Étude sur la différenciation in vitro de lymphocytes B
humains en plasmocytes
Microenvironnement et caractérisation des plasmocytes générés
Mémoire
Rayelle Itoua Maïga
Maîtrise en biochimie
Maître ès sciences (M.Sc.)
Québec, Canada
Résumé
Les plasmocytes, cellules responsables de la sécrétion d’anticorps, sont au cœur de la réaction immunitaire humorale. Cette caractéristique en fait des cellules intéressantes en thérapie cellulaire pour offrir une protection humorale aux patients immunosupprimés suite à une transplantation de cellules souches hématopoïétiques. Afin d’obtenir ces cellules, notre équipe à Héma-Québec propose d’utiliser la capacité de différenciation des lymphocytes B mémoires in vitro. Notre hypothèse est qu’il serait possible de différencier ces derniers en contrôlant leur microenvironnement de culture. Ce projet porte donc sur l’étude de l’interaction cellulaire CD27-CD70 combinée aux facteurs de survie APRIL et CXCL12. Les résultats montrent que cette interaction induit une différenciation rapide des lymphocytes B tandis que l’addition des facteurs solubles n’a pas d’impact sur la survie cellulaire. De plus, l’utilisation des marqueurs de surface CD31 et CD39 a permis de mettre en évidence l’hétérogénéité des plasmocytes générés in vitro et ceux retrouvés in vivo.
Table des matières
Résumé ... III
Liste des figures ... IX
Liste des abréviations ... XI
Remerciements ... XIII
1. Introduction ... 1
1.1 Les lymphocytes B humains ... 1
1.1.1 Développement et transition ... 1
1.1.2 Maturation des lymphocytes B et rencontre de l’antigène ... 1
1.1.3 Activation extrafolliculaire des lymphocytes B ... 2
1.1.4 Activation folliculaire et formation du centre germinatif ... 2
1.1.5 Formation du centre germinatif ... 3
1.1.6 Différenciation terminale des lymphocytes B ... 5
1.1.7 Réponse immunitaire secondaire ... 6
1.1.8 Les populations de lymphocyte B du sang ... 6
1.2 Les plasmocytes ... 7
1.2.1 Les immunoglobulines ... 7
1.2.2 Le phénotype des lymphocytes B différenciés ... 8
1.2.3 Plasmocytes à courte vie ... 10
1.2.4 Les plasmocytes à longue vie ... 10
1.2.5 Niche de survie des plasmocytes ... 11
1.3 Les plasmocytes et la thérapie cellulaire ... 13
1.3.1 Greffe de cellules souches hématopoïétiques ... 13
1.3.2 Les applications de la transplantation ... 14
1.3.3 La reprise de greffe et la reconstitution du système immunitaire ... 14
1.3.4 Problèmes post-transplantation et vulnérabilité aux infections ... 15
1.3.5 Traitement des problèmes post-transplantations ... 15
1.3.6 Introduction des plasmocytes générés in vitro ... 16
1.4 Modèles de culture développés jusqu’à présent ... 17
1.5 Expansion et différenciation in vitro de lymphocytes B humains ... 17
1.5.1 Interaction CD40-CD154 ... 17
1.5.2 Interaction CD27-CD70 ... 18
2. Hypothèse ... 19
3. Objectifs ... 21
4.1 Cellules humaines ... 23
4.1.1 Participants à l’étude ... 23
4.1.2 Les cellules mononuclées du sang ... 23
4.1.3 Lymphocytes B mémoires IgG+, IgA+, ou IgE+ ... 23
4.1.4 Les cellules mononuclées de la moelle osseuse ... 24
4.2 Les lignées cellulaires humaines ... 24
4.2.1 Les cellules 3H7 et L4.5 ... 24
4.2.2 Ramos, RPMI-8226, U266, Jurkat ... 24
4.3 Culture de lymphocytes B humains ... 25
4.3.1 Expansion des lymphocytes B ... 25
4.3.2 Transition ... 25
4.3.3 Différenciation des lymphocytes B ... 26
4.3.4 Survie des lymphocytes B et des plasmocytes ... 26
4.3.5 Système de culture des lymphocytes B mémoires ... 26
4.4 Mesure de la sécrétion d’immunoglobulines ... 27
4.4.1 ELISA ... 27
4.4.2 Essai BioPlex ... 27
4.4.3 ELISPOT ... 28
4.5 Cytométrie en flux ... 28
4.5.1 Marquage extracellulaire ... 28
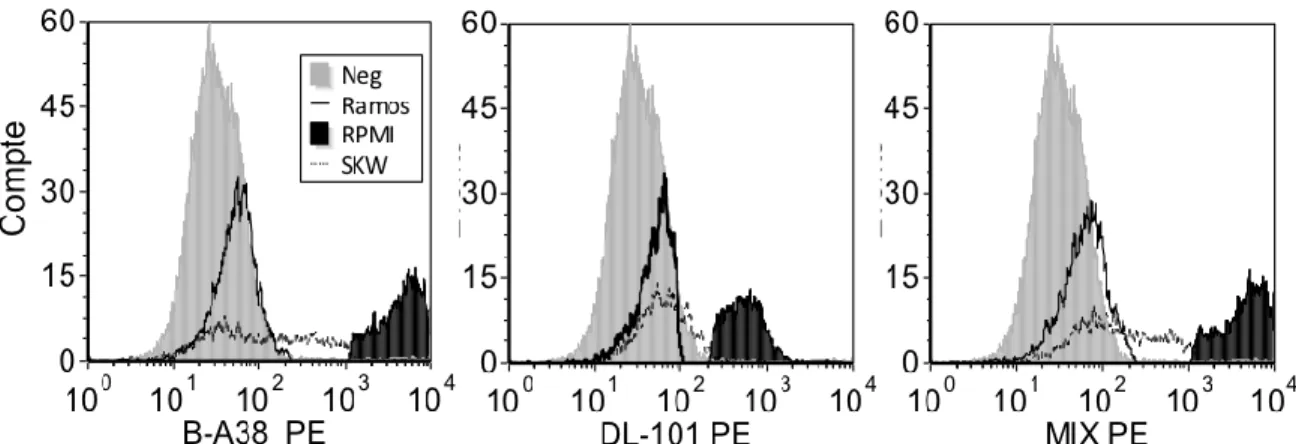
4.5.2 Comparaison des clones d’anticorps anti-CD138 ... 30
4.5.3 Prolifération cellulaire ... 30
4.5.4 Fluorescent Cell Barcoding ... 30
4.6 Immunofluorescence ... 31
4.6.1 Microscopie à fluorescence ... 31
5. Résultats ... 33
5.1 Étude comparative des anticorps monoclonaux anti-CD138 ... 33
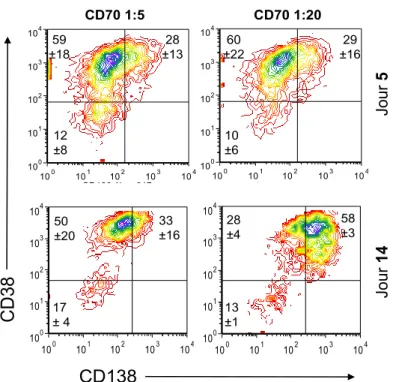
5.2 Différenciation des lymphocytes B avec l’interaction CD70 ... 37
5.2.1 Détermination du ratio de CD70 ... 37
5.2.2 Comparaison des interactions CD70 et CD154 ... 39
5.3 Amélioration de la viabilité des lymphocytes B ... 47
5.3.1 APRIL ... 48
5.3.2 CXCL12 ... 54
5.3.3 IGF-R3 ... 56
5.3.4 Instabilité de la viabilité ... 57
5.4 Caractérisation des plasmocytes générés in vitro ... 57
5.4.1 Patron de sécrétion des cellules différenciées ... 58
5.4.2 Morphologie cellulaire ... 59
5.4.3 Réponse intracellulaire des plasmocytes à des stimuli dans leur environnement ... 60
5.4.4 Expression de CD31 et CD39 ... 62
6.1 Le clone DL101 anti-CD138 détecte une nouvelle sous-population de plasmocytes in vitro
... 67
6.2 L’interaction cellulaire induit une différenciation plus rapide ... 69
6.3 Complexité du microenvironnement de survie des plasmocytes... 70
6.4 Vers une meilleure caractérisation des plasmocytes générés ... 70
7. Conclusion ... 73
Liste des figures
Figure 1.1 : Interaction cellulaire entre les lymphocytes T activés et cellules B avant le centre germinatif……..4
Figure 1.2 : Le centre germinatif………5
Figure 1.3 : Réponse des lymphocytes B mémoires lors de la réaction immunitaire secondaire………..6
Figure 1.4 : Niche de survie des plasmocytes à longue vie………12
Figure 1.5 : Reconstitution des cellules immunitaires après une transplantation de CSH………15
Figure 4.1 : Système de culture des lymphocytes B mémoires……….26
Figure 5.1 : Marquage des cellules CD138+ générées in vitro………..34
Figure 5.2 : Marquage de lignées cellulaires avec des clones anti-CD138……….35
Figure 5.3 : Marquage de plasmocytes in vivo……….35
Figure 5.4 : Essai de compétition des clones anti-CD138………..36
Figure 5.5 : Différenciation des lymphocytes avec l’interaction CD70………38
Figure 5.6 : Émergence de cellules CD38hiCD138+ ………30
Figure 5.7 : Sécrétion d’immunoglobulines……….41
Figure 5.8 : Expression de CD27………43
Figure 5.9 : Évolution de l’expression de CD27 et CD70………44
Figure 5.10 : Croissance des cellules………46
Figure 5.11 : Dose-réponse d’APRIL………48
Figure 5.12 : Effet d’APRIL sur la viabilité cellulaire en interaction CD70 et CD154…..………49
Figure 5.13 : Effet d’APRIL sur la fonction sécrétrice des lymphocytes B………50
Figure 5.14 : Effet de la présence simultanée d’APRIL et d’IL-10 sur la viabilité cellulaire………51
Figure 5.15 : Expression des récepteurs d’APRIL………52
Figure 5.16 : Expression de TACI et BCMA sur lignées cellulaires………53
Figure 5.17 : Dose réponse de CXCL12………54
Figure 5.18 : Influence de CXCL12 sur la viabilité cellulaire in vitro………55
Figure 5.19 : Expression de CXCR4………56
Figure 5.20 : Effet d’IGF-R3 sur la viabilité cellulaire………57
Figure 5.21 : Patron de sécrétion d‘IgG………58
Figure 5.22 : Patron de sécrétion d’IgG de plasmocytes de la moelle osseuse………59
Figure 5.23 : Morphologie des cellules CD138+ ………60
Figure 5.24 : Patron d’activation des plasmocytes générés in vitro………61
Figure 5.25 : Expression de CD31 et CD39 sur les lymphocytes B dans la phase d’expansion………62
