Insertion d’une mutation protectrice pour la maladie
d’Alzheimer dans le gène de la protéine précurseur de
l’amyloïde via le système CRISPR/Cas9
Thèse
Antoine Guyon
Doctorat en médecine moléculaire
Philosophiæ doctor (Ph. D.)
Insertion d’une mutation protectrice pour la
maladie d’Alzheimer dans le gène de la protéine
précurseur de l’amyloïde via le système
CRISPR/Cas9
Thèse
Antoine Guyon
Doctorat en médecine moléculaire
Philosophiæ doctor (Ph. D.)
Sous la direction de :
Résumé
La maladie d’Alzheimer est la plus commune des formes de démence qui touche presque cinquante millions de personnes dans le monde. Les symptômes les plus fréquents sont la perte de mémoire, la difficulté à planifier des tâches et des confusions temporelles et spatiales. Il n’existe à ce jour aucun traitement pour cette maladie.
La protéine précurseur de l’amyloïde (APP) est habituellement coupée par l’enzyme alpha-sécrétase, cependant une coupure anormale par la bêta-sécrétase conduit à la formation de peptides bêta-amyloïdes, qui forment des agrégats s’accumulant sous forme de plaques dans le cerveau des patients Alzheimer. De nombreuses mutations du gène APP sont à l’origine de changements dans la séquence d’acides aminés de la protéine, responsable d’une accumulation accrue de plaques. Ces mutations sont appelées formes familiales de la maladie d’Alzheimer ou FAD (Familial Alzheimer’s disease). Cependant il a été découvert qu’une forme de FAD d’APP (mutation islandaise A673T) présente dans une population d’Europe nordique, différant d’une seule paire de bases du gène normal dans l’exon 16, modifiant une alanine de la protéine en thréonine qui réduit de 40% sa coupure par la bêta-sécrétase. La découverte de la technologie CRISPR/Cas9 ouvre de nouvelles perspectives pour le développement de traitements préventifs ou curatifs des maladies génétiques et dans notre cas Alzheimer. L’endonucléase Cas9 peut couper l’ADN double brin du génome en étant guidée par un ARNg et en reconnaissant une séquence cible « protospacer » suivie d’un PAM (protospacer adjacent motif). Depuis 2016, une optimisation du système CRISPR appelée édition de base permet maintenant de modifier de façon très précise la base cytidine en thymine et les guanines en adénines sur le brin opposé de l’ADN.
Le premier article de cette thèse vise à démontrer que l’ajout de la forme FAD A673T en codominance avec une autre forme pathologique provoque ou non des répercussions bénéfiques sur la sécrétion de peptides amyloïde-beta. Les expériences ont été réalisées avec des plasmides, permettant de visualiser un effet maximum de la mutation A673T. Il était important de déterminer si cette mutation était protectrice en codominance pour assurer une approche thérapeutique la plus polyvalente possible. Nous avons confirmé cet effet bénéfique sur une majorité de formes FAD et en particulier sur la mutation London V717I.
Le deuxième article de cette thèse traite de l’introduction de la mutation A673T par un système dérivé de CRISPR/Cas9, l’édition de base. Plusieurs modèles d’éditeurs de base
ont été comparés et optimisés dans le but d’obtenir une modification du génome aussi efficace et précise que possible. Un candidat a été sélectionné après des tests sur cellules modèles HEK 293T et neuroblastomes SH-SY5Y.
La troisième partie de ce manuscrit présente les résultats obtenus lors de l’insertion de cet éditeur de base dans des vecteurs viraux dans le but d’infecter des modèles de neurones humains et murins présentant des formes FAD.
L’ensemble de cette démarche a permis d’ouvrir une nouvelle avenue à une potentielle thérapie pour la maladie d’Alzheimer.
Abstract
Alzheimer’s disease (AD) is the most common form of dementia in the world, with nearly fifty million people affected currently. The most common symptoms of this disease are memory loss, difficulties in task management, and temporal and spatial confusions. There is currently no treatment for this disease.
The amyloid precursor protein (APP) is usually cut by the alpha-secretase enzyme; however, abnormal cleavage by the beta-site APP cleaving enzyme 1(BACE1) leads to the formation of beta-amyloid peptides. These peptides in turn forms aggregates, which accumulate as plaques in the brains of Alzheimer patients. Many non-silent APP mutations cause changes to the amino acid composition of the protein and result in increased plaque accumulation. These mutations are called familial forms of Alzheimer’s disease (FAD). However, one of these mutations (Icelandic A673T mutation) has been shown to confer a protection against the onset and development of AD. This mutation of a single mutation in exon 16 changes an alanine into a threonine and has been shown to reduce the cleavage of the APP protein by BACE1 by 40%.
This kind of single point mutation is the perfect target for the newly discovered CRISPR/Cas9 technology, which opens new perspectives for the development of preventive or curative treatments for genetic diseases and in our case Alzheimer’s. The Cas9 endonuclease is a powerful tool for the modification of genetic data. The protein has been shown to cut double-stranded DNA with the help of a guide RNA (gRNA) to target a specified sequence adjacent to a PAM (protospacer adjacent motif). The base CRISPR system has been coopted by many different research teams; one of which used the technology to develop a technique they called base editing. This technique allows researchers to exchange cytidine bases for thymine and guanine bases for adenine with a strong accuracy.
The first article of this thesis aims to demonstrate that the addition of the A673T mutation in codominance with another pathological form of AD may have beneficial effects on the reduction of beta-amyloid peptides in patients’ brains. To determine if the mutation was protective, plasmids carrying the A673T mutation along with another random FAD mutation were used. Ultimately, we confirmed the beneficial effect for many forms of FAD, in particular the London V717I mutation demonstrated the greatest reduction in beta amyloid proteins.
The second article of this thesis deals with the insertion of the A673T mutation by the CRISPR/Cas9 derived system, base editing. Several base editor complexes were compared and optimized to achieve the most effective and accurate genome modification possible. A candidate was selected after testing on HEK293T cells and SH-SY5Y neuroblastoma.
The third part of this manuscript presents the results obtained when using lentiviral and AAV vectors to infect induced human and mouse neurons with a base editor complex and harvested mouse neurons with FAD forms.
This whole approach has opened up an avenue for a potential therapy for Alzheimer’s disease.
Table des matières
Résumé ... iii
Abstract ... v
Table des matières ... vii
Liste des figures ... xi
Liste des tableaux ... xiii
Liste des abréviations, sigles, acronymes ... xiv
Remerciements ... xix
Avant-propos ... xx
Introduction ... 1
Chapitre 1 : La maladie d’Alzheimer ... 1
1.1 Au niveau épidémiologique : la prévalence ... 1
1.2 Au niveau cérébral : l’étiologie ... 1
1.3 Les différents stades ... 2
1.4 Au niveau psychologique : La mémoire ... 3
1.5 Les différentes formes de la maladie d’Alzheimer ... 4
1.6 Les facteurs de risques ... 5
1.6.1 Les facteurs contrôlables ... 5
1.6.2 Les facteurs incontrôlables ... 5
1.7 Au niveau moléculaire : ... 5
1.7.1 Le précurseur de la protéine amyloïde (APP) ... 5
1.7.2 L’alpha-secrétase ... 7
1.7.3 La bêta-secrétase ... 8
1.7.4 La gamma-secrétase ... 9
