UNIVERSITÉ MOHAMMED V – AGDAL
FACULTÉ DES SCIENCES
Rabat
N° d’ordre 2574
THÈSE DE DOCTORAT D’ETAT
Présentée par
SALHAMEN Fatima
Discipline : Chimie
Spécialité : Chimie Minérale
Immobilisation de l’uranium et des terres rares par
l’hydroxyapatite et les sulfates de calcium : conditions,
quantifications et mécanismes.
Soutenue le 18/06/2012 à 16 h
Amphithéâtre Ibn Hayyan
Devant le jury :
Président :
Saidati Bouhlassa, Professeur, faculté des Sciences, Rabat
Examinateurs :
Abdelaziz EL JAZOULI, Professeur, Faculté des Sciences Ben M’sik
Casablanca
Hassan HANNACHE, Professeur, Faculté des Sciences, Ben M’sik
Casablanca
Ahmed ELYAHYAOUI, Professeur, Faculté des Sciences, Rabat
Said FAKHI, Professeur, Faculté des Sciences, Ben M’sik Casablanca
Mohammed HALIM, Professeur, Faculté des Sciences, Rabat
2
Immobilisation de l’uranium et des terres
rares par l’hydroxyapatite et les sulfates
de calcium : conditions, quantifications et
mécanismes
3 Résumé
L’utilisation des barrières ouvragées pour la dépollution, l’immobilisation des métaux lourds et le confinement des éléments radioactifs a connu un considérable regain d’intérêt. Dans le but de déterminer si des phases phosphatées peuvent retarder la migration de certains radioéléments, nous nous sommes attachés au cours de ce travail d’identifier et d’essayer de quantifier les processus gouvernant l’immobilisation de l’uranium par ces phases.
Lorsque les solutions initiales d’uranium sont de pH 5, l’hydroxyapatite se transforme en une phase apatitique déficiente contenant l’uranium, et retient de manière significative U(VI) lorsque sa concentration est égale à 4. 10-4 mol/l à pH neutre. Quand la concentration en uranium varie (8.10-4 mol/l à 12.10-4 mol/l), l’immobilisation des ions uranyles diminue. Le processus de rétention des cations uranyles par l’hydroxyapatite est vraisemblablement de type dissolution/précipitation. L’interaction entre les particules de HAP et les ions uranyles en solution a pH initial 4, conduit à une phase apatitique déficiente (avec HPO4--), probablement
faiblement uranifiée. Cette phase est obtenue dans les conditions ou la concentration de C[U(VI)] = 4.10-4 mol/l. Pour des concentrations en uranium supérieures à 4.10-4 mol/l, l’immobilisation de l’uranium est décrite par une réaction de dissolution du phosphate apatitique initial suivie de la précipitation de l’autunite.
Les sulfates de calcium peuvent être également envisagés comme matrice de conditionnement des éléments radioactifs ou encore comme additifs des barrières ouvragées assurant le confinement de ces éléments.
Lors de l’interaction des solutions d’uranium avec le gypse , l’oxyde d’uranium UO3 est coprécipité avec d’autres phases dont l’hémihydrate. Par contre lorsque
l’hémihydrate est le produit initial majeur au contact de U(VI) en solution, la phase condensée est essentiellement constituée de dihydrate. L’étude de la sorption de l’uranium dans ces milieux, semble favoriser la dissolution de la phase la plus soluble.
Dans le cas de l’hémihydrate synthétisée en milieux sulfophosphoriques, les ions uranyles seraient piégés dans une phase comparable au minéral rabejacite : Ca(UO2)4(SO4)2(OH)6. 6H2O.
Concernant les terres rares, leur coprécipitation avec le gypse est faible et régie par la substitution hétérovalente de Ca(II) par la terre rare. Le domaine de la solution solide reste cependant très limité.
Mots-clés : sorption, hydroxyapatite, uranium, dissolution-précipitation, sulfate de calcium, autunite.
4 Abstract
The use of the barriers worked for depollution, the immobilization of heavy metals and the containment of the radioactive elements knew a considerable renewed interest. With an aim of determining phosphatic phases so can delay the migration of certain radioelements, we are attached during this work to identify and try to quantify the processes controlling the immobilization of uranium by these phases.
When the initial uranium solutions are of pH 5, the hydroxyapatite is transformed into a defective apatitic phase containing uranium, and retains U(VI) significantly, when its concentration is equal to 4.10-4 mol/l at neutral pH. When the uranium concentration varies (8.10-4 mol/l to 12.10-4 mol/l), the immobilization of the ions uranyls decreases. The process of retention of the cations uranyls by the hydroxyapatite is probably of dissolution/precipitation type.
The interaction between the particles of HAP and the ions uranyls in solution has initial pH 4, led to a defective uranium - apatite phase (with HPO4--). This phase
is obtained under the conditions where the concentration of C[U(VI)] = 4.10-4 mol/l. For uranium concentrations higher than 4.10-4 mol/l, the immobilization of uranium is described by a reaction of dissolution of initial phosphate apatitic followed of precipitation of autunite.
The calcium sulphates can be also considered like matrix conditioning of the radioactive elements or like additives of the worked barriers ensuring the containment of these elements.
At the time of the interaction of the uranium solutions with the gypsum, the uranium oxide UO3 is coprecipity with other phases of which the hemihydrate. On the
other hand when the hemihydrate is the major initial product in contact with U(VI) in solution, the condensed phase primarily consists of dihydrate. The study of the sorption of uranium in these mediums, seems to support the dissolution of the most soluble phase.
In the case of the hemihydrate synthesized in mediums sulfophosphoric, the ions uranyls would be trapped in a phase comparable with the mineral rabejacite: Ca (UO2) 4 (SO4) 2 (OH) 6. 6H2O.
Concerning rare earths, their coprecipitation with the gypsum is weak and is governed by substitution heterovalente of Ca(II) by rare earth. The field of the solid solution remains however very limited.
Key words: sorption, hydroxyapatite, uranium, dissolution-precipitation, sulphate of calcium, autunite.
5
Remerciements
Les recherches qui font l’objet de ce mémoire ont été réalisées au laboratoire de Radiochimie de la Faculté de Sciences de Rabat.
Je remercie en premier lieu le Professeur S. Bouhlassa, de la faculté des sciences de Rabat, qui m’a encadré tout au long de ma thèse. Je suis entièrement reconnaissante des compétences et des encouragements que vous m’avez transmis pendant mon travail. Plus particulièrement, je vous remercie pour votre patience durant toutes ces années, votre gentillesse, votre aide précieuse et votre écoute permanente. Vous avez accepté de présider la soutenance de ce mémoire de thèse. Encore Merci
Je voudrais également exprimer ma reconnaissance aux membres de mon jury de thèse qui ont accepté d’évaluer mon travail :
Monsieur le Professeur A. Elyahyaoui, de la faculté des sciences de Rabat, a bien voulu siéger parmi les membres de jury, afin de juger ce modeste travail. Qu’il me soit permis de lui exprimer ma profonde estime et gratitude.
Mes vifs remerciements au professeur M. Halim de la faculté des sciences de Rabat, pour sa participation au jury de cette thèse.
J’adresse mes sincères remerciements au professeur A. Eljazouli, de la faculté des sciences Ben M’sik de Casablanca, pour avoir bien voulu juger ce travail en tant que rapporteur. Je le remercie également pour avoir participé à mon jury.
Que Monsieur le Professeur H. Hannanche, de la faculté des sciences Ben M’sik de Casablanca trouve ici l’expression de ma gratitude pour l’intérêt qu’il a porté à mes travaux en acceptant de participer au jury en tant que rapporteur.
6
Monsieur S. Fakhi, professeur à la faculté des sciences Ben M’sik de Casablanca, m’a fait l’honneur de donner son appréciation sur ce manuscrit. Qu’il trouve ici le témoignage de ma reconnaissance.
