T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Cancérologie
JURY
Dr. Gilles FREISS (Rapporteur) Dr. Serge ROCHE (Rapporteur) Pr. Philippe GAULARD (Examinateur) Dr. Christiane SUSINI (Examinateur) Pr. Georges DELSOL (Président du jury) Dr. Jeannie RAGAB-THOMAS (Directeur de Thèse)
Ecole doctorale : Biologie-Santé-Biotechnologies Unité de recherche :
INSERM U563-Centre de Physiopathologie Toulouse Purpan Equipe "mécanismes moléculaires de l'oncogenèse" Directeur(s) de Thèse : Dr. Jeannie RAGAB-THOMAS
Rapporteurs : Dr. Gilles FREISS et Dr. Serge ROCHE Présentée et soutenue par
Jean-François HONORAT Le 30 septembre 2008
Titre :
Rôle de la tyrosine phosphatase SHP1 dans la régulation de l'oncogène NPM-ALK des Lymphomes Anaplasiques à Grandes Cellules
Je tiens en premier lieu à exprimer toute ma gratitude envers le Pr Georges Delsol pour m’avoir accueilli dans son équipe et témoigné sa confiance pendant toutes ces années. J’avoue avoir un regret, qui est de ne pas avoir suffisamment communiqué et interagit avec vous. Impressionné par l’excellence de votre parcours qui m’a si souvent été conté et quelque peu inhibé par votre charisme, je suis conscient d’être en grande partie responsable de cet état de fait. Cela peut paraitre anecdotique mais je garde un excellent souvenir de cette matinée de samedi que nous avions passée au laboratoire d’anatomie pathologique à analyser des lames de TMA au microscope. Je pense que votre dimension humaine peu commune contribue grandement à votre succès auprès des étudiants. D’autre part, j’associe également à ces remerciements le Pr Pierre Brousset qui, avec Georges Delsol, m’ont tout deux soutenu et encouragé dans mon projet d’avenir. J’espère être à la hauteur de la confiance que vous avez placée en moi et être performant, entre autre, en Anatomie Pathologique.
Je suis particulièrement reconnaissant aux Docteurs Serge Roche et Gilles Freiss pour avoir accepté d’évaluer ce travail de thèse. Je remercie également le Pr Philippe Gaulard ainsi que le Dr Christiane Susini pour m’avoir fait l’honneur de participer à ce jury.
Comment ne pas remercier le « duo de choc » composé de Jany et Ashraf, qui m’a accompagné et soutenu tout au long de ce travail. Il est bien difficile de retranscrire par écrit tout ce pour quoi je vous suis reconnaissant. Je crois que vous savez déjà ce que je pense de vous, comme la réciproque est également vraie. Pour donner une idée de nos relations aux personnes qui liront ces remerciements, je pense que cela pourrait se résumer par l’idée suivante : si c’était à refaire, je le referais avec vous ! Merci pour tout ce que vous m’avez apporté ; sur le plan scientifique bien sûr mais aussi et surtout sur le plan humain. Votre gentillesse et votre compréhension n’ont d’égal que votre honnêteté morale. Merci à vous deux de m’avoir soutenu et rassuré dans les moments de doutes mais aussi de m’avoir canalisé dans mes moments de révolte...Jany, après m’avoir tant appris en biologie cellulaire et processus d’oncogenèse, je compte maintenant sur vous pour me transmettre votre savoir en biochimie clinique. Ashraf m’a révélé tous les secrets du travail à la paillasse et m’a également appris à appréhender les possibles ouvertures d’un sujet vers d’autres voies. Je lui dois aussi de savoir dégainer les pipettes plus vite que mon ombre ! Sa célèbre phrase ponctuant une intense réflexion sur le sujet et commençant toujours par : « je vais te dire
puisque l’on restera assurément en contact.
Comme de coutume, il est maintenant temps de dire une petite anecdote sur chacun de mes camarades de labo :
Alan : compagnon de première heure puisque tu étais déjà là alors que nous étions encore au CNRS ; tu as eu la bonne idée de revenir à Toulouse après ton interlude Montpelliérain (idée guidée par tes sentiments pour Séverine que j’associe également à ces remerciements) et je t’en remercie car ces années n’auraient certainement pas été les mêmes autrement. Les soirées ligue des champions, bière (une par mi-temps !), pizza (mycosée parfois…) resteront d’excellents souvenirs. Elles ont été parfois entrecoupées de quelques visionnages de ce drôle de sport, inconnu des Bretons, qui se joue avec un ballon aux rebonds capricieux et que tu as été curieux de découvrir et de partager avec moi. Le tout était bien sûr suivi de discussions savoureuses sur le thème « on refait le match » ! Enfin, comment ne pas avoir également une petite pensée pour ce brave Pépito qui nous a fait tant rire par sa particularité anatomique.
Julien : un zeste de lourdeur dans les blagues, une cuillérée d’humour répétitif, un soupçon de désinvolture, une louche de bonne humeur, une bolinette de « fétardise », un saladier de loyauté et une marmite de gentillesse. Mélangez tout ça à feu vif et vous obtenez : le Luc. Un gars aux mœurs bizarre, qui boit du sucre, est monté avec des genoux en carton et dont l’unique but est de faire disjoncter son chef (sans méchanceté aucune, je précise pour les grands chefs qui liraient ces quelques lignes). Je ne serai jamais celui que tu veux que je devienne (p.….logue) mais promis, le premier TR te sera dédié ! (comprenne qui pourra, ou plutôt, non ! n’essayez pas de comprendre…).
Etienne : autre Luc quasi indissociable du Luc précédent dans son entreprise de déstabilisation du boss. J’admire ta vivacité d’esprit et ton humour spontané et pertinent. J’espère que nos longues discussions pendant ton M2R auront un peu contribué à te rassurer. Plus de doute à avoir maintenant, tu as prouvé ta valeur et vas certainement continuer à le faire au fil du temps! Je te souhaite de trouver quantité de translocations, de faire les études fonctionnelles qui vont avec et tout ça devrait logiquement contribuer à l’excellente thèse que tu mérites.
tout en discrétion, sans faire de bruit. J’espère que tu vas t’épanouir dans le travail avec ton propre sujet mais aussi et surtout en tant que maman (ça fait tout drôle de dire ça). Pour la petite anecdote, saches que je ne te serai jamais assez reconnaissant de m’avoir accompagné pour ramener à l’aéroport la « lacrymale » Patricia dont j’avais du mal à gérer les hauts et bas (surtout les bas !).
Julie : camarade, au rapport ! Toujours souriante et de bonne humeur, tu débordes d’énergie et c’est très appréciable pour ton entourage à qui tu transmets un peu de tout ce cocktail détonnant. Tes phrases ponctuées de Tic ! Tac ! Tac ! et autres onomatopées me surprendront toujours. A l’affut du moindre ragot et accroc aux discussions « chiffons », te priver de Jaja te serait particulièrement néfaste. Malgré l’impression de petite fille sage qu’elle donne, la Julie est un chouillat baba cool ; pour preuve, elle écume les festivals Woodstock aux 4 coins de l’Europe et se délecte d’onctueux bains de boue. Au fait, qu’as tu pensé du dernier film Serbo-Hongrois sur la vie trépidante des éleveurs d’ornithorynques dans le piémont de l’Oural ? ;-)
Marina : après un parcours commun en DEA puis Thèse pendant lequel tu as fait main basse sur l’ordinateur que nous étions censés partager (mais je ne t’en veux pas, tu le sais), nos routes se séparent. Tu as choisi d’aller affronter ton « fear factor » à toi en allant t’immerger dans un labo occupé par tes nouveaux copains Li, Xang, Lu et Wu et ce, dans un pays dont tu maitrises la langue à merveille ! Je te souhaite bonne chance pour la suite de ton parcours mais je ne doute pas un instant que tout se passera bien pour toi.
Cyril : je te l’ai dit 50 fois mais je continue : mille fois merci pour m’avoir fait découvrir cette passerelle car sans toi, je ne pense pas que j’en aurais eu connaissance ! J’ai également beaucoup apprécié le soutien que tu m’as apporté quand je doutais ainsi que l’aide que tu m’avais proposée lorsque j’envisageais de « me vendre au grand capital » (ça va plaire au camarade Bergalet ça !) en rentrant dans l’industrie pharmaceutique. J’admire ta patience et comprends que tu sois grognon lorsqu’il s’agit de supporter les deux zigues que tu essaies de gérer tant bien que mal. Bon courage !!!
Sylvie : félicitations encore pour les deux heureux évènements récents que tu viens de vivre puisque te voilà maintenant maman et chercheuse statutaire ! J’espère que tu ne me licencieras pas de mes fonctions de jardinier estival maintenant que j’ai quitté le labo…et puis
stopper, ce serait dommage ;o). D’autre part, ta confiture de figues se marie à merveille sur du pain grillé au petit déjeuner.
