HA28-M1KB-83 = -3.66 kcal/mol, 3.6 A closest atom to CO to AlaA28), resulting in a AAAHA28 = -0.65
kcal/mol (Figure 2-6c-d). Interestingly, this is the only instance of opposing AAHWT and AAAH energies for any residue.
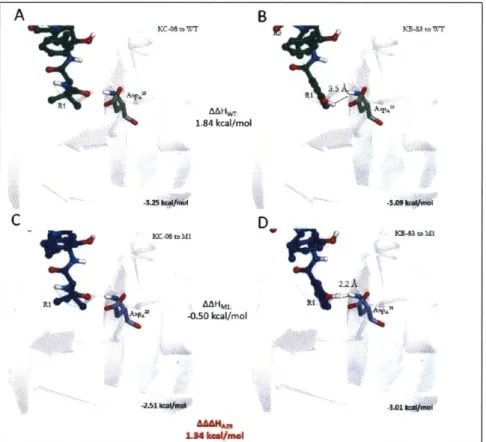
P~toWT ~-et~A1 B 1.49 kcaf/mol ' A C D MAHM 2.11 kcal/ntol U KCA4to sa
The hydroxyl group on the 3-hydroxyphenyl hydrogen-bonds to ASpA2 9 ( HA28-WTKB-8 3 = -5.09
kcal/mol, H-bond to 061=1.8 A) compared to the analogous substituent on KC-08 (HA2 9-WrKC- 8
= -3.25 kcal/mol, no H-bond) resulting in a AAHA29 = 1.84 kcal/mol (Figure 2-7a-b). In the M1 mutant, KB-83 makes an unfavorable electrostatic interaction between the backbone amine hydrogen (HA29-M1KB-8 3 = -3.01 kcal/mol, KB-83-hydroxy hydrogen to HA distance = 2.2
A),
which the R1 group of KC-08 does not make ( HA29-MlKC-08 = -2.51 kcal/mol). This differential
interaction results in a AAAHA29 = 1.34 kcal/mol (Figure 2-7c-d).
ZA
184 1.84kcal/mol _O- toM IAAHML -0.50 kcal/mol 1.34 kmmi/mel B D -5.0h5 ~~K- 5M IE3tf 22A -3Figure 2-7: KB-83 and KC-08 interaction with ASPA 2 resulting in AAAH of 1.29 kcal/mol A
C
RI
In contrast, the RI group from the susceptible binder KC-08 makes a stronger vdW interaction to IleA47 and IleB50 ( HA4 7-wTKC-O8= -1.67 kcal/mol, closest atom to CyI = 3.7
A;
HB5o-WTKC8 -3.92
kcal/mol, distance to C61 = 4.0 A) compared to the flat binder KB-83 ( HA47-WKB8 3 -- 1.67
kcal/mol, distance to Cyl = 4.5 A; HB50-WTKB8 3 = -3.10 kcal/mol, closest atom to C61 = 4.2 A)
resulting in a AAH of -0.56 kcal/mol and AAH of -0.82 kcal/mol respectively (Figure 2-8 and Figure 2-9). The susceptible binder KC-08 is more sensitive to the Ml mutant to both residues IleA47 and IleB50 ( HA4 7
-MlKC-8 -0.71 kcal/mol, closest atom to Cyl = 4.4
A;
HB50-M1 KC -- 3.20kcal/mol, distance to C1 = 4.3
A)
compared to the flat binder KB-83 ( HA47-MB 83 1.22kcal/mol, distance to Cyl = 5.0
A;
HB50-WTK 83 = -3.23 kcal/mol, closest atom to C61 = 4.3A).
These interactions result in a AAAHA47/ AAAHA47 of -1.05 kcal/mol/-0.83 kcal/mol (Figure 2-8 and Figure 2-9). Overall, the differential RI substitution results in a AAAHR1 of -1.19 kcal/mol,
A B KC- . AWTr D K11,4 C D 4.051ami4.4 - KKC-0 rIO I 0.49 kca/mIl .716mB"A"
B -0.82 kcal/mol 4.2 A A 4 . oA C A~MM,
Figure 2-9: KB-83 and KC-08 interaction with HeB"resulting in AAAH of -0.83 kcal/mol
which accounts for roughly half of the sensitivity of the susceptible binder compared to the flat binder.
