Étude de la (stéréo)sélectivité et d’interactions non
covalentes à l’échelle de la molécule unique sur le
Pt(111)
Thèse Guillaume Goubert Doctorat en Chimie Philosophiæ doctor (Ph.D.) Québec, Canada © Guillaume Goubert, 2014Résumé
Le travail décrit au sein de cette thèse a pour objectif l’exploration des phénomènes liés au transfert de chiralité à des sites uniques sur une surface catalytique. Plus précisemment, le but est d’effectuer des études dynamiques de la stéréochimie sur des molécules uniques. Au cours de ce travail, plusieurs problématiques ont été explorées :
1. La question du transfert de chiralité sur une surface de catalyseur, le Pt(111). En particulier, le cas du transfert d’information chirale d’un modificateur chiral vers un substrat prochiral est étudié, les deux molécules étant liées de façon non-covalente.
2. L’étude et la définition de sites catalytiques uniques sur un catalyseur métallique, le Pt(111). 3. Le comportement dynamique de réactifs à des sites catalytiques uniques.
Pour réaliser cette étude, on a utilisé le microscope à effet tunnel (STM), sur une surface de Pt(111) sous ultra haut vide (UHV), pour obtenir des images possédant une résolution moléculaire et sous-moléculaire des réactifs. Des images précises des assemblages entre modificateurs et réactifs peuvent ainsi être obtenues et les assemblages peuvent être classés en familles distinctes. L’acquisition de séries d’images dans le temps permet également d’obtenir une information dynamique sur la formation et la transformation des assemblages. Des expériences de spectroscopie vibrationnelle ont également été réalisées. Ces résultats expérimentaux en science des surfaces sont complétés par des calculs théoriques en théorie fonctionnelle de la densité (DFT) effectués par nos collaborateurs du groupe Hammer de l’Université d’Aarhus au Danemark. La synthèse de molécules et les tests en solution ont été effectués par les membres du groupe Boukouvalas du département de chimie de l’Université Laval.
Cette méthode expérimentale permet l’étude de sites catalytiques uniques. La problématique de l’ana-lyse des sites actifs sur un catal’ana-lyseur est au coeur de la recherche en catal’ana-lyse hétérogène moderne. Notre méthode est une des voies de recherche, complémentaire des études in situ et operando sur les catalyseurs en conditions réelles. En effet, le STM permet de caractériser des molécules uniques et de séparer espèces actives et spectatrices. Il ne fournit cependant pas d’information chimique directe. En effet, le courant tunnel ne donne pas d’information sur la source des électrons ou des atomes à travers lesquels les électrons ont pu passer. C’est pour cette raison que les calculs théoriques et les mesures de spectroscopie vibrationnelle sont importantes pour la compréhension des phénomènes de surface.
En premier lieu, on a suivi dans le temps la déshydrogénation de l’ethylnaphtalène (chapitre2) puis le bris d’une liaison C−N dans le pantoyl-naphtyléthylamine (PNEA) pour former une aminolactone chirale, décrite au chapitre3. Cette dernière réaction n’était auparavant pas connue dans la litérature et l’effet de cette aminolactone sur le transfert de chiralité n’avait donc pas été envisagé. La dernière réaction étudiée est l’hydrogénation partielle du 2,2,2-trifluoroacétophénone (TFAP) au chapitre5. Ce dernier cas a permis d’isoler un intermédiaire dans l’hydrogénation du TFAP, le produit final de l’hydrogénation, l’alcool, n’a pas été observé. La détermination de la structure chimique de cet inter-médiaire a été possible en observant un changement dans la géométrie des assemblages bimoléculaires formés en fonction de la température, avec le support de calculs théoriques. Ce dernier cas montre que la géométrie des assemblages intermoléculaires est un marqueur utile de l’état chimique des espèces en surface.
