© Youssef Bouhout, 2019
Étude clinique randomisée contrôlée de l’efficacité du
laser comme adjuvant au surfaçage radiculaire
conventionnel dans le traitement de la parodontite
chronique
Mémoire
Youssef Bouhout
Maîtrise en sciences dentaires - parodontie - avec mémoire
Maître ès sciences (M. Sc.)
ii
Résumé :
La valeur ajoutée du laser dans le traitement de la parodontite chronique est encore sujette à controverse et reste à prouver. Le but de cette étude est de vérifier le bénéfice de la combinaison des lasers Nd : YAG et Er : YAG utilisés conjointement au surfaçage radiculaire en comparaison avec le traitement conventionnel de la parodontite chronique.
Méthode : 12 patients atteints de parodontite chronique sévère furent recrutés. Les
dents du côté gauche et droit furent aléatoirement répartis dans le groupe test (traitement avec le laser suivi d’un surfaçage radiculaire) et le groupe contrôle (surfaçage radiculaire seul). Les signes cliniques de l’inflammation, la profondeur de sondage, le saignement au sondage, le niveau d’attache clinique, et la récession gingivale furent comparés à 8 semaines et 6 mois.
Résultats : amélioration significative des signes cliniques de l’inflammation, de la
profondeur des poches parodontales, du saignement au sondage et de la perte d’attache, pour chacun des traitements. Cependant, aucune différence significative ne fut remarquée entre les deux types de traitement offert.
Conclusion : dans les limites de cette étude, l’ajout du laser au traitement de
surfaçage conventionnel n’a pas démontré d’avantage significatif lorsque comparé au traitement de surfaçage conventionnel seul.
iii
Abstract
The exact added value of the laser in the non-surgical treatment of chronic periodontitis is still controversial and remains to be proven. The purpose of this study is to evaluate the benefit of Nd: YAG and Er: YAG lasers combined used in conjunction with root scaling compared to conventional treatment of chronic periodontitis.
Method: 12 patients with severe chronic periodontitis were recruited. The left and
right sides were randomly distributed in the test group (laser treatment followed by root scaling) and the control group (root scaling only). Clinical signs of inflammation, probing depth, bleeding on probing, level of clinical attachment, and gingival recession were compared at 8 weeks and 6 months.
Results: Significant improvement in clinical signs of inflammation, periodontal
probing depth, bleeding, and loss of attachment was observed for each treatment. However, no significant difference was noticed between the two types of treatment offered. Conclusion: Within the limits of this study, the addition of the laser to the conventional root scaling did not demonstrate a significant advantage when compared to the conventional surfacing treatment alone.
iv
Table des matières
RÉSUMÉ : ... II ABSTRACT ... III LISTE DES TABLEAUX ... VII LISTE DES FIGURES ... VIII LISTE DES ABRÉVIATIONS, SIGLES ET SYMBOLES ... X REMERCIEMENTS ... XIII
INTRODUCTION ... 1
1.1 PARODONTITE ... 1
1.1.1. Définition ... 1
1.1.2. Classification de la parodontie chronique ... 2
1.1.3. Prévalence de la parodontite chez l’adulte ... 3
1.1.4. Mécanisme destructeur de la parodontite chronique ... 7
1.1.4.1. Histopathogénèse de la gingivite et parodontite ... 7
1.1.4.2. Microbiologie de la parodontite chronique ... 10
1.1.4.2.1. Théories de l’étiologie microbienne ... 10
1.1.4.2.2. Complexes bactériens associés à la parodontite chronique ... 12
1.1.4.2.3. Étapes de la pathogenèse bactérienne ... 14
1.1.4.3. Réaction inflammatoire et immunitaire ... 17
1.1.4.3.1. Réponse immunitaire non spécifique ... 18
1.1.4.3.2. Réponse immunitaire spécifique ... 20
1.1.4.3.3. Médiateurs d’inflammation et réponse immuno-destructrice ... 23
1.1.5. Traitement de la parodontite chronique ... 28
UTILISATION DES LASERS EN PARODONTIE ... 38
1.2 ... 38
1.2.1 Principes de bases des lasers ... 38
Physique des lasers ... 41
1.2.1.3 ... 41
1.2.1.3.1 Fonctionnement du laser ... 41
1.2.1.3.1.1 Structure d’un atome ... 41
1.2.1.3.1.2 Absorption ... 41 1.2.1.3.1.3 Émission spontanée ... 42 1.2.1.3.1.4 Émission stimulée ... 43 1.2.1.3.1.5 Inversion de population ... 43 1.2.1.3.1.6 Pompage ... 44 1.2.1.3.1.7 Amplification ... 44
1.2.1.3.1.8 Composants et fonctionnement du laser ... 45
1.2.1.3.2 Propriétés de la lumière ... 46
1.2.1.3.2.1 Propriétés de la lumière blanche ... 47
1.2.1.3.2.2 Propriétés du laser ... 48 1.2.1.3.2.2.1 Monochromie ... 48 1.2.1.3.2.2.2 Cohérence ... 49 Directionnalité ... 49 1.2.1.3.2.2.3 ... 49 Fluence énergétique ... 49 1.2.1.3.2.2.4 ... 49
1.2.1.4 Interactions et effets des lasers sur les tissus ... 50
v
1.2.1.4.1.1 Réflexion ... 50
1.2.1.4.1.2 Absorption ... 51
1.2.1.4.1.3 Transmission ... 53
1.2.1.4.1.4 Diffusion ... 54
1.2.1.4.2 Principaux effets du laser sur les tissus ... 54
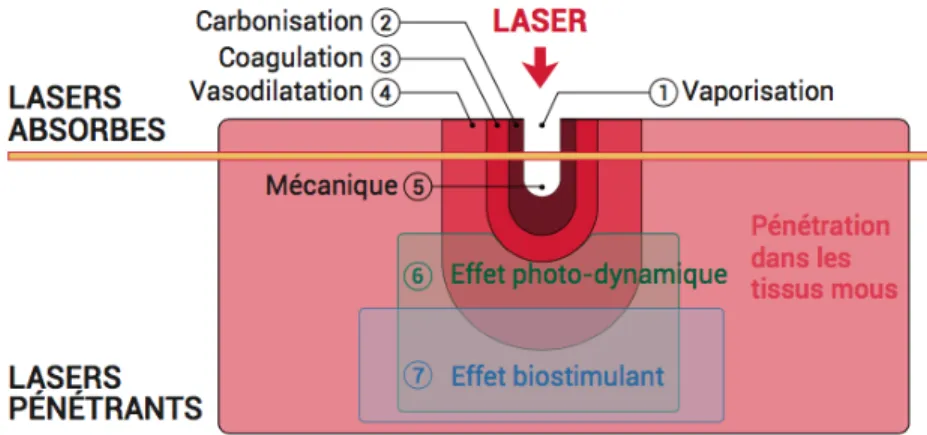
1.2.1.4.2.1 Effet photo-ablatif ... 54 1.2.1.4.2.2 Effets thermiques ... 54 1.2.1.4.2.2.1 Vaporisation ... 55 1.2.1.4.2.2.2 Coagulation ... 55 1.2.1.4.2.2.3 Vasodilation ... 55 1.2.1.4.2.2.4 Carbonisation ... 56 Effet mécanique ... 56 1.2.1.4.2.3 ... 56
1.2.1.4.2.4 Effets photochimiques ou photo-dynamiques ... 56
1.2.1.4.2.5 Effets biostimulants ... 57
1.2.1.4.3 Paramètres d’utilisation des lasers ... 58
1.2.1.4.3.1 Mode d’émission ... 58
1.2.1.4.3.2 Puissance ... 59
1.2.1.4.4 Principaux lasers utilisés en parodontie et leurs caractéristiques ... 60
1.2.1.4.4.1 Laser Er : YAG ... 60
1.2.1.4.4.2 Laser Nd : YAG ... 61
1.2.1.4.4.3 Laser CO2 ... 61
1.2.1.4.4.4 Laser à diode ... 62
1.2.2 Traitement non chirurgical de la parodontite par laser ... 63
1.3 BUTS ET OBJECTIFS DU PROJET ... 70
1.4 HYPOTHÈSE DE RECHERCHE ... 71 1.5 PERTINENCE DU PROJET ... 71 CHAPITRE 1 ... 73 MATÉRIEL ET MÉTHODE ... 73 2.1 PROCÉDURE EXPÉRIMENTALE ... 73 2.1.1 Design expérimental ... 73 2.1.2 Stratégie de recrutement ... 74 2.1.3 Critères d’inclusion ... 75 2.1.4 Critères d’exclusion ... 75 2.1.5 Variables mesurées ... 75
2.1.6 Collecte des données et stockages de l’information ... 76
2.1.7 Randomisation des patients ... 77
2.1.8 Traitements effectués ... 77 2.2 ANALYSE STATISTIQUE ... 78 CHAPITRE 2 ... 79 RÉSULTATS ... 79 3.1 PÉRIODE DE RECRUTEMENT ... 79 3.2 PÉRIODE DE SUIVI ... 79 3.3 PATIENTS RECRUTÉS ... 79
3.4 DIAGRAMME DU FLUX DE PARTICIPANTS ... 80
3.5 RÉSULTATS CLINIQUES ... 80
3.5.1 Saignement au sondage ... 81
3.5.2 Profondeur de sondage (PS) ... 81
3.5.3 Récession gingivale (RG) ... 91
vi
CHAPITRE 3 ... 100
DISCUSSION ... 100
CONCLUSION ... 109
BIBLIOGRAPHIE ... 110
ANNEXE II : DIMINUTION DE LA PROFONDEUR DE POCHE (PDP) EN FONCTION DE LA PROFONDEUR DE POCHE INITIALE ... 122
ANNEXE III : DIMINUTION DE LA PROFONDEUR DE POCHE (PDP) EN FONCTION DU GROUPE DE DENT ... 123