Figure 5.27 : Expression de CD31 et CD39 sur les plasmocytes in vivo………..………64 Figure 5.28 : Expression de CD31 et CD39 sur les plasmocytes………65 Figure 7.1 : Modèle proposé pour l’évolution de la différenciation des lymphocytes B mémoires en
Liste des abréviations
ACD : Acid–Citrate-DextroseAPRIL: A Proliferation Inducing Ligand ARN: Acide ribonucléique
ARNm: Acide ribonucléique messager BAFF-R : B cell activating factor receptor BAFF: B-cell activating factor
Bcl-2 : B-cell lymphoma protein 2 Bcl-6 : B-cell lymphoma protein 6 BCMA: B-cell maturation antigen
BCR: B-cell receptor
Blimp-1 : B-lymphocyte-induced protein 1 BSA : Bovine serum albumin
CAR cells : CXCL12-abundant reticular cells
CD: Cluster of differentiation, marqueurs de surface CMH : Complexe majeur d’histocompatibilité CSH: Cellule souche hématopïétique CXCL: CXC chemokine ligand CXCR: CXC chemokine receptor DMSO : Diméthysulfoxyde
DZ : Dark zone
ELISA: Enzyme-linked immunosorbent assay ELISPOT: Enzyme-linked immunosorbent spot
ENTPD1, NTDPase-1 : ecto-nucleoside triphosphate diphosphohydrolase-1 ERK1-2 : Extracellular signal-regulated kinases
FBS : Fetal bovine serum FCB : Fluorescent cell barcoding
G-CSF: Granulocyte-Colony Stimulating Factor ICOS : inducible co-stimulator
Ig: Immunoglobuline
IGF-1 : Insulin-like growth factor IL : Interleukine
iNOS : inducible-nitric oxide synthase IRF: Interferon-regulatory factor JAK : Janus tyrosine kinase
LFA-1 : Leukocyte function-associated antigen-1 LPS : Lipopolysaccharide
LZ : Light zone
MALT : Mucosa-associated lymphoid tissues MAPK : Mitogen-activated protein kinases MFI: Median fluorescence intensity NF-B : Nuclear factor-B
NK: Natural Killers
Pax5 : Paired protein box 5
PBMC: Peripheral blood mononuclear cells PBS: Phosphate buffered saline
PE : Phycoerythrin
PECAM-1 : Platelet-endothelial cell adhesion molecule-1 PSG-1 : P-selectin glycoprotein ligand-1
RPMI : Roswell Park Memorial Institute SDF-1 : Stromal derived factor-1
STAT : Signal transducer and activator of transcription
TACI: Transmembrane activator and calcium modulator and cyclophilin ligand interactor
TGF : Tumor growth factor
TMB : Tétraméthylbenzidine TNF : Tumor necrosis factor
TRANCE : Tumor-necrosis factor-related-TNF- activation-induced cytokine Treg : T régulateurs
UPR : Unfolded protein response
VEGF : Vascular endothelial growth factor VLA-4 : Very late activation antigen-4 XBP-1 : X-box binding protein
Remerciements
Déjà deux ans qu’a débuté ma belle aventure chez Héma-Québec. Je profite de cette occasion pour remercier toutes les personnes qui ont contribué de près ou de loin à la réussite de ce projet de maitrise. Je tiens tout d’abord à dire un énorme merci à ma directrice Dr Sonia Néron, pour m’avoir acceptée dans son laboratoire, tout d’abord comme stagiaire en 2010 et ensuite comme étudiante graduée. Je lui suis reconnaissante pour sa grande disponibilité ainsi que pour nos discussions si enrichissantes. Je la remercie également du fond du cœur pour son grand intérêt non seulement pour mon projet de maitrise, mais aussi pour mes projets futurs et de m’avoir donné l’occasion d’accomplir plusieurs tâches qui me seront d’une grande utilité dans ma carrière scientifique.
Je tiens à remercier chaleureusement mon co-directeur Dr André Darveau pour ses conseils, ses commentaires ainsi que sa disponibilité qui m’ont été fort utiles durant ma maitrise et qui le seront surement dans le futur. Je remercie également les autres membres de mon comité d’encadrement, soit les Drs Renée Bazin et Daniel Grenier pour leur disponibilité, leurs commentaires constructifs, ainsi que l’intérêt qu’ils ont montré envers mon projet.
Je voudrais également remercier la formidable équipe du Dr Néron, qui a rendu mon séjour chez Héma-Québec si enrichissant et agréable, soit Marc Cloutier, Carl Simard, Guillaume Bonnaure, Catherine Gervais-St-Amour ainsi que Jennifer Lemieux. J’ai grandement apprécié votre esprit d’équipe, votre disponibilité ainsi que votre support. Je tiens également à remercier Annie Roy et Josiane Tremblay Rochette de m’avoir initiée à la culture cellulaire et appris les rudiments de nombreuses méthodes d’immunologie.
Je remercie également tout le personnel d’Héma-Québec pour leur gentillesse et disponibilité durant tout mon parcours au sein de l’organisme. J’ai une pensée particulièrement pour les étudiants de la R&D, que je remercie pour les discussions aussi enrichissantes qu’hilarantes.
Je ne peux terminer sans remercier ma famille et mes amis, d’ici ou de l’autre côté de l’Atlantique, pour leur support constant et inconditionnel depuis le début de cette aventure.
1. Introduction
1.1 Les lymphocytes B humains
1.1.1 Développement et transition
La réponse immunitaire humorale est la ligne de défense de l’hôte faisant intervenir les anticorps ou immunoglobulines présents dans le plasma et la lymphe. Elle permet une protection contre les agents pathogènes et implique les lymphocytes B qui sont les seules cellules capables de se différencier en cellules sécrétrices d’anticorps. Chez les humains, le développement de ces cellules se déroule dans la moelle osseuse, à partir des cellules progénitrices lymphoïdes [1, 2]. Cette différenciation des cellules souches donne lieu aux pro-B. Un réarrangement réussi des segments des gènes des chaines lourdes (H) des immunoglobulines (Ig) permet à ces cellules pro-B de devenir des cellules pré-B. Ces dernières expriment la forme transmembranaire de la chaine lourde d’immunoglobuline (M). L’expression de cette protéine constitue le premier point de contrôle pour la sélection des lymphocytes B sains. Les cellules ayant passé ce point subissent une expansion clonale et le réarrangement des chaines légères des Ig [1, 3]. Elles acquièrent aussi l’expression à leur surface d’un BCR (B cell receptor) complet de type IgM. Les cellules subissent ensuite un second point de contrôle constitué d’une sélection négative des cellules qui auraient à leur surface des IgM reconnaissant des auto-antigènes. À ce stade, les cellules auto-réactives peuvent être éliminées, se retrouver dans un état anergique ou subir une réédition de leur récepteur. Dans ce dernier cas, elles subissent à nouveau des réarrangements génétiques permettant de modifier leur récepteur afin que celui-ci ne reconnaisse plus d’anticorps du soi [4]. À la suite de cette étape, les lymphocytes B, dits immatures ou transitionnels, quittent la moelle osseuse vers la rate, où ils vont terminer leur maturation. Ces cellules dans la circulation sanguine représentent environ 2 % des lymphocytes B [5]. Une fois arrivées à destination, elles subissent des étapes supplémentaires de maturation en plus d’acquérir l’expression d’IgD [6, 7]. Pendant ces étapes, elles peuvent encore subir des sélections négatives afin d’écarter les cellules autoréactives. Une partie de ces cellules demeure dans la zone marginale de la rate en tant que cellules naïves non circulantes tandis que l’autre se développe en lymphocytes B matures naïfs (folliculaires) circulant dans le sang à travers les ganglions lymphatiques, jusqu’à ce qu’elles rencontrent un antigène spécifique à leur BCR [3, 8].