Gilles : je pense qu’il me restera longtemps de bons souvenirs de récits épiques de la jeunesse tourmentée de « l’immunologiste rock’n roll » du labo. Comment ne pas avoir un œil critique lors de la lecture d’articles quand on a côtoyé « monsieur contrôle » pendant plusieurs années ? Merci pour ton implication dans les réunions de laboratoire avec des questions et critiques, qui, bien qu’étant parfois un peu brut de décoffrage, ont le mérite de faire avancer le schmilblick.
Estelle, Fabienne et Laurence : même si j’ai moins partagé avec vous qu’avec votre collègue immuno-rocker, j’ai beaucoup apprécié de travailler à vos côtés durant ces années. Laurence, après avoir travaillé et signé un article ensemble, je vais maintenant devenir ton étudiant en m’immergeant dans le monde de l’anatomie pathologique. A bientôt à la fac…
Michelle : je vous remercie pour l’intérêt que vous avez porté à mon travail tout au long de ces années ainsi que pour les judicieux conseils que vous m’avez donnés quelques jours avant la soutenance.
Jeannine aussi nommée « Jaja » ! Célèbre pour ses « bonjouuuuur » si souvent imités mais jamais égalés. Qu’est ce que j’ai pu vous en dire des choses en 1,2,3…6 ans ! C’est avec vous que j’ai découvert le monde fabuleux du laboratoire et de la culture cellulaire alors que j’avais encore des cheveux et un peu d’acné (non j’exagère), c’est dire si ça remonte. Confidente, soutien moral, qu’est ce que j’aurais fait sans vous? Je me le demande…Merci pour tous ces bons moments passés ensemble au laboratoire bien sûr, mais aussi en dehors comme par exemple à Paris chez Florence, à Limoges chez Nath ou encore chez vous à Leucate. Et ne vous découragez pas, vous finirez par mettre la main sur l’individu fantomatique qui laisse les cartons vides en salle PCR…
Une petite pensée pour les « anciens » (dont je fais partie maintenant), qui m’ont accueilli, ont facilité mon insertion dans le laboratoire et plus que ça encore :
Florence et son enthousiasme débordant.
Nathalie et sa douceur et gentillesse légendaires.
Virginie la déjantée et son fétichisme pour la race porcine.
Vous m’aviez offert un super cadeau d’anniversaire que je ne suis pas prêt d’oublier.
Je remercie également toutes les personnes de l’équipe avec qui j’ai partagé un peu de quotidien au laboratoire: Pr Eric Delabesse, Cécile, Naïs, Jahouar, Jérôme, Emilie, Laurie et Catherine. Un grand merci aux « Gillettes » (par analogie aux Claudettes) : Emmanuelle (madame propre) et Céline, avec qui j’ai partagé un coin de ce laboratoire si prisé où seuls quelques privilégiés ont le droit de pénétrer après avoir montré patte blanche au maitre des lieux. Un petit clin d’œil aux étudiantes que j’ai encadrées et à qui j’ai essayé de transmettre un peu de mon savoir : Claire, Stella (l’arracheuse de dents) et Patricia (from Brazil). Enfin je tiens à remercier le Dr Talal Al Saati, Florence et Aurore qui m’ont accompagné lors de mes premiers pas au laboratoire pendant mes stages en licence et maitrise.
Impossible d’oublier les membres de la « team Guy Laurent » qui supportent jour après jour les bruyants « Delsol » dans le bureau commun : Samar (une autre fan de SHP1) et la triplette d’Emilie ; Christine et Hélène avec qui j’avais passé une super journée au ski mais aussi les « anciens » : Ludivine, Fabien et Cindy. Je remercie également l’ensemble des membres des équipes du département (équipes Guy Laurent, Bernard Payrastre et Jean-Jacques Fournié) avec qui j’ai eu l’occasion de partager quelque chose à un moment ou à un autre au cours de ces longues années mais aussi toutes les personnes de l’administration et des différents plateaux techniques.
Camille : ce mois de septembre 2006 restera décidément l’un des meilleurs. Tout juste de retour d’un somptueux voyage en Polynésie, j’ai eu le bonheur de te rencontrer. Pour commencer et en dehors de toute considération sentimentale, il faut que je te dise que j’ai eu énormément de plaisir à travailler avec une personne enthousiaste, (ultra)motivée et ayant soif d’apprendre, ce qui fut très gratifiant pour l’encadrant en herbe que j’étais. Je me souviens qu’un jour tu m’as dit que j’étais en partie responsable de l’envie que tu avais de poursuivre dans cette voie et c’est pour moi une grande fierté. Je suis fier de toi et de ton brillant parcours (2 bourses !!!). Les Toulousains n’ont pas voulu de toi en M2R pour d’obscures raisons et tu leur as fait comprendre de la plus belle des manières à quel point ils étaient dans l’erreur ; belle revanche (jubilatoire)! Aujourd’hui, tu es toujours à mes côtés, tu contribues à mon
qui me fait cruellement défaut. Nous ne sommes qu’au début d’une route que j’espère longue. Je remercie bien sûr mes parents et ma famille qui m’aiment, me font confiance et m’apportent un soutien sans faille me permettant d’avancer et de devenir celui que je suis.
Enfin, je souhaite conclure ce « chapitre » en adressant mes plus sincères remerciements aux donateurs des diverses associations de lutte contre le cancer et plus particulièrement au comité du Gers de la ligue contre le cancer et à l’Association pour la Recherche sur le Cancer, qui m’ont permis de réaliser ce travail.
1
Table des matières
Introduction
I. Les tyrosine phosphatases : 4
I .1. Généralités : 4
I.2. Classification des PTPs : 5
I.3. Mécanisme d’action des PTPs : 8
I.4. Modifications post-traductionnelles : 9
I.5. Régulation des PTPs : 9
I.5.1. Régulation négative des PTPs récepteurs par homodimérisation : 9
I.5.2 Régulation par phosphorylation : 9
I.5.3 Régulation par clivage protéolytique : 10
I.5.4 Régulation par oxydation réversible du résidu cystéine du site actif : 10
I.5.5 Régulation par repliement intramoléculaire : 11
I.6. Rôles des PTPs dans les phénomènes de cancérisation des cellules : 11
I.6.1. Les PTPs suppresseurs de tumeurs (tableau 1) : 11
I.6.1.1. Mutations inactivatrices des PTPs et cancer du colon : 11
I.6.1.2. Mutations de PTEN : 12
I.6.1.3. Mutation et perte d’expression de DEP1 : 12
I.6.1.4. Perte d’expression de GLEPP1 et de SHP1 : 13
I.6.1.5. Rôle de PTPL1 : 13
I.6.2. Les PTPs oncogéniques (Tableau 2) : 15
I.6.2.1. PRLs et métastases des cancers colorectaux : 15
I.6.2.2. Mutations de SHP2 : 16
I.6.2.3. Autres PTPs oncogéniques : 17
I.6.3. Les PTPs comme potentielles drogues anti-tumorales : 18
II. Les PTPs à domaines SH2 : SHP1 et SHP2 (SHPs) : 20
II.1. Structure et expression cellulaire : 20
II.2. Localisation subcellulaire : 21
II.3. Régulation de l’activité des SHPs : 22
II.3.1. repliement intramoléculaire : 22
II.3.2. phénomènes de phosphorylation en C-terminal : 23
II.3.3. Activation par les analogues de la somatostatine : 26
II.4. Rôle biologique de SHP1 : 28
2
II.4.1.1 Rôle de SHP1 dans les lymphocytes B : 30
II.4.1.2. Rôle de SHP1 dans les lymphocytes T : 34
II.4.1.3. Rôle de SHP1 dans les cellules NK : 36
II.4.1.4. Rôle de SHP1 dans les cellules myéloïdes : 37
II.4.2. Rôle de SHP1 dans les processus tumoraux : 39
II.4.2.1. Rôle de SHP1 comme régulateur de kinases oncogéniques : 39
II.4.2.1.1. SHP1 et FLT3/ITD : 40
II.4.2.1.2. SHP1 et c-kit : 40
II.4.2.1.3. SHP1 et TRK-T3 : 41
II.4.2.1.4. SHP1 et Ret : 42
II.4.2.1.5. SHP1 et Bcr-Abl : 42
II.4.2.2. Mécanismes mis en jeu dans la modulation de l’expression de SHP1 dans les tumeurs : 44
II.4.2.3. SHP1 et autres types de cancer : 47
III. Les Lymphomes Anaplasiques à Grandes Cellules (LAGC) : 50
III.1. Généralités : 50
III.2. Caractéristiques morphologiques : 51