Il en résulte qu’une grande partie des résultats de cette thèse concerne l’identification et la caracté-risation des liaisons non covalentes qui mènent aux assemblages intermoléculaires, en particulier les liaisons hydrogène faibles qui peuvent se former entre un carbonyle et les hydrogènes d’un groupe-ment aryle ou alkyle dans le chapitre2. La capacité de ces interactions à créer des assemblages inter-moléculaire et à contrôler leur géométrie même en présence d’interactions fortes entre les molécules organiques et la surface métallique a également été étudiée. Dans le cas d’adsorbat chiraux, l’étude des assemblages intermoléculaires permet de comprendre les mécanismes de transfert de chiralité. En effet, dans le cas d’une réaction asymétrique sur une surface métallique les réactifs sont immobilisés dans une géométrie particulière par un modificateur chiral. Il est donc possible de découper la réaction à un site asymétrique comme suit :
1. Adsorption des réactifs
2. Rencontre du réactif avec le modificateur et formation d’assemblages énantiosélectifs.
3. Réaction au sites asymétrique et formation d’un énantiomère du produit de façon préférentielle.
Il en découle que l’étude des assemblages énantiosélectifs permet de mieux comprendre le transfert de chiralité. C’est ce qui a été réalisé au chapitre3pour l’étude des assemblages entre l’aminolactone chirale formée in-situ avec le TFAP et le cétopantolactone (KPL). On a ainsi montré qu’en présence de KPL, des assemblages multimoléculaires sont formés. Ce mode d’interaction n’avait jamais été ob-servé ni postulé dans la littérature pour des systèmes catalytiques asymétriques. Dans le chapitre4, les assemblages entre le méthylebenzoyl formate (MBF) et le (R)-1-(1-naphtyléthyleamine) (R)-NEA) ont été étudiés. On a examiné s’il était possible généraliser les résultats obtenus précédemment sur les assemblages entre le (R)-NEA et le TFAP [1] puis le méthyle trifluoropyruvate (MTFP) [2], pour expli-quer le rôle des liaisons hydrogène, de la répulsion stérique et des sites d’adsorption sur la géométrie des assemblages.
La dernière partie de mes travaux concerne l’étude de la diffusion des réactifs sur la surface, en par-ticulier le (R)-NEA) et le TFAP aux chapitres6 et7. Nous avons montré que la diffusion des deux
conformères du (R)-NEA) est très différentes, ce qui montre que c’est un marqueur précis de la struc-ture chimique. La diffusion peut être considérée comme une autre façon d’attaquer le problème du transfert de chiralité. En effet la liberté de diffusion des réactifs contrôle l’accès aux différents sites catalytiques et le passage de l’un à l’autre comme on le montre au chapitre7.
Table des matières
Résumé iii
Table des matières vii
Liste des tableaux xi
Table des figures xiii
Table des schémas xxi
Remerciements xxv
Avant-propos xxvii
Introduction 1
0.1 Chiralité . . . 1
0.2 Catalyse . . . 4
0.3 Sélectivité et mécanismes en catalyse hétérogène . . . 6
0.4 Catalyse hétérogène asymétrique : hydrogénation de cétones activées sur le Pt sup-porté . . . 12
0.5 Étude de la dynamique en surface . . . 19
1 Partie expérimentale 27 1.1 Introduction . . . 27
1.2 UHV. . . 28
1.3 Microscopie à effet tunnel . . . 31
1.4 Analyse de films STM . . . 36
1.5 Spectroscopie RAIRS . . . 37
1.6 Calculs théorique par théorie fonctionnelle de la densité (DFT) . . . 38
2 Weak interactions in the assembly of strongly chemisorbed molecules 43 2.1 Résumé . . . 43
2.2 Abstract . . . 43
2.3 Weak interactions in the assembly of strongly chemisorbed molecules . . . 44
2.4 Acknowledgements . . . 48
3 Aminolactone Chiral Modifiers for Heterogeneous Asymmetric Hydrogenation: Cor-rected Structure of Pantoyl-Naphthylethylamine, In-Situ Hydrogenolysis, and Scan-ning Tunneling Microscopy Observation of Supramolecular Aminolactone Substrate
Assemblies on Pt(111) 51
3.1 Résumé . . . 51
3.2 Abstract . . . 52
3.3 Introduction . . . 52
3.4 Results and discussion . . . 54
3.5 Conclusion . . . 61
3.6 Experimental section . . . 62
3.7 Acknowledgments. . . 64
3.8 Supplementary information . . . 64
4 Surface Diastereomeric Complexes Formed by Methyl Benzoylformate and (R)‐1-(1-naphthyl)ethylamine on Pt(111) 73 4.1 Résumé . . . 73
