354 Rev Med Liege 2017; 72 : 7-8 : 354-357 Résumé : Nous présentons le cas d’une patiente souffrant
d’une baisse d’acuité visuelle associée à des céphalées. L’inves-tigation a permis de mettre en évidence un syndrome de Vogt-Koyanagi-Harada. Il s’agit d’une pathologie multisystémique rare, affectant les organes présentant une haute concentration de mélanocytes. Il est important d’en poser rapidement le dia-gnostic pour administrer une corticothérapie. L’étude de ce cas est complétée par une revue de la littérature sur le sujet. mots-clés : Syndrome de Vogt-Koyanagi-Harada - Uvéite bila-térale - Méningite aseptique
Vogt-Koyanagi-HaRada syndRome
summaRy : We present the case of a patient who had a decrease of the visual acuity associated with headaches, diagnosed as Vogt-Koyanagi-Harada syndrome. This is a rare multisyste-mic pathology that affects organs with high concentration of melanocytes. This syndrome needs to be identified on time as corticotherapy has to be administrated urgently. This paper also summarizes the literature on this disease.
KeywoRds : Vogt-Koyanagi-Harada syndrome - Bilateral uveitis - Aseptic meningitis
A.-C. C
hApelle(1), e. D
uChAteAu(2), B. l
oCht(2)
LE SYNDROME DE VOGT-KOYANAGI-HARADA
intRoduction
La maladie (ou syndrome) de Vogt-Koya-nagi-Harada (VKH) est une pathologie auto-immune granulomateuse multisystémique affectant les organes caractérisés par une haute concentration en mélanocytes. Elle touche prin-cipalement l’œil, le système nerveux central, l’oreille interne et la peau. Une uvéite bilaté-rale associée à une méningite aseptique et des céphalées sont les signes les plus fréquemment rencontrés chez les patients souffrant de ce syn-drome.
descRiption du cas clinique
Une patiente afro-caucasienne, âgée de 20 ans, est adressée au service d’ophtalmolo-gie pour mise au point d’une baisse d’acuité visuelle à la lecture associée à des céphalées depuis une semaine.
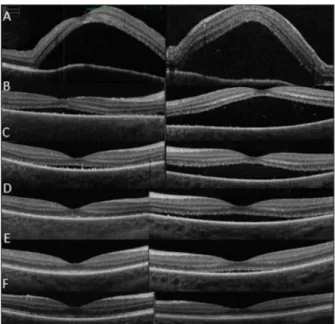
A l’examen ophtalmologique, l’acuité visuelle est chiffrée à 6/10 et Parinaud 4 bila-téralement. L’examen biomicroscopique du segment antérieur révèle un tyndall bilatéral correspondant à une inflammation. Le fond d’œil met en évidence une dépigmentation choroïdienne et un œdème papillaire. L’ima-gerie par tomographie en cohérence optique (OCT) objective un volumineux décollement séreux rétinien maculaire (Figure 1). L’exa-men artériographique par fluorescéine, étudiant les vaisseaux rétiniens, met en évidence de multiples points hyperfluorescents au niveau de l’épithélium pigmentaire (EP) confirmant l’existence d’une choroïdite multifocale. L’hy-perfluorescence s’étend progressivement au
sein du décollement séreux rétinien au cours de cet examen. Une hyperfluorescence papil-laire est également observée (Figure 2). L’an-giographie au vert d’indocyanine, permettant l’analyse des vaisseaux choroïdiens, montre une hypofluorescence précoce des vaisseaux choroïdiens signant un retard de perfusion. Ce signe est positif dès la phase prodromale. Aux temps intermédiaires apparaissent de nombreux spots hypofluorescents. Ceux-ci pourraient cor-respondre à des nodules inflammatoires cho-roïdiens. Ils sont plus nombreux que les points hyperfluorescents perçus en fluoangiographie (Figures 3 et 4).
Au niveau systémique, l’anamnèse révèle également une hypoacousie gauche et des acou-phènes d’apparition récente.
