Impact de la lipoprotéine(a) sur les
maladies cardiovasculaires en fonction d
u
sexe
Mémoire
Jakie Guertin
Maîtrise en sciences cliniques et biomédicales - avec mémoire
Maître ès sciences (M. Sc.)
Impact de la lipoprotéine(a) sur les maladies
cardiovasculaires en fonction du sexe
Jakie Guertin
Mémoire
Sous la direction de :
Dr Benoit Arsenault
Résumé
Les maladies cardiovasculaires (MCV) constituent la principale cause de décès au monde. Chez la femme, les MCV représentent un fardeau puisque les manifestations et la pathophysiologie des MCV sont en partie différentes de celles des hommes. La lipoprotéine(a) (Lp[a]), une lipoprotéine semblable à une lipoprotéine de faible densité (LDL), est l’un des facteurs de risques génétiques associés aux MCV. Cependant, les études portant sur l’impact de la Lp(a) sur les MCV en fonction du sexe sont rares.
Nos hypothèses étaient que les niveaux sanguins élevés de Lp(a) génétiquement déterminés et mesurés sont associés aux à la sténose aortique (SA), l’accident vasculaire cérébral (AVC) ischémique et aux maladies coronariennes (MC), autant chez les hommes que chez les femmes, et que l’association entre les niveaux sanguins de Lp(a) élevés génétiquement déterminés et chacune des trois maladies était indépendante des niveaux sanguins de LDL spécifique au sexe.
Nos résultats révèlent qu’il y a une association entre les niveaux de Lp(a) mesurés et le risque de SA et de MC chez l’homme et la femme, mais pas pour l’AVC ischémique. Les résultats d’associations entre les concentrations de Lp(a) génétiquement déterminées et le risque de ces trois maladies vont dans la même direction que les résultats d’analyses observationnelles. Finalement, nous avons déterminé qu’il existe une relation de cause à effet probable entre les niveaux élevés de Lp(a) et le risque de SA chez la femme et de SA et de MC chez l’homme indépendamment des niveaux de LDL.
L’ensemble de ces résultats suggère qu’il existe une relation causale probable entre les concentrations élevées de Lp(a) et le risque de SA et de MC pour les hommes et les femmes. Notre étude démontre que l’inhibition de la Lp(a) pourrait avoir des effets bénéfiques sur le risque de plusieurs MCV autant chez l’homme que chez la femme.
Abstract
Cardiovascular diseases (CVD) are the leading cause of death globally. CVD represents an important burden in women because clinical manifestations, mechanisms and risk factors for CVD may be different from those of men. Lipoprotein(a) (Lp[a]), similar to low density lipoprotein (LDL), is one of the genetic risk factors associated with CVD. Unfortunately, sex-specific studies of the impact of Lp(a) impact on CVD sex-specific are rare. Our hypotheses were that genetically elevated Lp(a) levels and plasma Lp(a) levels are associated with aortic stenosis (AS), ischemic stroke (IS) and coronary heart disease (CAD) in men and women, and the association between genetically-elevated Lp(a) plasma levels and each disease was independent of LDL plasma levels in men and women.
Our results suggest that the association between Lp(a) plasma levels and the risk of AS and CAD in men and women separately, but not for IS. The results of the association between genetically-elevated Lp(a) plasma levels and the risk of these three diseases point in the same direction as the results of observational analyzes. Finally, we determined that there is a causal effect relationship between elevated concentration of Lp(a) and the risk of AS in women and AS and CAD in men regardless of LDL plasma levels.
Together these results suggest that there is a causal relationship between high Lp(a) plasma levels and the risk of AS and CAD for men and women. Our study shows that Lp(a)-lowering therapy could be useful in reducing CVD risk in both men and women with high Lp(a) levels.
Table des matières
Résumé ...ii
Abstract ... iii
Table des matières ...iv
Liste des tableaux ...vi
Liste des figures ... vii
Liste des abréviations ... viii
Remerciements ... x Avant-propos ...xi Introduction ... 1 1. Maladies cardiovasculaires ... 1 1.1 Généralités ... 1 1.2. Prévalence et incidence ... 2 1.3. Étiologie et pathophysiologie ... 3 1.3. Facteurs de risque ... 3 1.4. Lipoprotéines et athérosclérose ... 7 2. Lipoprotéine(a) ... 9 2.1. Généralité ... 9 2.2. Mécanismes pathophysiologiques... 12
2.3. Thérapies ciblant la lipoprotéine(a) ... 13
3. Les maladies cardiovasculaires chez la femme... 17
3.1. Le fardeau des maladies cardiovasculaires chez la femme ... 17
3.2. Comorbidités et facteurs de risque spécifiques au sexe ... 18
4. La sténose aortique ... 24
4.1. Généralités ... 24
4.2. Physiopathologie sexe-spécifique de la sténose aortique ... 26
4.3. Implication de la lipoprotéine(a) dans la sténose aortique ... 26
5. Les accidents vasculaires cérébraux ischémiques ... 29
5.1. Généralités ... 29
5.2. Physiopathologie sexe-spécifique de l’accident vasculaire cérébral ischémique... 31
5.3. La lipoprotéine(a) et l’accident vasculaire cérébral ischémique ... 31
6. Les maladies coronariennes ... 33
6.1 Généralités ... 33
6.2. Physiopathologie des maladies coronariennes sexe-spécifiques ... 34
6.3. Implication de la Lp(a) dans les maladies coronariennes ... 35
7.1. Principe et utilisation de la randomisation mendélienne ... 37
7.2. Score de risque génétique ... 39
8. Problématiques, hypothèses et objectifs ... 40
Chapitre 1: Association sexe spécifique de l’exposition à long terme à des taux élevés de lipoprotéine(a) et les maladies cardiovasculaires athérosclérotiques : Analyses observationnelles et de randomisation mendélienne ... 43 RÉSUMÉ ... 44 ABSTRACT ... 45 INTRODUCTION ... 47 METHODS ... 48 Study populations ... 48
Exposures ascertainment and definitions ... 48
Outcomes ascertainment and definitions ... 49
Statistical analyses ... 49
RESULTS... 50
Genetically-elevated Lipoprotein(a) and ACVD in men and women of the UK Biobank ... 50
Genetically-elevated Lipoprotein(a) and low-density lipoprotein cholesterol and ACVD in men and women of the UK Biobank ... 50
Lipoprotein(a) levels and ACVD in men and women of the EPIC-Norfolk study ... 51
DISCUSSION ... 51 ACKNOWLEDGEMENTS ... 52 REFERENCES ... 53 TABLE ... 56 FIGURE LEGENDS ... 59 FIGURE... 60 Conclusion ... 64 Bibliographie ... 67
Liste des tableaux
Tableau 1. Impacts des facteurs de risques traditionnels modifiables sur la santé cardiovasculaire des femmes
... 19
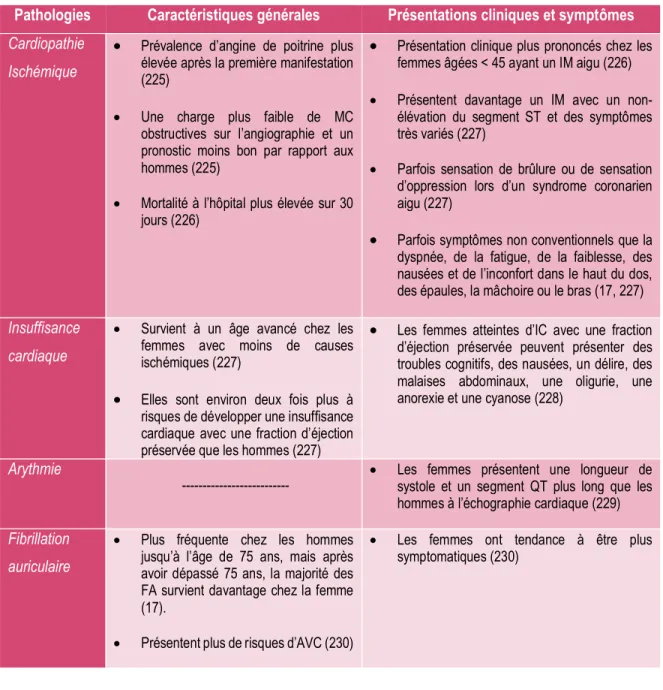
Tableau 2. Exemple de caractéristiques générales, de présentations cliniques et de symptômes pour la cardiomyopathie ischémique, l’insuffisance cardiaque, l’arythmie et la fibrillation auriculaire observés chez la femme... 24
Chapitre 1
Table 1. Sex-specific cardiovascular disease risk associated with elevated lipoprotein(a) levels. ... 56Table 2. Sex-specific risk of coronary artery disease associated with very high lipoprotein(a) levels. ... 57
Table 3. Sex-specific risk of ischemic stroke associated with very high lipoprotein(a) levels. ... 57
Liste des figures
Introduction
Figure 1. Structure de la paroi vasculaire des vaisseaux. ... 2 Figure 2. Résumé de la pathophysiologie de l'athérosclérose.. ... 9 Figure 3. Illustration de la lipoprotéine(a).. ... 10 Figure 4. Représentation d'une valve aortique normale et d'une valve aortique calcifiée après plusieurs étapes du développement de la sténose aortique. ... 25 Figure 5. Résumé des mécanismes pathophysiologiques de la Lp(a) et des LDLs oxydés dans la sténose aortique... 29 Figure 6. Représentation d'un AVC ischémique et d'un AVC hémorragique.. ... 30 Figure 7. Représentation d'une artère coronaire normale, avec athéroslcérose seulement et avec athéroslécrose et caillot sanguin. ... 33 Figure 8. Association entre le score de risque du niveau de Lp(a) et le risque de maladies coronariennes dans la cohorte PROCARDIS. ... 36 Figure 9. Représentation schématique de l'analogie entre les essais cliniques randomisés et la randomisation mendélienne. ... 38
Chapitre 1