1.1.2 Maturation des lymphocytes B et rencontre de l’antigène
Les cellules B naïves qui entrent dans un ganglion lymphatique migrent vers la zone des lymphocytes T et y restent environ 24h, avant de retourner dans la circulation sanguine [9] si elles ne rencontrent pas d’antigène pour lequel leur BCR est spécifique. Un lymphocyte B naïf peut ainsi rester en circulation de 80 à 120 jours,
après quoi il mourra par apoptose [10]. La rencontre de l’antigène se fait dans les ganglions lymphatiques [3]. Les antigènes de petite taille (<70 kDa) sont acheminés vers les follicules des ganglions lymphatiques à travers des canaux folliculaires. Les petites molécules peuvent aussi entrer dans les follicules à travers les espaces au niveau du sinus du ganglion et se lier par la suite aux lymphocytes B réactifs. Un autre modèle de contact implique les molécules plus grosses (>70 kDa). Celles-ci sont présentées aux lymphocytes B par des macrophages de la région subcapsulaire du sinus des ganglions lymphatiques. Il a aussi été suggéré que les cellules dendritiques circulantes peuvent transporter des antigènes dans les organes lymphoïdes secondaires [11][12][13][14].
1.1.3 Activation extrafolliculaire des lymphocytes B
Les lymphocytes B se trouvant dans la région extrafolliculaire des ganglions lymphatiques sont activés indépendamment des lymphocytes T. Les antigènes qui sont impliqués dans ce type de réaction sont appelés des antigènes T indépendants. Ils sont divisés en deux catégories, soit les types 1 et 2 [1][15]. Les premiers agissent indépendamment du BCR et induisent la prolifération et la différenciation des cellules B. Le LPS (lipopolysaccharide) retrouvé dans la membrane externe des bactéries à Gram négatif fait partie de ce groupe d’antigènes [16]. Le deuxième type d’antigène T-indépendants agit à travers le BCR. Ces antigènes sont souvent de haut poids moléculaire. Il peut s’agir notamment de polysaccharides provenant de la paroi cellulaire de bactéries encapsulées telles que le Streptococcus pneumoniae [17]. La différenciation induite par ces antigènes est plus complexe et nécessite plusieurs signaux en plus de la multimérisation du BCR. Ces signaux proviennent notamment de la reconnaissance de l’antigène à la surface des lymphocytes B par des molécules telles que les TLR (Toll-like receptors). L’antigène T indépendant de type 2 peut aussi induire la sécrétion de cytokines par d’autres cellules de l’environnement du lymphocyte B. Ces facteurs solubles agissent sur la prolifération et la différenciation de la cellule B. La survie et la différenciation des cellules B se trouvant dans la zone extrafolliculaire sont dépendantes de la présence de facteurs tels que BAFF (B cell
activating factor), sécrété notamment par les cellules dendritiques [18]. Les lymphocytes B naïfs rencontrant
ces types d’antigènes prolifèrent rapidement et se différencient tout aussi rapidement en cellules sécrétrices d’anticorps. Le stade final de différenciation des cellules B est le plasmocyte. Dans le cadre de l’activation T-indépendante, les plasmocytes générés sont dits à courte vie et sécrètent des anticorps, surtout de type IgM, ayant une faible affinité pour l’antigène. Une petite population de ces plasmocytes sécrète aussi des IgG [11][1].
1.1.4 Activation folliculaire et formation du centre germinatif
La plupart des antigènes induisant la production d’anticorps ont besoin de la collaboration des lymphocytes T. Ces antigènes sont connus comme antigènes T dépendants et sont de nature protéique [15]. Les lymphocytes B dans les follicules sont activés par la présentation antigénique par des macrophages du sinus subcapsulaire
des ganglions lymphatiques [19]. Les cellules dendritiques peuvent aussi présenter des antigènes aux lymphocytes B naïfs [20]. En fait, dans les ganglions lymphatiques, on retrouve deux régions : l’une concentrée en lymphocytes T (zones des T) et l’autre en lymphocytes B (zones des B) [21]. Les lymphocytes sont activés dans leur zone respective par la rencontre d’une cellule présentatrice d’antigène (CPA) présentant à sa surface l’antigène spécifique au lymphocyte T à travers son complexe d’histocompatibilité de type II. Les cellules T CD4+ ainsi activées prolifèrent et expriment le récepteur de chimiokine CXCR5. Cela a
comme conséquence d’attirer ces cellules à l’interface de la zone des lymphocytes B. Ces derniers, une fois qu’ils ont été activés par le même antigène dans leur zone respective, expriment à leur tour le récepteur de chimiokine CXCR7 et se retrouvent à l’interface des deux zones [21][15]. Suite à leur activation, les deux types de lymphocytes expriment plusieurs molécules de surface, permettant une meilleure interaction lors de leur rencontre. Les lymphocytes T expriment le CD154 ou encore ICOS (inducible co-stimulator). Ils peuvent aussi sécréter des cytokines telles que l’interleukine (IL-4), l‘interféron- ou encore l’IL-21 [22][23] (Fig. 1.1). C’est à cet endroit qu’a lieu la rencontre des lymphocytes B et T activés par un antigène spécifique. L’interaction avec les lymphocytes T induit l’activation des lymphocytes B. Ces cellules peuvent ensuite rester dans la zone extrafolliculaire ou former un centre germinatif. Cette décision est dépendante de la force d’interaction entre l’antigène et le BCR. Une forte liaison entre le BCR et l’antigène induit une différenciation des lymphocytes B en dehors du centre germinatif tandis que les lymphocytes ayant une plus faible affinité pour l’antigène formeront un centre germinatif [24].
1.1.5 Formation du centre germinatif
Le centre germinatif est une structure transitoire et dynamique dans laquelle on retrouve des lymphocytes B, T ainsi que des cellules dendritiques folliculaires [25]. Des études ont aussi révélé la présence de lymphocytes B naïfs dans cette structure [26].
Figure 1.1: Interaction cellulaire entre les lymphocytes T activés et cellules B avant le centre germinatif. Cette figure a été traduite de [27] où TAF sont les lymphocytes T auxiliaires folliculaires et GC est
le centre germinatif.
L’interaction de l’ensemble de ces cellules à travers des interactions cellulaires ainsi que la sécrétion de divers facteurs solubles induit la prolifération et la différenciation des lymphocytes B présents. Parmi ces interactions, on retrouve celui entre le récepteur CD40, exprimé par les lymphocytes B et son ligand CD154 exprimé par les lymphocytes T activés. Cette interaction est d’autant plus importante qu’il a été rapporté que les personnes ayant une mutation dans la molécule CD154 ou CD40 étaient incapables de former des centres germinatifs, menant au syndrome d’hyper IgM (X-linked hyper IgM syndrome) [28]. De plus, l’interaction entre le CD27 et le CD70 est aussi connue comme ayant un rôle dans la formation du centre germinatif et la différenciation des lymphocytes B [29][30].
La structure du centre germinatif est composée de deux régions, soit la zone claire (Light zone, LZ) et la zone foncée (dark zone, DZ). Cette dernière est située près de la zone des cellules T. On y retrouve principalement des lymphocytes B en prolifération, appelés centroblastes. Il a été rapporté que c’est à cet endroit que les cellules subissent des hypermutations somatiques ayant pour but d’augmenter leur spécificité pour l’antigène. Le cycle cellulaire de ces cellules est par la suite arrêté et elles migrent vers la zone claire. À cet endroit, les lymphocytes B testent leur capacité à reconnaitre de manière spécifique l’antigène en question. Les cellules subissent une sélection négative pour le recrutement de celles ayant la meilleure affinité pour l’antigène. Les cellules dont le BCR ne reconnait pas l’antigène reçoivent des signaux induisant leur apoptose [31][32]. Il a été rapporté récemment que les cellules pouvaient circuler entre la zone claire et foncée [25]. En effet, suite à la sélection positive dans la zone claire, les cellules favorisées retournent dans la zone foncée, après leur interaction avec des lymphocytes T auxiliaires, où elles subissent une seconde ronde de prolifération et
Cellule TAF pré-GC Cellule B activée
Récepteur de cytokine
Peptide CMH-II
d’hypermutation somatique [25][31]. Ce processus de sélection favorise la production de lymphocytes ayant une très grande affinité pour l’antigène et à la sortie du centre germinatif, on retrouve des plasmablastes CD38++, CD138+/- et des lymphocytes B mémoires (CD19+, CD27+, CD38+/-) [33][34].