III.3. Les marqueurs de diagnostic des LAGC : 52
III.4. Les marqueurs de pronostic des LAGC : 53
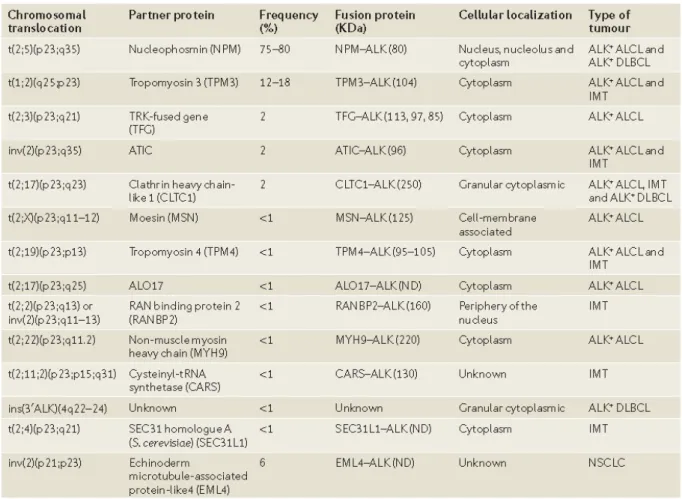
III.5. Les différents variants moléculaires impliquant ALK (tableau 3): 54
III.5.1. NPM-ALK, translocation (2 ;5)(p23 ;q35) : 55
III.5.2. TPM3-ALK, translocation (1 ;2)(q25 ;p23) : 58
III.5.3. TPM4-ALK, translocation (2 ;19)(p23 ;p13.1) : 58
III.5.4. TFG-ALK, translocation (2 ;3)(p23 ;q21) : 58
III.5.5. ATIC-ALK, inv(2)(p23 ;q35) : 58
III.5.6. CLTC-ALK, translocation (2 ;17)(p23 ;q23) : 59
III.5.7. MSN-ALK, translocation (2 ;X)(p23 ;q11-12) : 59
III.5.8. MYH9-ALK, translocation (2 ;22)(p13 ;q11.2) : 60
III.5.9. ALO17-ALK, translocation (2 ;17)(p23 ;q25) : 60
III.6. Expression de ALK dans d’autres pathologies hématologiques : 62 III.7. Expression de ALK dans les pathologies non hématologiques : 62 III.7.1. Les tumeurs myofibroblastiques inflammatoires (IMT) : 63
III.7.2. Les neuroblastomes : 63
III.7.3. Les glioblastomes : 63
III.7.4. Les Rhabdomyosarcomes : 64
3
III.7.6. Les cancers du poumon : 64
III.8. Les modèles animaux : 65
III.9 La signalisation de NPM-ALK : 67
III.9.1. NPM-ALK et prolifération : 67
III.9.2. NPM-ALK et survie : 71
III.9.3. Migration cellulaire et réarrangement du cytosquelette : 76 III.9.4. Modulation de l’immunophénotype des LAGC par NPM-ALK : 77
III.10. Génomique et protéomique des LAGC : 80
III.11. Traitements des LAGC : 82
III.11.1. Traitements actuels : 82
III.11.2. Les anticorps anti-CD30 : 82
III.11.3. ALK comme cible thérapeutique : 83
III.11.3.1. Utilisation de RNAi ou d’oligonucléotides antisens (ASO) dirigés contre ALK : 83 III.11.3.2. Inhibiteurs de l’activité kinase de ALK : 83 III.11.3.3. Vaccination contre les lymphomes présentant l’antigène ALK : 85
Objectifs
Objectifs du travail de thèse : 87
Résultats expérimentaux
I. La tyrosine-phosphatase SHP1 régule négativement l’oncogène NPM-ALK : 89 II. Stimulation de l’activité phosphatase de SHP1 par un analogue de la somatostatine : 95 III. Implication de tyrosine-phosphatases dans l’apoptose induite par l’acide ursolique dans
les cellules de LAGC ALK positives : 109
Discussion
Discussion : 124
Références bibliographiques
4
I. Les tyrosine phosphatases :
I .1. Généralités :
Les cascades de signalisation intracellulaire sont régies par des phénomènes de phosphorylations réversibles résultant des actions opposées mais complémentaires de protéines kinases et de protéines phosphatases. Les protéines peuvent être phosphorylées sur des résidus sérine, thréonine et tyrosine. Ces phénomènes de phosphorylation interviennent dans de nombreux processus physiologiques tels que les communications inter- et intra-cellulaires, la forme et la motilité des cellules, la différenciation, la prolifération ou l’apoptose, la régulation de la transcription des gènes ou encore le trafic moléculaire intracellulaire.
Contrairement aux protéines kinases qui sont issues d’un ancêtre commun, les protéines phosphatases ont évolué dans des familles structurellement et fonctionnellement distinctes. Ainsi l’on distingue deux grandes familles de protéines phosphatases :
- Les protéines tyrosines phosphatases : Elles sont caractérisées structurellement par une combinaison de domaines d’interactions protéiques et par un ou deux domaines catalytiques conservés avec un site actif sensible à l’oxydation et contenant un résidu cystéine.
- Les protéines sérine/thréonine phosphatases : Elles présentent une organisation totalement différente des protéines tyrosine-phosphatases. En effet, elles sont constituées de sous unités multiples qui s’associent entre elles. Les possibilités combinatoires sont donc très grandes, ce qui permet une plus grande diversité et flexibilité au détriment d’une spécificité stricte et d’une régulation fine.
L’équilibre entre l’action des PTKs (Protéines Tyrosine Kinases) et celle des PTPs (Protéines Tyrosines Phosphatases) est un mécanisme fondamental de la physiologie cellulaire. Aussi, lorsque cet équilibre est dérégulé, il participe à la pathogenèse de nombreuses maladies allant des déficiences immunitaires au cancer. L’étude des PTPs dans la régulation des signaux de transduction a fait l’objet d’un nombre plus limité de travaux que celle des PTKs. Cela est en partie du à des raisons historiques puisque la première PTP a été
5 purifiée1 et clonée2 10 ans après la première PTK3. Néanmoins, depuis cette date, de nombreux travaux ont mis en évidence le rôle majeur des PTPs dans la régulation des processus physiologiques.
I.2. Classification des PTPs :
Chaque PTP possède une fonction qui lui est propre, comme le montrent les expériences de délétions géniques chez les souris qui se traduisent par un phénotype unique. De la même manière qu’un « Kinome » a été défini pour les PTKs, le « PTPome » permet de regrouper et de classer les PTPs. On distingue 4 familles de PTPs classées sur la base de leurs fonctions, de leurs structures et de la séquence en acide aminé de leurs domaines catalytiques4 (figure 1) :
- Class I Cys-based PTPs (99 gènes) : Elles ont toutes évolué à partir d’un ancêtre commun.
o PTPs classiques (38 genes) :
PTPs transmembranaires de type récepteur (21 gènes). PTPs non récepteurs (17 genes).
o Dual specificity (DSPs) ou VH1-like (61 gènes) : Elles déphosphorylent les phospho-Tyr, phospho-Ser et phospho-Thr mais également des substrats non protéiques tels que les lipides.
MAP-kinase phosphatases ou MKPs (11 gènes) : Elles sont spécifiques des MAP kinases (Mitogen-Activated Protein kinase) telles que Erk, Jnk et p38.
DSPs atypiques (19 gènes) : Elles sont moins bien caractérisées et sont de plus petite taille (moins de 250 acides aminés). Elles ne présentent pas de domaines ciblant les MAP kinases bien que certaines déphosphorylent Erk ou Jnk. Un membre de cette famille PIR (DUSP11) déphosphoryle les ARNm.
Slingshots (3 gènes).
Phosphatases of Regenerating Liver ou PRLs (3 genes).
CDC14s (4 gènes) : Elles interviennent dans la déphosphorylation sur threonine de la boucle d’activation des « cyclin dependant kinases » (cdk) mais aussi dans l’inactivation des cdk et lors de la sortie de mitose.
6 PTENs (5 gènes) : Elles déphosphorylent spécifiquement le
phosphate en postion D3 des phosphoinositides et plus particulièrement le PI(3,4,5)P3 en PI(4,5)P2.
Myotubularines (16 gènes) : Elles ont la même spécificité que les PTENs mais déphosphorylent le PI(3)P en PI.
- Class II Cys-based PTPs (1 gène) : Ce sont des PTPs de bas poids moléculaire (LMPTP). Elles sont spécifiques des tyrosines et sont plus anciennes que celles de la classe I. En effet, des représentants de cette famille ont été retrouvés chez les plantes et les procaryotes.
- Class III Cys-based PTPs (3 gènes) : Elles sont spécifiques des tyrosines et thréonines et ont évolué à partir d’une enzyme bactérienne. Ces PTPs sont représentées par 3 régulateurs du cycle cellulaire : CDC25A, CDC25B et CDC25C. Elles déphosphorylent les cdk au niveau de leur extrémité inhibitrice N-terminale doublement phosphorylée sur thréonine et tyrosine. Cette réaction est indispensable pour l’activation de ces kinases afin de permettre la progression dans le cycle cellulaire. Elles sont elles mêmes régulées par phosphorylation. - Asp-based PTPs (4 gènes) : Ce sont les PTPs EyA. Elles utilisent un mécanisme
catalytique différent basé sur un acide aspartique. Elles sont spécifiques des tyrosines et des sérines. Elles jouent un rôle important au cours de l’embryogenèse.