ANNEXE IV : DIFFÉRENCE DES PROFONDEURS DE POCHE (PDP) EN FONCTION DU STATUS TABAGIQUE ... 124
ANNEXE V : ÉVOLUTION DES RÉCESSIONS GINGIVALES EN FONCTION DES TRAITEMENTS ... 125
ANNEXE VI : ÉVOLUTION DES RÉCESSIONS GINGIVALES EN FONCTION DU GROUPE DE DENTS ET DES GROUPES DE TRAITEMENTS ... 126
ANNEXE VII : ÉVOLUTION DU NIVEAU D’ATTACHE CLINIQUE EN FONCTION DES TRAITEMENTS ... 127
ANNEXE VIII : ÉVOLUTION DU NIVEAU D’ATTACHE CLINIQUE EN FONCTION DU GROUPE DE DENTS ET DES GROUPES DE TRAITEMENTS ... 128
vii
Liste des tableaux
Tableau I. Sévérité de la parodontite chronique selon la perte d’attache (mm) et la profondeur de poche (mm). (Tiré de AAP Task Force Report 2015) ... 2 Tableau II. Classification de la parodontite en fonction des stades. (Tiré de
Papapanou et coll. (7)) ... 5 Tableau III. Classification de la parodontite en fonction des grades. (Tiré de
Papapanou et coll. (7)) ... 6 Tableau IV. Comparaison des propriétés du faisceau laser et de la lumière blanche . 51 Tableau V. Profondeur de poches chez les fumeurs et non-fumeurs en fonction des suivis. ... 89 Tableau VI. Diminution de la profondeur de poche (mm) en fonction de la profondeur de poche initiale ... 90 Tableau VII. Moyennes des récessions gingivales pour les 3 groupes de dents. ... 94 Tableau VIII. Niveau d’attache clinique (mm) en fonction du temps. ... 96
viii
Liste des figures
Figure 1. Représentation des complexes bactéries de Socransky. (Tiré de Socransky et
coll. [18]) ... 12
Figure 2. Représentation de la succession bactérienne entrainant la formation d’inflammation gingivale. (Tiré de Socransky et coll. [18]) ... 14
Figure 3. Modèle contemporain de la pathogenèse de la parodontite et gingivite (Tiré de Chapple et coll. (25)) ... 18
Figure 4. Représentation schématique d’un atome selon le modèle de Bohr. (Tiré de Chimie.net (102)) ... 42
Figure 5. Représentation schématique d’une émission spontanée. (Tiré de Coluzzi et coll. (64)) ... 42
Figure 6. Représentation schématique d’une émission stimulée. (Tiré de Coluzzi et coll. (65)) ... 43
Figure 7. Processus de pompage, amplification et production de la lumière laser (tiré de Freitas et coll. (62)) ... 44
Figure 8. Composants et mécanisme du laser. (Tiré de Coluzzi et coll. (65)) ... 46
Figure 9. Propriétés de la lumière. (Tiré de Coluzzi et coll. (65)) ... 47
Figure 10. Spectre électromagnétique. (Tiré de KhanAcademy (70)) ... 48
Figure 11.Quatre types d’interactions avec les tissus. (Tiré de Convissar et coll.(67)) ... 52
Figure 12. Coefficient d’absorption (1/cm) en fonction de la longueur d’onde des différents types de lasers. (Tiré de Coluzzi et coll.(63)). ... 53
Figure 13. Principaux effets du laser sur les tissus. (Adapté de Rey et coll.(76)) ... 58
Figure 14. Profondeur de sondage (mm) en fonction du traitement. ... 82
Figure 15. Réduction de la profondeur de poche en fonction des rendez-vous de suivis. ... 82
Figure 16. Profondeur de sondage des différents groupes de dents en fonction du temps ... 83
Figure 17. Diminution de la profondeur de poche pour les molaires ... 84
Figure 18. Diminution de la profondeur de poche pour les prémolaires ... 85
Figure 19. Diminution de la profondeur de poche pour les incisives/canines ... 85
Figure 20. Diminution de la profondeur de poche pour chacun des traitements par groupes de dents ... 86
Figure 21. Profondeur de poche des différentes surfaces en fonction du temps ... 87
Figure 22. Réduction de la profondeur de poche des différentes surfaces en fonction des suivis ... 88
Figure 23. Diminution de la profondeur de poche en fonction du statut tabagique. ... 89
Figure 24. Diminution de la profondeur de poches (PDP) en fonction de la profondeur de poche initiale. ... 90
Figure 25. Récession gingivale en fonction du traitement ... 92
Figure 26. Augmentation des récessions gingivales en fonction du temps. ... 92
Figure 27. Augmentation des récessions gingivales en fonction du groupe de dents pour les 2 traitements. ... 93
Figure 28. Augmentation des récessions gingivales pour les molaires à 8 semaines et 6 mois ... 94
ix
Figure 29. Augmentation des récessions gingivales pour les prémolaires à 8 semaines et 6 mois ... 95 Figure 30. Augmentation des récessions gingivales pour les incisives et canines à 8 semaines et 6 mois ... 96 Figure 31. Niveau et gain d’attache clinique en fonction des traitements pour les molaires. ... 97 Figure 32. Niveau et gain d’attache clinique en fonction des traitements pour les prémolaires. ... 98 Figure 33. Niveau et gain d’attache clinique en fonction des traitements pour les incisives et canines. ... 99
x
Liste des abréviations, sigles et symboles
º C Degré Celsius
∆ Difference
AAP American Academy Of Periodontology A.A Aggregatibacter actinomycetemcomitan BCR B-cell receptor
C. rectus Campylobacter rectus CD4 Cluster de différenciation 4 CD8 Cluster de différenciation 8
CMH Complexe majeur d'histocompatibilité COX-1 Cyclo-oxygénase 1
COX-2 Cyclo-oxygénase 2 CO2 Dioxyde de carbone
Er, Cr : YSGG Erbium, chromium-doped yttrium-scandium-gallium-garnet; Er:YAG, Erbium-doped yttrium-aluminium-garnet;
FDA Food and Drug Administration
h heure
H2O Eau
H2O2 Peroxyde d’hydrogène
Hb Hémoglobine
hBDs ß-defensines humaines He-Ne Hélium-néon
Hz Hertz
ICAM-1 InterCellular Adhesion Molecule IFN-y Interferon gamma
IGM Immunoglobulines M IGA Immunoglobulines A IGG Immunoglobulines G IL-1 Interleukine 1 IL-6 Interleukine 6 IL-8 Interleukine 8 IL-12 Interleukine 12 J/m2. Joule par mètre carré
J/s Joule par seconde
kd kilodalton
LANAP Laser-assisted new attachment procedure LPS Lipopolysaccharides
MMP Métalloprotéinase matricielle
NADPH oxydase Nicotinamide adenine dinucleotide phosphate oxidase NAC Niveau d’attache clinique
Nd:YAG, Neodymiumdoped yttrium-aluminium-garnet; Nd :YAP, Neodymium- doped yttrium-aluminum-perovskite
OHo. Radical hydroxyle
PDP Profondeur de poche PGE2 Prostaglandine E2
xi P.gingivalis Porphyromonas gingivalis PI Plaque index (indice de plaque) PRR pattern recognition receptors PS Profondeur de sondage RG Récession gingivale SAS Saignement au sondage SRP Surfaçage radiculaire T. denticola Treponema denticola TCR T cell receptor Th1 T helper cell type 1 Th2 T helper cell type 2
TK Tissu kératinisé
TLR4 Toll Like Receptor 4
TNF-𝛼 Facteur de nécrose tumorale alpha TNF -ß Facteur de nécrose tumorale bêta Tx1 Traitement 1 : Surfaçage + laser Tx 2 Traitement 2 : Surfaçage seul
W Watt
xii
« Osez! le progrès est à ce prix » Victor Hugo
xiii
Remerciements
Je tiens à remercier tout d’abord mon directeur de recherche, le Dr Goncalves pour sa disponibilité et son dévouement tout au long de ce projet de recherche. Merci de m’avoir fait confiance pour ce projet, ce fut une expérience cliniquement enrichissante.
