T H E S E
en vue de l’obtention du
DOCTORAT DE L'UNIVERSITE DE TOULOUSE délivré par l’Université Toulouse III – Paul Sabatier
Ecole doctorale : Sciences de la Matière Spécialité: Physique et Nanophysique
présentée et soutenue par
Saioa COBO SANTAMARIA
le 23 novembre 2007
Dépôt en couches minces et nano-structuration de
complexes bistables à transition de spin et à transfert de
charge : élaboration et propriétés physiques
_______
Directeurs de thèse : Azzedine BOUSSEKSOU Gabor MOLNAR
______
JURY
M. Gero DECHER, Professeur à l’Université Louis Pasteur, Strasbourg, Président M. Didier ASTRUC, Professeur à l’Université de Bordeaux I, Rapporteur
M. Talal MALLAH, Professeur à l’Université de Paris XI, Rapporteur
M. José Antonio REAL, Professeur à l’Université de Valence, Espagne, Examinateur M. Michel ETIENNE, Professeur à l’Université Paul Sabatier, Toulouse, Examinateur
M. Azzedine BOUSSEKSOU, Directeur de recherche au CNRS, Toulouse, Directeur de thèse
Laboratoire de Chimie de Coordination du C.N.R.S. associé à l’Université Paul Sabatier 205 route de Narbonne, 31077 Toulouse Cedex
Je remercie Monsieur Jean-Jacques Bonnet, initialement Directeur du Laboratoire de Chimie de Coordination, de m’avoir accueilli au sein de celui-ci et de m’avoir permis de mener à bien mes travaux de recherche.
Je tiens également à remercier Messieurs Bruno Chaudret et Denis Neickbeker, actuels directeurs du Laboratoire de Chimie de Coordination, de m’avoir permis de poursuivre ce travail.
Ce travail de thèse a été réalisé dans le groupe « Propriétés Physiques Moléculaires » dirigé par Azzedine Bousseksou, Directeur de Recherche au CNRS, à qui j’exprime toute ma gratitude. D’abord pour m’avoir accueilli il a déjà quatre ans, et ensuite pour s’être battu afin que je puisse réaliser cette thèse. Je veux aussi exprimer ma plus sincère reconnaissance au Docteur Gabor Molnar, Chargé de Recherche au CNRS, et co-directeur de ma thèse, pour avoir crû en moi et m’avoir encadrée dans la juste mesure. Je voulais vous dire à les tous deux : MERCI.
Je tiens à remercier vivement Monsieur Didier Astruc, Professeur à l’Université de Bordeaux I, et Monsieur Talal Mallah, Professeur à l’Université de Paris XI, de m’avoir fait l’honneur d’accepter de juger ce travail.
J’exprime ma profonde gratitude à Monsieur Gero Decher, Professeur à l’Université Louis Pasteur (Strasbourg) et Michel Etienne, Professeur à l’Université Paul Sabatier pour leur participation au jury de thèse.
Que José Antonio Real, Professeur de l’Université de Valencia, participe à ce jury est un honneur pour moi. Sa contribution a été fondamentale à l’avancée de ce travail. Je lui adresse mes remerciements les plus sincères. Gracias José Antonio !
Je voudrais remercier toutes les personnes que j’ai connues lors de mes differents stages : un grand merci au Professeur John McGarvey à Belfast et à l’ensemble de l’équipe de Valencia pour l’accueil qui m’a été réservé lors de mes séjours là bas. Mes remerciements vont aussi à Christophe Vieu, Professeur à l’INSA de Toulouse, pour son aide précieuse.
Au sein du laboratoire, je souhaite remercier les personnes qui ont participé, de près ou de loin, à ce travail de recherche.
Auprès de Monsieur Lionel Salmon, Chargé de Recherche au CNRS, j’ai pu trouver, en plus de conseils avisés, une atmosphère chaleureuse et détendue. Un grand merci pour tout Lionel !
C’est avec le plus grand plaisir que je remercie Lionel Rechignat, ITA, pour avoir su me supporter pendant ces années et pour toutes les mesures Raman et SQUID réalisées. Je ne voudrais pas oublier Nicolas Bréfuel à qui je dois une grande partie de cette thèse : pour m’avoir donné l'envie de l’entreprendre, pour ses précieux conseils et pour toutes les journées passées à corriger les fautes d’orthographe de ce manuscrit…
J’adresse aussi mes remerciements à tous mes camarades, amis du laboratoire et d’ailleurs pour leur soutien, leur bonne humeur et leur sympathie : Merci Sébas, Thomas, Amaia, Laurent, Carlos, Denis, Raquel, Villö, Petra, Nawel, Mouri…
Enfin, même loin de mon cher Pays Basque, j’ai eu le soutien de l’ensemble de mes proches, en particulier ma mère et mon père, toujours à mes cotés. Gracias Aita y Ama!
Sommaire
Introduction générale p. 1
Partie A - Transition de spin : Clathrates de Hofmann
A.1 Introduction à la Transition de spin p. 5
A.1.1 Le phénomène de la transition de spin p. 6 A.1.2 Déclenchement de la transition de spin p. 9 A.1.2.1 Origine entropique de la transition de spin thermo-induite p. 10 A.1.2.2 Transition de spin photo-induite p. 13 A.1.3 Principales méthodes d’étude de la transition de spin p. 16
A.1.4 Références p. 19
A.2 Introduction aux Clathrates de Hofmann p. 22
A.2.1 Composé {Fe(pz)[Pt(CN)4]}·n H2O p. 27
A.2.1.1 Description de la structure cristalline p. 27 A.2.1.2 Influence du degré de solvatation sur les propriétés magnétiques p. 29
A.2.1.3 Spectroscopie Raman p. 30
A.2.1.4 Etudes des effets de Pression p. 32 A.2.1.5 Photo-commutation réversible par impulsion laser p. 33
A.2.2 Références p. 35
A.3 Dépôt en couches minces des complexes Fe(pz)[M(CN)4] (M=Pt, Pd, Ni) p. 36
A.3.1 Synthèse : dépôt en couches minces du complexe p. 39
A.3.1.1 Le substrat p. 39
A.3.1.2 La couche d’accrochage p. 39 A.3.1.3 Assemblage du complexe p. 40
A.3.2 Etude des propriétés p. 40
A.3.2.2 Etude des propriétés physiques : Micro-Spectroscopie Raman p. 44 A.3.3 Conclusions et perspectives p. 47
A.3.4 Références p. 49
A.4 Dépôt en couches minces du complexe Fe(azpy)[M(CN)4] (M=Pt, Pd, Ni) p. 51
A.4.1. Synthèse des complexes Fe(azpy)(M(CN)4)]·H2O p. 51 sous forme de poudre
A.4.2. Synthèse en couches minces de la famille Fe(azpy)(M(CN)4)]·H2O p. 53 A.4.3 Comparaison des propriétés physico-chimiques p. 54 de la poudre et des dépôts
A.4.3.1 Techniques d’imagerie p. 54
A.4.3.2 Propriétés physiques p. 57
A.4.4 Conclusions et perspectives p. 64
A.4.5 Références p. 65
A.5 Structuration de la surface p. 66
A.5.1 Introduction p. 66
A.5.2 Lithographie électronique et lift off p. 67 A.5.3 Caractérisation et étude des propriétés p. 71
A.5.3.1 Techniques d’imagerie p. 71
A.5.3.2 Détection Raman p. 78
A.5.4. Structuration par Sélectivité chimique p. 81
A.5.4.1 µ-contact printing (µCP) p. 82
A.5.4.2 Lift-off p. 84
A.5.4 Conclusion p. 88
Partie B - Transfert de charge : Bleu de Prusse et analogues
B.1 Introduction p. 94
B.1.1 Composes de formule Ax[B(CN)6]y·zH2O p. 94 B.1.2 Composés de formule DAII[BIII(CN)6] ·xH2O p. 96
B.1.3 Résultats : le Bleu de Prusse et ses analogues p. 96
B.1.3.1 Tautomères de valence p. 96 B.1.3.2 Les aimants moléculaires à haute température critique p. 98 B.1.3.3 De nouveaux analogues p. 99
B.1.4 Références p. 101
B.2 Dépôt en couches minces du bleu de Prusse p. 104
B.2.1. Etat de l’art p. 104
B.2.2. Dépôt en couches minces du complexe du bleu de Prusse: p. 105
B.2.2.1. Le substrat p. 105
B.2.2.2 Assemblage du complexe p. 105 B.2.3. Caractérisation des couches minces du bleu de Prusse p. 106 B.2.3.1. Propriétés physiques des dépôts KxFe[Fe(CN)6] y p. 111 B.2.4. Structuration de la surface du BP p. 117 B.2.4.1 Lithographie optique p. 118 B.2.4.2 Caractérisation microscopique p. 119 B.2.4.3 Lithographie électronique et lift off p. 123
B.2.4.4 µ-contact printing p. 126
B.2.5 Conclusion p. 128
B.2.6 Références p. 129
B.3 Dépôt en couches minces des analogues du bleu de Prusse : AxCo[Fe(CN)6]y·zH2O
B.3.1. Etat de l’art. p. 131
B.3.2.1. Couche d’accrochage p. 133 B.3.2.2. Dépôt en couches minces p. 135
B.3.3. Caractérisation des couches minces du NaxCo[Fe(CN)6]y·zH2O p. 136 B.3.2.1. Etudes en fonction de la concentration de sodium p. 140
B.3.2.2. Etude Raman en fonction de la température p. 142 B.3.2.3. Etude des propriétés magnétiques des dépôts p.144 de AxCo[Fe(CN)6]y·zH2O
B.3.2.4. Etude du photomagnétisme p. 146 B.3.4 Conclusion perspectives p. 149
B.3.5 Références p. 151
B.4 Synthèse en forme de poudre : RbMn[Fe(CN)6] p. 152
B.4.1 Introduction p. 152 B.4.2Synthèse p. 154 B.4.3 Stœchiométrie p. 154 B.4.4 Mesures magnétiques p. 156 B.4.5 Changement d’entropie lors de la transition de phase p. 160 B.4.6 Conclusions et perspectives p. 164 B.4.7 Références p. 165