1.7.5 Des clivages particuliers de l’APP ... 9
1.7.6 Du peptide Aβ à la plaque amyloïde ... 10
1.7.7 La protéine Tau ... 12
1.8 Les modèles de souris transgéniques de la maladie d’Alzheimer ... 12
1.9 Les différentes hypothèses ... 13
1.9.1 Théorie de la cascade d’amyloïde ... 13
1.9.2 Hypothèse revisitée : la maladie Alzheimer serait initiée par Aβ mais toxique de façon Tau dépendante – L’axe de Tau ... 15
1.10 Traitements pour la maladie d’Alzheimer ... 16
1.10.1 Diagnostic : les biomarqueurs ... 16
1.10.2 Traitements actuels ... 17
2.1 Les systèmes d’enzymes endonucléases programmables ... 20
2.1.1 La première coupure ciblée de l’ADN ... 20
2.1.2 Les nucléases à doigts de zinc (ZFN) ... 20
2.1.3 Les nucléases effectrices de type activateur de transcription... 22
2.2 Le système CRISPR/Cas9 ... 23
2.2.1 Les origines du système CRISPR ... 23
2.2.2 Système CRISPR/Cas de type II ... 24
2.2.3 L’édition de base ou CRISPR 2.0 ... 27
2.3 Les effets hors-cibles ... 28
2.3.1 Créer des Cas9 plus précises ... 29
2.3.2 Une Cas9 inductible ... 29
2.4 Méthode de livraison pour les endonucléases programmables ... 30
2.4.1 Les méthodes physiques ... 30
2.4.2 Les vecteurs viraux ... 31
2.4.3 Les vecteurs non viraux ... 35
2.5 Cibles thérapeutiques ... 37
Chapitre 3 : Projet de doctorat ... 40
Problématique ... 40
Hypothèse ... 40
Objectifs ... 41
Résultats ... 42
Chapitre 4 : The protective mutation A673T in Amyloid Precursor Protein gene decreases Aβ peptides production for 14 forms of Familial Alzheimer’s Disease in SH-SY5Y cells ... 42
4.1 Résumé ... 43
4.2 Abstract ... 44
4.3 Introduction ... 45
4.4 Results ... 47
4.4.1 Amyloid-β peptide quantification ... 47
4.4.2 Aβ42/Aβ40 ratio ... 48
4.4.3 The London mutation (APP V717I) ... 48
4.5 Discussion ... 48
4.6 Methods ... 52
4.6.1 Construction of plasmid libraries containing an FAD mutation ... 52
4.6.2 Transfection in SH-SY5Y of plasmid libraries ... 52
4.6.3 Culture medium analysis ... 53
4.6.4 Statistical analysis ... 53
4.9 References ... 53
4.10 Figures and tables ... 57
Chapitre 5 : Base editing strategy for insertion of the A673T mutation in the APP gene to prevent the development of AD in vitro ... 64
5.1 Résumé ... 65
5.2 Abstract ... 66
5.3 Introduction ... 67
5.4 Results ... 69
5.4.1 Deaminase design ... 69
5.4.2 APP deamination: A673T editing ... 70
5.4.3 Aβ peptides concentration decreases in APP SH-SY5Y cell lines ... 72
5.5 Discussion ... 74
5.6 Material and methods ... 77
5.6.1 Deaminase variants description and construction ... 77
5.6.2 Co-transfection in HEK293T and SH-SY5Y cells of Cas9n/Cytidine deaminase plasmid and pBSU6 sgRNA... 79
5.6.3 Cell harvesting ... 80
5.6.4 DNA extraction ... 80
5.6.5 Stable SH-SY5Y APP cell lines generation ... 80
5.6.6 Supernatant analysis ... 81
5.6.7 Deep sequencing analysis ... 81
5.6.8 Bioinformatics analysis of the deep sequencing results ... 81
5.7 Statistical Analysis ... 82
5.8 Author contributions ... 82
5.9 Conflicts of interest ... 82
5.10 Acknowledgments ... 82
5.11 Bibliography ... 82
5.12 Figures and tables ... 86
5.13 Supplemental results ... 91
Chapitre 6 : Optimization of in vitro base editing strategy delivery targeting neurons afflicted with Alzheimer’s disease ... 101
6.1 Résumé ... 102
6.2 Abstract ... 103
6.3 Introduction ... 104
6.4 Results ... 106
6.4.1 Induced neuron generation... 106
6.4.3 Neuron transduction ... 107
6.4.4 Cas9 protein detection ... 108
6.4.5 Reduction of A42 peptides following cell transduction ... 108
6.4.6 Virus optimization ... 109
6.5 Discussion ... 110
6.6 Methods ... 113
6.6.1 Isolation and culture of NL/F/G hippocampal neurons ... 113
6.6.2 Cell culture and cell lines ... 114
6.6.3 Viral vectors and virus transduction ... 114
6.6.4 Lentivirus production ... 115
6.6.5 Direct neuronal reprogramming ... 116
6.6.6 Immunofluorescence staining protocol ... 116
6.6.7 Western Blot ... 117
6.6.8 Transfection in HEK293T cells of AAVs and lentivirus constructs ... 118
6.6.9 Culture medium analysis ... 118
6.6.9 Statistical analysis ... 118
6.7 Acknowledgment ... 119
6.8 References ... 120
6.9 Figures and tables ... 125
Conclusion ... 130
Discussion générale et perspectives ... 130
Bilan des résultats obtenus ... 130
La pertinence de développer une thérapie génique ciblée contre la maladie d’Alzheimer. ... 137
Nouvelles technologies disponibles pour l’édition du génome ... 138
Les différents défis à relever ... 139
Perspectives ... 142
Liste des figures
Figure 1. Suivi de l’atrophie corticale dans le développement de la Maladie
d’Alzheimer. ... 3
Figure 2. Métabolisme protéolytique de la protéine APP ... 6
Figure 3. Clivages globaux de la protéine APP. ... 10
Figure 4. Phase d’agrégation du peptide bêta. ... 11
Figure 5. Structure d’un dimère de ZFN. ... 21
Figure 6. Structure d’un dimère de TALEN ... 23
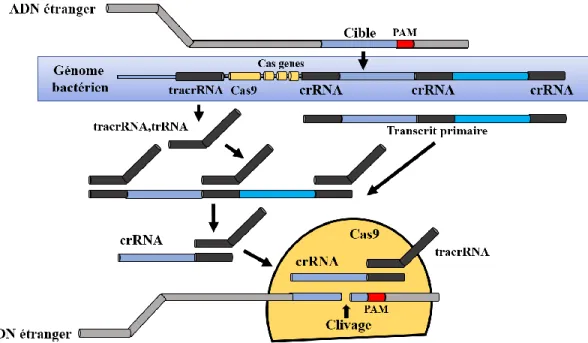
Figure 7. Description du mécanisme du système CRISPR de type II ... 25
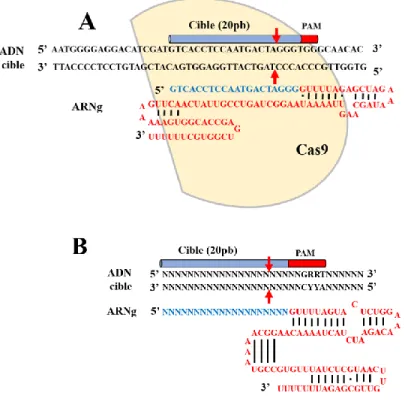
Figure 8. Représentation schématique de CRISPR/SpCas9 ... 26
Figure 9. Représentation schématique d’éditeurs de base de cytosine ... 27
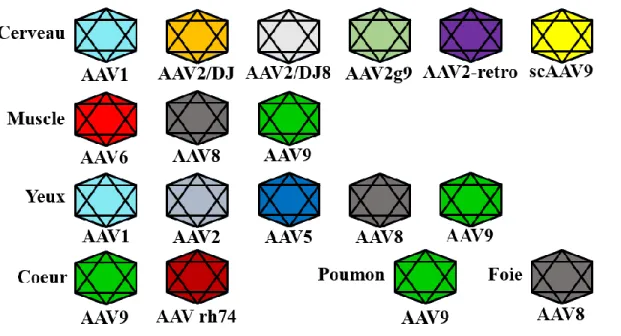
Figure 10. Les sérotypes d’AAV et leurs tropismes ... 34
Figure 11. APP proteolytic pathways. ... 57
Figure 12. Concentrations of Aβ40 and Aβ42 in the neuroblastoma supernatant using the MSD Elisa test. ... 58
Figure 13. Aβ peptide concentrations in culture medium... 59
Figure 14. Variations of Aβ40 and Aβ42 peptide concentrations with A673T mutation ... 60
Figure 15. Aβ42/Aβ40 ratios. ... 61
Figure 16. Aβ peptide concentrations with wild type or London APP gene with or without A673T mutation. ... 62
Figure 17. Structures of BE3, BE4 and Target-AID variants. ... 86
Figure 18. Amino acid modifications produced by base editing of the various cytidines. ... 87
Figure 19. Percentages of cytidine deamination produced by various enzymes and sgRNAs. ... 88
Figure 20. Deamination efficiencies using various Cas9n-deaminases and sgRNAs targeting various numbers of nucleotides. ... 89
Figure 21. Deamination and Aβ peptide quantification in APPWT and SH-APPV717I cell supernatant... 90
Figure 22. Percentages of cytidine deamination produced by various enzymes and sgRNAs. ... 94
Figure 23. Percentages of cytidine deamination produced by various enzymes and sgRNAs. ... 95
Figure 24. Percentages of cytosine deamination produced by BE3_SpCas9nVQR, BE4_SpCas9nEQR, BE3_SpCas9nEQR, BE3_SaCas9nKKH, Target-AID_SpCas9nEQR, Target-AID_SaCas9nKKH enzymes. ... 96
Figure 25. Deamination efficiencies using various Cas9n-deaminases and sgRNAs targeting various numbers of nucleotides. ... 97
Figure 26. Deamination efficiencies using various Cas9n-deaminases and sgRNAs targeting various numbers of nucleotides. ... 98 Figure 27. Off target analysis performed with Benchling.com interface. ... 99 Figure 28: Deep sequencing file analysis scripts. ... 100 Figure 29. Conversion of Alzheimer’s London V717I patient’s and mouse model fibroblasts to induced neurons over time. ... 125 Figure 30. Culture of cells isolated from the hippocampus of a NL/G/F prenatal
mouse. ... 126 Figure 31. Cas9 delivery immunofluorescence results in hippocampal neurons from NL/F/G prenatal mice infected with the lentivirus CBE... 126 Figure 32. Aβ42 peptide concentration of transduced neurons medium. ... 127 Figure 33. Comparison of CBh and EF1 promoters on the expression of Target-AID SpCas9n-VQR protein in HEK293T cells. ... 127 Figure 34. Anti-Cas9 immunofluorescence results in HEK293T cells. ... 128 Figure 35. CBE expression comparison between lentivirus CBE 2U6 and dual AAV AID-BE4max transfected in HEK293T cells. ... 129 Figure 36. Base editing comparison between Target-SpCas9-VQR and AID-BE4max transfected in HEK293T cells. ... 129