J’aimerais aussi remercier les membres de mon comité d’encadrement, Dr Simon Gauthier et Dr Yanik Roussy. Je vous remercie de votre disponibilité, vos avis et vos conseils ont été extrêmement utiles pour le développement de ce projet.
Merci aussi au Dr Luc Giasson, pour ses précieux conseils en méthodologie et en rédaction. L’ensemble des notions apprises durant vos cours nous facilitent grandement l’écriture de notre mémoire.
Merci à mon statisticien Mr Gaétan Daigle pour son travail et son intérêt pour ce projet de maitrise.
J’aimerais finalement remercier ma famille et mes amis pour leurs soutiens inconditionnels durant ces 3 années de maitrise, mais aussi pendant ces 9 années d’études. Merci d’avoir cru en moi. Finalement, un merci particulier à Mina, ma partenaire de vie et de rédaction, qui n’a pas hésité à m’accompagner durant mes heures d’écriture en alternant des périodes d’écoutes et d’encouragements.
1
INTRODUCTION
1.1 Parodontite1.1.1. Définition
La parodontite est définie comme une inflammation, chronique ou aiguë, de la gencive qui s’étend aux tissus parodontaux adjacents (1). Elle est caractérisée par la formation de poches parodontales associée à une perte d’attache et d’os alvéolaire. Les signes cliniques de cette maladie sont généralement un changement de couleur de la gencive, de l’œdème, du saignement au sondage, l’apparition de poche parodontale profonde et une perte de support osseux (2). Dans la majorité des cas, cette maladie est d’origine bactérienne suite à l’accumulation de tartre et d’une flore bactérienne sous gingivale anaérobe, principalement à Gram négatif (3).
Les caractéristiques cliniques de la parodontite chroniques sont (4) :
• Altération de la couleur, texture et volume de la gencive marginale • Saignement au sondage
• Résistance réduite des tissus mous marginaux au sondage • Perte d’attache au sondage
• Récession de la gencive marginale • Perte d’os alvéolaire
• Exposition de furcations
• Augmentation de la mobilité dentaire • Migration ou exfoliation dentaire
2
1.1.2. Classification de la parodontie chronique
La parodontite chronique est la forme la plus commune de maladie parodontale destructive chez l’adulte. Elle possède généralement une progression modérée à lente avec quelques fois des périodes de progression rapide. La parodontite chronique peut être décrite en fonction de son étendue et de sa sévérité. Ainsi, selon la classification d’Armitage en 1999 (5), la parodontite chronique est dite localisée lorsque moins de 30 % des sites ont démontré une perte d’attache et osseuse. Lorsque le nombre de sites est supérieur à 30 %, elle est dite généralisée. De même, cette maladie peut être classifiée en fonction de sa sévérité : débutante (1 à 2 mm de perte d’attache), modérée (3 à 4 mm de perte d’attache) ou sévère (plus de 5 mm de perte d’attache). La sévérité de la parodontite chronique est résumée dans le Tableau 1.
Tableau I. Sévérité de la parodontite chronique selon la perte d’attache (mm) et la profondeur de poche (mm). (Tiré de AAP Task Force Report 2015)
En 2018, L’Académie Américaine de Parodontie (AAP) propose en partenariat avec un comité d’expert, une mise à jour de la classification des maladies parodontales d’Armitage de 1999 (5,6). Parmi les changements apportés, on note l’introduction du diagnostic de santé parodontale sur parodonte intact ou réduit, ou encore des maladies ou conditions péri-implantaire (absente de l’ancienne classification).
D’autre part, la littérature des 20 dernières années a permis de mieux comprendre la maladie parodontale et l’impact des facteurs de risques environnementaux et systémiques sur celle-ci. Ces avancées ont amené l’AAP et ce comité d’expert à proposer une nouvelle classification de la parodontite (6).De ce fait, cette dernière est fondée sur deux systèmes diagnostiques : le stade et le grade (7).
Sévérité Perte d’attache (mm) Poche parodontale
(mm)
Débutante 1 à 2 ]3 - 5 [
Modérée 3 à 4 [5-7 [
3
Le stade dépend de la sévérité et de la complexité du cas au moment de l’examen. On peut ainsi caractériser la parodontite en 4 stades (I à IV) en prenant en considération la perte d’attache clinique, le pourcentage de perte osseuse, le sondage parodontal, la présence de défauts osseux, les atteintes de furcation, la mobilité et la quantité la perte dentaire associée à la maladie parodontale (7). Les caractéristiques de chacun de ces stades sont résumées dans le Tableau 2.
Par ailleurs, le grade apporte des informations supplémentaires sur la progression de la maladie parodontale, parmi lesquelles on retrouve la vitesse de destruction, le risque de progression, le pronostic des traitements et l’effet possible sur la santé générale. La parodontite peut donc être classifiée en 3 grades (A, B et C) en fonction d’évidences direct ou indirect de progression de la maladie, et des facteurs de risque comme le statut tabagique ou encore le contrôle du diabète (7). Les caractéristiques de chacun des grades sont rapportées dans le Tableau 3.
Il est important aussi de noter la disparition dans la nouvelle classification de la parodontite agressive anciennement définie comme une maladie distincte de la parodontite chronique en raison de sa nature agressive (vitesse de progression), de la localisation des lésions (incisives et molaires pour la forme localisée), de la prédisposition familiale et de la finesse de son biofilm sous-gingival (8). Cependant, la difficulté clinique à différencier la parodontite chronique et de l’agressive a amené la fusion de ces deux diagnostics en une seul catégorie : la parodontite. La forme anciennement agressive est alors caractérisée dans la nouvelle classification par une parodontite à vitesse de progression rapide (grade C).
1.1.3. Prévalence de la parodontite chez l’adulte
Dès 1950, de nombreux auteurs s’attardent sur la prévalence de la parodontite chronique au sein de la population. Ainsi, en utilisant la perte osseuse alvéolaire comme outils de distinction entre la gingivite et la parodontie, Marshall-Day et ses collaborateurs rapporte une prévalence de 100 % de maladie parodontale chronique destructrice chez des habitants de Boston de plus de 46 ans avec une perte moyenne de 12 dents par sujet (9). La moyenne de perte dentaire était de 19 dents pour les plus de 60 ans (9).
