LA REGULATION DE LA MORT CELLULAIRE PAR
LA DYNAMIQUE DE L'ACTINE
Leçons de la protéine E4orf4 de Fadenovirus
Thèse présentée
à la Faculté des études supérieures de l'Université Laval
dans le cadre du programme de doctorat en Biologie cellulaire et moléculaire pour l'obtention du grade de Philosophiae Doctor (Ph.D)
DEPARTEMENT DE BIOLOGIE MEDICALE FACULTÉ DE MÉDECINE
UNIVERSITÉ LAVAL QUÉBEC
2007
La mort cellulaire programmée assure l'élimination des cellules représentant une menace pour l'organisme. Des lésions génétiques permettant de résister à la mort par apoptose contribuent à la progression tumorale et à la résistance aux thérapies anticancéreuses classiques. Les travaux présentés dans cette thèse visent une meilleure compréhension des programmes alternatifs de mort cellulaire agissant efficacement dans les cellules tumorales. La protéine E4orf4 de l'adénovirus active un tel programme indépendant de p53 et de l'activité des caspases. Des données suggèrent que E4orf4 redirige l'activité oncogénique de Src vers la réorganisation de l'actine au détriment des voies de survie cellulaire. L'objectif de cette thèse est d'étudier la contribution de la dynamique de l'actine dans la mort cellulaire induite par E4orf4.
Les résultats indiquent que E4orf4 induit deux programmes alternatifs de mort cellulaire selon sa localisation dans les cellules transformées. E4orf4 induit un programme indépendant de Src à partir du noyau et un programme dépendant de Src et de l'activité des calpaïnes à partir d'un compartiment membranaire. Une étude approfondie du programme dépendant de Src a révélé que la translocation de E4orf4 dans le cytoplasme et les membranes active la myosine II et stimule une nouvelle polymérisation de l'actine, laquelle est associée avec le recrutement et l'ancrage des endosomes de recyclage dans une région juxtanucléaire. Ce remodelage de l'actine requiert l'association de E4orf4 avec Src et une activation locale des voies de signalisation des petites GTPases RhoA/Rho kinase, Racl et Cdc42/N-Wasp. Certains résultats suggèrent que la présence de E4orf4 dans les radeaux lipidiques pourrait favoriser le recrutement de la machinerie de régulation de l'actine par la production de PIP2. E4orf4 détournerait le trafic endosomal pour permettre l'assemblage d'un anneau contractile juxtanucléaire ancré à la périphérie de la cellule par des fibres de stress. Des résultats solides indiquent que l'activité cytotoxique de E4orf4 relève de son effet sur la dynamique de l'actine. Cette thèse propose qu'un dérèglement de la dynamique de l'actine qui interfère avec le trafic membranaire normal peut déclencher un programme de mort cellulaire alternatif à partir des voies de sécrétion.
J'entreprenais mon premier stage d'été dans le laboratoire du Docteur Josée N. Lavoie il y a maintenant presque huit ans. J'ai ressenti sa fougue scientifique dès notre première entrevue. Merci Josée de m'en avoir demandé plus lorsque je croyais m'être dépassée et pour tes encouragements les fois où j'étais moins fière de moi. Merci pour tes conseils, ton soutien et ta confiance. J'ai mené dans ton laboratoire un projet fascinant imprégné par ta créativité et ton avant-gardisme. Je n'oublierai jamais les moments d'émerveillement partagés avec toi devant le microscope à contempler une cellule qui bourgeonne ou des endosomes propulsés par des comètes.
J'aimerais également saluer tous les membres présents et passés du labo Lavoie avec qui j'ai partagé une bonne partie de ma vie pendant cette aventure. Un gros merci à ma personne ressource depuis le début, Claudia Champagne, pour son enseignement à mon arrivée, pour son dévouement au bon fonctionnement du labo et pour son aide précieuse pendant les révisions d'article. Merci à ma grande sœur de labo, Marie-Claude Gingras dont la détermination est un exemple pour nous tous et à Marie-Claude Landry, ma première étudiante d'été avec qui j'ai très vite développé une belle complicité. Je ne pourrais passer sous silence le seul homme ayant persisté dans le laboratoire, Nicolas Lamère avec qui j'ai plongé dans le merveilleux monde des petites GTPases et qui se fait toujours une joie d'ouvrir nos bouteilles vissées trop serrées. Merci à Marie-Chloé Boulanger pour ses fous rires et à Andréanne Sicotte, notre relève. Merci également à Mélanie Roy avec qui j'ai fait ma maîtrise et qui est restée une bonne amie, à Isabelle Turcotte qui a travaillé avec minutie dans le laboratoire durant quelques mois et aux étudiants d'été qui ont eu le don de me faire réfléchir par leurs questions.
Merci aux Docteurs Côté, Huot, Landry et Marceau ainsi qu'aux membres de leur laboratoire pour le prêt de matériel et leurs conseils et à tous les chercheurs du centre qui m'ont évaluée et m'ont permis de progresser pendant mes études graduées. J'aimerais également souligner la rigueur avec laquelle le Docteur Jacques Landry s'est appliqué à la prélecture de cette thèse avec la plus grande générosité. Je salue l'équipe de soccer du CRHDQ, tous mes amis du troisième étage et je lève mon chapeau à tous ceux qui
s'impliquent dans la vie sociale et scientifique du centre de recherche. Le CRHDQ est un environnement riche et stimulant où j'ai adoré passer toutes ces années.
Je remercie mes parents, Lucie et Donald, pour leur amour et leur confiance. Merci de m'avoir toujours encouragée à faire mes propres choix et de les avoir respectés. Votre générosité et votre éternelle détermination à donner le meilleur de vous-mêmes dans tout ce que vous faites sont un exemple pour moi. Merci à mes deux grands frères François et Vincent et à leur blonde, Isabelle et Mélanie, qui sont devenues mes grandes sœurs. Vos conseils et votre écoute lors de mes nombreuses remises en questions m'ont beaucoup aidée à cheminer, tant sur le plan professionnel que personnel. La curiosité et la vitalité de vos enfants Marc-Antoine, Léa et Alexandra ensoleillent les jours plus sombres. Merci aussi à ma tante Johanne qui a toujours été présente dans ma vie.
Merci Jean-François, pour tes encouragements ton immense compréhension et ta façon de voir la vie et de rendre la mienne plus simple. Ta vision de la science est très inspirante. Tu es mon amoureux, mon équilibre.
Finalement, j'aimerais remercier les organismes subventionnaires qui m'ont octroyé des bourses pendant mes études graduées : d'abord le CRHDQ, puis le Conseil de recherches en sciences naturelles et en génie du Canada (CRSNG) et bien sûre les Instituts de recherche en santé du Canada (IRSC) qui en plus de supporter mon salaire pendant mon doctorat, m'ont permis de participer à de nombreux congrès nationaux et internationaux. Merci également aux IRSC à la Fondation canadienne pour l'innovation (FCI) pour les subventions octroyées au laboratoire.
qu 'ils ont de s'investir dans tout ce qu 'ils entreprennent.