Malgré leur similarité dans les mécanismes catalytiques et dans la structure de leur site actif, les PTPs des classes I, II et III ont évolué indépendamment.
7
Figure 1 : Représentation schématique de l’ensemble des membres des quatre familles de PTPs (Source : Alonso, A, Cell, 20044).
Une des caractéristiques des PTPs est qu’elles sont composées pour la plupart d’une combinaison de domaines modulaires. Au moins 79 des 107 PTPs présentent au moins un domaine supplémentaire à leur domaine catalytique. Ces domaines peuvent être des sites de liaisons à d’autres protéines ou bien à des phospholipides. Comme pour les PTKs, ces domaines servent à la fois à la régulation des PTPs mais aussi au ciblage des substrats et à leur localisation subcellulaire. Alors que de nombreuses PTKs présentent une combinaison de domaines SH2 et SH3, seulement deux PTPs humaines (SHP1 et SHP2) contiennent des domaines SH2 en tandem. A ce jour, aucune PTPs contenant de domaine SH3 n’a été décrite. Par contre, certaines contiennent des domaines riches en proline leur permettant de s’associer
8 avec des protéines possédant des domaines SH3. Certaines PTPs utilisent les domaines FERM (Four point 1 ezrin radixin mosein) pour être localisées à l’interface entre le cytosquelette et la membrane plasmique par l’intermédiaire d’un phosphoinositide.
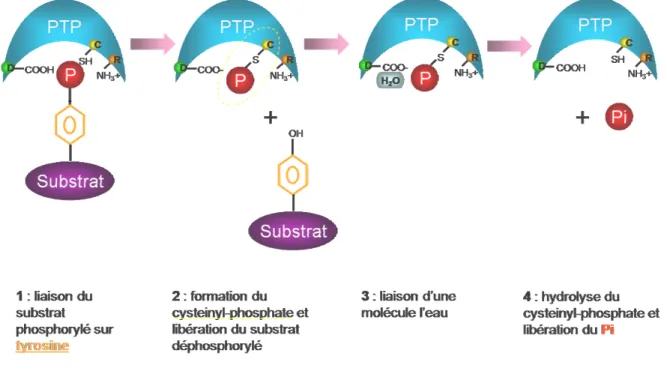
I.3. Mécanisme d’action des PTPs :
Il s’agit d’un mécanisme catalytique basé sur un résidu cystéine5. La « boucle PTP » formée par le motif caractéristique des PTPs (H/V)C(X)5R(S/T) et un acide aspartique sont
indispensables à l’activité catalytique et sont conservées dans les trois principales familles de PTPs. Le site actif cystéine est utilisé comme un nucléophile lors de la formation d’un état intermédiaire phospho-cystéinyl. Le résidu arginine agit comme un « substrat binding » et participe à la stabilisation de cet état transitoire intermédiaire. L’acide aspartique joue un rôle dans l’hydrolyse de la liaison thiophosphoryl (figure 2). La spécificité stricte des PTPs pour les phosphotyrosines est due à une poche catalytique de 9 Angstroms de profondeur6 alors que celle qui reconnait les phosphoserines et phosphothreonines mesure seulement 6 Angstroms.
9
I.4. Modifications post-traductionnelles :
De nombreuses PTPs subissent des modifications post-traductionnelles4. Les PTPs transmembranaires sont soumises à des phénomènes de glycosylation sur leurs parties extra-cellulaires alors que certaines DSPs peuvent être myristoylées ou farnésylées en N- et C-terminal respectivement. Cependant la modification la plus courante est la phosphorylation sur sérine, thréonine ou tyrosine. Il existe de nombreux exemples qui seront développés par la suite, dans lesquels la phosphorylation est impliquée dans la régulation de l’activité.
I.5. Régulation des PTPs :
Comme toutes les familles d’enzymes jouant un rôle majeur dans la signalisation intracellulaire, l’activité des PTPs est étroitement régulée in vivo par de nombreux mécanismes. Ces régulations sont hautement dynamiques dans le temps et dans l’espace.
I.5.1. Régulation négative des PTPs récepteurs par homodimérisation :
La structure cristallographique des domaines PTP a permis d’améliorer la compréhension des mécanismes de régulation des PTPs récepteurs (RPTPs) par dimérisation. La plupart des RPTPs contiennent deux domaines PTP intracellulaires : D1 et D2. On a pu observer une organisation des domaines PTPs en dimères symétriques dans lesquels un motif particulier (helix-turn-helix wedge) d’un domaine bloque le site actif du domaine partenaire7,
8
. Ainsi, contrairement au PTKs, dont la dimérisation induite par la fixation du ligand provoque une activation, l’état de dimére des RPTPs se traduit par une diminution de l’activité catalytique par occlusion des sites actifs.
I.5.2 Régulation par phosphorylation :
Certaines PTPs sont régulées par phosphorylation par l’action des PTKs, des Ser/Thr kinases mais aussi d’autres PTPs. Parfois des réactions de déphosphorylation par autocatalyse peuvent avoir lieu. La phosphorylation sur tyrosine reste difficile à étudier parce que difficilement détectable (stochiométriquement faible). Il existe néanmoins certaines PTPs pour lesquelles le rôle de la phosphorylation sur tyrosine dans la régulation a été démontré. Par exemple, la régulation par phosphorylation de l’activité des PTPs possédant des domaines SH2 (SHP1 et SHP2) sera développée dans le chapitre correspondant à ces PTPs (SHPs).
10
I.5.3 Régulation par clivage protéolytique :
Certaines PTPs sont activées par clivage protéolytique. C’est par exemple le cas de la TC-PTP (T cell protein tyrosine phosphatase) qui se présente sous la forme d’une protéine inactive de 45kDa (TC45) à localisation nucléaire et une autre de 48kDa (TC48) associée à la membrane du réticulum endoplasmique. L’isoforme TC45 est clivé en un fragment actif de 33kDa par la trypsine au niveau d’une région sensible aux protéases dans la partie non catalytique C-terminale. Ce clivage permet d’augmenter l’activité enzymatique de 20 à 200 fois par rapport à la protéine pleine taille9. Un mécanisme équivalent existe pour l’isoforme TC48. Le fragment C-terminal semble être important pour la localisation de la protéine puisque cette PTP est relocalisée dans la fraction soluble du cytoplasme suite au clivage par la trypsine10. Un autre exemple d’activation de PTP par clivage protéolytique est celui de la PTP1B. En effet, PTP1B est clivée en un fragment de 42kDa par la calpaine et est ainsi activée11.
I.5.4 Régulation par oxydation réversible du résidu cystéine du site actif :
Les PTPs de classes I et II sont connues pour être très sensibles à l’oxydation12. La production d’espèces réactives de l’oxygène (ROS) lors de la stimulation cellulaire par les facteurs de croissance, dans le cancer, l’inflammation ou les maladies neurodégénératives a donc un effet majeur sur la régulation des PTPs. De plus, cette régulation réversible des PTPs par l’équilibre redox apparait comme un phénomène intervenant également dans les cellules normales. En effet, il a été montré que les PTPs pouvaient être oxydées transitoirement en réponse à différents stimuli cellulaires. Suivant le niveau d’oxydation, le site actif cystéine des PTPs peut être transformé en acide sulphenique (SOH), sulphinique (SO2H) ou
sulphonique (SO3H). Pour que l’oxydation soit réversible il ne faut pas que le site actif soit
oxydé au-delà du niveau acide sulphenique au risque de devenir irréversible. L’oxydation de la cystéine nucléophile en acide sulphenique provoque par exemple pour PTP1B, un profond changement conformationnel qui se traduit par une perte de l’interaction avec son substrat et une exposition de la cystéine oxydée à l’environnement cellulaire. Cela permet à la fois de prévenir une oxydation irréversible mais aussi de permettre une réduction aisée pour un retour de la PTP à un état actif13, 14. D’autres PTPs comme CDC25C, PTEN ou encore LMPTP sont également sensibles à l’oxydation. Contrairement aux PTPs classiques, elles disposent d’un second résidu cystéine au niveau du site actif, qui va servir à réaliser un pont disulfure avec
11 l’autre cystéine au moment de l’oxydation, empêchant alors un niveau d’oxydation supérieur et donc une irréversibilité dans la réaction15. Le pont S-S peut également être très facilement réduit pour un retour à un état actif de ces PTPs.
I.5.5 Régulation par repliement intramoléculaire :
SHP1 et SHP2 sont les principaux exemples de ce mode de régulation des PTPs. Les régions non catalytiques se replient ou se déplient de façon dynamique afin d’empêcher ou de libérer l’accès des substrats au site catalytique des enzymes. Cet aspect sera décrit plus en détail dans la partie consacrée à ces SHPs.