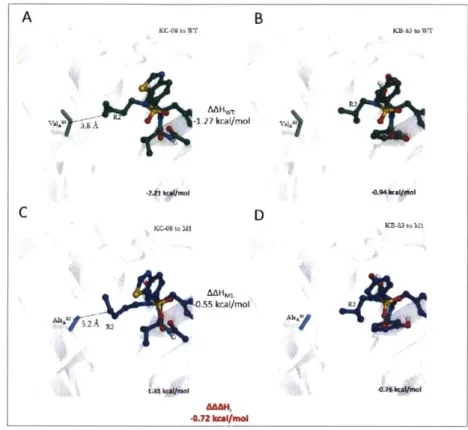
R2 Group
The susceptible binder KC-08 has an n-pentyl at the R2 position which is chemically and sterically larger than the isobutyl group at the R2 position of the flat binder KB-83 (Figure 2-10), which results in differential AAH and AAAH for ProA81 and ValA82.
0 0 HO KB-83 IN ,HN N N H OH S KC-08
Figure 2-10: The R2 group for both KB-83 and KC-08
Kr-M to MI 0.03 kcal/mol 4 3 l -3I0mbaUnI D -K.2kafmoi
For both residues, the vdW interaction for KC-08 (HA81-WTKC-08 = -2.22 kcal/mol, closest atom to CP = 3.9
A;
HA82-WTKC 8 = -2.21 kcal/mol, closest atom to C62 = 4.6A
) is more favorable compared to the flat binder KB-83 ( HA81-WTKB-8 3 = -1.27 kcal/mol, closest atom to CP = 5.3 A; HA82-WTKB83 = -0.94 kcal/mol, closest atom to Cy2 = 5.0A
) (Figure 2-11 a-b & Figure 2-12a-b).B KB-3 o WT -0.95 kcal/mol 4~ -2.22 kcali/mD KC-09 to M1 -0.12 kcal/mol PW U'--V -1.M kxI/M ol -1.22 kwi/mol C D AA&H, -.83 kcal/mol
Figure 2-11 KB-83 and KC-08 interaction with ProA5' resulting in AAAH of -0.83 kcal/mol
The M1 mutant substitutes an alanine to a valine, which also results in the proline conformation shifting 0.75 A away from the active site. These structural differences result in greater sensitivity of the susceptible binder KC-08 ( HA8-M IKC-08 = -1.32 kcal/mol, closest atom to CE = 5.0
A;
HA82-MIKC-08 = -2.21 kcal/mol, lost interaction in VAL-+ALA mutation) compared to the flat binderKB-83 ( HA81-M1KB-8 3= -1.22 kcal/mol, closest atom to CP = 5.4
A;
HA82-M1KB-8 3 = -0.76 kcal/mol,-1.27 "rg/mot K-3to MI A A NO 3.9A
residues account for a AAAHA81/AAAHA82 of -0.83 kcal/mol/-0.72 kcal/mol and the differential R2 substitution accounts for a AAAHR2 of -1.55 kcal/mol.
Put together, the varying RI and R2 groups between KC-08 and KB-83 account for the differential resistance profile of the inhibitors to the M1 mutant.
K"cs to r -1.27 kcal/mol KcC-O to M2 .55 kcal/mol' B D
1'
KB ola wr KB-83 to Ml R2--MWt -0.72 kCe.moIFigure 2-12: KB-83 and KC-08 interaction with VaIA2resulting in AAAH of -0.82 kcal/mol
2.2.3 Generalizing sources of differences for susceptible and flat
inhibitors
The first part of this study aimed to understand the sources of difference for one susceptible inhibitor and one flat inhibitor by computing the difference between the WT and mutant for the pair. Component analysis suggests that the susceptible inhibitors make stronger interaction energy with multiple residues in the active site, which also collectively results in a higher total interaction
A
energy with the wild-type HIV-1 protease. Furthermore, in the MI mutant, the susceptible inhibitor is more sensitive to these residues (Figure 2-13).
-5 Enthalpy Energies For Flat/Susceptible Inhibitors
Flat Binder Susceptible -AA >~i 0 -4 -3 Z -2 Uj
Figure 2-13: Residues that contribute to the AAAH difference between
KC-08 and KB-83.
