Impact du derme et d'une irradiation chronique aux
rayons ultraviolets sur la réparation des dimères
cyclobutyliques de pyrimidines dans les kératinocytes
humains
Thèse
Marie Dorr
Doctorat en biologie cellulaire et moléculaire
Philosophiæ doctor (Ph. D.)
Impact du derme
et d’une irradiation chronique aux rayons ultraviolets
sur la réparation des dimères cyclobutyliques de
pyrimidines dans les kératinocytes humains
Thèse
Marie Dorr
Sous la direction de :
Résumé
La lumière solaire constitue le principal facteur de risque des cancers de peau non mélanocytaires (NMSC). L'effet génotoxique de la lumière solaire est dû aux dommages dans l'ADN induits par les rayonnements ultraviolets (UV). Les rayons UVB longs (290-315 nm) sont les principaux responsables de l'initiation et de la promotion des NMSC qui prennent naissance dans les kératinocytes épidermiques. En effet, l’absorption directe des photons d’UVB par l’ADN conduit à la génération de deux principaux types de dommages, les dimères cyclobutyliques de pyrimidines (CPD) et les photoproduits de pyrimidine (6-4) pyrimidone (6-4PP). Les CPD sont les plus abondants et sont hautement mutagènes. Ils sont responsables des mutations de transitions C → T au niveau des sites dipyrimidiniques, les mutations signatures observées dans les cancers de peau. Les cellules possèdent différents mécanismes pour éviter la conversion des CPD en mutations, à savoir, l’arrêt du cycle cellulaire, la réparation des dommages dans l'ADN par le système de réparation par excision de nucléotides (NER) et la mort cellulaire par apoptose. L’importance de la NER dans la prévention des cancers de peau est bien démontrée par le fait qu’une déficience en protéines de la NER, comme chez les patients atteints de Xeroderma Pigmentosum (XP), entraîne une incidence jusqu’à 2000 fois plus élevée de cancers de peau.
De nombreux facteurs influencent la NER et une meilleure compréhension de ces derniers pourrait conduire au développement de nouvelles stratégies de prévention contre les cancers de peau. La peau est un assemblage complexe de cellules et de matrice dans lequel la communication entre les composants épidermiques et dermiques est essentielle pour de nombreux mécanismes cutanés. En utilisant des peaux reconstruites dérivées uniquement de fibroblastes et de kératinocytes primaires humains, nous avons analysé l’impact des composants dermiques sur l’efficacité de réparation des CPD épidermiques. Nous avons montré que l’élimination des CPD dans les kératinocytes est positivement influencée par la présence d'un derme et nous avons déterminé que cet effet du derme sur les kératinocytes proviendrait de molécules sécrétées. En étudiant le sécrétome, nous avons découvert que la cytokine CXCL5 (ou ENA78 - Epithelial neutrophil-activating peptide 78) possède un patron d'expression unique : elle est pratiquement absente du milieu de culture des peaux reconstruites, comparativement au milieu de culture de fibroblastes et de kératinocytes seuls. En modulant les niveaux de CXCL5 dans les milieux de culture de kératinocytes, nous avons montré que CXCL5 était un inhibiteur de la réparation des CPD. Cette première étude décrit l'impact des molécules sécrétées par le derme sur la réparation
des CPD épidermiques et met en lumière un nouveau rôle de CXCL5 dans la réparation des dommages induits par les rayons UV.
L’environnement immédiat des kératinocytes n’est pas le seul facteur qui peut influencer la réparation des CPD, le régime d’irradiation a également un impact sur cette efficacité d’élimination des lésions. Jusqu’à présent, l'efficacité de la NER a été largement étudiée après une seule exposition aiguë aux rayons UV. Cependant, l'utilisation d’une irradiation unique n'est pas représentative de l'exposition solaire humaine, qui est plutôt constituée d’une multitude d'irradiations répétées. Dans ce travail, nous avons donc exposé des cellules épidermiques à un régime d’irradiation chronique composé de faibles doses d’UVB (CLUV) afin de déterminer l’impact de cette irradiation sur la réparation NER. Nous avons montré que le traitement CLUV entraîne l’accumulation de CPD résiduels, qui ne sont pas réparés mais plutôt tolérés et dilués lors de la réplication de l’ADN. Nous avons également constaté que le prétraitement CLUV réduisait la capacité d'élimination des nouveaux dommages sans induire de sensibilité accrue à la mort cellulaire. Enfin, en utilisant nos données expérimentales, nous avons élaboré un modèle théorique pour prédire l’induction, la dilution et la réparation des CPD épidermiques lors d’une irradiation chronique aux rayons UVB. Nos résultats montrant que les kératinocytes accumulent des dommages dans l'ADN après des irradiations chroniques, constituent un facteur important à prendre en compte, car l'accumulation de CPD non réparés pourrait entraîner une augmentation des mutations dans les kératinocytes.
Dans l’ensemble, ces travaux soulignent l’importance d’utiliser des modèles plus complexes, visant une meilleure représentation physiologique, pour mieux comprendre les réponses de la peau à l’exposition solaire.
Abstract
Skin exposure to solar light is the main risk factor for non-melanoma skin cancers (NMSC). The genotoxic effect of sunlight is attributed to DNA damage induced by ultraviolet (UV) radiations. Long UVB wavelengths (290-315 nm) are the main responsible for NMSC initiation and promotion that occur in epidermal keratinocytes. Indeed, the direct absorption of UVB photons by DNA leads to the generation of the two main types of UV-induced DNA damage, i.e. cyclobutane pyrimidine dimers (CPD) and (6-4) pyrimidine-pyrimidone photoproducts (6-4PP). CPD are the most abundant and are highly mutagenic. They are responsible for the C → T transition mutations at dipyrimidine sites, the signature mutation found in sun-related skin cancers. Skin cells use different mechanisms to avoid the conversion of UVB-induced CPD into skin cancer driver mutations, i.e. cell cycle arrest, DNA damage removal by nucleotide excision repair (NER) pathway and cell death by apoptosis. The importance of NER for skin cancer prevention is well demonstrated by the fact that a deficiency in NER proteins, such as in Xeroderma Pigmentosum (XP) patients, leads to an increase of up to 2,000-fold in skin cancer occurrence.
Many factors influence NER and a better understanding of those factors might lead to new prevention strategies against skin cancer. Skin is a complex assembly of cells and matrix in which a crosstalk between epidermal and dermal components is essential for many cutaneous mechanisms. Using self-assembled tissue-engineered skin equivalents derived from human primary fibroblasts and keratinocytes, we have analyzed the impact of dermal components on epidermal CPD repair efficiency. We showed that CPD repair in keratinocytes is positively influenced by the presence of the dermis and we brought evidence that this dermal effect comes from secreted molecules. We then investigated the secretome and found that the cytokine CXCL5 (also known as ENA78 - Epithelial neutrophil-activating peptide 78) has a unique expression pattern, i.e. is virtually absent in the culture medium of reconstructed skin, when compared to the media from fibroblasts and keratinocytes alone. By modulating CXCL5 levels in keratinocytes culture medium, we have shown that CXCL5 is an inhibitor of CPD repair. This work outlines the impact of the secreted dermal components on epidermal UV-induced DNA damage repair and shed light on a novel role of CXCL5 in CPD repair.
The immediate environment of the keratinocytes is not the only factor that can influence the CPD repair, the irradiation protocol also has an impact on this damage removal. Until now,
NER efficiency has been extensively studied after a single acute UVB exposure. However, the use of single UVB irradiation is not representative of the human solar exposure, which is rather a multitude of repeated irradiations than a single acute one. In this work, we thus exposed keratinocytes to a chronic low-dose of UVB (CLUV) protocol to determine the impact of this irradiation procedure on CPD removal. We showed that the CLUV treatment leads to the accumulation of residuals CPD. Those residual CPD are not repaired but rather tolerated and diluted through DNA replication. We also found that a CLUV pre-treatment reduces CPD removal rate of newly generated damage without inducing a higher sensitivity to UV-induced cell death. Finally, using our experimental data, we derived a theoretical model to predict CPD induction, dilution and repair that occur in keratinocytes when chronically irradiated with UVB. These results showing that keratinocytes accumulate DNA damage after chronic irradiations is an important factor to consider since the accumulation of unrepaired CPD might lead to an increase of skin cancer driver mutations formation. Taking together, this work outlines the importance of more relevant and physiological models to study the skin response to solar exposure.