4
À partir des années 1980s, la prévalence de la parodontite n’était plus simplement établie par analyse de patients atteints ou non atteints par la maladie parodontale. Les études commencent à éclaircir certains aspects de la maladie comme l’étendue, la sévérité et la vitesse de progression (4). De ce fait, Löe et coll.(10), réalisent une étude longitudinale au Sri Lanka et démontent différents modèles de progression de parodontite non traitée. Ainsi, 480 cueilleurs de thé, jamais exposés aux mesures d’hygiène orale conventionnelles, furent recrutés en 1970 à des fins d’examens et suivis. En 1985, 161 de ces individus furent réexaminés et ont permis d’établir 3 modèles de progression de la parodontite non traités chez une population de 14 à 46 ans. Le premier groupe de cueilleurs de thé a démontré une progression modérée évaluée à 0,05 à 0,5 mm de perte osseuse par an. Il regroupait près de 81 % de ces cueilleurs. Un autre groupe d’approximativement 11 % n’a démontré aucune progression de la maladie parodontale destructrice malgré la présence de gingivite. Enfin, le dernier groupe représentant 8 % de la population, manifesta une progression rapide de la destruction parodontale (0,1 à 1 mm par an). À travers cette étude, les auteurs démontrent une grande variabilité dans la progression de la maladie parodontale destructrice dans une population relativement homogène malgré la présence d’accumulation de plaque et d’inflammation gingivale. Ces résultats furent aussi confirmés par l’étude de Baelum et coll.(11), en 1986, chez des adultes tanzaniens. Malgré l’absence de mesure d’hygiène adéquate, 31 % des sujets seulement présentaient près de 75 % des sites avec une perte d’attache de plus de 7 mm. Ainsi, la parodontite avancée n’est donc pas équitablement répartie dans la population et ne semble pas forcement lié à la quantité de plaque et tartre présente. Au travers d’une étude épidémiologique récente (12), la prévalence de la parodontite chez les plus de 30 ans était de 46%, avec une prévalence plus élevée chez les Hispaniques (63,5 %) et les non hispaniques noirs (59,1%). La moyenne de perte d’attache rapportée chez les adultes était de 1,72 mm. Par ailleurs, en fonction de la classification de la parodontite, la prévalence de la parodontie sévère était de 8,9% à 12%. Cette étude épidémiologique a permis d’établir aussi certaines conditions où la prévalence de la parodontite sévère était plus élevée. Ainsi, les adultes de plus de 50 ans, les hommes, les hispaniques et non hispaniques noirs, les personnes n’ayant pas
5
terminés le secondaire, les fumeurs et les personnes vivant à 200% sous le niveau de pauvreté ont présenté une plus grande prévalence de la parodontite sévère (12).
Tableau II. Classification de la parodontite en fonction des stades. (Tiré de Papapanou et coll. (7))
*Interdentaire au site ayant la plus grande perte
Stade de la parodontite Stade I Stade II Stade III Stade IV Sévérité d’attache Perte
clinique* 1 à 2 mm 3 à 4 mm ³5 mm ³5 mm Perte osseuse radiologiqu e 1/3 coronaire (<15%) 1/3 coronaire (15 à 33%) Extension au 2/3 de la racine ou au-delà Extension au 2/3 de la racine ou au-delà Perte
dentaire Aucune perte dentaire causée par parodontite dentaire Perte causée par parodontite de £4 dents Perte dentaire causée par parodontite de ³5 dents
Complexité Locale maximale au Profondeur sondage £4 mm Perte osseuse principaleme nt horizontale Profondeur maximale au sondage £ 5 mm Perte osseuse principalement horizontale En plus de la complexité du stade II : Profondeur au sondage ³6 mm Perte osseuse verticale ³ 3 mm Atteinte de furcation classe II ou III Défaut de crête modéré En plus de la complexité du stade III Dysfonction masticatoire Traumatisme occlusale secondaire Défaut de crête sévère Effondrement de l’occlusion Migration pathologique Moins de 20 dents restantes Ampleur et répartition À ajouter au stade
Pour chaque stade, décrire l’ampleur :
Localisé (< 30% dents impliquées), généralisée (>30%) ou molaires>incisives
6
Tableau III. Classification de la parodontite en fonction des grades. (Tiré de Papapanou et coll. (7))
Grade de la parodontite Progression Grade A lente Grade B Progression modérée Grade C Progression rapide Critère primaire Évidence directe de progression Perte osseuse radiologique ou perte d’attache clinique Aucune perte après plus de 5 ans < 2 mm après plus de 5 ans ≥ 2 mm après plus de 5 ans Évidence indirecte progression % perte osseuse/âge < 0,25 0,25 à 1,0 > 1,0 Phénotype du cas Dépôts épais sur le biofilm avec bas niveaux de destruction Destruction importante avec dépôts sur le biofilm Destruction plus grande qu’attendu considérant les dépôts de biofilm Configurations cliniques spécifiques suggérant des périodes de progression rapide et/ou une maladie à début précoce Manque de réponse attendue au traitement standard de contrôle bactérien) Modificateurs
du grade Facteurs de risque
Tabagisme Non-fumeur Fumeur < 10 cigarettes/jo ur Fumeur ≥ 10 cigarettes/jour Diabète Normoglycé mique/ aucun diagnostic de diabète HbA1c < 7.0% chez les patients atteints de diabète HbA1c ≥ 7.0% chez les patients atteints de diabète
7
1.1.4. Mécanisme destructeur de la parodontite chronique
La maladie parodontale est la conséquence d’interactions entre le biofilm sous gingival et la réponse inflammatoire et immunitaire de l’hôte. En présence d’une gingivite, l’inflammation est limitée au tissu gingival seulement et peut être réversible. Tandis que lors d’une parodontite, le processus inflammatoire atteint les tissus de support parodontaux, à savoir le ligament parodontal et l’os alvéolaire. Ce processus inflammatoire entraine la destruction irréversible des fibres du ligament parodontal et la résorption d’os alvéolaire associé à une perte d’attache clinique (11). Malgré que dans de nombreux cas la gingivite précède la parodontite, il est important de noter que toutes les gingivites ne progressent pas en parodontite (12).
Durant plusieurs années, la plaque dentaire fut considérée comme la cause principale de l’installation de la maladie parodontale. Cependant, ce modèle n’explique pas pourquoi certains individus, avec un faible niveau d’hygiène, ne développent pas de maladie parodontale, alors que d’autres patients malgré une bonne hygiène orale continuent à avoir une progression de la maladie parodontale. Cette variabilité amène les chercheurs à considérer d’autres modèles histopathologiques, dans lesquels la susceptibilité et le degré de progression de la maladie parodontale sont influencés par la nature de la réponse inflammatoire et immunitaire (13). L’ensemble de la pathogenèse de la parodontite chronique sera détaillé dans cette section.
1.1.4.1. Histopathogénèse de la gingivite et parodontite
Le rôle de la plaque dentaire comme principal facteur étiologique dans le développement de la gingivite fut démontré dans les études de gingivite expérimentale de Löe et ses collaborateurs durant les années 1960s (14). Cependant la variation de la réponse d’un individu à l’autre à cette plaque bactérienne affirme qu’il existe d’autres facteurs contribuant à cette réponse qui ne sont pas encore parfaitement compris.
Par ailleurs, Page est Schroeder, en 1976, (12) décrivent les changements histologiques observés dans les tissus gingivaux lors de l’installation de la maladie gingivale. Ils divisent ces changements en 4 types de lésions : initiale, débutante,
8
établie et avancée. La lésion avancée correspond à la transition vers une parodontite chronique marquée par une perte d’attache et une résorption osseuse.
Lésion initiale : La lésion initiale se développement typiquement entre 2 à 4 jours
suivant l’accumulation de la plaque autour de la dent. Il s’agit d’une réponse inflammatoire de leucocytes résidents du tissu gingival et de cellules endothéliales au biofilm bactérien. Cliniquement, aucun signe d’inflammation n’est visible, mais des changements histologiques sont perceptibles. Elle est caractérisée par une dilatation et une augmentation de la perméabilité vasculaire facilitant la migration des neutrophiles et monocytes vers l’épithélium de jonction et le sulcus gingival. Aussi, l’augmentation des mouvements des fluides à partir des vaisseaux sanguins entraine l’augmentation de la pression hydrostatique et par le même effet du flux du liquide créviculaire. À ce stade, la composition du liquide créviculaire est similaire à celle du liquide interstitiel. Les tissus atteints par cette lésion inclues une portion de l’épithélium de jonction et la portion coronaire du tissu conjonctif, rarement plus de 5-10 % (12).