CHAPITRE 1 1 Introduction 1
1.1 La mort cellulaire programmée 3 1.1.1 L'apoptose classique 3 1.1.2 Les processus de mort cellulaire alternatifs 12
1.1.3 Les nouveaux acteurs de la mort cellulaire 17 1.1.4 Rôle potentiel de la dynamique de l'actine dans l'initiation de la mort cellulaire
28
1.2 La dynamique de l'actine 30 1.2.1 Le principe du tapis roulant 30 1.2.2 Contrôle de l'actine par les petites GTPases de la famille Rho 31
1.2.3 La déformation des membranes par la force de l'actine 39 1.2.4 Rôle de l'actine dans les voies de transport membranaire 47 1.3 La mort cellulaire par la protéine E4orf4 de l'Adénovirus 57
1.3.1 E4orf4 dans son contexte naturel d'infection virale 57
1.3.2 E4orf4 en dehors du contexte du virus 63 1.3.3 Signalisation de la mort cellulaire programmée déclenchée par E4orf4 dans les
cellules transformées 65 1.4 Hypothèses et objectifs du projet de recherche 72
1.4.1 Mise en contexte et hypothèse générale 72
1.4.2 Objectifs du projet de recherche 72
1.4.3 Aperçu de la thèse 73
CHAPITRE 2 75 Deux voies de mort cellulaire distinctes induites par la protéine E4orf4 75
2.1 Résumé 76 2.2 Article 77 2.2.1 Abstract 77 2.2.2 Introduction 78 2.2.3 Results 79 2.2.4 Discussion 92 2.2.4 Material and Methods 96
2.2.5 Acknowledgments 99
2.2.6 Références 99 CHAPITRE 3 103 La dynamique de l'actine actionne les changements morphologiques de type apoptotiques
induits par la protéine E4orf4 103 3.1 Des agents chimiques qui interfèrent avec la dynamique de l'actine modifient
l'activité de E4orf4 104 3.1.1 Le bourgeonnement cellulaire induit par E4orf4 requiert la réorganisation
dynamique du cytosquelette d'actine 105 3.1.2 La dynamique de l'actine participe à la condensation nucléaire induite par
E4orf4 109 3.2 E4orf4 recrute les Rho GTPases dans les endomembranes 112
3.3 Discussion et conclusions 116
La protéine E4orf4 tue les cellules transformées en exploitant la fonction des Rho GTPases
sur la dynamique de l'actine associée aux endosomes 118
4.1 Résumé 119 4.2 Article 120
4.2.1 Abstract 121 4.2.2 Introduction 121 4.2.3 Materials and Methods 123
4.2.4 Results 127 4.2.5 Discussion 147 4.2.6 Acknowledgments 151 4.2.7 Références 151 4.2.8 Supplemental data 156 CHAPITRE 5 159 La dynamique de l'actine induite par E4orf4 est associée avec la perturbation du trafic
membranaire 159 5.1 E4orf4 perturbe le compartiment endosomal de recyclage 160
5.1.1 E4orf4 modifie le trafic endosomal normal 161 5.1.2 E4orf4 ralentit le recyclage de la transferrine et de son récepteur 167
5.2 Rôle potentiel pour Arf6 dans la coordination de la dynamique de l'actine induite par
E4orf4 avec le trafic endosomal 172 5.2.1 Une fraction de E4orf4 insoluble au Triton X-100 est localisée dans des
endosomes tabulaires où la polymérisation d'actine est induite 172
5.2.2 E4orf4 induit une augmentation de PI(4,5)P2 175 5.3 Lien entre l'actine et le trafic membranaire : un rôle potentiel pour la dynamine en
avaldeE4orf4 179 5.4 Conclusions 182 CHAPITRE 6 183 Discussion générale 183
6.1 E4orf4 génère des changements dans la dynamique de l'actine à partir des
membranes cellulaires 183 6.1.1 Localisation membranaire de E4orf4 183
6.1.2 Recrutement de complexes de polymérisation d'actine par E4orf4 et Src au
niveau des membranes cellulaires 185 6.1.3 La dynamique de l'actine dictée par E4orf4 affecte le trafic endosomal 187
6.1.4 Consolidation du modèle d'interrelation entre la dynamique de l'actine et le
trafic endosomal 190 6.2 L'initiation la mort cellulaire par la dynamique de l'actine 197
6.2.1 L'effet sur le recyclage endosomal et les voies de survie 198
6.2.2 Effet sur l'intégrité d'autres organelles 200 6.2.3 Amplification possible de la réponse par la voie intrinsèque de l'apoptose 203
CHAPITRE 7 206 Conclusion générale 206 CHAPITRE 8 208 Méthodologie 208 Bibliographie 212 ANNEXE 237 Article de synthèse 237
A488 Alexa Fluor 488 A594 Alexa Fluor 594 A647 Alexa Fluor 647 Abi Abl interactor Actine-F actine filamenteuse Actine-G actine globulaire
Ad adénovirus
Ad2 adénosérotype 2
ADF actin depolymerizing factor ADN acide désoxyribonucléique ADP adénovirus death protein AIF apoptosis-inducing factor AKT protéine kinase B
AP-1 activating protein-1 AP-2 adaptor protein 2 complex
Apaf-1 apoptotic protease activating factor-1 APC/C anaphase promoting complexe/cyclosome Arf ADP-ribosylation factor
ARN acide ribonucléique
ARNm ARN messager
ARNO un facteur d'échange pour Arf6 Arp2/3 actin-related protein
ATG autophagy-relatad gène
ATP adénosine triphosphate
Bad bcl-XL/bcl-2 associated death promoter homolog Bak Bcl-2 homologous antagonist/killer
Bap31 BCR-associatedprotein 31
BAPTA 1,2-bis (0-aminophenoxy)ethane-N,N,N,N-tetraacetic acid) BAX Bcl-2 associated Xprotein
Bcl-2 B-cell lymphoma 2 Bcl-w Bcl-2-like 2
BC1-XL longer alternatively spliceform ofbcl-2 homolog X protein from avian
lymphocyte development
BH Bcl-2 homology
BHPvFl homologue de Bcl-2 chez le virus Epstein-Barr Bid BH3 interacting death domain agonist
Bik bcl-2 interacting killer
Bim bcl-2 interacting mediator ofcell death Bmf bcl-2 modifying factor
BRK baby rat kidney
BSA bovine sérum albumin
Btf bcl-2-associated transcription factor C. elegans Caenorhabditis elegans
Cdc42 Cdc55 Cdkl CED c-Fos CHUL CMH 1 COP CRE CRIB CrmA CSK c-src cytoD DAPI DH DISC DMEM DMSO DOCKl 80/ELMO DRF DRP-1 Dyn2 E1A E1B E2 E3 E4orf4 E40RF6 EGFR EM EndoG EPP ERK FAK F as GAP GDI GDP GED GEF GFP GPI-AP Gr Grb2 GTP Ha
cell division cycle 42
homologue de PP2A chez la levure cyclin-dependent kinase 1
cell death abnormal
cellular FBJ osteosarcoma oncogene centre hospitalier de l'Université Laval
complexe majeur d'histocompatibilité de classe 1 coatomer protein
compartiment de recyclage endosomal Cdc42 and Rac interactive binding dytokine response modifier A c-terminale Src kinase cellular Src
cytochalasine D
4 ' -6-diamidino-2-phenylindole Dbl homology
death-inducing signaling complex dulbecco 's modified eagle médium dimethyl sulfoxide
dedicator of cytokinesis protein 180/Engulment and cell motility diaphanous relatedformins dynamin-related protein 1 dynamine 2 early région 1A early région 1B early région 2 early région 3
early 4 open readingframe 4 early 4 open readingframe 6 epidermal growth factor receptor endomembranes
endonucléase G
endosomes précoces périphériques extracellular-regulated kinase focal adhésion kinase
fibroblast associated GTPase activating protein GDP dissociation inhibitor guanosine diphosphate GTPase effector domain
guanosine nucleotide exchange factor green fluorescent protein
glycosylphosphatidylinositol -anchor protein granzyme
growth factor receptor-bound protein-2 guanosine triphosphate
Hrk harakiri/DP5
HSP heat shock protein
HSPC haematopoetic stem/progenitor cell protein Htra2/Omi high température requirement protein A2 IAP inhibitor of apoptosis
ICAD inhibitor ofcaspase activated DNAse IL-2R interleukin 2 receptor
JNK c-jun N-terminale kinase kDa kilo Dalton
LEI leukocyte elastase inhibitor LIMK lin-1 l/Isll/Mec-3 kinase
MAP kinase mitogen activated protein kinase Mcl-1 myeloid cell leukemia 1 protein MDM2 murine double minute 2
MEM minimum essential médium
MLC myosin light chain
MP membrane plasmique
MRCK myotonic-dystrophy-related cdc42-binding kinase mPvFP monomeric red fluorescent protein
MTOC microtubules organising center