Table des matières
Résumé ... ii
Abstract ... iv
Table des matières ... vi
Liste des figures ... x
Liste des abréviations ... xii
Remerciements ... xvi
Avant-propos ...xviii
Introduction ... 1
1.1 La lumière et le spectre électromagnétique solaire ... 1
1.1.1 Les rayons infrarouges ... 2
1.1.2 La lumière visible ... 2
1.1.3 Les rayons ultraviolets ... 3
1.2 La peau, principal organe humain exposé à la lumière ... 4
1.2.1 L’épiderme ... 5
1.2.2 Le derme ... 7
1.2.3 L’hypoderme ... 9
1.3 Les dommages induits par l’absorption directe des rayons UV ... 9
1.3.1 Les photoproduits de pyrimidine (6-4) pyrimidone ... 11
1.3.2 Les Dewar ... 11
1.3.3 Les dimères cyclobutyliques de pyrimidines ... 12
1.3.4 Le potentiel mutagène des dimères cyclobutyliques de pyrimidines ... 13
1.4 Les réponses aux dommages induits par les rayons UV ... 15
1.4.1 L’arrêt du cycle cellulaire ... 17
1.4.2 La réparation des dommages ... 18
1.4.3 La mort cellulaire ... 19
1.4.3.1 Via les dommages dans l’ADN ... 19
1.4.3.2 Via les récepteurs de mort cellulaire ... 20
1.4.3.3 Via les espèces réactives de l’oxygène ... 21
1.5 La réparation par excision de nucléotides (NER) ... 22
1.5.1 Les étapes de la réparation par excision de nucléotides ... 22
1.5.1.1 La reconnaissance du dommage ... 24
1.5.1.1.1 La réparation globale du génome (GG-NER) ... 24
1.5.1.2 La vérification du dommage... 27
1.5.1.3 L’excision du fragment d’ADN contenant la lésion ... 27
1.5.1.4 La synthèse du nouveau brin ... 28
1.5.1.5 La ligature ... 28
1.5.2 Les facteurs régulant et influant la NER ... 28
1.5.2.1 Les types de dommages, leurs localisations et leurs accessibilités ... 29
1.5.2.2 Les modifications post-traductionnelles ... 30
1.5.2.3 Les autres facteurs régulant la NER ... 31
1.5.2.3.1 p53 ... 31
1.5.2.3.2 BRCA1 ... 32
1.5.2.3.3 Le rythme circadien ... 32
1.5.3 Les conséquences cliniques d’une déficience en NER ... 33
1.5.3.1 Le Xeroderma Pigmentosum ... 33
1.5.3.2 Le syndrome de Cockayne ... 34
1.5.3.3 La trichothiodystrophie ... 35
1.6 Les effets physiopathologiques des rayons UV sur la peau ... 35
1.6.1 La pénétration de la lumière dans la peau ... 35
1.6.2 Les conséquences d’une exposition aiguë aux rayons UV ... 35
1.6.2.1 L’érythème ... 36
1.6.2.2 La mélanisation ... 36
1.6.2.3 La synthèse de vitamine D3 ... 37
1.6.3 Les conséquences d’une exposition chronique aux rayons UV ... 38
1.6.3.1 L’immunosuppression... 38
1.6.3.2 Les cancers cutanés... 39
1.6.3.2.1 Les mélanomes cutanés ... 39
1.6.3.2.2 Les cancers non mélanocytaires ... 40
1.6.3.3 La photocarcinogénèse ... 41
1.6.3.4 Le photovieillissement ... 42
1.6.3.4.1 Les mécanismes d’induction du photovieillissement ... 42
1.6.3.4.2 Les changements cliniques et histologiques induits par le photovieillissement ... 44
1.7 La communication entre le derme et l’épiderme ... 44
1.8 Les modèles d’études en photobiologie ... 46
1.8.1 Les modèles de peaux ... 46
1.8.1.1.1 Les kératinocytes et fibroblastes primaires ... 46
1.8.1.1.2 Les lignées cellulaires ... 47
1.8.1.2 Les peaux reconstruites ... 48
1.8.1.3 Les peaux humaines normales ... 50
1.8.1.4 Les modèles animaux... 50
1.8.2 Les protocoles d’irradiation ... 52
1.9 Contexte et objectifs des travaux de recherche ... 53
1.9.1 Comprendre l’impact des différents composants dermiques sur l’efficacité de réparation des CPD épidermiques ... 54
1.9.2 Étudier l’effet d’une irradiation chronique avec de faibles doses de rayons UVB sur des kératinocytes humains ... 54
Chapitre 1 ... 56
2.1 Résumé ... 57
2.2 Abstract ... 58
2.3 Introduction ... 59
2.4 Material and methods ... 61
2.5 Results and discussion ... 64
2.6 Acknowledgments ... 69
2.7 Figures ... 70
2.8 References ... 77
Chapitre 2 ... 84
Chronic UVB-irradiation of keratinocytes leads to reduced DNA damage repair efficiency and residual damage accumulation ... 84
3.1 Résumé ... 85
3.2 Abstract ... 86
3.3 Introduction ... 87
3.4 Material and methods ... 88
3.5 Results and discussion ... 90
3.6 Conclusions... 95
3.7 Acknowledgments ... 95
3.8 Figures ... 96
3.9 References ...104
Discussion ...108
4.1 L’impact des composants dermiques sur la réponse de l’épiderme au stress UV ..109
4.1.1 La peau reconstruite par génie tissulaire comme modèle d’étude ...109
4.1.2.1 La matrice extracellulaire dermique ...111
4.1.2.2 Les molécules diffusibles ...111
4.1.2.2.1 CXCL5 ...111
4.1.2.2.2 La famille des CXC et les substrats du récepteur CXCR2 ...116
4.1.2.2.3 Les autres familles de molécules diffusibles...118
4.1.3 Les techniques alternatives pour étudier l’influence du derme sur la réparation des CPD épidermiques ...118
4.1.4 La mort cellulaire ...121
4.2 Les effets d’une irradiation chronique aux rayons UVB sur la réponse des kératinocytes ...122
4.2.1 Les modèles d’études d’une irradiation chronique aux rayons UVB ...122
4.2.2 L’impact d’une CLUV sur la réparation des CPD ...124
4.2.3 La mort cellulaire induite par les rayons UV ...126
4.2.4 L’arrêt du cycle cellulaire ...127
4.2.5 La localisation et la nature des CPD résiduels ...127
4.2.6 L’importance du modèle théorique de prédiction de l’induction, la dilution et la réparation des CPD épidermiques ...128
4.3 Perspectives...129
Conclusion ...131
Bibliographie de l’introduction et de la discussion...132
Annexe 1 ...151
Faster DNA repair of ultraviolet-induced cyclobutane pyrimidine dimers and lower sensitivity to apoptosis in human corneal epithelial cells than in epidermal keratinocytes ...151
A 1.1 Résumé ...152
A 1.2 Abstract ...153
A 1.3 Introduction ...154
A 1.4 Material and methods...155
A 1.5 Results ...160
A 1.6 Discussion ...165
A 1.7 Acknowledgments ...170
A 1.8 Figures ...171
Liste des figures
Introduction
Figure 1.1 Représentation graphique des spectres solaires au zénith ... 1
Figure 1.2 Représentation schématique des rayons ultraviolets solaires ... 3
Figure 1.3 Représentation schématique d’une coupe histologique de peau humaine et de la pénétrance des rayons ultraviolets dans celle-ci ... 4
Figure 1.4 Représentation schématique de la formation d’un CPD, d’un 6-4PP et d’un isomère de Dewar par les rayons ultraviolets ... 10
Figure 1.5 Représentation schématique simplifiée de la DDR après induction de dommages dans l’ADN par les rayons ultraviolets. ... 16
Figure 1.6 Représentation schématique de la NER chez l’humain ... 23
Figure 1.7 Représentation schématique des différents modèles de peaux reconstruites . 51 Figure 1.8 Représentation schématique des différents protocoles d’irradiation aux rayons UVB ... 53
Chapitre 1
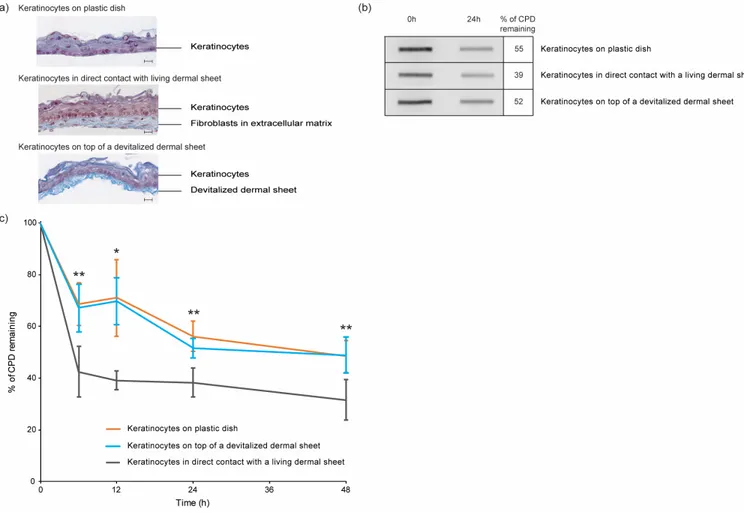
Figure 2.1 Emission spectrum of the UVR lamp (RPR-3000) after filtering through a Kodacel TA-407 clear 0.015” filter ... 70Figure 2.2 CPD repair kinetics in keratinocytes cultured on 3 different supports ... 71
Figure 2.3 Cytokine content of fibroblasts, keratinocytes and reconstructed skin culture media... 73
Figure 2.4 Effect of CXCL5 on UVR-induced CPD repair efficiency in human keratinocytes and fibroblasts ... 75
Chapitre 2
Figure 3.1 Repair kinetics of UVB-induced CPD in keratinocytes ... 96Figure 3.2 Accumulation of residual CPD in CLUV irradiated keratinocytes ... 98
Figure 3.3 CPD fate from day 2 to day 7 and proliferation of keratinocytes ... 99
Figure 3.4 Sensitivity to UVB-induced cell death after a CLUV pre-stimulation ...101
Figure 3.5 Theoretical model for CPD removal by dilution and repair during a chronic UVB irradiation ...102