Conclusion générale p. 166
Abréviations Phen : 1,10- Phénantroline Ptz : 1-n-Propyl-tétrazole pm-bia : N-2’-pyridilméthylèn-4-aminobiphényl pz : pyrazine py : pyridine azpy : 4,4’-azopyridine TBA : Tétrabutylammonium PMMA : poly(méthyl méthacrilate) IPA : IsoPropylic Alcool
BP: bleu de Prusse
PDMS : poly(diméthylsiloxane) OTS : trichloro(octadécyl)silane ODT octadécanethiol
APTES : 3-amino-propyl triéthoxy silane HDMS : hexaméthyldisiloxane
PAH : poly(allylamine hydrochloride) PSS : poly(styrène sulfonate)
AFM : Microscopie à Force Atomique ATG : Analyse Thermogravimétrique BT : Basse Température
BS : Bas Spin
µCP : µ-Contact Printing
DSC : Differential Scanning Calorimetry EBL : Electron-Beam Lithography EDX: Energy Dispersive X-ray analysis FC : Field-Cooled magnetization HS : Haut Spin
HT : Haute Température LbL : Layer by Layer
LD-LISC : Ligand-Driven Light Induced Spin Change LIESST : Light Induced Excited Spin State Trapping
LITH : Light Induced Thermal Hysteresis MEB : Microscopie Electronique à Balayage MLCT : Metal to Ligand Charge Transfer MSA : Multiple Sequential Adsorption PIPT : Photo-Induced-Phase Transition
RPE : Résonnance Paramagnétique Electronique SA: Self-Assembled
SAM : Self-Assembled Monolayers
SERS: Surface enhanced Raman scattering TS : Transition de Spin
XANES : X-ray Absorption Near Edge Structure XES : X-ray Emission Spectroscopy
1
Introduction générale
De nos jours, l’idée qu’une molécule ou un ensemble de molécules (matériau moléculaire) puissent servir comme un élément actif dans un dispositif électronique stimule de plus en plus l’activité scientifique des laboratoires à l’échelle de la planète. Une des stratégies des plus prometteuses est basée sur le concept de bistabilité moléculaire, qui se traduit par le basculement entre deux états électroniques, de la même manière qu’un interrupteur binaire : il est ainsi possible de passer de façon réversible et détectable d'un état (OFF = 0) à un autre (ON = 1) sous l'effet d'une perturbation extérieure contrôlée.
Dans ce contexte, le phénomène de la transition de spin constitue un exemple typique de cette propriété. Il est possible dans ce cas de commuter le système entre ses états de spin haut et bas. Un autre exemple est celui des tautomères de valence dans lesquels la commutation est liée à un transfert d’électron. La présence d’hystérésis du phénomène pour certains de ces composés en fait des matériaux de choix pour les dispositifs de stockage de l’information à très petite échelle.
En effet, dans le domaine informatique, les demandes technologiques en termes de capacité de stockage de l’information augmentent de manière exponentielle et sont à l’origine du développement des nanosciences : l’objectif est de stocker toujours plus dans un volume le plus petit possible et le plus rapidement possible. De plus, la miniaturisation d’autres composants tels que les capteurs, les dispositifs de l’optique est aussi un domaine en plein expansion.
L’élaboration d’objets de taille nanométrique est, outre leur intérêt intrinsèque pour des applications données, un enjeu majeur du point de vue du fondamental : l’objectif est de maîtriser des systèmes modèles pour progresser dans la compréhension d’effets spécifiques aux objets de petite taille. Par exemple, dans le cas des complexes à transition de spin, une des questions des plus importantes est de savoir comment la réduction de taille influencera la largeur de l’hystérésis observée à l’échelle macroscopique ainsi que l’établissement même du phénomène via les processus coopératifs.
2
Le travail de thèse que je présente dans ce manuscrit s’inscrit, pour la première fois, dans le cadre d’une combinaison de ces deux thématiques : la bistabilité moléculaire et les nanosciences, tant au niveau de la synthèse des objets qu’au niveau de leur analyse par les différents méthodes existantes. Un premier objectif de cette thèse est la synthèse de couches minces avec maintien de la bistabilité autour de la température ambiante et, si possible, avec hystérésis. Un deuxième objectif principal est la nanostructuration de ces couches minces avec maintien de la propriété. Le mariage de ces deux objectifs donne lieu à la stratégie de recherche ‘‘de la molécule au device’’, en passant par toutes les études fondamentales physico-chimiques que nécessite le passage du macroscopique au nanoscopique.
En effet, le dépôt en forme de couches minces des complexes à transition de spin est une étape incontournable dans la conception de tout dispositif, mais aussi dans la fabrication des nano-objets par les méthodes lithographiques. Cependant, les méthodes de dépôt proposées jusqu’à présent sont peu adaptées aux procédés de la microélectronique. L’obstacle majeur rencontré lors du dépôt de ces matériaux est la perte de la propriété de transition de spin ou la non-régularité de la surface obtenue ou encore la dilution du produit actif dans une matrice inactive.
La première partie de ce manuscrit concernera les complexes à transition de spin et sera divisée en cinq sous-chapitres. Le premier sous-chapitre sera consacré à la présentation générale des propriétés de la transition de spin des composés du Fe(II), en rappelant brièvement le phénomène. Dans le deuxième sous-chapitre, nous présenterons une introduction aux matériaux de type « clathrates d’Hofmann » et leurs propriétés. Les troisième et quatrième sous-chapitres aborderont le dépôt en couches minces par la méthode d’assemblage séquentielle des « clathrates d’Hofmann » Fe(pyrazine)[M(CN)4] et Fe(4,4’-azopyridine)[M(CN)4] à transition de spin (M : Ni(II), Pt(II), Pd(II)). Enfin, le cinquième
sous-chapitre abordera la nano-structuration de ces couches minces à transition de spin par les techniques lithographiques.
La deuxième partie du mémoire sera dédiée aux complexes à transfert de charge et est décomposée en quatre sous-chapitres. Le premier sous-chapitre donnera un aperçu des systèmes à transfert de charge de la famille du bleu de Prusse. Dans le deuxième sous- chapitre sera détaillée la synthèse des couches minces du bleu de Prusse par la méthode d’«assemblage électrostatique séquentielle» ainsi que sa nanostructuration et sa
3
caractérisation. Ces méthodes élaborées seront utilisées dans le troisième sous-chapitre pour la fabrication des dépôts des analogues Co-Fe du bleu de Prusse, susceptibles de posséder un transfert charge. Enfin, dans le quatrième sous-chapitre, nous présenterons l’étude de l’analogue Rb-Mn-Fe du bleu de Prusse possédant un transfert de charge. Cette dernière étude a été achevée sur les échantillons massifs. Leur mise en forme de couches minces est encore en cours.
Enfin, nous conclurons ce manuscrit en rappelant les principales avancées que nous avons apportées aux différentes questions abordées puis nous proposerons quelques perspectives de recherche.
5
A.1 Introduction à la transition de spin
Au début du XXème siècle, la synthèse par Delépine[1] d’un nombre important de complexes du Fe(III), donna lieu à plusieurs publications de Cambi et al., entre 1931 et 1937.[2-4] Ces travaux incluaient l’étude des propriétés magnétiques de ces complexes à base de ligands dérivés de dithiocarbamates (Figure A.1.1). Vingt ans plus tard, après la consolidation de la théorie du champ de ligands et de la chimie de coordination, Martin et White font des apports décisifs sur l’étude détaillée des propriétés magnétiques des complexes tris(dithiocarbamate) du Fe(III).[5] Ces complexes présentent en fonction de la température des variations du moment magnétique. Les états haut-spin (HS) et bas-spin (BS) peuvent coexister en équilibre thermodynamique. Ceci établit les bases conceptuelles de l’équilibre de la transition spin (TS).
Figure A.1.1. Anion dithiocarbamate
Depuis, de tels équilibres ont été observés pour un grand nombre de complexes du Fe(II) et Fe (III),[6-8] du Co(II) [9-11] et Co(III),[12-14] du Cr(II), [15, 16] du Mn(II) [17, 18] et Mn(III) [19, 20] qui ont été étudies tant à l’état solide qu’en solution.