7
SOMMAIRE
Introduction générale
……… 18Références……… 23
Chapitre I synthèse bibliographique
……….25I. Généralités sur les apatites……… 28
I.1 Structure cristallographique……….. 28
I.2 Substitution dans le réseau apatitique………. 34
I.2.1 Substitution des ions Ca2+………35
I.2.2 Substitution des ions PO43……… 35
I.2.3 Substitution de OH- ………36
I.2.4 Substitution des ions OH- ou PO43- par des ions CO32 -………..38
I.2.5 Substitution des ions Ca2+ par les terres rares, et même par des ions fortement chargés (Sn4+ par exemple)………...39
I. 3 Propriétés des hydroxyapatites……….40
I.3.1 Stabilité thermique……….40
I.3.2 Stabilité en milieu neutre ou alcalin………...41
I.3.3 Stabilité sous rayonnements radioactifs………45
I.4 Synthèse et étude physico-chimique de l’hydroxyapatite phosphocalcique…... 45
I.4.1 Synthèse de l’hydroxyapatite phosphocalcique……….45
I.4.1.1 Synthèse par voie sèche………..46
8
I.4.1.2 Synthèse par voie humide………...46
I.4.1.2.1 synthèse par neutralisation………46
I.4.1.2.2 synthèse par double décomposition ……… .47
I.4.2 Autres méthodes de synthèse de l’hydroxyapatite phosphocalcique……49
I.4.2.1 Réactions sol-gel………...49
I.4.2.2 Réactions solide-solide………50
I.4.2.3 Synthèse hydrothermale………..50
I.4.2.4 Réaction liquide/solide : la voie des ciments………50
I.4.2.5 Réactions en sels fondus……….51
I.5 Caractérisation de l’hydroxyapatite phosphocalcique……….51
I.5.1 Diffraction des rayons X sur poudre………51
I.5.2 Spectrométrie infrarouge………...53
I.6 Conclusion……….54
II Uranium, propriétés………55
II.1 Définition………..55
II.2 Découverte………..55
II.3 L’uranium dans la nature………..56
II.4 Propriétés chimiques de l’uranium………..56
II.5 Conclusion………...57
III Notion de sorption……….59
III.1 Définitions………...59
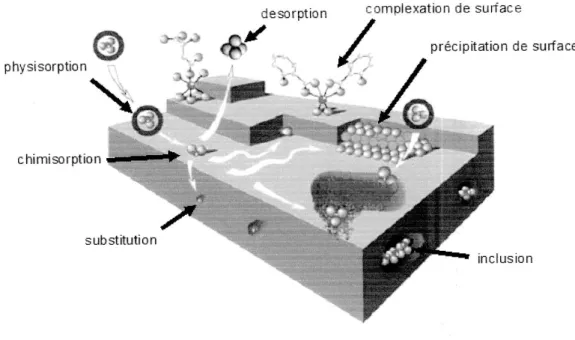
III.2 Rappels sur les différents mécanismes de rétention………...59
9
III.2.1 Adsorption non spécifique physisorption……….60
III.2.2 Adsorption spécifique chimisorption……….60
III.2.3 Echange d’ions………60
III.2. 4 Substitution isomorphe………...60
III.2.5 Dissolution – précipitation………61
III.2.6 Complexation de surface……… 62
III.2.7 Inclusion ou piégeage mécanique………...63
III.3 Conclusion………63
Références……….64
Chapitre II Synthèse et caractérisation de l’hydroxyapatite
phosphocalcique
………..73I. Etude physico-chimique des hydroxyapatites obtenues par la méthode de double décomposition ……….74
I.1 Mode opératoire……….74
I.2 Analyse par la diffraction des rayons X……….76
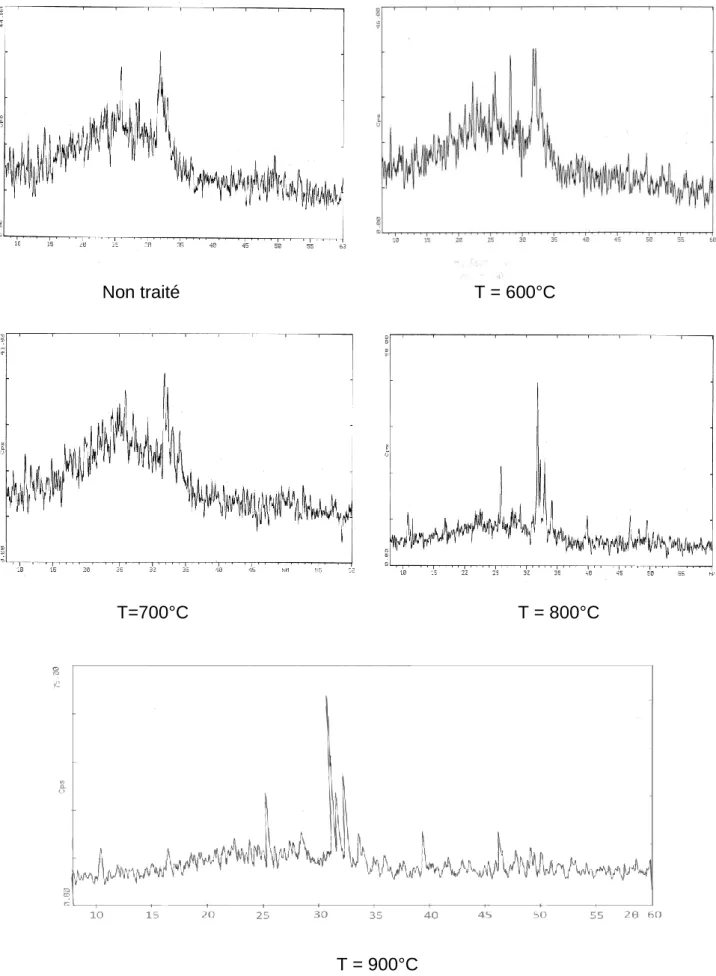
I.2.1 Produit non traité………76
I.2.2 Calcination à 600,700, 800 et 900°C………..76
I.2.3 Calcination à 1000°C……….78
I.3 Analyse par la spectroscopie d’absorption IR………..80
II. Examen des produits longuement calcinés………82
II.1 Analyse par la diffraction des rayons X……….83
10
II.1.2 T = 900°C, durée de calcination 6heures ………83 II.2 Analyse par la spectroscopie d’absorption IR, T = 900°C, durée de calcination 6heures………. 84
II.3 Conclusion………..84
III Effet de l’ajout de l’ammoniaque 25% (0,91) à la solution (NH4)2HPO4 :
0,06M……….85
III.1 40% en NH3 (0,91) ………85
III.1.1 T = 80°C………85
III.1.1.1 Analyse par la spectroscopie IR………..85 III.1.1.2 Analyse par la diffraction des RX………88
III.1.2 Calcination à 900°C et analyse par la spectroscopie d’absorption IR………..…88
III.2 60 en % NH3 (0,91) ………89
III.2.1 Analyse par la spectroscopie IR, après traitement à T=80°C………89 III.2.2 Analyse par la spectroscopie IR, du composé calciné à T=900°C………..90
IV Conclusion……….91 Références……….92
Chapitre III Hydroxydes d’uranium et Phosphates d’uranium :
11
I. Les hydroxydes d’uranium………..94
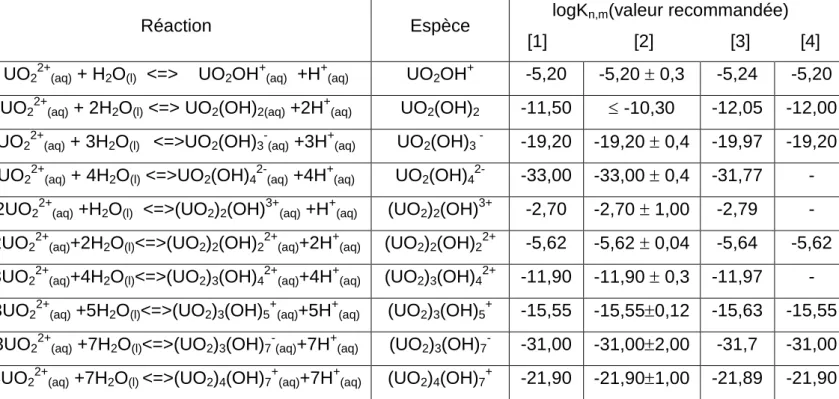
I.1 Données thermodynamiques sur les espèces uranyles aqueuses………94
I.2 Hydroxydes d’uranium en solution………...95
I.3 Sorption de l’uranium (VI) sur les tubes en polypropylène : UO2(OH)2.H2O ……….100
I.4 Spectroscopie IR, formation des hydroxydes d’uranyle……… 101
II. Les complexes de phosphate d’uranium……… 102
II.1. Espèces complexantes : H2PO4-, H3PO4………102
II.2. Ions complexants : HPO42-, PO43- ……….. 105
II.3 Minéraux des phosphates d’uranium……… 108
II.3.1 Différents minéraux de phosphate d’uranium……… 108
II.3.2 Synthèse de l’autunite………109
II.3.3 Sorption de l’uranium sur différents minéraux……… 111
III. Conclusion……….113
Références……… 114
Chapitre IV Etude physico-chimique de la sorption de l’uranium
sur l’hydroxyapatite phosphocalcique (Ca
10(PO
4)
6(OH)
2)…....