Discussion
Figure 4.1 Influence de l’inhibition de CXCL5 sur l’expression génique de XPC et DDB2 ...113 Figure 4.2 Influence de l’inhibition de CXCL5 sur le niveau protéique de DDB2 et XPC .114 Figure 4.3 Niveau relatif des cytokines de la famille CXC dans les différents modèles cellulaires de peau ...117 Figure 4.4 Cinétique de réparation des CPD dans des kératinocytes cultivés dans du milieu de culture conditionné ou en co-culture avec des feuillets de fibroblastes. ...119 Figure 4.5 Spectre de transmission de la lumière UVB filtrée ou non par le transwell utilisé pour la co-culture. ...120
Annexe 1
Figure A 1.1 More CPD are found in corneal explants than in skin explants following a same dose of UV-irradiation but similar levels of CPD are detected in cultured NHEK and HCEC. ...171 Figure A 1.2 NHEK are more sensitive to UV-induced cell death when compared to HCEC. ...173 Figure A 1.3 CPD repair, but not 6-4PP, is more efficient in HCEC than NHEK. ...174 Figure A 1.4 DDB2, XPC and p53 protein levels are higher in HCEC than NHEK. ...176 Figure A 1.5 DDB2, XPC and p53 follow the same induction/translocation pattern after UV exposure in NHEK and HCEC. ...177 Figure A 1.6 DDB2 and XPC mRNA levels are lower in HCEC than NHEK. ...179 Figure A.1.7 NER damage recognition proteins DDB2 and XPC are longer-lived in HCEC than NHEK...180
Liste des abréviations
% Pourcentage ° Degré 1,25 [OH] 2D 1,25-dihydroxyvitamine D 25 [OH] D 25-hydroxyvitamine D 5mC 5 méthyl cytosine6-4PP Photoproduit de pyrimidine (6-4) pyrimidone 8-oxo-dG 8-oxo-2'-désoxyguanosine
A Adénine
ADN Acide désoxyribonucléique AP-1 Activator protein 1
APAF-1 Apoptotic peptidase activating factor 1 ARN Acide ribonucléique
ARNm Acide ribonucléique messager ARNPII Acide ribonucléique polymérase II ATM Ataxia telangiectasia mutated ATP Adénosine triphosphate
ATR Ataxia telangiectasia and Rad3-related ATRIP ATR interacting protein
BCC Carcinome basocellulaire
Bcl Beta-cell lymphoma 2
BHD Beta hairpin domain
BRCA1 Breast cancer 1
C Cytosine
C4 Carbone 4
CAK Cyclin activating kinase CCL Chemokine (C-C motif) ligand
Cdk Cyclin-dependent kinase
CDKN2 Cyclin-dependent kinase inhibitor 2
CETN2 Centrin2
Chk Checkpoint kinase
CLUV Chronic low doses of UVB
CoFS Syndrome cérébro-oculo-facio-squelettique CPD Dimère cyclobutylique de pyrimidines CRL Cullin ring ligase
CS Syndrome de Cockayne
CtBP Carboxy-terminal binding protein
CUL4A Cullin 4A
CXCL5 C-X-C motif chemokine ligand 5 ou ENA78 CXCR2 C-X-C motif chemokine receptor 2
CYR61 Cysteine-rich angiogenic inducer 61
DC Dendritic cells
DDB2 DNA damage-binding protein 2
DDR DNA damage response
DR-3 Death receptor 3
ELISA Enzyme-linked immunosorbent assay
ENA78 Epithelial-neutrophil activating peptide ou CXCL5 FADD Fas-associated protein with death domain
GAG Glycosaminoglycan
GGNER Réparation globale du génome par excision de nucléotides
h Heure
H2O2 Peroxyde d’hydrogène
HGF / SF Hepatic growth factor-scatter factor
HIPK2 Homeodomain-interacting protein kinase 2
HMGN1 High-mobility group nucleosome-binding protein 1 hTERT Human telomerase reverse transcriptase
IGF-1 Insuline like growth factor 1
Il Interleukine
IR Infrarouge
JDE Jonction dermo-épidermique Jm-2 Joules par mètre carré
kDa Kilodalton
LC Cellules de Langerhans
m Mètre
m2 Mètre carré
MAPK Mitogen-activated protein kinase MDM2 Mouse double minute 2 homolog MEC Matrice extracellulaire
MIF Macrophage migration inhibitory factor
mm Millimètre
MMP Métalloprotéinase matricielle
MRN Complexe Mre-Rad50-Nbs1
NER Système de réparation par excision de nucléotides NF-κB Facteur de transcription nucléaire kappa B
nm Nanomètre
NMSC Cancer cutané non mélanocytaire
O2- Anion superoxyde
OH- Radical hydroxyle
PARP Poly (ADP-ribose) polymérase PCNA Proliferating cell nuclear antigen pRB Retinoblastoma protein
PTM Modifications post-traductionnelles RFC Replication factor C
ROC1 Regulator of cullins 1
ROS Espèces réactives de l'oxygène RPA Protéine de réplication A
RT-QPCR Réaction en chaîne par polymérase quantitative à partir d'ARNm SCC Carcinome spinocellulaire
sedADN ADN simple brin excisé contenant le dommage SUMO Small ubiquitin-related modifier
SV40 Simian virus 40
T Thymine
TCNER Réparation par excision de nucléotides couplée à la transcription TFIIH Facteur de transcription II humain
TGD Transglutaminase-like domaine
TGF-b Facteur de croissance transformant beta TNF Facteur de nécrose tumorale
TRAIL TNF alpha-related apoptosis inducing ligand trans-UCA Acide trans-urocanic
Tregs Lymphocytes T régulateurs TTD Trichothiodystrophy
U Uracile
USP7 Ubiquitin-specific protease 7
UV Ultraviolet
UV-DDB Ultraviolet radiation–DNA damage-binding protein UVsS Syndrome de sensibilité aux ultraviolets
UVSSA UV-stimulated scaffold protein A
VEGF Facteur de croissance vasculaire endothéliale XAB2 XPA-binding protein 2
XP Xeroderma pigmentosum
α-MSH α-melanocyte stimulating hormone
δ Delta ε Epsilon ζ Zeta η Eta κ Kappa μm Micromètre
Parce que mon doctorat a été un voyage au sens propre comme au sens figuré.
« Travel isn't always pretty. It isn't always comfortable. Sometimes it hurts, it even breaks your heart. But that's okay. The journey changes you; it should change you. It leaves marks on your memory, on your consciousness, on your heart, and on your body. You take something with you. Hopefully, you leave something good behind. »
Remerciements
J’aimerais remercier les personnes qui de près ou de loin m’ont accompagnée dans ce voyage qu’est mon doctorat.
Je voudrais en premier lieu et tout particulièrement, remercier mon directeur Patrick Rochette. Votre laboratoire est une belle destination de recherche. Merci d’avoir toujours eu votre porte grande ouverte et de m’avoir consacré d’innombrables heures à me questionner, à m’expliquer et à répondre à mes interrogations. Je vous suis aussi reconnaissante de m’avoir laissé travailler sur un projet risqué parce qu’il me tenait à cœur. Avec un peu de chance, cette randonnée difficile nous mènera à notre premier British Journal of Dermatology ! Merci d’avoir cru en moi. J’aimerais également souligner votre aide précieuse pour poser convenablement mes valises : le prêt de voiture pour déménager, d’outils pour bricoler et surtout le don d’orchidée à ressusciter ! Merci pour votre implication.
Je tiens à exprimer ma reconnaissance aux membres de mon comité de thèse. Dr Laurent Marrot, Dr Jean-Yves Masson et Dre Stéphanie Proulx, c’est un honneur pour moi de pouvoir compter sur vos commentaires. Merci beaucoup d’avoir accepté de prendre du temps pour examiner ma thèse.
Je voudrais également remercier mes collègues du CUO-recherche, et plus particulièrement les membres, passé et présent, avec qui j’ai le plus partagé mon périple doctoral : Sébastien G., Marie-Catherine, Sébastien M., Marie-Christine, Corinne, Gaétan et Léo. Justin, merci pour ta motivation et ton écoute, j’ai beaucoup apprécié travailler à tes côtés. Un grand merci également à Alicia, Line et Anne-Sophie qui ont su ensoleiller mon voyage grâce à leur bonne humeur, leur sourire et leur aide.
Un merci particulier à Sarah qui a, depuis le début, été dans le même bateau que moi. Lors de notre croisière doctorale nous n’avons pas que traversé des mers tranquilles mais les tempêtes que nous avons croisées ont été plus faciles à gérer grâce à toi. Plus qu’une collègue, j’ai trouvé en toi une amie.