4.2 Abstract . . . 73
4.3 Introduction . . . 74
4.4 Experimental section . . . 77
4.5 Results and discussion . . . 77
4.6 Conclusions . . . 85
4.7 Ackknowledgements . . . 85
5 Isolating a Reaction Intermediate in the Hydrogenation of 2,2,2-Trifluoroacetophenone on Pt(111) 87 5.1 Résumé . . . 87
5.2 Abstract . . . 88
5.3 Introduction . . . 88
5.4 Methods . . . 89
5.5 Results and discussion . . . 91
5.6 Conclusion . . . 93
5.7 Supplementary Information: determination of s-AP and l-AP population . . . 94
6 Walking-like diffusion of two-footed asymmetric aromatic adsorbates on Pt(111) 95 6.1 Résumé . . . 95
6.2 Abstract . . . 96
6.3 Introduction . . . 96
6.4 Methods . . . 98
6.5 Results and Discussion . . . 100
6.6 Concluding comments . . . 112
7 Résultats préliminaires sur la dynamiques des sites catalytiques (R)-NEA/TFAP 113 7.1 Introduction . . . 113
7.2 Diffusion TFAP . . . 114
7.3 Dynamique des complexes . . . 115
Conclusion 125 7.4 Mise en perspective . . . 125
7.5 Ouverture . . . 127
Annexe : Liste des publications 129
Liste des tableaux
4.1 Relative Abundances of Left-, Top- and Right-hand Side Binding Geometries in a
Sam-ple of 350 MBF/(R)-NEA ComSam-plexes on Pt(111) . . . 79
4.2 Prochiral Ratios (pro-R:pro-S) for MBF Binding to the Left, Right, Top of the
Table des figures
0.1 Illustration de la chiralité, deux mains sont images miroir l’une de l’autre et les deux
énantiomère d’un acide aminé également. . . 1
0.2 Règle de Cahn, Ingold et Prelog pour la détermination de la configuration absolue des
énantiomères. . . 2
0.3 Les deux formes miroir des cristaux d’acide tartrique. . . 3
0.4 Profil énergétique d’une réaction sans catalyseur (ligne pleine) et avec un catalyseur (ligne pointillée). La barrière d’activation est réduite de 𝐸∗à 𝐸∗
𝑐 . . . 5
0.5 Illustration de la structure macroscopique, microscopique et nanoscopique (A) d’un catalyseur oxyde mésoporeux et (B) d’un catalyseur métallique supporté [18]. Repro-duit depuis Buurmans, I.L.C ; Weckhuysen, B.M. ; Nature Chemistry, 2012, 4, 873-886
©Nature Publishing Group 2012. . . 7
0.6 Différents sites rencontrés à la surface d’un cristal outre les terrasses atomiques : (1) atome supplémentaire (2) et (3) atomes manquants à une marche et dans une terrasse
atomique (4) Coin et (5) marche atomique. . . 7
0.7 Le 2,2,2-trifluoroacétophénone (TFAP) peut être adsorbé de deux façons sur la surface selon le côté de la molécule (énantioface) en contact avec la surface. On défini alors
deux prochiralités [77]. . . 11
0.8 La prochiralité du réactif contrôle la chiralité du produit puisque l’hydrogène est ajouté au réactif depuis la surface. Un réactif proR produit un énantiomère R et un réactif proS
produit un énantiomère S.. . . 12
0.9 Sélection de réactifs étudiés pour la réaction d’Orito avec le meilleur excès
énantiomé-rique répertorié [68, 76, 83–90]. . . 13
0.10 Trois modificateurs chiraux, (A) la cinchonidine, (B) le NEA, (C) le pantoyl-naphtyléthylamine (PNEA). Le chiffre 1 indique le groupement aromatique, 2 indique l’amine et 3 indique
le carbone chiral entre les deux groupes précédents. . . 14
0.11 Les deux conformères de NEA sur le Pt(111), (A) NEA-1, la forme exo et (B) NEA-2,
la forme endo. . . 15
0.12 Comparaison des images STM et des structures calculées par DFT pour les deux
com-plexes les plus stables sur le Pt(111) dans le cas de (A) NEA/TFAP et (B) NEA/MTFP. 16
0.13 Complexe entre la cinchonidine (2) et le KPL (1) sur le Pt(111) incluant une liaison hydrogène avec la fonction OH et une liaison hydrogène avec l’amine protonée [103]. Reproduit depuis Meemken, F ; Maeda, N ; Hungerbühler, K ; Baiker, A Angewandte
International Edition, 2012, 51, 8212-8216 ©Wiley-VCH 2012. . . . 18
0.14 Disparition des TFAP sous hydrogène alors que les NEA restent sur le Pt(111) [77]. Reproduit depuis Demers-Carpentier, V. ; Goubert, G. ; Masini, F. ; D, Y. ; Ras-mussen, A. M. H. ; Hammer, B. ; McBreen, P. H. The Journal of Physical Chemistry