Liste des tableaux
Tableau 1. Percentage variations in Aβ40 and Aβ42 peptide concentrations due to the A673T insertion. ... 63 Tableau 2. List of sgRNA used in the study. ... 91 Tableau 3. Example of Deep-Sequencing analysis. ... 92 Tableau 4. Percentage of reduction of A40 and A42 peptides secretion induced by the addition of the A673T mutation to wild type APP gene or to an APP gene
Liste des abréviations, sigles, acronymes
AAV : « adeno-associated virus » ou virus adéno-associés ABE: « Adenosine Base Editor »
AChEI : « Acetylcholinesterase inhibitor » ou inhibiteurs de l'acétylcholinestérase AD : « Alzheimer’s disease »
ADAM: A disintegrin and metalloprotease ADN : acide désoxyribonucléique
Adv : adénovirus
AEP : « asparagine endopeptidase »
AGD : « agyrophilic grain disease » ou la maladie des grains argyrophiles AICD: « APP intracellular domain »
ANOVA: « Analysis of variance »
APBA1: « Amyloid beta precursor protein binding family A member 1 » APBB1: « Amyloid beta precursor protein binding family B member 1 » APH-1 : « Anterior pharynx defective-1 »
APLP1/2 : « APP-like protein ½ » APOE4 : apolipoprotéine isoforme 4
APP : « Amyloid precursor protein » ou précurseur de la protéine Aβ ARN : acide ribonucléique
Ascl1: « Achaete-scute homolog 1 » Aβ : Bêta-amyloïde
BACE1: Beta-site APP cleaving enzyme 1 BACE2: Beta-site APP cleaving enzyme 2 BBB : « Blood-brain barrier »
BCS : « bar code sequence »
BE: « Base Editing » ou édition de base BE3: «Base Editing Version 3 »
BPF : bonnes pratiques de fabrication
Brn2: « brain-specific homeobox/POU domain protein 2 » Cas : CRISPR-associated protein
Cas9 : CRISPR-associated protein Cas9n : Cas9 nickase
CBE : « Cytidine Base Editor »
CDB : « corticobasal degeneration » ou la dégénération corticobasale CjCas9 : Campylobacter jejuni
CMV : cytomegalovirus
Cpf1: « CRISPR from Provotella and Francisella 1 »
CPP : « Cell Penetrating Peptides » ou peptides de transduction
CRISPR: « Clustered Regularly Interspaced Short Palindromic Repeat” CSF ou LCR: « Cerebrospinal fluid »
DBD : « DNA binding domain » ou domaine de liaison de l’ADN dCas9 : « dead Cas9 »
DFTP-17 : la démence fronto-temporale avec syndrome parkinsonien liée au chromosome 17
DMD : Dystrophie Musculaire de Duchenne DMEM: « Dulbecco’s modified Eagle’s medium » DMEM: « Dulbecco′s Modified Eagle′s Medium » DSB : « double strand break » ou coupure double brin EOAD: « Early Onset Alzheimer’s Disease »
EP : électroporation
FAD : « Familial Alzheimer’s disease » FDA: « Food and Drug Administration » FS : Feldan Shuttle
GFP: « Green fluorescent protein »
gRNA (ARNg): « guide RNA » ou ARN guide HC-Ad: « High capacity Adv »
HD : « Huntington Disease » ou maladie d’Huntington
HDR : « Homologous Directed Repair » ou réparation homologue dirigée INDEL : Insertion-Délétion
iNs : « induced neurons »
iPSC: « induced pluripotent stem cells » IRM : l’imagerie à résonnance magnétique
ITR : « Inverted Terminal Repeat » ou répétitions terminales inversées IV : intraveineuse
kDa : kiloDalton
KPI: « Kunitz-type protease inhibitor » LOAD: « Late Onset Alzheimer’s Disease » LTD: « Long-term depression »
LTP: « Long-term potentiation » LV : lentivirus
MA : maladie d'Alzheimer MEC : matrice extra-cellulaire MOI: « multiplicity of infection » MSD: Meso Scale Discovery
MT5-MMP : « membrane-bound matrix metalloproteinases »
NFTs : « Neuroneurofibrillary tangles » ou enchevêtrements neurofibrillaires NHEJ : « Non Homologous End-Joining »
NMDA : N-méthyl-D-aspartique
OX-2: OX-2 antigen of thymus derived lymphoid cells PAGE: « polyacrylamide gel electrophoresis »
Pb : paire de base
PBS : « Phosphate-buffered saline » PBS: « primer binding sequence »
PCR : « polymerisation chain reaction », réaction de polymérisation en chaine pegRNA: « PRIME editing guide RNA »
PEN-2: « Presenilin enhancer 2 » PET: « Positron emission tomography » PiB: « Pittsburg compund B »
PiD : « Pick disease » ou la maladie de Pick PmCDA1 : désaminase de Petromyzon marinus PSEN1 : Préséniline 1
PSEN2 : Préséniline 2
PSP : paralysie supra nucléaire progressive rAAV : « recombinant AAV
REST: RE1-silencing TF
RTT: « reverse transcriptase template » RVD: « Repeat Variable di-Residues » SaCas9 : Cas9 de Staphylococcus aureus SAD: « Sporadic Alzheimer’s disease » sAPPα : APP soluble alpha
sAPPβ : APP soluble bêta
SDS: « sodium dodecyl sulphate » shRNA: « short hairpin RNA »
SIDA : Syndrome de l’Immunodéficience Humaine Acquise SpCas9 : Cas9 de Streptococcus pyogenes
StCas9 : Streptococcus thermophilus TadA : adénosine désaminase mutante
TAHA : traitement antirétroviral hautement actif
TALEN: « Transcription Activator-Like Effector Nucleases » TCL : Troubles cognitifs légers
TF: « transcription factors » THR668 : « Thréonine 668 »
Ubx : homéodomaine de Drosophila Ultrabithorax
UGI : « Uracile Glycosylase Inhibitor » ou inhibiteur de la glycosylase de l’uracile UNG: Uracil N-glycosylase
ZFN : « zinc finger nuclease » ou nucléase à doigts de zinc ZFP : « zinc finger protein » ou protéines à doigts de zinc α-secrétase : Alpha-secrétase
β-secrétase : Beta-secrétase γ-secrétase : Gamma-secrétase δ-secrétase : Delta-secrétase
ε-secrétase : Epsilon-secrétase ζ-secrétase : Zêta-secrétase η-secrétase : Êta-secrétase θ-secrétase : Thêta-secrétase
« Les choses changent. Mais si vite... Est-ce que les habitudes des hommes pourront suivre ? »
Isaac Asimov
« Adventurers beware: do not begin unless you intend to finish. The exciting consequences of the game / will vanish only when a player has reached PhD and called out its name. »
Remerciements
Je commencerais par remercier mille fois le Dr Tremblay, qui m’a engagé dans son laboratoire en 2016 alors que la situation économique du labo était alors incertaine. J’ai pu passer un doctorat dans les meilleures conditions possibles avec une liberté qui m’a permis de développer tous mes projets à mon rythme. Merci pour votre confiance, de m’avoir envoyé à l’étranger pour de supers congrès et d’avoir toujours corrigé mes papiers à la vitesse de l’éclair. Meilleur directeur de recherche de l’univers!
Il ne peut pas y avoir de bonnes ambiances de laboratoire sans collègues de laboratoire! Merci à Benjamin, Joël, Cathy, Khadija, Jean-Paul, Dominique, Nathalie, Daniel, Pouiré, Solange, Malek, Cédric, Chantale, Vanessa, Gabriel, Francis, Guillaume. Remerciement spécial à Gabriel qui a passé de longues heures à corriger mes papiers dans la langue de Shakespeare.
Merci à tous mes stagiaires que j’ai encadré qui ont toujours été agréables et qui m’ont permis de me perfectionner dans le mentorat : Francis, Monica, Simon, Tom, Meredith, Guillaume.
Merci au Dr Serhat, pour tous tes sages conseils et m’avoir ouvert les yeux sur beaucoup de choses pendant mon doctorat.
Merci à ma famille pour les bons moments passés lors de mes retours à chaque Noël. Merci à mes parents pour le soutien et surtout Maman qui a relu ma thèse contentieusement jusqu’à la fin. Merci à Un merci spécial à Isa qui m’a permis de m’installer au Québec sereinement.
Parce qu’on ne le fait jamais assez, je remercie mes amis : Marion, Maxime, Flora, Emma. Maud, Victor, Margaux, Flore, Stéphane, Michael. Vous êtes loin des yeux toute l’année mais toujours près du cœur. Un merci spécial à Alex, qui a sa place particulière.