Merci aux membres du LOEX pour la culture et l’irradiation des 3t3 et merci à Rina pour ces précieux conseils sur la technique de peau reconstruite par auto-assemblage.
Un grand merci à ma famille qui m’a apporté une force inestimable. Papa, maman et Catherine, un immense merci pour votre soutien sans faille et votre amour inconditionnel. C’est grâce à votre confiance que je suis partie sereine au Canada et que j’ai pu accomplir ce beau projet. Merci de m’avoir toujours laissé le choix et de m’avoir encouragé dans mes études, même si je pense que vous n’auriez jamais imaginé qu’elles soient si longues ! Merci aussi à Oma d’avoir pris le temps de m’écrire des belles lettres manuscrites si régulièrement.
Finalement, j’aimerai remercier celui qui partage ma vie et qui a choisi notre destination doctorale. Thierry, merci de m’encourager et de me rendre heureuse depuis si longtemps. Notre séjour au Canada n’a pas toujours été facile mais ensemble nous avons surmonté les épreuves, passé de magnifiques moments et accomplis de très beaux projets. Je ne sais pas encore où elle nous mènera, mais j’espère que la vie nous permettra de réaliser tous nos projets et de profiter des joies de la vie commune tous les jours de la semaine. J’ai hâte de vivre ces prochaines étapes avec toi.
Avant-propos
Contributions
Cette thèse se présente sous forme d’insertion d’articles scientifiques. Les chapitres 1 et 2 ainsi que l’annexe 1 sont des articles revus par des pairs ou en cours de publication. Cette section décrit ma contribution ainsi que celle des co-auteurs à ces ouvrages scientifiques.
Chapitre 1
Le chapitre 1 présente les travaux du manuscrit « The use of tissue-engineered skin to demonstrate the negative effect of CXCL5 on epidermal UV-induced CPD repair efficiency » dont les auteurs sont les suivants Marie M. Dorr, Rina Guignard, François A. Auger et Patrick J. Rochette. Cet article a été accepté avec révisions mineures le 10 juin 2019 dans la revue scientifique British Journal of Dermatology.
Ma contribution à cet article a été de mettre au point les différents protocoles, d’exécuter la grande majorité des expériences, d’analyser les résultats et de préparer les figures. La réalisation, la coloration et la prise de photo des coupes histologiques des différents modèles de peaux ont été confié à Rina Guignard (Figure 2.2.a), qui m’a également appris la technique de peaux reconstruites par auto-assemblage. J’ai effectué la rédaction complète de la première version de l’article ainsi que contribué à la révision des versions suivantes. Le Dr Patrick J. Rochette a financé la recherche et participé à la conception des protocoles d’expériences ainsi qu’à l’analyse des données. Il a également corrigé toutes les versions de l’article.
Pour l’ensemble de cet article, ma contribution relative est donc d’environ 80%.
Chapitre 2
La chapitre 2 est composé de l’article « Chronic UVB-irradiation of keratinocytes leads to reduced DNA damage repair efficiency and residual damage accumulation » dont je suis première auteure (Auteurs : Marie M. Dorr et Patrick J. Rochette). Ces travaux ont été soumis pour publication dans la revue scientifique Scientific Reports en juillet 2019.
Dans cet article, j’ai préparé et mis au point les différents protocoles, exécuté toutes les expériences, analysé les résultats et préparé les figures. J’ai également rédigé la totalité de la première version de l’article, ainsi que participé à la révision de ses différentes versions. Le Dr Patrick J. Rochette a financé la recherche et participé à la conception des protocoles d’expériences ainsi qu’à l’analyse des données. Il a aussi contribué de manière significative à la correction et à la révision de toutes les versions de l’article.
Ma contribution à cet article est donc d’environ 80%.
Annexe 1
L’annexe 1 présente les travaux publiés en 2016 dans la revue PLoS One sous le titre « Faster DNA repair of ultraviolet-induced cyclobutane pyrimidine dimers and lower sensitivity to apoptosis in human corneal epithelial cells than in epidermal keratinocytes », dont les auteurs sont Justin D. Mallet, Marie M. Dorr, Marie-Catherine Drigeard Desgarnier, Nathalie Bastien, Sébastien P. Gendron et Patrick J. Rochette.
Justin D. Mallet, en tant que premier auteur, a préparé les protocoles, exécuté la majorité des expériences (Figures A1.1, A1.2 A, A1.3 B et D, A1.7) et analysé les résultats. Il a effectué la rédaction de la première version de l’article. L’exécution de certaines manipulations a été confié à Marie-Catherine Drigeard Desgarnier (Figures A1.3 A et C ainsi que A1.6), à Nathalie Bastien (contribution à la Figure A1.3 A et C) ainsi qu’à Sébastien P. Gendron (matériel non publié). Tous ces auteurs ont contribué à la révision critique de l’article. Le Dr Patrick J. Rochette a financé la recherche et contribué de manière significative aux révisions de l’article avant publication. Dans celui-ci, j’ai réalisé ou participé à la réalisation et à l’analyse des expériences des Figures A1.1 A, A1.2, A1.4 et A1.5 A-F. De plus, j’ai contribué à la révision de l’article.
Introduction
1.1 La lumière et le spectre électromagnétique solaire
La lumière est un phénomène physique présentant deux aspects complémentaires décrit par la physique quantique comme la dualité onde-particule. En effet, la lumière est à la fois ondulatoire, d’où le concept de longueur d’onde, et particulaire, comme en témoignent les photons qui la composent. La lumière peut être décrite selon sa longueur d’onde et son énergie, et deux grands principes la caractérisent. Un premier principe, la relation de Planck-Einstein qui démontre que l’énergie de la lumière est inversement proportionnelle à sa longueur d’onde. Et un second principe, qui énonce que la profondeur de pénétration de la lumière est proportionnelle à sa longueur d’onde.
Spectres solaires à l’extérieur de l’atmosphère (Courbe bleue) et atteignant la surface terrestre (Courbe rouge) (Adaptée de l’American Society for Testing Materials, 1998).
La principale source de lumière à laquelle les hommes sont exposés provient du soleil. Le soleil émet un spectre continu de radiations électromagnétiques sources de lumière et de chaleur. Cependant, seule une partie de ce spectre arrive à la surface terrestre car les composants stratosphériques comme l’ozone, l’oxygène et l’azote bloquent le passage de certaines radiations (Figure 1.1). Le spectre solaire peut être divisé en trois grandes catégories de rayons : les rayons infrarouges (IR ; 800-1000 nm), la lumière visible (400-800 nm) et les rayons ultraviolets (UV ; 100-400 nm) (Coblentz, 1932, Standardization, 2007).
1.1.1 Les rayons infrarouges
Les IR représentent 31,9 % du rayonnement solaire atteignant la surface terrestre (Sage, 1993). Ils sont caractérisés par une grande longueur d’onde, supérieure à 800 nm, et donc par une faible énergie. Ces rayons sont responsables de la transmission de l’énergie thermique du soleil à la surface du globe. En effet, ils peuvent être absorbés par des molécules d’eau qui entrent alors dans un état excité et qui pour revenir à leur état originel émettront de la chaleur (Svobodova and Vostalova, 2010).
1.1.2 La lumière visible
La lumière visible ou lumière blanche correspond environ à 62,7 % du rayonnement électromagnétique terrestre (Sage, 1993). Elle peut être divisée en 6 régions : le violet (400 à 450 nm), le bleu (450 à 500 nm), le vert (500 à 570 nm), le jaune (570 à 591 nm), le orange (591 à 610 nm) et le rouge (610 à 800 nm) (Standardization, 2007, Svobodova and Vostalova, 2010). La rétine de l’œil humain a la capacité d’absorber la lumière visible et de la transformer en signal visuel, permettant ainsi à l’homme de distinguer le contraste des objets et donc de voir son environnement.
1.1.3 Les rayons ultraviolets
Le spectre UV, contenant les longueurs d’onde de 100 à 400 nm correspond environ à 5,4 % du spectre solaire. Il peut être divisé en trois grandes régions : les UVA de 315 à 400 nm, les UVB de 280 à 315 nm et les UVC de 100 à 280 nm (l’Éclairage, 2014). Les UVA peuvent être divisés en 2 sous régions : les UVA-1 de 340 à 400 nm et les UVA-2 de 315 à 340 nm (Sklar et al., 2013). L’ozone stratosphérique bloque le passage des longueurs d’ondes inférieures à 290 nm, à savoir les UVC et les UVB courts, de sorte que celles-ci ne se rendent pas à la surface terrestre (Figure 1.2). Le ratio UVB / UVA atteignant la terre dépend de nombreux facteurs comme l'angle du soleil, la latitude, mais aussi la saison et l'heure de la journée. Cependant, le soleil est principalement une source d'UVA, qui compose 95 % des rayons UV à la surface terrestre. Les UVA et les UVB représentent ainsi respectivement 5,1 % et 0,3 % du rayonnement solaire (Diffey, 2002, Sage, 1993, Young, 2006). Les UVB bien que moins représentés sont les rayonnements les plus énergétiques à atteindre la surface terrestre.