118I. Conditions et techniques expérimentales………..119
I.1 Mode opératoire……….. 119
I.2 Appareillages de mesures………...120
I. 2. 1 Mesures de la radioactivité……… 120
I. 2. 2 pH-mètre……….. 120
12
II. Analyse par D.R.X. et I.R.des phases solides résultant de l’interaction de U(VI) avec HAP………..120
II.1 Sorption de l’uranium sur l’hydroxyapatite : le pH de la solution initiale d’uranium est égal à 5………. 120
II.1.1 Sorption de l’uranium 4. 10-4 mol/ l sur l’hydroxyapatite………..120
II.1.2 Influence de la concentration du sorbat……… 122
II.1.2.1 Analyse par la diffraction des rayons X………..122
II.1.2.2 Analyse par la spectroscopie infrarouge………125
II.1. 2. 3 Conclusion………... 127
II.1.3 Influence du temps d’agitation……….127 II.2 Sorption de l’uranium sur l’hydroxyapatite : le pH de la solution d’uranium initiale est égal à 4……….131
II.2.1 Sorption de l’uranium 4. 10-4 mol/ l……….131
II.2.2 Effet de la concentration des ions U (VI)………132
II. 2. 2. 1 [U (VI)] = 8.10-4 mol/l...132
II. 2. 2. 2 [U (VI)] = 10.10-4 mol/l……….134
II. 2.3 Effet de la durée du temps d’agitation………...135
II. 2. 4 Influence de la température……….137
II. 2. 4.1 Analyse par la diffraction des rayons X ……….137
II. 2.4.1.1 T = 50°C……….137 II. 2.4.1.2 T = 80°C………138 II.2.4.2 Analyse par la spectroscopie d’absorption infrarouge……138 II.2.4.3 Conclusion……… .139
III. Distribution de U(VI) entre les phases solides et liquides………141
13
III.1 Sorption de l’uranium sur l’hydroxyapatite : le pH de la solution initiale d’uranium est proche de 5………141 III.1.1 C[U(VI)] : 4. 10-4 mol/l, 8. 10-4 mol/l, 10. 10-4 mol/l et 12. 10-4
mol/l……….. 141
III.1.2 Effet de la durée d’agitation : C[U(VI)] = 4. 10-4 mol/l, t = 69heures………..142
III.2 Sorption de l’uranium sur l’hydroxyapatite : le pH de la solution initiale d’uranium est aux environs de 4………..143
III.2.1 C[U(VI)] : 4. 10-4 mol/l, 8. 10-4 mol/l, et 10. 10-4 mol/l………….143 III.2.2 Influence de la durée d’agitation : C[U(VI)] = 4. 10-4
mol/l, t = 78heures………...144
Références………..145
Chapitre V Interprétation des résultats et mécanismes de
sorption de l’uranium
………..146I.Synthèse bibliographique des mécanismes de sorption de l’uranium par l’hydroxyapatite………147
II Interprétation des résultats et mécanismes……… 149
II.1 pH de la solution initiale d’uranium = 5……….149
II.1.1 Analyse par la diffraction des rayons X(DRX)………150
II.1.2. Etude par la spectroscopie d’absorption infrarouge……….151 II.1.2.1 C[U(VI)] = 4.10-4 M………151 II.1.2.2 8.10-4 ≤ C[U(VI)] ≤ 12.10-4M...………152 II.1.3 Mécanismes………..153 II.2 pH de la solution initiale d’uranium = 4………..156
14
II.2.1 Diffraction des rayons X………..156
II.2.2 Spectroscopie d’absorption infrarouge………159
II.2.3 Mécanisme………160
II.2.4 Effet de la durée d’agitation : 78 heures………...162
II. 2. 5 Effet de la température………..163
Conclusion………..163
Références………..165
CHAPITRE VI Rappels sur les sulfates de calcium et leurs
synthèses en présence de certaines impuretés………...
168I. Différentes formes de sulfates de calcium……….170
I.1 Gypses ou sulfates de calcium dihydratés : CaSO4,2H2O………..170
I.2 Semihydrates………..171
I.3 Anhydrite III ou anhydrite soluble………173
I.4 Anhydrite II, dite insoluble...174
I.5 Anhydrite I...174
II. Solubilité des sulfates de calcium……... 175
III. Déshydratation et réhydratation des sulfates de calcium ………..176
III.1 Hydratation………. 176
III.2 Déshydratation………….………..179
IV. Synthèse et caractérisation du sulfate de calcium………..181
IV.1 Diffraction des rayons X……… 181
IV.2 Spectroscopie d’absorption infrarouge………183
15
IV.3.1 Etude par la diffraction des RX et la spectroscopie IR………186 IV.3.2 Effet des impuretés : ions phosphates et Gd(III)………188
V. Effet des impuretés Gd(III), F-, Si(IV), PO43- et SO4-- et de la température sur
la nature du sulfate de calcium précipité et mécanismes de séquestration des terres rares(REE) par les sulfates………190
Références………..194
Chapitre VII Sorption de l’uranium sur le sulfate de
calcium……….
196I. Synthèse et caractérisation du sulfate de calcium………...197
I.1 Synthèse des produits……….197
I.1.1 Préparation du sulfate de calcium bihydraté………....197 I.1.2 Préparation du sulfate de calcium semihydraté ……….198
I.2 Identification des produits………...198
I.2.1 Milieu sulfurique………... 198 I.2.1.1 Diffraction des rayons X……….198 I.2.1.2 Spectroscopie d’absorption infrarouge………200
I.2.1 Milieu sulfophosphorique……….200 I.2.1.1 Diffraction des rayons X……….200 I.2.1.2 Spectroscopie d’absorption infrarouge………202
II. Etude physico-chimique de la sorption de l’uranium sur le sulfate de calcium……….202
II.1 Mode opératoire………... 202
16
II.2 Sorption par les sulfates de calcium dihydrate et hémihydrate préparés
dans les milieux sulfuriques : CH2SO4 = 0,09 mol/l ……….202
II.2.1Sorption de l’uranium U(VI) sur le sulfate de calcium bihydraté :
CaSO4, 2H2O………. 202
II.2.1.1 Analyse par la diffraction des rayons X………202
II.2.1.2 Etude par la spectroscopie d’absorption infrarouge….203
II.2.2 Sorption de l’uranium sur le mélange de sulfate de calcium semihydraté et dihydraté...204
II.2.2.1 Analyse par la diffraction des rayons X……… 204 II.2.2.2 Etude par la spectroscopie d’absorption infrarouge…..205
II.2.3 Discussion……….. 207
II.3 Sorption par les sulfates de calcium dihydrate et hémihydrate préparés dans les milieux sulfophosphoriques : CH2SO4 = 0,09 mol/ l ; CH3PO4 = 0,02
mol/ l……...208 II.3.1 Sorption des ions U(VI) sur le gypse : CaSO4, 2H2O…………208
II.3.1.1 Analyse par la diffraction des rayons X……… …208
II.3.1.2 Analyse par la spectroscopie d’absorption infrarouge…208
II.3.2 Sorption des ions U(VI) sur le sulfate de calcium dihydraté et hémihydraté ……….………209 II. 3. 3 Discussion………210 Conclusion………...212 Références………..214
Conclusions générales
………...215Annexe
………...22017
18
Introduction générale
Dans les installations de retraitement des combustibles nucléaires irradiés, il reste en fin de traitement, des actinides à vie longue et certains lanthanides qui doivent être conditionnés, en vue d'un stockage à long terme, dans des matrices résistantes et étanches.
Plusieurs concepts proposent l'emplacement des barrières artificielles, constituées par des phases minérales, disposées autour des dépôts de déchets radioactifs, afin d'affaiblir la mobilité des radionucléides. Les matériaux utilisables comme phase minérale doivent présenter des performances très élevées de stabilité chimique, de stabilité aux rayonnements et de stabilité à la température, pour isoler les éléments radioactifs de l'environnement et les maintenir dans un état isolé pendant des durées très longues, en raison de leurs périodes radioactives. Cette étape permet de réduire la biodisponibilité et par conséquent la radiotoxicité de ces éléments.
Les céramiques apatitiques sont des matériaux qui présentent un grand intérêt comme matrice de conditionnement des déchets industriels et en particulier les déchets radioactifs. Plusieurs travaux récents ont montré que les phases apatitiques présentaient des propriétés particulièrement adaptées à un stockage à long terme et
pouvaient être utilisées comme matrice de confinement. Cette utilisation dépend de
leur capacité d’immobilisation et du mode de piégeage des éléments divers, en particulier des cations tels que Sr2+, Cd2+, Cs+, U(+IV,+VI) et/ou des groupements anioniques tels que VO43-, UO42- etc…
Ainsi donc, des connaissances quantitatives sur la sorption des radioéléments par ces phases solides semblent très importantes. Le processus de la sorption permet en particulier, la limitation et/ou le retard du transport des éléments radioactifs dans l'environnement. Parmi ces derniers, nous avons choisi l'uranium. C'est l'élément trace le plus étudié à cause de son importance économique et scientifique. Son intérêt scientifique est du à son utilisation dans les travaux de datation [1] comme indicateur dans la diagenèse des phosphorites [2]. C'est le métal lourd prédominant dans les combustibles nucléaires. Et puisque l'uranium est le
19
constituant le plus important des déchets nucléaires, la prédiction de sa sorption a donc un grand intérêt. En outre, la chimie aqueuse de l’U(VI) est relativement bien connue ; c'est une chimie nécessaire pour interpréter les résultats expérimentaux et pour élucider les différentes réactions de sorption.