Parce qu’immigrer c’est aussi refaire sa vie et trouver de nouveaux piliers pour se construire, je veux remercier mes amis du Québec : Anna, Sébastien, Stéphane, Caleb, Pier-Luc, Noémie, Loïc, Lysis, Guillaume, Irène, Marine, Régis.
Et le meilleur pour la fin avec Kévin et sa famille qui m’ont adopté et m’ont vraiment fait découvrir l’essence du Québec. Merci, Kari, Soun et Serge, Isa et Enrico, Mathieu, les enfants et les poules!
Avant-propos
Dans ce manuscrit, le chapitre 4 est un article intitulé « The protective mutation A673T in Amyloid Precursor Protein gene decreases Aβ peptide production for 14 forms of Familial Alzheimer’s Disease in SH-SY5Y cells » qui a été accepté pour publication dans la revue PLOS One le 10 Décembre 2020. En tant que premier auteur, j’ai élaboré et réalisé la majorité des expériences de cette étude ainsi que rédiger le manuscrit, analysé les données et préparer les figures. Dr Jacques P Tremblay a relu et corrigé le manuscrit avec Gabriel Lamothe. Joël Rousseau m’a assisté dans mes expérimentations.
Le chapitre 5 est un deuxième article soumis au journal Molecular Therapy Nucleic Acid et accepté le 25 Février 2021 ayant pour titre « Base editing strategy for insertion of the A673T mutation in the APP gene to prevent the development of AD in vitro ». Je suis également le premier auteur de cette étude. J’ai réalisé la majorité des expériences avec Joël Rousseau ainsi qu’analysé les données et préparé les figures. Francis-Gabriel Bégin et Tom Bertin ont aidé à la construction des plasmides d’éditeur de base CRISPR. Gabriel Lamothe et Jacques P. Tremblay ont révisé le manuscrit.
Le chapitre 6 est un troisième article en préparation ayant pour titre « Optimization of in vitro base editing strategy delivery targeting neurons afflicted with Alzheimer’s disease ». Je suis le premier auteur de ce manuscrit. J’ai également réalisé la majorité des expériences avec Joël Rousseau et j’ai été assisté par le docteur Janelle Drouin-Ouellet pour la conversion des fibroblastes en neurones induits. Gabriel Lamothe et Jacques P. Tremblay ont révisé le manuscrit.
Les trois manuscrits ont été légèrement adaptés pour faire correspondre les numéros de figures avec la numérotation globale de la thèse.
Introduction
Chapitre 1 : La maladie d’Alzheimer
1.1 Au niveau épidémiologique : la prévalence
La maladie d'Alzheimer (MA) a été pour la première fois discutée en 1906 par le psychiatre éponyme Alois Alzheimer dans une conférence de psychiatrie en Allemagne (1, 2). Depuis sa découverte et le perfectionnement des tests diagnostics, on remarque que la MA est en réalité la démence la plus commune et que sa prévalence ne cesse d’augmenter (3). Actuellement, il y a plus de 50 millions de personnes atteintes. En raison de l’augmentation des cas chaque année, on estime qu’il y aura 150 millions de nouveaux patients à l’horizon 2050 (3). Cette maladie se développe très rarement avant 65 ans, mais à partir de cet âge la prévalence double tous les 5 ans (4). En moyenne, on constate que l’âge de déclaration de la maladie est de 80 ans chez l’homme et de 83 ans chez la femme (5, 6). Malheureusement, l’apparition des premiers symptômes engendre un taux de survie de 9 ans en moyenne.
1.2 Au niveau cérébral : l’étiologie
La pathologie principale de la MA est l’atrophie progressive du cerveau due à la mort neuronale du cortex et des ventricules commençant principalement dans l’hippocampe et par la suite dans le cortex (7). Ceci résulte en un agrandissement des ventricules. Après de nombreuses observations, le psychiatre Alzheimer avait listé des critères neurologiques clés pour identifier la maladie. Il décrira aussi par la suite la présence de dépôts de plaques amyloïdes au niveau des neurones et des vaisseaux sanguins (angiopathie amyloïde cérébrale), des enchevêtrements neurofibrillaires (« neurofibrillary tangles » ou NFTs) liés à la protéine tau. Il observe également des traces de glioses, prolifération des cellules gliales faisant office de cicatrice, ainsi qu’une accumulation de lipides intracellulaires (2).
L’apparition des symptômes liés à ces caractéristiques de neurodégénérescence est en général trop tardive pour pouvoir espérer une récupération du patient. Voilà pourquoi de nombreux laboratoires de recherche se spécialisent dans l’élaboration de tests diagnostics précoces, la maladie ayant une phase prodromale et asymptomatique d’environ 20 ans (7).
Sur un plan moléculaire, les deux marqueurs principaux du développement de la maladie admis à ce jour sont d’une part, la protéine bêta-amyloïde (« Amyloid beta » ou Aβ), composante essentielle de la plaque amyloïde s’accumulant à l’extérieur des neurones et d’autre part, la protéine Tau, qui, en étant hyperphosphorylée, forme des NFTs désorganisant les microtubules des synapses. La protéine Aβ est en réalité un peptide qui est un sous-produit du clivage de la protéine précurseur de l’amyloïde (« Amyloid precursor protein » ou APP), une protéine membranaire neuronale.
Malgré leur implication significative dans la MA, ces deux molécules ne parviennent pas encore à expliquer formellement tous les symptômes de la maladie ; c’est pourquoi la MA est aujourd’hui considérée comme un désordre neurodégénératif multifactoriel (8).
1.3 Les différents stades
La maladie d'Alzheimer a été décrite suivant trois stades d’évolution. Le premier stade dit pré-symptomatique, suivi du stade prodromal qui présente des symptômes bénins et, enfin, le stade symptomatique incluant la perte de mémoire et la démence (9, 10). Les troubles épisodiques de la mémoire surviennent en premier et s’ensuivent des troubles du langage et de la coordination (dysphasie/dyspraxie). Puis, le patient ressent le déclin musculaire et une perte importante de ses facultés cognitives. La progression des symptômes correspond à celle de l’atrophie corticale. La première zone touchée est celle avoisinant l’hippocampe, le siège de la mémoire, plus précisément le cortex transenthorinal, suivi du cortex enthorinal et enfin l’hippocampe. L’atrophie s’étend ensuite au cortex temporal et préfrontal pour ensuite se généraliser. Le cortex visuel primaire est parfois épargné (11-14).
Figure 1. Suivi de l’atrophie corticale dans le développement de la Maladie d’Alzheimer.
Illustration de la propagation de l’atrophie aux différents stades de la maladie. Image produite par Antoine Guyon modifiée de Braak & Braak,1991.
Les troubles cognitifs légers (TCL) peuvent affecter la mémoire des individus touchés sans pour autant nuire à leurs activités quotidiennes (15). Ces TCL pourraient être des précurseurs de la MA. Certaines études démontrent qu’un patient atteint de TCL a environ a une chance sur trois de développer la MA (16-18).
1.4 Au niveau psychologique : La mémoire
La perte de la mémoire est le problème qui cause le plus de tort aux patients touchés par la MA et à leurs proches. La mémoire se divise en plusieurs catégories : la mémoire sensorielle, automatique et instantanée, qui entre en jeu lorsqu’on utilise nos sens.
La mémoire à court terme est utilisée lorsqu’on effectue des tâches requérant de l’attention, comme lire ou conduire une voiture par exemple. La mémoire à long terme est la plus importante car elle conserve les souvenirs récents encore fragiles et ceux de longues dates. Elle peut être divisée en deux parties selon les informations stockées : la mémoire explicite ou déclarative nécessite une volonté pour récupérer le souvenir voulu. Cette mémoire se divise d’une part en mémoire épisodique qui est liée aux évènements vécus et d’autre part, en mémoire sémantique, qui conserve tous les mots ou les notions apprises ainsi que la perception du monde. La deuxième partie de la mémoire à long terme est la mémoire implicite ou non-déclarative qui ne peut pas être explorée consciemment. Elle réunit toutes les capacités de l’individu ou ses conditionnements émotionnels sans pour autant se rappeler de l’expérience qui leur est associée (comme par exemple savoir lacer ses chaussures) (19, 20).
Deux mécanismes très importants sont impliqués dans la mémorisation ou dans l’oubli d’un souvenir impliqué dans la mémoire explicite. La potentialisation synaptique à long terme (« Long-term synaptic potentiation » ou LTP) est un renforcement des synapses du lobe temporal médial et de la plasticité synaptique de l’hippocampe qui accroit la capacité d’apprentissage et la fixation d’un souvenir. Son phénomène opposé est appelé dépression
synaptique à long terme (« Long-term synaptic depression » ou LTD) (20, 21). La MA, favorisant la LTD et perturbant la LTP en modifiant la distribution du glutamate par exemple, touche en premier la zone de l’hippocampe et explique ainsi pourquoi les mémoires épisodique et sémantique sont les premières touchées par la dégénérescence (14, 22-26). C’est pour cela qu’un malade va d’abord oublier un évènement particulier ou certains mots avant d’oublier comment changer une ampoule par exemple.