Interestingly, the susceptible binder makes stronger interactions with a subset of residues, which can be entirely defined by analyzing known resistance mutants to current therapies. Mutation sites were defined by considering resistance mutants in patients on HIV protease inhibitor therapies from the Stanford HIV Database. All neighboring residues to these mutations were added to the subset.
Interaction energy for both KC-08 and KB-83 to this subset is shown below (Figure 2-14). Interestingly, the D30N resistance mutation is only observed for patients on neflanavir, and not for any other therapies including darunavir. Compared to KB-83, KC-08 makes stronger interaction
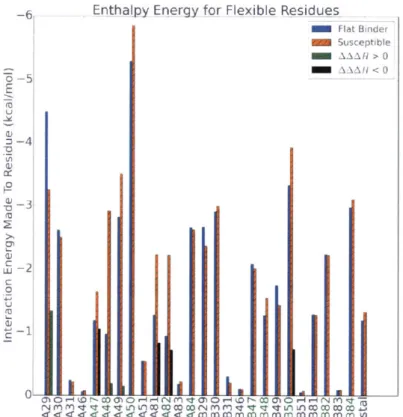
-6 Enth ajpy Energy for Flexible Residues Flat Binder Susceptible AA HX >O0 -AA.A H <0 -6-U a) 0
4-Figure 2-14: Residues that contribute to sources of difference between KB-83 and KC-08. The residues highlighted in green have been identified as resistance mutants for multiple inhibitors, as identified via the Stanford Resistance Database to most flexible residues including ILEA 7, GLYA 4', GLY 49, PROA8, VALA82, and ILEB50.
Collectively, KC-08 forms stronger contacts with these residues (-49.3 kcal/mol) compared to the flat binder KB-83(-44.5 kcal/mol).
Put together, the data suggest that differential interaction with flexible/mutable residues along with total interaction energy to wild-type HIV-1 protease could be a key driver of sensitivity. We next wanted to determine if this driver of susceptibility in KC-08 could be generalized for a larger set
2.2.4 Testing set inhibitor selection
We were careful in selection of inhibitors for cross-validation. Importantly, we wanted inhibitors that had been measured both precisely and accurately, as the differences in binding energy between WT and mutant are relatively small. Therefore, we selected compounds were designed in our group previously and assayed by our collaborators.
A set of 7 HIV- 1 protease inhibitors that were previously designed in our group and with known
resistance profiles were selected in our testing set (Table 2-4). All binders share the common darunavir scaffold and differ in their functional groups (Figure 2-15). The clinically approved
Inhibitor Affinity (nM) Worst Fold Loss Designation
KC-08 0.002 113 S AF-78 0.44 98 S AF-69 0.07 264 S AD-93 0.02 60 B AF-77 0.54 6 F AG-23 0.021 18 F AF-68 0.135 10 F KB-83 0.19 9 F
Table 2-4: Inhibitors studied, with the wild-type affinity and worse fold loss. Each inhibitors is designated as susceptible (S), flat (F), and borderline (B)
darunavir, a known flat binder, was also included in the testing set. In total, the set consisted of 6 flat binders, 3 sharp binders, and one binder that is borderline with a 55-fold affinity loss.
Flat Inhibitors HO NH OH KB-83 NN" H 0 H AG-23 HO OH N H N AF-68 AF-77 N HO N NO KB-98 Borderline Inhibitor H -HO - N AD-93 Susceptible Inhibitors N. 0, 0 0 . S HOH H' H N b OH KC-08 AF-69 AF-78
Interaction with HIV-protease fully predicts binder sensitivity
The binding free energy (AG) to the wild-type HIV-1 protease was computed for all 10 inhibitors and compared with experimentally measured AG (Figure 2-16a). The computed AG had high correlation (r2 = .89) to the experimental measurements. Analogously, the easily decomposable AH was also computed and had high correlation with experimental AG. Note that both the susceptible and flat inhibitors in this set span a relatively large experimental affinity range, which suggests that affinity and susceptibility are not complexly correlated. This point is addressed in more detail in the final section.