Figure 1.2 : Le centre germinatif. Résumé de différentes étapes menant à la génération de lymphocytes B
mémoires et de plasmablastes. Tirée de [27].
1.1.6 Différenciation terminale des lymphocytes B
Les plasmocytes représentent le stade final de différenciation des lymphocytes B en cellules sécrétrices d’anticorps [11][1]. Cette différenciation débute par une activation du gène PAX5 (Paired Box Protein 5). Cette étape marque un état de développement permettant l’expression de quelque 170 gènes nécessaires à la signalisation, à la différenciation, à la migration et à l’adhésion des lymphocytes B matures [1][33, 35]. Plusieurs facteurs de transcription sont aussi impliqués dans cette progression complexe dont les facteurs IRF4, IRF8, STAT3, STAT5, Blimp-1 et Bcl-6. Ces changements sont induits dès l’initiation du centre germinatif et à la sortie, on retrouve les lymphocytes B à mémoire et les plasmablastes sécrétant des anticorps, mais qui, contrairement aux plasmocytes, sont capables de proliférer [15, 36]. Une partie de cette population de plasmablastes se différenciera en plasmocytes à courte vie tandis qu’une autre migrera vers des niches de survie se trouvant notamment dans la moelle osseuse et dans les organes lymphoïdes secondaires où ils deviendront des plasmocytes à longue vie. Ces plasmocytes à longue vie survivront durant plusieurs années ou même la vie entière de l’individu [37, 38] grâce à des facteurs solubles ou des interactions intercellulaires se trouvant dans leur environnement. Cette séquence assure donc une production maximale d’anticorps au pic de la réponse immunitaire tout en maintenant une concentration protectrice d’anticorps dans le sérum [39][40].
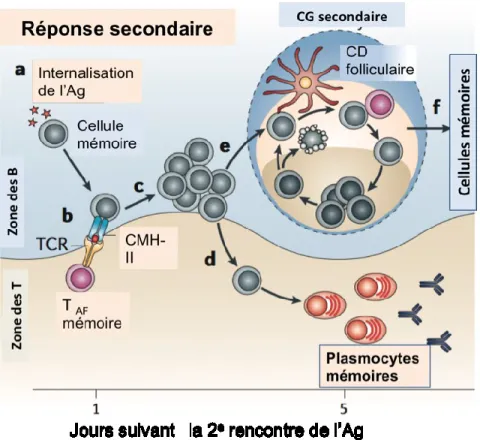
1.1.7 Réponse immunitaire secondaire
Comme il a été mentionné précédemment, en plus de la génération de cellules sécrétrices à la sortie du centre germinatif, les lymphocytes B se différencient aussi en cellules mémoires. Ces dernières sont en fait des cellules non sécrétrices, mais ayant une forte affinité pour l’antigène étant donné qu’elles ont subi les différentes mutations somatiques dans le centre germinatif pour améliorer leur affinité. Suite à leur sortie du centre germinatif, ces cellules demeurent en périphérie du centre germinatif ou se retrouvent dans la circulation sanguine [41][42]. Tel que résumé à la figure 1.3, lors d’une seconde rencontre avec l’antigène, ces cellules l’internalisent rapidement et le présente dans le contexte de leur CMH-II aux lymphocytes T mémoires préalablement activés. Cette interaction induit la prolifération massive des lymphocytes B. Une partie de ces cellules forme un centre germinatif (CG secondaire) comme lors de la première rencontre de l’antigène tandis que l‘autre subit une différenciation extrafolliculaire en cellules sécrétrices d’anticorps [27].
Figure 1.3: Réponse des lymphocytes B mémoires lors de la réaction immunitaire secondaire. Image
traduite de [27], où CG est le centre germinatif et CD sont les cellules dendritiques.
1.1.8 Les populations de lymphocyte B du sang
Les lymphocytes B représentent environ 5 à 15 % des lymphocytes du sang. Ils sont caractérisés par la présence d’immunoglobulines à leur surface qui sont les récepteurs spécifiques à un antigène. Chez l’humain, les lymphocytes B du sang se divisent en deux sous-populations : soit les lymphocytes B naïfs (60 %) et les lymphocytes B mémoires (40 %) [43]. Ces deux populations sont hétérogènes et se distinguent toutefois par la
présence de marqueurs de surface. Les B naïfs sont identifiables par leur phénotype IgD+IgM+CD27- tandis
que les B à mémoire peuvent être de type IgD+IgM+CD27+, IgD-IgM+CD27+ ou suite à un changement de
classe, IgG+/IgA+CD27+ [43]. De plus, des études ont démontré l’existence d’une sous-population,
représentant environ 20 %, de lymphocytes B à mémoire IgG+ ou IgA+, « switched-memory », n’exprimant pas
le CD27 [44]. La présence des cellules mémoires IgE est longtemps demeurée un mystère. Un modèle chez la souris avait été proposé selon lequel, ces cellules proviendraient de la différenciation supplémentaire des cellules mémoires IgG1+ [45]. Une étude récente a toutefois été capable de démontrer la présence de cellules
mémoires IgE+ dans le centre germinatif, dont la présence n’avait aucun lien avec des cellules IgG1+ [46, 47].
1.2 Les plasmocytes
Les plasmocytes sont au cœur de la réaction immunitaire humorale, car il s’agit des cellules sécrétrices d’anticorps. Leurs précurseurs, les plasmablastes, sécrètent aussi des anticorps tout en gardant la capacité de proliférer [48][49]. Dans la circulation, on retrouve très peu de ces cellules, soit environ 2/L [50]. La moitié (49 %) des plasmocytes et plasmablastes se trouvant dans le sang ont des IgA à leur surface. La fréquence des cellules IgM+ est de 18 % et seulement 11 % expriment les IgG. Environ 14 % des plasmocytes et
plasmablastes en circulation n’ont aucune immunoglobuline à leur surface [47, 50, 51]. La majorité des plasmocytes se retrouvent dans la muqueuse gastro-intestinale, où 80 à 90 % des cellules sécrétrices d’anticorps produisent des IgA tandis que le reste produit des IgM et des IgG. Ce dernier est sécrété par 0 à 2 % des cellules dans cette région [52]. En plus de la sécrétion d’immunoglobulines, il a récemment été rapporté que les plasmocytes IgA+ des muqueuses pouvaient sécréter des molécules pro-inflammatoires telles
que le TNF- [tumor-necrosis factor-] et iNOS [inducible-nitric oxide synthase], afin d’assurer l’homéostasie
au niveau des muqueuses de l’intestin [53]. Le reste des cellules sécrétrices sont localisées dans les organes lymphoïdes et la moelle osseuse.
1.2.1 Les immunoglobulines
Les immunoglobulines sont formées par l’assemblage de deux chaines légères de type kappa ou lambda et de deux chaines lourdes de types , , , ou , reliées par des ponts de disulfure et donnant respectivement les isotypes : IgG, IgA, IgM, IgD et IgE. Chez l’humain, les IgG forment environ 85 % des immunoglobulines présentes dans le sang d’un adulte alors que les IgA et IgM composent chacun entre 5 à 10 %, le reste étant des IgE et des IgD en quantités négligeables (<1 %) Les IgG ont une très grande affinité pour l’antigène et ont une demi-vie de 23 jours. Les IgG peuvent être divisées en sous-classes soit IgG1, IgG2, IgG3 et IgG4 où les IgG1 sont les plus abondants [54-56]. Les IgA quant à eux sont les immunoglobulines prédominantes dans les muqueuses. On retrouve deux sous-classes de cette immunoglobuline, soit les IgA1 et IgA2, dont la fréquence
ou l’autre d’immunoglobuline dépend des signaux qu’ils reçoivent de leur environnement lors de leur différenciation, que ce soit dans le centre germinatif ou dans la zone extrafollicullaire [58]. L’IL-4 est connu pour réguler le changement de classe des lymphocytes B vers IgG1 et IgE. L’interféron- quant à lui stimule le changement de classe vers IgG2 [59]. L’IL-21 promeut l’apparition de cellules la classe IgG1 et IgG3 [60] tandis que l’IL-10 est un facteur important pour le changement en IgG1 et IgG2 [61]. Le changement de classe vers IgA est favorisé par la présence de TGF [62].