Lésion débutante : La lésion débutante apparait, généralement, 4 à 7 jours après
l’accumulation continue de plaque et correspond aux signes cliniques débutants de la gingivite. La gencive apparait cliniquement érythémateuse en raison de la prolifération capillaire, de l’ouverture de lit vasculaire dormant, d’une augmentation de la perméabilité vasculaire et d’une vasodilatation continue. On assiste, de ce fait, à une augmentation du flux du liquide réticulaire qui à ce stade se transforme en exsudat inflammatoire. Ce stade se caractérise aussi par le passage d’une lésion constituée principalement de neutrophiles à une lésion avec une augmentation du nombre de lymphocytes et macrophages. Initialement, la lésion se développe sous forme de petits infiltrats périvasculaires qui augmentent progressivement en taille. Au jour 21, les lymphocytes représentent près de 70 % de l’infiltrat inflammatoire qui représente en lui-même 5 à 15 % du tissu conjonctif (15). Cependant, même si l’infiltration reste assez restreinte à ce stade, 60 à 70 % du collagène de la zone infiltré est dégradé (12).
Lésion établie : La lésion établie correspond cliniquement à la « gingivite
9
de plaque. Même si la lésion est encore limitée au fond du sulcus et confinée à une petite potion du tissu conjonctif gingival, les cellules plasmatiques ne sont plus limitées à cette région, on les retrouve au niveau des vaisseaux sanguins et entre les fibres de collagènes profondes. Page et Schroeder, définissent cette lésion comme étant dominée par des cellules plasmatiques. On assiste à une production massive d’immunoglobulines de type IgG, quelques IgA et rarement des IgM. La majorité des lymphocytes sont des cellules B portant des immunoglobulines, même si jusqu’à 30 % des lymphocytes peuvent être des lymphocytes T.
La destruction des fibres de collagènes continue dans la zone d’infiltration. La rupture du tissu conjonctif entraîne une attache du tissu conjonctif à la dent et, par conséquent, l’épithélium de jonction et l’épithélium sulculaire prolifèrent et migrent vers le tissu conjonctif, formant ainsi une poche parodontale.
Lésion avancée : La lésion avancée présente les mêmes composants cellulaires et
caractéristiques que la lésion établie. La différence principale réside dans la perte d’attache visible cliniquement et histologiquement. La lésion n’est plus localisée, on assiste à une progression apicale et latérale de l’infiltrat inflammatoire pour former une bande autour du collet et racine de la dent. La destruction du collagène s’étend à ce stade au ligament parodontal et à l’os alvéolaire. Les neutrophiles sont prédominants au niveau de l’épithélium de la poche tandis que les cellules plasmatiques et les lymphocytes B sont prédominants dans le tissu conjonctif. Les macrophages ne sont pas les composants majeurs de la lésion avancée, puisqu’ils ne constituent que 5 % des cellules immunitaires. Les fibroblastes, lorsque stimulés par les cytokines inflammatoires IL-1, IL-6, TNF- 𝛼 et prostaglandine PGE2, produisent des métalloprotéinases matricielles responsables de la dégradation de la matrice extracellulaire, dont le collagène. L’épithélium de jonction et l’épithélium sulculaire continuent leur migration vers le tissu conjonctif. Plus la profondeur de poche augmente, plus la plaque bactérienne migre apicalement et se retrouve dans un environnement favorable à la prolifération de bactéries anaérobes parodontopathogènes.
Cette lésion marque donc la transition de la gingivite vers la parodontite. Une lésion gingivale peut rester au stade établi pendant plusieurs mois ou années, voir ne jamais
10
progresser vers un stade avancé (12). Cette transition est déterminée par plusieurs facteurs qui restent pour certains encore inconnus. Parmi ces facteurs on retrouve la composition et l’importance du biofilm, la réponse de l’hôte et les facteurs de susceptibilité, y compris les facteurs de risque environnementaux et génétiques (11).
1.1.4.2. Microbiologie de la parodontite chronique
La parodontite est une maladie infectieuse mixte nécessitant la participation de plusieurs espèces bactériennes agissant en coopération et synergie. Le rôle aujourd’hui des bactéries dans l’étiologie de la maladie parodontale est démontré et reconnu (16). En effet, l’induction de gingivites expérimentales, la corrélation positive entre la sévérité de la gingivite et le degré d’accumulation de plaque, l’amélioration de la condition suite aux contrôles de la plaque, et le fort potentiel pathogène des bactéries de la plaque sous gingivale sont des arguments qui soutiennent le lien entre la plaque bactérienne et la maladie parodontale.
1.1.4.2.1. Théories de l’étiologie microbienne
Au cours de l’histoire, 3 théories principales sont émises sur l’étiologie microbienne de la maladie parodontale : les théories de la plaque non spécifique et spécifique ainsi que la théorie écologique (15). Selon la théorie non spécifique, surtout reconnue avant les années 1960s, toutes les bactéries de la plaque dentaire possèdent un ou plusieurs facteurs de virulence participant à l’inflammation gingivale et la destruction parodontale. L’inflammation est liée à l’augmentation de la charge bactérienne plutôt qu’à la présence d’espèces microbiennes spécifiques. Le passage de la gingivite à la parodontite s’explique alors par une déficience au niveau de la réponse de l’hôte et est indépendante au type de bactéries présentes dans le site parodontal. Cependant, cette théorie n’explique pas comment certains individus avec une quantité importante de plaque et tartre ne développent jamais de maladie parodontale destructrice. Elle ne permet pas aussi de montrer pourquoi certains sites chez le même individu demeurent non atteints, tandis que d’autres sites présentent une atteinte parodontale avancée. Ces questionnements amènent les scientifiques à chercher des agents pathogènes spécifiques à la maladie parodontale. Même si aujourd’hui la théorie non spécifique
11
fut écartée en faveur des théories spécifique et écologique, la modalité de traitement de la maladie parodontale est restée la même, à savoir l’élimination complète de la plaque bactérienne et du tartre (11).
La théorie spécifique, apparue à partir de 1960, repose sur la présence d’espèces bactériennes spécifiques aux différentes maladies parodontales. La qualité de la flore bactérienne est donc plus importante que la quantité. La perte d’attache est reliée à la présence ou à l’augmentation en nombre de certaines espèces bactériennes spécifiques. Cibler ces bactéries spécifiques permettrait de guérir ses maladies parodontales (13). Cependant la parodontite, étant une condition dynamique avec des périodes de progression et d’inactivé, il est donc difficile de déterminer la ou les bactéries spécifiques dans la destruction parodontale. Il faudrait, pour cela, avoir le bon échantillon du bon site au moment de l’activité parodontale de destruction (11). Finalement, Marsh et ses collaborateurs (16) établissent durant les années 1990s, la théorie écologique de la plaque. Selon cette hypothèse, la quantité ainsi que la composition spécifique de la plaque dentaire seraient à l’origine de l’apparition de la maladie parodontale. Cette théorie repose sur le fait que les pathogènes associés à la maladie parodontale sont initialement des composants mineurs de la flore bactérienne de sites en santé. Cette flore bactérienne de sites en santé serait contrôlée par l’organisme hôte grâce à une réponse immunitaire modérée et cela pendant plusieurs années. Cependant, une perturbation de cet équilibre par une accumulation excessive de plaque ou encore par d’autres facteurs indépendants comme une déficience immunitaire, malnutrition ou habitudes de vie, pourrait être à l’origine de changement de la composition de la flore bactérienne (11). Ainsi, des changements environnementaux comme l’inflammation, la formation de tissus de granulation et/ou l’augmentation du flux du liquide créviculaire, entrainent la prolifération de bactéries parodontopathogènes aux dépens des bactéries bénéfiques de la flore bactérienne. L’environnement sous-gingival dicte et sélectionne ainsi la flore microbienne spécifique à la maladie parodontale.