La recherche diagnostique débute par une bio-logie sanguine qui met en évidence essentielle-ment une hyperleucocytose (14 000/mm3). Le bilan auto-immun n’est pas contributif (ACE et anticorps anti-nucléaires négatifs). Les analyses de sérologie infectieuse (CMV, Herpès simplex, Herpès zoster, syphillis, maladie de Lyme) sont négatives. La ponction lombaire montre une hyperleucocytose associée à une hypergammaglo-bulinémie. La culture du liquide céphalorachidien se révèle négative. Le bilan ORL démontre une perte neurosensorielle gauche.
Sur base de ces différents éléments, le dia-gnostic de syndrome de VKH incomplet, vu l’absence d’atteinte cutanée, est posé.
Un traitement par mégadose de méthylpre-dnisolone (Solumedrol®) en intra-veineux (1 g
pendant 3 jours), suivi d’un schéma dégressif per os pendant 15 semaines, est administré à la patiente. Une amélioration des céphalées, ainsi que des symptômes ophtalmologiques et ORL, est observée après une semaine, tandis que l’acuité visuelle est chiffrée à 10/10 et Pari-naud 2 après un mois de traitement.
(1) Assistante, (2) Médecin spécialiste, Service d’Oph-talmologie, CHU de Liège, site Sart Tilman, Liège, Bel-gique.
Syndromede Vogt-Koyanagi-Harada
355 Rev Med Liege 2017; 72 : 7-8 : 354-357
discussion
Le syndrome de VKH a été décrit par Vogt en 1906 et Koyanagi en 1929 comme une inflam-mation oculaire associée à un vitiligo, ainsi qu’à une perte des cheveux et des cils (1, 2). Par ailleurs, Harada rapporte, en 1926, le cas d’un patient souffrant d’une uvéite postérieure bilatérale s’accompagnant d’un décollement séreux rétinien et d’une pléocytose au niveau du liquide céphalo-rachidien (3). Ce n’est que des années plus tard qu’il a été suggéré que ces différents signes et symptômes étaient l’expres-sion d’une seule et même pathologie (4, 5).
La maladie de VKH représenterait 4 à 11 % des uvéites endogènes (6). Ce syndrome s’ob-serve plus fréquemment chez des patients à la peau pigmentée, âgés de 20 à 50 ans; la pré-valence de cette maladie varie donc en fonc-tion de l’ethnie et des régions. Les asiatiques, les américains natifs et les hispaniques sont plus fréquemment touchés que les individus de race blanche ou d’origine africaine (7). Les femmes sont généralement plus touchées que les hommes, bien que le ratio soit inversé dans la population japonaise (8). Une augmenta-tion de l’incidence des phénotypes HLA DR4, DR53, DR1, DRB1*0405 est observée chez les patients présentant la maladie de VKH (9). Figure 2. Fluoangiographie.
A : Multiples zones hypofluorescentes donnant un aspect pommelé aux temps précoces.
B : Multiples points hyperfluorescents au niveau de l’épithélium pigmen-taire aux temps intermédiaires.
C : Aspect bilobé correspondant au remplissage hyperfluorescent du décol-lement séreux rétinien.
Figure 1. OCT maculaire. Visualisation de la régression du décollement séreux rétinien :
(A) avant traitement, (B) après 1 semaine, (C) après 2 semaines, (D) après 4 semaines et (E) après 6 semaines de traitement.
Notons la résolution complète du DSR après 3 mois de traitement (F).
Figure 3. Angiographie au vert d’indocyanine : retard de perfusion cho-roïdienne caractérisé par une hypofluorescence aux temps intermédiaires.
Figure 4. Angiographie au vert d’indocyanine. Visualisation au temps tar-dif de multiples points hypofuorescents correspondant probablement à des nodules inflammatoires et une zone d’aspect bilobé liée au volumineux DSR.
A.C. ChApelleetColl.