Les complexes à TS font l’objet d’un important regain d’intérêt ces vingt dernières années,[21] du fait que : (i) d’une part, les perturbations utilisées pour modifier l’équilibre se diversifient notamment avec la découverte des effets photo-induits dans ces systèmes ; (ii) d’autre part, parmi les systèmes synthétisés présentant des transitions thermo-induites, certains sont le siège d’une hystérésis ouvrant la voie à des applications potentielles, en particulier dans le domaine du stockage de l’information ou de l’affichage numérique.[21-23a]
Dans ce chapitre, nous présenterons les concepts les plus importants du phénomène de la TS.
N
S
S R
6
A.1.1. Le phénomène de la transition de spin
Certains métaux de la première série de transition, de configuration électronique 3dn (4< n < 7) peuvent exister dans deux états électroniques stables, selon la force du champ cristallin : l’état HS et l’état BS.
Pour ces systèmes, l’effet d’un champ de ligand octaédrique provoque l’éclatement des niveaux d’énergie des orbitales d (dégénérés dans le cas de l’ion libre) en deux niveaux d’énergie : un premier niveau comportant trois orbitales t2g non liantes et triplement dégénéré (dxy, dyz, dzx) et un second composé de deux orbitales eg antiliantes et doublement dégénéré
(dz2, dx2-y2). Ces deux niveaux sont séparés par un éclatement de 10 Dq, caractérisant la force
du champ de ligand. Cet éclatement dépend de la nature de l’ion et des ligands qui l’entourent.
Figure A.1.2: Levée de dégénérescence et configuration électronique des états HS et BS dans le cas d’un complexe octaédrique du FeII.
Pour des systèmes comptant plus d’un électron d, la répulsion électron-électron (énergie d’appariement Π) doit être aussi considéré en même temps que la force du champ de ligands.
e
ge
gt
2gt
2g10Dq
HSΠ>10Dq
S=2
5T
2gΠ<10Dq
S=0
1A
1gIon libre
10Dq
BSe
ge
gt
2gt
2ge
ge
gt
2gt
2g10Dq
HSΠ>10Dq
S=2
5T
2gΠ<10Dq
S=0
1A
1gIon libre
10Dq
BS7
Dans le cas du Fe(II) de configuration électronique d6, deux cas limites sont envisageables en fonction de la force du champ de ligands (Figure A.1.2), conduisant aux états fondamentaux 1A
1 et 5T2g comme le montre le diagramme de Tanabe-Sugano (Figure A.1.3).
- Quand Π > 10 Dq (champ cristallin faible) : les électrons d se répartissent sur les deux niveaux t2g et eg en respectant la règle de Hund du maximum de spins parallèles. Le spin total est S = 2 et la configuration électronique énergétiquement la plus favorable est celle de l’état HS paramagnétique 5T2g (t42ge2g).
- Dans le cas où Π < 10 Dq (champ cristallin fort) : les électrons s’apparient dans les orbitales t2g de plus basse énergie, en violant la règle de Hund et en suivant cette fois la règle de Pauli. Le spin total est S = 0 et le complexe est dans l’état BS diamagnétique 1A
1g(t62g).
Le diagramme de Tanabe-Sugano de la figure A.1.3 montre l’ensemble de termes spectroscopiques correspondant aux niveaux fondamentaux et excités d’un ion d6 en environnement octaédrique en fonction de la force du champ du ligand 10Dq. Pour des complexes avec un champ ligand proche de la valeur critique (∆crit), une TS provoque le changement du paramètre 10Dq.
Figure A.1.3 : Diagramme de Tanabe-Sugano pour un ion de configuration d6 en symétrie octaédrique.
De plus, les liaisons de coordination métal-ligand des complexes FeII HS sont substantiellement plus faibles et leur longueur plus grande que pour les complexes BS (1.9-2.0 Ǻ dans l’état BS et 2.1-2.2 Ǻ dans l’état HS, soit un changement d’environ 10%).
8
Figure A.1.4 : Variation de l’énergie potentielle des niveaux HS et BS pour des complexes à TS du FeII en fonction de la distance métal-ligand.
Comment évolue la force du champ de ligand lorsqu’un complexe passe de l’état HS à l’état BS ?
Le rapport des forces de champ de ligand entre les deux états de spin est donné par l’équation:
où rHS et rBS sont respectivement les distances métal-ligand moyennes dans les états HS et BS
et n = 5-6. En considérant les valeurs moyens rBS = 2.0 Ǻ et rHS = 2.2 Ǻ et n = 6, ce rapport est estimé à ~1.77.
En se basant sur le diagramme de la figure A.1.3 et A.1.4, du fait que 10Dq dépende de la longueur de liaison métal-ligand et comme Π varie peu, la différence d’énergie ∆E°= E°HS -E°LS peut être estimée en fonction de 10DqHSou 10DqBS :
- Pour 10DqHS < 10000 cm-1, ∆E° < 0 et l’état fondamental est HS qui est donc thermodynamiquement stable à toute température.
- Pour 10DqBS> 23000 cm-1, ∆E° > 2000 cm-1et l’état fondamental est BS qui reste stable pour des très hautes températures.
( )
HSBS n HS BSr
r
Dq
Dq =
10
10
9
Pour l’intervalle 10DqHS= 11000-12500 cm-1et 10DqBS= 19000-22000 cm-1, ∆E° = 0-2000 cm-1 et correspond à l’intervalle de champ de ligand pour lequel le phénomène de TS thermo-induite peut avoir lieu (Figure A.1.5).
Figure A.1.5 : Régions de stabilité des états HS et BS en fonction de la force du champ de ligand.[23]
A.1.2. Déclenchement de la transition de spin
Il est possible de contrôler la configuration HS ou BS d’un système à TS au moyen de perturbations extérieures telles qu’une variation de température (T), d’une pression hydrostatique (P), d’un champ magnétique intense (H), ou d’une irradiation lumineuse (hν). Dans le cas de la pression, l’état BS est favorisé. Ceci conduit à déplacer l’équilibre du système biphasique à TS à plus haute température. Cet effet peut s’expliquer en tenant compte de la réduction de volume qui accompagne le changement de l’état HS vers l’état BS : la pression favorise l’état de plus petit volume.
L’application d’un champ magnétique (H) provoque un glissement de son cycle d’hystérésis thermique vers les basses températures. Ce déplacement thermique négatif est rigoureusement opposé à l’effet d’une pression et peut s’expliquer par l’effet Zeeman : le champ magnétique stabilisant l’état de plus haut spin ici l’état HS.
Dans ce chapitre, les effets d’une variation de température (TS thermo-induite) et d’une irradiation lumineuse (TS photo-induite) seront décrits plus en détail plus loin.
10
A.1.2.1. Origine entropique de la transition de spin thermo-induite
Parmi les moyens d’induire une transition de spin, la variation de température est historiquement le premier et sans conteste le plus utilisé, servant même le plus souvent de référence et de comparaison par rapport aux autres stimuli. Du point de vue thermodynamique, l’énergie libre de Gibbs G est composée d’un terme entropique TS et d’un terme enthalpique H. A pression constante, la variation d’énergie libre ∆G vérifie à chaque instant la relation :
∆G = ∆H − T∆S
avec dans le cas d’une TS : ∆G = GHS −GBS, ∆H = HHS −HBS et ∆S = SHS − SBS.
Les deux quantités ∆H et ∆S sont positives. A basse température, le terme enthalpique domine, ∆G est positive et l’état fondamental est BS. A haute température au contraire, le terme entropique devient dominant, ∆G est négative et l’état fondamental est HS. Enfin, à la température d’équilibre T1/2 pour laquelle la moitié des molécules se trouvent dans l’état HS et l’autre dans l’état BS, ∆G = 0 et T1/2=∆H/∆S.
La variation de l’entropie peut être estimée en prenant en compte les contributions électroniques, vibrationnelles, configurationnelles, rotationnelles et translationnelles. Ces deux dernières pouvant être exclues si on considère le complexe à l’état solide. De plus, en absence de désordre d’orientation, le terme configurationnel est aussi exclu. L’entropie est composée alors en deux contributions, une électronique (∆Sél) et une vibrationnelle (∆Svib) :
∆S =∆Sélec + ∆Svib + ∆Sconf + ∆Srot + ∆Strans
∆Sélec est elle-même la somme de contributions dues à des changements d’état de spin (∆Sélecspin) et de moment orbital (∆Sélecorb). Cependant, la dégénérescence orbitale n’est à prendre en compte qu’en cas de symétrie octaédrique parfaite, ce qui n’est jamais le cas pour les systèmes moléculaires complexes. En conséquence, dans la plupart des cas :
⎥ ⎦ ⎤ ⎢ ⎣ ⎡ + + = ∆ = ∆ LS HS élec élec S S R S S spin ) 1 2 ( ) 1 2 ( ln
11
où (2S+1)HS et (2S+1)BS sont respectivement les multiplicités de spin de l’état HS et BS. De ce fait, pour un ion Fe(II) où la conversion de spin a lieu entre les états 1A1 et 5T2, ∆Sélspin = R ln 5/1 = 13,38 J K-1 mol-1.