Myc v-myc myelocytomatosis viral oncogene homolog MYPT myosin phosphatase
NA numerical aperture
Napl25 Nck-associated protein 125 NES nuclear exclusion signal NLS nuclear localization signal
nM nanomolaire
Noxa BH3-only protein
p53 cellular tumor antigen p53
p62Dok p62 protein downstream oftyrosine kinases Pak2 p21-activated protein kinase 2
PARP Poly(ADP-ribose)polymerase PBS phosphate buffered saline
PH pleckstrin homology
Phase S phase de synthèse
PI (4,5) P2 phosphatidylinositol 4,5-biphosphate PI3K phosphatidylinositol 3-kinase
PIP5K phosphatidylinositol phosphate 5-kinase de classe I PIR121 p53-inducible messenger RNA
PLC phospholipase C PLD phospholipase D
PP2A protéine phosphatase 2A pTyr phosphorylation sur tyrosine
Puma p53-upregulated modulator of apoptosis
Rac Ras substrate ofC3
Rb rétinoblastome
RBD Rho binding domain
RE réticulum endoplasmique Rho GTPase GTPase de la famille Rho Rho Ras homolog gène family Rock rho- kinase
ROS espèces réactives de l'oxygène S. cerevisiae Saccharomyces cerevisiae SD écart type (standart déviation)
sec seconde(s)
SFK kinases de la famille Src (Src family Kinase)
SH1 Src homology domain 1
SH2 Src homology domain 2
SH3 Src homology domain 3
SIDA syndrome d'immunodéficience acquise SLs sphingolipides
Smac/DIABLO second mitochondrial-derived activator of caspases/direct IAP binding protein with low pi
SR splicing repressor
Stat3 signal transducer and activator of transcription SUG1 supressor ofGal4D lésions
SV40 Simian virus 40 Tf transferrine
TfR récepteur de la transferrine (Transferrin Receptor)
TGN trans Golgi network
TNFR1 tumor necrosis factor receptor 1
Toca-1 transducer of cdc42-dependent actin assembly
TRAIL tumor necrosis factor-related apoptosis-inducing ligand v-src viral Src
VV vecteur vide
Wasp Wiskott-Aldrich syndrome protein
WAVE/Scar WASP family verprolin homologous/suppressor ofcAMP receptor WIP WASP interacting protein
YFP yellow fluorescent protein
Yndlp yeast nucleoside diphosphatase-1 zVAD-fmk Z-Val-Ala-Asp-CH2F
Figure 1-1. Principaux régulateurs de l'apoptose classique p. 11 Figure 1-2. Communication entre les voies de signalisation de la mort cellulaire
programmée p.27 Figure 1-3. Cycle GTP/GDP des petites GTPases de la superfamille Ras p.33
Figure 1-4. Régulation des deux mécanismes majeurs de polymérisation d'actine par les
Rho GTPases p.36 Figure 1-5. Exploitation du complexe Arp2/3 par les pathogènes comme moyen de
propulsion intracellulaire p.40 Figure 1-6. Le transport endosomal p.48 Figure 1-7. Transformation de la cellule hôte par les oncogènes viraux E1A et ElB..p.59
Figure 1-8. Rôles de E4orf4 au cours de l'infection virale p.62 Figure 1-9. Représentation schématique des domaines fonctionnels de E4orf4 p.70
Figure 1-10. Relation bidirectionnelle entre E4orf4 et Src p.71
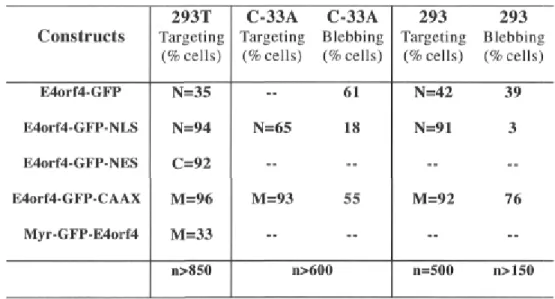
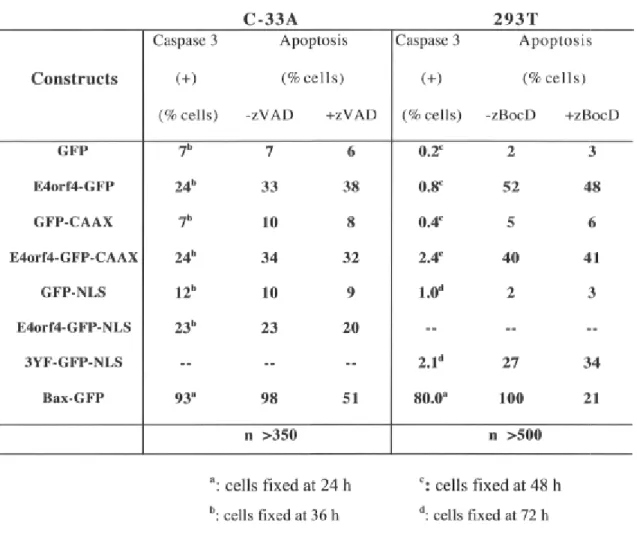
Figure 2-1. GFP fusions and protein targeting p.80 Figure 2-2. Functional activities of the E4orf4-GFP proteins p.82
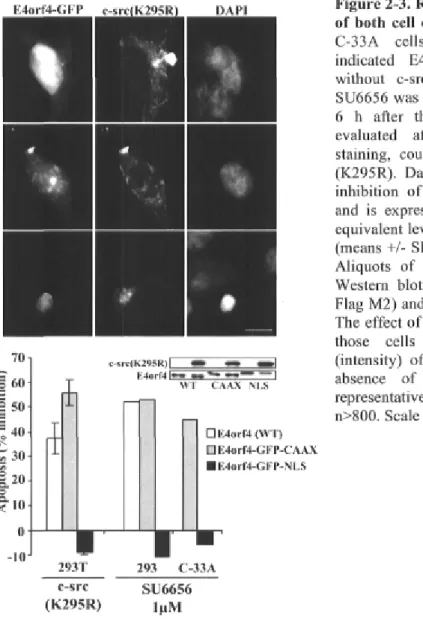
Figure 2-3. Rôle of Src kinases in régulation of both cell death pathways p.85 Figure 2-4. Effect of caspase inhibitors and caspase activation in cells expressing
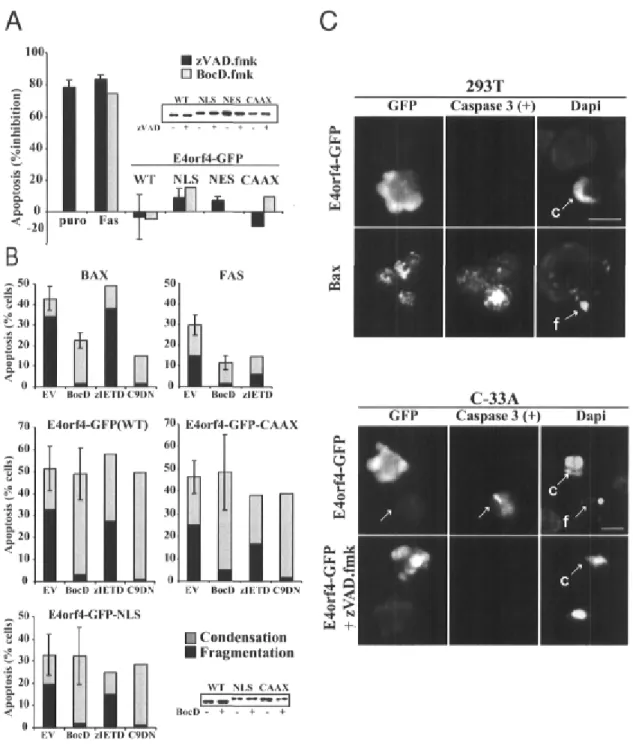
E4orf4-GFP proteins p.87 Figure 2-5. Caspase-independent apoptosis induced by E4orf4-GFP proteins is not
inhibited by Bcl-2 p.90 Figure 2-6. Inhibition of E4orf4-mediated caspase-independent apoptosis by
calpastatin p.91 Figure 2-7. Working model for the mechanisms of Ad2 E4orf4-induced cell death.. .p.94
Figure 3-1. Modèle classique d'induction du bourgeonnement membranaire p. 105 Figure 3-2. Le bourgeonnement cellulaire induit par E4orf4 est associé avec une
réorganisation du cytosquelette d'actine p. 106 Figure 3-3. La stabilisation des filaments d'actine inhibe la rétraction des bourgeons
induits par E4orf4 p. 108 Figure 3-4. L'élongation des filaments d'actine participe à la condensation nucléaire
induite par E4orf4 p. 111 Figure 3-5. E4orf4 induit le recrutement membranaire de Cdc42, Racl et Rho p. 114
Figure 4-1. The cytoplasmic translocation of E4orf4 is associated with dynamic changes in actin p. 129 Figure 4-2. E4orf4 induces myosin II activation in juxtanuclear régions and polarized
blebbing p. 131 Figure 4-3. E4orf4 induces de novo actin polymerization in the perinuclear region..p. 133 Figure 4-4. E4orf4-induced actin nucleation is not associated with Golgi
Figure 4-5. E4orf4 promûtes the nucleation of dynamic actin particles associated with endosomes motility p. 136
Figure 4-6. Spatial activation of Rho proteins by E4orf4 p.139 Figure 4-7. Distinct contributions of Rho proteins to E4orf4-induced de novo actin
polymerization p. 142 Figure 4-8. RhoA, but not Cdc42 or Racl, is required for E4orf4-induced myosin II
activation p. 144 Figure 4-9. E4orf4-induced actin dynamics trigger apoptotic-like cell death and cell
killing p. 146 Figure 4-10. Working models p. 149 Figure 4-S1 p. 156 Figure 4-S2 p. 157
Figure 4-S3 p.158
Figure 5-1. Localisation intracellulaire des petites GTPases de la famille Rab p. 160 Figure 5-2. E4orf4 perturbe le compartiment de recyclage péricentriolaire p. 162
Figure 5-3. E4orf4 recrute les endosomes le long des fibres d'actine p.164
Figure 5-4. E4orf4 modifie la mobilité des endosomes p.166 Figure 5-5. E4orf4 ralentit le recyclage du récepteur de la transferrine p. 169
Figure 5-6. E4orf4 n'interfère pas avec l'identité des endosomes visités par le récepteur de la transferrine p. 171 Figure 5-7. E4orf4 est enrichie dans des radeaux lipidiques qui s'alignent le long des
fibres d'actine p. 174 Figure 5-8. La polymérisation d'actine au niveau périnucléaire induite par E4orf4 est
associée avec la production de PI(4,5)P(2) p. 177 Figure 5-9. Le mutant de la dynamine 2 (dyn2) K44A interfère avec la réorganisation
d'actine induite par E4orf4 p. 181
Figure 6-1. Modèle reliant la dynamique de l'actine induite par E4orf4 avec la
détournement du trafic endosomal p. 190 Figure 6-2. Modèle d'initiation de la mort cellulaire par la dynamique de l'actine régulée
par E4orf4 p.203
Tableau 2-1. Targeting efficiencies and Blebbing-inducing activities of the E4orf4-GFP proteins p. 81 Tableau 2-II. The tyrosine phosphorylation of Ad2 E4orf4 régulâtes its
cytoplasmic-membrane activity but not its nuclear activity p.83 Tableau 2-III.Analysis of activated caspase-3 in cells expressing E4orf4-GFP
Introduction
La mort cellulaire programmée est un processus physiologique qui assure l'élimination de cellules superflues lors du développement pour sculpter les organes, et l'élimination des cellules endommagées ou infectées pouvant constituer une menace pour l'organisme. Une régulation serrée des mécanismes d'induction de la mort cellulaire programmée est nécessaire au développement embryonnaire normal, au bon fonctionnement du système immunitaire et au maintien de l'homéostasie tissulaire dans les organismes multicellulaires. Une induction accrue de la mort cellulaire dans un tissu est associée avec différentes conditions pathologiques, notamment les maladies neurodégénératives et le SIDA (pour synthèse, voir ). À l'opposé, la résistance face aux mécanismes de mort cellulaire contribue au développement du cancer de même qu'à la réplication virale2'3.