Figure 1A. Sex-specific association between genetically-elevated lipoprotein(a) levels and the risk of coronary artery disease. ... 60 Figure 1B. Sex-specific association between genetically-elevated lipoprotein(a) levels and the risk of ischemic stroke. ... 60 Figure 1C. Sex-specific association between genetically-elevated lipoprotein(a) levels and the risk of calcific aortic valve stenosis. ... 61 Figure 2A. Sex-specific association between genetically-elevated lipoprotein(a) levels and the risk of coronary artery disease with or without genetically-elevated low-density lipoprotein cholesterol levels... 62 Figure 2B. Sex-specific association between genetically-elevated lipoprotein(a) levels and the risk of ischemic stroke with or without genetically-elevated low-density lipoprotein cholesterol levels. ... 62 Figure 2C. Sex-specific association between genetically-elevated lipoprotein(a) levels and the risk of calcific aortic valve stenosis with or without genetically-elevated low-density lipoprotein cholesterol levels. ... 63
Liste des abréviations
Abréviations
Mot ou expression
Anglais
Français
apo Apolipoprotein Apolipoprotéine
ARN Ribonucleic acid Acide ribonucléique
ARNm Messenger ribonucleic acid Acide ribonucléique messager
ATX Autotaxin Autotaxine
AVC Stroke Accident vasculaire cérébral
BMP2 Bone morphogenetic protein 2 Protéine osseuse morphogénique 2
CE Endothelial cell Cellule endothéliale
CETP Cholesteryl ester transfer protein Protéine de transfert des esters de
cholestérol
CRP C-protein reactive Protéine C-réactive
CT Computed tomography Tomographie axiale
eQTL Expression quantitative trait loci Étude de la quantification de l’expression de variant
FA Auricular fibirilation Fibrillation auriculaire
GRS Genetic risk score Score de risque génétique
GWAS Genome-wide association study Étude d’association pangénomique
HF Familial hypercholesterolemia Hypercholestérolémie familiale
IC Heart Failure Insuffisance cardiaque
ICAM Intracellular adhesion molecule Molécule d’adhésion intracellulaire
IL Interleukine Interleukine
IM Myocardial infarction Infarctus du myocarde
IMC Body mass index Indice de masse corporelle
LDL Low density lipoprotein Lipoprotéine de faible densité
LDLR LDL receptor Récepteur LDL
LOX-1 Lectine-like oxidized receptor 1 Récepteur oxydé de type lectine 1
Lp-PLA2 Lipoprotein-associated phospholipidase A2 Phospholipase A2 associée aux lipoprotéines
Lp(a) Lipoprotein(a) Lipoprotéine(a)
LPC Lysophosphatidylcholine Lysophosphatidylcholine
LysoPA Lysophosphatidic acid Acide lysophosphatidique
MC Coronary heart disease Maladie coronarienne
MCV Cardiovascular disease Maladie cardiovasculaire
MVP Peripheral vascular disease Maladie vasculaire périphérique
NF-κB Nuclear factor-kappa B Facteur nucléaire kappa B
NO Nitric oxide Oxyde nitrique
OMS World Heath Organization Organisation mondiale de la santé
oxLDL Oxidized LDL LDL oxydé
OxPL Oxidized phospholipids Phospholipides oxydés
PAM-1 Plasminogen activator inhibitor 1 Inhibiteur d’activateur du plasminogène 1
PCSK9 Proprotein convertase subtilisin/kexin type
9 Proprotéine convertase subtilisine/kexine de type 9
PET Positron emission tomography/computed
tomography Tomographie par émission de positons
RUNX2 Runt-related transcription factor 2 Facteur de transcription RUNX2
SA Aortic stenosis Sténose aortique
SMC Smooth muscle cells Cellules des muscles lisses
SNP Single nucleotide polymorphism Polymorphisme nucléotidique unique
SOPK Polycystic ovary syndrome Syndrome des ovaires polykystiques
THR Hormone replacement therapy Traitement hormonal de remplacement
TWAS Transcriptome-wide association study Étude d’association pantranscriptomique
VCAM Vascular cell adhesion molecule Molécule d’adhésion aux cellules
vasculaires
Remerciements
Avant de faire mes remerciements, je tiens à mentionner que ce manuscrit est, pour moi, un symbole de détermination et de dépassement. Avant le début de mes études de deuxième cycle, je n’avais jamais pensé que j’aurais entrepris des études graduées. Mon baccalauréat était l’objectif que je m’étais fixé pour conclure mon parcours académique. Ce mémoire se situe au-delà de mes ambitions et de toutes mes attentes que j’avais envisagées. Non seulement je me suis surpassé au niveau académique, mais je me suis surpassé également en tant qu’athlète. Les mots me manquent à quel point je suis fière et reconnaissante d’avoir fait partie de l’équipe de cheerleading du Rouge et Or. Moi qui croyais que ma carrière de gymnaste était terminée depuis longtemps, faire partie de cette équipe a été une occasion en or de me surpasser encore une fois et m’a permis de m’épanouir tout au long de ma maîtrise.
Bien sûr, mes études graduées et mes travaux de recherches n’auraient pas pu avoir lieu sans l’aide et le soutien de nombreuses personnes. Benoit, merci de m’avoir accueilli dans ton équipe de recherche et de m’avoir donné cette belle opportunité. Dès le début, tu avais déjà confiance en mes capacités et en ce que je pouvais apporter au sein de l’équipe. J’ai eu la chance d’avoir un directeur de recherche extrêmement présent, surtout lorsque mon projet de maîtrise a complètement changé de direction.
Merci à toute l’équipe, Raphaëlle, Marjorie, Audrey-Anne, Nicolas, Hasanga et William, qui a été, pour moi, une deuxième famille. Je garderais de précieux souvenirs des activités d’équipe, sans oublier les 5 à 7. Merci, Nicolas, de m’avoir accompagné dans les petits joggings du midi et de m’avoir donné du beurre de peanuts quand je n’en avais plus. Merci à Trish et Sylvain pour vos conseils et de votre soutien technique dans la première année malgré le fait que les expériences n’ont pas tourné comme nous l’avons souhaité. Merci, Christian, pour l’aide que tu m’as apportée dans la réalisation de ce projet.
J’aimerais remercier mes entraîneurs, Cédric, Jay et Geneviève, qui ont cru en moi et qui m’ont motivé à vouloir constamment me surpasser. Je n’ai jamais fait autant de vrilles dans une routine de cheerleading de toute ma vie! Merci également à toute l’équipe de cheerleading du Rouge et Or pour ces deux belles années.
Je remercie mes parents qui m’ont suivi et qui m’ont soutenu tout au long de mon parcours académique. Merci à mon amoureux, Maxime, de faire partie de ma vie et de m’avoir été présent durant mes études graduées, ainsi que mes deux petites lapines, Athéna et Zelda, qui m’ont tenu compagnie lors de la crise du COVID et de l’écriture de ce mémoire.
Avant-propos
L’intérêt pour la biologie médicale a débuté lors de mes études collégiales en technique d’analyse biomédicale. Je n’ai pas hésité une seconde d’entreprendre mon baccalauréat en biologie médicale à l’Université de Québec à Trois-Rivières afin d’approfondir mes connaissances, et ce, tout en exerçant ma profession de technologiste médicale. À la fin de mes études de premier cycle, j’ai eu l’opportunité d’effectuer un stage en recherche. Parmi toutes les offres de stages, seulement une seule a attiré mon attention. C’est à l’été 2018 que j’ai entrepris mon stage dans l’équipe du Dr Arsenault et que j’ai découvert le monde de la recherche. Ce stage a été révélateur et c’est à ce moment que mon intérêt pour la recherche a débuté. Durant ce stage, j’ai commencé à être impliquée dans le projet du protéome de la lipoprotéine(a) (Lp[a]) et de son implication dans le développement de la sténose aortique. J’ai entrepris le processus de développement d’une méthode immuno-enzymatique ELISA afin de doser une protéine d’intérêt, PCSK9, sur la Lp(a). À la fin de ce stage, les résultats s’avéraient prometteurs et j’ai décidé de vouloir poursuivre ce projet dans le cadre de ma maîtrise. Malheureusement, après une multitude d’essais et de travail acharné, les analyses se sont avérées incertaines. Nous avons dû envisager une autre option concernant mon projet de maîtrise. Toutefois, j’ai pu contribuer au projet de doctorat de ma collègue Raphaëlle en effectuant des prises de sang et en traitant les échantillons des participants de la cohorte ATLAS, en vue d’extraire la Lp(a). J’ai également participé aux dosages de plusieurs biomarqueurs dans diverse cohorte dont nous avons la chance de collaborer, soit SALTIRE II, CARDS et ATLAS.