I.6. Rôles des PTPs dans les phénomènes de cancérisation des cellules :
Les rôles des PTPs dans la physiopathologie cellulaire sont multiples. Nous nous limiterons ici à développer leur implication dans le cancer. Les PTPs ont des effets inhibiteurs et stimulateurs sur de nombreuses voies de signalisation impliquées dans l’oncogenèse. Il est connu que la dérégulation de la fonction des PTPs peut être associée avec l’apparition de tumeurs dans différents types de tissus. Certaines PTPs ont été identifiées comme étant des suppresseurs de tumeur alors que d’autres sont considérées comme oncogéniques.
I.6.1. Les PTPs suppresseurs de tumeurs (tableau 1) :
Depuis leur découverte, certaines PTPs sont considérées comme étant des suppresseurs de tumeurs du fait de leur effet antagoniste sur la signalisation induite par les PTKs oncogéniques. Nous allons illustrer ce concept par quelques exemples de PTPs connues pour jouer un rôle important dans le cancer.
I.6.1.1. Mutations inactivatrices des PTPs et cancer du colon :
Wang, Z et al.16 ont recherché les mutations des PTPs dans 180 échantillons de tumeurs de cancer du colon. Ils ont ainsi mis en évidence 83 mutations somatiques différentes dans 26% des cas. Dans la plupart des cas, ces mutations provoquent une perte de fonction des protéines. Elles concernent à la fois les RPTPs et les PTPs non récepteurs et touchent le domaine catalytique dans moins de 1/3 des cas seulement. Par exemple, les mutations affectant les RPTPs sont toutes situées dans la partie extracellulaire. Toutes les PTPs non récepteurs mutées appartiennent au sous groupe de PTPs contenant un domaine FERM, qui
12 connecte le cytosquelette de filaments d’actine avec les membranes. Cette donnée indique que la perte de fonction de telles PTPs affecte les propriétés adhésives et migratoires des cellules cancéreuses. Cette étude a également montré que les mutations inactivatrices des PTPs étaient la plupart du temps associées avec des mutations activatrices des PTKs.
I.6.1.2. Mutations de PTEN :
L’action de PTEN est antagoniste à celle de la PI3 kinase (PI3-K) car elle déphosphoryle le PI(3,4,5)P3 (PIP3) en PI(4,5)P2 (PIP2) sur la face interne de la membrane plasmique. PTEN exerce donc un rôle de régulateur négatif sur la voie PI3K/Akt qui stimule la progression dans le cycle, la prolifération, l’adhésion, la migration et la survie cellulaire17,
18
. PTEN est régulée par la phosphorylation d’un cluster de résidus sérine et thréonine situé dans sa partie C-terminale. La phosphorylation de PTEN affecte sa liaison à la membrane plasmique et sa stabilité19. Elle semble résulter d’une boucle de retro-contrôle négatif puisqu’elle dépend du niveau de PIP3 dans la cellule20. Le gène PTEN est muté avec une fréquence élevée dans une grande variété de cancers (poumons, cerveau, prostate). En ce sens, il est considéré, avec le gène p53, comme l’un des plus relevants dans l’oncologie clinique. L’absence de PTEN fonctionnelle s’accompagne d’une augmentation d’activité de la voie de survie PI3K/Akt/mTOR ainsi que d’un mauvais pronostic21. L’efficacité de certaines thérapies anticancéreuses dépend de PTEN22. Enfin, la déplétion de PTEN dans les cellules souches hématopoïétiques se traduit par une perte de ces cellules indifférenciées auxquelles se substituent des cellules souches leucémiques23.
I.6.1.3. Mutation et perte d’expression de DEP1 :
La surexpression de DEP1 dans des cellules de cancer du sein a permis de réduire de 5 à 10 fois l’indice de croissance tumorale24. De tels résultats ont également été mis en évidence dans des cellules cancéreuses de pancréas, de thyroïde25 et de colon. Il a été montré chez la souris que le gène Ptprj, qui code pour DEP1, possède un locus conférant la susceptibilité au cancer du colon et est fréquemment délété ou muté dans plusieurs types de cancer26.Plusieurs travaux ont montré le rôle de régulateur négatif de DEP1 sur les signaux de prolifération via la déphosphorylation directe de plusieurs récepteurs des facteurs de croissance (PDGFR, VEGFR et HGFR). Il a également été montré que DEP1 intervient dans la stabilisation de p27, une protéine inhibitrice de Cdk qui provoque un arrêt du cycle en G127. La majorité de ces fonctions tendent à conférer à DEP1 un statut de suppresseur de tumeur.
13
I.6.1.4. Perte d’expression de GLEPP1 et de SHP1 :
La perte d’expression par modifications épigénétiques telle que l’hyperméthylation du promoteur est un mécanisme général d’inactivation de suppresseurs de tumeur. Ce phénomène d’hyperméthylation du promoteur a été particulièrement étudié pour les PTPs GLEPP1 et SHP1. GLEPP1 (Glomerular Epithelial Protein 1) est une PTP structurellement similaire à DEP1 avec un seul domaine catalytique intracellulaire et une partie extracellulaire composée exclusivement de fibronectine de type III. La méthylation du promoteur de GLEPP1, PTPRO, a été mise en évidence dans 50% des cas de cancer du poumon28 et de cancer du colon associé à une instabilité microsatellitaire29. La réexpression de GLEPP1 dans des cellules de cancer du poumon dans lesquelles le promoteur est méthylé se traduit par une diminution de la croissance indépendante de l’ancrage, de la prolifération et de la résistance à l’apoptose28.
Comme nous le verrons plus en détail dans le chapitre suivant concernant les SHPs, SHP1 est un antagoniste des signaux des facteurs de croissance dans les cellules épithéliales et hématopoïétiques. La perte d’expression de SHP1 par hyperméthylation de son promoteur a dans un premier temps été décrite dans des lignées cellulaires issues de lymphomes T30 dans lesquels l’expression de SHP1 peut être restaurée par l’utilisation d’agent déméthylants. Cette hyperméthylation du promoteur du gène codant SHP1 (PTPN6) a également été mise en évidence dans les lymphomes anaplasiques à grandes cellules31, différents types de leucémies et dans les myélomes multiples32. Le rôle de SHP1 dans ces pathologies sera développé dans le chapitre consacré à cette phosphatase.
I.6.1.5. Rôle de PTPL1 :
La PTP cytoplasmique PTPL1 a été clonée et caractérisée pour la première fois en 1994 par l’équipe de Gonez, LJ33. Elle est caractérisée par la présence d’un domaine FERM. Elle possède également un motif « leucine zipper », cinq domaines PDZ d’interaction protéique et un domaine catalytique C-terminal. Il a été montré qu’elle pouvait se lier via ses domaines PDZ à la partie C-terminale du récepteur de mort Fas34 mais aussi à la protéine PARG1 (PTPL1-associated RhoGap1) qui joue un rôle de régulateur négatif sur Rho et, par l’intermédiaire de PTPL1, sur les protéines phosphorylées impliquées dans la signalisation de Rho35. Il a également été montré que son domaine FERM permet sa localisation à la membrane plasmique via sa liaison au PI(4,5)P236. L’expression de PTPL1 est régulée par les
14 anti-œstrogènes. Ainsi, le traitement de cellules de cancer du sein par l’hydroxitamoxifène induit de façon temps et dose dépendante l’expression de PTPL1 et augmente l’activité PTP globale37. PTPL1 possède également des propriétés pro-apoptotiques en affectant la voie PI3-K/Akt. La protéine IRS-1 est également un substrat de PTPL1. Cette PTP est donc capable d’inhiber la voie IRS-1/PI3-K/Akt ainsi que la survie cellulaire induite par l’IGF-1 en induisant l’apoptose38. Une étude cristallographique a montré que PTPL1 est capable de déphosphoryler des peptides bis-phosphorylés plus facilement que des peptides mono-phosphorylés. Elle peut donc agir sur des récepteurs aux facteurs de croissance présentant des tyrosines phosphorylées en tandem39. Une recherche de mutations de PTPL1 dans les cancers colorectaux a mis en évidence 19 mutations somatiques dont 8 sont responsables de la délétion complète du domaine d’activité PTP, 6 sont des mutations ponctuelles localisées en dehors du domaine catalytique, et 5 à l’intérieur de celui-ci16. Le gène codant PTPL1 apparait donc comme étant un candidat suppresseur de tumeur puisque des mutations somatiques, une perte allélique ou des méthylations de son promoteur ont été reportées dans certaines tumeurs. Cependant, des propriétés oncogéniques de PTPL1 ont également été décrites dans certaines pathologies. Ainsi, un facteur de transcription aberrant résultant de la translocation t(11 ;22) cible la réexpression de PTPL1 dans les sarcomes de Ewing’s. PTPL1 y est surexprimée et sont taux d’expression augmente avec les stades de la pathologie40.
15
Tableau 1 : PTPs ayant un rôle suppresseur de tumeur (Source : Tonks, NK, Mol Cell Biol, 20068).