12
1.1.4.2.2. Complexes bactériens associés à la parodontite chronique
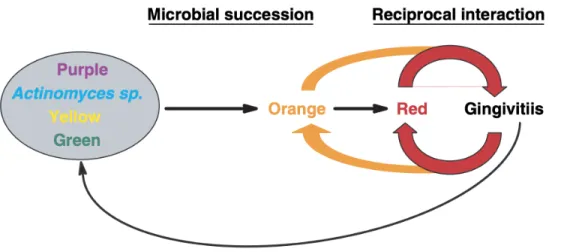
Les nombreuses avancées techniques dans l’identification et l’isolation des bactéries, dans le domaine de la microbiologie, ont permis d’effectuer différentes associations entre certaines espèces bactériennes de la flore sous-gingivale et les maladies parodontales destructrices (17). Ainsi, Socransky et ses collaborateurs (3) proposent, en 1998, la notion de complexe bactérien permettant de regrouper schématiquement les différentes espèces bactériennes parodontopathogènes. Cette notion est fondée sur le principe suivant : lorsqu’une bactérie est détectée, d’autres bactéries du même complexe seront probablement aussi détectées. Ce concept fut testé par l’analyse de données provenant de plus de 13 000 échantillons de biofilm sous-gingival de 185 sujets avec différents états de santé et de maladie parodontales (3). Ainsi six complexes furent proposés par Socransky et ses collaborateurs : rouge, orange, jaune, vert, pourpre et bleu ainsi détaillé dans la Figure 1.
Figure 1. Représentation des complexes bactéries de Socransky. (Tiré de Socransky et coll. [18])
13
Le complexe rouge est composé des espèces bactériennes : Porphyromonas gingivalis, Tannerella forsythia et Treponema denticola. Ce complexe est fortement corrélé avec les sites atteints de pathologies parodontales tels que la gingivite et la parodontite. Ce complexe est souvent associé avec le complexe orange, mais peu avec les complexes jaune, vert, pourpre et bleu.
On retrouve parmi le complexe orange notamment les espèces bactériennes : Fusobacterium, Prevotella, Campylobacter, P. micros. Streptococcus constellatus. Comme le complexe rouge, il est souvent détecté au niveau des sites avec une perte d’attache importante.
Les complexes jaune, vert, pourpre et bleu sont composés principalement d’espèces bactériennes compatibles avec la santé gingivale. Certaines sont même bénéfiques. Les espèces du complexe pourpre et jaune sont, quant à elle, pour la plupart des colonisateurs primaires. L’ensemble de ces complexes sont plutôt associés entre eux. On parle de succession bactérienne lorsque certaines espèces bactériennes, dites pionnières (ou encore colonisateurs primaires), sont remplacées par d’autres espèces après avoir modifié l’environnement qui devient favorable aux espèces suivantes. Cette succession est aussi retrouvée dans la gingivite et parodontite. Cette succession est schématisée par la Figure 2. En effet, les coques et les bâtonnets Gram positif, présents en premier, sont remplacés tout d’abord par des coques et des bâtonnets Gram négatif puis par des filaments et fusobactéries. Ces dernières sont finalement remplacées par des spirochètes (14). Ainsi, la complexification des espèces bactériennes présentes dans la plaque, vers un biofilm majoritairement composé de bactéries anaérobes à Gram négatif, marquerait le début de la maladie parodontale. Les différents complexes de Sokransky se succèdent alors jusqu’à l’obtention des complexes orange et rouge de bactéries, qui se stimulent et maintiennent un état inflammatoire à l’origine de la maladie gingivale (3).
14
Figure 2. Représentation de la succession bactérienne entrainant la formation d’inflammation gingivale. (Tiré de Socransky et coll. [18])
Ainsi, les sites atteints de maladies parodontales destructrices démontrent une flore bactérienne hétérogène majoritairement dominée par des bactéries anaérobies asaccharolytique comme les bactéries du complexe rouge et orange. De ce fait, l’analyse microbiologique de 100 sites avec de récentes pertes d’attaches en comparaison avec des sites non actifs, démontre des concentrations plus élevées en Porphyromonas gingivalis, Prevotella intermedia, Fusobacterium nucleatum et Tannerella forsythia (19). De plus, l’élimination de ces complexes de bactéries est accompagnée d’une amélioration des paramètres cliniques associés à la parodontite chronique (20).
Finalement, les formes les plus sévères et agressives de parodontite semblent être associées à la présence en forte concentration des bactéries possédant la capacité d’envahir les tissus comme les bactéries Porphyromonas gingivalis et Aggregatibacter actinomycetemcomitans (22,23).
1.1.4.2.3. Étapes de la pathogenèse bactérienne
Quatre étapes sont importantes dans le développement de la maladie parodontale chronique par les bactéries parodontopathogènes : la colonisation des sites sous-gingivaux, la multiplication, la résistance au système de défense de l’hôte et
15
finalement l’invasion et la destruction des tissus parodontaux (4). La production de facteurs de virulence spécifiques par ces bactéries facilite chacune de ces étapes. Parmi ces facteurs de virulences, nous retrouvons les adhésines, les protéases, les leucotoxines, et lipopolysaccharides.
La colonisation des sites sous-gingivaux est une étape importante dans l’initiation de la parodontite (24). Certaines bactéries parodontopathogènes, peuvent produire des facteurs de virulence comme les fimbriaes et les adhésines qui favorisent l’adhérence aux fibres de collagène, aux cellules épithéliales, aux fibroblastes du tissu hôte et à d’autres espèces bactériennes. En effet, la surface de certaines bactéries, comme le P. gingivalis sont recouvertes de fins filaments, de 3 à 10 µm de diamètre, formés de sous unités protéiques appelées fimbrilline. Ces fimbriaes permettent à la bactérie d’établir un contact avec la surface de contact, même si elle est à distance de cette surface (24).
D’autres mécanismes peuvent aussi être utilisés par les bactéries pour faciliter leur colonisation. Ainsi, la production de protéases par ces bactéries entraine tout d’abord l’exposition de récepteurs sur les cellules épithéliales, appelés cryptitopes, habituellement recouvert par des protéines ou glycoprotéines de surface. Les bactéries peuvent alors adhérer facilement à la surface épithéliale (25). C’est le mécanisme utilisé par les bactéries P. gingivalis et T. denticola (complexe rouge) avec leurs protéases gingipaine et chymotrypsine respectivement (26).
La croissance bactérienne constitue aussi un élément essentiel de la pathogenèse de la maladie parodontale (24). Pour cela, ces bactéries doivent obtenir les éléments nutritifs et les conditions environnementales idéales pour leur croissance. La plupart des bactéries parodontopathogènes sont des bactéries anaérobes, à Gram (-) et asaccharolytiques. Elles obtiennent leur énergie à partir peptides et d’acides aminés. Ainsi la dégradation de protéines plasmatiques et tissulaires du tissu cible par des protéases génère les nutriments nécessaires à leur croissance. C’est aussi l’une des fonctions des gingipaines produites par la bactérie P. gingivalis pour l’acquisition de nutriments suite à la dégradation des protéines en peptides (26).
D’autre part, certaines bactéries ont la capacité de résister au mécanisme de défense de l’hôte par l’action de certains facteurs de virulence (24). Ainsi, la présence d’une
16
capsule, constituée principalement de polysaccharides, empêche la fixation de cellules phagocytaires ou encore de facteurs opsonisant à la bactérie. Cette capsule joue aussi un rôle de résistance contre les agents antimicrobiens agissants sur la membrane cellulaire. De plus, les bactéries parodontopathogènes peuvent aussi produire des protéases impliquées dans la dénaturation des immunoglobulines A et G, paralysant ainsi la réponse humorale. D’autres protéases sont aussi impliquées dans la destruction de protéines du système du complément, altérant ainsi le mécanisme de phagocytose (25). C’est le cas des Arg-gingipaines et Lys-gingipaines produites par la bactérie P. gingivalis, ou encore la trypsine et chymotrypsine produites par la bactérie T. denticola (24).
Les bactéries A. actinomycetemcomitans et C. rectus produisent aussi des toxines sans activité enzymatique, appelées leucotoxines, dirigées contre les monocytes et les neutrophiles. Les leucotoxines, comme leur nom l’indique, provoquent ainsi une destruction des cellules immunitaires et libèrent un ensemble de chémokines, dont des facteurs pro-inflammatoires. L’ensemble de ces mécanismes permettent aux bactéries d’éviter le contact avec les cellules du système immunitaire et de pouvoir ainsi envahir les tissus sans une forte résistance (24).