En étudiant la littéraire, nous nous sommes aperçus que les données concernant les effets de la Lp(a) sur les maladies cardiovasculaires de manière sexe-spécifique étaient rares et incohérentes. J’étais très enthousiaste d’entreprendre ce nouveau projet qui est au cœur de ce mémoire. J’ai laissé les pipettes de côté et j’ai plongé dans l’univers de la bio-informatique. Pour réaliser mon projet, nous avons fait appel à la randomisation mendélienne et au score de risque génétique puisque ce sont des méthodes qui permettent d’analyser une multitude de données rapidement et qui permettent de contourner les facteurs confondants des niveaux de Lp(a), notamment le sexe. Nous avons également utilisé des données génétiques de libre accès telles que des variants génétiques associés aux niveaux élevés de Lp(a) bien connu et des données des concentrations de Lp(a) dans deux cohortes du Royaume-Uni. J’ai effectué les analyses nécessaires afin de répondre à notre question avec l’aide de notre professionnel de recherche. J’ai finalement participé à l’interprétation des résultats et à la rédaction de l’article présenté au chapitre 1 : « Sex-Specific Associations of Long-Term Exposure to Elevated Lipoprotein(a) Levels with Atherosclerotic Cardiovascular Diseases: Observational and Mendelian Randomization Analyses ». Cet article a été soumis dans le journal « Circulation: Genomic and Precision Medicine » le 12 novembre 2020. Les coauteurs sont : Yannick Kaiser, Nicolas Perrot, Raphaëlle Bourgeois, Christian Couture, Nicholas J. Wareham, Yohan Bossé, Philippe Pibarot, Erik S.G. Stroes, Patrick Mathieu, Marie-Annick Clavel, Sébastien Thériault, S. Matthijs Boekholdt, et Benoit J. Arsenault.
Introduction
1. Maladies cardiovasculaires
1.1 Généralités
Le cœur est un organe musculaire constituant le moteur central de l’appareil circulatoire. Les vaisseaux sanguins, c’est-à-dire les artères, les veines et les capillaires, reliés au cœur, sont aussi des structures essentielles à la circulation sanguine. L’ensemble de ces structures forme le système cardiovasculaire et permet le transport de l’oxygène et à tous les nutriments nécessaires aux maintiens de l’homéostasie vers les extrémités du corps et les organes. Un dysfonctionnement ou l’altération d’une ou plusieurs de ses structures peuvent avoir de graves conséquences sur la santé de l’individu et voir même sur sa survie. Le cœur comporte quatre cavités, soit le ventricule gauche, l’oreillette gauche, le ventricule droit et l’oreillette droite. Le myocarde, composé de fibres musculaires striées, forme le muscle cardiaque. Ce tissu musculaire est irrigué par les artères coronaires qui permettent d’alimenter le cœur en nutriments et en oxygène. Le cœur dispose également de gros vaisseaux sanguins dont chacun d’entre eux est relié à une cavité du cœur. Ses principaux vaisseaux sont l’aorte, l’artère pulmonaire, la veine cave inférieure et supérieure ainsi que les veines pulmonaires. Quatre valves cardiaques ont pour principalement rôle d’assurer la circulation sanguine dans un seul sens. Deux de ces valves se situent entre les oreillettes et les ventricules, dont la valve mitrale et la valve tricuspide. Les deux autres valves siègent entre les ventricules et l’artère correspondante, soit la valve aortique et la valve pulmonaire.
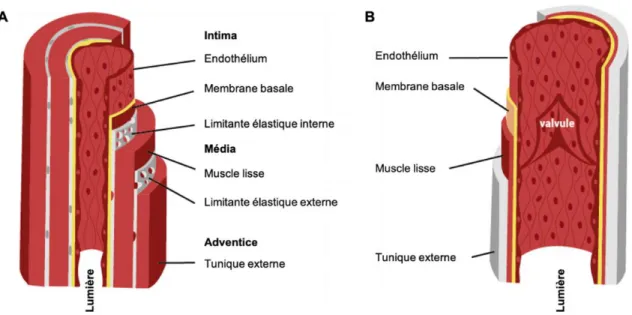
Le réseau vasculaire, pour sa part, est composé d’artères, d’artérioles, de capillaires, de veines et de veinules. Chacun de ses vaisseaux a un calibre différent et une composition histologique différente. Les artères sont composées de trois couches, de l’extérieur vers la lumière, en coupe transversale, l’adventice (tunique externe), la média et l’intima. L’adventice contient du tissu conjonctif, des fibres élastiques et des fibres nerveuses. Il est également composé d’un réseau de capillaires qui vascularise la paroi artérielle appelée le vasa vasorum et séparé de la média par la limitante élastique externe. La média, la couche se trouvant au milieu est composée de collagène, d’élastine et peut contenir des fibres musculaires lisses pour des artères de petit et moyen calibre. L’intima est formée par l’endothélium vasculaire qui repose sur une couche de tissu conjonctif. Les veines sont également composées de ses trois structures, cependant, la limitante élastique externe entre l’adventice et la média est absente. Les capillaires, pour leur part, sont composés d’une simple couche de cellules endothéliales et d’une lame basale afin d’assurer une perméabilité à l’eau, aux électrolytes, aux gaz et aux nutriments.
Figure 1. Structure de la paroi vasculaire des vaisseaux. A) Structure d’une artère. B) Structure et
composition d’une veine. Image adaptée de la Fédération Française de Cardiologie : (https://www.fedecardio.org/Je-m-informe/Le-coeur/les-arteres-et-les-veines)
Les maladies cardiovasculaires (MCV) constituent donc un ensemble de pathologies de du système circulatoire dont le cœur et les vaisseaux sanguins. Bien que ce système soit complexe, il existe un large éventail de problèmes pouvant survenir. Les MCV font références aux cardiopathies ischémiques ou maladies coronariennes (MC), aux accidents vasculaires cérébraux (AVC), aux maladies vasculaires périphériques (MVP) et à l’insuffisance cardiaque (IC) (1). Les valvulopathies, le rhumatisme cardiaque, les cardiopathies congénitales ainsi que plusieurs autres troubles cardiaques et vasculaires font également partie des MCV. Ces maladies ont des répercussions majeures sur la qualité de vie et imposent un lourd fardeau financier à la société.
1.2. Prévalence et incidence
Malgré les avancés en médecine dans les traitements et la gestion des maladies, les MCV sont de plus en plus présentes dans notre société moderne et représentent la première cause de mortalité dans le monde. En 2017, environ 17,8 millions d’individus sont décédés d’une MCV, ce qui représente 31% de tous les décès dans le monde (2, 3). Selon l’Organisation mondiale de la Santé (OMS), parmi ces décès, 85% sont dus à une crise cardiaque ou à un AVC (3). Selon les projections, près de 24 millions de personnes mourront de MCV d’ici 2030 et ces maladies resteront la première cause de mortalité (3). Plus de 80% des décès reliés aux MCV surviennent principalement dans les pays à revenu faible ou intermédiaire (4, 5). La prévalence des MCV augmente chez les personnes de plus de 65 ans, en particulier chez les personnes de plus de 80 ans (6). Au Canada, elles sont la deuxième cause de décès et 51 134 Canadiens sont décédés en 2018 d’une
maladie du cœur et 13 480 sont décédés de maladies cérébrovasculaires (7). La cardiopathie ischémique désigne l’accumulation de plaque dans les artères coronaires pouvant provoquer un IM, un AVC ou une insuffisance cardiaque, est l’affection cardiaque la plus courante au Canada (8). Les données les plus récentes de 2012-2013 concernant la prévalence montrent que 8,5% (2,4 millions) des Canadiens âgés de 20 ans ou plus ont un diagnostic de cardiopathie ischémique, dont 2,1% (578 000) ont subi une crise cardiaque (8). De plus, 3,6% (669 600) des Canadiens âgés de 40 ans ou plus ont été diagnostiqués d’IC (8). Au Québec, selon des données de 2016, les maladies du cœur et les AVC étaient la deuxième et la quatrième cause de décès respectivement (9). Environ 730 000 Québécois âgés de 20 ans et plus ont été diagnostiqués de maladies vasculaires, c’est-à-dire de cardiopathies ischémiques, d’AVC ou de MVP, représentant une prévalence brute de 11,3% (9). Dans la même année, plus de 47 000 individus ont reçu un diagnostic de MCV pour la première fois, avec une incidence brute de 8,3% et 36 000 personnes diagnostiquées de MCV sont décédées (9). Ce qui représente un taux brut de mortalité de 5%.