1.2.2 Le phénotype des lymphocytes B différenciés
Les lymphocytes B, les plasmablastes et les plasmocytes possèdent à leur surface plusieurs marqueurs, dont les CD « clusters of differentiation », les molécules d’adhésion ou les récepteurs de chimiokines qui permettent de les distinguer les uns des autres. Le phénotype des plasmocytes est souvent basé sur la présence de CD138. À ce marqueur s’ajoute une expression élevée de CD27 et CD38 et une faible expression de CD19 [50]. Cependant, plusieurs autres marqueurs dont CD20, CD31, CD39, CD126 et les récepteurs de chimiokines CCR2 et CCR1 peuvent aider à les distinguer des cellules matures et des plasmablastes [50][63][64][65]. Les méthodes de cytométrie en flux permettant la visualisation de plusieurs marqueurs de surface en simultanée ont fait avancer nos connaissances du phénotype des plasmocytes du sang, des organes lymphoïdes et de la moelle osseuse, mais ne permettent cependant pas encore de distinguer les plasmocytes à courte vie des plasmocytes à longue vie. Cependant, il existe tout de même des marqueurs intéressants pour la distinction des plasmocytes des autres populations de lymphocytes B.
1.2.2.1 CD138
Jusqu’à présent, le CD138 [syndecan-1 ou SDC-1] est l’unique marqueur de surface permettant la reconnaissance de plasmocytes matures sains ou malins [66], bien que l’existence de plasmocytes CD138- ait
été rapportée dans la littérature [50]. Il s’agit d’un protéoglycane transmembranaire de type 1, dont la majorité du poids moléculaire provient des molécules de sulfates d’héparane qui y sont liées. Cette molécule joue un rôle dans l’adhésion cellulaire en provoquant une agrégation homotypique des cellules [67]. En plus de cela, le CD138 est un facteur dans plusieurs voies de signalisation où il se lie à des facteurs liant des sulfates d’héparane, ce qui a pour conséquence d’augmenter la disponibilité de ces molécules pour la cellule l’exprimant. Plusieurs molécules connues comme étant importantes pour la survie ou l’engagement des lymphocytes B dans la voie lymphoïde telles que le CXCL12, le CXCL13, l’IL-4 et l’IL-6 possèdent des domaines de liaison aux protéoglycanes de sulfate d’héparane [68][69][70]. Récemment, il a été montré que APRIL (A Proliferation Inducing Ligand) était aussi séquestrée par le CD138. En fait, le CD138 agirait comme plateforme de multimérisation de cette molécule et faciliterait ainsi son cross-linking avec les récepteurs de cette dernière [71].
Ces caractéristiques du CD138 assurent également la survie de plasmocytes cancéreux dans le cas des myélomes multiples [68]. Il s’agit d’un cancer incurable caractérisé par une expansion non contrôlée des plasmocytes au niveau de la moelle osseuse [72]. Ces cellules cancéreuses perturbent l’homéostasie de la moelle osseuse et sécrètent diverses cytokines et facteurs solubles indispensables pour leur survie, leur prolifération et leur rétention dans la moelle osseuse tels que l’IL-6, IGF-1 (Insulin-like Growth Factor), TRANCE (Tumor-necrosis Factor-related-TNF-activation-induced cytokine), VEGF (Vascular Endothelial
Growth Factor), TNF, CXCL12, APRIL ou BAFF [68]. Ces molécules agissent en induisant l’activation de diverses voies de signalisation à l’intérieur des cellules comme NFB, AKT, ou encore les MAPK [73][68]. D’ailleurs, une étude récente porte sur le diagnostic de cette maladie à travers la capacité des cellules cancéreuses à répondre à divers stimuli par l’activation de certaines voies de signalisation [74].
1.2.2.2 CD31 et CD39
Le CD31 ou PECAM-1 (Platelet-endothelial cell adhesion molecule-1) est une glycoprotéine appartenant à la superfamille des Ig. Il est impliqué dans plusieurs processus tels que la migration cellulaire, l’angiogenèse et l’adhésion cellulaire [75]. Il accomplirait notamment ces fonctions par sa liaison au CD38 qui est un de ces ligands [76]. Il a aussi été montré que cette molécule pourrait être un acteur important dans la régulation de l’activation cellulaire induite par un antigène chez les lymphocytes [77]. Il est exprimé par les plaquettes, les neutrophiles, les monocytes, les lymphocytes T naïfs ainsi que les cellules endothéliales [78]. Ce marqueur est aussi retrouvé sur les lymphocytes B naïfs et les plasmocytes dans les amygdales humaines [75, 79].
Le CD39 (ENTPD1, NTDPase-1, ecto-nucleoside triphosphate diphosphohydrolase-1) quant à lui est une ectoenzyme membranaire métabolisant l’ATP et l’ADP en AMP [80][81][82]. Il est retrouvé à la surface des cellules NK (Natural Killers), de certaines sous-populations de lymphocytes T, sur des cellules dendritiques, des macrophages ainsi que des cellules endothéliales vasculaires [82]. Chez les leucocytes, cette molécule est connue comme ayant un rôle dans la modulation de sécrétion de cytokines, dans la réponse inflammatoire ainsi que dans l’adhésion cellulaire [83]. Une étude menée sur les lymphocytes du sang de volontaires adultes en santé a démontré la présence de CD39 sur 94.5 % des lymphocytes B en circulation [83].
Une autre étude menée par plusieurs groupes travaillant sur les plasmocytes a décortiqué à l’aide du microarray, la présence différentielle d’ARN messager de plusieurs molécules exprimées par les populations de lymphocytes B différenciés, soit les B mémoires, les plasmablastes ainsi que les plasmocytes [64]. Ces groupes ont répertorié le niveau d’expression de plusieurs dizaines de cibles incluant des facteurs de transcription, des marqueurs de surface, des récepteurs de chimiokines, des sélectines et des molécules d’adhésion, des facteurs de croissance et des récepteurs de facteurs de croissance, des marqueurs d’apoptose ainsi que des gènes liés à la réponse de protéines mal repliées (UPR, unfolded protein response). Cette étude exhaustive a montré la présence différentielle des marqueurs de surface CD31 et CD39 dans les lymphocytes B mémoires, les plasmablastes et les plasmocytes. L’ARNm de CD31 était en plus grande
quantité dans les plasmocytes comparativement aux autres sous-populations. Quant au CD39, la quantité relative d’ARNm codant pour cette protéine était la plus élevée dans les plasmablastes [64].
Malgré l’identification de plusieurs marqueurs tels que ceux décrits ci-haut, la distinction entre les plasmocytes à longue vie et à courte vie demeure toujours un mystère.
1.2.3 Plasmocytes à courte vie
Tel que décrit plus haut (section 1.1.6), cette sous-population de plasmocytes peut provenir de la réponse T dépendante ou indépendante, sécrétant des anticorps ayant beaucoup ou peu d’affinité pour l’antigène. Les plasmocytes générés lors de la réponse T-indépendante sécrètent principalement des anticorps de type IgM, ayant peu d’affinité pour l’antigène [3][84]. Ces cellules demeurent principalement dans les ganglions lymphatiques, près de leur lieu de formation [3]. La principale caractéristique de ces cellules est leur grande capacité de sécrétion d’immunoglobulines. Cette grande sécrétion et donc de production de protéines induit un stress au niveau du réticulum endoplasmique des cellules et une réponse de protéine mal repliée (UPR,
unfolded protein response) chez les cellules en activant des facteurs de transcription tels que XBP-1, ce qui
résulte en l’apoptose de la cellule [85]. Une étude récente a démontré qu’il y avait un blocage actif des caspases apoptotiques, ce qui avait pour conséquence d’assurer la survie des plasmocytes à courte vie face à la réponse du réticulum endoplasmique. Ce même mécanisme permet donc la régulation de la durée de vie des plasmocytes à courte vie [86].
1.2.4 Les plasmocytes à longue vie
Une autre sous-population de cellules sécrétrices d’anticorps émerge lors de la réponse T dépendante, soit les plasmocytes à longue vie. Le centre germinatif donnant lieu à l’émergence des cellules mémoires et des plasmablastes, une partie de ces cellules se différencie en plasmocytes à courte vie tandis que l’autre prend la circulation sanguine, à la recherche d’une niche où ils pourront survivre. La quantité de cellules générées de cette population à la suite de la réaction du centre germinatif est estimée entre 10 à 20 % [35]. Ces cellules, malgré le stress endoplasmique causé par la sécrétion pouvant aller jusqu’à 10 000 à 20 000 molécules d’immunoglobulines par seconde, réussissent tout de même à survivre plusieurs années et parfois même, toute la vie de l’individu [87][88]. Ainsi, les plasmocytes à longue vie continuent à sécréter des anticorps bien après la rencontre de l’antigène afin de garder un niveau d’anticorps protecteur dans le sérum. Un bel exemple de ce phénomène se trouve dans une étude sur le suivi de la vaccination. En effet, la persistance ans le sang d’un niveau protecteur d’anticorps spécifiques contre le tétanos est de 11 ans, de 19 ans contre la diphtérie et de 3013 ans pour la rougeole [37]. Plusieurs modèles ont été proposés pour expliquer la capacité de sécrétion des plasmocytes durant d’aussi longues périodes. Le modèle le plus accepté est basé sur la compétition pour les niches de survie [37][39]. En effet, pour assurer leur survie, les plasmocytes doivent se retrouver dans un microenvironnement spécialisé qui se trouve dans des organes précis tels que la moelle
osseuse. Ce type d’organe fournit au plasmocyte un nid protecteur. Étant donné que chaque nouvelle infection menant à l’émergence de plasmocytes à longue vie génère de 10 000 à 100 000 cellules, l’espace dans la moelle osseuse devient un facteur limitant pour la survie de ces cellules sécrétrices [15]. Ce modèle propose donc une compétition entre les nouveaux plasmablastes et les plasmocytes résidents des niches. Les plasmocytes ainsi dégagés se retrouvent dans la circulation sanguine et finissent par mourir, faute d’environnement adéquat [37][15][89].