L'uranium est concentré dans les phosphates naturels, à des quantités proches de 100 ppm [3]. D'ailleurs, un facteur caractéristique des apatites sédimentaires est relié au pourcentage élevé de l'uranium. La quantification de cette matière dans les phosphorites montre que l'uranium se trouve sous deux états d'oxydation différents : +IV et +VI, avec des proportions variables 2/3 et 1/3.
Le travail effectué par Atschuler et coll. [3] sur le mode d'incorporation de l'uranium dans les phosphorites indique que la majorité est sous forme tétravalente +IV. Plusieurs auteurs [1,3,4] proposent que l'uranium +IV substitue le calcium à l'intérieur de la maille d'apatite à cause de la similarité de leur rayon ionique (rU (IV) = 0,97 Å et rCa2+ = 0,99 Å).
Quant à l'association de l'uranium hexavalent avec les phosphorites, la nature exacte de cette incorporation reste moins claire. Il a été démontré expérimentalement [5] que les ions UO2++ peuvent être fixés à l'intérieur de l'apatite sans une préalable réduction à l'état d'oxydation + IV, mais la dimension large des ions uranyles exclue la substitution directe de UO2++ au calcium. D'autres travaux [6] affirment que l'existence des ions U(VI) dans les phosphorites peut être interprétée comme un résultat dû soit à la chimisorption des ions uranyles sur les cristallites de l'apatite, soit à une oxydation des ions U(IV) piégés. L'incorporation de l'U(VI) dans la matrice apatitique peut être favorisée par un désordre dans la maille cristalline.
Finalement, certains chercheurs [7,8] suggèrent que dans les phosphorites, l'uranium se trouve sous forme de phases d'uranium distinctes.
Pour examiner la rétention de l’U(VI), nous avons utilisé l'hydroxyapatite comme phase sorbante. Cette phase minérale accepte de nombreuses substitutions mais sa tolérance pour l'uranium hexavalent reste assez limitée.
L'hydroxyapatite de formule idéale Ca10(PO4)6(OH)2 n'est pas seulement le constituant minéral principal des phosphates, mais est aussi le constituant inorganique principal des os et des dents, et l'incorporation des éléments radioactifs à l'intérieur de ce minéral, ainsi que les interactions à la surface de l'hydroxyapatite
20
avec plusieurs composés organiques tels que les protéines [9] ou inorganiques, dans les milieux aqueux, sont des sujets intéressant divers domaines : biologie, géologie, science des sols, chromatographie, environnement.... Il est possible que ces interactions affectent les processus de la croissance et de la dissolution de l'hydroxyapatite [10,11].
A cause de son insolubilité, son abondance et son habilité à la fixation d'un grand nombre d'éléments, l'hydroxyapatite est utilisée comme un milieu pratique pour l'élimination de nombreux polluants. Plusieurs travaux montrent que l'hydroxyapatite est capable de retenir les métaux lourds à partir des solutions aqueuses, soit par coprécipitation, soit par sorption. D'autres recherches indiquent que l'hydroxyapatite peut fixer 1,2 mol d'uranium par mol d'apatite [12].
Le but de ce travail est d'étudier la rétention de l'uranium à l'état de valence +VI par l'hydroxyapatite synthétique, préparée au laboratoire par la méthode de double décomposition. L'influence des paramètres tels que le pH, la température, la concentration de l’uranium sur la quantité retenue, est examinée dans le but d'élucider le mécanisme de la sorption. Ce faisant, et au cours de l’avancement de nos travaux nous avons constaté l’état embryonnaire des connaissances sur le comportement de l’élément en milieu sulfurique ou en présence de sulfates. Nous avons ainsi été naturellement conduits à examiner un deuxième sorbant : le sulfate de calcium.
Globalement cette étude comprend donc deux parties :
Partie A. Sorption de l’uranium sur l’hydroxyapatite Partie B. Sorption de l’uranium sur le sulfate de calcium
La première partie se décline en cinq chapitres. Dans le premier chapitre, nous donnerons quelques rappels bibliographiques sur les propriétés physico-chimiques de l’hydroxyapatite phosphocalcique. Il présente également des généralités sur le phénomène de sorption et les différentes propriétés chimiques et radioactives de l’uranium. L’analyse des échanges lors des expériences d’immobilisation, nécessite de connaître parfaitement la synthèse du sorbant utilisé. Dans cette optique, le second chapitre présente la préparation et la caractérisation de la poudre d’hydroxyapatite calcique spécialement synthétisée pour cette étude.
21
Une synthèse bibliographique sur les hydroxydes et les phosphates d’uranium, présentée dans le troisième chapitre est nécessaire pour examiner les résultats expérimentaux, décrits en quatrième chapitre. Enfin, le cinquième chapitre interprète et discute l’ensemble des résultats afin de préciser les mécanismes de rétention des ions UO22+ par l’hydroxyapatite phosphocalcique.
Une étude bibliographique sur l’incorporation de l’uranium dans la structure des composés sulfatés en particuliers les sulfates de calcium montre que ce sujet reste peu documenté. Des expériences concernant le piégeage de ce radioélément par les sulfates de calcium ont été réalisées dans la seconde partie. Le sixième chapitre aborde brièvement quelques généralités sur les sulfates de calcium et précise les conditions de formation et de synthèse des différents sulfates de calcium ainsi que les effets de certaines impuretés du milieu sur leurs précipitations. Des résultats intéressants relatifs à la coprécipitation des terres rares dans ces milieux sont aussi annexés à ce chapitre. La sorption des ions uranyles par le dihydrate CaSO4, 2H2O et l’hémihydrate CaSO4,1/2H2O est examinée dans le septième
chapitre.
22
Références
[1] Y. Kolodny et I. R. Kaplan. Uranium isotopes in sea floor phosphorites. Geochim. Cosmochim. Acta, vol. 34, pp. 3-24 (1970).
[2] A. Starinnsky, A. Katz et Y. Kolodny. The incorporation of uranium into diagenetic phosphorite. Geochemica et Cosmochimica Acta, vol. 46, pp. 1365-1374 (1982).
[3] Z. S. Altschuler, R. S. Jr. Clarke et E. J. Young. The geochemistry of uranium in apatite and phosphorite. U. S. Geol. Surv. Prof. Paper, 314-D, pp. 45-90(1958).
[4] Z. S. Altschuler. The bearing of geochemistry on the recovery of uranium and rare earths in phosphorites. Proc. 2nd Internat. Congr. Phosphorus Compounds, Boston, April, pp. 605-625(1980).
[5] R.P. Sheldon. Geochemistry of uranium in phosphorites and black shales of the phosphoria formation. U.S. Geol. Surv. Bull., 1084-D, p. 83-115 (1959).
[6] W. C. Burnett et H. H. Veeh. Uranium – series disequilibrium studies on phosphorite nodules from the west coast of America. Geochim. Cosmochim. Acta, vol. 41, p.755-764 (1977).
[7] R.T. Runnels, J.A. Schleicher et H.S.V. Nortwick. Composition of some uranium – bearing phosphate nodules from Kansas shales. Kansas Geol. Surv. Bull., 102, pt.3, p. 93- 104 (1953).
[8] G.N. Baturin et V.T. Dubinchuk. On uranium forms in oceanic phosphorites. Okeanologiya, 8, p. 1036 -1041(1978).
[9] M. R. Christoffersen, J. Christoffersen, P. Ibsen et H. Ipsen.Adsorption of ovalbumin on calcium hydroxyapatite crystals. Colloids and Surfaces, vol. 18, pp. 1-8(1986).
23
[10] Ph. Schaad, Ph. Germain, F. Gorce et J. C. Voegel. Dissolution of Synthetic Hydroxyapatite in the Presence of Lanthanum Ions. J. Chem. Soc. Faraday Trans., vol. 90, 22, pp. 3405-3408 (1994).
[11] J. Christoffersen, M. R. Christoffersen, R. Larsen, E. Rostrup, P. Tingsgaard, O. Andersen et P. Grandjean. Interaction of Cadmium Ions with Calcium Hydroxyapatite Crystals : A Possible Mechanism Contributing to the Pathogenesis of Cadmium-Induced Bone Diseases. Calcif. Tissue Int., 42, p. 331-339 (1988).
[12] J.J. Jeanjean, J.C. Rouchaud, L. Trains, M. Fedoroff. "Sorption of uranium and other heavy metals on hydroxyapatite". J. Radioanal. Nucl. Chem. Lett; 201, pp. 529-539 (1995).
24
CHAPITRE I
SYNTHESE BIBLIOGRAPHIQUE
25
Chapitre I Synthèse bibliographique
Les composés phosphocalciques constituent le minéral phosphaté le plus abondant sur terre, et jouent un rôle important dans divers domaines.
Les principaux avantages des céramiques phosphocalciques sont leur similarité aux composés minéraux osseux [1], leur bioactivité, leur biocompatibilité et leur capacité à former une interface fortement liée à l’os.