1.5 Les différentes formes de la maladie d’Alzheimer
La MA existe sous deux formes : la forme sporadique (« Sporadic Alzheimer’s Disease » ou SAD) également appelée forme tardive (« Late Onset Alzheimer’s Disease » ou LOAD) et la forme familiale (« Familial Alzheimer’s Disease » ou FAD) ou encore forme précoce (« Early Onset Alzheimer’s Disease » ou EOAD. Les FAD sont rares (environ 2% de la population). Un patient atteint d’une FAD va développer des symptômes similaires à une SAD mais bien avant 65 ans, pouvant même débuter vers 30 ans (6, 27). Cette neurodégénérescence accrue et assurée vient du fait que la maladie est provoquée par une mutation héréditaire autosomale dominante dans l’un des trois gènes clés de la MA identifiés à ce jour (27). Les trois gènes sont : l’APP, localisé dans le chromosome 21, la préséniline 1 (PSEN1) dans le chromosome 14 et la préséniline 2 (PSEN2) situé dans le chromosome 1. Il existe 68 mutations FAD répertoriées dans le gène APP à ce jour, 322 pour la PSEN1 et 64 pour PSEN2 (28). Toutes ces mutations ne sont pas forcément pathogéniques. Pour prendre l’exemple d’APP, seules 28 des 68 mutations ont été confirmées pathogéniques et elles sont toutes localisées entre les exons 16 et 17 du gène où sont situés les sites de clivages de la protéine. La majorité des FAD d’APP sont nommées d’après l’endroit où elles ont été mises en évidence, suivi du code de l’acide aminé modifié, sa position dans le gène et l’acide aminé obtenu (ex : mutation London V717I, première FAD découverte dans le gène APP) (29, 30). Les FAD ont généralement pour effet d’augmenter la production du peptide Aβ ou de favoriser son agrégation en plaque, ce qui expliquerait le développement précoce (27, 31-33). Contre toute attente, une des variantes du gène APP, la mutation Islandaise A673T, s’est révélée provoquer l’effet inverse et apporter un effet protecteur à son porteur (34).
La majorité des cas de MA sont donc sporadiques. Bien que les mécanismes de la maladie n’aient pas encore été élucidés, les chercheurs ont tout de même réussi à identifier la plupart des facteurs de risque pouvant favoriser l’apparition de la maladie.
1.6 Les facteurs de risques
1.6.1 Les facteurs contrôlablesLa MA peut être favorisée par les conditions suivantes : une forte pression artérielle, le tabagisme, le diabète, un taux de cholestérol augmenté, une forte consommation d’alcool, un manque de stimulation cérébrale, la dépression, une blessure à la tête, etc. (35)
1.6.2 Les facteurs incontrôlables
Un des facteurs de risques que l’on ne peut pas éviter est l’âge, comme le montre l’augmentation de la prévalence après l’âge de 65 ans. Le sexe a également un impact car la diminution d’œstrogène liée à la ménopause augmente également les risques (6, 36).
Le facteur de risque génétique principal est de posséder l’isoforme 4 de l’apolipoprotéine (ApoE4) (37, 38). Cette protéine du chromosome 19 est impliquée dans le transport du cholestérol, notamment entre les vaisseaux sanguins et les neurones. L’isoforme 4 de cette protéine diminue son efficacité. La protéine possède 4 isoformes ApoE1,2,3 et 4 et est produite principalement dans les astrocytes (39). Le lien avec la MA serait que ApoE jouerait également un rôle dans l’évacuation du peptide Aβ vers le système sanguin (40).
D’autres facteurs de risques génétiques sont dus à des mutations d‘autres gènes. On peut noter le gène SORL1, impliqué dans la production de l’amyloïde ; TREM2, qui est un récepteur microgliale de l’Aβ et ABCA7, impliqué dans le transport membranaire dans le système nerveux central (41-43).
1.7 Au niveau moléculaire :
1.7.1 Le précurseur de la protéine amyloïde (APP)
La protéine APP a été identifiée comme la source de l’Aβ en 1984 (44). Son gène est localisé dans le chromosome 21 qui est transcrit dans tout le corps mais n’est exprimé en protéine que dans le cerveau et moindrement dans le foie, le pancréas, le tractus gastro-intestinal et les tissus reproductifs masculins (45). Cette protéine se retrouve
remarquablement conservée chez tous les mammifères (46). La fonction d’APP n’est pas complètement établie mais pourrait être impliquée dans beaucoup de voies métaboliques dont, entre autres, la synaptogénèse, la prolifération cellulaire, la différenciation cellulaire, le développement des neurites et l’adhésion cellulaire (47). C’est une protéine transmembranaire de type I localisée près des synapses et appartenant à la famille des « APP-like 1 and 2 » (APLP1 et APLP2) qui sont impliqués dans l’homéostasie du glucose et de l’insuline (48, 49). L’APP subit des épissages alternatifs car elle existe sous trois isoformes dont les deux minoritaires expriment des domaines inhibiteurs de protéases et de reconnaissance cellulaire appelés respectivement domaine KPI (« Kunitz-type protease inhibitor ») et OX-2 (antigène lymphoïde). L’isoforme majoritaire APP695 qui est principalement trouvée dans le cerveau ne contient aucun des deux domaines (50). L’APP est une protéine fréquemment clivée par différentes sécrétases donnant lieu à deux voies qu’on appelle non-amyloïdogénique ou amyloïdogénique (Figure 2).
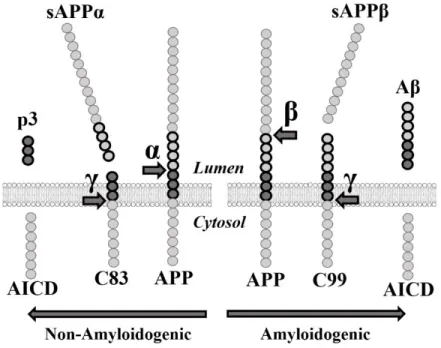
Figure 2. Métabolisme protéolytique de la protéine APP
Profil des différents clivages. Image produite par Antoine Guyon et tirée de Guyon et al. 2020. AICD : « APP intracellular domain » ; APP : précurseur de la protéine amyloïde ; α : alpha-secrétase ; β : bêta-secrétase ; γ : gamma-secrétase ; Aβ : bêta-amyloïde ; C83 : «
C-terminal fragment alpha » ou CTFα ; C99 : « C-C-terminal fragment bêta » ou CTFβ ; sAPPα : APP soluble alpha ; sAPPβ : APP soluble bêta.
Le clivage non-amyloïdogénique débuté par l’α-sécrétase à la surface de la cellule est la voie majoritaire (51). Le clivage par l’α-sécrétase est suivi par celui de la γ-sécrétase, ce qui va libérer au final trois peptides : le peptide APP soluble alpha (sAPPα), le P3 et le domaine intracellulaire de l’APP (AICD). La voie amyloïdogénique en revanche, initiée par la bêta-sécrétase (« Beta-site APP cleaving enzyme 1 » ou BACE1) va libérer le peptide Aβ ainsi que le sAPPβ et l’AICD.
Si on fait abstraction d’Aβ, les peptides sécrétés ont leur importance. sAPPα et sAPPβ seraient impliqués dans la croissance cellulaire, le développement neural, la formation synaptique, la survie neuronale et le rétablissement cellulaire (49, 52, 53). Pendant longtemps, on a pensé que P3 ne jouait pas de rôle particulier mais des études ont finalement montré qu’il était retrouvé dans des plaques amyloïdes diffuses de patients atteints de trisomie 21 et qu’il renforcerait l’agrégation d’Aβ (54, 55). Le rôle de l’AICD est resté flou durant des années avec des études contradictoires sur son effet bénéfique ou non. Il contient huit sites de phosphorylation parmi lesquels celui de la thréonine 668 (Thr668) particulièrement étudié pour les interactions qu’il pourrait créer avec l’APP et d’autres protéines. En effet, la phosphorylation du site Thr688 rendrait l’APP moins vulnérable au clivage par la caspase 18 et diminuerait la toxicité intracellulaire (56). Cependant, une autre étude démontrait que Thr668 favoriserait la voie amyloïdogénique en interagissant avec APP (57). Finalement, ce n’est que cette année en 2020 qu’une équipe a démontré que le rôle d’AICD est, en réalité le maintien l’homéostasie lors du développement du cerveau via une interaction avec le facteur de transcription FOXO3a (58).
1.7.2 L’alpha-secrétase
Cette enzyme importante dans le clivage de l’APP appartient à une famille d’enzymes protéolytiques transmembranaires appelée ADAM (« A disintegrin and metalloprotease domain »). Plusieurs des enzymes de cette famille ont une activité α-sécrétase et notamment ADAM10 (59). Elle clive APP entre les résidus Lys17 et Leu17 dans la zone correspondant au peptide Aβ et ce clivage s’effectue très rapidement lorsque APP atteint la surface de la
cellule (60). Ceci pourrait expliquer que la voie non-amyloïdogénique soit favorisée. L’α-sécrétase est impliquée dans plusieurs voies métaboliques dont la régulation des contacts entre cellules et entre les cellules et la matrice extra-cellulaire (MEC), ainsi que dans toutes les voies reliées à la sécrétion du sAPPα (49, 61). La famille de protéine ADAM a surtout pour vocation de cliver des protéines dans le but de les activer comme des protéines d’adhésion ou des facteurs de croissance (61).