0.15 Population relative des principaux complexes pour NEA/TFAP sur le Pt(111) et énergie de stabilisation des complexes trouvés par DFT correspondants [1]. Reproduit depuis Demers-Carpentier, V. ; Goubert, G. ; Masini, F. ; Lafleur-Lambert, R. ; Dong, Y. ; La-voie, S. ; Mahieu, G. ; Boukouvalas, J. ; Gao, H. ; Rasmussen, A. M. H. ; Ferrighi, L. ; Pan, Y. ; Hammer, B. ; McBreen, P. H. Science 2011, 334, 776–780. ©the American
Association for the Advancement of Science 2011. . . 19
0.16 Les trois types de sites principaux sur une surface 111 d’un cristal fcc : simplement lié (on-top) en vert, ponté (bridge) en rouge et triplement lié (threefold) en bleu. Le disque
blanc représente la position d’un adsorbat simple dans chaque site. . . 20
0.17 En haut, barrière à la diffusion entre deux sites stables (bleu) en passant par l’état de transition instable (rouge). En bas, illustration de la géométrie de ces sites, pour passer d’un site stable au-dessus d’un atome à un autre voisin, l’adsorbat doit passer dans un
creux, une géométrie moins stable. . . 22
0.18 (A) Le naphtalène tourne sur lui-même sur la surface. (B) On mesure le temps passé par chaque molécule entre deux rotations. (C) La distribution de ces intervalles donne
le taux de diffusion par comparaison avec une loi exponentielle.. . . 25
1.1 Photographie du système UHV à deux chambres, une chambre de préparation (à droite) et une chambre expérimentale (à gauche) dans laquelle le STM est monté. Un bras de transfert permet de transférer l’échantillon entre les deux chambres. Il permet
égale-ment le chauffage de l’échantillon par e-beam. . . 30
1.2 Exemples d’images STM . . . 31
1.3 Effet tunnel en 1D pour un potentiel en créneau. E est l’énergie de l’électron incident
et V la hauteur du potentiel.. . . 32
1.4 Schéma de fonctionnement d’un microscope à effet tunnel. Un courant de consigne est spécifié et le moteur piézoélectrique déplace la pointe dans la direction perpendiculaire à la surface pour maintenir le courant constant grâce à une boucle de contre-réaction. Ce mouvement du moteur donne une cartographie de la densité électronique sur la surface. Le courant provient principalement des quelques atomes à l’extrémité de la
pointe. . . 33
1.5 Schéma de la pointe STM en cours de balayage sur une surface métallique présentant des marches atomiques. La ligne rouge représente le mouvement de la pointe et donc
le signal fourni par le microscope. . . 34
1.6 Description de l’effet tunnel entre une pointe STM et une surface sous une tension tun-nel 𝑈𝑇. 𝐸𝐹𝑒𝑟𝑚𝑖désigne l’énergie du niveau de Fermi dans la pointe et la surface. 𝜙𝑒𝑐ℎ
et 𝜙𝑝𝑜𝑖𝑛𝑡𝑒 représentent le travail de sortie de la surface et de la pointe, respectivement. 35
1.7 Explication de la méthode d’analyse semi-automatisée des images. . . 37
1.8 Illustration des règles de sélection en spectroscopie infrarouge pour des molécules sur
une surface métallique. Seule la composante perpendiculaire à la surface est active. . 38
1.9 Comparaison entre différentes fonctionnelles pour l’énergie de différents complexes entre NEA et TFAP, représentés par un angle θ défini au chapitre 7. La fonctionnelle optB88-vdW inclus une correction pour les forces de van der Waals. [138]. Repro-duit depuis Groves, M. N. ; Goubert, G. ; Rasmussen, A. M. H. ; Dong, Y. ; Lemay, J-C. ; Demers-Carpentier, V. ; McBreen, P. H. ; Hammer, B. Surface Science, DOI :
1.10 En haut : images STM entre le NEA et le MTFP (A, B) et en bas : la simulation Tersoff-Hamann de la structure DFT la plus proche (C, D). Reproduit depuis Demers-Carpentier, V. ; Rasmussen, A. M. H. ; Goubert, G. ; Ferrighi, L. ; Dong, Y. ; Lemay, J.-C. ; Masini, F. ; Zeng, Y. ; Hammer, B. ; McBreen P. H. Journal of the American
Chemical Society 2013, 135, 9999–10002 ©ACS 2013 . . . 41
2.1 STM images of naphthalene, MN and EN on Pt(111) at room temperature. Naphtha-lene forms an oval shape (A). MN forms a majority dark species (B) and a minority short-lived bright species (C). EN forms a majority dark species (D), a minority dim species (E) and a transient bright species (F). Schematic representations of the molec-ular structures of the substituted aromatics are given below the images. The position of the alkyl moiety in (B) and (D) is determined by finding the smallest protusion in the molecular images. Image F is attributed to intact EN and image D to vinylnaphthalene.