La biologie des plasmocytes soit encore peu connue, plusieurs études sont menées afin de déterminer des marqueurs de surface propres à ces cellules ou d’autres caractéristiques qui pourraient les distinguer des autres populations de lymphocytes B [90]. Il a récemment été rapporté que le CD28, bien qu’exprimé sur les plasmocytes à courte et longue-vie, était un facteur important pour la survie des plasmocytes à longue vie de la moelle osseuse [91].
Les niches permettant la survie des plasmocytes à longue vie se trouvent dans les tissus lymphoïdes associés aux muqueuses (MALT, mucosa-associated lymphoid tissues), dans les amygdales ainsi que dans la moelle osseuse [92][93][94]. Les plasmocytes et les niches retrouvés dans la moelle osseuse sont cependant les plus étudiés étant donné que cet organe contient 80 à 90% de la réserve de plasmocytes dans le corps [15].
1.2.5 Niche de survie des plasmocytes
1.2.5.1 Composition des niches
La moelle osseuse est à la croisée des chemins du sang et de l’immunité [95]. Malgré qu’il s’agisse du site par excellence où on retrouve des plasmocytes, ces cellules ne représentent pas plus de 0,5-1 % des cellules mononuclées de cet organe [15][39][96][65][97]. Il a été démontré que la sécrétion de la chimiokine CXCL12 au niveau de la moelle osseuse avait pour cause d’attirer les plasmablastes nouvellement générés dans la moelle osseuse [98]. La niche des plasmocytes est en fait un microenvironnement, où la synergie de plusieurs facteurs dont les cytokines, chimiokines, et molécules d’adhésion assurent la survie de ces cellules centrales de la mémoire immunologique humorale [99]. La composition des niches des plasmocytes est illustrée dans la figure 1.4. Les plasmocytes de la moelle osseuse sont en contact direct avec les cellules stromales [3][100][101] qui sécrètent la chimiokine CXCL12, dont le ligand CXCR4 est exprimé par les plasmocytes [100]. Les plasmocytes sont aussi retrouvés à proximité dès des cellules CAR (CXCL12-abundant reticular
cells), sécrétrices de CXCL12. Les souris déficientes en CXCR4 ont une déficience dans la migration de leur
plasmablastes vers la moelle osseuse [102] ce qui démontre le rôle clé de cette chimiokine dans le microenvironnement des plasmocytes. Il a aussi été démontré que les éosinophiles et les mégacaryocytes faisaient également partie de la niche des plasmocytes. Le rôle de ces cellules consiste principalement en la sécrétion des facteurs de survie tels IL-6 et APRIL. Plusieurs autres cellules telles que les ostéoclastes, les ostéoblastes, les monocytes, les neutrophiles ainsi que les cellules épithéliales joueraient un rôle dans les
niches des plasmocytes de la même façon à travers la sécrétion d’IL-6, d’APRIL ou encore de BAFF [95][103][101]. En plus de la présence de ces différents facteurs solubles, les plasmocytes possèdent à leur surface plusieurs molécules d’adhésion telles que VLA-4 (very late activation antigen-4), LFA-1 (leukocyte
function-associated antigen-1), l’antigène CD44 ou encore PSG-1 (P-selectin glycoprotein ligand-1), qui leur
permettent d’interagir avec les cellules présentes dans ce microenvironnement et d’assurer leur survie [103][104].
Figure 1.4 Niche de survie des plasmocytes à longue vie. Cellules, CAR (CXCL12-abundant reticular cells)
cellules réticulaires sécrétrices de CXCL12; MGK : mégacaryocytes. Figure traduite de [101][103]. 1.2.5.2 Principaux facteurs de survie des plasmocytes
APRIL est l’une des plus importantes molécules connues pour la survie des plasmocytes. Cette molécule de la famille des TNF est sécrétée dans la zone sous-épithéliale des muqueuses lors d’une infection et de façon constitutive dans la moelle osseuse où elle permet la survie des plasmocytes à longue vie [105][106]. Dans la moelle osseuse, cette molécule est sécrétée par de nombreuses cellules, dont les cellules dendritiques, les cellules endothéliales, les mégacaryocytes, les neutrophiles et les éosinophiles [101]. APRIL possède trois récepteurs, soit TACI (transmembrane activator, calcium modulator and cyclophilin ligand interactor), BCMA (B cell maturation antigen) et BAFF-R (B cell activating factor receptor) ; l’affinité de APRIL est cependant plus faible sur BAFF-R comparativement aux deux autres récepteurs [107][108]. APRIL a aussi été rapporté comme liant les sulfates d’héparane sur des molécules telles que le CD138 [71]. Le niveau d’expression des récepteurs de APRIL sur les lymphocytes B varie selon le degré de maturité de la cellule et son stade de différenciation. En effet, la progression des lymphocytes B vers l’état de plasmablaste et de plasmocyte, est caractérisée par une diminution de l’expression de TACI et de BAFF-R à leur surface contrastant avec une augmentation pour BCMA [108].
Dans le microenvironnement permettant la survie des plasmocytes, on retrouve aussi la chimiokine CXCL12 ou SDF-1 (Stromal derived factor-1) principalement produite par les cellules stromales de la moelle
osseuse, mais aussi par les ostéoblastes, les cellules endothéliales ainsi que les cellules réticulaires de la moelle osseuse [109]. Son interaction avec son récepteur principal (CXCR4) est connue pour être importante pour le développement des lymphocytes B, la colonisation de la moelle osseuse par les cellules souches hématopoïétiques ainsi que leur rétention à cet endroit [110]. La sécrétion et la concentration de CXCL12 dans cet organe sont contrôlées par le système nerveux central et suivent un rythme circadien qui permet la libération de certaines cellules souches dans la circulation sanguine [111]. Le CXCL12 possède également une grande affinité pour le CXCR7, mais sa liaison avec ce dernier ne semble pas induire une signalisation menant à la migration cellulaire induite telle qu’observée lors de sa liaison avec le CXCR4 [112].
Cette survie des plasmocytes au niveau de la moelle est primordiale pour le système immunitaire humoral et offre une protection pendant plusieurs années contre des pathogènes de toutes sortes. Leur habilité à sécréter des anticorps sans contact supplémentaire avec l’antigène en font des cellules ressources pour la protection des personnes immuno-supprimées.
1.3 Les plasmocytes et la thérapie cellulaire
1.3.1 Greffe de cellules souches hématopoïétiques
La transplantation de cellules souches hématopoïétiques (CSH) est une intervention médicale consistant en l’injection intraveineuse de cellules souches autologues ou allogéniques à un receveur. Indiqué autant chez les enfants que les adultes, ce traitement permet le rétablissement du système hématopoïétique chez les patients dont le système serait endommagé ou absent. Les cellules utilisées peuvent provenir de la moelle osseuse, du sang de cordon ombilical ou de la mobilisation de CSH dans la circulation sanguine [113]. En fait, la majorité des transplantations de cellules souches hématopoïétiques sont réalisées avec des cellules mobilisées, car elles induisent une meilleure reconstitution immunitaire en plus de faire appel à l’aphérèse comme processus de prélèvement, ce qui est moins invasif. [114]. Le G-CSF est injecté au donneur allogénique ou au patient 4 à 5 jours précédents le prélèvement [109] et peut augmenter la quantité de CSH dans la circulation sanguine de 44 fois [115]. Le G-CSF peut activer simultanément le système nerveux central et les macrophages de la moelle osseuse qui agiront de concert pour diminuer la sécrétion de CXCL12 dans la moelle osseuse [109]. Il peut aussi induire le clivage de CXCL12 et d'autres molécules d'adhésion par des protéases produites par des neutrophiles de la moelle osseuse activés par le G-CSF [116]. Cette action libère les CSH et induit aussi leur prolifération.