1.7.3 La bêta-secrétase
L’activité β-sécrétase est presque entièrement assurée par BACE1 (62). L’enzyme possède deux sites de clivage bêta : entre Met671 et Asp672 ainsi qu’entre Thr681 et Gln682, produisant respectivement C99 et C89 (62, 63). Le C89 n’est pas présenté dans la Figure 2 car il est minoritaire chez l’humain. Toutefois, une étude montre que ce clivage β’ dix acides aminés plus bas que le site original libère un peptide beaucoup moins toxique pour les cellules (63). Une mutation chez la souris dans son gène APP H684R favorise justement la coupure de BACE1 au site β’ et expliquerait pourquoi les souris ne développent jamais la MA naturellement (63). Chez l’homme, c’est la mutation islandaise A673T qui favorise la coupure en β’ (63). BACE1 est une protéase aspartique et fonctionne donc mieux en milieu acide (pH 4.5), ce qui ne correspond pas au pH extracellulaire mais plutôt à celui des endosomes. Il a été démontré que la majorité de son activité se déroulait dans ces structures (51, 64). Le gène de BACE1 est exprimé majoritairement dans le cerveau et le pancréas mais est aussi exprimé dans pratiquement tous les tissus (65). On ne connait pas de mutation dans la BACE1 impliquée avec une aggravation ou une amélioration de la MA.
On sait cependant que APP n’est pas son seul substrat. BACE1 est également impliqué dans la transmission synaptique et l’endocytose (66-69).
La fonction de BACE2, qui a 59% d’homologie avec la BACE1, n’est toujours pas formellement déterminée. Les dernières recherches l’impliquent dans la pigmentation de la peau via une interaction avec les mélanocytes et des polymorphismes de cette enzyme pourraient être de nouveaux facteurs de risque pour la MA (70, 71). BACE2 serait également capable de cliver l’APP deux résidus plus bas que l’α-sécrétase en Phe19 pour former le fragment C80. BACE2 a été renommée theta-sécrétase (θ) (72).
1.7.4 La gamma-secrétase
La γ-sécrétase, localisée dans la membrane cellulaire et dans les endosomes/lysosomes, est composée d’un complexe protéique regroupant principalement PSEN1 et PSEN2, la Nicastrine (Nct), l’APH-1 (« Anterior pharynx defective-1 ») et PEN-2 (« Presinilin enhancer PEN-2 ») (51, 73-75). La PSEN1 et la PSENPEN-2 contiendraient le domaine catalytique de la γ-sécrétase grâce à deux aspartates dans le domaine transmembranaire, ce qui explique l’importance de ces deux protéines dans la MA (76). Capable de coupures de tailles multiples, la γ-sécrétase relâche in fine des peptides Aβ variant de 34 à 50 acides aminés, la majorité des peptides sécrétés contenant 40 et 42 acides aminés (49, 77). Outre l’APP, l’enzyme possède également d’autres substrats tels que Notch1, ErbB4, E-cadhérine et N-cadhérine ainsi que CD44 (78-80).
1.7.5 Des clivages particuliers de l’APP
De nouvelles activités sécrétases, impliquant APP, ont été découvertes au fil des années. Une activité delta-sécrétase a été mise en évidence chez une endopeptidase d’asparagine (AEP) (81). Capable de cliver APP aux résidus N373 et N585 en N-terminal du site β, elle serait un facteur aggravant de la MA en facilitant la coupure par le clivage de la β-sécrétase (81). L’abolition de AEP dans un modèle de souris 5xFAD provoquerait une nette diminution de la génération d’Aβ et permettrait à la souris de recouvrer ses capacités cognitives (81). Cette sécrétase coupe également Tau en deux endroits (N255 et N368) et favoriserait l’agrégation et l’hyperphosphorylation. Une déplétion du gène AEP dans un modèle de souris Tau P301S restaure les capacités cognitives et diminue les NFTs (82).
Des nouveaux sites de clivage zêta (ζ) et epsilon (ε) ont également été observés dans la région zonale de la γ-sécrétase (83). Les clivages aux différents sites, tous réalisés par la γ-sécrétase, forment respectivement les peptides Aβ40/42, 46 et 49 (83). L’étude démontre que la formation C-terminale du peptide Aβ serait séquentielle, en commençant par un clivage epsilon, zêta, puis gamma (83).
Une eta-sécrétase (η) a également été mise en évidence, coupant l’APP entre le résidu N504 et N505 (84). Cette enzyme appelée MT5-MMP (« membrane-bound matrix
metalloproteinases ») serait pro-amyloïdogénique car son fragment Aη-α sécrété suite au clivage Eta/Alpha inhiberait fortement la potentialisation synaptique à long terme LTP (84).
Les activités sécrétases et les sites de clivages sont résumés dans la Figure 3. Finalement, il semblerait que l’APP soit également clivé par certaines caspases, ce qui pourrait avoir un effet amyloïdogénique (51, 85).
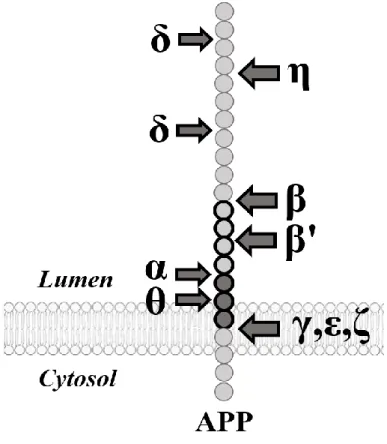
Figure 3. Clivages globaux de la protéine APP.
Description de tous les sites de clivages connus de l’APP. Image produite par Antoine Guyon. APP : précurseur de la protéine amyloïde ; α : alpha-secrétase ; β : bêta-secrétase ou BACE1 ; β’ : site de clivage bêta prime ; γ : gamma-secrétase ; δ : delta-secrétase ; ε : site de clivage epsilon ; ζ : site de clivage zêta ; η : eta-secrétase ; θ : théta-secrétase ou BACE2.
1.7.6 Du peptide Aβ à la plaque amyloïde
La majorité des peptides Aβ sécrétés dans le milieu extracellulaire sont en majorité des peptides Aβ40 suivis par les Aβ42 (77). Même si les Aβ40 représentent 90% des peptides totaux formés, les plaques amyloïdes sont pourtant constituées en majorité de peptides Aβ42
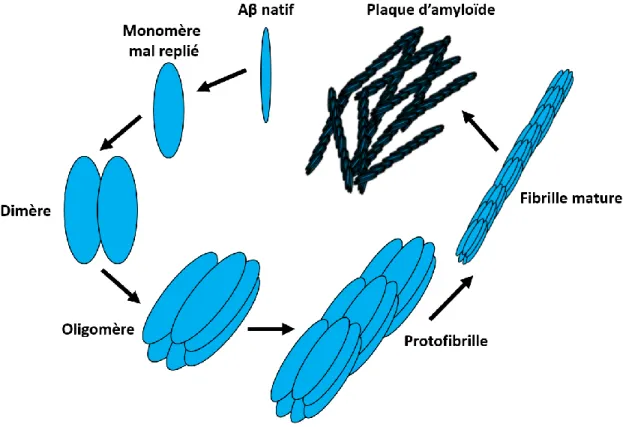
à cause de plus grande propension à s’agréger (86, 87). Les plaques formées existent sous forme diffuses ou séniles. Les plaques diffusent, comme leur nom l’indique, ne sont pas compactes et tout indique qu’elles sont les précurseurs des plaques séniles repliées en feuillet-bêta (88). Les plaques séniles sont en général entourées par des neurites dystrophiques (88, 89). Cependant, il existe plusieurs intermédiaires solubles entre le peptide Aβ fraichement clivé et la plaque diffuse. Le peptide est capable de se replier en monomères et de s’assembler en dimères, oligomères, protofibrilles, fibrilles matures et en plaques (Figure 4).
Figure 4. Phase d’agrégation du peptide bêta.
Illustration de l’agrégation amyloïde. Image produite par Antoine Guyon,
Tous ces éléments ont démontré de la toxicité in vitro dans des modèles de souris (25, 90). Chez des patients autopsiés, il a été démontré que la concentration d’oligomères Aβ était plus corrélée à la sévérité de la maladie qu’à la quantité de plaques séniles (91-94). Ces éléments Aβ solubles sont connus pour inhiber la LTP et la signalisation calcique dans les synapses (95-97). Des cellules environnantes, les microglies notamment sont considérées comme un autre acteur majeur de la formation des plaques qui, dans un souci de protection, vont encercler les plaques diffuses et joueront ainsi un rôle dans leur compaction (98). Il faut
se rappeler que le gène TREM2, qui est un récepteur amyloïde, est un facteur de risque s’il est muté car il empêche les microglies d’évacuer les peptides Aβ (42).