Le soleil est la principale source d’émission de rayons UV auxquels les hommes sont exposés. Mais ces derniers peuvent également être exposés à des rayons UV artificiels provenant essentiellement de lampes UV comme dans les cabines de bronzage, mais aussi des lampes à vapeur de mercure et d’outils de soudure à l'arc.
Figure 1.2 Représentation schématique des rayons ultraviolets solaires
Seules les longueurs d’ondes supérieures à 290 nm atteignent la surface terrestre (Adaptée de JD Mallet).1.2 La peau, principal organe humain exposé à la lumière
Seuls deux organes du corps humain sont directement exposés au rayonnement solaire : la peau et les yeux. La peau est le plus grand organe du corps, elle représente jusqu’à 15 % du poids total de l’individu avec une superficie d’environ 2 m2. C’est un tissu complexe et
dynamique responsable de nombreuses fonctions vitales comme les fonctions de barrière contre les agressions extérieures, de thermorégulation, de perception de signaux, de sécrétion, d’immunité et de synthèse de vitamine D3. C’est la structure élaborée de la peau,
qui associe de nombreux composants et types cellulaires, qui rend possible ces diverses activités. La peau est constituée de trois principaux compartiments cellulaires, de la surface vers l’intérieur : l’épiderme, le derme et l’hypoderme (Figure 1.3). Elle contient également plusieurs appendices : des follicules pileux, des glandes sébacées et sudoripares et des ongles (Voir revues dans (Kanitakis, 2002, Tobin, 2006)).
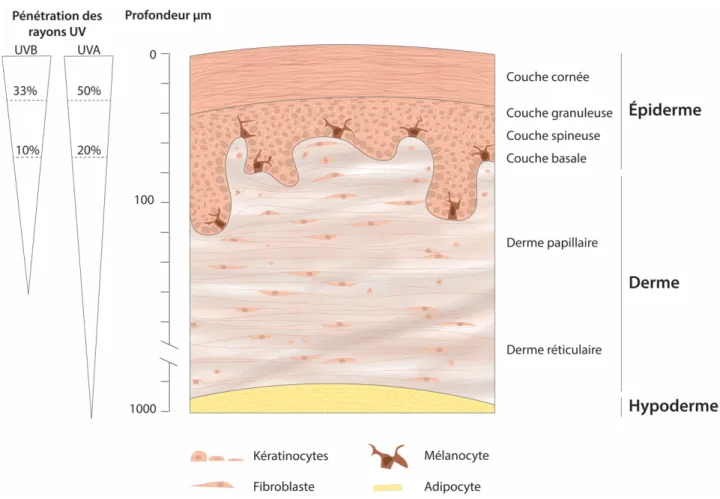
Figure 1.3 Représentation schématique d’une coupe histologique de peau
humaine et de la pénétrance des rayons ultraviolets dans celle-ci
1.2.1 L’épiderme
L’épiderme, la couche la plus superficielle de la peau, joue un rôle primordial dans la protection de l’organisme contre les agressions externes. C’est un épithélium pluristratifié d’une épaisseur moyenne de 100 μm. L’épiderme étant non vascularisé, ses besoins nutritionnels sont comblés par la diffusion de nutriments en provenance du derme sous-jacent. L’épiderme est très majoritairement composé de kératinocytes (90-95 %) qui selon leur état de différentiation forment des couches distinctes à savoir, du plus profond au plus superficiel : la couche basale, la couche spineuse, la couche granuleuse et la couche cornée (Kanitakis, 2002, Koster, 2009).
La couche basale de l’épiderme est principalement constituée de kératinocytes souches et de leurs cellules filles, les cellules amplificatrices transitoires, qui sont responsables du renouvellement de l’épiderme et qui sont reliées entre elles par des desmosomes. La couche spineuse ou épineuse contient des kératinocytes avec une capacité limitée de division cellulaire ainsi que des cellules de Langerhans. La strate suivante est la couche granuleuse, constituée de kératinocytes produisant des granules de keratohyaline composés d’involucrine, de loricrine et de profillagrine. Ces cellules s'aplatissent sous la pression des cellules sous-jacentes qui se divisent et les poussent jusqu'à la surface de la peau. Les organelles et noyaux de ces cellules se décomposent et leurs membranes cellulaires deviennent de plus en plus imperméables. La couche cornée fournit la principale barrière à l'environnement extérieur et est essentielle au maintien d'une hydratation cutanée optimale. Sa structure a été décrite par un modèle « en briques et mortier » avec des cornéocytes imbriqués dans une matrice de céramides, de cholestérol et d'acides gras. Les cornéocytes sont des kératinocytes terminalement différentiés, dépourvus de noyau et d'organelles, composés d'une matrice dense de kératine et d'une enveloppe cornée épaisse formée de protéines réticulées et de protéolipides. Les cellules de cette couche desquament en raison des faibles liens intercellulaires résiduels et doivent être constamment remplacées par de nouveaux cornéocytes pour assurer la stabilité de la fonction de barrière cutanée (Kanitakis, 2002, Tobin, 2006).
L’épiderme se renouvèle en 40 à 56 jours environ (Halprin, 1972, Iizuka, 1994). Ce renouvellement cellulaire est assuré par une prolifération, une migration et une différenciation des kératinocytes. Les cellules souches, présentes dans la couche basale,
se divisent de façon asymétrique engendrant une autre cellule souche et une cellule amplificatrice transitoire qui se divise elle-même à plusieurs reprises avant d’entamer le processus de différenciation terminale (Koster, 2009). La différentiation terminale se caractérise par une série de changements biochimiques et morphologiques accompagnés d’une migration vers la surface de la peau. Au sein de l'épiderme, les cellules souches interfolliculaires et leurs descendances différenciées sont organisées en colonnes nommées unités de prolifération épidermique. De telles unités ont été proposées pour la première fois il y a plus de 30 ans et leur existence a été confirmée plus récemment en traçant génétiquement la progéniture de cellules souches épidermiques (Allen and Potten, 1974, Ghazizadeh and Taichman, 2001). En plus des cellules souches interfolliculaires, l'épiderme contient au moins deux autres populations de cellules souches: les cellules souches du follicule pileux et les cellules souches des glandes sébacées (Fuchs and Horsley, 2008).
D’autres cellules composent l’épiderme comme les cellules de Langerhans (2 %) qui sont des cellules dendritiques présentatrices d’antigènes impliquées dans le système immunitaire. Les cellules de Merkel (0,5 %), elles, contribuent à la perception cutanée par une action mécanoréceptrice. Alors que les mélanocytes (3 %) produisent et distribuent aux kératinocytes la mélanine, le pigment responsable de la couleur de la peau (voir section 1.6.2.2). Les mélanocytes sont répartis régulièrement entre les kératinocytes de la couche basale à raison de 1 mélanocyte pour 4 à 10 kératinocytes basaux (Tobin, 2006).
L’épiderme est ancré au derme par la jonction dermo-épidermique (JDE), une région acellulaire jouant le rôle de support mécanique mais aussi de barrière sélective régulant les échanges moléculaires et cellulaires entre ces deux compartiments. Cette jonction est principalement composée de laminine, de collagène de type IV, de nidogène qui fait le lien entre les deux molécules précédentes et de perlécan. Au niveau des kératinocytes, ce sont les hémidesmosomes qui font le lien entre l’intérieur des cellules et la jonction dermo-épidermique. Pour solidifier l’attachement du derme à la JDE, des filaments d’ancrage constitués de collagène VII se projettent vers le derme et s’enchevêtrent dans les fibres des collagènes de type I et III. Cette jonction indique leur polarité et donc leur organisation spatiale aux kératinocytes basaux et sert de support à la migration des kératinocytes lors de la guérison de plaies. La JDE peut être traversée par différents types cellulaires (comme des cellules de Langerhans ou des lymphocytes) lors de processus immunitaires ou inflammatoires (Breitkreutz et al., 2013, Breitkreutz et al., 2009, Tobin, 2006).
1.2.2 Le derme
Le derme est un tissu conjonctif fibreux, vascularisé et innervé, principalement composé d’une matrice extracellulaire (MEC) et de fibroblastes qui en synthétisent les composants. Il contient également des cellules immunitaires, des mécanorécepteurs et des appendices comme des glandes sudoripares et des unités pilo-sébacées. Le derme est un tissu de soutien, compressible et élastique dont l’épaisseur varie grandement en fonction de sa localisation anatomique (environ 6 mm à la plante des pieds et 1 mm aux paupières). Il peut être divisé en deux régions qui se distinguent par l’organisation des fibres de collagène et d’élastine et par le nombre de fibroblastes qui les composent. Le derme papillaire, le plus en surface, est caractérisé par un tissu lâche aréolaire contenant de nombreux fibroblastes et le derme réticulaire, en profondeur, est lui caractérisé par un tissu dense moins riche en cellules (Tobin, 2006). Le derme papillaire forme des papilles dermiques entre les crêtes épidermiques, qui augmentent la surface de contact et permettent une meilleure adhésion et un échange moléculaire entre le derme et l’épiderme (Figure 1.3).