1.3. Étiologie et pathophysiologie
Il existe différentes étiologies pour les pathologies incluses dans les MCV. Par exemple, l’AVC ischémique peut survenir lors d’un évènement thrombotique ou embolique, provoquant une diminution du flux sanguin vers le cerveau (10). L’évènement thrombotique est souvent secondaire à une maladie athérosclérotique, provoquant une obstruction et un dysfonctionnement du vaisseau (10). Un caillot ou un débris qui bloque le flux sanguin provenant d’ailleurs dans le corps sera responsable, pour sa part, d’une embolie (10). Une pathologie du système cardiovasculaire pourrait également être d’origine congénitale. Une cardiopathie peut également être une réaction immunitaire suite à une infection bactérienne par le streptocoque du Groupe A, causant une cardiopathie rhumatismale (11). D’autres pathologies peuvent être d’origine génétique. C’est le cas notamment pour l’hypercholestérolémie familiale, l’hyperbêtalipoprotéine, la cardiomyopathie hypertrophique, quelques formes d’hypertensions artérielles, le syndrome de Marfan, etc (12). Cependant, le dénominateur commun dans la physiopathologie de la plupart des MCV est l’athérosclérose. Ce phénomène résulte d’une accumulation de plaque, composé de cholestérol, qui entraîne le durcissement et le rétrécissement des artères au fil du temps. C’est pourquoi il est important de s’attarder aux facteurs de risques qui sont associés à l’athérosclérose.
1.3. Facteurs de risque
Les MCV peut être le résultat d’une combinaison de facteurs génétiques et de facteurs liés au mode de vie dits modifiables. L’âge et le sexe sont les principaux facteurs de risques non-modifiables. Il y a des facteurs de risque cardiovasculaire liés à des maladies tels que le diabète, l’hypertension artérielle et
l’hypercholestérolémie. Parmi les facteurs de risque modifiables, c’est-à-dire lié aux habitudes de vie, il y a le tabagisme, la sédentarité, une mauvaise alimentation et l’obésité.
1.3.1. L’âge
L’âge est l’un des facteurs de risques les plus dominants des MCV (6, 13, 14). Dans une dizaine d’années, il est estimé que 23% de la population canadienne aura 65 ans ou plus (15). De plus, 40% de tous les décès dans ce groupe d’âge seront attribués aux MCV (14). Le vieillissement a un effet remarquable sur le cœur et les vaisseaux, et est associé à un déclin progressif de nombreux processus physiologiques (14). Ce qui entraîne une augmentation de l’athérosclérose et des MCV.
1.3.2. Le sexe
Le sexe de l’individu est également un facteur de risque éminent des MCV puisque les hommes sont plus susceptibles de développer une MCV à un âge plus jeune que chez la femme (16, 17). Ce phénomène résulte des changements hormonaux qui suivent après la ménopause chez la femme et du fait que les symptômes et les manifestations des maladies semblent différer des hommes (17). Le diagnostic est donc parfois plus difficile pour les femmes et elles ont tendance à être sous-estimée et sous-traitée.
1.3.3. L’histoire familiale
L’histoire familiale et les facteurs génétiques sont des facteurs important à considérer. L’histoire familiale est en fait un facteur de risque indépendant des MCV et reflète la présence d’un ou de plusieurs gènes pouvant être responsables d’une pathologie (18). Ces gènes peuvent être également plus ou moins exprimés selon l’environnement avec lequel ils interagissent. L’hypercholestérolémie familiale (HF) est un bon exemple de pathologie héréditaire, où le niveau de cholestérol LDL (low density lipoprotein) est particulièrement élevé dû majoritairement à une mutation dans le gène du récepteur LDL, LDLR, empêchant la clairance des particules de LDL de la circulation (19, 20). D’autres mutations existent pour l’HF dont un défaut dans le gène APOB et
PCSK9 qui affectent aussi le métabolisme et la clairance des LDLs (12, 19). Un niveau élevé de Lp(a), une lipoprotéine qui ressemble à une particule de LDL, est un facteur de risque héréditaire considérable pour les MCV (21, 22).
1.3.3. Le mode de vie
Le mode de vie a un impact non négligeable sur la santé cardiovasculaire. Le manque d’activité physique et une mauvaise alimentation peuvent avoir un impact sur l’augmentation du risque de MCV et ont pour effet d’augmenter la glycémie, la pression artérielle, le niveau de lipides sanguins et d’entraîner un surpoids et l’obésité (23). Il est donc clairement démontré que la pratique régulière d’activité physique et la saine alimentation réduisent les risques de MCV (24, 25).
1.3.4. Le tabagisme
Le tabagisme est également bien établi comme étant un facteur de risque des MCV athérosclérotiques et des preuves solides démontrent que la cessation de l’usage du tabac améliore la santé cardiovasculaire et prolonge l’espérance de vie (26-28) Le tabagisme favoriserait et induirait l’athérosclérose via plusieurs mécanismes d’actions tels que le dysfonctionnement endothélial, l’inflammation, la formation de thrombose, la modification du métabolisme lipidique, la résistance à l’insuline et l’équilibre de l’oxygène du myocarde (26).
1.3.5. Le diabète de type 2
Le diabète de type 2, pour sa part, est une maladie multifactorielle impliquant des facteurs génétiques et environnementaux et se caractérise par un désordre métabolique engendrant un taux de sucre élevé dans le sang (hyperglycémie). Il en résulte d’une résistance à l’insuline entraînant une hyperinsulinémie compensatoire par les cellules bêta du pancréas pour finalement mener au dysfonctionnement de ces cellules (29). Les personnes atteintes du diabète de type 2 sont deux à quatre fois plus susceptibles de souffrir de MC et d’AVC ischémique que les personnes non diabétiques (30). Des études antérieures ont démontré que la résistance à l’insuline des cellules vasculaires joue un rôle important dans la progression de l’athérosclérose (31, 32).
1.3.6. L’obésité
Un autre facteur de risque indépendant des MCV est l’obésité, qui se définit généralement par un excès de masse grasse corporelle. L’indice de masse corporelle (IMC), exprimant le poids corporel en fonction de la taille corporelle, est principalement utilisé comme outil permettant d’estimer l’excédent de graisse corporelle. Un IMC entre 18,9 et 25,0 kg/m2 est considéré normale chez l’adulte, tandis que l’obésité est définie comme
un IMC supérieur ou égal à 30,0 kg/m2 (33). D’autres mesures sont de plus en plus utilisées en clinique telle
que le tour de taille et le rapport taille/hanches. Selon des études cliniques et épidémiologiques, ces mesures ont démontré des associations plus fortes avec les facteurs de risques de MCV et les MCV (34-36). Le développement de l’obésité est influencé par plusieurs facteurs, notamment par l’équilibre entre la consommation de calories et la dépense énergétique (37). Cet excès de masse grasse corporelle au niveau abdominal est l’une des principales causes du risque de dyslipidémie, de résistance à l’insuline, d’hypertension et d’athérosclérose, autant chez l’adulte que chez l’enfant (38). Le tissu adipeux est en fait un organe endocrinien associé à des systèmes métaboliques inflammatoires. L’association entre l’obésité abdominale et le risque de MCV pourrait par ailleurs s’expliquer par le fait que ce dernier a la capacité de sécréter différentes protéines et hormones agissant non seulement sur la régulation biologique et physiologique, mais joue également un rôle important dans la résistance à l’insuline, l’inflammation et l’immunité, ainsi que sur l’athérosclérose (39).
1.3.7. L’hypertension artérielle
L’hypertension artérielle se définit comme une valeur de pression artérielle systolique ≥140 mmHg et/ou une valeur de pression artérielle diastolique ≥90mmHg pour un adulte (40). Une valeur normale se situe plutôt à une pression artérielle systolique de 120 mmHg et une pression artérielle diastolique de 60 mmHg. L’hypertension contribue à l’athérosclérose en agissant à différents niveaux soit au développement de la dysfonction en endothéliale en favorisant le stress oxydatif et en générant des ROS, en participant à la formation des stries lipidiques et des plaques d’athérome précoce et en favorisant la progression de la plaque et de l’enlèvement de la plaque via divers mécanismes (41). D’ailleurs, il existe une association linéaire entre l’ampleur de la réduction de la tension artérielle et le risque de MCV et de mortalité toutes causes confondues. Par exemple, en abaissant la pression artérielle systolique de 10 mmHg pour atteindre l’objectif de traitement de 120 à 124 mmHg, le risque de MCV a été réduit de 29%. Pour une diminution de 20 mmHg, le risque de MCV était réduit de 42% (42). Selon un méta-analyse de 19 essais clinique un traitement intensif d’hypotenseur (pression artérielle moyen de 133/76 mmHg atteinte) a été associé à une réduction de 14% pour les MCV majeurs, 13% pour les IM et 22% pour l’AVC (43).