I.6.2. Les PTPs oncogéniques (Tableau 2) :
Certaines PTPs sont connues pour réguler positivement les signaux de prolifération cellulaire en inhibant par déphosphorylation des protéines régulant négativement ces voies de signalisation. Aussi, des mutations conférant un gain de fonction à certaines PTPs peuvent être à l’origine d’effets oncogéniques.
I.6.2.1. PRLs et métastases des cancers colorectaux :
Il a été montré que la « phosphatase of regenerating liver-3 » (PRL-3) est surexprimée dans les cancers colorectaux métastatiques comparativement aux tumeurs primaires non métastatiques ou aux tissus normaux41. Le niveau d’expression de PRL-3 est élevé quelque soit la localisation des métastases (foie, poumon, cerveau, ovaires) dérivées de cancers colorectaux. Cependant, PRL-3 est peu voire pas du tout exprimée dans les lésions
16 métastatiques dérivées d’autres types de cancer (pancréas, estomac, œsophage). La PTP est isoprénylée et localisée à la membrane plasmique42. PRL-3 apparait donc comme étant un bon marqueur pronostique des métastases des cancers colorectaux. Pour cela, des anticorps monoclonaux dirigés contre PRL-3 ont été produits43. Les travaux de Kato, H et al.44 ont montré par l’utilisation d’un siRNA ciblant l’ARNm de PRL-3, que l’extinction transitoire de la PTP se traduisait par une forte diminution de la motilité cellulaire in vitro et de la colonisation hépatique in vivo. Cependant, aucun effet sur la prolifération cellulaire n’a été observé. PRL-3 joue donc un rôle majeur dans la dissémination métastatique des cancers colorectaux, notamment lorsque les cellules quittent la circulation sanguine pour pénétrer dans le tissu cible. PRL-3 parait donc être une cible thérapeutique intéressante pour le traitement des cancers colorectaux45.
I.6.2.2. Mutations de SHP2 :
L’exemple de mutations gain de fonction le plus caractéristique est celui de la PTP SHP246. SHP2 est connue pour être un médiateur positif de la signalisation des protéines kinases activées. Les capacités transformantes de plusieurs récepteurs à activité tyrosine kinase constitutivement activés comme EGFR, FGFR3, Ret, ERBB2 et Bcr-Abl semblent dépendantes de SHP2. SHP2 est recrutée au niveau des complexes protéiques présents à la membrane plasmique grâce à ses domaines SH2 et ce, soit directement, soit indirectement via des protéines adaptatrices telles que IRS1 ou Grb2. Les substrats majeurs de SHP2 sont des protéines dont la phosphorylation régule négativement la voie Ras-Erk ou des protéines phosphorylées qui recrutent un régulateur négatif de Ras : « Ras-GTPase-Activating protein » (RasGAP)47. La déphosphorylation du site de liaison de RasGAP empêche son recrutement à la membrane plasmique et de ce fait, augmente l’activité de Ras. SHP2 agit également sur les PTKs de la famille Src en déphosphorylant le site de liaison de Csk (protéine inhibitrice de Src) sur l’adaptateur PAG/Cbp48. Cela se traduit par une augmentation de l’activité de Src et de la voie Ras-Erk qui en découle. L’activation de la voie Ras-Erk est le principal effet transformant des mutants de SHP247. SHP2 peut également activer d’autres voies de signalisation impliquant Akt et STAT5. En 2001, Tartaglia, M et al.49 ont identifié une mutation gain de fonction dans le gène PTPN11 qui code pour SHP2 comme étant la cause de 50% des syndromes de Noonan. Le syndrome de Noonan est une maladie complexe impliquant un nanisme, des anomalies faciales et cardiaques parfois associée à un risque accrue de développement de désordres hématologiques comme la leucémie juvénile
17 myélomonocytique (LJMM). Ces mutations de PTPN11 sont aussi retrouvées dans 1/3 des cas sporadiques de LJMM, 6% des patients atteints de leucémie lymphoblastique aigue et à 5% des patients développant une leucémie aigue myéloïde50. Enfin, ces mutations ont également été trouvées avec des fréquences moins élevées dans des tumeurs solides de cancer du poumon ou du colon, des neuroblastomes et des mélanomes51. La plupart des mutations de
PTPN11 sont des mutations faux sens se traduisant par le changement unique d’un acide
aminé. Le mécanisme d’activation de SHP2 intervient par la liaison de ses domaines SH2 N- et C-terminaux à d’autres protéines et par la levée de l’inhibition du domaine catalytique par le dépliement du domaine SH2 N-terminal. En effet, de nombreuses mutations de SHP2 interviennent dans la région qui lie le domaine SH2 N-terminal au domaine catalytique empêchant ainsi l’inhibition de ce dernier et augmentant de ce fait l’activité basale de SHP2. Les mutations de SHP2 peuvent aussi modifier la sélectivité de SHP2 par rapport à ses substrats.
I.6.2.3. Autres PTPs oncogéniques :
Des fonctions oncogéniques ont été démontrées pour d’autres PTPs. Leur action passe majoritairement par une activation des PTKs de la famille Src. C’est par exemple cas des PTPα, PTPε, PTP1B et SHP1 (tableau 2).
18
Tableau 2 : PTPs ayant un rôle oncogénique (Source : Tonks, NK, Mol Cell Biol, 20058).
I.6.3. Les PTPs comme potentielles drogues anti-tumorales :
Contrairement aux PTKs, pour lesquelles de nombreux inhibiteurs sont utilisés en clinique, les PTPs sont des nouvelles venues dans le domaine du développement des drogues anti-tumorales. Leur émergence comme protéines suppresseurs de tumeur ou oncogéniques a conduit à les considérer comme des cibles thérapeutiques potentielles52, 53. Il existe des challenges techniques à relever pour mettre au point des activateurs ou des inhibiteurs de
19 PTPs. En effet, il faut identifier des composés suffisamment discriminants dans leur action sur des PTPs aux domaines catalytiques proches et aux fonctions différentes. Du fait de leur site actif très conservé, il est difficile de mettre au point des drogues spécifiques. Néanmoins, ces problèmes ont également été soulevés lors du développement des inhibiteurs des PTKs visant la poche à ATP et, depuis lors, de nombreux inhibiteurs spécifiques ont vu le jour. Les études de structure permettent de mettre en évidence à proximité des sites actifs, la présence d’autres sites propres à chaque PTP pouvant constituer des cibles intéressantes pour la mise au point de composés spécifiques ciblant à la fois le site actif et ce site particulier54. De plus, une PTP peut réguler plusieurs voies de signalisation, de même qu’une voie de signalisation peut être régulée par plusieurs PTPs. Cela pose donc le problème d’éventuels effets secondaires provoqués par l’inhibition ou l’activation d’une PTP. Une approche intéressante pour le design de drogues est celle qui utilise pour seule caractéristique la structure topologique de la poche catalytique des différentes PTPs. En effet, on observe une grande diversité au niveau de ces structures qui pourrait être bénéfique au développement de drogues très spécifiques et efficaces. Du fait de son rôle potentiellement oncogénique, SHP2 pourrait représenter la cible la plus évidente pour un traitement anti-tumoral. Ainsi, de récents travaux ont montré l’efficacité et la spécificité d’un nouvel inhibiteur de SHP2, le phenylhydrazonopyrazolone sulfonate (PHPS1)55. D’autres approches visent à cibler les domaines extracellulaires des PTPs récepteurs afin de moduler leur activité. Les avancées dans la compréhension des mécanismes moléculaires par lesquelles les PTPs interviennent dans la physiopathologie des maladies ainsi que le rôle majeur des PTPs comme régulateurs des phénomènes de phosphorylation/déphosphorylation, devraient permettre aux inhibiteurs/activateurs de PTPs d’être des thérapies cliniquement relevantes.
Les activateurs de PTPs peuvent également représenter des traitements intéressants dans le cas de ciblage de PTP ayant une fonction suppresseur de tumeur. Si peu d’activateurs sont à ce jour connus, il a été démontré que les analogues de la somatostatine permettent d’activer certaines PTPs comme SHP1 et DEP1. Ces analogues sont déjà utilisés en clinique (Octréotide, Lanréotide) dans le traitement des tumeurs neuroendocrines et seront développées plus en détail dans les chapitres suivants.