La reconnaissance du rôle crucial de défauts génétiques affectant les mécanismes de mort cellulaire dans la promotion tumorale et la résistance face aux agents chimiothérapeutiques a été un tremplin important pour notre compréhension de la biologie du cancer. D'après les connaissances actuelles, le dérèglement des points de contrôle de la prolifération cellulaire et l'inhibition des mécanismes de mort cellulaire programmée sont deux événements majeurs qui constituent la plateforme minimale nécessaire à l'apparition du phénotype tumoral 4. En dépit des défauts sévères dans les voies de signalisation de la mort par
apoptose, les cellules cancéreuses n'ont pas perdu entièrement leur capacité d'induire la machinerie de mort cellulaire programmée. À preuve, l'induction spontanée de la mort cellulaire est communément observée dans les tumeurs, et certains types de cancer répondent très bien à la chimiothérapie, à la radiothérapie ou à l'hormonothérapie 5. Les
recherches actuelles visent à réactiver les voies d'apoptose classique ou à caractériser des voies de signalisation alternatives de la mort cellulaire dans le but de mieux les cibler par de nouveaux outils thérapeutiques.
Des travaux récents dans le domaine de la mort cellulaire programmée ont mis en évidence de nouveaux concepts qu'on se doit de considérer. D'une part, un même stimulus, dommage ou stress, peut induire plus d'un type de mort cellulaire programmée. Or, seul le programme le plus efficace se manifestera. C'est donc dire que plusieurs voies de signalisation sont activées en même temps et communiquent entre elles par des mécanismes moléculaires encore très peu caractérisés. D'autre part, l'efficacité relative des différents programmes de mort cellulaire dépend du contexte génétique, incluant le statut des gènes surpresseurs de tumeur. Par exemple, une cellule cancéreuse qui résiste à l'apoptose classique peut demeurer sensible à l'activation de certaines voies de signalisation alternatives. D'où l'intérêt d'élucider les mécanismes impliqués et surtout, les liens moléculaires qui unissent les différents programmes de mort cellulaire de façon à maximiser l'impact des nouveaux agents anticancéreux.
C'est dans cette optique que les travaux présentés dans cette thèse ont été réalisés. Ces travaux s'inscrivent dans l'objectif principal du laboratoire qui vise à comprendre les bases moléculaires de l'activité toxique de la protéine E4orf4 de l'adénovirus. Cette protéine, qui a évolué dans un contexte viral très semblable à celui d'une cellule cancéreuse, active une voie de signalisation alternative de la mort cellulaire dans les cellules transformées.
Ce chapitre d'introduction a pour but de définir le contexte des travaux décrits dans cette thèse. Premièrement, les différents mécanismes de mort cellulaire programmée seront présentés. Ensuite, la régulation du cytosquelette d'actine ainsi que son importance dans le trafic membranaire seront décrites, ceci afin d'apprécier la contribution de la dynamique de l'actine dans la mort cellulaire programmée induite par E4orf4.
1.1 La mort cellulaire programmée
Pendant longtemps, la mort cellulaire programmée a été synonyme d'apoptose. Le terme apoptose a été choisi au début des années 1970, par Kerr, Wyllie et Curie pour définir un type de mort cellulaire physiologique s'opposant à la nécrose, une mort cellulaire accidentelle incontrôlée 6. La distinction entre les deux types de mort cellulaire est alors
basée sur des critères morphologiques précis. L'apoptose se distingue par le rétrécissement du volume cellulaire, la préservation des organelles, la condensation et la marginalisation de la chromatine, et la formation de fragments de cellule et de noyau (corps apoptotiques) pouvant être éliminés par phagocytose. À l'opposé, la nécrose entraîne l'œdème cellulaire et le démantèlement de la membrane plasmique menant au déversement du contenu cellulaire et à une réponse inflammatoire du tissu 6' . Les découvertes récentes ont révélé
que cette classification dichotomique de la mort cellulaire opposant apoptose et nécrose n'était guère réaliste vu la diversité des mécanismes de mort cellulaire programmée. Outre l'apoptose, on parle maintenant de processus de mort cellulaire alternatifs comme l'apoptose non classique, la mort par autophagie, la catastrophe mitotique et la nécrose programmée. Tous ces programmes de mort cellulaire sont régulés et impliquent des processus cellulaires actifs pouvant être contrecarrés par l'inhibition de certaines voies de signalisation intracellulaires.
1.1.1 L'apoptose classique
De tous les mécanismes de mort cellulaire, l'apoptose demeure de loin le mieux caractérisé et le plus efficace. Les études pionnières menées par Horwitz dans les années 1970 à l'aide du nématode Caenorhabditis elegans comme modèle d'étude ont contribué grandement à la démonstration du caractère génétique de la mort cellulaire programmée. En 1976, Sulston et Horwitz démontraient qu'environ 13 % des cellules somatiques de l'embryon de C.elegans sont prédestinées à mourir 8. Par la suite, des études génétiques menées chez le
nématode ont permis d'identifier trois composantes majeures de la machinerie apoptotique. Les gènes CED-3 et CED-4 permettent la mort cellulaire tandis que CED-9 inhibe la mort cellulaire. Les grands régulateurs de l'apoptose classique chez les mammifères ont été
identifiés par homologie avec ces gènes de nématode. Ainsi, CED-3, 4 et 9 correspondent respectivement aux membres de la famille des caspases, au facteur proapoptotique APAF-1 (apoptotic protease-activating factor 1) et aux membres anti-apoptotiques de la famille Bcl-2 chez les mammifères 9.
1.1.1.1 Les caspases
Les caspases sont des protéases à cystéine qui clivent leurs substrats après un résidu acide aspartique (Asp). Au moins 14 caspases ont été identifiées chez les mammifères et leurs orthologues sont retrouvés chez plusieurs espèces animales allant du nématode à l'humain (pour synthèse, voir 10). Il existe aussi une métacaspase nommée YCA1 (Yeast Caspase-1)
chez la levure " . L a première caspase identifiée chez les mammifères, par homologie de séquence à CED-3, est ICE (Interleukin 1 (3-converting enzyme), ou caspase-1 12. Elle a été
identifiée comme un régulateur clé de la réponse inflammatoire. Cependant, au moins 8 des 14 caspases connues jouent un rôle important au cours de l'initiation ou de l'exécution de l'apoptose .
Mécanisme d'activation des caspases
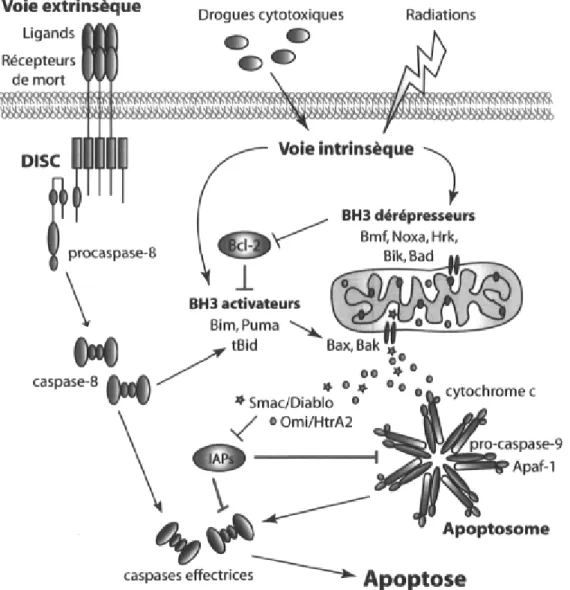
Les caspases impliquées dans l'apoptose se divisent en deux catégories. Les caspases initiatrices regroupent les caspases-2, 8, 9 et 10, tandis que les caspases exécutrices regroupent les caspases-3, 6 et 7. Pour permettre une réponse rapide à un stimulus apoptotique, les caspases sont présentes dans les cellules sous forme de zymogènes nommés procaspases. Classiquement, deux voies de signalisation majeures permettent l'activation des caspases initiatrices : la voie extrinsèque et la voie intrinsèque de l'apoptose. La voie extrinsèque est déclenchée par l'activation des récepteurs de mort à la surface de la cellule. La liaison d'un ligand à son récepteur, comme Fas L (Fas ligand) à Fas ou TNF (tumor necrosis factor) à TNFR (TNF receptor), induit la formation d'un complexe de signalisation de la mort cellulaire nommé DISC (death-induced signalling complex). Le DISC recrute et favorise la maturation protéolytique de la procaspase-8 dans le cas de Fas, ou de la procaspase-10 dans le cas de TNFR. Quant à elle, la voie intrinsèque est activée par une grande variété de stress extracellulaires et intracellulaires, comme la privation en facteurs de croissance, l'hypoxie, les dommages à l'ADN et l'activation des
oncogènes. Les signaux déclenchés par ces stress convergent vers la mitochondrie où un changement de perméabilité de la membrane externe cause la libération d'un certain nombre de facteurs pro-apoptotiques dont le cytochrome c. En présence d'ATP, le cytochrome c cytosolique permet la formation de l'apoptosome, composé de cytochrome c, de APAF-1 et de la procaspase-9. Ce complexe protéique permet l'autoactivation de la caspase-9 par oligomérisation 13. L'activité des caspases est régulée par les membres de la
famille des lAPs (Inibitor of apoptosis proteins) u. Entre autres, XIAP (X-linked IAP)
inhibe la maturation de la caspase-9 et l'activation de la caspase-3 au niveau de l'apoptosome 15' 16. Afin de permettre l'activation des caspases en aval du cytochrome c,
d'autres facteurs proapoptotiques comme Smac/Diablo et Omi/HtrA2 sont libérés de la mitochondrie et neutralisent l'action des IAPs 17'l8.
Les caspases initiatrices activées clivent les procaspases exécutrices pour les activer à leur tour. Ensuite, les caspases exécutrices s'attaquent à des molécules anti-apoptotiques pour les inactiver et clivent des molécules pro-apoptotiques pour les activer. Les caspases exécutrices possèdent une panoplie de substrats, dont des protéines impliquées dans la signalisation, dans la régulation de l'apoptose et dans la réparation de l'ADN, mais aussi plusieurs protéines liées au cytosquelette comme l'actine, la gelsoline, la (3-caténine, et la FAK {focal adhésion kinase) .