Plus particulièrement l’hydroxyapatite, phase minérale majeure des tissus durs des vertébrés [2,3], est utilisée en chirurgie osseuse ou dentaire comme substitut osseux synthétique [4-9], sous formes de granules, de blocs ou d’enduits pour implants dentaires (figure I.1).
Outre leur importance biologique, dans l’industrie, les minerais apatitiques sont la source principale des engrais phosphatés et servent à préparer l’acide phosphorique et différents dérivés phosphatés [10]. Le processus de production de l’acide phosphorique est basé sur l’attaque des apatites naturelles par l’acide sulfurique (processus humide) :
Ca10(PO4)6X2 + 10H2SO4 +10 yH2O 10CaSO4,yH2O + 6H3PO4 + 2HX (1)
Roche phosphatée Phosphogypse
X = OH-, Cl-, F- … Y = 2, ½ ou 0
Les hydroxyapatites sont également, étudiées pour leurs propriétés électroniques, utilisées dans les lampes fluorescentes [11] ou les matériaux pour laser [12]. A moindre échelle, elles sont aussi utilisées en pharmacie (excipient) et en chromatographie (colonne) [13,14].
26
Depuis quelques années, de nouvelles applications dans le domaine de l’environnement sont apparues. Les apatites sont notamment étudiées en tant qu’électrolyte dans les piles à combustible à oxyde solide [15], comme dépolluant des eaux ou des sols contaminés par les métaux lourds [16], ou encore comme matériaux de remblayage entrant dans la composition des barrières construites, pour stocker et piéger les éléments radioactifs contenus dans les déchets solides [17,18] et les effluents liquides des installations nucléaires.
L’hydroxyapatite, à laquelle nous nous intéressons dans le présent travail, constitue en effet un adsorbant de métaux lourds le plus étudié récemment, comme en témoignent les nombreuses publications traitant de ce sujet.
Ce chapitre décrit dans une première partie, les propriétés générales des apatites, et plus spécifiquement celles de l’hydroxyapatite phosphocalcique. Dans une deuxième partie sont présentées quelques notions chimiques essentielles sur l’uranium choisi comme sorbat. Enfin, dans la troisième partie nous rappellerons avec concision les principaux mécanismes qui gouvernent le phénomène de sorption ou rétention des éléments.
Figure I.1 : Différentes formes disponibles de substituts osseux de synthèse (MBCPTM, Biomatlante, France)
27
I Généralités sur les apatites
Le terme apatite provient d’un terme grec « apatân» signifiant «déception» ou «tromperie». Cette déception touchait les minéralogistes, abusés par la multiplicité des formes et des couleurs, parfois très belles, des apatites cristallisées naturelles qui pouvaient être ainsi confondues avec d’autres minéraux précieux [19].
Les apatites les plus répandues dans le milieu naturel sont les apatites phosphocalciques fluorées, Ca10(PO4)6F2.
I.1 Structure cristallographique de l’hydroxyapatite
Il est bien connu que les apatites ont pour formule générale : Me10(XO4)6Y2
Me : représente des cations trivalents Al3+, Eu3+…, bivalents Ca2+, Sr2+, Ba2+, Mn2+, Eu2+… ou partiellement monovalents Na+, K+…
Y : peuvent être des anions bivalents O2-, S2-, CO32-… ou monovalents OH-, Cl-, Br-…
XO4 : représente outre PO43- des groupements tétravalents SiO44-, CrO44-… trivalents
VO43-, AsO43-…ou des anions divalents CO32-, SeO42-, HPO42-, SO42-…
Les apatites phosphocalciques cristallisent en général dans le système hexagonal (figure I.2) avec un groupe spatial de symétrie P63/m [20]. Elles sont
caractérisées par des axes sénaires hélicoïdaux et des axes ternaires perpendiculaires au plan de base de la maille hexagonale.
Figure I.2 : Caractéristiques du système hexagonal [20]. En noir : les nœuds du réseau
28
Les paramètres cristallographiques de la maille a, b et c dépendent de la nature des ions Me, XO4, et Y. Selon Kay [21], a = b = 9,432Å et c = 6,881Å dans le
cas de l’hydroxyapatite.
Dans cette structure, les atomes de phosphore occupent des sites de coordinence quatre, générés par l’arrangement de quatre anions oxygène premiers voisins (figure 1.3).
Figure I.3 : Site tétraédrique des atomes phosphores [22]
La formule Ca10(PO4)6(OH)2 représente la maille élémentaire de
l’hydroxyapatite synthétique (figure I.4) [22]. La structure peut alors se décrire comme un arrangement d’anions (PO4)3- stabilisé par des cations Ca2+.
29
Figure I.4 : Maille élémentaire de Ca10(PO4)6(OH)2 [22]
Dans cette maille, les anions (OH-) sont localisés sur l’axe cristallographique c (figure I.5)
30
Dans cette structure, les groupements XO4 sont isolés les uns des autres et
ne sont pas liés par les atomes d’oxygène. Leur arrangement compact donne naissance à deux types de tunnels, parallèles à l’axe c (Figure I.6) [23]. Le premier, étroit de diamètre égal à 2,5 Å renferme les cations Me(I) associés aux axes ternaires. Le second de section plus large, a un diamètre qui varie entre 3 à 4,5 Å. Il est occupé par Y- ou OH- et les bords matérialisés par les atomes d’oxygène du tétraèdre XO4. Les ions Me++(II) sont situés à coté des ions OH- aux niveaux ¼ et ¾
suivant l’axe c.
La projection de la maille d’apatite sur son plan de base, montre nettement ces larges tunnels bordés par des atomes d’oxygène et des ions Me++
(II) ; deux atomes d’oxygène d’un tétraèdre XO4 sont superposés sur la projection (Figure I.6).
La taille des tunnels confère aux apatites des propriétés d’échangeur d’ions et d’accepteur de petites molécules, dont on peut citer l’oxygène, l’eau et la glycine (NH2-CH2-COOH), premier composé de la série des aminoacides, et
31
Les cations Me2+ se répartissent dans ces deux sites cristallographiques. Les sites Me(I), aux nombres de 4 par maille et de symétrie C3h, ont une coordinence 9.
Ils sont entourés de neuf atomes d’oxygène (trois O(1), trois O(2) et trois O(3)) (figure I.7). Le second site contient les cations Me(II), au nombre de 6 par maille et présente une symétrie de site Cs. Ils forment des triangles équilatéraux décalés de
60°C autour de l’axe sénaire hélicoïdal (axe c de la maille hexagonale). Leur coordinence est égale à 7 (un O(1), un O(2), quatre O(3) et un anion Y) (Figure I.8).
Figure I.7: Environnement du Figure I.8: Environnement du calcium des sites I calcium des sites II
• Positions des dix atomes de calcium :
Quatre atomes occupent la position Ca(I) dont deux sont à la position z = 0 et les autres à z = 0,5. Ils forment ainsi les colonnes parallèles à c, respectivement en x = 1/3, y = 2/3 et x = 2/3, y = 1/3. Les six autres atomes occupent la position Ca(II) avec trois d’entre eux formant un triangle à z = ¼ et les autres en z = ¾ (figure I.9), d’où la formule détaillée de l’HAP :
Ca(I)4Ca(II)6(PO4)6(OH)2
• Positions des hydroxydes :
Ils sont disposés selon une colonne sur l’axe parallèle à c en x = 0, y = 0 et z = ¼ et z = ¾ (fig.I.9).
32 • Positions des ions phosphates :
Les tétraèdres PO4 occupent les niveaux z = ¼ et z = ¾ (figure I.9). Les tétraèdres
XO4 définissent le squelette de la structure apatitique. Ce sont ces motifs qui donnent
sa stabilité à l’apatite.
Figure I.9 : Arrangement des ions autour de l’axe c dans l’hydroxyapatite [27] Une caractéristique fondamentale de la structure apatitique est l’existence des tunnels qui contiennent les ions Y-. Cette structure permet l’introduction, d’un nombre considérable d’ions [28-33], soit pendant la formation de l’apatite soit après, par le processus d’échange d’ions ou de substitution ou par le phénomène de la sorption de molécules ou d’ions.
33
Les ions Y- peuvent être substitués par les anions fluorures, chlorures et bromures. Leur disposition suivant l’axe sénaire hélicoïdal, varie avec la nature de l’anion Y
-. Ainsi quand Y = F-, les atomes du fluor viennent se placer aux cotes ¼ et ¾ au centre des triangles équilatéraux formés par les ions Me(II). Dans le cas où Y = OH- ou Cl-, ces ions sont décalés par rapport aux triangles équilatéraux (Figure I.10) [34].
Fig. I.10 : Position des ions F-, Cl- et OH- par rapport au triangle Ca(II) le long de l’axe sénaire hélicoïdal [34].