Finalement, les bactéries peuvent aussi entrainer la destruction directe ou indirecte des tissus parodontaux (25). En effet, les protéases, produites par les bactéries, induisent la destruction directe de collagène et fibronectine des tissus cibles. Les bactéries peuvent aussi activer des métalloprotéinases matricielles (MMP) latentes impliquées dans la dégradation de la plupart des composants de la matrice extracellulaire comme le collagène, protéoglycane, laminine, élastine, et fibronectine (24). Ces mécanismes d’action donnent alors un plus grand pouvoir d’invasion tissulaire aux bactéries. Les protéases bactériennes produites peuvent aussi induire l’inactivation des molécules régulatrices plasmatiques et tissulaires et accentuent ainsi le phénomène destructeur. Ces mécanismes sont retrouvés encore une fois chez la bactérie P. gingivalis grâce à l’Arg-gingipaines et la Lys-gingipaines (26).
Les lipopolysaccharides et les vésicules sont aussi des facteurs de virulences impliqués dans l’invasion et la destruction des tissus parodontaux (25). En effet, le lipopolysaccharide (LPS) est une endotoxine retrouvée dans le feuillet externe de la
17
membrane des bactéries Gram (-). L’acide gras, appelé lipide A et inséré dans la membrane externe de la bactérie, est responsable de la toxicité du LPS. En se liant au récepteur TLR4, présent à la surface cellulaire, le LPS induit la libération de cytokines pro-inflammatoires qui participent au processus de destruction tissulaire (24). Les vésicules, quant à elle, sont des excroissances de la membrane externe bactérienne. Ces dernières sont constituées d’un ensemble de protéase et d’endotoxine pouvant être libéré à distance. La faible dimension de ces vésicules leur permet d’être diffusés au travers des barrières épithéliales et donc d’accéder aux tissus sous-jacents (25).
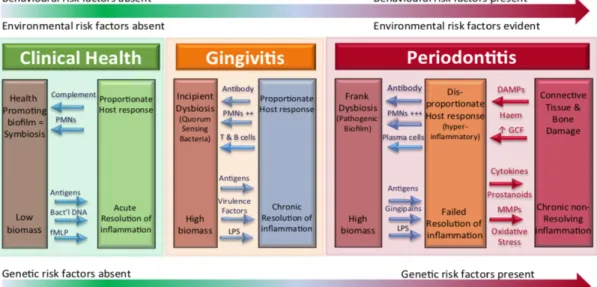
Le phénomène de destruction tissulaire n’est pas seulement lié à l’action destructrice directe des bactéries. Elle est aussi la conséquence d’une réponse immunodestructrice maintenue par une agression constante de la part des bactéries. Meyle et Chapple (28) proposent, en 2015, un modèle contemporain de la pathogenèse de la maladie parodontale dans lequel on retrouve une relation circulaire entre le biofilm parodontal et la réponse inflammatoire immunitaire. Ce modèle est résumé dans la Figure 3. Le passage d’une santé gingival à une gingivite ou parodontite résulte toute d’abord d’une dysbiose ou on assiste à l’évolution d’un biofilm compatible avec la santé gingivale vers un biofilm pathogénique. Au même moment, la réponse inflammatoire de l’hôte, qui était initialement proportionnée, se transforme graduellement en une réponse inflammatoire disproportionnée et destructrice. Cette réponse immunitaire semble aussi participer à la fonction, l’évolution, et au maintien du biofilm pathogénique (24).
1.1.4.3.Réaction inflammatoire et immunitaire
L’être humain ne peut survivre dans un environnement entouré de bactéries sans un mécanisme de défense efficace. L’immunité est ainsi constituée d’un ensemble de mécanismes biologiques permettant d’assurer l’intégrité de l’organisme en éliminant les substances étrangères et les agents infectieux (29). Ce système immunitaire humain peut être divisé en 2 catégories : l’immunité innée et l’immunité acquise. L’immunité innée est un mécanisme de défense naturel, immédiat, présent dès la naissance, non spécifique, et constitue la première ligne de défense. Alors que
18
l’immunité acquise, dite spécifique, constitue une réponse adaptative suite au développement d’une mémoire immunologique (30).
Figure 3. Modèle contemporain de la pathogenèse de la parodontite et gingivite (Tiré de Chapple et coll. (25))
DAMPs damage-associated molecular patterns; fMLP, N-formylmethionyl-leucyl-phenylalanine; GCF, gingival crevicular fluid; LPS, lipopolysaccharide; MMPs, matrix metalloproteinases; PMNs, polymorphonuclear neutrophils.
1.1.4.3.1. Réponse immunitaire non spécifique
La réponse immunitaire non spécifique est constituée d’un vaste éventail de barrière mécanique ou physiologique qui évite l’invasion des tissus et des cellules par les éléments pathogènes. Ainsi la salive, le flux du liquide créviculaire et les cellules épithéliales interviennent comme première ligne de défense en empêchant l’adhésion et l’invasion des bactéries (30). De plus, la présence dans le liquide créviculaire et dans la salive d’anticorps, de protéases et d’agent antimicrobien (tel que la mucine et la lactoferrine) a un effet inhibiteur sur la croissance bactérienne et un effet bactéricide. Les cellules épithéliales jouent quant à elle un rôle clé dans la réponse innée dans le cas de maladie parodontale puisqu’elles sont le site d’interactions entre
19
les bactéries pathogènes et l’organisme hôte en plus d’assurer la fonction de barrière mécanique contre l’invasion bactérienne. Ces cellules épithéliales peuvent aussi produire certains peptides antimicrobiens tels que les ß-defensines humaines (hBDs) capables de rompre la membrane bactérienne et ainsi tué la bactérie parodontopathogène (31).
Cependant, la relative perméabilité de l’épithélium jonctionnel parodontal ainsi que l’augmentation des espaces intercellulaires en présence d’inflammation facilitent la colonisation et l’invasion tissulaire par certaines bactéries parodontopathogènes comme P. gingivalis (32).
D’autre part, la reconnaissance des microorganismes pathogènes et le recrutement de cellules effectrices ou molécules jouent un rôle central dans l’immunité innée. Ainsi, lorsqu’une bactérie et ses produits atteignent les tissus parodontaux, des macrophages et cellules dendritiques les reconnaissent via des récepteurs de reconnaissance appelés « pattern recognition receptors » (PRRs) et déclenchent une réponse immunitaire médiée par un ensemble de cytokines et chemokines pro-inflammatoires (30). Parmi ces cytokines pro-inflammatoires, nous retrouvons l’interleukine IL-1ß à l’origine d’un ensemble de mécanismes d’installation de la réponse immunitaire non spécifique et impliquée dans le recrutement des neutrophiles via la production de la chemokine IL-8. Son effet est amplifié par l’action d’autres cytokines comme le facteur de nécrose tumorale TNF-𝛼. En plus de la réponse pro-inflammatoire, l’interleukine IL-1ß stimule aussi la production de prostaglandine PGE2, inducteur puissant de la sécrétion de MMP à leur tour responsable de la destruction du tissu conjonctif (4,33).
Les neutrophiles sont les cellulaires phagocytaires les plus abondantes et les premières cellules immunitaires à migrer dans le site d’infection ou de traumatisme. Ils sont attirés par différents facteurs de chimiotactisme comme les protéines du complément C5a, les leucotriènes B4, ou encore la chemokine IL-8 (30). Les neutrophiles migrent à partir du plexus sanguin gingival vers le tissu conjonctif extravasculaire, puis vers l’épithélium de jonction par la membrane basale. Cette migration est assurée par l’interaction de molécules d’adhésion présentes à la surface
20
des cellules épithéliales, comme l’ICAM-1, avec l’intérgrine ß-2 des neutrophiles (29).
Une fois dans le site inflammé, les principales fonctions des neutrophiles sont la migration, la phagocytose, la dégranulation et l’explosion oxydative (34). Ainsi, les neutrophiles possèdent des récepteurs capables de reconnaitre directement les lipopolysaccharides et polysaccharides de certains pathogènes et ainsi de les enrober et les emprisonner dans une vacuole appelée phagosome. Ils peuvent aussi phagocyter des micro-organismes opsonisés, c’est-à-dire entourés par des produits humoraux de la réponse immunitaire (immunoglobulines ou fragments du complément activés) en se liant la partie Fc des anticorps via leurs récepteurs Fcy.