Remodelage de l'actine par les caspases durant la phase extranucléaire de l'apoptose Les changements morphologiques importants qui caractérisent les cellules apoptotiques témoignent des grands bouleversements qui surviennent au niveau du cytosquelette d'actine au cours de la mort cellulaire. La phase extranucléaire de l'apoptose se définit par les changements morphologiques qui surviennent dans le cytoplasme en trois étapes subséquentes (l'arrondissement cellulaire, le bourgeonnement membranaire et la diminution du volume cellulaire), auxquelles a été associé le clivage d'un nombre important de protéines structurales et régulatrices impliquées dans la dynamique de l'actine (pour synthèse, voir 19).
L'arrondissement cellulaire implique le détachement partiel de la matrice extracellulaire et la réorganisation des points focaux d'adhérence (FA). Le clivage par les caspases de la
FAK et d'une protéine de structure des FAs, la pl30CAS favorise le démantèlement des FAs lors de l'apoptose 20' 21. Ce démantèlement est associé avec une baisse de la
phosphorylation de la paxiline, une autre composante des FAs 20. Avec la réorganisation
des FAs, qui normalement ancrent les fibres de stress à la périphérie de la cellule, le réseau d'actine est réorganisé en un anneau périphérique.
Les caspases sont également tenues responsables de l'une des plus spectaculaires manifestations cytoplasmiques associées à plusieurs programmes de mort cellulaire, le bourgeonnement membranaire. Ce processus dynamique au cours duquel les événements de sortie et d'entrée de bourgeons membranaires se produisent simultanément sur toute la surface cellulaire est causé par un déséquilibre des forces contractiles, engendré par une augmentation soutenue de la phosphorylation de la chaîne légère de la myosine (MLC) qui stimule l'activité de la myosine II 22'23. Pour permettre la sortie d'un bourgeon par la force
de contraction actine-myosine, le lien entre l'anneau cortical d'actine et la membrane plasmique doit être brisé localement 19. La protéolyse de la fodrine par les caspases
participe à ces bris pour permettre la sortie des bourgeons. 2 "27. Certains effecteurs des Rho
GTPases impliqués dans la régulation de l'actine comptent également parmi les substrats des caspases. Le fragment de clivage de la Rho kinase I (Rock I) généré par la caspase-3 possède une activité constitutive qui serait responsable de la phosphorylation soutenue de la MLC durant le bourgeonnement 28' 29. Un fragment actif de la LIMK-1, une autre kinase
impliquée dans la régulation du cytosquelette d'actine, serait également libéré sous l'action des caspases. La forme clivée de la LIMK-1 induit le bourgeonnement tandis la réduction de la LIMK-1 par interférence à l'ARN (ARNi) diminue le bourgeonnement membranaire
30. Finalement, la cellule apoptotique cessera de bourgeonner et la cellule apparaîtra sous la
forme d'un seul corps hypercondensé ou en fragments de plusieurs petits corps apoptotiques où l'actine et les microtubules sont désassemblés ou dégradés 22. Puisque
l'actine elle-même et plusieurs composantes du cytosquelette sont clivées par les caspases, on assume que le bourgeonnement cesse et que la condensation cellulaire survient lorsque le cytosquelette est complètement démantelé 19.
Les caspases ne sont pas les seules responsables de ces changements morphologiques. Les calpaïnes, des protéases dépendantes du calcium, participeraient à la phase
d'arrondissement cellulaire en clivant certaines protéines de structure qui relient l'actine à la membrane plasmique (a-actinine, fodrine, taline, spectrine) 27>31. Aussi, il semble que
dans certaines circonstances, le bourgeonnement membranaire soit insensible à l'inhibition des caspases par le zVAD-fmk mais plutôt sensible à l'inhibition de la petite GTPase RhoA, l'activateur de la ROCK I 22. D'ailleurs, la forme active de RhoA induit du
bourgeonnement membranaire lorsqu'elle est surexprimée 32. Chez des cellules non
apoptotiques, le bourgeonnement est une manifestation toxique réversible nécessitant aussi la force de contraction générée par la liaison de la myosine II avec l'actine 23. On peut donc
supposer qu'au cours des processus de mort cellulaire où les caspases ne sont pas activées, le bourgeonnement pourrait dépendre de l'activité de RhoA sur la Rock. Il semble que la seule modification extranucléaire entièrement dépendante des caspases soit la formation de corps apoptotiques. D'ailleurs, lorsque la formation de corps apoptotiques est bloquée par l'inhibition des caspases, la cellule est maintenue dans une phase interminable de bourgeonnement membranaire 22,33.
Il n'en demeure pas moins qu'au cours de l'apoptose classique, les changements dans la dynamique de l'actine sont perçus comme des événements tardifs conséquents à l'activation des caspases. Ces changements sont considérés comme un sous-programme de l'apoptose ayant pour but de faciliter la reconnaissance et l'élimination des cellules par phagocytose. D'ailleurs, vu la grande efficacité avec laquelle les caspases déclenchent les événements protéolytiques irréversibles et induisent la mort cellulaire, leurs mécanismes d'induction nécessitent une régulation serrée. Une grande partie de cette régulation revient aux protéines de la famille Bcl-2, qui contrôlent également la grande majorité des programmes de mort cellulaire alternatifs qui seront discutés plus loin.
1.1.1.3 La régulation de l'apoptose par les protéines de la famille Bcl-2
Lors de l'exécution de la voie intrinsèque de l'apoptose, la perméabilité de la membrane mitochondriale constitue le point de non-retour du programme de mort cellulaire. La régulation fine de cette étape critique est assurée par les membres de la famille Bcl-2 34. Le
membre fondateur de la famille Bcl-2 (B-cell lymphoma 2), a été clone à partir d'une translocation chromosomique retrouvée chez la majorité des patients atteints d'un lymphome folliculaire 35. La surexpression de Bcl-2 associée avec cette translocation
augmente les chances de survie des cellules tumorales en conditions sous-optimales en prévenant l'apoptose. Favorisant la survie cellulaire plutôt que la prolifération, Bcl-2 est qualifié de protooncogène non conventionnel36. La famille Bcl-2 regroupe aujourd'hui plus
d'une vingtaine de protéines anti-apoptotiques et pro-apoptotiques qu'on peut diviser en trois sous-classes selon le nombre de domaines d'homologie partagés avec Bcl-2 (BH, Bcl2 homolgy 1-4) 37. Les protéines anti-apoptotiques, incluant Bcl-2, Bcl-XL, MLC-1, Al et
Bcl-w, partagent les domaines BH1 à BH4. Les molécules proapoptotiques sont sous-divisées entre les membres à domaines BH multiples (Bax et Bak) possédant les domaines BH1 à BH3 et les molécules contenant uniquement le domaine BH3 (BH3 unique).
Les protéines pro-apoptotiques à domaines BH multiples
Les cellules déficientes pour Bax et Bak sont résistantes à l'activation de la voie intrinsèque de l'apoptose par divers stimuli3 ' .En condition basale, les protéines pro-apoptotiques à
domaines BH multiples existent sous forme de monomères inactifs au cytosol (Bax) ou à la mitochondrie (Bak). En réponse à un stimulus apoptotique, Bax s'insère sous forme d'oligomères actifs dans la membrane externe de la mitochondrie, où Bak s'oligomérise également, permettant une augmentation de la perméabilité de la membrane externe de la mitochondrie 40' 41. Ceci favorise la libération de facteurs intermembranaires
pro-apoptotiques dans le cytosol, dont le cytochrome c, par un mécanisme moléculaire encore controversé (pour synthèse, voir 42). Un premier modèle est basé sur l'homologie de
séquence entre Bax et la toxine diphtérique. Comme cette toxine, Bax et Bak formeraient des pores membranaires au niveau de la mitochondrie qui permettraient la libération de facteurs proapoptotiques. Ce modèle est appuyé par des études réalisées in vitro, montrant que Bax forme des canaux suffisamment larges dans des liposomes pour laisser passer le cytochrome c 43. Un deuxième modèle suggère que Bax pourrait interagir avec des canaux
anioniques mitochondriaux préexistants, par exemple le VDAC {voltage-dependent anion channel), pour en modifier la perméabilité 44. Cependant, aucune étude n'a pu confirmer ou
Les protéines pro-apoptotiques à domaine BH3 unique
Les protéines pro-apoptotiques à domaine BH3 unique agiraient en amont des molécules à domaines multiples, puisqu'elles sont incapables de tuer les cellules en absence de Bax ou de Bak 45' 46. Chez les mammifères, on en retrouve au moins 9 (Bad, Bik, Blk, Bid, Bim,
Bmf, Hrk, Noxa et Puma). Elles sont considérées comme des sentinelles pouvant déclencher une réponse apoptotique en réaction à divers stimuli d'ordre développemental ou cytotoxique (pour synthèse, voir 47). Certaines molécules, notamment Hrk, Noxa et
Puma, sont contrôlées au niveau transcriptionnel 48~51. D'autres protéines à BH3 unique
sont présentes sous forme inactive chez les cellules saines et sont activées en réponse à des stimuli particuliers. Par exemple, Bmf est séquestrée au cytosquelette d'actine par association avec la myosine V. Son activation se produit lors de l'anoïkose, une forme d'apoptose induite par le détachement cellulaire et la perte de la signalisation par les intégrines 52. Par ailleurs, Bad est séquestrée dans le cytoplasme par la protéine 14-3-3,
suite à sa phosphorylation par Akt ou par la protéine kinase A qui sont activés par des signaux de survie. L'action de Bad n'est efficace qu'en absence de ces signaux 53. La
molécule Bid, quant à elle, est synthétisée sous la forme d'un zymogène pouvant être clivé par la caspase-8 54 ou par d'autres protéases dont il sera question plus loin. Le fragment
actif de Bid (tBid) sert de mécanisme d'amplification reliant la voie extrinsèque induite par les récepteurs de mort, à la voie intrinsèque en stimulant l'action de Bax et de Bak 55. Les
mécanismes exacts en amont de l'activation des caspases qui régulent l'activation et la translocation des protéines à domaine BH3 unique sont encore peu connus.