Expérimentalement les valeurs de ∆S pour des TS complètes de complexes du Fe(II) se situent entre 30 et 80 kJ mol−1[29]. Ces valeurs expérimentales sont très supérieures aux 13,38 J K-1 mol-1 correspondants à la variation de l’entropie d’origine électronique. La contribution restante vient alors des variations des fréquences de vibration moléculaires. Ainsi, la différence de la valeur totale de l’entropie obtenue expérimentalement et l’entropie d’origine électronique déterminent la contribution vibrationnelle dont Sorai et Seki ont montré les premiers le rôle prépondérant.[30] Étant donné que ∆Sélec et ∆Svib sont toujours positives, ∆Stotal peut être considérée comme la force motrice de la TS, le terme T∆Stotal modulant la différence d’énergie entre GHS et GBS.
Les paramètres thermodynamiques ∆H et ∆S associés à la TS sont obtenus directement à partir des mesures calorimétriques pour lesquelles une anomalie de Cp est observée autour de T1/2 :
Figure A.1.6: capacité calorifique (à p constante) pour le complexe [Fe(Phen)2(NCSe)2].30
Depuis la découverte du premier complexe à TS, la compréhension du phénomène et des propriétés associées à celui-ci ont considérablement progressé, tant du point de vue physique que de la synthèse chimique.
En phase liquide, le phénomène de changement (transition) de spin s’effectue toujours selon une statistique de Boltzmann : la TS est par conséquent très graduelle.
A l’état solide, les effets de réseau cristallin, résultant des interactions entre molécules, conduisent à une grande variété de comportements : la TS peut être graduelle (complète
12
(A.1.7a) ou incomplète (A.1.7e)), correspondant à des systèmes présentant de très faibles interactions ou à des systèmes fortement dilués. Chaque centre métallique subit alors le phénomène de TS indépendamment de son voisin. A l’inverse, une TS abrupte (A.1.7b) est caractéristique d’un système beaucoup plus coopératif possédant des interactions fortes. Chaque centre métallique est fortement associé aux autres et subit l’influence de la modification d’état de spin de ses voisins. Il est important de noter que ces interactions entre les molécules ont une origine élastique et sont donc directement liées au changement de volume des molécules lors de la TS.[27]
Figure A.1.7 : Les différents types de transition de spin possibles :
(a) graduelle ; (b) abrupte ; (c) avec hystérésis ; (d) en deux étapes ; (e) incomplète.
La TS peut être aussi accompagnée d'une boucle d’hystérésis (A.1.7c), celle-ci conférant au système un effet mémoire sur un intervalle de températures qui pourrait être mis à profit dans l’élaboration de dispositifs de stockage d’information, d’affichage et de commutation moléculaire. Les transitions avec hystérésis (figure A.1.7c) sont définies par deux températures, une en mode de refroidissement (T↓1/2) et l’autre en mode de chauffage (T↑1/2). La TS peut aussi s’effectuer en deux étapes (A.1.7d) en présence de deux sites cristallographiques distincts dans le cristal mais également dans les systèmes mono ou binucléaires où l’environnement des atomes métalliques est identique. Dans ces systèmes, des
13
interactions de courte portée rentrent en compétition avec des interactions de longue portée et vont stabiliser les paires HS-BS conduisant à une transition de spin en deux étapes. Dans le langage des modèles phénoménologiques, cette stabilisation peut être vue comme la manifestation d’une interaction type « antiferro ». On peut citer comme exemple le complexe binucleaire {Fe(bt)(NCS)2]2bpym} dans lequel l’augmentation du volume d’un des deux centres métalliques lors de la TS donne lieu à une augmentation de la barrière d’énergie entre l’état HS et BS pour le deuxième centre métallique. Cet effet dû à une pression intra-moléculaire générée par le premier centre métallique conduit à un décalage de la température de transition du deuxième centre vers de plus hautes températures.
A.1.2.2. Transition de spin photo-induite
Il existe deux principaux moyens d'induire la TS par la lumière (UV, visible ou proche IR) : soit directement sur l’ion à basse température (effet LIESST), soit sur le ligand (LD-LISC). Plus récemment, un autre phénomène appelé transition de phase photo-induite (PIPT- de l’anglais photo-induced-phase transition) a été également décrit : il s’agit alors d’irradier le complexe dans la boucle d’hystérésis thermique.
a) L’effet LIESST
:
McGarvey et al. [31] furent le premier en 1982 à montrer qu'une irradiation laser pulsée dans la bande de transfert de charge métal-ligand (MLCT en anglais) de certains complexes du Fe(II) à TS induisait la population transitoire d'un état HS.L’effet LIESST (abréviation de l'expression anglaise: "Light Induced Excited Spin State Trapping") a été découvert dans l’état solide il y a une vingtaine d’années par Descurtins et al.[32] Il correspond au piégeage quantitatif de composés dans un état HS métastable par pompage optique à température cryogénique (typiquement au dessous de 50 K), c'est-à-dire quand la relaxation HSÆBS, rapide à haute température, ralentit. Par exemple, une excitation lumineuse à 530 nm transforme l’état BS d’un composé, [Fe(ptz)6](BF4)2, en un état HS métastable dont la durée de vie à 20 K est supérieure à 106 s. Le mécanisme présumé de ce piégeage photo-induit est schématisé dans la figure 2.1.8 :
14 E r HS LS 1A 1 5T 2 1T 1 5E 3T 1 530 nm 820 nm 980 nm E r HS LS 1A 1 5T 2 1T 1 5E 3T 1 530 nm 820 nm 980 nm E r HS LS 1A 1 5T 2 1T 1 5E 3T 1 530 nm 820 nm 980 nm E r HS LS 1A 1 5T 2 1T 1 5E 3T 1 530 nm 820 nm 980 nm
Figure A.1.8: Diagramme de Jablonski représentant les effets LIESST direct et inverse: cas du complexe [Fe(ptz)6](BF4)2.[33] Les flèches ondulées représentant des relaxations
non-radiatives (intersystem crossing)
Ce processus implique initialement une transition depuis l’état fondamental 1A
1 vers l’état excité 1T
1 qui possède une durée de vie courte (<1 ps) [47] et relaxe vers l’état HS métastable 5T
2. Si la température est assez basse, le système reste piégé dans cet état, puisque la barrière d’énergie potentielle associée au passage vers l’état 1A1 est supérieure à l’énergie thermique du système, le processus de relaxation étant alors gouverné par l’effet tunnel.
Si ce phénomène a pu être reproduit pour un grand nombre de complexes à TS du FeII,[34] il est par contre beaucoup moins fréquent dans le cas du FeIII[35]. Dans ce dernier cas, la variation de la distance métal-ligand ∆rHL est plus faible et, par conséquent, la barrière d’énergie pour la relaxation HSÆBS est faible aussi.
Le phénomène inverse (reverse LIESST) a été mis en évidence par A. Hauser en 1986 sur le même matériau et correspond au basculement de l'état HS métastable vers l'état BS par irradiation dans le proche infra rouge (820 nm).[33,36]
Par ailleurs, Létard et al. [37] ont montré en 1998 la possibilité de générer une hystérésis thermique sous irradiation permanente, à basse température : c’est l’effet LITH (Light Induced Thermal Hysteresis) (figure A.1.9). Cet effet résulte de la compétition entre la population de l’état HS (par irradiation) et la dépopulation de celui–ci (par relaxation) en présence des interactions fortes.
15
Figure A.1.9 : Irradiation permanente à 647 nm du composé [Fe(PM-BiA)2(NCS)2]. Evolution en température (◊) avant irradiation et (■) pendant irradiation.
b) L’effet LD-LISC (Ligand Driven - Light Induced Spin Crossover) a été introduit par Zarembowitch et al.[38] il y a une quinzaine d’années avec l’ambition de mener à la synthèse de matériaux photo-commutables à des températures plus élevées que celles permises par l’effet LIESST. L’irradiation d’un composé à la longueur d’onde correspondant à la réaction d’isomérisation chimique du ligand permet de modifier le champ du ligand et donc le comportement magnétique du complexe.[39] Ces études utilisent la photo-isomérisation cis-trans de l’entité C=C incorporée à un ligand coordonné à un centre FeII.[40]
Par exemple, le composé [Fe(trans-stpy)4(NCS)2] présente une TS graduelle centrée autour de 180 K alors que le composé [Fe(cis-stpy)4(NCS)2] reste HS sur toute la gamme de température (10-300 K). Cette différence de propriété magnétique est attribuée à un champ de ligand plus fort dans le cas du ligand trans que du ligand cis. (Figure A.1.10)
16
Figure A.1.10: a) Structure cristallographique du
complexe [Fe(trans-stpy)4(NCS)2] (Ct) et
[Fe(cis-stpy)4(NCS)2] (Cc), b)χMT vs. T pour les isomères Ct
(trans) et Cc (cis).
c) PIPT dans la boucle d’hystérésis : La première transition BSÆHS photo-induite à haute température n’a pu être observée que très récemment dans le cas du complexe [Fe(pm-bia)2(NCS)2] à la température de 170 K, c'est-à-dire à l’intérieur de la boucle d’hystérésis thermique à l’aide d’un laser pulsé (8 ns).[17] Le changement inverse HSÆBS, n’a par contre pas pu être observé. Dans notre équipe, dans le cadre de la thèse de S. Bonhommeau[11] par application d’un laser pulsé de 8 ns a été mise en évidence par spectroscopie Raman une TS photo-induite et bidirectionnelle (BSÆHS et HSÆBS) à température ambiante à l’intérieur de la boucle d’hystérésis du complexe [Fe(pz)Pt(CN)6]. Cette observation ouvre des perspectives d’applications intéressantes et sera décrite plus en détail dans le chapitre A.2.