Les fibroblastes, les cellules majoritaires du derme, sont responsables de la sécrétion de molécules de signalisation et de la synthèse de nombreuses macromolécules bioactives formant la MEC. La MEC est constituée d’un échafaudage de collagènes sur lequel se lient de l’élastine, des glycoprotéines comme la laminine et des protéoglycans. Ces composants interagissent avec des cellules dans ou à proximité de la matrice via des récepteurs cellulaires dont les principaux sont les intégrines. La MEC n’est pas statique, mais constamment remodelée notamment par une famille d’enzymes: les métalloprotéinases matricielles (MMP). La MEC joue un rôle essentiel d’organisation morphologique et structurale du derme. Elle a également de nombreuses fonctions physiologiques de par sa capacité de liaison à de nombreuses molécules et d’interaction avec les cellules (Casu and Lindahl, 2001, Hildebrand et al., 1994, Kim et al., 2011, Patel et al., 2007, Whitelock et al., 2008). La MEC permet ainsi, via l’activation de voies de signalisation, la transduction de signaux et la régulation de la transcription (Bosman and Stamenkovic, 2003).
La grande majorité des fibres dermiques sont des collagènes, principalement de types I (85-90 %) et III (8-11 %), responsables de la résistance mécanique de la peau. Le collagène représente environ 75 % de la masse sèche totale du derme. Ces fibres sont disposées en faisceaux lâches dans le derme papillaire et deviennent plus épaisses dans le derme
profond. D'autres collagènes se retrouvent aussi dans le derme : le collagène IV (dans la JDE), le collagène V, le collagène VII (fibres d'ancrage de la JDE) et le collagène XVII. La biosynthèse du collagène est complexe et implique la synthèse de polypeptides de procollagène qui sont sécrétés et clivés à l’extérieur de la cellule par des enzymes afin de générer une fibre de collagène mature et fonctionnelle (Bosman and Stamenkovic, 2003, Tobin, 2006).
Les fibres élastiques sont des macromolécules de MEC comprenant un noyau d'élastine entouré de microfibrilles riches en fibrilline. Elles confèrent une élasticité et une résilience aux tissus conjonctifs, permettant une déformabilité et un retour à l’état initial sans apport d'énergie. La synthèse des fibres élastiques nécessite le dépôt de tropoélastine, le précurseur soluble de l'élastine mature, sur une matrice de microfibrilles. Diverses enzymes comme les métalloprotéinases matricielles et les sérine protéases sont capables de cliver les molécules de fibres élastiques (Kielty et al., 2002).
La substance fondamentale est la troisième composante de la MEC. C’est une substance amorphe et hydroscopique, biologiquement très active et diversifiée. Elle se compose de glycosaminoglycans, de protéoglycans, de glycoprotéines et d'eau. Les glycosaminoglycans (GAG) sont des chaines de polysaccharides chargés négativement. Ces polysaccarides attirent des cations osmotiquement actifs qui à leur tour attirent des molécules d’eau, ce qui induit une turgescence permettant la résistance à la compression. Il existe cinq groupes de GAG : l’acide hyaluronique, les chondroïtines et dermatanes sulfates, les héparanes sulfates et les kératanes sulfates. Les protéoglycans sont formés par la liaison de glycosaminoglycans à une protéine. Le versican, un protéoglycan produit par les fibroblastes, s’associe aux fibres élastiques et à l'acide hyaluronique ce qui permet la formation de la tension de la peau. La famille des syndécans sont des protéoglycans multifonctionnels impliqués dans l’adhésion des cellules à la MEC. Ces composants de la substance fondamentale ont la capacité de lier des molécules de signalisation, comme des facteurs de croissance ou des cytokines, contrôlant ainsi leur diffusion à travers la matrice, leur portée d’action et leur durée de vie tout en stimulant ou inhibant leur activité de signalisation. La bioactivité de ce réseau complexe de macromolécules permet la régulation de nombreuses fonctions cellulaires (Tobin, 2006).
1.2.3 L’hypoderme
C'est un tissu adipeux représentant la partie la plus profonde de la peau. Il joue un rôle important dans la thermorégulation, l'isolation, l'apport d'énergie (réserve nutritionnelle) et la protection contre les blessures mécaniques. Les principales cellules de l'hypoderme sont les adipocytes, de grandes cellules avec un cytoplasme chargé en lipides (triglycérides, acides gras). Les adipocytes sont disposés en lobules primaires et secondaires, séparés par des cloisons de tissu conjonctif contenant des cellules (fibroblastes, dendrocytes, mastocytes), la partie la plus profonde des glandes sudoripares, ainsi que des vaisseaux et des nerfs contribuant à la formation des plexus dermiques correspondants (Kanitakis, 2002).
1.3 Les dommages induits par l’absorption directe des rayons UV
L’exposition de la peau à la lumière solaire et donc aux rayons UV qui la compose a des conséquences. En effet, les rayons UV sont responsables de la formation de dommages dans l’ADN. Cette formation de dommages peut être de deux types : directe ou indirecte. Les lésions dans l’ADN induits de façon indirecte ne seront pas abordées en détails ici. De façon simplifiée, elles sont formées lorsque certains chromophores de la cellule absorbent l’énergie des rayons UV et passent à un état excité. Ces molécules sont instables dans cet état et retournent dans un état fondamental en transférant de l’énergie aux molécules d’oxygène, ce qui induit la formation d’espèces réactives de l’oxygène (ROS), comme l’anion superoxyde O2-, le peroxyde d’hydrogène H2O2 ou le radical hydroxyle OH-. Toutesces ROS peuvent endommager les protéines, les lipides et l’ADN. Au niveau de l’ADN, les ROS peuvent créer différents types de dommages comme des altérations de bases ou des cassures simple brin. Les guanines sont les bases les plus sensibles à l’oxydation et le 8-oxo-2'-désoxyguanosine (8-oxo-dG) est un des dommages oxydatifs les plus communs (Cadet et al., 2015, Cadet et al., 2012, Kvam and Tyrrell, 1997).
L’induction de dommages dans l’ADN peut aussi être directe, de par la capacité de l’ADN à absorber l’énergie de certains rayonnements. L’absorption maximale de l’ADN se situe à 260 nm, dans la région des UVC qui ne se rendent pas à la surface terrestre (Figure 1.2). Cependant, l’absorption de l’ADN reste significative dans la région des UVB, avec des valeurs de 20 % et 3 % d’absorption à respectivement 290 nm et 300 nm comparativement à 260 nm (Cadet and Douki, 2018). Les photons provenant des UVA sont moindrement
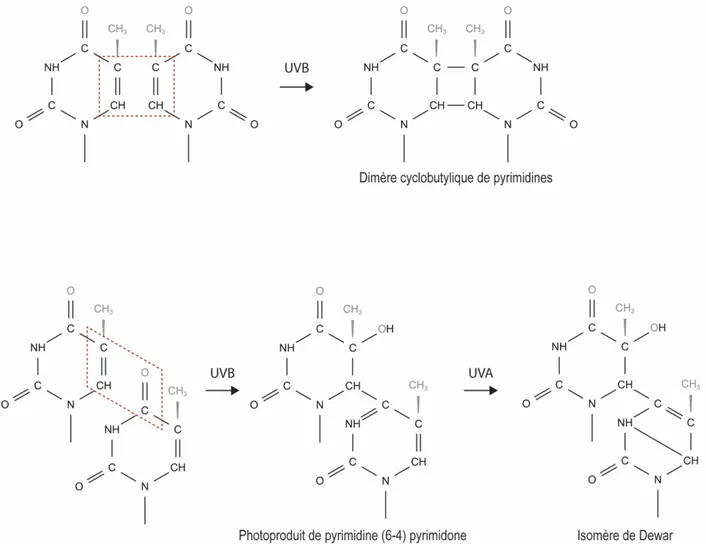
absorbés par l’ADN mais peuvent tout de même induire des dommages directs (Douki et al., 2003, Mouret et al., 2006, Rochette et al., 2003). L’absorption directe des photons UV provoque la formation de liaisons covalentes entre deux pyrimidines adjacentes, constituant les dommages les plus fréquents induits par les rayons UV que sont les dimères de pyrimidines (Sage, 1993). Identifiés il y a presque 60 ans, les dimères de pyrimidines regroupent trois principaux types de dommages : les dimères cyclobutyliques de pyrimidines (CPD), les photoproduits de pyrimidine (6-4) pyrimidone (6-4PP) et leurs isomères de Dewar (Figure 1.4) (Beukers and Berends, 1960).
Figure 1.4 Représentation schématique de la formation d’un CPD, d’un 6-4PP
et d’un isomère de Dewar par les rayons ultraviolets
CPD : Dimère cyclobutylique de pyrimidines, 6-4PP : Photoproduit de pyrimidine (6-4) pyrimidone (Adaptée de JD Mallet).