Deux mécanismes bactéricides sont utilisés par les neutrophiles pour l’élimination des microorganismes ingérés : la voie non oxydative et la voie oxydative. La voie oxydative ou l’explosion oxydative consiste à produire des formes réactives d’oxygène à partir d’anions superoxydes formés par une enzyme, appelée NADPH oxydase, située à la surface externe de la membrane cytoplasmique des neutrophiles. Cette surface externe de la membrane plasmique, en entourant le corps étranger, devient alors la face interne du phagosome et la NADPH peut ainsi jouer son effet bactéricide. L’anion superoxyde est ensuite transformé en d’autres formes d’oxygène fortement réactives et plus toxiques pour le pathogène comme le peroxyde d’hydrogène (H2O2), le radical hydroxyle (OHo) et l’oxygène singulet (34). La voie non oxydative intervient lorsque des lysosomes fusionnent avec le phagosome pour former le phagolysosome. Les agents lysosomiaux, les acides et protéases vont agir avec les différentes formes d’oxygène réactif pour obtenir un effet bactéricide sur les agents pathogènes.
1.1.4.3.2. Réponse immunitaire spécifique
Lorsque l’invasion bactérienne n’est pas résolue, la réponse inflammatoire devient chronique, ce qui peut être considéré comme non physiologique ou pathologique (30). La réponse immunitaire adaptative est alors activée avec l’implication des mécanismes cellulaires et humoraux de l’immunité acquise. Les lymphocytes T, B et les plasmocytes sont les cellules immunitaires principalement
21
impliquées dans la réponse immunitaire spécifique. Chaque lymphocyte porte un récepteur membranaire lui permettant d’identifier un déterminant antigénique ou épitope (29).
Un élément essentiel de l’activation des lymphocytes T et B est la présentation de l’antigène par les cellules présentatrices d’antigène, plus précisément les cellules dendritiques, les macrophages et les cellules B. Ces cellules ont la capacité de capter l’antigène dans le milieu extracellulaire par phagocytose ou par endocytose (par un récepteur membranaire), de le dégrader, et de le coupler à un complexe majeur d’histocompatibilité de classe II (CMH II). L’ensemble est alors exprimé au niveau de la membrane cellulaire et prêt à être présenté à une cellule de l’immunité spécifique. Ces cellules présentatrices d’antigène font alors le lien entre la réponse innée et la réponse adaptative (30).
Les lymphocytes T jouent un rôle important dans l’orchestration de la réponse immunitaire adaptative à médiation cellulaire ainsi que dans l’activation des lymphocytes B. Ces lymphocytes T présentent à leur surface un récepteur membranaire pour antigène appelé TCR, ce dernier est incapable de reconnaître directement l’antigène. Il nécessite la présentation de l’antigène grasse au complexe majeur d’histocompatibilité. En fonction de l’origine intracellulaire ou extracellulaire de l’antigène, le type de cellules présentatrices d’antigène, la molécule CMH et le type lymphocyte T sont différents (30).
Il existe ainsi deux types de lymphocytes T : les lymphocytes T helper ou auxiliaire (dit CD4) et les lymphocytes T cytotoxiques ou tueurs (CD8). Les lymphocytes T CD4, sont essentiels à la réponse immunitaire acquise et innée, puisqu’ils initient et amplifient l’activation de plusieurs autres cellules immunitaires comme les lymphocytes B, lymphocytes T CD8, macrophages et cellules Natural Killer (NK). Les lymphocytes T CD8, quant à eux, jouent un rôle important dans l’élimination de cellules infectées par un virus (antigène endogène) grâce à la reconnaissance de l’antigène couplé au complexe d’histocompatibilité de classe I.
Le type de lymphocytes T auxiliaire (CD4) produit détermine la polarisation de la réponse immunitaire. Elle peut être par exemple caractérisée par une production d’anticorps majoritaire avec peu d’activation de cellules cytotoxiques et de
22
macrophages ou l’inverse (29). Ainsi les lymphocytes Th1, libèrent des cytokines avec un profil pro-inflammatoire, comme les TNF -ß et IFN-y. Alors que les lymphocytes Th2 ont un profil non inflammatoire avec par exemple les interleukines IL-6 et IL-10 (30).
Les cytokines présentent lors de la première rencontre avec l’antigène ainsi que le type de cellule présentatrice d’antigène influencent la transformation d’un lymphocyte T CD4 en Th1 ou Th2 (cellule dendritique souvent en Th1 et lymphocyte B souvent en Th2). De plus, il existe un contrôle réciproque entre les Th1 et Th2. Les cytokines de type Th2 inhibent le développement des réponses de type Th1 et vis versa. Grâce à ces mécanismes, les lymphocytes T peuvent ainsi réguler la réponse humorale et cellulaire de l’immunité adaptative (30).
Le lymphocyte B est responsable de la réponse immunitaire humorale spécifique grâce aux anticorps qu’il produit et qui serviront à la reconnaissance spécifique de l’agent pathogène. Ils représentent 5 à 15 % des lymphocytes circulants et reconnaissent l’antigène grâce à des récepteurs membranaires spécifiques appelés BCR (B cell receptor)(35). Ces BCR sont des immunoglobulines membranaires (IgM) caractérisées par leur diversité combinatoire. Ainsi une immunoglobuline est formée de deux chaînes lourdes et de deux chaînes légères liées entre elles de manière covalente par des ponts disulfures. Les différentes combinaisons des régions variables (site de fixation de l’antigène) et invariables de ces chaines permettent la production d’immunoglobulines spécifiques à chacun des épitopes antigéniques (32).
Les lymphocytes B après activation se transforment en plasmocytes qui sécrètent des immunoglobulines (anticorps) de la même spécificité que leur BCR. Les différentes combinaisons de chaines lourdes déterminent les classes ou isotypes d’immunoglobulines. Ces dernières vont avoir un effet bactéricide ou bactériostatique par agglutination bactérienne, inhibition d’adhérence, inactivation des microorganismes, neutralisation des toxines, opsonisation ou encore par activation du complément. De ce fait, les IgG facilitent la phagocytose des bactéries par les neutrophiles dans les crevasses gingivales et les poches parodontales par
23
opsonisation, alors que les IgA inhibent l’adhérence des bactéries aux surfaces dentaires ou aux cellules épithéliales.
Les lésions parodontales sont caractérisées par la prédominance de cellules plasmatiques et lymphocyte B (14). Ces lymphocytes B, producteurs d’anticorps, peuvent être activés par un antigène spécifique directement ou par d’autres activateurs comme les lymphocytes T ou cellules présentatrices d’antigène. Les immunoglobulines principalement retrouvées dans les tissus parodontaux sont les IgG, suivi par les IgM et certains IgA (30).
D’autre part, certains auteurs décrivent la réaction inflammatoire suite à l’accumulation de plaque comme une réaction d’hypersensibilité tardive (36). La réponse immunitaire innée efficace entraine la production d’interleukine IL-12, par les neutrophiles et macrophages, qui activent à son tour des cellules avec un profil cytokinique Th1. Plusieurs hypothèses et théories furent proposées, quant au rôle, des profils Th1 et Th2 des lymphocytes lors de parodontite chronique. Toutefois, il est maintenant généralement accepté que la parodontite chez l’homme est médiée par un équilibre entre les cellules Th1 et Th2 qui tend ensuite vers un profil à prédominance Th2 dans le cas de progression de la maladie parodontale destructrice (4). Ainsi, la prédominance de cellule B dans le cas de lésion avancée ou progressive suggère un rôle important des cellules à profil Th2. Dans le cas où la réponse innée est pauvre, la production d’IL-12 est réduite, une réponse Th1 pauvre est induite et la réponse inflammatoire inefficace. Par ailleurs, si les anticorps produits par ces cellules B sont efficaces et hautement spécifiques, la progression de la maladie sera freinée. Cependant, si ces derniers sont peu spécifiques et peu avides, la lésion persistera et l’activation continue de lymphocytes B mènera à la production continue d’IL-1(4).
Il est donc clair que la balance entre les cytokines produites par les cellules Th1/Th2 détermine si la maladie parodontale reste stable ou progresse.
1.1.4.3.3. Médiateurs d’inflammation et réponse immuno-destructrice
Les modèles récents de pathogenèse de la parodontite mettent l’emphase sur le rôle important de la réponse immunitaire dans la destruction parodontale (28). Ainsi, les