Image E is tentatively attributed to EN that has undergone one dehydrogenation step. 45
2.2 RAIRS spectra of ethylnaphthalene (EN) at 130 and 300 K on Pt(111). The 130 K
spectrum is attributed to intact EN. . . 47
2.3 Representative STM images for TFAP coadsorbed with naphthalene (A), MN (B,C) and EN (D,E) on Pt(111) at room temperature. (A), (D), (E) were acquired at a tun-nelling current of 0.7 nA whereas (B) and (C) were acquired a tuntun-nelling current of 0.3 nA. This explains the different appearance of TFAP between these images. The TFAP complexes are formed to the dim MN and EN species shown in Figure 2.1. The schematic structures shown below the images place the emphasis on whether TFAP is in interaction with the naphthyl group or with the substituent. TFAP is located at the aromatic group in structures (A), (B) and (D) and close to the substituent in structures
(C) and (E). . . 47
2.4 Representation of the π+σ-bonded η9-naphthyl adsorption geometry of the
methylnaph-thalene species after dehydrogenation. . . 49
3.1 Originally reported [101, 167] and revised structures of PNEA and its epimer. . . 54
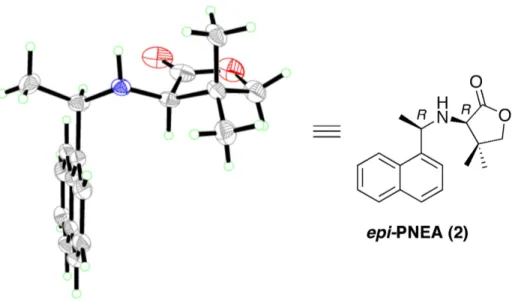
3.2 X-ray structure of epi-PNEA (2). . . . 55
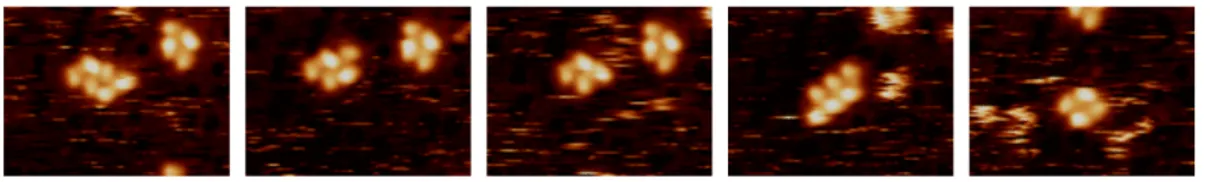
3.3 STM images for PNEA adsorbed on Pt(111) at room temperature. (A−D) Images at-tributed to adsorbed conformers of PNEA; (E, F) Images atat-tributed to two adsorbed
dissociation fragments. Imaging conditions: bias, 1−1.1 V; current, 0.3−0.4 nA. . . . 56
3.4 Proposed attribution of the protrusions in the STM image of PNEA/Pt(111) shown in
Figure 3.3 B: (1) lactone moiety, (2) ethylamine moiety, (3) naphthyl moiety.. . . 56
3.5 STM images obtained following (S)-AF adsorption on Pt(111) at room temperature:
(A) monomer, (B) dimer, (C) trimer. . . 57
3.6 STM images of supramolecular assemblies formed by the coadsorption of (S)-AF and
KPL on Pt(111) at room temperature. . . 58
3.7 Selected assemblies formed through the coadsorption of KPL and PNEA on Pt(111) at room temperature. The assemblies shown in A−E are attributed to the interaction of KPL and aminolactone species resulting from the dissociative chemisorption of a fraction of chemisorbed PNEA. Complex F is attributed to the interaction of a PNEA
molecule with one KPL. . . 59
3.8 Time-lapsed images (0, 80, 160, 240, 520 s) of supramolecular assemblies of KPL and (S)-AF in a single area (6.8 by 5.1 nm2 ) of the Pt(111) surface. The measurements
3.9 STM images of complexes formed by coadsorbed (S)-AF and TFAP on Pt(111) at room
temperature: (A) Large scan image, (B−C) (S)-AF/TFAP complexes. . . . 60
4.1 (A, B) Previously published [1] STM images and DFT calculated structures of
(R)-NEA-1 and (R)-NEA-2 on Pt(111). . . . 76
4.2 STM images of MBF on Pt(111) at room temperature. (A) Monomer MBF; (B-D) MBF
dimers. . . 77
4.3 A time-lapsed sequence of images of a MBF dimer showing interconversion between different geometries (a-f). The numbers within the colored segments of the bar indicate the number of sequential images in which the corresponding dimer configuration is seen. The measurements were performed at room temperature and each image was