1.3.8. L’hypercholestérolémie
Enfin, un niveau de cholestérol LDL élevé est fortement démontré par des études épidémiologiques et de randomisation mendélienne comme étant un facteur risque notable des MCV athérosclérotiques (44, 45). Ce type d’études ont également démontré que des variants génétiques associés à des niveaux de cholestérol LDL réduits sont associées un risque plus faible de MCV. Ce qui fournit également des preuves que le cholestérol LDL est causalement associé au risque de MC (46-49). De plus, une relation causale a bien été établie entre les lipoprotéines contenant l’apolipoprotéine B-100 (apoB) et les MCV athérosclérotique (44, 50). L’apoB est un composant structurel clé de toutes les particules de lipoprotéines athérogènes, y compris les LDL, les VLDL (very low density lipopoprotein), les IDL (interdmediate density lipoprotein) et la Lp(a). Chacune de ces particules ne porte qu’une seule molécule d’apoB, ainsi, le niveau total d’apoB représente le nombre total de particules de lipoprotéines athérogènes. Ce qui fournit une image plus précise di risque d’événements cardiovasculaire d’un individu. D’ailleurs, lorsque les concentrations d’apoB et de cholestérol LDL sont discordants chez certains individus, l’apoB a une association plus forte avec le risque de MCV que le cholestérol LDL (51). Les particules contenant l’apoB, notamment les LDL modifiés, ont la capacité d’entrer et de sortir facilement de l’intima artérielle. Pour un niveau physiologique de cholestérol LDL, soit de 0,5-1,0 mmol/L (20-40 mg/dl) qui est retrouvé chez les nouveau-nés et plusieurs mammifères, les probabilités de rétentions dans l’intima et le risque de former une plaque d’athérome est plutôt faible (52, 53). Cependant, l’augmentation du cholestérol LDL circulant peut accroître la possibilité de leur rétention à l’intérieur de l’intima et ainsi initier la formation d’athérosclérose, et ce, de façon dose dépendante (54).
1.4. Lipoprotéines et athérosclérose
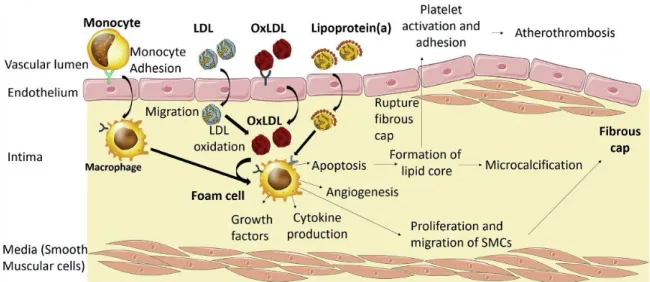
La formation de plaque d’athérome est très complexe nécessitant une combinaison de multitude de facteurs environnementaux et de facteurs génétiques et individuels. Un des évènements déclencheurs primaires dans la formation de l’athérosclérose est l’accumulation de LDLs au niveau de la matrice sous-endothéliale (55). Le LDL est en fait une particule sphérique circulante qui transporte des lipides, plus particulièrement le cholestérol estérifié. En effet, un niveau de LDL circulant élevé favorise davantage cette accumulation, surtout dans les sites favorables à la formation de lésions tels que les sites de ramifications des vaisseaux. De plus, la force de cisaillement du fluide qui a un effet sur la morphologie des cellules endothéliales (CE) et sur leur perméabilité rend ces régions plus propices à la formation de lésions (56). À ce moment, les LDLs et autres lipoprotéines athérogènes comme la lipoprotéine(a) (Lp[a]) peuvent diffuser de façon passive entre les jonctions des CE. L’apolipoprotéine B-100 (apoB) que contiennent les LDLs et la Lp(a) interagirait avec les protéoglycans de la matrice extracellulaire et entraînerait la rétention et l’accumulation des lipoprotéines athérogènes dans l’intima (57). D’ailleurs, les particules de LDL sont sensibles à l’oxydation et forment des LDLs oxydées (oxLDL) qui seraient athérogènes et favoriseraient la production de molécules pro-inflammatoires. Plusieurs mécanismes d’actions qui entrent dans le développement de l’athérosclérose via les oxLDLs ont été proposés : a) dysfonction endothéliale, b) formation de cellules spumeuses, c) migration et prolifération des cellules musculaires lisses et d) l’induction de l’adhésion et de l’agrégation plaquettaire.
1.4.1. Dysfonction endothéliale
L’absorption de oxLDLs par les récepteurs de type lectine (LOX-1; lectine-like oxidized receptor 1) des cellules des vaisseaux induirait un dysfonctionnement des CE et des cellules musculaires lisses (SMC; smooth muscle
cells) (58, 59). Il a été rapporté que les oxLDLs induisent l’expression de molécules d’adhésion intercellulaire (ICAM-1; intercellular adhesion molecule-1) et de molécules d’adhésion aux cellules vasculaires (VCAM-1;
vascular cell adhesion molecule-1) des cellules vasculaires, permettant le roulement et l’adhésion des cellules de l’immunité telles que les monocytes et certains lymphocytes (60). Ces leucocytes sont attirés vers des chimiokines sécrétés par les CE et les SMC et vont migrer vers l’intima. Il a également été établit que la production et la sécrétion d’oxyde nitrique (NO; nitric oxide) par les CE, une molécule protectrice cardiovasculaire importante empêchant l’adhésion des leucocytes et des plaquettes, seraient altérées (61). Enfin, les oxLDLs auraient la capacité de provoquer la mort cellulaire des CE et des SMC via différentes voies de signalisation, aggravant le dysfonctionnement endothélial et accélérant la formation de la plaque d’athérome (62).
1.4.2. Formation de cellules spumeuses
À la suite de leur intégration intégré la matrice sous-endothéliale, les monocytes se différencient en macrophages, portant à leur surface, différents récepteurs éboueurs (aussi appelés récepteurs scavengers), dont le récepteur de différenciation CD36 et les récepteurs scavengers SR-AI/II et SR-BI (62). Les oxLDLs seront capturés via ces récepteurs pour être ensuite internalisés dans les macrophages, entraînant la formation de cellules spumeuses (63, 64). Ces cellules gorgées de lipides sont propices au développement des stries lipidiques et par la suite des lésions athérogènes (65). Les cellules spumeuses sont propices à créer un milieu pro-inflammatoire en sécrétant des cytokines, des chimiokines et des espèces réactives de l’oxygène (ROS; reactive oxygen species), et ainsi former un cercle vicieux (63). De récentes études ont démontré que les SMCs peuvent également devenir des cellules spumeuses (66, 67). Une fois que les cellules spumeuses ont accumulés trop de oxLDLs, elles sont susceptibles de s’auto détruire, un processus qui porte le nom d’apoptose, ou de nécroser. Les débris cellulaires se déposent au cœur de la plaque athérosclérotique et forment le cœur nécrotique, contribuant à l’inflammation (68).
1.4.3. Migration et prolifération des cellules musculaires lisses
Les SMCs sont aussi essentielles dans la progression de la formation de la plaque. Après avoir migré vers l’espace sous-endothélial, en réponse à des facteurs de croissance, elles vont proliférer. Les oxLDLs participent à la prolifération des SMCs en induisant la sécrétion d’une multitude de facteurs de croissance (69). Il a été démontré que le phénotype des SMCs peut changer en présence d’oxLDLs pour produire quantités importantes de matrices extracellulaires, dont le collagène interstitiel et l’élastine (69, 70). Ces molécules vont contribuer à la génération d’une chape fibreuse recouvrant la plaque d’athérome et participer à la formation du noyau nécrotique (71). Les oxLDLs auraient aussi la capacité d’induire l’expression de LOX-1 dans les SMCs. D’ailleurs, une augmentation de ROS induite par les oxLDLs dans les SMCs serait médiée par LOX-1, ce qui contribue à la mort cellulaire, à l’instabilité de la plaque et finalement à sa rupture en stade très avancé de l’athérosclérose (69).
1.4.4. Induction de l’adhésion et de l’agrégation plaquettaire
La rupture de la plaque va favoriser la formation d’un thrombus, c’est-à-dire l’agrégation plaquettaire et l’activation du système de coagulation. La production NO altérée des CE serait associé à une augmentation de la sécrétion de prostaglandine (une cytokine) et donc à l’activation de l’agrégation plaquettaire (72). Le CD36 serait impliqué dans l’activation plaquettaire puisqu’il est retrouvé dans les plaquettes au repos et interagirait avec les oxLDLs (73). Non seulement les oxLDLs induisent l’activation des plaquettes, mais peuvent également promouvoir le changement de forme des plaquettes grâce à la voie de signalisation de Rho (74). Les plaquettes ont la capacité de libérer des chimiokines lorsqu’elles sont exposées aux oxLDLs, ce qui favorise le développement de la plaque. Le récepteur LOX-1 est également retrouvé sur les plaquettes
et participerait à la dysfonction endothéliale (75). Cela dit, les oxLDLs semblent être importants de l’activité plaquettaire en ce qui concerne le rôle des plaquettes dans le développement de la plaque athérosclérotique. L’élément déclencheur de l’athérosclérose est donc principalement les LDLs qui sont oxydés dans les régions favorables à la formation de plaque. Cependant, d’autres facteurs liés au mode de vie ou à la génétique participent également au développement de l’athérosclérose.
Figure 2. Résumé de la pathophysiologie de l'athérosclérose. Tiré de Arsenault et al, 2017.