20
II. Les PTPs à domaines SH2 : SHP1 et SHP2 (SHPs) :
II.1. Structure et expression cellulaire :
Les SHPs pour Src Homology-2 (SH2) domain-containing phosphatases appartiennent au groupe des PTPs classiques non récepteurs de classe I. On trouve deux SHPs chez les vertébrés : SHP1 et SHP2 qui ont, chez Drosophila et Caenorhabditis elegans, des orthologues nommés respectivement Corkscrew (Csw) et Ptp-256. Elles sont codées chez l’homme par les gènes PTPN6 pour SHP1 et PTPN11 pour SHP257. Le gène de SHP1 est localisé sur le chromosome 12p1358. Il contient 17 exons et présente une taille de 17kb ; la taille du transcrit étant comprise entre 2.4 et 2.6 kb. Il code pour deux formes de SHP1 résultant de deux codons d’initiation de la traduction localisés sur les exons 1 et 2 alors que le codon stop est présent sur l’exon 16. Les exons 3 et 4 codent pour le domaine SH2 N-terminal, les 5 et 6 pour le domaine SH2 C-terminal et les exons 8,9 et 10 pour le domaine catalytique. Deux promoteurs tissu spécifiques P1 et P2 sont actifs respectivement dans les cellules non hématopoïétiques et hématopoïétiques. Le promoteur P1 est situé approximativement 7 kb en amont de P2. Ils contiennent tous deux des séquences reconnues par les facteurs de transcription SP1 et/ou AP259. Les différences majeures entre les deux formes de SHP1 apparaissent en N-terminal avec la séquence en acide aminé MLSRG pour la forme 1 contre MVR pour la forme 2. Ainsi le gène PTPN6 code quatre isoformes de SHP1 constitués de trois variants présentant des modifications dans la partie N-terminale et une forme plus longue de SHP1, nommée SHP1L, dans laquelle la partie N-terminale est conservée et la partie C-terminale étendue60, 61. SHP2 compte quant à elle, seulement un isoforme (figure 3).
21
Figure 3 : Structures des quatre isoformes de SHP1 et de SHP2 (Source : Poole, AW, Cell Signalling, 200561).
De part la présence dans leur structure de deux domaines SH2 capables de lier des tyrosines phosphorylées, les SHPs participent à de nombreuses voies de signalisation et présentent de ce fait un grand intérêt. Elles sont structurellement très proches et sont constituées, en plus des domaines SH2 dans la partie N-terminale, d’un domaine catalytique PTP et d’une queue C-terminale comprenant les sites de phosphorylation sur tyrosine et sur serine (Figure 4)61. Alors que SHP2 présente un domaine riche en proline capable de lier les domaines SH3 d’autres protéines, SHP1 en est dépourvue mais présente une séquence basique fonctionnant comme une séquence NLS (Nuclear Localisation Sequence)62. SHP2 est exprimée de façon ubiquitaire alors que l’expression de SHP1 est restreinte aux cellules hématopoïétiques et épithéliales.
II.2. Localisation subcellulaire :
Dans les cellules hématopoïétiques, SHP1 est exprimée exclusivement dans le cytoplasme où elle exerce un rôle de régulateur négatif. On peut également la retrouver au niveau nucléaire dans les cellules épithéliales où elle peut réguler positivement la signalisation cellulaire63. Il a été montré qu’elle posséde une NLS bipartite avec une séquence
22 KVKK dans la partie N-terminale et un motif KRK en C-terminal. Ce motif KRK, qui est indispensable à la localisation nucléaire de SHP1, n’existe pas chez SHP2 qui est donc exclusivement cytoplasmique. L’hypothèse selon laquelle la séquence KRK en C-terminal serait mise sous silence dans les cellules hématopoietiques par la phosphorylation de résidus à proximité, pourrait expliquer que SHP1 soit maintenue dans le cytoplasme de ces cellules62, 64. Dans le lymphocyte T, il a été montré que SHP1 est localisée au niveau des micro-domaines membranaires (rafts) où elle est recrutée suite à l’engagement du TCR (T cell receptor) et où elle participe à la déphosphorylation de protéines de signalisation telles que la protéine adaptatrice LAT (Linker for Activation of T-cells)65. Il semble que la localisation de SHP1 aux rafts (20 à 30% du pool de SHP1) dépende de son association avec des protéines membranaires telles que, par exemple, CD3 ou CD5. En effet, aucune modification post-traductionnelle (acylation, palmitoylation) pouvant être responsable d’une localisation aux « rafts », n’a été décrite pour SHP1. Néanmoins, Fawcett, VC et al.66 ont identifié la partie C-terminale de SHP1 comme étant responsable de sa localisation au niveau des « rafts ». De plus, dans cette partie C-terminale, un motif de 6 acides aminés (SKHKED) a été décrit comme étant nécessaire et suffisant pour localiser SHP1 aux « rafts »67. Ce motif, n’est pas retrouvé dans SHP2, ni dans aucune autre protéine. Cette séquence semble également être nécessaire à la liaison de SHP1 aux phospholipides à acides gras saturés. Les travaux de Su, MW et al.68 ont décrit deux sous populations de SHP1, une cytosolique phosphorylée sur tyrosine et une autre non phosphorylée localisée au niveau des rafts. Chacune de ces populations participe à la régulation négative des voies de signalisation induites par l’engagement du TCR et inhibe la production d’IL-2 (Interleukine 2).
II.3. Régulation de l’activité des SHPs:
II.3.1. repliement intramoléculaire :
Des études ont montré que l’activité catalytique de SHP1 était régulée par le réarrangement structurel des deux domaines SH269. Ainsi, la suppression du domaine SH2 N-terminal provoque une forte augmentation de l’activité catalytique suggérant que cette activité est réprimée par une interaction intramoléculaire impliquant ce domaine70. Ces données ont été confirmées par l’activation de SHP1 à l’aide d’un peptide bis-phosphotyrosyl. Ce peptide est capable de se lier simultanément aux deux domaines SH2 de SHP1 et d’augmenter un niveau d’activité initialement bas, probablement en levant l’inhibition du domaine SH2
N-23 terminal69. Des études de cristallographie sur la protéine SHP2, structurellement similaire à SHP1, ont montré qu’à l’état inactif, le domaine SH2 N-terminal était lié par interaction de charges au domaine catalytique, empêchant ainsi toute liaison au substrat. Lors de ce blocage du site actif, le site de liaison du domaine SH2 est exposé vers l’extérieur. La liaison du domaine SH2 N-terminal à une autre protéine provoque un switch allostérique au sein du domaine catalytique qui passe d’un état inactif à un état actif. Ce changement de conformation va rompre l’interaction entre le domaine SH2 et le domaine catalytique, laissant ce dernier libre d’accès aux substrats de la PTP71. Une SHP2 mutée dans son domaine SH2 N-terminal montre une activité basale accrue tout en gardant la faculté de se lier aux substrats72. L’analyse cristallographique de SHP1 dans différentes conformations a permis de préciser les mécanismes d’inhibition et d’activation observés pour SHP273. Comme pour SHP2, l’auto-inhibition de SHP1 a été mise en évidence avec le repliement du domaine SH2 N-terminal sur le domaine catalytique. Le domaine SH2 C-terminal très mobile a été défini comme étant une « antenne » cherchant le phosphopeptide activateur. Ainsi la fixation du domaine SH2 C-terminal à un phosphopeptide se traduit par un profond changement conformationnel de SHP1, notamment au niveau du domaine SH2 N-terminal voisin qui va ouvrir sa poche de liaison à un phosphopeptide pour lier une autre protéine et lever l’inhibition du domaine catalytique. Ces évènements diminuent l’auto-inhibition de la protéine et libèrent le site actif du domaine catalytique73. Le site actif du domaine catalytique de SHP1 contient trois acides aminés importants : la cystéine 455 se comportant comme un nucléophile pour attaquer le substrat, l’arginine 459 stabilisant la charge négative du peptide phosphorylé sur tyrosine et l’acide asparatique 421 servant de donneur ou d’accepteur de proton pour le relargage du substrat. L’activité du domaine catalytique est déterminée par la flexibilité de la boucle WPD qui contient le résidu actif Asp421. Les données biochimiques et cristallographiques ont mis en évidence une spécificité de substrat pour SHP1. Elle serait due à une séquence consensus (D/E)X(L/I/V)X1-2pYXX(L/I/V) que reconnait préférentiellement le domaine catalytique de
SHP174, 75.