Les protéines anti-apoptotiques
Parmi les molécules anti-apoptotiques, Bcl-2 et ses deux plus proches homologues, BC1-XL
et Bcl-w, inhibent la plupart des réponses apoptotiques. Ces molécules anti-apoptotiques possèdent 4 domaines d'homologie hautement conservés (BH1 à BH4), et une région hydrophobe à leur extrémité C-terminale permettant leur ciblage à la membrane côté cytoplasmique de trois organelles : la mitochondrie, le réticulum endoplasmique (RE) et l'enveloppe nucléaire56. Bcl-2 est retrouvé de façon constitutive au niveau des membranes
signaux proapoptotiques. La séquence de Mcl-1 et A-l, diverge davantage de celle de Bcl-2 et leur activité anti-apoptotique semble moins efficace (pour synthèse, voir37).
Modèle de la régulation de la voie intrinsèque de l'apoptose par la famille Bcl-2
Plusieurs mécanismes ont été proposés pour expliquer la régulation de l'apoptose par les membres de la famille Bcl-2. Selon un modèle récent, une régulation hiérarchique existerait entre les différents membres selon laquelle différents types de protéines à domaine BH3 unique agiraient à deux niveaux 57 (figure 1). Les BH3s unique tBid, Bim et Puma
formeraient la catégorie des activateurs directs des protéines pro-apoptotiques à domaines multiples Bax et Bak. En condition normale, ces BH3s activateurs seraient séquestrés par les molécules anti-apoptotiques telles que Bcl-2, Bcl-XL et MLC-1. Les autres BH3s unique
(Bad, Bmf, Bik, Hrk, Noxa) seraient des « dérépresseurs » capables de lever la répression des molécules anti-apoptotiques sur les BH3s activateurs en réponse à un stimulus apoptotique. Chaque stimulus activerait au moins une protéine BH3 de chaque catégorie pour assurer la neutralisation de Bcl-2 et l'activation de Bax et de Bak. Par exemple, les dommages à l'ADN ou l'activation d'oncogène induisent à la fois NOXA (dérépresseur) et PUMA (activateur) par l'intermédiaire de p53 50'58. Selon cette théorie, l'effet antagoniste
Voie extrinsèque Drogues cytotoxiques Radiations
Figure 1-1. Principaux régulateurs de l'apoptose classique. 1) La voie extrinsèque est déclenchée par le recrutement de la procaspase-8 par le complexe de signalisation de la mort cellulaire DISC formé à la membrane plasmique suite à l'activation des récepteurs de mort par leur ligand. 2) Les drogues cytotoxiques, les radiations ou l'activation d'oncogènes stimulent la voie intrinsèque de l'apoptose. Cette voie nécessite la libération du cytochrome c et l'activation subséquente de la procaspase-9 au niveau de l'apoptosome, un complexe protéique formé du cytochrome c, APAF-1 et la procaspase-9. Les caspases initiatrices 8 ou 9 peuvent lier et activer par clivage les caspases exécutrices 3, 6 et 7. Les caspases actives peuvent être inhibées par les IAPs
{inhibitor of apoptosis proteins), elles-même neutralisées par Smac ou Omi qui sont libérées de la
mitochondrie avec le cytochrome c. La libération des facteurs proapoptotiques à partir de la mitochondrie est régulée par les membres de la famille Bcl-2. Bax et Bak augmentent la perméabilité de la membrane externe de la mitochondrie. Cette fonction serait stimulée par les molécules à domaine BH3 unique de type activateur (Bid, Bim, PUMA). Les BH3 activateurs sont neutralisés par les molécules anti-apoptotiques Bcl-2 ou Bcl-XL. Cette effet est antagonisé par les molécules à domaine BH3 unique de type dérepresseur. Le clivage de Bid par la caspase-8 relie la voie extrinsèque avec l'activation de la voie intrinsèque de l'apoptose. Les actions anti-apoptotiques sont représentées par les traits rouges.
En somme, la famille Bcl-2 régule l'activation des caspases au niveau de la mitochondrie. Or, vers le milieu des années 1990, des études à l'origine très controversées, ont démontré que Bax peut provoque la mort cellulaire même lorsque l'activité des caspases est inhibée
5 . À l'ère où l'activation des caspases était considérée nécessaire à toutes formes de mort
cellulaire programmée, cette découverte a levé le voile sur l'existence d'une multitude de voies de signalisation alternatives régulées en majorité par la famille Bcl-2. Il n'est donc pas étonnant d'observer la suractivation de Bcl-2 chez plusieurs tumeurs, puisque celle-ci inhibe la majorité des processus de mort cellulaire programmés. Certains virus profitent également de cet avantage en codant pour des homologues viraux de Bcl-2, notamment ElB19k chez l'adénovirus 60 et BHRF1 chez le virus Epstein-Barr . L'activité
anti-apoptotique de ces homologues viraux préviendrait la mort précoce de la cellule hôte lors de l'infection (pour synthèse, voir 62). On tente actuellement de cibler ce point de
convergence des programmes de mort cellulaire en développant des molécules qui miment les protéines à domaine BH3 unique qui pourraient antagoniser l'effet anti-apoptotique de Bcl-2 dans les cellules cancéreuses. Nous verrons que même si la famille Bcl-2 régule la majorité des processus de mort alternatifs qui impliquent la mitochondrie, certaines données, dont celles présentées dans cette thèse, soulèvent l'existence de processus de mort cellulaire programmée qui sont insensibles à l'effet protecteur direct de Bcl-2.
1.1.2 Les processus de mort cellulaire alternatifs
Une série d'études réalisées avec différents modèles, dont la protéine virale E4orf4 qui fait l'objet de cette thèse63, ont révélé l'existence de programmes de mort cellulaire
indépendants des caspases (pour synthèse, voir 64). Ces premiers travaux ont soulevé la
controverse puisqu'ils étaient basés sur l'utilisation d'un inhibiteur chimique de caspases à large spectre (zVAD-fmk). En effet, l'efficacité mitigée de cet inhibiteur contre certaines caspases atypiques, notamment la caspase-2, laissait planer le doute sur la validité du caractère « indépendant des caspases» de ces nouveaux programmes de mort.
Cependant, des expériences réalisées in vivo, d'abord chez C. elegans puis chez la souris appuient l'existence des programmes de mort cellulaire indépendants des caspases (pour synthèse, voir 65). Entre autres, la surexpression de CED-4 (homologue de APAF-1) induit
la mort cellulaire chez des neurones de C. elegans déficients pour CED-3 (seul homologue des caspases) 66. De plus, la mort cellulaire a été observée chez des cellules du pharynx
d'un nématode déficient pour CED-3 67. Chez les mammifères, certaines molécules à BH3
unique comme tBid, Bim et Bad induisent la mort cellulaire chez des cellules MEF (mouse embryonic fibroblasi) déficientes pour Apaf-1 chez lesquelles les caspases ne sont pas activées 45. L'action cytotoxique indépendante des caspases de Bax ou des molécules à
BH3 unique a été attribuée à la libération de facteurs cytotoxiques mitochondriaux dans le cytosol, notamment l'endonucléase G ou AIF (apoptosis inducing factor) qui mènent à la condensation de l'ADN en absence de l'activité des caspases 68'69.
1.1.2.1 Différents types de mort cellulaire alternatives
Il est désormais bien accepté que la mort cellulaire programmée adopte différentes formes. Cependant, les processus de mort cellulaire alternatifs sont souvent identifiés par des critères d'exclusions par rapport aux mécanismes de régulation de l'apoptose classique. Ces programmes sont qualifiés de: « indépendants des caspases », « indépendants de p53 » ou « insensibles à l'expression de Bcl-2 ». Le défi actuel est de caractériser les facteurs impliqués dans ces nouveaux processus de mort cellulaire. Devant l'augmentation du nombre de programmes de mort cellulaire alternatifs identifiés, un effort considérable de classification s'est mis en branle. Or, vu la compréhension très limitée des mécanismes moléculaires impliqués, on se base principalement sur des critères morphologiques pour tenter une classification 70"72. Les termes les plus couramment utilisés pour désigner les
processus de mort cellulaire alternatifs sont l'apoptose non classique, la mort par autophagie, la catastrophe mitotique et la nécrose programmée.
L'apoptose non classique
L'apoptose classique se distingue par des critères morphologiques qui sont strictement conséquents à l'activation des caspases, soit la dégradation de l'ADN en fragments internucléosomaux et la fragmentation cellulaire en corps apoptotiques. On qualifie d'apoptose non classique les processus de mort caractérisés par une condensation irrégulière de la chromatine et par l'absence de dégradation de l'ADN en petits fragments
comme l'externalisation des phospholipides membranaires nécessaires à la reconnaissance et à l'élimination des cellules par phagocytose, la diminution du volume cellulaire et le bourgeonnement membranaire. Or, lors de Vapoptose non classique, ces changements morphologiques sont indépendants de l'activation des caspases par la voie extrinsèque ou intrinsèque. Par contre, certains processus durant lesquels interviennent l'activation de caspases atypiques, comme la caspase-2, sont habituellement classés dans cette catégorie. Parmi les exemples d'inducteurs de l'apoptose non classique, notons la mort cellulaire induite par la déplétion de HSP70 (Heat shock protein 70) 73 et par la protéine adénovirale
E4orf4 63.