2. Lipoprotéine(a)
2.1. Généralité
2.1.1. Structure
La Lp(a) est composé d’une particule de LDL et partage donc plusieurs caractéristiques communes avec celle-ci. D’abord, ces deux lipoprotéines contiennent un cœur lipidique, constitué principalement d’ester de cholestérol et de triglycérides, et une surface polaire contenant des phospholipides et du cholestérol libre. À leur surface, elles ont une apoB qui assure le transport et diverses actions métaboliques de la Lp(a) et du LDL. Ce qui distingue ces deux lipoprotéines, c’est que la Lp(a) contient une autre protéine de surface bien spécifique à elle, l’apolipoprotéine(a) [apo(a)] (figure 3). L’apo(a), qui est en fait une glycoprotéine polymorphique constituée de domaines Kringle codée par le gène LPA, est liée à l’apoB par un pont disulfure (76). Le nom Kringle fait référence à une brioche au beurre d’origine scandinave qui signifie « petit cercle ». Les domaines Kringle sont constitués d’une série de dix différents types de Kringle IV (KIV), dont la structure est homologue à la proenzyme fibrinolytique plasminogène, ainsi qu’un domaine Kringle V et d’une région protéase inactive (77-78). Chaque domaine comporte une seule copie, sauf pour le KIV de type 2 (KIV-2) qui
est présent en plusieurs exemplaires, soit de 3 copies à plus de 30 (79). Le nombre de copies de KIV-2 va définir la taille de l’apo(a) et va hautement varier entre chaque individu (80). La taille de l’apo(a) est inversement associée aux concentrations plasmatiques de Lp(a), c’est-à-dire que les individus ayant de grandes tailles d’apo(a) ont des concentrations plus faibles que ceux ayant de petites tailles d’apo(a) (81). Le taux de production plus important de la petite isoforme expliquerait, en partie, cette association négative entre la taille de l’apo(a) et les concentrations plasmatiques de Lp(a) (82).
Figure 3. Illustration de la lipoprotéine(a). A) Structure de la lipoprotéine(a) contenant une particule
d’apo(a) et d’apoB. B) Structure de l’apo(a) et sa similarité avec le plasminogène. Adapté de Boffa et al. Nature. 2019.
2.1.2. Aspects génétiques et épidémiologie
La distribution des concentrations plasmatiques de Lp(a) est plutôt asymétrique et varie plus de 1000 fois entre les individus d’une même population (83). Des variations au gène LPA, situé sur le chromosome 6, codant pour l'apo(a), seraient responsables jusqu'à 90% de la variation de la concentration de Lp(a) (81). Le polymorphisme de la taille des KIV de l'apo(a) serait responsable entre 20 à 80% de la variabilité de la concentration de Lp(a) dépendamment de l'ethnicité (81). De plus, il existe des variations génétiques à l’intérieur du domaine KIV-2 et qui seraient responsables, en partie, de la disparité des concentrations de Lp(a) chez les personnes porteuses d’isoformes d’apo(a) de même taille (78). Le site de restriction DraIII dans les introns est l’une des variations qui a été identifiée dans certaines copies du KIV -2 (84). Une mutation dans le premier exon de KIV-2 qui entraîne une protéine tronquée a été identifiée et représente un allèle rare d’un polymorphisme nucléotidique (SNP; single nucleotide polymorphism) (85). Un variant du domaine KIV-2 a récemment été identifié chez des individus ayant un phénotype discordant entre la concentration plasmatique de Lp(a) et la taille de l’apo(a). Il s’agit d’un variant dans le site d’épissage du domaine KIV-2 et qui est associé à des niveaux très faibles de Lp(a). Ce qui explique
environ 20% la variation de Lp(a) chez les individus avec un petit isoforme (86). Dans la région promotrice du gène LPA, une répétition pentanucléotique serait associée avec la concentration de Lp(a) (87). Même si cette lipoprotéine a été découverte en 1963, ce n'est seulement que dans les dernières décennies qu'elle devient de plus en plus attrayante aux yeux de plusieurs chercheurs (88). D'ailleurs, durant cette période, plusieurs études épidémiologiques, méta-analyses ainsi que des études de randomisation mendélienne ont démontré une forte association avec une concentration élevée de Lp(a) et les MCV (22, 89, 90-93). Bien que 90% de la variation des taux de Lp(a) serait due à des facteurs génétiques, le risque d’être atteint d’une MCV est élevé lorsque la concentration de Lp(a) atteint 30 mg/dL (94-95). De plus 20% des individus ont une concentration de Lp(a) supérieure à 50 mg/dl serait nettement plus associée au risque de MCV (90, 96). Des études ont démontré que la variation du nombre de copies du domaine KIV-2 à l’intérieur du gène LPA, de plus que les SNPs rs10455872 et rs3798220, étaient associés à la concentration plasmatique de Lp(a) et aux MC (91-93, 97-99). D’ailleurs, le SNP rs10455872 lui-même explique environ 25 à 30% de la variation des concentrations de Lp(a), alors que cette proportion augment à 36% lorsque le SNP rs37982290 s’ajoute (91).
2.1.3. Métabolisme
Il est bien clair que l’apo(a) est synthétisé et sécrété exclusivement par les cellules du foie. Cependant les sites et les mécanismes d’assemblages de la Lp(a) sont encore incertains. Les hypothèses sont que l’assemblage de la Lp(a) se produit probablement à la surface des hépatocytes ou dans le plasma, et se fait en deux étapes. La première étape consiste à l’accostage de l’apo(a) au LDL et la seconde étape est qu’une liaison disulfure se forme entre le KIV de type 9 de l’apo(a) et l’apoB du LDL (100). Le rôle de l’apoB nouvellement synthétisée semble critique dans la formation de la Lp(a) puisque qu’un inhibiteur de la synthèse d’apoB abaissant le taux de LDL plasmatique réduit également Lp(a) (101). Par contre, l’inhibition de la synthèse de cholestérol via les inhibiteurs HMG-CoA réductase (statines) ne diminue pas la Lp(a) (102). Bien qu’il soit connu que l’apo(a) et l’apoB s’assemblent dans le foie, les études sont plutôt contradictoires en ce qui concernent le lieu précis d’assemblage. Certaines études effectués sur des hépatocytes en culture indiquent que l’apo(a) et l’apoB se lient à la surface de l’hépatocyte, plutôt que dans la voie de sécrétion intracellulaire (103-104). Toutefois, d’autres études soutiennent l’assemblage dans le milieu intracellulaire (105-106). Au niveau de la clairance de la Lp(a) de la circulation sanguine, il n’y a aucun mécanisme clair qui a été élucidé pour le moment. Néanmoins, elle semble se produire principalement au foie et plusieurs récepteurs de Lp(a) ont pu être identifiés, en particulier le récepteur LDL (LDLR), les récepteurs scavenger B1 (SCARB1; scavenger receptor class B type 1) et différents récepteurs du plasminogène (107-112). D’autres récepteurs de la famille des LDLR semblent également jouer un rôle dans la clairance de la Lp(a), dont les récepteur LRP1 (low density lipoprotein receptor-related protein 1) et le récepteur LRP2 (low density
lipoprotein receptor-related protein 2) ainsi que le récepteur de lipoprotéines de très faible densité (VLDLR;
very low density lipoprotein receptor) (113-116). D’ailleurs, les inhibiteurs de PCSK9 diminuent légèrement le taux de Lp(a) circulant, soit environ 20-30%, en augmentant l’abondance du LDLR à la surface des hépatocytes (117-118). Ce qui peut expliquer l’implication du LDLR dans le processus de catabolisme de la Lp(a). Il est évident que le foie est indispensable dans le métabolisme de la Lp(a). Toutefois, des évidences solides soutiennent que les reins soient impliqués dans la clairance de la Lp(a). Les individus atteints d’insuffisance chronique, les niveaux plasmatiques de Lp(a) sont augmentés et diminuent après une transplantation rénale (119-120). De plus, l’augmentation de la concentration de Lp(a) est inversement corrélée avec le débit de filtration glomérulaire (121).