1.3.1 Les photoproduits de pyrimidine (6-4) pyrimidone
Les 6-4PP représentent environ 20 % des dommages induits par les rayons UV (Besaratinia et al., 2011, Courdavault et al., 2005). Ils sont formés par une cycloaddition [2+2] impliquant la création d’une liaison entre les carbones C5 et C6 de la pyrimidine à l’extrémité 5’ et le groupe carbonyle en C4 de la pyrimidine à l’extrémité 3’. Cela conduit à la formation d'un intermédiaire instable à savoir un oxetane ou un azetidine dépendamment de la nature de la base à l’extrémité 3’. Cet intermédiaire se réarrange spontanément et donne lieu à l’adduit de 6-4PP (Figure 1.4) (Ravanat et al., 2001, Yokoyama and Mizutani, 2014). Il existe 4 types de 6-4PP qui sont formés à des fréquences différentes après l’exposition d’ADN nu aux rayons UVB: 81 % des dommages sont formés sur des TC, 13 % sur des TT, 5 % sur des CC et moins de 1 % sur des CT (C; cytosine et T; thymine) (Douki and Cadet, 2001). Les 6-4PP modifient la structure de l’hélice d’ADN en induisant une distorsion de 44° par rapport à la conformation B, conformation native de l’hélice d’ADN chez l’homme. Cette déformation engendre le blocage des machineries de réplication et de transcription traditionnelles mais aussi un important ralentissement des ADN polymérases translésionnelles (Kim et al., 1995, Tissier et al., 2000). Ces dommages sont très rapidement réparés par le système de réparation par excision de nucléotides (NER) (Voir la section 1.5 pour plus de détails). Environ 80 % des 6-4PP sont réparés dans les 5 heures suivant l’irradiation aux rayons UV (Courdavault et al., 2005, Mitchell et al., 1985, Mitchell and Nairn, 1989). S’ils persistent dans l’ADN, les 6-4PP empêchent la réplication et induisent la mort cellulaire par apoptose comme décrit dans la section 1.4.3 (Lo et al., 2005, You et al., 2001).
1.3.2 Les Dewar
Les 6-4PP peuvent, par absorption d'un photon d’UVA, subir une deuxième réaction de photoisomérisation et donner lieu à un second type de photoproduit connu sous le nom d'isomère de Dewar (Figure 1.4). Il a été montré que 20 % des 6-4PP sont convertis en Dewar dans l'ADN de micro-organismes exposés à la lumière naturelle du soleil (Meador et al., 2014). Les isomères de Dewar sont responsables de l’induction d’un angle moins important (21°) que les 6-4PP dans l’hélice d’ADN mais sont réparés avec une efficacité similaire (Voir revue dans (Douki and Sage, 2016)).
1.3.3 Les dimères cyclobutyliques de pyrimidines
Les CPD représentent environ 80 % des dommages dans l’ADN induits par les rayons UV (Besaratinia et al., 2011, Courdavault et al., 2005, Yoon et al., 2000). Ils sont formés par une photocycloaddition [2+2] des doubles liaisons des carbones C5-C6 de deux pyrimidines adjacentes. Cette saturation des doubles liaisons induit la formation d’un anneau cyclobutane (Figure 1.4). En raison de contraintes stériques, seuls les isomères cis-syn peuvent être générés dans l'ADN (Ravanat et al., 2001). Il existe donc 4 types de CPD produits aux fréquences suivantes après une exposition de cellules humaines aux rayons UVB: 27 % des dommages sur les TT, 27 % sur les CC, 25 % sur les TC et 21 % sur les CT (Bastien et al., 2013).
Il a été longtemps admis que les CPD étaient formés quelques picosecondes après l’absorption d’un photon ultraviolet par une pyrimidine. Cependant, une seconde voie de formation des CPD a plus récemment été démontrée dans les mélanocytes puis dans les kératinocytes. Les dommages induits par cette seconde voie de formation sont appelés « dark CPD » car ils sont formés jusqu’à 3 heures après la fin de l’exposition aux rayons UVA. Ces « dark CPD » apparaissent lorsque des espèces réactives de l'oxygène et de l'azote, induites par les rayons UV, se combinent pour exciter un électron contenu dans des produits de dégradation de la mélanine. Cela crée un état de triplet qui a l'énergie d'un photon UV et qui induit des CPD par transfert d'énergie à l'ADN d'une manière indépendante du rayonnement. Les CPD peuvent ainsi être induit par les rayons UV de deux façons distinctes directement pendant l’exposition et indirectement après celle-ci (Delinasios et al., 2018, Premi et al., 2015).
Dans des cultures cellulaires irradiées aux rayons UVC (254 nm), en moyenne, 0,45 CPD sont produits par 105 bases par Jm-2. Pour une irradiation avec des rayons UVB, le
rendement est inférieur d'un ordre de grandeur avec en moyenne 0,05 CPD produit par 105
bases par Jm-2 (Voir revue dans (Cadet and Douki, 2018)). Après une exposition aux rayons
UVA, des CPD sont également détectés bien qu'ils soient produits 103 et 105 fois moins
efficacement que par les rayons UVB et UVC, respectivement (Kuluncsics et al., 1999). Les rayons UVC et les rayons UVB courts étant bloqués par la couche d’ozone, les rayonnements UVB supérieurs à 290 nm sont les principaux responsables de la formation
des CPD. L’utilisation des rayons UVB pour étudier l’induction et les conséquences des CPD dans la peau humaine est donc physiologiquement pertinente.
Les CPD induisent une moins grande déstabilisation de la structure de l’hélice d’ADN que les 6-4PP. Cette distorsion par un CPD est seulement de 7° à 9° par rapport à la conformation B de l’hélice d’ADN (Kim et al., 1995). Ces dommages, sont donc plus difficilement détectables par la machinerie de réparation NER et seront réparés beaucoup plus lentement que les 6-4PP. Seul 10 % environ des CPD sont réparés dans les 5 heures suivant l’irradiation UV et 50 % après 24h (Courdavault et al., 2005, Ferguson-Yates et al., 2008, Mallet et al., 2016).
1.3.4 Le potentiel mutagène des dimères cyclobutyliques de
pyrimidines
La probabilité qu’un dommage soit converti en mutation dépend de sa fréquence, de sa vitesse de réparation et de son potentiel mutagène (Thilly, 1983).
Pour ce qui est de la fréquence, les CPD sont les dommages les plus fréquemment formés. Ils sont 20 à 40 fois plus fréquents que tout autre photoproduit dans l’ADN lorsque celui-ci est irradié avec un simulateur solaire (Yoon et al., 2000). Pour ce qui est de la vitesse de réparation, la faible distorsion qu’ils induisent dans l’hélice d’ADN rend leur détection difficile et leur réparation lente.
Le potentiel mutagène d’un dommage dans l’ADN est caractérisé par la probabilité qu’il soit mal interprété par une ADN polymérase lors de la réplication. Une base incorrecte est alors insérée en face du site de dommage et peut ainsi induire une mutation irréversible. Parmi les lésions induites par les UV, les CPD possèdent le potentiel mutagène le plus élevé. Ce fort potentiel mutagène des CPD peut mener à la formation de mutations de transition C → T ou CC → TT. Ces transitions lorsqu’elles sont situées dans des sites dipyrimidiques sont les mutations signatures induites par les rayons UV et les principales responsables de l’initiation des cancers de peau non-mélanocytaires (Brash, 2015, Jiang and Taylor, 1993, Miller, 1985). Une mutation signature étant une mutation unique à un mutagène et permettant la déduction du mutagène à partir de la nature de la mutation. Deux processus peuvent mener à ces mutations dans l’ADN : la lecture infidèle de l’ADN par les polymérases translésionnelles ou la désamination de base.
Lors de la réplication, les ADN polymérases traditionnelles (delta δ et epsilon ε) sont bloquées au niveau des CPD. Des ADN polymérases translésionnelles comme les polymérases eta η, zeta ζ et kappa κ peuvent alors être recrutées aux sites de dommages, car ces dernières sont capables de poursuivre la réplication même en présence de CPD. Cependant, certaines sont peu fidèles et insèrent régulièrement une adénine (A) en face d’un CPD. L’incorporation d’une adénine en face d’une thymine (T) n’induit pas d’erreur. Alors que l’insertion d’une adénine en face d’une cytosine (C) donne elle, lors de la réplication suivante, lieu à une incorporation d’une thymine en face de l’adénine. C’est ainsi que se forment les mutations de transition C → T. Ce sont donc les CPD contenant des cytosines qui sont les plus mutagènes (Choi and Pfeifer, 2005, Yoon et al., 2009).
La désamination de bases de l’ADN est une réaction chimique très lente (demi-vie de 20 000 ans) transformant, entre autres, les cytosines en uraciles (Frederico et al., 1990). Cette désamination spontanée des cytosines peut être corrigée par le retrait des uraciles par l'uracile-ADN glycosylase (Slupphaug et al., 1996). Mais si la correction n’est pas effectuée avant la réplication, une adénine sera insérée en face de l’uracile. Adénine qui à son tour code pour une thymine dans la réplication suivante, menant ainsi à une transition C → T. Dans le contexte d’un CPD, la désamination des cytosines est accélérée d’un facteur de six ordres de grandeur, passant d’un temps de demi-vie de 20 000 ans à 5 jours (Peng and Shaw, 1996). Ce qui rend le potentiel mutagène des CPD encore plus important.