Avant la transplantation des CSH, le receveur peut subir plusieurs traitements permettant de réduire les risques de développement de la maladie du greffon contre l’hôte, le rejet de la transplantation ou encore l’éradication de la maladie pour laquelle la transplantation a lieu. Ces traitements consistent notamment en
l’irradiation complète du patient, en administration de médicaments chimiothérapeutiques ou les deux en même temps [117].
1.3.2 Les applications de la transplantation
La transplantation de CSH est indiquée principalement pour le traitement de maladies hématologiques. Les transplantations autologues sont indiquées pour des maladies telles que les myélomes multiples, les lymphomes hodgkiniens et non-hodgkiniens, la leucémie myéloïde aigue ou encore les neuroblastomes. Les transplantations allogéniques peuvent être aussi effectuées dans les maladies précédentes en plus des leucémies lymphatiques chroniques, d’anémie aplasique, de thalassémie, d’anémie drépanocytaire ou de désordres génétiques [113][118]. Le traitement est aussi indiqué pour les tumeurs solides telles que le cancer des testicules [119]. Une étude internationale d’envergure a révélé qu’il y a eu 50 417 transplantations en 2006, dont 43 % étaient des transplantations allogéniques et 57 % autologues. La majorité (54 %) des interventions effectuées était pour le traitement de désordres lymphoprolifératifs. Les leucémies représentaient 34 % des interventions alors que le traitement de tumeurs solides représentait 6 % des cas [113].
1.3.3 La reprise de greffe et la reconstitution du système immunitaire
Suite à la transplantation, les CSH doivent migrer vers la moelle osseuse du receveur. Ce processus implique plusieurs molécules dont les intégrines 4 et 5, le CD44 ainsi que le CXCL12 et son ligand CXCR4 [120]. Une
fois ce processus complété, la régénération du système hématopoïétique peut débuter. Selon la source des CSH (moelle osseuse, sang de cordon, sang périphérique), le délai de régénération des sous-populations cellulaires peut varier [121]. Dans le cas des cellules du système immunitaire inné, les neutrophiles sont les premières cellules à apparaitre, environ 14 jours après la transplantation de CSH du sang, 21 jours après la transplantation de cellules de la moelle osseuse et environ 30 jours suite à la transplantation de cellules de sang de cordon. La reconstitution totale des cellules NK (Natural Killers) a lieu 1 à 2 mois après la transplantation [118]. Les monocytes et les macrophages font ensuite leur apparition durant la même période [121]. Dans le cas de transplantations de cellules provenant du sang de cordon, la reconstitution des lymphocytes T peut prendre de 9 à 12 mois tandis que celle des lymphocytes B va de 3 à 6 mois [122]. La lymphopoïèse des cellules B requiert un environnement spécialisé, qui est souvent influencé dans la maladie du greffon contre l’hôte ou par les traitements pour contrer cet état [123]. D’ailleurs, la reconstitution complète de ces cellules peut prendre de 1 à 2 ans suivant la transplantation [121]. La présence d’anticorps du receveur plusieurs années après la transplantation de CSH a toutefois permis de prouver la capacité de chimio-radiorésistante des plasmocytes, plus précisément ceux résidents dans la moelle osseuse [124][125]. Ce sont ces plasmocytes qui sont à l’origine de la proportion de cellules observée à la figure 1.5.
Figure 1.5 Reconstitution des cellules immunitaires après une transplantation de CSH. La quantité
approximative des populations cellulaires est exprimée comme le pourcentage du compte cellulaire normal avant la transplantation et avant la radiation complète du patient. Figure traduite de [121].
1.3.4 Problèmes post-transplantation et vulnérabilité aux infections
Une fois la transplantation effectuée, plusieurs problèmes peuvent survenir, dont le développement de la maladie du greffon contre l’hôte, une rechute dans les cas de cancer ou encore des infections. La moitié des décès suivant une transplantation provenant de sang de cordon sont dus à des infections [126]. Dans les cas où il y a irradiation du patient avant la transplantation, cela peut endommager les surfaces des muqueuses du patient et donc laisser place à la libération de pathogènes commensaux dans la circulation sanguine [118]. Dans les 100 premiers jours suivant la transplantation, les patients sont surtout victimes d’infections virales et fongiques [117]. Pour ce qui est de l’immunité humorale, l’absence de lymphocytes B mémoires et d’immunoglobulines spécifiques expose les patients à des infections par des pathogènes tels que le
Streptococcus pneumonia ou encore Haemophilus influenzae [117]. Les patients greffés sont donc
vulnérables et plusieurs moyens sont envisagés afin de leur procurer une meilleure protection immunitaire.
1.3.5 Traitement des problèmes post-transplantations
Plusieurs traitements post-transplantation de thérapies cellulaires ont été développés durant les dernières années. Beaucoup de ces thérapies consistent en l’expansion ex vivo de cellules immunomodulatoires telles que les cellules T régulateurs (Treg), les cellules NK/ Treg, les cellules NK provenant du donneur, les cellules
souches mésenchymateuses ou encore au transfert de cellules T spécifiques à un antigène viral ou tumoral. Tous ces traitements visent l’amélioration de la reconstitution immunitaire ainsi que la protection face aux infections [117]. Dans le cadre des transplantations de CSH du sang de cordon, une thérapie consistant en
C el lu le s im m u n it ai re s (% d u c o m p te n o rm al )
Semaines Mois Années
Transplantation - Neutrophiles, monocytes, cellules NK - Cellules T CD4+ - Cellules B, T CD8+ - Plasmocytes, cellules dendritiques
l’injection de lymphocytes T provenant du sang de cordon a été proposée [127]. Les lymphocytes T mémoires sont aussi des ressources intéressantes pour la prévention des infections. Ces cellules sont expansionnnées in vitro en présence de cellules présentatrices d’antigènes ayant internalisé l’antigène spécifique pour laquelle les cellules mémoires sont spécifiques. Ces cellules une fois injectées au patient suite à la transplantation, aident à la protection contre les infections de cytomégalovirus, du virus Epstein-Barr ou encore d’adénovirus [128]. Étant donné que la quantité de lymphocytes T mémoires dans le sang de cordon est assez faible, cette thérapie n’est pas la plus appropriée. Des études sont présentement en cours pour induire l’activation in vitro de cellules T naïves provenant de cette source, afin de réduire les risques d’infections chez les patients [129]. Dans le cas des transplantations effectuées pour le traitement des leucémies, le risque de rechute peut être diminué par l’utilisation de lymphocytes T génétiquement modifiés pour être dirigés vers les cellules tumorales [130].
1.3.6 Introduction des plasmocytes générés in vitro
Les lymphocytes B sont les cellules au coeur de l’immunité humorale. Tel que vu précédemment, leur reconstitution peut nécessiter plusieurs mois, ce qui expose les patients à plusieurs dangers. Lorsqu’un régime pré-transplantation exigeant une irradiation est nécessaire, cela entraîne la perte des lymphocytes en périphérie ainsi que la mémoire immunologique du patient. La plupart des thérapies développées jusqu’à présent servent principalement à la reconstitution des lymphocytes T pour le rétablissement de l‘immunité cellulaire. L’introduction de cellules procurant une immunité humorale pourrait être un traitement supplémentaire administré aux patients afin de les aider à combattre les infections. Par ailleurs, une étude menée par le groupe du Dr Klein a démontré que la mobilisation de CSH de la moelle osseuse vers le sang périphérique à l’aide du G-CSF [115] induit aussi une mobilisation des plasmocytes de cet organe. Ces cellules étaient de phénotype CD38hi, dont seulement 38% exprimaient le marqueur CD138 et leur quantité
dans la circulation sanguine était de 6 fois supérieure à la normale suite à un traitement avec le G-CSF. Sachant qu’un individu en santé possède environ 1x109 plasmocytes, la quantité de cellules ainsi mobilisée et
injectée au receveur représenterait seulement 3% de son nombre initial de plasmocytes. Les auteurs de cette étude suggèrent tout de même que ces cellules pourraient se réfugier dans la moelle osseuse du patient et contribuer à la reconstitution de sa mémoire immunologique humorale [115]. Cette observation ouvre une voie vers une thérapie qui serait principalement basée sur l’utilisation de plasmocytes générés in vitro, qui pourraient être injectés au patient au moment de la transplantation, comme il se fait naturellement lors des transplantations de cellules souches mobilisées. Ces plasmocytes obtenus de la différenciation de lymphocytes B mémoires du patient pourraient sécréter des anticorps provenant de sa propre mémoire immunologique.
L’équipe de Dr Sonia Néron travaille actuellement au développement d’un microenvironnement in vitro permettant l’expansion et la différenciation de lymphocytes B mémoires provenant de donneurs en santé en plasmocytes sécréteurs d’immunoglobulines. À la lumière des observations de Caraux et coll, il était logique de proposer d’utiliser ce microenvironnement pour la préparation ex vivo de plasmocytes pour les patients greffés.