2.2. Mécanismes pathophysiologiques
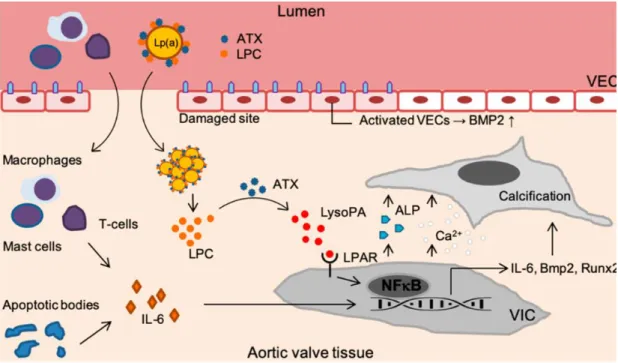
Actuellement, le rôle physiologique de la Lp(a) n’est pas tout à fait clair. Il se trouve que les individus ayant des niveaux extrêmement faibles de Lp(a) n’ont pas de carence ou de maladies quelconques (122). Quelques travaux antérieurs s’entendent pour dire que la Lp(a) fourni un avantage aux humains en accélérant la cicatrisation des plaies et la réparation des tissus (122-123). Cependant, bien que la Lp(a) soit très similaire aux LDLs, ces deux lipoprotéines partagent divers risques athérogènes, y compris la capacité de s’oxyder dans la paroi des vaisseaux et de devenir hautement immunogène et pro-inflammatoire (124). Non seulement la Lp(a) contient les propriétés athérogènes des LDLs, mais elle contient une composante pro-thrombotique via l’apo(a) (125). In vitro, il a été observé que la Lp(a) peut interférer avec de nombreuses réactions clés de coagulation/fibrinolyse et se dépose dans les plaques d’athérosclérose (126-127). Un des facteurs clés de la Lp(a) dans le développement des MCV athérosclérotique et de la SA calcifiante est le fait que la Lp(a) transporte des OxPLs qui peuvent être liés à l’apo(a) et à l’apoB, et être libres dans la fraction lipidique (128). Les OxPLs colocalisent avec la Lp(a) dans les lésions des artères et des valves aortiques et participent directement à la pathogénèse de la SA calcifiante et des MCV athérosclérotique en favorisant le dysfonctionnement endothélial, l’inflammation, le dépôt de lipidique et la calcification (128-130). D’ailleurs, les niveaux plasmatiques de Lp(a) et d’OxPLs sont fortement corrélés, ce qui suggère que les individus ayant des niveaux élevés de Lp(a) ont une capacité de liaison plus élevée pour les OxPLs et des niveaux de OxPLs plus élevés. Il existe également une association entre les niveaux d’OxPLs et les MCV (131-132). De plus, les mécanismes pro-athérogènes des LDLs peuvent également être attribués à la Lp(a), c’est-à-dire l’augmentation de la capacité de liaison aux CE et aux protéoglycans de la matrice, stimuler la prolifération de cellules musculaires lisses et favoriser la formation de cellules spumeuses, du cœur nécrotique et des lésions (124). Parmi les mécanismes pro-inflammatoires, la Lp(a) induit une activité chimioattracttante des monocytes et l’expression de l’interleukine (IL) 8 par les macrophages (133). Les mécanismes pathogènes
de la Lp(a) sur la SA calcifiante, l’AVC et les MC seront abordés un peu plus en profondeurs dans les sections ultérieures.
2.3. Thérapies ciblant la lipoprotéine(a)
Il est bien documenté que la concentration de Lp(a) est principalement modulée par des facteurs génétiques. Toutefois, des études ont été réalisées afin d’évaluer si l’alimentation ou l’activité physique avait un impact sur le taux de Lp(a). En ce qui concerne l’impact des interventions diététiques sur les niveaux de Lp(a), les résultats rapportés sont plutôt incohérents, probablement dus à l’hétérogénéité des modèles d’études utilisés, des populations d’études, de la composition du régime alimentaire, des interventions diététiques et des résultats. Finalement, l’impact de l’alimentation sur la concentration de Lp(a) s’avère non cliniquement significatif (134-139). Du côté de l’activité physique, les études semblent plutôt contradictoires, mais la majorité d’entre elles s’entendent qu’il n’y a aucune amélioration des niveaux de Lp(a) (140-146). Même si le mode de vie n’a pas vraiment d’influence sur la Lp(a), une étude prospective effectuée dans la cohorte EPIC (European Prospective Investigation of Cancer) Norfolk suggère que parmi les personnes ayant des niveaux élevés de Lp(a), la gestion des facteurs liés au mode de vie peut réduire le risque de MCV jusqu’à 75% (147). Quelques traitements pharmacologiques et une intervention non pharmacologique ont démontré des effets sur la réduction des niveaux de Lp(a).
2.3.1. L’aphérèse
L’aphérèse est un processus non pharmacologique qui vise à éliminer les lipoprotéines extracorporelles contenant une apoB. Elle est principalement conçue pour traiter les individus atteints d’HF homozygote, soit une forme sévère d’hypercholestérolémie. Cependant, l’aphérèse est autant efficace pour les individus ayant une hyperlipoprotéinémie(a). Un seul traitement peut réduire jusqu’à 75% des niveaux de Lp(a) et demeure, jusqu’à ce jour, le meilleur traitement approuvé et efficace pour abaisser significativement la concentration de Lp(a) (148). Dans une étude longitudinale multicentrique de 120 patients atteints de MCV et ayant une concentration de Lp(a) élevée se situant dans le 95e percentile ou plus, un traitement à l’aphérèse plus un
traitement médical toléré au maximum ont entraîné une réduction médiane de la Lp(a) de 73%. De plus, cette étude a démontré que la polythérapie combinée à l’aphérèse réduisait les évènements cardiaques majeurs de 88% sur une période de 10 ans (149). Ces résultats ne sont pas à négliger, mais malheureusement les niveaux de lipoprotéines contentant l’apoB tendent à revenir au point de départ quelques semaines après le traitement. Bien que l’aphérèse semble efficace, il y a quand même des inconvénients, notamment qu’il est dispendieux et que son accès soit limité. C’est également un traitement qui demande beaucoup de temps et qui doit être répété plusieurs reprises par année. Enfin, les effets connus de l’aphérèse sont généralement sur plusieurs lipoprotéines et les données concernant l’effet de l’élimination spécifique de la Lp(a) seul sont rares.
2.3.2. Les statines
Les statines sont l’une des classes de médicaments les plus prescrites au Canada et elles sont utilisées pour baisser le cholestérol sanguin afin d’améliorer la survie et de réduire le risque d’évènements cardiovasculaires chez les personnes susceptibles de souffrir d’une MCV (150). Cependant, elles ne démontrent aucun effet bénéfique sur les niveaux de Lp(a) plasmatiques, mêmes que les études à ce sujet suggèrent une augmentation de 10% à 20% des taux de Lp(a). Une méta-analyse effectuée par Yeang et al. incluant 7 essais cliniques sur les statines et 1688 participants a démontré une augmentation moyenne d’environ 11% de la concentration de Lp(a) après le traitement aux statines (atorvastatine, la rosuvastatine, la pitavastatine et la pravastatine) (151). Dans l’étude ILLUMINATE (Investigation of Lipid Level Management to
Understanding its Impact in Atherosclerotic Events), les chercheurs ont évalué l’impact du traitement au torcetrapib sur les résultats cardiovasculaires chez des individus traités par 4 doses d’atorvastatine ont été testés (10, 20, 40 et 80 mg/jour). Les résultats ont révélé que les niveaux de Lp(a) augmentaient en fonction de la dose d’atorvastatine (152). Il est important de noter que la réduction des concentrations de lipoprotéines athérogènes à la suite d’un traitement aux statines sur la réduction du risque cardiovasculaire l’emporte significativement sur l’augmentation de la Lp(a). Hopewell et al. ont très bien documenté dans leur étude
Heart Protection Study les avantages du traitement par simvastatine par rapport au placebo chez les patients avec ou sans SNP associé au niveau élevé de Lp(a) (153). Les résultats démontrent une réduction du risque relatif associé au traitement par simvastatine de 27,6% et 20,2% chez les porteurs et les non porteurs de l’allèle associée à une Lp(a) élevée respectivement. La réduction du risque absolu correspondante était de 6,5% et de 4,3%.
4.3.3. Niacine
La niacine est l’un des seuls médicaments pouvant diminuer les niveaux de Lp(a) circulants de manière significative. En fait, les mécanismes pouvant affecter le métabolisme de la Lp(a) ne sont pas tout à fait clairs. Cependant, il est suggéré que la niacine peut altérer la production et le taux cataboliques d’apo(a). Croyal et
al. ont démontré qu’un traitement de 8 semaines à la niacine a entraîné une réduction de 50% de l’apo(a) nouvellement synthétisé, qui a été, en partie, compensé par une diminution de la clairance catabolique. Dans cette étude, l’effet net de la niacine sur la baisse de la concentration plasmatique de Lp(a) était de 20% (154). Ceci est compatible avec la répression du gène LPA via la niacine. Dans une analyse post-hoc de l’étude HPS2-THRIVE (Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events), une étude effectuée chez les individus à haut risque cardiovasculaire et qui compare l’impact de la niacine combiné à laropirant (un antagoniste de la prostaglandine) à un placebo, la réduction moyenne des taux de Lp(a) circulants était de 31%. Les participants ayant une plus grande taille d’isoforme d’apo(a) et un niveau plus faible de Lp(a), une réduction de 50% a été observée, tandis que les individus qui ont une petite isoforme et un taux élevé de Lp(a) avaient une réduction de seulement 16% (155). De plus, une méta-analyse
comprenant 14 études a démontré que le traitement à la niacine a été associée à une diminution moyenne du niveau de Lp(a) de 23% (156). Deux études publiées dernièrement, soit AIM-HIGH (Atherothrombosis
Intervention in Metabolic Syndrome with Low HDL/High Triglycerides : Impact on Global Health Outcomes) et HPS2-THRIVE, ont obtenus des résultats négatifs concernant la réduction des évènements cardiovasculaires et d’importants effets secondaires ont également été rapportés (157-158). D’ailleurs, l’impact de la niacine sur les évènements cardiovasculaires chez les individus avec un niveau élevé de Lp(a) n’a pas encore été documenté.