356 Rev Med Liege 2017; 72 : 7-8 : 354-357
70 % des patients présentent une baisse d’acuité visuelle (13). Une choroïdite multifocale peut être visualisée à la fluoangiographie et un décollement séreux rétinien à l’OCT. L’angio-graphie au vert d’indocyanine montre un retard de perfusion choroïdien global, associé à des petites taches hypofluorescentes. Une uvéite antérieure ainsi que des précipités rétro-desce-métiques peuvent également être objectivés.
Enfin, la troisième phase, ou phase de convalescence, s’étalant parfois sur plusieurs mois, voire des années, est caractérisée par la dépigmentation cutanée ainsi que par la perte des cheveux, des cils et des sourcils.
Une phase chronique récurrente, caractéri-sée par une panuvéite et des épisodes d’uvéites antérieures résistant à la corticothérapie, est également décrite. Dans ce cas, des nodules iriens, des membranes rétiniennes et des glau-comes liés à des angles irido-cornéens syné-chiés peuvent également apparaître (14).
L’angiographie au vert d’indocyanine se normalise au cours du traitement. Par contre, il peut persister une alternance d’atrophie et d’hyperpigmentation de l’épithélium pigmen-taire, donnant un aspect mité à la rétine à l’an-giographie. Des complications néovasculaires peuvent également être rencontrées.
L’utilisation précoce et agressive de corti-coïdes systémiques en intraveineux a pour but de diminuer la réponse inflammatoire initiale. Un relais per os à dose décroissante lente évite le passage à la chronicité et permet de diminuer les complications extra-oculaires. Dans certains cas de corticorésistance ou lorsqu’un patient ne supporte pas les effets secondaires de la cor-ticothérapie, un traitement par immunosup-presseur peut être proposé (cyclophosphamide, cyclosporine, aziathoprine, par exemple) (14). Certains auteurs suggèrent l’utilisation d’im-munomodulateurs (interféron alpha) comme alternative aux immunosuppresseurs (15).
conclusion
Le syndrome de Vogt-Koyanagi-Harada est une uvéite bilatérale granulomateuse associée à des manifestations neurologiques, auditives et cutanées. La fluoangiographie ainsi que l’OCT maculaire sont les examens clés du diagnostic. Un traitement précoce et agressif est conseillé pour améliorer le pronostic visuel et diminuer les complications extra-oculaires.
L’étiologie exacte de ce syndrome reste inconnue. Il est probable qu’il s’agisse d’un mécanisme auto-immun dirigé contre les méla-nocytes des yeux, de l’oreille interne, des méninges et de la peau. Cette réponse auto-immune pourrait être provoquée par un agent infectieux chez un patient prédisposé généti-quement (9).
Le syndrome de VKH peut être qualifié de complet, incomplet ou probable selon que le patient présente ou non l’ensemble des critères diagnostiques repris dans le Tableau I. Il sem-blerait que la forme incomplète soit liée à un traitement plus précoce dont résulterait une expression restreinte des symptômes autres qu’oculaires.
La maladie de VKH comporte trois phases successives : la phase prodromale, la phase uvéitique et la phase de convalescence. La première phase, ou phase prodromale, d’une durée de 3 à 5 jours, s’apparente à un syndrome grippal se manifestant, principalement, par des céphalées (49 % des cas), des nausées (13 %), de la fatigue (21 %), de la photophobie (48 %), et des troubles de l’audition (32 %) (10). Dans certains cas, des neuropathies optiques, des hémiparésies et certaines atteintes des nerfs crâniens sont décrites (11). Enfin, la ponction lombaire met en évidence une pléocytose, qui peut s’observer pendant 8 semaines (12).
Au cours de la deuxième phase, ou phase uvéitique, pouvant durer plusieurs semaines,
Tableau I. CrITèresdedIagnosTICdusyndromede VKH del’amerICan uVeITIs soCIeTy
réVIsésen 2001 (15)
Syndrome de VKH complet
Les critères de 1 à 5 doivent être présents :
1. Pas d’antécédents de traumatisme ou de chirurgie oculaire. 2. Pas d’anomalie clinique ou biologique évoquant d’autres pathologies oculaires.