A.1.3. Principales méthodes d’étude de la transition de spin
La TS, qu’elle soit activée thermiquement, par application d’une pression, d’un champ magnétique ou par la lumière, peut être suivie par des techniques très variées, rendant compte de la structure électronique du centre métallique. Parmi ces techniques, nous citerons :
Ct
17
- La mesure de la susceptibilité magnétique en fonction de la température, χ(T), est sans doute la principale technique de caractérisation des complexes à TS.[26, 27] Le produit χT pour un matériau à TS est déterminé par la dépendance en température des contributions χHS et χBS, selon l’équation : χ(T)=γHSχHS+(1-γHS)χBS. Connaissant les susceptibilités de l’état HS et BS purs, la fraction molaire de l’état HS (resp. BS), γHS, (resp. γBS) peut être facilement déterminéeet représentée en fonction de T pour obtenir la courbe de la TS ; cependant, le plus souvent on représente χT vs. T (quand χHS et χBS ne sont pas connus, ou déterminés de façon imprécise). Notons par ailleurs, que pour un paramagnétique χT est une constante à toute température (loi de Curie). Cette représentation permet donc pour un composé à TS de mieux localiser la température de transition correspondant à un saut entre deux constantes lorsque la transition est abrupte.
- La spectrométrie Mössbauer [41] est la méthode spectroscopique de choix pour l'étude de la TS dans le cas du fer. Elle permet de suivre la proportion atomique d'un état de spin donné et elle est ainsi souvent utilisée pour calibrer les courbes γHS (T) obtenues à partir des mesures magnétiques où γHS est la proportion de l’état HS dans le matériau. Elle fournit aussi de précieuses informations structurales locales du matériau.
- Les mesures cristallographiques (diffraction des rayons X sur monocristaux principalement) permettent de suivre l’évolution structurale des composés dans la gamme de température de la TS. De manière générale, on observe une augmentation des distances métal-ligand ainsi que des déformations d’angles lors d’une conversion BSÆ HS.43
- Les paramètres thermodynamiques comme les changements d’enthalpie et d’entropie peuvent être estimés par mesures calorimétriques. Ces méthodes permettent de remonter à la température de transition et à l’ordre de la transition.44
- Les mesures de réflectivité ou d’absorption optique sont basées sur les changements de propriétés optiques du matériau lors de la TS.45 En effet, pour de nombreux systèmes, les bandes d’absorption sont dues aux transitions entre les états électroniques des centres métalliques. A cause des règles de sélection de spin, différentes bandes apparaissent dans les états HS et BS : ainsi, par exemple, l’état HS du Fe(II) est généralement peu coloré, au contraire dans l’état BS de l’ion métallique, la couleur du matériau est plus foncée. Cependant, ces changements peuvent être occultés (à l’œil) par l’absorption des ligands même. Notons ainsi que les transitions MLCT dépendent aussi de l’état de spin du système.
18
- Le passage de deux électrons d’orbitales liantes à des orbitales anti-liantes lors d'une transition BSÆHS conduit à un changement de polarisabilité moléculaire qu’il est possible de suivre par des mesures de la constante diélectrique du matériau.46
- Les mesures spectroscopiques vibrationnelles comme l’absorption infra rouge23c ou la diffusion Raman sont aussi très utilisées car la force des liaisons de coordination change spectaculairement d’un état de spin à l’autre.
19
A.1.4. Références
[1] M. Delpine, Bull. Soc. Chim. France, 1908, 3, 643. [2] L. Cambi, L. Szego, Ber., 1931, 64, 167.
[3] L. Cambi, L. Szego, Ber., 1933, 66, 656. [4] L. Cambi, L. Malatesta, Ber., 1937, 70, 2067.
[5] A. H. Ewald, R. L. Martin, I. Ross, A. H. White, Proc. Roy. Soc., 1964, A 280, 235; A. H. White, R. Roper, E. Kokot, H. Waterman, R. L. Martin, Austral. J. Chem., 1964, 17, 294; R. M. Golding, W. C. Tennant, C. R. Kanekar, R. L. Martin, A. H. White , J. Chem. Phys., 1966, 45, 2688.
[6] K. Reeder, E. Dose, and L. Wilson, Inorg. Chem., 1978, 17, 1071.
[7] M. Haddad, W. Federer, M. Lynch, and D. Hendrickson, Inorg. Chem., 1981, 20, 131. [8] S. Schenker, A. Hauser, and R. Dyson, Inorg. Chem., 1996, 35, 4676.
[9] R. Stoufer, B. Busch, and W. Hadley, J. Am. Chem. Soc., 1961, 83, 3732. [10] J. Zarembowitch and O. Kahn, Inorg. Chem., 1984, 23, 589.
[11] K. Heinze, G. Huttner, L. Zsolnai, and P. Schober, Inorg. Chem., 1997, 36, 5457. [12] P. Gütlich and W. K. B.R. McGarvey, Inorg. Chem.,1980, 19, 3704.
[13] W. Eberspach, N. E. Murr, and W. Klaui, Angew. Chem. Int. Ed., 1982, 21, 915. [14] W. Klaui, W. Eberspach, and P. Gütlich, Inorg. Chem., 1987, 26, 3977.
[15] D. Halepoto, D. Holt, L. Larkworthy, G. Leigh, D. Povey, and W. Smith, J. Chem. Soc. Chem. Comm., 1989, 1322.
[16] M. Sorai, Y. Yumoto, D. Halepoto, and L. Larkworthy, J. Phys. Chem. Solids, 1993, 54, 421.
[17] J. Ammeter, R. Bucher, and N. Oswald, J. Am. Chem. Soc., 1974, 96, 7883. [18] M. Switzer, R. Wang, M. Rettig, and A. Maki, J. Am. Chem. Soc., 1974, 96, 7669. [19] P. Sim and E. Sinn, J. Am. Chem. Soc., 1981, 103, 241.
[20] L. Kaustov, M. Tal, A. Shames, and Z. Gross, Inorg. Chem., 1997, 36, 3503. [21] O. Kahn, J. Kröber, C. Jay, Advanced Materials 1992, 4, 718.
[22] O. Kahn and C. Martinez, Science 1998, 279, 44.
[23] Spin Crossover in Transition Metal Compounds, Topics in Current Chemistry 2004, Vol. Ed. P. Gütlich, H. A. Goodwin: aJ.-F. Létard, P. Guionneau, and L. Goux-Capes, Towards spin crossover applications, III, 221. bA. Hauser, Ligand Field Theoretical Considerations, I, 59; cW. Linert, M. Grunert, A. B. Koudriavtsev, Isokinetic and Isoequilibrium Relationships in Spin Crossover Systems, III, 104; dJ. –P. Tuchagues, A. Bousseksou, G. Molnar, J. J.
20
McGarvey, F. Varret, The Role of Molecular Vibrations in the Spin Crossover Phenomenon, III, 85;
[24] H.A. Goodwin, Coord.Chem.Rev.1976, 18, 293. [25] P. Gütlich, Structure and Bonding, 1981, 44, 83. [26] E. König, Structure and Bonding;. 1991, 76, 51.
[27] P. Gütlich, A. Hauser, H. Spiering, Angew. Chem. Int. Ed. Engl. 1994, 33, 2024.
[29] E. König, Nature and dynamics of the spin-state interconversion in metal complexes, in Structure and Bonding, volume 76, page 51, Springer-Verlag, Berlin Heidelberg New York, 1991.
[30] a) M. Sorai and S. Seki, J. Phys. Soc. Jpn 1972, 33, 575, b) M. Sorai and S. Seki, J. Phys. Chem. Solids 1974, 35, 555.
[31] J.J. McGarvey, I. Lawthers, J. Chem. Soc., Chem. Comm. 1982, 906.
[32] S. Descurtins, P. Gütlich, C.P. Kohler,, H. Spiering, A. Hauser, Chem. Phys. Lett. 1984, 105, 1.
[33] A. Hauser, Chem. Phys. Lett. 1986, 124, 543.
[34] A. Hauser, Comments Inorg. Chem. 1995, Vol 17, 1, 17.
[35] S. Hayami, Z.-Z. Gu, M. Shiro, Y. Einaga, A. Fujishima, O. Sato, J. Am. Chem. Soc.
2000, 122, 7126.
[36] A. Hauser, Coord. Chem. Rev. 1991, 111, 275.
[37] A. Desaix, O. Roubeau, J. Jeftic, J. G. Haasnoot, K. Boukhedaden, E. Codjovi, J. Linares, M. Nogues, F. Varret, Eur. Phys. J. B. 1998, 6, 183.