2.3.4. Inhibiteurs de PCSK9
La proprotéine subtilisine/kexine de type 9 (PCSK9) joue un rôle important dans le métabolisme du cholestérol LDL en augmentant la clairance des LDLR, ce qui a pour conséquence l’augmentation des niveaux de cholestérol LDL. Sous le nom d’alirocumab et d’évolucumab, ces deux anticorps monoclonaux humains ont récemment été approuvés par Santé Canada puisqu’ils ont prouvé leur efficacité ainsi que leur innocuité. Les inhibiteurs de PCSK9, en augmentant l’expression des LDLR à la surface des hépatocytes, réduisent les niveaux de Lp(a) circulants. Une étude a évalué l'effet de l'alirocumab sur le niveau de Lp(a) à partir de données regroupées provenant de trois études randomisées à double aveugle, en phase II, chez des patients avec une hypercholestérolémie et ayant pris des traitements hypolipémiants. Suite au traitement à l'alirocumab, le taux de Lp(a) a diminué de 30% par rapport aux valeurs de départs et ce résultat est significatif comparer au placebo (0,3%) (117). Ces résultats ont pu être confirmés dans l’étude de phase III incluant 10 essais randomisés (159). Une seconde étude de phase II, cette fois-ci avec un traitement par l’évolucumab, a obtenu des résultats similaires (118). Par contre, les mécanismes biologiques par lesquels les inhibiteurs de PCSK9 agissent sur la Lp(a) ne sont pas encore tout à fait élucidés aujourd’hui. Quelques hypothèses sont toutefois suggérées, bien qu’elles soient contradictoires. En premier lieu, certaines théories proposent que le LDLR contribue à la clairance des particules de Lp(a) circulantes. D’un autre côté, les statines augmentent la densité des LDLR à la surface des hépatocytes, mais ne réduisent pas la concentration de Lp(a). Chez 20 hommes en bonne santé ne recevant aucun hypolipémiant, une tendance de l’augmentation du taux de la fraction catabolique de l’apo(a) de 24,6% après l’inhibition de PCSK9 avec l’alirocumab a été observée (160). Par la suite, Watts et al. ont étudié l’effet des anticorps monoclonaux évolocumab, en monothérapie et en bithérapie avec l’atorvastatine, sur la Lp(a) chez 63 hommes en santé. En monothérapie, ils ont constaté que l’évolocumab abaissait de manière significative, soit de 36% la production de la production de l’apo(a), mais n’avait aucun effet sur la fraction catabolique d’apo(a). Inversement, la portion catabolique de l’apo(a) a été augmenté de 59% par la bithérapie en n’ayant eu aucun impact sur la production d’apo(a) (161). Un des faits saillants de leur étude est que le taux catabolique de l’apo(a) n’a pas été augmenté ni par la statine ni par l’évolucumab seuls.
2.3.5. Oligonucléotides anti-sens anti-apo(a)
Les oligonucléotides anti-sens ciblant l'acide ribonucléique messager (ARNm) de l'apo(a) hépatique sont la première classe d'agent pharmacologique le plus spécifique à la Lp(a). Ces oligonucléotides inhibent la production de l’apo(a) directement dans les hépatocytes, ce qui représente la source d’environ 99% de la Lp(a) plasmatique (162). AKCEA-APO(a)-LRx est un médicament de deuxième génération qui portait le nom IONIS- APO9(a)-LRx auparavant. Un oligonoculéotide anti-sens est un fragment d’acide ribonuclique (ARN)
qui se lie à l’ARNm ciblé afin d’inhiber la production de protéines, soit l’apo(a) dans ce cas-ci. AKCEA-APO(a)-LRx présente une conjugaison de N-acétylgalactosamine (GalNAc), facilitant l’absorption de ce composé par
les hépatocytes via les récepteurs asialoglycoprotéine de type 1, ce qui en fait un inhibiteur de Lp(a) très puissant. Dans une étude clinique en phase IIa, 58 volontaires avec une concentration de Lp(a) élevée ont été assignés au traitement AKCEA-APO(a)-LRxinjecté de façon sous-cutanée, soit par simple dose, multiple dose ou par un placebo. Une réduction dose-dépendante significative de la concentration de Lp(a) a été rapportée dans le groupe ayant reçu la simple dose et une réduction entre 66 et 92% a été observée dans le groupe qui a reçu la multiple dose (163). De plus, une étude très récente incluant des individus avec MCV et une concentration de Lp(a) supérieure à 50mg/dl, l’AKCEA-APO(a)-LRx a entraîné une diminution des niveaux
de Lp(a), et ce, de manière dose-dépendante. Une dose de 20 mg toutes les 4 semaines a réduit la Lp(a) de 35% en moyenne, 40 mg toutes les 4 semaines correspondaient à une diminution de 56%, de 58% avec 20 mg toutes les semaines, 72% pour 60 mg administrés toutes les 4 semaines et 80% avec une dose de 20 mg toutes les semaines (164). De plus, 98% des participants de cette étude ont atteint des niveaux "normaux" de Lp(a). Une grande étude internationale de phase III sera lancée prochainement afin de documenter les potentiels bénéfices de cette thérapie sur les évènements cardiovasculaires.
2.3.6. Autres thérapies
Le mipomersen est un oligonucléotide anti-sens qui cible l’apoB qui a été approuvé par la FDA pour traiter les individus ayant une HF homozygote. Il diminue les niveaux de cholestérol LDL de 25% à 39% et réduit également le taux de Lp(a) de 21% à 39% (165). Cependant, le mipomersen peut provoquer certains effets indésirables tels que des symptômes pseudo-grippaux et des réactions aux site de l’injection (douleur, démangeaisons, gonflement, érythèmes, etc.). Son utilisation est très restreinte en raison du risque d’hépatoxicité, ce qui exclut l’utilisation généralisée pour l’hyperlipoprotéinémie(a) (166).
Les inhibiteurs de la protéine de transfert des esters de cholestérol (CETP; cholesteryl ester transfer protein) ont également démontré qu’ils diminuent les niveaux de Lp(a). L’inhibiteur de CETP anacétrapib est associé à la diminution dose-dépendante de la concentration de Lp(a) plasmatique lorsqu’il est administré en monothérapie (167). Dans l’essai DEFINE (Determining the Efficacy and Tolerability of CETP inhibition With
a été associé à une réduction de 23,8% de la Lp(a) après 24 semaines et 17,1% après 76 semaines (168). Ensuite, une étude exploratoire menée par Thomas et al. pour étudier le mécanisme d’abaissement de la Lp(a) par l’anacétrapib, les niveaux de Lp(a) dans toute la cohorte ont été réduits de 34,1%. La diminution de la concentration de Lp(a) était similaire entre le groupe statine et le groupe anacétrapib (34,5%), et le groupe placebo et le groupe anacétrapib (32,6%) (169). Dans cette même étude, la réduction du niveau de Lp(a) reflète en fait de la diminution de la production de l’apo(a). Finalement, l’impact des inhibiteurs de la CETP sur le risque de MCV sont plutôt divergent, c’est-à-dire que les études cliniques se sont avérées nuisibles, neutres ou légèrement positifs (170).
Un dernier agent thérapeutique ayant des effets sur la concentration de Lp(a) est la famille des fibrates. Ces molécules sont des hypolipémiants qui peuvent être utilisés chez les patients atteints d’HF et qui sont intolérants aux statines. Les études de l’impact des fibrates sur la Lp(a) se sont révélées contradictoires. Toutefois, dans une méta-analyse, les fibrates avaient un effet significativement plus important dans la réduction des niveaux de Lp(a) comparativement aux statines (171). La diminution de la concentration de Lp(a) de 2,70 mg/dl demeure cependant non cliniquement significative.
3. Les maladies cardiovasculaires chez la femme
3.1. Le fardeau des maladies cardiovasculaires chez la femme
Les MVC ont longtemps été considérées comme étant des maladies qui affectent davantage les hommes. Malheureusement, ce n’est que dans les dernières années que la science et la médecine ont commencé à accorder une importance plus importante à la santé cardiovasculaire de la femme. Pourtant, une femme sur trois décède d’une MCV chaque année, ce qui en fait la principale cause de décès au monde chez la femme (172). Les crises cardiaques et les AVC coûtent la vie de 3,3 millions et 3,2 millions respectivement à tous les ans, tandis que 2,1 millions de femmes décèdent des autres formes de MCV dans la population mondiale (173). Ces maladies sont la cause principale de décès prématuré chez les femmes canadiennes (174). Les trois principales causes de décès reliés aux MCV chez la femme au Canada sont les MC, l’AVC et l’IC (17). Bien qu’il ait reconnu que les œstrogènes ont un impact sur la réponse vasculaire chez la femme, il existe des évidences que les réponses cardiovasculaires physiologiques et physiopathologiques sont affectées de manière unique chez la femme. De plus, les femmes présentent un risque plus faible de MCV que les hommes de même âge au cours de leurs années de procréation. Dans l’étude INTERHEART, une étude de cas-contrôle standardisée de MC au niveau mondial chez 27 098 participants dans 52 pays différents, les femmes subissent leur premier infarctus du myocarde en moyenne 9 ans plus tard que les hommes (175). Malgré que les différences des manifestations et des conséquences des MCV commencent à être généralement mieux