1.4 Modèles de culture développés jusqu’à présent
La culture in vitro de lymphocytes B est pratiquée depuis plusieurs années. Plusieurs équipes ont élaboré des systèmes de culture permettant leur différenciation in vitro en cellules sécrétrices d’anticorps [65][131][132][133][134]. Dans la majorité des cas, il s’agit d’une combinaison de cytokines ainsi que l’implication de l’interaction CD40-CD154 (revue par Néron et coll., 2011) [135] . Les cytokines utilisées sont principalement les interleukines 2, 4, 6, 10, 12, 15, 21 et l’IFN-. Ces modèles en une [131], deux [65] ou trois phases [134][133] permettent la génération de plasmablastes et de plasmocytes. Toutefois, aucun de ces systèmes de culture in vitro ne permet une grande expansion des cellules ni le maintien de la culture au-delà d’une dizaine de jours.
Une seule étude a récemment décrit la génération de plasmocytes à longue vie à partir de lymphocytes B mémoires [132]. Le système de culture développé par cette équipe consistait en la mise en culture de cellules B mémoires dans un milieu additionné d’IL-21, d’anti-IgM et IgG en présence de cellules CD154+ pendant 3
jours. À la suite de cette étape, les cellules-supports étaient retirées et les lymphocytes B étaient cultivés dans un milieu additionné d’IL-2, d’IL-21, de facteur de croissance pour hybridome (HybridoMax) pendant 3 jours. Pour le reste de la culture des cellules B, un milieu composé d’IL-6, d’IL-21 d’IFN- en présence d’une lignée de cellules stromales était utilisé. La première phase de ce système de culture avait pour but d’activer les lymphocytes B, la deuxième d’induire leur différenciation tandis que la troisième permettait la survie des plasmocytes générés.
1.5 Expansion et différenciation in vitro de lymphocytes B
humains
1.5.1 Interaction CD40-CD154
L’interaction entre les molécules CD40 et CD154 est importante pour l’activation des lymphocytes B naïfs ainsi que celle des cellules mémoires. Son utilisation in vitro a été introduite par Banchereau et coll en 1991 et depuis, c’est l’interaction la plus utilisée pour l’activation in vitro de lymphocytes B [136]. Plusieurs études ont été menées en utilisant cette interaction cellulaire sous différentes formes, soit à travers des lignées cellulaires exprimant le CD154, des anticorps anti-CD40, des membranes cellulaires CD154+ ou encore la molécule
L’équipe du Dr Néron a démontré que l’interaction entre les molécules CD40 et CD154 agissait comme un rhéostat sur la régulation de l’activation lymphocytes B suite à une réponse immunitaire primaire ou secondaire [47][48] Brièvement, une forte interaction CD40-CD154 (environ 5000 à 10 000 molécules de CD154 par lymphocyte B) favorise la prolifération plus importante des lymphocytes B naïfs (CD27-, IgM+IgD+),
tandis qu’une faible interaction (500 à 1000 molécules CD154 par lymphocyte B) favorise plutôt la prolifération et la différenciation des cellules naïves et à mémoire (CD27+, IgG+IgA+) [48]. Ce modèle de culture est basé
sur l’interaction entre les lymphocytes B venant du sang périphérique et d’une lignée cellulaire adhérente (L4.5) exprimant le CD154 [137]. Ce modèle a permis la mise au point de conditions permettant une grande expansion de lymphocytes B mémoires provenant du sang de participants en santé. La forte interaction entre les lymphocytes B et la lignée cellulaire CD154+ ainsi que la présence des interleukines 2, 4 et 10 permettent
une expansion de 1x106 après 50 jours de culture. Les cellules générées par cette expansion secrètent des Ig
polyclonaux et la distribution des sous-classes est proportionnelle à celle retrouvée dans le sérum humain [138]. De plus, les travaux de maîtrise de Josiane Tremblay-Rochette ont permis le criblage de plusieurs facteurs solubles permettant d’induire une différenciation des cellules B ainsi expansionnées, en contact avec une faible interaction avec CD154. Il s’est révélé que c’était la combinaison de l’IL-6 et IL-10 qui induisait la meilleure différenciation des lymphocytes B mémoires en plasmocytes [139]. Ces cytokines sont donc à la base de la phase de différenciation in vitro.
1.5.2 Interaction CD27-CD70
L’interaction cellulaire entre CD27 et CD70 est aussi impliquée dans la différenciation cellulaire in vivo et est étudiée également in vitro [29][140][141]. CD27 est habituellement présent sur les lymphocytes B mémoires et CD70 est présent sur les lymphocytes T activés et peut aussi être présent sur les lymphocytes B activés [142]. Le groupe de Avery et coll. a également observé que cette interaction augmentait la génération de plasmocytes in vitro [143]. De plus, la culture simultanée de cellules B naïves (CD27-) et mémoires (CD27+)
activées en présence de l’interaction CD40-CD154 a permis de montrer que ces deux sous-populations avaient une réponse différentielle des cellules naïves et mémoires par rapport à l’interaction avec CD154 [63]. Une étude ciblant l’interaction entre CD27 et CD70 a montré que les deux populations cellulaires avaient en fait la capacité de s’influencer via la liaison entre les cellules naïves CD70+ qui poussaient les cellules B
mémoires CD27+ à se différencier [144]. De plus, la forte expression de CD27 sur les plasmocytes indique un
rôle potentiel de cette protéine au niveau de la différenciation ou de la survie cellulaire, suite à son interaction avec son ligand, le CD70 [50]. Un microenvironnement favorable aux plasmocytes pourrait donc inclure une interaction entre CD27 et CD70.
2. Hypothèse
Il serait possible de différencier des lymphocytes B mémoires en plasmocytes en contrôlant leur microenvironnement de culture in vitro.
3. Objectifs
1. Comparer l’effet des interactions CD40-CD154 et CD27-CD70 sur la différenciation des lymphocytes B afin de déterminer si la liaison CD27-CD70 serait un meilleur choix.
2. Dans les conditions établies, optimiser la viabilité cellulaire par l’utilisation de facteurs de survie. 3. Procéder à la caractérisation des plasmocytes générés in vivo.
4 Méthodologie
4.1 Cellules humaines
4.1.1 Participants à l’étude
La trentaine de participants de cette étude ont signé un consentement éclairé. Le processus de recrutement, le protocole de recherche et les formulaires de consentement étaient tous approuvés par le comité d’éthique de recherche d’Héma-Québec.
4.1.2 Les cellules mononuclées du sang
Lors du don de plaquettes, celles-ci sont récoltées avec des instruments d’aphérèse (Trima Accel, Gambro BCT, Lakewood, CO, É.-U.) et sont leucoréduites pendant la procédure. Les leucocytes sont en fait retenus dans une chambre de leucoréduction qui est utilisée afin d’isoler les cellules mononuclées du sang (PBMC,
peripheral blood mononuclear cells) [145]. Après le processus, les chambres de leucoréduction ont été
gardées à température pièce jusqu’à la récupération des PBMC. Pour l’isolement, les chambres ont été vidées de leur contenu et rincées avec une solution saline tamponnée au phosphate (PBS) composée de 10 mM de K2PO4, 136 mM de NaCl (Invitrogen, Burlington, ON, Canada), 8 g/L de glucose (Sigma Aldrich, Oakville, ON,
Canada) additionnée de 10 % d’ACD (acid–citrate-dextrose) (Baxter Healthcare Corp., Deerfield, IL, É.-U.), soit le PBS-Glucose avec ACD. Pour chaque chambre, un volume de rinçage de 45 mL était injecté avec une seringue de 60 mL et une aiguille de taille 22. La suspension de PBMC a ensuite été déposée sur un coussin de Ficoll-Hypaque (GE HealthCare Biosciences, Baie-d’Urfé, QC, Canada) et a été centrifugée pendant 8 min à 1000 g sans frein. L’interface composée des cellules mononuclées du sang a ensuite été récupérée, lavée avec du PBS-glucose sans ACD et les cellules ont été mises en suspension à 100x106 cellules/mL dans du
milieu de congélation composé à 50 % d’IMDM (Iscove’s modified Dulbecco’s medium), 40 % de FBS (fœtal
bovin sérum) (tous les deux d’Invitrogen) et à 10 % de DMSO (diméthysulfoxyde) (Sigma-Aldrich) et
congelées à -80 °C dans un système de congélation CoolCell LX (Biocision, Mill Valley, CA, É.-U.) pour être transférées ensuite dans de l’azote liquide.
4.1.3 Lymphocytes B mémoires IgG
+, IgA
+, ou IgE
+Pour l’isolement des lymphocytes B mémoires à partir des PBMC, deux étapes de sélections négatives sont nécessaires. Les PBMC ont rapidement été décongelés en les déposant goutte à goutte dans une solution de PBS contenant 50 % de FBS. Cette suspension cellulaire a ensuite été centrifugée à 1000g pendant 6 min. Les cellules ont été remises en suspension dans du PBS-glucose contenant 2 % FBS à une concentration de