[38] J. Zarembowitch, C. Roux, Brevet Français 9205928, 1992.
[39] a) J. Zarembowitch, C. Roux, M.-L. Boillot, R. Claude, J.-P. Itie, A. Polian, M. Bolte, Mol. Cryst. Liq. Cryst. 1993, 234, 247; b) M.-L. Boillot, A. Sour, P. Delhaès, C. Mingotaud, H. Soyer, Coord. Chem. Rev. 1999, 190, 47.
[40] a) C. Roux, Thèse de doctorat, Université de Paris-Sud, Centre d'Orsay, 1992; b) C. Roux, J. Zarembowitch, B. Gallois, T. Granier, R. Claude, Inorg. Chem. 1994, 33, 2273. [41] M. N. Greenwood, T. G. Gibb, Mössbauer Spectroscopy, Chapman and Hall Ltd, London, 1971
[42] A. Bousseksou, J. McGarvey, F. Varret, J.A. Real, J.-P. Tuchagues, A.C. Dennis, M.L. Boillot, Chem. Phys. Lett. 2000, 318, 409-416.
[43] B. Gallois, J. A. Real, C. Hauw, J. Zarembovitch, Inorg. Chem. 1990, 29, 1152.
[44] M. Sorai, S. Seki, J. Phys. Japan. 1972, 33, 575 ; M. Sorai, S. Seki, J. Phys. Chem. Solids. 1974, 35, 555.
21
[45] W. Morscheidt, J. Jeftic, E. Codjovi, J. Linares, A. Bousseksou, H.Constant-Machado, F. Varret, Meas. Sci. Technol. 1998, 9, 1311.
[46] A. Bousseksou, G. Molnar, P. Demont, J. Menegotto, J. Mat. Chem. 2003, 13, 2069 [47] C. Brady, J-J. McGarvey, J-K. McCusker,H. Toftlund, D-N. Hendrickson, Top Curr Chem, 2004, 235, 1.
22
A.2 Introduction aux Clathrates d’Hofmann
Tout récemment, de remarquables progrès ont été accomplis dans le domaine des complexes à TS : des polymères de coordination constitués de réseaux infinis mono- (1D), bi- (2D), ou tridimensionnels (3D), mettant en jeu des liaisons covalentes fortes entre centres métalliques, ont permis d’augmenter la coopérativité de ces matériaux, en particulier la largeur de la boucle d’hystérésis. Ainsi, de nouveaux systèmes polymères fortement coopératifs récemment rapportés dans la littérature présentent une bistabilité (température, pression ou irradiation lumineuse), parfois à température ambiante.[9]
L’histoire des complexes de métaux de transition avec des ligands cyanures débute par la découverte accidentelle par Diesbach du complexe connu comme « Bleu de Prusse ». Ce complexe a été très utilisé aux XVIII et XIX siècles comme pigment dans l’industrie des peintures. Depuis, il fait l’objet de très nombreuses études. Cependant, sa composition chimique exacte, Fe4III[FeII(CN)6]3·15H2O et sa structure cristallographique détaillée n’ont été élucidées que depuis 40 ans par Buser et al.[1],à cause de difficultés de synthèse des cristaux. Les clathrates d’Hofmann font partie de cette grande famille de complexes. Les premiers ont êtes synthétisés par Hofmann et Höchtlen en 1903 [2a, 2b] et présentent la formule générale du type [Ni(NH3)2Ni(CN)4·2G] où G ( = benzène, pyrrol, thiofène, ou furane) est une molécule ‘‘invitée’’ (de l’anglais Guest). Leur structure, résolue par Powell et Rayner dans les années 50 [3a, 3b, 3c] est formée de 2 types d’ions nickel (II) : (i) l’anion diamagnétique plan-carré [Ni(CN)4]2- ; (ii) l’autre ion paramagnétique coordonné octaédriquement à quatre atomes d’azote des groupes CN appartenant à 4 unités [Ni(CN)4]2-, les deux sites restants étant occupés par des atomes d’azote des molécules d’ammoniaque (figure A.2.1). En résumé, les deux types d’ions nickel liés par le groupe cyanure forment un réseau bidimensionnel infini : ces plans sont empilés dans la direction z avec un espacement d’environ 8 Å, ce qui permet l’introduction des molécules ‘‘Guest’’.
23
Figure A.2.1 : Structure du Clathrate de Hofmann [Ni(NH3)2Ni(CN)4·2G] où G= benzène
Au début des années 60, Schwarzenbach et al. [4] ont synthétisé de nouveaux complexes de type clathrates d’Hofmann en substituant dans la structure originale les atomes de nickel (II), qui occupent l’environnement octaédrique, par des ions cadmium (II), cuivre (II) et zinc (II). Dans les années 80, Iwamoto et col. [5,6,7] ont poursuivi ces travaux en développant la famille de formule générale : [M(NH3)M’(CN)4]·2G (M= Mn(II), Fe(II), Co(II), Ni(II), Zn(II), Cd(II), ou Cu(II) ; M’= benzène, pyrrol, thiofène, dioxane, aniline, ou biphényle). De plus, ces auteurs ont substitué les molécules d’ammoniaque par des ligands bis-monodentes tels que l’éthylènediamine, la diaminobutane ou la méthanolamine dans le but d’étudier la possibilité du changement de dimensionnalité (de deux dimensions (2D) à trois dimensions (3D)) (Figure A.2.2).
24
Plus tard, au cours de la dernière décennie, Kitazawa et col. [8] ont synthétisé le système polymère 2D {Fe(py)2[Ni(CN)4]} (1) où les ligands axiaux d’ammoniaque ont été substitués par deux pyridines. La structure de ce complexe est similaire aux clathrates d’Hofmann classiques, mais la taille beaucoup plus grande de la pyridine (par rapport à l’ammoniaque), conduit à un déplacement des plans formés par les atomes de fer et les groupes cyano, de sorte que l’atome de fer d’un plan donné se place verticalement par rapport aux atomes de nickel des plans immédiatement supérieur et inférieur. Ceci conduit à un espace restreint entre ces plans d’où la plus grande difficulté d’accueillir des molécules ‘‘Guest’’ dans ce type de matériaux par rapport aux structures poreuses précédentes (Figure A.2.3).
Figure A.2.3: Structure du complexe [Fe(py)2Ni(CN)4](1)
L’étude des propriétés magnétiques du composé 1 montre une TS centrée à 191 K, avec une boucle d’hystérésis de 10 K (T1/2↓=186 K et T1/2↑=196 K). Plus récemment Niel et al. [9a, 9b], ont observé des résultats similaires pour des composés homologues à base de [Pd(CN)4]2-(2) et[Pt(CN)4]2- (3). Ces complexes présentent aussi une TS avec une boucle d’hystérésis (2 : de 5K, T1/2↓=208K, T1/2↑=213K ; 3 : de 8K, T1/2↓=208 K, T1/2↑=216 K).
Pour ces trois systèmes {Fe(py)2[M(CN)4]} (M= Ni, Pd, Pt), un changement de couleur caractéristique est observé avec la TS : les complexes sont respectivement orange, jaune pale ou blanc dans l’état HS et rouge orangé dans l’état BS.
25
Ces travaux constituent un premier pas vers une nouvelle stratégie de synthèse pour des nouveaux systèmes polymères à TS. En ce sens, le remplacement du ligand pyridine monodente par le ligand pyrazine (pz) bidente conduit à une nouvelle famille de composés tridimensionnels {Fe(pz)[M(CN)4]}·2H2O (M= Ni (4), Pd (5), Pt (6)) où le ligand pyrazine joue le rôle de pont entre deux atomes de fer de couches consécutives (Figure A.2.4).
Figure A.2.4: Structure du complexe {Fe(pz)[Pt(CN)4]}(6)
Le comportement magnétique des complexes 4-6 met en évidence une TS du premier ordre avec une boucle d’hystérésis marquée (4 : T1/2↓=280 K et T1/2↑=300 K ; 5 : T1/2↓=239 K et T1/2↑=262 K ; 6 : T1/2↓= 222 K et T1/2↑= 240 K). Le changement d’état de spin s’accompagne également d’une modification prononcée de la coloration du matériau : jaune (platine, palladium) ou orange (nickel) dans l’état HS, il devient rouge dans l’état BS.
Les propriétés magnétiques des complexes [9a, 9b] 1-3 (ligand pyridine) et 4-6 (ligand pyrazine) ont été comparées sur la figure A.2.5. Les dérivés 3D présentent des TS plus coopératives que leurs homologues 2D, avec hystérésis très large (entre 20-40 K, alors qu’elle est au maximum de 9 K pour les dérivés de la pyridine) et à plus haute température. L’augmentation significative des valeurs T1/2 observées pour les dérivés de pyrazine, comparés aux homologues de pyridine, ne peut pas être expliquée par la théorie du champ de ligand, puisque la pyridine est un ligand à champ plus fort que la pyrazine. Par contre, la pression interne induite par la structure 3D plus rigide, peut expliquer la stabilisation de l’état BS dans le cas de la pyrazine.