3. Atteinte oculaire bilatérale : précoce ou tardive se manifestant soit par une uvéite antérieure, une hyalite, une hyperhémie papillaire, une dépigmentation rétinienne ou un décollement séreux rétinien. 4. Atteinte neurologique à type de syndrome méningé ou de dysacousie ou pléocytose du liquide céphalorachidien.
5. Atteinte cutanée survenant après les signes oculaires et neuroméningés.
Syndrome de VKH incomplet
Les critères de 1 à 3 doivent être présents associés au critère 4 ou 5.
Syndrome de VKH probable
Présenté par une atteinte oculaire isolée ; les critères 1 à 3 doivent être présents.
Syndromede Vogt-Koyanagi-Harada
357 Rev Med Liege 2017; 72 : 7-8 : 354-357
11. Lubin J, Loewenstein J, Frederick A, et al.— Vogt-Koyanagi-Harada syndrome with focal neurologic signs. Am J Ophtalmol, 1981, 91, 332-341.
12. Rajendram R, Evans M, Khurana R, et al.— Vogt-Koyanagi-Harada disease presenting as optic neuritis.
Int Ophtalmol, 2007, 27, 217-220.
13. Surgira S.— Vogt-Koyanagi-Harada disease. Jpn J
Ophtalmol, 1978, 22, 9-35.
14. Moorthy RS, Imonata H, Rao N.— Vogt-Koyanagi-Harada syndrome. Surv Ophtalmol, 1995, 39, 265-292. 15. Toitou V, Escande C, Bodaghi B, et al.— Prise en
charge diagnostique et thérapeutique du syndrome de Vogt-Koyanagi-Harada. J Fr Ophtalmol, 2005, 28, 9-16.
16. Russel W, Gary N, Narsing A, et al.— Revised dia-gnostic criteria for Vogt-Koyanagi-Harada disease. Am
J Ophtalmol, 2001, 131, 647-652.
BiBliogRapHie
1. Vogt A.— Fruhzeitiges ergaruen der zilien und bemer-kungen uber den sogenaten plotzlichen eintreitt dieser veranderung. Klin Monalsble Augenheilkd, 1906, 44, 228-242.
2. Koyanagi Y.— Dysakusis, alopecia und poliosis bei schwerer uveitis nicht traumatischen ursprugs. Klin
Monalsble Augenheilkd, 1929, 82, 194-211.
3. Harada E.— Acute diffuse choroiditis. Acla Soc
Oph-talmol Jpn, 1926, 30, 356-372.
4. Babel J.— Syndrome de Vogt- Koyanagi (Uveite bila-teral, poliosis, alopecie, vitiligo et dysacousie). Schweis
Med Wochenschr, 1932, 44, 1136-1140.
5. Bruno MG, McPherson SD.— Harada’s disease. Am J
Ophtalmol, 1939, 32, 513-522.
6. Limon S, Girard P, Bloch-Michel E, et al.— Les aspects actuels du syndrome de Vogt-Koyanagi-Harada. J Fr
Ophtalmol, 1985, 8, 29-35.
7. Read RW, Holland GN, Rao NA, et al.— Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international commitee of nomenclature.
Am J Ophtalmol, 2001, 131, 647-652.
8. Tabbara KF, Chavis PR, Freeman WR, et al.— Vogt-Koyanagi-Harada in childrens compared to adults.
Acta Ophtalmol Scand, 1998, 76, 723-726.
9. Greco A, Fusconi M, Gallo A, et al.— Vogt-Koyanagi-Harada syndrome. Autoimmun Rev, 2013, 12, 1033-1038.
10. Rao N, Gupta A, Dustin L, et al.— Frequency of dis-tinguishing clinical features in Vogt-Koyanagi-Harada disease. Ophtalmology, 2010, 117, 591-599.
Les demandes de tirés à part doivent être adressées au Dr E. Duchateau, Service d’Ophtalmologie, CHU de Liège, site Sart-Tilman, 4000 Liège, Belgique. Email : [email protected]