-15 -14 Experimental Energy (kcal/mol
B
-74 -78 -84 -88 -88 -SC -13 -12 e n Ener ,!Aqq __ K. shpen'' 44Figure 2-16: (A): The computed AG is consistent with the experimental free energy with an r2 =0.89. (B): The ensemble averaged interaction energy separates the susceptible and flat binders, with the borderline in the middle.
The ensemble averaged interaction energy was next computed for each inhibitor in the testing set
A _63
-64 -65 0 -68 -69Computed A G vs Experimental Energy [ . Flat Binders 4 4 Sharp Binders EU0 Bo rde rlin e 8 k Ier
-7/ 4
4
S
protease with an interaction energy range of -89.2 kcal/mol to -88.3 kcal/mol compared to the flat inhibitors with a range of -71.3 kcal/mol to -81.9 kcal/mol. These results are consistent with the findings in the first part of the study. Interestingly, the borderline forms an interaction energy
(-85.1 kcal/mol) that is in the middle compared to the flat and susceptible inhibitors. Given the separation of flat and susceptible binders along with the lack of correlation between affinity and resistance profile, the ensemble interaction energy is not itself a proxy for binding free energy.
The pairwise-additive contributions from each flexible residue (as defined above) was next computed for all inhibitors. For each susceptible inhibitor, we compared the interaction energy to that residue to the average of the flat inhibitors (Figure 2-17). The susceptible inhibitors AF-78, KC-08, and AF-69 bind the flexible residues with an interaction of -7.4,-7.7, and -7.3 kcal/mol more compared to the flat inhibitor average. This, along with the total interaction energy, provide evidence that susceptible inhibitors form excessive interaction in the binding site.
Interaction Energy to Subset of Residues
E
U.1
Borderline Inhibitor ad93-wt
M fat Average M ad93wt -31 2!
!MZ :all
-/, Fla Interaction: -39.3 kcal/mol Susceptible:af69-wt FlRat Average Susceptibleaf7B-wt M Flat Average m vflA-wt Int raction: -41.2 kcal/mol 4 s1s3e tii22 Sq~gptjIekc8-wt I FIatAverageii m afA-wt kcOS wt Interaction Interaction: -- 42.7 kcal/mo -41.5 kcal/molL Binders Residue (Flexible) Average: -34.5 kcal/mol
Std Dev: 2.2 kcal/mol
Figure 2-17: Interaction energy to flexible residues for the susceptible and borderline inhibitors. Compared to the flat binders (statistics shown in green), these inhibitors make more interaction energy with the flexible residues which cross-validates our
hypothesis.
2.2.5
Binding Affinity Theory
Next, theory was developed to reconcile the apparent discrepancy between the ensemble interaction energy and free energy of binding. For any inhibitor, high affinity is paramount for clinical efficacy.
The free energy of binding can be expressed as the sum of two discrete conformational events: docking, and preconformation. Docking free energy accounts for interaction between the receptor and ligand in the bound complex while the preconformation energy quantifies the change in energy between the ground state and the bound state in solution (Figure 2-18).
to be =0.
funfavoroble)
Figure 2-18: Cartoon representation of ligand binding. The left panel has the ligand in free solution. The middle panel is the ligand adaption a conformation to bind, which is depicted in the right panel
The equations that govern these are shown below. Pu and Pb are the ensemble weights for the
unbound and bound state respectively.
AGbind = AGdock + AGpreconforin (1)
AGDock 'Dc -vdw(R-L) = PB(G docke + GdockeA coul (R-L) + G dcked2)Dfe.solvation)
AGpr conform PB [Gdocked r dw(L-L) + Gdocked cout(L-L) Gdo*ked solvation(L-L) + Gdocked]geom (3)
p- U[relax- vdw(L-L) +Grelax cout(L-L) "solvation(L-L) (relax + Grelaxgeoml
By the definition of a ground state, AGpreconform is bounded by zero. Furthermore, the results from this study suggest that the coulombic and vdW subset of AGdock should be bounded for each
residue: this implies that the total interaction energy should be bounded as well. The desolvation component of the docking free energy is not strictly positive, but in general is an unfavorable term for small molecule inhibitors. A highly hydrophobic ligand could have a favorable desolvation
penalty, but would likely exhibit negative pharmacokinetic and formulation properties, both of which are paramount for a successful drug.