1.7.7 La protéine Tau
La protéine Tau a été découverte en 1986 et identifiée comme la source des NFTs (99, 100). Bien qu’étant directement liée à la MA, aucune mutation de la protéine Tau n’a encore été démontrée comme responsable d’une FAD (6). Cette protéine dont le gène MAPT se trouve dans le chromosome 17 existe sous 6 isoformes présents dans le cerveau (6). Son rôle est clé dans l’assemblage des microtubules composant le cytosquelette des axones (101). En effet, Tau joue un rôle de maintien des polymères de tubulines ainsi que dans leur élongation (102-104). Une des propriétés principales de Tau est son grand nombre de sites de phosphorylation s’élevant à 85. Semblant joué un rôle dans le développement embryonnaire du cerveau, l’hyperphosphorylation de la protéine Tau dans des neurones matures s’avère extrêmement délétère (101, 105). En effet, ce phénomène a pour résultat de la dissocier des microtubules, ce qui aura pour effet de disloquer complètement la synapse et de stopper l’influx nerveux. La protéine va former des pelotes dans la région somatodendritique et des NFTs.
Une des capacités malheureuses de cette protéine est de pouvoir propager la désorganisation microtubulaire aux cellules voisines via les axones. C’est un comportement de protéine prion (106). Une étude montre que cette capacité est plus ou moins développée selon l’isoforme de Tau concerné (107). Les maladies liées à la protéine Tau et au NFTs, appelées tauopathies, sont nombreuses. Les plus connues sont la MA, la paralysie supra nucléaire progressive (PSP), la dégénération corticobasale (« corticobasal degeneration » ou CDB), la maladie des grains argyrophiles (« agyrophilic grain disease » ou AGD), la maladie de Pick (« Pick disease » ou PiD), la maladie d’Huntington (HD), et la démence fronto-temporale avec syndrome parkinsonien liée au chromosome 17 (DFTP-17) (108).
1.8 Les modèles de souris transgéniques de la maladie d’Alzheimer
Les modèles de souris sont un élément capital de la recherche en général, mais d’autant plus pour la MA, les modèles reproduisant l’environnement neuronal étant
quasiment impossible à reproduire in vitro. Les souris ne peuvent pas développer naturellement la MA, probablement, grâce à leur mutation H684R (63) ; c’est pourquoi les chercheurs ont dû créer des lignées transgéniques possédant des copies humanisées des gènes clés. Les premiers modèles se basaient sur les mutations provoquant des FAD (109). Il y a aujourd’hui plus de 200 modèles de souris modèle pour la MA, disponibles avec une grande variété de gènes insérés tels que APP, MAPT, BACE1, APOE, PSEN1, etc. (110). Il est possible de choisir son modèle en fonction de la caractéristique que l’on veut observer comme, par exemple, le dépôt de plaque, les NFTs, la gliose, les changements LTP/LTD ou la perte cognitive. Certaines souris sont meilleures que d’autres pour exprimer des symptômes de façon précoce, ce qui économise du temps et permet de savoir rapidement si la thérapie fonctionne par exemple (111). Il existe beaucoup de modèles qui cumulent les FADs pour tenter d’aggraver la maladie le plus vite possible comme les modèles APPPS1 (APP KM670/671NL (Swedish), PSEN1 L166P), 5xFAD (APP KM670/671NL (Swedish), APP I716V (Florida), APP V717I (London), PSEN1 M146L (A>C), PSEN1 L286V) ou encore NL/F/G (APP KM670/671NL (Swedish), APP I716F (Iberian), APP E693G (Arctic)) (112-114).
Un fait intriguant observé dans les modèles APP+PSEN1 est une hyperphosphorylation du Tau murin ne menant jamais à la formation de NFTs, ; ce qui a contraint les chercheurs à intégrer une copie du gène MAPT humain pour pouvoir les observer (38).
1.9 Les différentes hypothèses
1.9.1 Théorie de la cascade d’amyloïdeLe fait que le peptide Aβ se voit débalancé par toutes les mutations FAD d’APP, PSEN1 ou PSEN2 qui a fait émettre l’hypothèse à des chercheurs qu’il pourrait être à la source de la MA (6, 44, 115). L’exemple le plus révélateur est cette étude qui a démontré qu’il suffisait d’être porteur de la mutation protectrice islandaise A673T pour ne jamais développer la maladie (34, 38).
Il y a toujours eu un débat sur cette hypothèse sachant que les plaques amyloïdes avaient toujours été observées avec des NFTs, et il était bien difficile d’estimer lequel était
le précurseur. L’hypothèse est qu’un individu sain métabolisera toute sa vie le peptide Aβ de façon optimale et qu’il n’en accumulera jamais ou très peu dans son cerveau. Un malade Alzheimer en revanche, présentera un problème dans la production ou l’élimination de ce peptide et ce déséquilibre mènera au développement de la MA (115). L’accumulation des peptides Aβ prendrait forme de dépôts observables, créant un stress cellulaire dû à l’inflammation déclenchée par les astrocytes et microglies qui produiraient une grande quantité de radicaux libres. Ce phénomène altèrerait les phosphatases et kinases, provoquant l’hyperphosphorylation de la protéine Tau et donc des NFTs. Ce changement désorganiserait les synapses et induirait l’inflammation généralisée et la dégénérescence. Cette neuro-dégénération provoquerait alors des troubles cognitifs et mémoriels (38, 116).
Cette hypothèse est soutenue par plusieurs faits solides, comme, par exemple, le fait qu’il n’existe aucune FAD liée au gène de la protéine Tau (6). Une autre observation vient des individus touchés par la Trisomie 21 (syndrome de Down). Possédant trois chromosomes 21, ils ont également trois copies du gène APP. Bien qu’étant non altéré par aucune mutation, les personnes trisomiques vont développer à coup sûr des pathologies très semblables à la MA. L’autopsie de jeunes patients trisomiques a démontré une accumulation précoce de plaques diffuses dans leur cerveau, et ce, dès la fin de l’adolescence, certaines plaques ayant même été détectées chez un enfant de 8 ans (38, 117). L’absence de NFTs ou d’inflammation dans leur cerveau appuie le fait que les plaques se forment les premières et que la surexpression du gène APP suffit à provoquer la MA.
Les études sur modèles cellulaires apportent aussi de nombreux arguments à la théorie de l’amyloïde. L’apparition des modèles de souris Alzheimer et des cellules souches pluripotentes induites (« induced pluripotent stem-cell » ou iPSC) a permis d’obtenir de très bons modèles d’études de neurones. Plusieurs expériences marquantes ont été réalisées. Des neurones de souris exprimant Tau humaine in vitro ont été supplémentés avec des oligomères d'Aβ humain, ce qui a provoqué l’hyperphosphorylation de Tau (118). Si, par contre, des anticorps anti-Aβ sont incorporés en même temps, le phénomène ne se produit jamais (119). Avec les neurones issus d’iPSC ou avec les neurones induits (« Induced Neurons » ou iNs) de patients, l’observation des phénotypes montrent que l’accumulation d’Aβ précède la formation de NFTs (38, 120, 121).
Il y a pourtant un élément important qui vient contrer cette théorie. En effet, les travaux de Braak décrivant la progression de la maladie fait corréler la propagation de l’atrophie cérébrale avec la propagation des NFTs, et non pas celle de la plaque amyloïde (11-13). Il est vrai que cette échelle très largement utilisée montre que les plaques ne suivent pas la dégénérescence. Cependant, il est important de se rappeler qu’aujourd’hui il est plutôt démontré que ce sont les oligomères d’Aβ solubles qui sont liés à la toxicité et qu’ils suivent également parfaitement l’atrophie neuronale (38, 92, 109). Un exemple intéressant est la fameuse « étude des nonnes » effectuée dans les années 1980 dans un couvent aux Etats-Unis (122). L’étude avait démontré que certaines nonnes présentaient des signes de démence mais ne présentaient pas de dépôt de plaques problématiques alors que d’autres en avaient de façon bien plus importante et gardaient néanmoins une capacité cognitive normale.
1.9.2 Hypothèse revisitée : la maladie Alzheimer serait initiée par Aβ mais toxique de façon Tau dépendante – L’axe de Tau
Il semblerait que Aβ et Tau travaillent en binôme dans la MA. En effet, une inhibition de la phosphorylation de Tau causé par GSK3 empêcherait la mort cellulaire même si les cellules étaient supplémentées en Aβ (123). Des neurones primaires de souris en culture dépourvus de Tau (Tau -/-) ne subiront aucun déficit induit normalement par l’Aβ (101, 124). Cette expérience a été reproduite avec succès in vivo (125). On peut donc se demander quel élément de Tau donnerait sa toxicité à Aβ. Ittner et Götz ont formulé une théorie appelée l’axe de Tau (101). La toxicité d’Aβ serait déclenchée par l’hyperphosphorylation de la protéine Tau, avant son agrégation en NFTs. Tau interagirait avec une protéine kinase Fyn pouvant phosphoryler une sous-unité du récepteur NMDA (N-méthyl-D-aspartique) en changeant sa localisation cellulaire vers la partie somatodendritique (126). Une fois transloquée, Fyn phosphoryle le récepteur NMDA, qui est hyper-stimulé et augmente donc la signalisation glutamate. Il est connu que cette action augmente la toxicité neuronale et les peptides Aβ s’accumulant aux jonctions synaptiques, leur effet cumulé à l’activité glutamate créerait la toxicité (101, 127-129).