II.3.2. phénomènes de phosphorylation en C-terminal :
D’autres mécanismes de régulation de l’activité des SHPs ont été proposés. C’est par exemple le cas des phénomènes de phosphorylation intervenants au niveau de la partie C-terminale de la protéine. En effet, des expériences de délétions de cette partie C-C-terminale ont permis de révéler son importance dans la régulation de l’activité des SHPs. Ainsi, la perte des
24 35 derniers acides aminés de SHP1 se traduit par une forte augmentation de l’activité PTP in
vitro70
. Une protéolyse de la partie C-terminale au niveau d’un site de clivage par la thrombine se traduit, elle aussi, par une augmentation de l’activité76. De façon intéressante, les parties C-terminales des SHPs comprotent des résidus tyrosines et sérines pouvant être phosphorylés (figure 4). Ainsi, SHP1 et SHP2 sont toutes deux soumises à des phénomènes de phosphorylation sur des résidus tyrosines dans leur partie C-terminale. Néanmoins, la signification de ces modifications reste controversée. Deux fonctions majeures ont été proposées pour ces tyrosines phosphorylées : elles peuvent servir de sites de recrutement de protéines adaptatrices comme Grb2 ou avoir un rôle dans la régulation de l’activité PTP. En 1994, Lorenz, U et al.77 ont montré que SHP1 est phosphorylée sur les résidus tyrosines 536 et 564 et que la phosphorylation de la Y564 dépend de la tyrosine kinase Lck. Dans le même temps, Bouchard, P78 et al. ont montré in vitro que la Y536 est soumise à un processus d’autodéphosphorylation puisque la phosphorylation peut être maintenue grâce à un traitement à l’orthovanadate (inhibiteur à large spectre des PTPs) ou par une mutation du résidu cystéine dans le site catalytique. Ils ont également défini cette tyrosine 536 comme étant le site de recrutement de la protéine Grb2. Cependant, dans les deux études, les auteurs n’ont pas relevé de modification de l’activité phosphatase de SHP1 lors de modifications de l’état de phosphorylation de cette Y536. D’autres études ont montré que la phosphorylation des résidus tyrosines de la queue C-terminale permettait à SHP1 de revêtir une fonction adaptatrice et de recruter des substrats de la PTP79. Cependant, des travaux ont montré que la phosphorylation sur tyrosine de SHP1 se traduisait par une augmentation de l’activité PTP. C’est par exemple le cas lorsque le récepteur à l’insuline ou encore les tyrosines kinases de la famille Src, phosphorylent SHP180. Par ailleurs, la mutation des Y536 et 564 en phénylalanine conduit à une diminution de l’activité PTP81. Pour SHP2, les tyrosines 542 et 580 ont également été définies comme sites de recrutement de protéines adaptatrices et notamment de Grb2 qui permet ainsi un couplage avec la voie des MAP kinases82-84. Cependant une controverse existe également quant à leur importance dans la fonction de la PTP puisque des mutations de ces tyrosines en phénylalanines n’ont pas eu d’effet majeur sur la signalisation cellulaire85. Il a été montré par ailleurs que la phosphorylation sur tyrosine participait à l’activation de SHP286, 87. Pour palier à la nature très labile de la phosphorylation sur tyrosine qui rend complexe l’étude de son rôle sur l’activité, une stratégie visant à mimer les phosphotyrosines par des analogues non hydrolysables (phosphonate) a été employée. Avec cette approche expérimentale, une légère stimulation de l’activité PTP de SHP2 a été relevée. En effet, les tyrosines 542 et 580 lient respectivement les domaines SH2 N- et C-terminaux,
25 levant ainsi l’inhibition du domaine N-terminal sur le domaine catalytique88. Pour SHP1, la substitution de la tyrosine 536 par un résidu phosphonate provoque une nette augmentation de l’activité alors que la même modification au niveau de la tyrosine 564 a un effet moindre89.
Figure 4 : Représentation schématique des parties C-terminales de SHP1 et SHP2 avec les sites de phosphorylation sur tyrosine, serine et thréonine (Source : Poole, AW, Cell Signalling, 200561).
En plus de la phosphorylation sur tyrosine des SHPs, d’autres travaux ont montré que ces PTPs pouvaient être phosphorylées sur sérine, toujours dans leur partie C-terminale. Ainsi, il a été montré que SHP2 pouvait être phosphorylée sur serine (positions 576 et 591) par la PKC (Protéine Kinase C) ou par les MAPK90-92. Les résultats concernant l’incidence de ces phosphorylations sur l’activité de SHP2 restent controversés puisque certaines études n’ont pas décelé de modification alors que d’autres ont mis en évidence une diminution de l’activité phosphatase. SHP1 est elle aussi phosphorylée sur serine77 en position 591 par la PKC93. La phosphorylation sur serine de SHP1 semble provoquer une diminution de l’activité de la PTP. Lors de l’activation cellulaire, la PKC phosphoryle sur serine SHP1 qui voit alors son activité décroitre transitoirement. Cela permet ainsi d’augmenter le niveau de phosphorylation des protéines participant à l’activation cellulaire94 (figure 5). Enfin, une étude récente a montré que l’activation du TCR permet une phosphorylation séquentielle de SHP1 sur sérine 591 (1min) puis sur tyrosine 536 (30min). La phosphorylation sur S591 inhiberait la phosphatase et modifierait sa localisation subcellulaire alors que la phosphorylation plus tardive sur Y536, l’activerait95.
26
Figure 5 : Représentation schématique de la régulation des SHPs par repliement intramoléculaire et phosphorylation en C-terminal par la PKC (Source : Poole, AW, Cell Signalling, 200561) :
II.3.3. Activation par les analogues de la somatostatine :
L’activation des PTPs via la stimulation des récepteurs à la somatostatine (SSTR) représente un des principaux mécanismes intracellulaires impliqués dans les effets antiprolifératifs de la somatostatine et de ses analogues. Principalement, trois PTPs ont été identifiées comme effecteurs des SSTRs : SHP196, 97, SHP298, 99 et DEP-1100-102. SHP1 reste à ce jour la PTP la plus couramment impliquée dans les effets antiprolifératifs de la somatostatine. L’activation de SHP1 et l’arrêt de la prolifération qui en résulte, a été décrite dans de nombreuses lignées de cancer du sein, du pancréas, de la thyroïde ou encore d’adénomes pituitaires. La co-transfection de SHP1 et SSTR2 dans des cellules CHO montre une association des deux protéines dans un complexe régulé par la sous unité Gi3α103
. L’activation de SSTR2 par un analogue de la somatostatine : l’octréotide, provoque l’activation de SHP1 et sa dissociation du complexe. Une fois SHP1 activée, elle va aller s’associer à ses substrats et les déphosphoryler. SHP1 va par exemple déphosphoryler le récepteur à l’insuline (IR) ainsi que IRS1 et Shc afin d’inhiber la prolifération induite par
27 l’IR104. L’effet antiprolifératif est reflété par un blocage des cellules en phase G1 et une surexpression de l’inhibiteur p27kip1105. Il a aussi été montré que la sous unité p85 de la PI3-K est associée à la fois à SHP1106 et à SSTR2. Dans une lignée de cancer du pancréas, l’activation de SHP1 via SSTR2 provoque la déphosphorylation de la p85, sa dissociation du récepteur et l’inhibition de la PI3-K107. Un phénomène identique a été identifié dans les cellules pituitaires108. Dans cette étude, l’inhibition de la PI3-K via SHP1 se traduit par une inhibition des activités de PDK1 et Akt qui provoquent l’activation de la glycogen synthase kinase 3β (GSK3β). Cette augmentation d’activité induit la surexpression du suppresseur de tumeur Zac1, provoquant ainsi un arrêt du cycle cellulaire. Dans les cellules de cancer du sein, la stimulation de SSTR induit l’apoptose par le biais de l’activation de SHP1 et de la caspase 8 mais aussi d’une acidification intracellulaire109, 110. SHP1 est également impliquée dans l’apoptose dans les cellules de cancer du pancréas111. Récemment, un nouveau mécanisme d’induction de l’apoptose par le biais d’une activation de SHP1 conséquente à une stimulation de SSTR ; a été découvert dans les cellules NIH3T3. SHP1 serait responsable de l’activation du facteur de transcription NKκB qui inhiberait les effets anti-apoptotiques de la MAPK JNK, induisant une activation de la caspase 8 et de l’apoptose112. De plus, il a été montré dans des cellules de cancer du pancréas, que l’activité kinase de Jak2 est nécessaire à l’effet antiprolifératif des analogues de la somatostatine (SST). En effet, SHP1 et Jak2 sont liés à SSTR2 au repos et lors de la stimulation, Jak2 est activée et participe à l’activation de SHP1 en la phosphorylant113. Enfin, il semble que l’effet cytostatique des analogues de SST, passe par une activation séquentielle de kinases et phosphatases. Ainsi, l’activation de SHP2 par Src est un élément indispensable à l’association de SHP1 au récepteur SSTR2 et à son activation (figure 6)99.
28
Figure 6 : Représentation schématique du mécanisme d’activation de SHP1 par le récepteur à la somatostatine SSTR2 (Source : Ferjoux, G, Mol Biol Cell, 200399) : A l’état basal (1), Src et SHP2 sont associées au récepteur SSTR2. La liaison de la somatostatine au récepteur permet l’activation rapide de Src et SHP2 (2a) puis le recrutement et l’activation de SHP1 (2b). Les protéines se dissocient alors du récepteur (3) et SHP1 activée peut déphosphoryler ses substrats et médier les effets inhibiteurs du récepteur SSTR2.
II.4. Rôle biologique de SHP1 :
L’essentiel du travail de thèse ayant porté sur SHP1, nous nous limiterons ici à décrire ses fonctions en physiologie et en pathologie. Les grandes fonctions de SHP2 en situation normale et pathologique ne seront pas développées dans ce chapitre mais sont résumées dans de nombreuses revues bibliographiques47, 114, 115.
Les connaissances concernant le rôle biologique de SHP1 ont été considérablement accrues grâce au modèle murin motheaten portant des mutations perte de fonction dans le