Put together, the strategy to designing high affinity binders would be to have a AGdock to ensure specificity, but bounded to limit susceptibility. To gain affinity, the AGpreconform term should be
minimized, with the objective being AGpreconform=. If AGpreconform is zero, then the only other term
that can be optimized to improve free energy of binding is the docking free energy, which can affect susceptibility.
The AHpreconform was computed for all ten HIV-1 protease inhibitors studied (Figure 2-19). Interestingly, the flat binders had a lower AHpreconform compared to susceptible binders. The flat
inhibitor with the tightest experimental binding free energy, darunavir, is the binder with the lowest preconformation energy. In contrast, the susceptible inhibitors that are relatively weak affinity
Computed HJPreconformation vsExgpri mentalFree Energy
*
O Flat Binders 13 4 A Sharp Binders U N Borderline Binder 12 EGood Affinity Lower affinity,
S10i
too much high
g
9 preconform preconform ~0 E 8 c TI 6 (UU 4) 0. E3 3 e 0 2i -17 -16 -15 -14 -i3 -12Experimental Free Energy (kcal/mol)
Figure 2-19: Computed preconformation energy for the inhibitors studied. The susceptible inhibitors have notably higher preconformation energy. These binders are still nM binders, and therefore gain too much of the energy from the docking.
2.2.6
Improvement to design
The overall goal of this study was to understand drivers of susceptibility, which could eventually
result in a new design framework to be broadly applicable to rapidly mutating targets including
HIV- 1 protease, cancer targets, etc. Our results suggest that susceptible inhibitors generally form excessive interaction with the binding site compared to the flat inhibitors. Furthermore, our results
suggest that a subset of residues contribute more to driving sensitivity of inhibitors. Therefore, a new design framework must incorporate these principles.
An overly conservative approach to minimize interaction energy would result in a low affinity binder, which is clinically irrelevant. In the section above, we reconciled the apparent discrepancy between affinity and interaction energy and observed that interaction energy is controlled by only one term (AGdock) while affinity is controlled by two (AGdock, AGpreconfonm). Therefore, while AGdock
should be bounded, AGpreconform should be minimized to yield a high-affinity binder.
Overall Docking Energy Docking Energy Subset
Flat Binders
Cutoff AND
Potentially - Drives
Susceptible specificity
MIN(AGpreconform) Drives affinity
Figure 2-20: Some key factors that drive specificity and sensitivity (to avoid mutation resistance) and also affinity.
2.2.7
Future Work
There are multiple areas for future work that would build upon the results presented in this chapter. The natural extension is to develop and implement a new design framework with HIV-1 protease
Separately, a similar analysis could be done for other rapidly mutating target to demonstrate the robustness of the findings in this chapter. For example, HIV reverse transcriptase inhibitors or hepatitis C protease would be ideal candidates for future study. More data and studies such as these will help our overall understanding of mutation resistance from a mechanistic and biophysical standpoint.
2.3Methods
2.3.1 Receptor Structure Preparation
Crystal structure of HIV protease bound to darunavir was obtained from the Protein Data Bank (PDB) (CODE: 1T3R)56. The receptor was separated from the inhibitor and both were treated separately. Additionally, all waters were renumbered with a new segid.
Terminal side chain dihedrals were considered for a 180 degree flip for all the HIS, ASN, and GLN if a flipped dihedral would yield a hydrogen bond. Any crystal water that made 2 hydrogen bonds was retained; furthermore, the conserved flap water was retained. Hydrogens were added using the
HBUILD module in CHARMM, using the CHARMM22 parameter force field. Missing
sidechain atoms were built using CHARMM and the CHARMM22 parameter force field. Hydrogen atoms and computationally added heavy atoms were minimized using a steepest descent minimization routing for 1000 steps followed by 250 rounds of Newton-Raphson energy minimization. All heavy atoms from the PDB were taken directly from the initial structure. Partial charges for standard amino acids were assigned based on the PARSE parameters.
2.3.2 Scaffold Preparation/Ligand Optimization
From the bound darunavir structure, a subset of the atoms were kept as the scaffold (Figure 2-21), with the appropriate hydrogens renamed to indicate attachment points. All other hydrogens were also renamed to indicate that they cannot be expanded.