La méthylation de l’ADN est une marque fréquente permettant de réguler la transcription. Une cytosine méthylée (5mC) peut elle aussi désaminer, cette fois en thymine. Ce qui forme
également une transition C → T. La désamination d’une 5mC (demi-vie de 2 000 ans) est
plus rapide qu’une simple cytosine. Et lorsque la 5mC se trouve dans un CPD cette
désamination est encore plus rapide (Cannistraro and Taylor, 2009). De plus, la méthylation d’une cytosine augmente la probabilité de formation d’un CPD par les rayons UVB d’un facteur 1,7 fois, car la méthylation d’une cytosine décale son pic d’absorption maximal de 267 nm à 273,5 nm et le rend donc plus proche des rayons UVB (Rochette et al., 2009, Tommasi et al., 1997).
Ainsi les CPD sont des dommages UV-induits à fort potentiel mutagène et sont responsables de l’induction des mutations signatures des rayons UV, les transitions C → T.
1.4 Les réponses aux dommages induits par les rayons UV
Les dommages dans l'ADN sont des événements courants dans la vie d'une cellule qui peuvent conduire à une déstabilisation du génome. La présence de ces lésions engendre donc plusieurs réponses qui permettent à la cellule de les éliminer, d’y faire face, ou d'activer un processus de mort cellulaire programmée afin d’éliminer les cellules contenant des mutations potentiellement trop nombreuses. Ces différentes réactions, appelées réponses aux dommages dans l'ADN (DNA Damage Response; DDR) sont hautement régulées, inter-connectées et comprennent:
(1) L’activation de points de contrôle, qui arrêtent la progression du cycle cellulaire pour permettre la réparation et prévenir la transmission de chromosomes endommagés ou incomplètement répliqués.
(2) La réparation des dommages dans l'ADN.
(3) L'apoptose, qui élimine les cellules fortement endommagées ou dérégulées.
Ces différentes réponses permettent ainsi la préservation de la stabilité du génome (Figure 1.5).
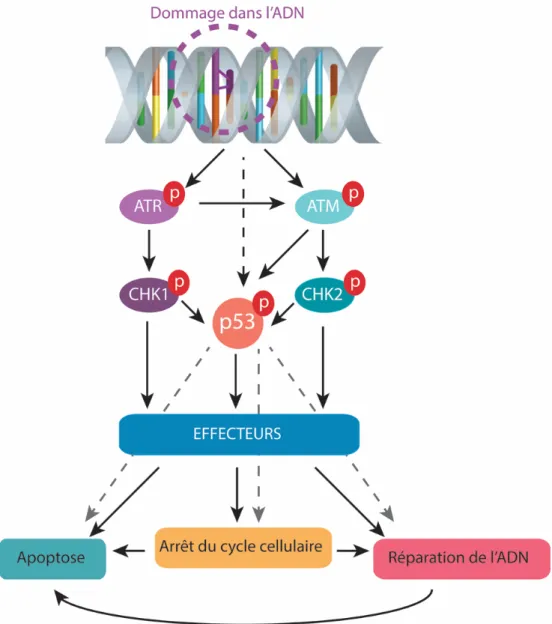
Figure 1.5 Représentation schématique simplifiée de la DDR après induction
de dommages dans l’ADN par les rayons ultraviolets.
Les dommages à l'ADN activent les kinases ATM et ATR, des transducteurs de signaux clés de la voie DDR. Une fois phosphorylées, ces dernières déclenchent l'activation des régulateurs de cycle cellulaire en aval, Chk1, Chk2 et p53, qui activent à leur tour des effecteurs en aval qui agissent sur la progression du cycle cellulaire, l’apoptose ou la réparation des dommages à l’ADN. (Adaptée de (Tsuiko et al., 2019))
1.4.1 L’arrêt du cycle cellulaire
Pour répondre efficacement au stress induit par une irradiation aux rayons UV, les cellules peuvent arrêter transitoirement leur croissance à différents points du cycle cellulaire. Ce cycle cellulaire est divisé en 4 phases : la phase G1, dans laquelle la cellule croit en taille et se prépare à dupliquer son génome; la phase S, durant laquelle l’ADN est répliqué; la phase G2, où la cellule croit et se prépare à la division cellulaire et finalement la phase M, durant laquelle a lieu la mitose. Le passage de l’une à l’autre de ces phases est hautement régulé et ponctué de points de contrôle. De façon très simplifiée, le mécanisme d’arrêt de la progression du cycle cellulaire est basé sur une cascade d'événements de phosphorylation et comprend trois composants principaux: les capteurs (principalement ATM et ATR), les transducteurs de signaux (principalement Chk1 et Chk2) et les effecteurs (principalement Cdc25 et p53) (Smith et al., 2010).
ATM (ataxia telangiectesia mutated) est la principale kinase responsable de l'activation des points de contrôle en réponse aux rayonnements ionisants et aux cassures bicaténaires. ATM est recrutée au niveau des cassures doubles brins par le complexe MRE11-RAD50-NBS1 (MRN). L’autophosphorylation d’ATM convertit l'oligomère en monomères, qui semblent être la forme active de l'enzyme dans cette cascade (Bakkenist and Kastan, 2003). ATM activée va alors phosphoryler de nombreuses protéines dont Chk2, MDM2 et p53. Chk2 activée se dissocie du site de dommage et agit comme un transducteur diffusible de signal induisant la phosphorylation de cibles en aval (Lukas et al., 2003). Les substrats de Chk2 incluent entre autres des protéines de point de contrôle telle que BRCA1 et, dans certains contextes, p53. Chk2 peut également contribuer à la phosphorylation de la phosphatase CDC25 qui, en temps normal, favorise la progression du cycle cellulaire en déphosphorylant et en activant les kinases cycline-dépendantes (CDK) (Cai et al., 2009). La phosphorylation de CDC25 par Chk2 induit sa dégradation ou son exclusion du noyau. Les kinases cycline-dépendantes ne sont alors plus activées et la progression du cycle cellulaire est arrêtée en phase G2 (Sorensen et al., 2003). La phosphorylation de p53 par Chk2 ou ATM induit sa stabilisation et son accumulation, ce qui permet l’augmentation de la transcription de gènes dont p21, un inhibiteur de kinases cycline-dépendantes. p21 inhibe alors la cycline E/Cdk2 et cause l’arrêt du cycle cellulaire en G1 (Blackford and Jackson, 2017, Kastan and Bartek, 2004, Smith et al., 2010).
ATR (ATM and Rad3 related) en complexe avec ATRIP (ATR Interacting Protein) est la principale kinase responsable de l'activation des points de contrôle en réponse aux lésions induites par les rayons UV. ATR est recrutée dans les régions d'ADN simple brin qui sont formées lorsque les fourches de réplication sont bloquées ou pendant la réparation des lésions. L’accumulation d’ADN simple brin recouvert de protéines RPA permettrait le recrutement du complexe ATR-ATRIP (Sertic et al., 2012, Zou and Elledge, 2003). La kinase ATR semble constitutivement prête à phosphoryler ses substrats, mais son activité est largement contrôlée par sa localisation subcellulaire (Kastan and Bartek, 2004). ATR est ainsi recrutée au site d’arrêt de la réplication de l’ADN ou aux sites de dommages et peut alors phosphoryler ses substrats comme Chk1 et RAD17 (Kastan and Bartek, 2004). Chk1 activée peut à son tour phosphoryler CDC25 et ainsi arrêter la progression du cycle en G2 comme décrit précédemment.
De plus en plus d’études décrivent une interaction et une communication encore plus complexes entre ATR et ATM pour l’induction de l’arrêt de la progression du cycle cellulaire. Ainsi, ATM peut être phosphorylée par ATR après un traitement aux rayons UV, indépendamment de la formation des cassures doubles brins (Voir revues dans (Hurley and Bunz, 2007, Sertic et al., 2012)). La DDR serait également activée en réponse à un stress oxydatif pour coordonner la réparation, l’arrêt du cycle cellulaire, la transcription et l’apoptose (Voir revue dans (Yan et al., 2014)). Les protéines de détection de dommages de la NER : DDB2 et XPC permettraient elles aussi le recrutement d’ATR et d’ATM aux sites de dommages (Ray et al., 2013).
1.4.2 La réparation des dommages
L’arrêt du cycle cellulaire décrit précédemment accorde plus de temps à la cellule afin de lui permettre de réparer les dommages dans l’ADN avant le prochain cycle de réplication. Il existe 5 principaux systèmes de réparation dans les cellules eucaryotes, chacun spécifique à l’élimination d’une classe distincte de lésion dans l’ADN. Ces systèmes comprennent : la réparation par recombinaison homologue, la réparation par recombinaison non homologue, la réparation de mésappariements, la réparation par excision de base et la réparation par excision de nucléotides. Chez certaines espèces, il existe également des enzymes de photoréversion des dommages induits par les rayons UV, mais pas chez l’humain, chez qui, le principal système permettant d’éliminer ces dommages UV-induits est la réparation par