26
Figure A.2.5 : Propriétés magnétiques des polymères 1-6.
Les complexes décrits ci-dessus constituent avec les systèmes monodimensionnels (1D) à base de triazole développés par Kahn et al.[10], des exemples typiques de systèmes polymères à TS. Ces matériaux dotés d’une coopérativité accrue par rapport aux systèmes monomères font l’objet d’un intérêt croissant de la part de la communauté scientifique, en raison de leurs potentielles possibilités d’utilisation dans les dispositifs applicatifs. Aussi, afin de pouvoir contrôler à volonté leurs propriétés (T1/2 et la largeur de l’hystérésis en particulier), il est nécessaire de synthétiser de nouveaux complexes polymères 3D. En ce sens, une nouvelle famille de clathrates d’Hofmann a été obtenue en remplaçant le ligand pyrazine par le ligand azopyridine : [Fe(4,4’-azopyridine)M(CN)4]·nH2O (M = Ni(II) (7), Pd(II) (8) et Pt(II) (9)). Ceci fera l’objet du chapitre A.4.
Dans la suite de ce chapitre, nous décrirons les études physico-chimiques réalisées précédemment sur la famille Fe(pz)[M(CN)4] (M = Pt, Pd, Ni) en particulier le complexe {Fe(pz)[Pt(CN)4]}·nH2O. La thématique principale de ce manuscrit s’inscrit dans la suite de ces travaux : il s’agit de l’élaboration de couches minces dans un premier temps puis la micro-nano-structuration de ces composés ainsi que leur caractérisation.
(5) (2) (4) (1) (3) (6)
27
A.2.1. Composé {Fe(pz)[Pt(CN)
4]}·nH
2O.
A.2.1.1 Description de la structure cristalline [9a, 9b]
La structure cristalline du complexe Fe(pz)[Pt(CN)4]·2H2O (6) a été déterminée par V. Niel par calcul « ab initio » à partir du diffractogramme de poudre, mesuré à température ambiante. Elle se présente sous forme d’un système tétragonal (groupe d’espace P4/m) des paramètres de maille a = b = 7.4144(5) Å et c = 7.2141(6) Å, avec V = 396.58 Å3, ρcalc = 1.955 g cm-3 et Z = 1.
Chaque atome de FeII est lié à quatre groupes [Pt(CN)
4]2- à travers les ponts CN, et chaque atome de platine entouré de quatre atomes de FeII situés à une distance de 5.243 Å ; la distance entre deux atomes de Fe du même plan est de 10.486 Å, formant ainsi un réseau infini dans les directions x et y. Les atomes de Fe de deux plans consécutifs sont liés à deux molécules de pyrazine, à une distance 7.214 Å donnant lieu à un réseau tridimensionnel très stable.
Récemment, nous avons pu synthétiser des monocristaux (figure A.2.6) en forme d’aiguilles jaunes du complexe Fe(pz)[Pt(CN)4]·nH2O par une méthode similaire à celle développée par E. Velterman. A température ambiante, un flacon (i) de 8 ml est placé à l’intérieur d’un autre flacon (ii) de 60 ml rempli d’une solution aqueuse 0.5 M de pyrazine jusqu’à environ 0.5 cm en dessus de la limite supérieure. 2ml d’une solution aqueuse 0.5 M de Fe(BF4)2·6H2O (refroidi dans la glace) est injectée au fond de (ii), et 2ml d’une solution aqueuse 0.5 M de K2Pt(CN)4 (refroidi dans la glace) est injecté au fond de (i). (i) est fermé et placé dans un bain d’eau à 45°C pendant 3 jours. Après 3 jours des cristaux jaunes en forme d’aiguilles apparaissent, ceux-ci sont filtrés et séchés à l’air pendant un jour. Le produit est déshydraté par la suite à 150°C (30 min) dans une étuve.
Bain d’eau 45 °C Pyrazine
K2[Pt(CN)4]
28
Figure A.2.6: a) Montage expérimental pour l’obtention des cristaux, b) cristaux du complexe Fe(pz)Pt(CN)4 dans l’état HS (droite) et BS (gauche).
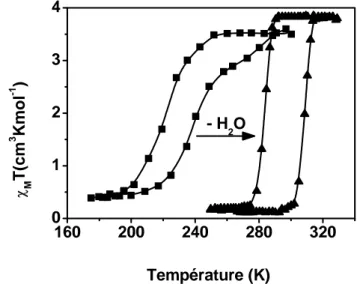
Les mesures de susceptibilité magnétique montrent une transition de spin avec une boucle d’hystérésis de 14 K centrée à température ambiante pour le produit dans sa forme déshydratée (Figure A.2.7). L’influence du degré d’hydratation est décrite dans le paragraphe qui suit.
Figure A.2.7 : Evolution du produit χMT en fonction de la température pour les cristaux du complexe 6 après traitement thermique à 430 K.
La structure cristalline obtenue par diffraction de rayons X montre que les paramètres de maille à température ambiante sont proches de ceux obtenus par calcul « ab initio » dans la poudre non-déshydratée: groupe d’espace P4mmm (tétragonal), a = b = 7.4314(6) Å, c = 7.2468(18) Å, V = 400.21(11) Å3, ρcalc = 1.789 g cm-3 et Z = 1. Les paramètres obtenus dans l’état BS sont : a = b = 7.1710(6) Å et c = 6.7700(18) Å, avec V = 348.14(10) Å3, ρcalc = 2.056 g cm-3 et Z = 1. Notons le changement de volume de 13%, le plus grand observé pour des complexes à TS. (voir annexe E)
250 260 270 280 290 300 310 320 330 0 1 2 3 4 χ M T ( cm 3 K mol -1 ) T (K)
29
A.2.1.2 Influence du degré de solvatation sur les propriétés magnétiques
Le composé Fe(pz)[Pt(CN)4] (6) a été synthétisé pour la première fois sous sa forme polycristalline hydratée(9a), puis des études complémentaires ont montré que le comportement de ce matériau dépend fortement du nombre de molécules d’eau que le réseau cristallin contient.[12] En effet, les premières synthèses de ce type de clathrates montraient des TS très coopératives avec des boucles d’hystérésis assez larges autour de 30-40 K mais leur reproductibilité (températures critiques T1/2↑ et T1/2↓ et largeur de la boucle d’hystérésis) n’est pas garantie.[12]
A titre d’exemple, le comportement magnétique du composé 6 avant et après chauffage à 430K est présenté sur la figure A.2.8. La TS de la forme hydratée s’étale sur un large intervalle de températures (110K), avec une hystérésis de largeur 20 K centrée autour de 260K. Le traitement thermique améliore notablement les caractéristiques de cette transition, celle-ci devenant alors plus rectangulaire et centrée à la température ambiante. De plus, la largeur du cycle d’hystérésis augmente à 24 K et les propriétés magnétiques restent reproductibles après plusieurs cycles thermiques.
Des analyses thermo-gravimétriques ont révélé qu’un chauffage des échantillons à plus de 473 K est nécessaire pour éliminer les deux molécules d’eau de cristallisation, mais en contact avec l’air l’échantillon récupère rapidement une molécule d’eau. Le matériau déshydraté possède des propriétés magnétiques reproductibles, identiques à celle de l’espèce monohydratée. [12] On peut obtenir une reproductibilité des propriétés magnétiques suite à un traitement thermique entre 430-450 K pendant 10-30 minutes qui permet d’obtenir l’espèce monohydratée.
Figure A.2.8 : Evolution du produit χMT en fonction de la température pour le complexe 6 avant et après traitement thermique à 430 K.
160 200 240 280 320 0 1 2 3 4 Température (K) χ M T( cm 3 Km ol -1 ) - H2O
30
A.2.1.3. Spectroscopie Raman
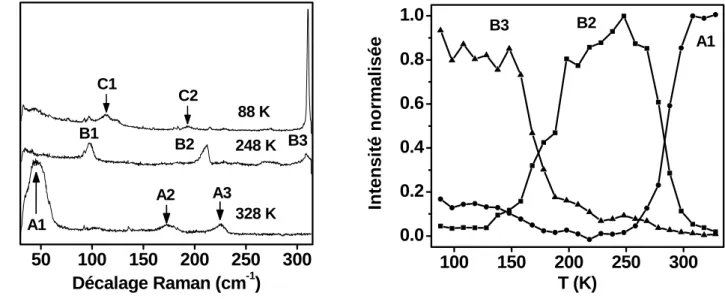
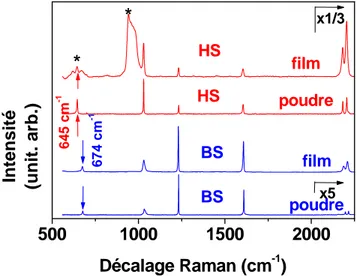
Les spectres Raman de l’échantillon déshydraté du complexe 6 ont été enregistrés dans la gamme de fréquence 30 - 2250 cm−1 à 88, 248 et 328K (figures A.2.9 et A.2.10) [13]. Dans la zone 600 - 1700 cm−1(figure 6a), toutes les raies Raman peuvent être attribuées aux modes intramoléculaires de la molécule de pyrazine. Les modes situés autour de 2100 - 2200 cm−1 correspondent aux modes d’élongation CN;[13] ces derniers ne subissent toutefois que de faibles déplacements de fréquence lors de la TS.[14, 15] Par contre, les pics autour de 650 cm−1 constituent de bons marqueurs pour suivre la transition du fait de leur déplacement fréquentiel de 32 cm−1(de 645 cm−1(HS) à 677 cm−1(BS)). Il est ainsi possible de reproduire les courbes de magnétisme en reportant l’évolution de l’intensité d’un de ces modes par rapport à l’autre (I(645 cm−1) / [I(645 cm−1) + I(677 cm−1)]), en fonction de la température.