La nécrose programmée
Les cellules mortes par nécrose ont longtemps été considérées comme les victimes d'un stress intense qui provoque une mort subite et passive, c'est-à-dire non contrôlée par la signalisation intracellulaire. Or, des études récentes indiquent que la nécrose peut parfois être régulée par la signalisation intracellulaire et contribuer au développement et au maintien de l'homéostasie, au même titre que les autres modes de mort cellulaire programmée (pour synthèse, voir 7 4'7 5). La nécrose programmée implique les produits de
gènes qui induisent une déficience irréversible en ATP ou qui seront relâchés sélectivement dans l'environnement extracellulaire pour déclencher une réaction chez l'organisme 75. La
nécrose programmée se manifeste par un gonflement des organelles et une perte précoce de l'intégrité de la membrane plasmique, sans condensation de la chromatine. On connaît encore très peu les mécanismes régulant la nécrose programmée, dont la signalisation peut être inhibée entre autres par les bloqueurs de radicaux libres 76'77, par l'inhibition de la
PARP ipoly (ADP) ribose polymerase) 78 ou par des chélateurs de calcium 79. L'avantage
évolutif conféré par la nécrose programmée serait de permettre aux régions d'un organisme multicellulaire ayant subi une invasion massive ou de graves lésions de stimuler activement une réponse de défense ou de réparation de la part de l'organisme en activant le système immunitaire .
La mort cellulaire par autophagie
L'autophagie est un processus au cours duquel il y a formation d'une double membrane autour d'organelles entières. Ces vésicules autophagiques, appelées autophagosomes, fusionnent avec d'autres vésicules puis avec les lysosomes qui causent la dégradation de leur contenu. Les acteurs de ce processus sont codés par les gènes reliés à l'autophagie (ATGs, autophagy-related gènes). Normalement, l'autophagie est un processus de survie déclenchée en condition de carence nutritionnelle pour fournir les nutriments nécessaires à la cellule 80. Or, un excès d'autophagie peut induire la mort cellulaire par autophagie, qui
se distingue de l'apoptose ou de la nécrose par des caractéristiques morphologiques très Q 1
particulières . Ces cellules contiennent une grande quantité d'autophagosomes, elles perdent leur capacité à former des colonies et leur membrane plasmique se désagrège 80.
Des études récentes soutiennent un rôle pour l'autophagie dans le contrôle de la mort cellulaire. En effet, la répression des gènes ATG 5, 6 ou 7 qui régulent la formation d'autophagosomes, protège les cellules de la mort cellulaire par autophagie 82'83. L'un de
ces gènes, ATG 6, code pour la protéine de suppression tumorale Beclin-1 84'85, ayant été
identifiée par sa liaison à Bcl-2 86. Une étude récente défend clairement que la liaison de
Bcl-2 avec Beclin-1 inhibe la mort cellulaire par autophagie, suggérant que les propriétés oncogéniques de Bcl-2 proviendraient non seulement de sa capacité à inhiber l'apoptose, mais également à inhiber la mort cellulaire par autophagie 87. Cependant, le rôle des
membres de la famille Bcl-2 dans l'autophagie reste controversé. En effet, des cellules MEFs déficientes pour Bax et Bak, insensibles à une grande variété de stimuli apoptotiques, meurent par autophagie lorsqu'elles sont exposées à l'étoposide, la staurosporine et la thapsigargine. Selon cette étude, la surexpression de Bcl-2 chez les MEFs sauvages stimulerait la mort par autophagie induite par ces drogues cytotoxiques 88.
Le rôle des caspases est également débattu. En effet, un traitement par l'inhibiteur de caspases zVAD-fmk induit la mort cellulaire par autophagie chez les fibroblastes L929 et chez certaines lignées de macrophages. . À l'opposé, une autre étude supporte le rôle clé des caspases en aval de l'autophagie pour la mort cellulaire causée par l'IFN-y chez certaines lignées cellulaires transformées et cancéreuses 82. Quoi qu'il en soit, l'autophagie
cellulaire dans certaines circonstances, et ainsi servir de mécanisme de suppression tumorale.
La catastrophe mitotique
La catastrophe mitotique est causée par un conflit au cours de la progression du cycle cellulaire, lorsque les mécanismes de contrôle du cycle cellulaire sont défectueux 89. Cette
forme de mort cellulaire n'est pas accompagnée des critères morphologiques typiques de l'apoptose. Elle est plutôt associée avec la formation de cellules géantes pourvues de micronoyaux dont la chromatine n'est pas condensée. La catastrophe mitotique peut être déclenchée par des agents chimiques qui stabilisent ou déstabilisent les microtubules, ou par les dommages à l'ADN 90. Elle peut dépendre ou non de p53, selon l'étape du cycle
cellulaire pendant laquelle elle est induite 89. L'implication des caspases dans l'une ou
l'autre des étapes menant à la catastrophe mitotique reste encore l'objet d'intenses débats. Dans certains systèmes, la catastrophe mitotique est associée avec l'activation de la caspase-2 en amont de la perméabilisation de la mitochondrie, suivie de l'activation de la caspase-3 91. Or, certains auteurs affirment que l'inhibition des caspases ou la surexpression
de Bcl-2 n'inhibent pas, et même exacerbent la prévalence des catastrophes mitotiques chez les cellules cancéreuses en réponse aux agents chimiothérapeutiques 90'92.
Les nombreuses connexions qui existent entre les différentes voies de signalisation menant à la mort cellulaire rendent la classification des programmes de mort cellulaire quasi impossible. De plus, comme il a été mentionné précédemment, il semble qu'un même stimulus active plusieurs des mécanismes qui viennent d'être décrits et que seul le programme de mort le plus efficace se manifeste dans une population cellulaire donnée 93' 94. Puisque la cascade d'activation protéolytique des caspases est très rapide, l'apoptose est
souvent prédominante sur les programmes alternatifs. Mais dans certains contextes cellulaires, l'efficacité relative des différentes voies de signalisation de mort cellulaire peut être différente 70'71-95. pa r exemple, une cellule cancéreuse ayant nécessairement développé
une résistance à l'apoptose, soit par la suractivation de molécules anti-apoptotiques comme Bcl-2 ou par l'inhibition de l'activité de suppresseurs de tumeur comme p53, peut demeurer sensible à l'activation de certaines voies de signalisation alternatives. Cependant, les interactions fonctionnelles entre les différents programmes de mort de même que les causes
exactes de la prédominance d'un programme sur un autre dans un contexte cellulaire donné restent à élucider. Une meilleure compréhension des mécanismes moléculaires qui dirigent et relient entre eux les différents programmes alternatifs est requise afin de mieux cibler ces mécanismes par des stratégies thérapeutiques novatrices visant l'élimination des cellules cancéreuses.
1.1.3 Les nouveaux acteurs de la mort cellulaire
L'intérêt grandissant pour les mécanismes régissant les voies alternatives de signalisation de la mort cellulaire a permis la découverte de nouveaux joueurs ou de nouveaux rôles pour des joueurs connus dans la régulation de la mort cellulaire. En plus des caspases, d'autres protéases ont révélé leur potentiel toxique. De plus, il semble qu'en plus de la mitochondrie, d'autres organelles dont certaines semblent être sous le contrôle des membres de la famille Bcl-2, puissent être le siège de l'initiation de la mort cellulaire programmée.
1.1.3.1 Les autres protéases impliquées dans la mort cellulaire programmée
Au cours de l'apoptose classique, plusieurs manifestations morphologiques sont reconnues comme étant une conséquence de l'action des caspases (voir section 1.1.1.1). Or, certains changements morphologiques dramatiques, associés aux différents processus de mort cellulaires classiques et alternatifs, se manifestent même en présence d'inhibiteurs de caspases à large spectre comme le zVAD-fmk. Il semble qu'en l'absence des caspases, d'autres protéases, notamment les calpaïnes, les cathepsines, et certaines serines protéases, auraient un rôle clé dans l'exécution des processus de mort cellulaire alternatifs 64. Bien
qu'une coopération entre ces protéases et les caspases existe au cours de l'apoptose classique, certaines études révèlent leur implication dans divers mécanismes de mort cellulaire programmée indépendants des caspases 6' 7.
Les calpaïnes
Les calpaïnes sont des protéases à cystéine activables par le calcium. Parmi les 14 membres de cette famille, la n-calpaïne et la m-calpaïne sont ubiquitaires dans les cellules humaines. En présence d'une concentration physiologique de calcium intracellulaire, les calpaïnes
sont maintenues inactives par leur inhibiteur naturel, la calpastatine. Lorsque la concentration du calcium intracellulaire augmente, la liaison du calcium libère le domaine catalytique de la calpaïne. L'activation de la calpaïne entraîne la protéolyse de la calpastatine et de divers autres substrats impliqués dans l'organisation du cytosquelette, le contrôle du cycle cellulaire, la signalisation intracellulaire et l'apoptose (pour synthèse, voir
98,99x
Certaines études supportent un rôle pour les calpaïnes en amont ou en aval de l'activation des caspases dans différents systèmes. Par exemple, les caspases et les calpaïnes agiraient en synergie lors de l'apoptose induite par le stress oxydant chez les cellules photoréceptrices de la rétine et les cellules de neuroblastome humain 100'I01. La calpastatine
est un substrat des caspases, et sa protéolyse mènerait à l'activation des calpaïnes dans i m i m
plusieurs types cellulaires ' . De plus, les caspases et les calpaïnes possèdent une grande variété de substrats en commun pouvant expliquer leur activité synergique lors de
97
certains programmes apoptotiques .