Plusieurs autres hypothèses viennent rajouter des pièces à ce qui pourrait être une hypothèse globale de l’apparition de la MA. Toutes ces théories avancent des arguments sur le fait qu’un facteur activerait la toxicité de Aβ, mais tous sont d’avis que le peptide reste le
noyau du problème. On trouve l’hypothèse cholinergique (130), l’hypothèse de la neuro-inflammation (131), l’hypothèse du calcium (132, 133), l’hypothèse du stress oxydatif ou de la cascade mitochondriale (134), l’hypothèse métabolique (135, 136) ou la très récente hypothèse lysosomale (137).
Toutes ces hypothèses pourraient très bien n’être que des pièces du tableau général qui expliquerait le mécanisme complet de la MA. Il ne faut pas oublier qu’étant multifactorielles, toutes ces hypothèses suppositions ou interprétations sont peut-être justes.
1.10 Traitements pour la maladie d’Alzheimer
1.10.1 Diagnostic : les biomarqueursDes tests ont été développés ces dernières années permettant de diagnostiquer une évolution de la MA de façon non invasive et même pré-symptomatique (9).
Ces biomarqueurs étudiés sont divisés en deux catégories : l’étude de l’accumulation Aβ et des marqueurs de neurodégénérescence.
La première catégorie de marqueurs est la détection des dépôts Aβ dans le liquide céphalorachidien (« Cerebrospinal fluid » ou CSF), liquide dans lequel baigne notre cerveau qui peut être extrait via une ponction lombaire de la moëlle épinière. En effet, une diminution de concentration du peptide Aβ42 et plus particulièrement le ratio entre le peptide Aβ42/ Aβ40 est un signe de progression de la maladie. L’hypothèse derrière cette diminution serait que le peptide Aβ42 commencerait à être séquestré dans le cerveau et ce peptide serait plus difficilement détectable dû à son agrégation (9). On peut également réaliser une tomographie par émission de positrons contre l’amyloïde (« positron emission tomography » ou PET), utilisant le composant B de Pittsburg (« Pittsburg Coumpound B » ou PiB) (9, 138). Ce composant est injecté par voie sanguine et va pouvoir traverser la barrière hémato-encéphalique pour se fixer aux fibrilles Aβ, et les rendre visualisables par imagerie (139). Ces deux biomarqueurs ont besoin d’être validés pour permettre un diagnostic d’Alzheimer.
La deuxième catégorie de marqueurs vise à déterminer si le cerveau du patient commence à dégénérer. Pour cela, on peut quantifier la protéine Tau dans le CSF de la même manière que pour Aβ. Une concentration de Tau qui augmente est un indice de dégénérescence mais pas forcément de la MA (140). On peut également utiliser une version du PET, le PET fluodeoxyglucose 18F ou FDG-PET, qui va quantifier la consommation du
glucose cérébral, donnant un bon indice des régions qui pourraient ou qui ont commencé à s’atrophier en cas de diminution d’absorption (9, 139, 141). On dispose aussi de l’imagerie à résonnance magnétique (IRM) pour observer le volume du cerveau et là aussi pour détecter une atrophie.
Des tests sanguins se développent de plus en plus, certains étant même capables de détecter des variations de l’Aβ plasmatique jusqu’à 20 ans avant l’apparition des symptômes (142, 143).
On peut se demander quel avantage il y a à détecter la progression d’une maladie qui ne possède pas de traitement connu. Les avantages sont qu’un patient avisé tôt aurait l’occasion de faire certains choix de vie qui pourraient diminuer partiellement la progression de la maladie (régime alimentaire, pratique cognitive, etc.). Il pourrait également être plus enclin à rejoindre une étude clinique proposant un traitement novateur. Les médicaments présentés dans la partie suivante sont tous plus efficaces à des stades précoces de la maladie, ce qui améliorerait de façon notable la qualité de vie. Un diagnostic dès la naissance peut être réalisé pour les personnes touchées par des FAD via un séquençage de leur génome.
1.10.2 Traitements actuels
Il n’existe aujourd’hui aucun traitement curatif pour la MA disponible sur le marché. Les seuls médicaments distribués, pour usage oral, ciblent des voies métaboliques secondaires voire tertiaires des causes de la MA. Ainsi, elles ne font qu’apporter un confort relatif au patient traité.
Un des très grands critères de la maladie est le manque d’acétylcholine présent chez les patients et l’hyperstimulation de leur récepteur NMDA (144). C’est pourquoi sur quatre médicaments disponibles, trois sont des inhibiteurs de l'acétylcholinestérase (« Acetylcholinesterase inhibitor » ou AChEI) qui éliminent l’acétylcholine et le dernier est un antagoniste des récepteurs NMDA (8). Au Canada, les quatre médicaments disponibles sont les AChEI Donepezil (AriceptMC), Galantamine (ReminyMC), Rivastigmine (ExelonMC) et l’inhibiteur des récepteurs NMDA Memantine (ExibaMD) (145). Ils sont remboursés au
Québec par la RAMQ à titre de médicaments d’exception, c’est-à-dire qu’il faut avoir un diagnostic bien établi pour y avoir accès. Ces médicaments ont cessé d’être remboursés en France en 2016 car aucune étude n’a su démontrer une réelle efficacité probante des quatre
molécules (39, 146). Une nouvelle qui a bien sûr attristé de nombreux patients et leurs familles qui, en dépit du peu d’efficacité et des nombreux effets secondaires, estimaient que ces médicaments pouvaient leur apporter une aide non négligeable (39, 144).
Parmi les autres angles d’approche de développement d’un traitement pour Alzheimer, on peut citer le premier « vaccin » historique contre l’Aβ en 1996 (147). L’idée était de présenter le peptide Aβ comme un antigène, poussant le système immunitaire à attaquer les dépôts dans le cerveau. Cette approche avait fonctionné in vivo sur des modèles de souris, mais avait également mis en lumière l’importance de traiter précocement, le traitement ne réagissant pas sur les plaques séniles (148, 149). L’étude clinique du vaccin AN1792 débutée en Décembre 1999 a rapidement été interrompue en 2002 car 6% des patients développaient des méningo-encéphalites dues à la réaction immunitaire violente contre l’Aβ cérébral (150). Les compagnies pharmaceutiques ont longtemps voulu inhiber la coupure d’APP par la protéine BACE1 en développant des inhibiteurs mais aucun des essais cliniques n’a été concluant, car inhiber BACE1 déclenche des effets adverses, étant donné son implication dans plusieurs voies métaboliques (67, 151).
Actuellement, il y a 132 études cliniques en cours qui testent pour la majorité des petites molécules ou des anticorps monoclonaux (152). La majorité de ces études teste l’efficacité de petites molécules sur des voies métaboliques impliquées dans la MA. Sur les 185 études menées au total, 69 sont déjà interrompues, 20 sont inactives, mais 58 sont au moins en phase II ou III. Les chercheurs n’arrêtent pas d’innover car 19 études ont débuté récemment et sont en phase I (153).
Il y a eu 43 études cliniques mises en œuvre en immunothérapie contre la MA. La majorité des anticorps développés ciblent l’Aβ sous différentes formes. Malgré tous les communiqués sur des études interrompues, on n’en compte que 9 qui ont été officiellement arrêtées et 3 seulement qui sont inactives (154). On peut citer le cas de l’étude sur l’Aducanamab de Biogen qui a été suspendue en Mars 2019 pour manque d’efficacité mais reprise en Octobre 2019 suite à des résultats positifs inattendus (155). La majorité des études sont en phases II ou III (17 études) (153).
Le souci majeur que l’on peut observer à travers toutes ces études, ce n’est qu’aucune propose réellement de guérir la maladie mais uniquement d’inhiber certains symptômes. Détruire de la plaque amyloïde ne l’empêchera pas de s’accumuler à nouveau. De plus, le
développement de petites molécules ou de l’immunothérapie implique une prise régulière du traitement et est donc synonyme d’inconfort et de dépense. Si un traitement venait à voir le jour proposant d’inhiber totalement la formation d’oligomère Aβ, il faudrait qu’il soit administré le moins de fois possibles, dans l’idéal en une seule fois. C’est dans cette perspective que notre laboratoire se spécialise dans l’élaboration de thérapies géniques qui modifieraient ou corrigeraient, en une seule injection et de façon définitive, les causes réelles d’une maladie, dont la MA. A l’heure actuelle, seules 2 études de Phase I sont en cours utilisant des traitements ADN/ARN. Une seule propose une thérapie génique permanente consistant à délivrer la protéine APOE2 dans le cerveau, aidant significativement l’élimination du peptide Aβ dans la circulation (156).