La modification d’état de spin entre 88 et 328 K provoque aussi des changements importants dans la zone de fréquences 315 - 585 cm−1(figure A.2.9b). Les spectres à 248 K présentent des raies Raman supplémentaires à ceux enregistrés à 88 K, indiquées par des flèches sur la figure 6b. Ces modes additionnels suggèrent l’existence des deux phases cristallines distinctes, correspondant chacune à des ions FeIIBS, indépendamment de la TS:[13] en effet à 88 K comme à 248 K, le complexe est dans l’état BS.
Figure A.2.9 : Spectres Raman du complexe 6 : a) 585-2250 cm-1 et b) 315-585 cm-1.
Entre 30 et 315 cm−1 (figure A.2.10a), trois modes caractéristiques de l’état HS (330 K) apparaissent autour de 50, 175 et 225 cm−1 comme indiqué par les notations A1, A2 et A3, respectivement. Les modes Raman A2 et A3 sont attribués à l’élongation de la liaison Fe-N
800 1200 1600 2000 677 cm-1 645 cm-1 x 1/15 x 1/15 Int e ns it é Décalage Raman (cm-1) 328 K 248 K 88 K 350 400 450 500 550 377 cm-1 509 cm -1 431 cm-1 248 K 88 K 328 K In te n s ité Décalage Raman (cm-1) a) b)
31 50 100 150 200 250 300 B3 A1 A3 A2 B2 B1 88 K 248 K 328 K In te n s ité Décalage Raman (cm-1) C1 C2
de l’état HS [14]. Puisque la raie Raman A1 disparaît au voisinage de la transition, elle peut également être attribuée à un mode Fe-ligand HS. Entre 300 et 250 K, les pics A1-A3 disparaissent et les modes Raman indiqués par B1 - B3 émergent.
Figure A.2.10 : a) Spectres Raman du complexe 6 entre 30-315 cm-1 et b) Intensité normalisée des modes Raman A1, B2, B3 en fonction de la température.
En poursuivant le refroidissement (entre 200 et 150 K), les modes B1 et B2 disparaissent et de nouveaux modes (C1 et C2) apparaissent. En même temps, l’intensité du mode B3, décrit comme un mode d’élongation Fe-N(pz) BS[14], s’amplifie. La figure A.2.10b montre la dépendance thermique des intensités Raman intégrées des pics A1, B2 et B3 en mode refroidissement. Ces intensités sont également normalisées par rapport à l’intensité maximale mesurée. Le mode A1 apparaît dans l’état HS, le mode B2 dans l’état BS mais exclusivement dans la gamme de températures intermédiaires entre 150 et 275 K et le mode B3 dans l’état BS en dessous de 275 K. Ce dernier présente une forte hausse en intensité en dessous de 150 K. Ces changements spectraux sont totalement reproductibles sur plusieurs cycles de chauffage et de refroidissement.
En résumé, il est possible de suivre la TS pour le complexe 6 à l’aide de ces différents marqueurs. Cependant, lorsque l’intensité du signal Raman est faible, il est préférable de suivre d’autres marqueurs plus intenses situés autour de 1028 et 1231 cm-1 permettant un tracé plus précis du rapport d’intensité.
100 150 200 250 300 0.0 0.2 0.4 0.6 0.8 1.0 B3 B2 A1 Int e n s it é n o rm alis é e T (K)
32 240 260 280 300 320 340 0 1 2 3 4 0.0 0.2 0.4 0.6 0.8 1.0 χ MT χ M T ( cm 3 mo l-1 K) T (K) Raman I nor m
A titre d’exemple, on présente la courbe obtenue en suivant le rapport d’intensité de ces derniers pics, en fonction de la température : cette courbe est superposable à celle obtenue à partir des mesures de susceptibilité magnétique. Il est donc possible de reproduire précisément le comportement des données magnétiques en traçant l’évolution de l’intensité d’un de ces modes par rapport à l’autre, I(1231 cm-1)/[I(1231 cm-1)+I(1028 cm-1)]. (Figure A.2.11)
Figure A.2.11 : Dépendance en température de l’intensité Raman normalisée
(I(norm)=I(1025 cm-1) / I(1230 cm-1)) et celle du produit χMT du complexe Fe(pz)[Pt(CN)4]
A.2.1.4. Etudes des effets de Pression
Des études Raman de la famille Fe(pz)[M(CN)4]·2H2O (M = Ni, Pd, Pt) à température ambiante ont été effectuées en fonction de la pression dans notre équipe. Il a été montré pour la première fois une boucle d’hystérésis piézo-induite à température constante (ambiante) pour le complexe de nickel, avec les pressions caractéristiques P1/2↑ = 1350 (±50) bar et P1/2↓ = 650 (±50) bar (figure A.2.12a). Pour les complexes de palladium et platine, la TS se produit autour de 1800 (±50) et 3500 (±50) bar, respectivement. La spectrométrie Raman a également montré pour ces deux derniers complexes que la TS est repoussée à de plus hautes pressions en raison d’une transition de phase structurale dans l’état HS, ce qui n’avait pas été observée dans le cas du nickel. En effet, à des pressions intermédiaires, des raies Raman différentes de celles de la phase HS et BS sont observées (par exemple autour de 660 cm-1 ou encore dans la région des élongations CN) qu’on peut attribuer à une transition de phase structurale différente de la TS (figure A.2.12b).
33
Figure A.2.12: a) Piezo-hystérésis expérimentale du complexe Fe(pz)[Ni(CN)4]·2H2O à TS ; b) Spectres Raman du complexe Fe(pyrazine)[Pt(CN)4]·2H2O à différentes valeurs de la
pression appliquée
A.2.1.5. Photo-commutation réversible par impulsion laser nano-seconde
L’effet LIESST dans ce composé a été démontré à 5K à l’aide d’un laser continu. Cependant, la durée de vie de l’état HS métastable déjà très courte à basse température décroit exponentiellement avec l’augmentation de la température. Pour palier à ce problème, nous avons eu l’idée de travailler à l’intérieur de la boucle d’hystérésis (centrée autour de la température ambiante) où la durée de vie des états métastables (macroscopiques) est infinie. Dans notre équipe, S. Bonhommeau[11] a mis en évidence par spectroscopie Raman une TS photo-induite à température ambiante et bidirectionnelle (BSÆHS et HSÆBS) dans le complexe 6 par application d’un laser pulsé.
Cette transition bidirectionnelle a été induite à l’aide d’impulsions de 5 ns, de longueur d’onde λ = 532 nm et d’énergie d’environ ~ 20 mJ cm-2 environ au niveau de l’échantillon. La figure A.2.12 indique la fraction HS, évaluée à partir de l’intensité des pics Raman, avant et après excitation laser. Les spectres Raman enregistrés entre 620 et 720 cm-1, avant et après l’application des impulsions, sont présentés dans les encarts de la figure. Les modifications spectrales, comme le changement de couleur observé après excitation lumineuse, indiquent clairement qu’une impulsion génère aussi bien la transition BSÆ HS que la transition HSÆBS, selon la position initiale sur le cycle d’hystérésis. Les effets thermiques (chauffage de l’échantillon) provoqués par cette irradiation laser très courte peuvent être négligés. En
T=295 K 0 50 0 10 00 15 00 20 00 25 00 0.6 0.8 1.0 n H S pressure (ba r) a b

![Figure A.4.3 : Images AFM de la couche mince du [Fe(azpy)Ni(CN) 4 ] (1b) après 4, 8, 12 et 16 cycles](https://thumb-eu.123doks.com/thumbv2/123doknet/2120887.8280/66.892.77.834.540.1044/figure-images-afm-couche-mince-fe-azpy-cycles.webp)

![Figure A.5.7. Images MEB des motifs du complexe [Fe(azpy)Ni(CN) 4 ] : a-20x20µm b – 80x30 nm, c - 80x80 nm, d – 670x60 nm](https://thumb-eu.123doks.com/thumbv2/123doknet/2120887.8280/87.892.108.836.130.682/figure-images-meb-des-motifs-complexe-azpy-m.webp)
![Figure A.5.8 : Hauteur des motifs obtenue par AFM pour des motifs du complexe [Fe(pz)Pt(CN) 4 ] : a – 500 nm (période 5 µm), b – 200 nm (période 2 µm), c – 30 nm](https://thumb-eu.123doks.com/thumbv2/123doknet/2120887.8280/88.892.127.742.90.554/figure-hauteur-motifs-obtenue-motifs-complexe-periode-periode.webp)