I.2 Substitution dans le réseau apatitique
Les hydroxyapatites ont une forte capacité d’immobiliser des ions quand elles sont mises en contact de solutions aqueuses. Les différentes possibilités de substitutions sont répertoriées ci-dessous :
34
Tableau I.1 : Substitutions possibles dans la maille apatitique
I.2.1 Substitution de Ca2+
Les cations bivalents Ca2+ de l’hydroxyapatite peuvent être remplacés par d’autres cations bivalents, monovalents, trivalents ou des lacunes [35-40]. L'incorporation de Mg2+ dans la structure apatitique, qui présente un grand intérêt en biologie et en médecine, est limitée mais elle peut diminuer le paramètre a de la maille ainsi que le taux de cristallinité. En raison des similitudes des tailles atomiques entre Ca2+ et Na+ l'incorporation de ce dernier ne change pas les paramètres cristallins.
I.2.2 Substitution de PO43-
Les groupements PO43- peuvent également être substitués par des
groupements anioniques tétravalents, trivalents [41] ou bivalents [42]. La substitution par CO32- conduit à un minéral défini comme apatite de type B, très importante dans
les apatites biologiques [43]. Elle se produit lors de la préparation des apatites soit par précipitation directe, soit par hydrolyse des autres phosphates de calcium en présence de carbonate. Le taux d'incorporation de carbonate dépend directement de la présence des autres cations. Par exemple, la présence de Na+ augmente le taux de substitution, tandis que Sr2+ le diminue. Cette substitution diminue la taille et la cristallinité du cristal et augmente sa solubilité.
La substitution par des ions carbonates diminue le paramètre a et augmente légèrement le paramètre c de l'apatite.
Ca2+ PO43- OH -Sr2+ Ba2+ Terres rares AsO43- SO42- CO32- F- O2 Pb2+ Mg2+ VO43- HPO42- S22- Cl- H2O Cd2+ Na+ SiO4 4- CO32- O22- ou O2- Br- N2 Mn2+ K+ Lacune Lacune I
-35 I.2.3 Substitution de OH-
Les anions OH- peuvent être substitués par des ions ou des groupements anioniques monovalents, bivalents ou des lacunes. Généralement les anions qui se substituent aux ions OH- se disposent dans les tunnels de l'apatite. L’ion CO32- peut
entrer dans les tunnels des apatites synthétiques préparées à haute température (1000°C) ou dans certaines apatites biologiques. Il y a alors augmentation du paramètre a et diminution du paramètre c. La substitution des OH- par F- augmente la stabilité structurale et la cristallinité et diminue donc la solubilité des cristaux d'apatite. Elle peut également diminuer le paramètre a sans changer le paramètre c. Lorsqu'un Cl- entre dans les tunnels, le paramètre a augmente et c diminue fortement.
Ces différentes substitutions se traduisent sur le plan physico-chimique par des modifications de la structure apatitique, cette dernière peut présenter un certain nombre de lacunes, soit dans les sites anioniques, soit dans les sites cationiques ; ceci permet l’existence d’hydroxyapatites non stoechiométriques. Le domaine d’existence de ces composés se situe entre le phosphate octocalcique apatitique (Ca/P = 1,33) et l’hydroxyapatite stoechiométrique (Ca/P = 1,67). Le tableau I.2 résume ces différentes apatites déficientes avec leurs rapports atomiques correspondants. Elles peuvent être sous forme d’hydrate, d’hydroxyde ou anhydre et de structures cristallines très variées. Il est à noter que le phosphate tétracalcique monoxyde n’existe qu’à haute température et n’est pas d’intérêt biologique ; c’est le phosphate de calcium basique le plus connu.
En résumé, cette non-stoechiométrie se traduit par : • La présence de lacunes en site cationique et OH-
• Une teneur en OH-
inférieure à 2 par maille
• Un état de cristallinité d’autant plus médiocre que l’hydroxyapatite est éloignée de la stoechiométrie
36
Notons qu’il peut exister des lacunes en ions calcium ou hydroxydes, mais aucune lacune de phosphates n’a été rapportée, les ions PO43- forment le squelette
de la structure.
Tableau I.2 : Les différentes orthophosphates de calcium [44]
Orthophosphates de calcium Formule chimique Symbole Ca/P
Phosphate tétracalcique Ca4(PO4)2O TTCP 2,00
Hydroxyapatite Ca10(PO4)6(OH)2 HAP 1,67
Phosphate tricalcique anhydre Phosphate tricalcique anhydre
3(PO4)2 3(PO4)2 -TCP -TCP 1,50 1,50 Phosphate tricalcique apatitique
Phosphate tricalcique amorphe
Ca9(HPO4)(PO4)5OH
Ca9(PO4)6,nH2O ACP
1,50 1,50
Phosphate octocalcique Ca8H2(PO4)6,5H2O OCP 1,33
Phosphate dicalcique dihydraté Phosphate dicalcique anhydre
Pyrophosphate de calcium CaH(PO4),2H2O CaH(PO4) Ca2P2O7 DCPD DCPA PPC 1,00 1,00 1,00 Phosphate monocalcique hydraté
Phosphate monocalcique anhydre
Ca(H2PO4)2,H2O Ca(H2PO4)2 MCPM MCPA 0,50 0,50
L'hydroxyapatite synthétique non stœchiométrique peut ainsi être décrite par la formule chimique plus détaillée Ca10-x(HPO4)x(PO 4)6-x (OH)2-x. Dans les composés
répondant à cette formule il existe des lacunes dans les sites calcium et dans les sites hydroxyle. Elles s'accompagnent de la substitution des groupements phosphate PO4 3- par les groupements hydrogénophosphate (HPO4)2- .
Les neutralisations successives des différentes acidités de l’acide orthophosphorique conduisent à des sels de calcium très peu solubles. Les constantes d’acidité de l’acide phosphorique dans l’eau à 25°C sont les suivantes [45] :
37
H3PO4 + H2O H2PO4- + H3O+ pka1 = 2,12 (2)
H2PO4- + H2O HPO42- + H3O+ pka2 = 7,21 (3)
HPO42-+ H2O PO43- + H3O+ pka3 = 12,7 (4)
La majorité des orthophosphates de calcium (tableau I.2), dérivent donc de la neutralisation de la première, de la seconde ou de la troisième acidité de l’acide phosphorique.
Les phosphates de calcium sont fréquemment classés selon le rapport atomique calcium/phosphore. Plus ce rapport augmente et plus la solubilité des phosphates de calcium diminue en milieu aqueux. En effet, le produit de solubilité des phosphates acides (contenant des protons) est beaucoup plus grand que celui des phosphates basiques [46]. Les sels acides sont d’autant plus stables que le pH du milieu est acide à l’inverse des sels basiques.
Certaines substitutions peuvent engager des groupements avec une charge électrique différente de celle habituellement rencontrée sur ce site, on peut citer les terres rares (REE3+) en substitution de Ca2+ et les ions carbonates (CO32-) en
substitution de OH-. L’éléctroneutralité de la structure est alors obtenue par des substitutions couplées ou par la création de lacunes cationiques (site Me) et/ou anioniques (Site Y). A titre d’illustration, deux exemples sont donnés ci-après :
* Substitution des ions OH- ou PO43- par des ions CO3
* Substitution des ions Ca2+ par les terres rares
I.2.4 Substitution des ions OH- ou PO43- par des ions CO32-
La substitution des ions OH- ou PO43- de l’hydroxyapatite par des ions CO3
2-conduit à des apatites carbonatées phosphocalciques respectivement de type A ou B. Les apatites carbonatées de type B sont non stoechiométriques (rapport atomique (Me/X) différent de 10/6 = 1,667) et lacunaires. Le composé peut être décrit par la formule suivante [46] :
38
Ca10-x+y (lacune) x-y (PO4)6-x (CO3)x(OH)2-x+2y (lacune) x-2y
avec 0 x 2, 0 2y x
I.2.5 Substitution des ions Ca2+ par les terres rares, et même par des ions fortement chargés (Sn4+ par exemple).
Lorsqu’on effectue une substitution couplée en échangeant un Ca2+
par une terre rare telle Nd3+ et un ion PO43- par un ion SiO44-, le minéral est dénommé
britholite. La formule générale de celle-ci peut s’écrire [18] :
Ca10-xNdx(PO4)6-x(SiO4)xF2 (avec 6 ≤ x > 0).
D’une manière générale, le remplacement de l’élément calcium divalent par une terre rare qui est un élément trivalent, peut se faire de plusieurs façons :
• (Ca2+, OH-) (Ln3+, O2-) • (Ca2+ , PO43-) (Ln3+, SiO44-) • (2Ca2+ ) (Ln3+, Na+)
Ces substitutions entraînent souvent des modifications profondes des propriétés physico-chimiques des apatites, et leur étude apporte aujourd’hui une meilleure compréhension du comportement des apatites présentes dans les os et les dents ; il en est ainsi des apatites partiellement carbonatées largement utilisées dans le domaine biomédical.
Les apatites complexes forment également les gisements de phosphates naturels.