Dans certains systèmes, les calpaïnes servent d'élément initiateur de l'apoptose en activant les caspases directement ou par l'intermédiaire des régulateurs de la famille Bcl-2. Par exemple, l'activation de la caspase-12 par la m-calpaine serait responsable de la cytotoxicité des plaques |3 amyloïdes et de l'ischémie neuronale causées par la maladie d'Alzeimer chez le rat104. Or, l'incorporation d'un nucléotide dans le gène de la caspase-12
chez la majorité des populations humaines, entraînerait la production d'une caspase-12 dépourvue d'activité enzymatique 105. Chez l'humain, la cytotoxicité des plaques |3
amyloïdes a plutôt été attribuée à l'activation de la caspase-4, mais l'effet des calpaïnes sur cette caspase reste inconnu 106. Également, les calpaïnes activent la molécule
proapoptotique Bid en réponse au cisplatin, dans les cellules de mélanomes humains 107.
Les calpaïnes peuvent aussi agir comme protéase exécutrice de la mort cellulaire en absence de l'activation des caspases. En effet, une forme active de la vitamine D provoque une mort cellulaire dépendante des calpaïnes, mais indépendante des caspases dans les cellules de cancer du sein 108.
Les cathepsines
La libération vers le cytosol et/ou le noyau des protéases lysosomiales à cystéine, cathepsines B et L, et de la protéase lysosomiale à aspartate, cathepsine D, a été associée avec l'induction de la mort cellulaire programmée (pour synthèse, voir 95' 109). La
translocation des cathepsines suite à la perméabilisation partielle des lysosomes a été observée en réponse à une grande variété de stimuli pouvant induire la mort cellulaire, notamment Fas L ou TNF, l'activation de p53, le stress oxydant, la privation en facteur de croissance et le traitement par des agents stabilisateurs des microtubules ou par des agents lysosomotropiques 109. De plus, des données génétiques supportent un rôle pour la
cathepsine B dans la mort cellulaire. En effet, des cellules hépatiques provenant de souris déficientes pour la cathepsine B seraient résistantes à l'apoptose induite par le TNF "°, tandis qu'une destruction massive des cellules serait observée au niveau du cerveau des souris nulles pour la cystatine B, l'inhibiteur naturel de la cathepsine B " '.
Comme les calpaïnes, les cathepsines B, L ou D peuvent intervenir en amont ou en aval des caspases, selon le contexte cellulaire et le stimulus impliqué. Entre autres, l'activation de la cathepsine B en aval des caspases est nécessaire à l'apoptose induite par les sels biliaires chez les hépatocytes de rats "2. Chez des cellules provenant de cancer de l'utérus, la
cathepsine B est un effecteur essentiel en aval des caspases initiatrices activées par le TNF
113. Notamment, la cathepsine B clive et active certaines caspases in vitro, suggérant un rôle
pour les cathepsines en amont des caspases n . Cependant, plusieurs études réalisées in vivo indiquent que l'activation des caspases par les cathepsines se produirait indirectement, via le clivage de Bid et l'induction de changements de la perméabilité mitochondriale et la libération du cytochrome c "5"'19.
Des travaux majeurs réalisés par Jââttelâ et collaborateurs indiquent que les cathepsines possèdent également une activité toxique indépendante des voies classiques de l'apoptose chez certaines cellules cancéreuses. Selon ces travaux, la mort des cellules de fibrosarcome induite par le TNF est indépendante des caspases et nécessite l'activité cytosolique de la cathepsine B 113. Ce groupe a également démontré qu'une déplétion de la protéine de choc
programme de mort cellulaire serait indépendant des caspases et résistant à la surexpression de Bcl-2 dans plusieurs lignées cellulaires provenant de cancer du sein humain 73.
Les serines protéases
Afin d'assurer l'élimination des cellules endommagées ou infectées, les lymphocytes cytotoxiques libèrent des granules qui sont captées par les voies d'endocytose puis libérées dans le cytosol de la cellule cible (pour synthèse, voir 121). Ces granules regroupent
plusieurs types de serine protéases, dont les plus étudiées sont les granzymes A et B. Le granzyme B (GrB) induit l'apoptose classique par le clivage de Bid 122124. Or, le GrB peut
également induire une forme de nécrose programmée en présence d'inhibiteurs de caspases
125. De plus, le GrB cliverait directement ICAD (Inhibitor of caspase activated DNase)
pour induire la fragmentation de l'ADN en absence de l'activation des caspases 126. Le
granzyme A (GrA) induit la mort cellulaire indépendante des voies classiques de l'apoptose . Son activité toxique implique sa capacité à dégrader les lamines A, B et C et à induire des bris simple-brin dans l'ADN en activant une DNAse associée au réticulum endoplasmique 129.
Dans un contexte différent, d'autres serines protéases, notamment Omi/HtrA2, se sont révélées importantes pour la mort cellulaire. Lors de l'induction de la voie intrinsèque de l'apoptose, Omi est relâchée de l'espace intermembranaire de la mitochondrie vers le cytosol 18. Dans le cytoplasme, Omi lie et clive les IAPs pour permettre l'activation des
caspases et l'apoptose. 130' . Omi induit également une voie de mort cellulaire
indépendante des caspases qui implique son activité serine protéase par un mécanisme inconnu 18'132. Il semble que dans certaines circonstances où les caspases sont inactivent,
l'activité serine protéase de Omi pourrait induire l'apoptose non classique sans être libérée de la mitochondrie . La protéine anti-apoptotique HAX-1 (HSl-associatedprotein-1), un substrat mitochondrial de Omi, pourrait y jouer un rôle '" .
D'après ces données récentes, la machinerie de mort cellulaire est beaucoup plus complexe que ce qui avait été décrit au départ. Considérant le rôle crucial des mécanismes de mort cellulaire programmée dans le maintien de l'homéostasie cellulaire, il apparaît évident que plusieurs mécanismes alternatifs de mort cellulaire aient émergé au cours de l'évolution des
organismes pluricellulaires. Ainsi, dans un contexte où l'activation des caspases est bloquée, d'autres protéases comme les calpaïnes, les cathepsines et les serine protéases peuvent agir comme deuxième ligne de défense pour assurer la protection de l'organisme contre les désordres d'ordre prolifératif. D'ailleurs, puisque ces protéases sont souvent fortement exprimées dans les tumeurs agressives 134, les cellules tumorales seraient
sensibilisées aux programmes de mort alternatifs qui impliquent les calpaïnes, les cathepsines ou les serines protéases 7 . À l'appui de cette hypothèse, des programmes de
mort cellulaire alternatifs se sont révélés sélectifs pour les cellules transformées. Par exemple, l'autophagie indépendante des caspases induite par Binl en présence de l'oncogène c-Myc est bloquée par un inhibiteur de serine protéases 135. Aussi, la déplétion
de HSP 70 de même que l'expression de la protéine adénovirale E4orf4 induisent l'apoptose non-classique dans les cellules transformées, mais pas dans les cellules normales 73,136 r ja n s u n contexte normal, ces protéases contribuent avec les caspases, à l'élimination
efficace des cellules potentiellement dangereuses. Une cascade d'activation entre les calpaïnes et les cathepsines a également été proposée dans des cas d'ischémie neuronale où l'activation des calpaïnes mène à la libération des cathepsines puis à la mort du neurone 137' 138. Ceci illustre clairement les possibilités de connexions qui relient non seulement les
processus alternatifs avec les voies classiques de l'apoptose, mais bien toute la machinerie exécutrice de la mort cellulaire programmée.
1.1.3.2 Les organelles impliquées dans la mort cellulaire programmée
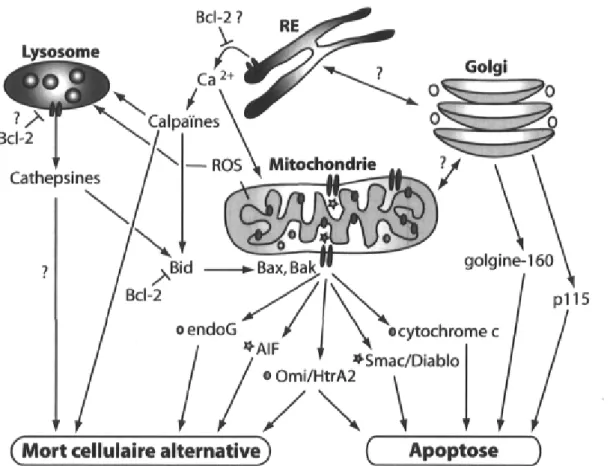
Les connexions entre les différentes voies de signalisation de la mort cellulaire programmée se traduisent également par une communication s'établissant entre les différentes organelles pour propager efficacement le signal de mort93. De nombreux stimuli
provoquent des changements de perméabilité de la membrane mitochondriale. Or, les données s'accumulent à l'effet que d'autres organelles comme le réticulum endoplasmique, les lysosomes et l'appareil de Golgi seraient en mesure de détecter des stimuli spécifiques et d'activer localement des voies de signalisation de mort cellulaire par l'intermédiaire de protéases ou par la libération de facteurs cytotoxiques. Certains de ces signaux sont transmis d'une organelle à une autre, tandis que d'autres peuvent induire directement la