Il ressort donc, qu’à l’échelle atomique, que ce soit par la création des lacunes et/ou pour des raisons d’encombrement stérique, les substitutions ioniques modifient les paramètres de maille de la structure. A plus grande échelle, les substitutions
39
engendrent, de manière générale, une baisse de cristallinité, une diminution de la stabilité thermique, ainsi qu’une augmentation de la solubilité. Les apatites apparaissent néanmoins comme des édifices particulièrement stables qui peuvent tolérer des écarts importants à la stoechiométrie.
I. 3 Propriétés des hydroxyapatites
I.3.1 Stabilité thermique
L’apatite stœchiométrique peut contenir 2 à 3% d’eau, qui peut être facilement éliminée par chauffage à 600°C. Néanmoins, une perte supplémentaire d’eau au sein des tunnels du réseau cristallographique peut se produire à partir de 850°C sous air par la réaction :
Ca10(PO4)6(OH)2 Ca10(PO4)6(OH)2-2x(O)x + xH2O (5)
Cet équilibre indique qu’un chauffage :
• sous vide en atmosphère sèche, favorise l’élimination d’eau et déplace l’équilibre à droite. Il y a donc une dégradation de HAP et formation d’une apatite ne contenant pas de OH-, appelée oxyapatite Ca10(PO4)6O.
• en milieu humide, déplace l’équilibre à gauche et stabilise l’HAP.
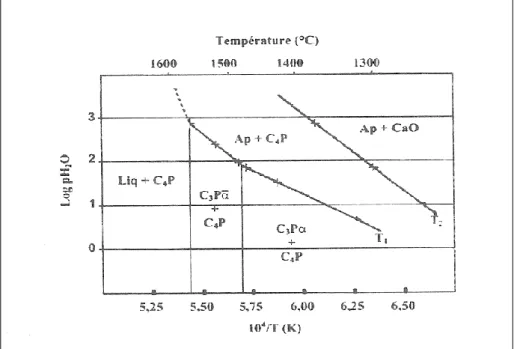
L’hydroxyapatite présente une structure très stable. De nombreux auteurs ont montré que sa décomposition, en phosphate tricalcique (TCP) et en phosphate tétracalcique (TTCP), n’intervient qu’à des températures élevées au-delà de 1300°C, et dépend de la pression de vapeur d’eau (fig. 1.11). L’HAP est d’autant plus stable à haute température que la pression partielle de vapeur d’eau dans l’environnement de l’échantillon considéré est élevée (fig. 1.11).
40
Figure I.11 : Evolution des températures de transition T1 et T2 en fonction de la
pression partielle de H2O dans le système de coordonnées logPH2O = f(1/T)
[47].
T1 et T2 étant les températures de décomposition de l’hydroxyapatite en absence et
en présence de chaux, respectivement pour une pression partielle de vapeur d’eau donnée.
I.3.2 Stabilité en milieu neutre ou alcalin
L’équilibre de solubilité de l’hydroxyapatite se généralise sous la forme : Ca10(PO4)6(OH)2 10Ca2+ + 6PO43- + 2OH- (7)
C3P : Phosphate tricalcique Ap : Apatite phosphocalcique avec
Ca/P = 1,67
C3P: Phosphate tricalcique CaO : Chaux, Liq : phase liquide
41
Cet équilibre de précipitation est régi par une constante d’équilibre thermodynamique, ksp appelée produit de solubilité.
ksp = (Ca)10 (PO43-)6(OH-)2
D’une manière plus générale dans le cas de la précipitation d’une espèce MnLm, il s’exprime sous la forme :
ksp = (Mm+)n (Ln-)m
où (Mm+) et (Ln-) représentent les activités des espèces dans la phase liquide (assimilées aux concentrations dans le cas de solutions diluées), et n et m leurs stœchiométries respectives.
Le produit de solubilité ksp est la constante numérique qui décrit la condition
d’équilibre d’une solution saturée par un soluté peu soluble. En d’autres termes ksp
est la valeur à saturation du produit des ions ou produit d’activité ionique kip. Pour
définir l’état de la solution, on introduit le produit d’activité ionique , tel que :
= kip/ksp
Lorsque > 1, la solution est dite sursaturée par rapport au sel solide. Dans ces conditions, le sel précipite jusqu’à ce que le maximum de solubilité dicté par le ksp soit rétabli. Inversement, une solution sous-saturée en sel solide ( ) va
provoquer la dissolution de celui-ci.
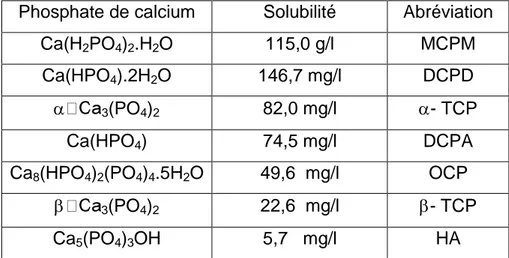
La propriété la plus importante des phosphates de calcium est probablement sa solubilité variable décroissante de MCPM, DCPD, - TCP, DCPA, OCP, -TCP à HA (Tableau I.3) [48].
42
Tableau I.3 Principaux phosphates de calcium rangés par ordre de solubilité décroissante du haut vers le bas.
L’hydroxyapatite phosphocalcique présente le produit de solubilité le plus faible des orthophosphates de calcium [49]. Il diminue quand la température augmente [50], on parle alors d’une solubilité rétrograde. La solubilité des apatites dépend également du pH. En effet, à 25°C de pH 5 à 7, des valeurs de pksp
comprises entre 115 et 117 ont été rapportées [51] ; l’apatite phosphocalcique est plus soluble en milieu acide qu’en milieu neutre ou basique.
Le tableau I.4 rassemble quelques valeurs de produits de solubilité proposées dans la littérature. Ces constantes varient selon les conditions d’étude, la méthode de préparation de la poudre et sa cristallinité.
Phosphate de calcium Solubilité Abréviation
Ca(H2PO4)2.H2O 115,0 g/l MCPM
Ca(HPO4).2H2O 146,7 mg/l DCPD
3(PO4)2 82,0 mg/l - TCP
Ca(HPO4) 74,5 mg/l DCPA
Ca8(HPO4)2(PO4)4.5H2O 49,6 mg/l OCP
3(PO4)2 22,6 mg/l - TCP
43
Tableau I.4 : Valeurs des produits de solubilité de l’hydroxyapatite disponibles dans la littérature.
Du point de vue physico-chimique, l’HAP est le composé le plus insoluble et le plus basique des orthophosphates de calcium si on ne tient pas compte du tétracalcique. C’est pourquoi la plupart des phosphates de calcium de rapport Ca/P inférieur à 1,67 évoluent en solution dans des conditions de température et de pH bien définis vers l’HAP [57].
Les différentes substitutions de l’apatite peuvent modifier le produit de solubilité du minéral. Par exemple, les ions silicates et carbonates ont tendance à augmenter la solubilité des apatites tandis que le fluor stabilise l’apatite (le produit de solubilité est de l’ordre de 10-120
).
Du fait de la faible solubilité des minéraux de phosphates de type apatitique, et de leur grande stabilité géochimique sur un large domaine de pH, l’immobilisation des métaux par des phosphates est de plus en plus étudiée. Les métaux piégés dans la structure cristalline ne peuvent être libérés que par la destruction du réseau cristallin [58]. Ksp Température (°C) Référence 6,62 10-126 25 [46] 9,24 10-118 5,52 10-118 25 37 [49] 2,70 10-152 1,76 10-152 37 25 [52] 5,01 10-105 - [53] 4,38 10-144 2,15 10-137 5,52 10-145 25 25 25 [54] 1,45 10-115 37 [55] 3,98 10-117 25 [56]
44
I. 3. 3 Stabilité sous rayonnements radioactifs
L’étude du réacteur naturel sur le site d’Oklo (au Gabon), a montré que les apatites contenant dans leur structure des radioéléments tels les actinides et/ou produits de fission, sont particulièrement stables thermiquement et chimiquement, même en milieu fortement irradiant [59, 60]. En effet, ces apatites présentent une structure telle que les dégâts d’irradiation sont instables. Elles résistent dans les conditions d’un stockage de déchets radioactifs jusqu’à plus de 1000°C.
Un ciment apatitique a été réalisé par J. Carpéna et coll.,[61]; ce composé a été élaboré et testé en tant que matériau de blocage, afin de freiner la dissémination des produits de fission labile : césium (135Cs) et iode (129I) de période longue, lors d’un contact du combustible irradié avec l’eau.
I.4 Synthèse et étude physico-chimique de l’hydroxyapatite phosphocalcique
I. 4. 1 Synthèse de l’hydroxyapatite phosphocalcique
En raison de nombreuses applications de l’hydroxyapatite dans le domaine médical comme substitut osseux ou dans le domaine environnemental comme adsorbant de métaux toxiques et radioactifs, plusieurs méthodes de synthèse ont été mises en œuvre. Les synthèses de l’HAP s’opèrent soit par voie sèche où les précurseurs sont mélangés à l’état solide, soit en milieu humide en utilisant des réactifs en solution.
La voie humide est caractérisée par l’emploi de solvants alors que la voie sèche n’emploie que des poudres. La pureté, la morphologie ainsi que le degré de cristallinité des produits obtenus diffèrent selon la méthode employée et les conditions expérimentales.
Ces diverses méthodes de synthèse ont été envisagées, pour la préparation d’une hydroxyapatite phosphocalcique stœchiométrique et stable à haute température [62,63]. Deux auteurs A. J. Christoffersen et M.R Chrisoffersen [64] ont réussi à préparer une hydroxyapatite bien définie à basse température, à partir des solutions aqueuses.
45
I.4.1.1 1 Synthèse par voie sèche
Il consiste à traiter à haute température, 1000°C, dans une atmosphère riche en vapeur d’eau, un mélange de phosphate tricalcique et de carbonate de calcium. La réaction mise en jeu est représentée par l’équation chimique suivante :
3Ca3(PO4)2 + CaCO3 Ca10(PO4)6(OH)2 + CO2 (8)
Dans ces conditions, l’apatite obtenue est mélangée à la chaux. Il est donc nécessaire de chauffer le mélange réactionnel, pendant plusieurs jours et effectuer des broyages intermédiaires. L’absence de la chaux est vérifiée par un test de basicité du produit obtenu après calcination, et la disparition des ultimes traces de phosphate tricalcique nécessite des temps de traitement très long, environ 1 à 2 mois.
I.4.1.2 Synthèse par voie humide
On distingue deux grands types de préparations par voie humide : une synthèse par neutralisation et une synthèse par double décomposition.
I.4.1.2.1 Synthèse par neutralisation
Cette méthode de synthèse a été mise en œuvre par Wallaeys [65] et améliorée par Trombe [66] :" On dilue à 500 cm3 d’eau distillée et décarbonatée, 7g d’acide orthophosphorique. On ajoute à froid le lait de chaux fraîchement préparé, exempt de CO2 en agitant jusqu’à virage de la phénophtaléine. On chauffe alors la
suspension à l’ébullition et on termine la saturation à l’eau de chaux de façon à obtenir un virage stable pendant 20 minutes. On filtre et on sèche à l’étuve à 80°C ".
10Ca(OH)2 + 6H3PO4 Ca10(PO4)6 (OH)2 + 18H2O (9)
L’hydroxyapatite obtenue est également mélangée à la chaux. Une calcination à 1000°C suivie d’un lavage, sont donc nécessaires. Ce traitement doit être répété plusieurs fois afin d’éliminer totalement la chaux.
46
I.4.1.2.2 Synthèse par double décomposition
L’hydroxyapatite que nous avons utilisée aux cours de nos essais expérimentaux, est préparée par la méthode de double décomposition [64] ; c’est une technique largement utilisée et susceptible de fournir une hydroxyapatite la plus proche de la stoechiométrie, le mode opératoire est le suivant :
Une solution ammoniacale de phosphate biammonique (0,06M) est versée goutte à goutte pendant 3 heures dans une solution de nitrate de calcium à l’ébullition (0,1M). Les proportions correspondent à la composition de l’hydroxyapatite stœchiométrique. Lorsque la précipitation est terminée, on ajoute lentement 200 ml d’ammoniaque concentrée (d = 0,91), pour porter le pH de la solution entre 11 et 12 ; c’est un pH basique nécessaire à la précipitation de la phase apatitique.
Le mélange réactionnel est porté à ébullition afin d’éliminer par évaporation l’excès d’ammoniaque.
On maintient la suspension à l’équilibre pendant environ une demi-heure. On filtre à chaud et on sèche le solide obtenu à l’étuve vers 80°C. Cette phase est ensuite calcinée pendant 3heures à différentes températures.
10Ca(NO3)2 ,4H2O+ 6(NH4) H2PO4 + 14NH4OH (10) Ca10(PO4)6(OH)2 + 20NH4NO3 + 52H2O
En synthétisant de l’HAP par précipitation à 35°C à partir de Ca(NO3)2 et
(NH4)2HPO4 à pH 10-11, Changheng Liu et al., [67], ont montré qu’au début de la
réaction le rapport Ca/P est inférieur à 1,5, valeur atteinte en 10 min, et qu’ un temps considérablement long était nécessaire pour obtenir le rapport stoechiométrique de 1,67. Dans la même étude les auteurs ont proposé la chronologie de la formation d’HAP suivante :
47
2. Transformation très rapide de l’OCP en phosphate de calcium amorphe (ACP) : Ca3(PO4)2, xH2O
3. Transformation progressive de l’ACP en hydroxyapatite déficiente en calcium (DCP) : Ca10-z(HPO4)z(PO4)6-z(OH)2-z, nH2O avec 0 < z < 1.
4. Finalement, la DCP se transforme en hydroxyapatite (HA) stable : Ca10(PO4)6(OH)2.
Changheng Liu et al., [67] ont étudié, par la suite, l’évolution du rapport atomique Ca/P avec le temps. Ce rapport évolue pour atteindre la valeur théorique de HA, 10/6 à l’équilibre. La figure I.12 présente les résultats obtenus.
Figure I.12 : Evolution du rapport Ca/P en fonction du temps [67]
Ces résultats sont en accord avec ceux antérieurement décrits par de nombreuses équipes. Jarcho et al.[68], en 1976, ont constaté que pour un temps de réaction supérieur à 24h, le rapport Ca/P est invariable et très proche de 1,67. Ils ont défini des temps minimum de maturation pour l’obtention de HA : à 25°C le temps doit être supérieur à 7h.
Outre le temps de réaction, la température est également un paramètre majeur. Elle modifie le temps de maturation nécessaire et affecte la morphologie des grains de HA.
Changheng Liu et al., [67] et L. Dean-Mo et al.,[69] ont étudié également ce facteur, la figure I.13 définit le temps nécessaire en fonction de la température de synthèse.
48
Lors d’une synthèse à 15°C, la taille des particules est inférieure à 10nm alors qu’à 60°C, il se forme un cristal aciculaire de longueur 100nm. La température peut ainsi permettre de contrôler la morphologie des grains.
FigureI.13 : Temps nécessaire à la formation d’hydroxyapatite en fonction de la température [69]
I.4.2 Autres méthodes de synthèse de l’hydroxyapatite
phosphocalcique
I.4.2.1 Réactions sol-gel
Dans le cas de la synthèse d’HAP par la méthode sol-gel, le vieillissement de la solution est un facteur très important. Dean-Mo Liu et al., [69] ont étudié cet effet lors de la préparation d’HAP par cette méthode, à température ambiante ainsi qu’à d’autres valeurs de la température. Les auteurs ont remarqué que la formation d’une hydroxyapatite pure à faible température nécessite un temps de vieillissement long.
Le procédé sol-gel est basé sur la polymérisation de précurseurs organométalliques de type alcoxydes M(OR)n. Après une hydrolyse contrôlée de ces alcoxydes en solution, la condensation des monomères conduit à des ponts oxo puis à un oxyde organique. La polymérisation progressive de ces précurseurs forme des oligomères puis des polymères en augmentant ainsi la viscosité. Ces solutions polymériques conduisent à des gels qui permettent une mise en forme aisée des
![Figure I.5 : Maille élémentaire de Ca 10 (PO 4 ) 6 (OH) 2 , projection sur le plan (010) [22]](https://thumb-eu.123doks.com/thumbv2/123doknet/2191527.11524/29.892.164.708.149.475/figure-maille-elementaire-po-projection-plan.webp)
![Fig. I.10 : Position des ions F - , Cl - et OH - par rapport au triangle Ca(II) le long de l’axe sénaire hélicoïdal [34]](https://thumb-eu.123doks.com/thumbv2/123doknet/2191527.11524/33.892.237.713.333.778/fig-position-ions-rapport-triangle-long-senaire-helicoidal.webp)
![Figure I.15 : Spectre infrarouge caractéristique de l’hydroxyapatite stoechiométrique [72]](https://thumb-eu.123doks.com/thumbv2/123doknet/2191527.11524/52.892.140.717.671.942/figure-spectre-infrarouge-caracteristique-l-hydroxyapatite-stoechiometrique.webp)
![Tableau 4 : Pourcentages des ions uranyles adsorbés sur les tubes de centrifugation [9]](https://thumb-eu.123doks.com/thumbv2/123doknet/2191527.11524/99.892.156.740.791.950/tableau-pourcentages-ions-uranyles-adsorbes-tubes-centrifugation.webp)
![Figure 3 : Spéciation des ions uranyles en solution et en phase solide, calculée avec le logiciel HYDRAQL, dans un système ouvert, à la concentration initiale C[U(VI)] = 10 -5 M](https://thumb-eu.123doks.com/thumbv2/123doknet/2191527.11524/100.892.207.750.282.569/speciation-uranyles-solution-calculee-logiciel-hydraql-systeme-concentration.webp)