acquired in 44 s. . . 78
4.4 STM images of (R)-NEA/MBF complexes formed on Pt(111) at room temperature. MBF forms complexes mainly on the right-hand side of the bright protrusion of (R)-NEA, as shown in (B-D and F) for (R)-NEA-1 and (G and I) for (R)-NEA-2. Complexes formed with MBF to the left-hand side of the bright protrusion are presented in (A and F) for (R)-NEA-1 and in (H and I) for (R)-NEA-2. Termolecular, (MBF)2/(R)-NEA, complexes are shown in (F and I). Complexes where MBF is located at the top of the
bright protrusion are presented in (E) for (R)-NEA-1 and in (J) for (R)-NEA-2. . . . . 78
4.5 Primary classification of complexes into structures formed by MBF binding to the left, top or right of (R)-NEA. Structures A-C (shown in both the top and bottom panels) are three different MBF/(R)-NEA-1 complexes. The bright protrusion locates the ethy-lamine group of the modifier. The top panel includes a color scheme as a guide to the eye. The red oval indicates the naphthyl group. The blue oval indicates the ethylamine group. The MBF molecule is indicated in green, with the narrow and wide ends of the image differentiated. The black arrow connecting the head of MBF and the ethylamine
group schematically indicates an NH···OC bond. . . 80
4.6 Comparison of published [2] DFT calculated structures and STM images for MTFP/(R)-NEA-2 complexes (A-D) with proposed structures (A’-D’) and STM images (C’, D’) for MBF/(R)-NEA-2 right-hand side complexes. The calculated complexation energies of the four MTFP/(R)-NEA-2 structures A (0.59 eV), B (0.54 eV), C (0.45 eV) and D (0.35 eV) [2]. A red X indicates a structure that is not observed in STM measurements. Proposed MBF/(R)-NEA-2 structures are drawn by replacing the CF3group of the cor-responding MTFP structure with a phenyl group. Structures A, A’, B and B’ differ from structures C, C’, D and D’, respectively, in terms of the enantioface of the substrate that
is directed toward the surface. . . 81
4.7 Comparison of STM images of right-hand side MBF/(R)-NEA-1 (A, B) complexes to structures extrapolated from previously published data [1] on TFAP/(R)-NEA-1
com-plexes (A’, B’). . . 83
4.8 Comparison of STM images of left-hand side NEA-1 (A’) and MBF/(R)-NEA-2 complexes (B’) to structures extrapolated from previously published data on
TFAP/(R)-NEA complexes (A, B). . . . 83
5.1 (A) Color-coded complexation energies of all relaxed TFAP/OH-TFAP dimer configu-rations. (B) Color-coded complexation energies of all relaxed TFAP/TFAP configura-tions. In both cases, the geometry optimisations were evaluated on two layer Pt(111)
5.2 (A-D) Representative STM images of bimolecular structures formed through TFAP adsorption on Pt(111). The images were acquired at 240 K following TFAP exposure
below 213 K. . . 90
5.3 (A) Parametrization of the geometry of bimolecular structures formed through TFAP adsorption on Pt(111). Three parameters are defined: the head-head (dhead) and body-body (dphenyl) distances, and the angle (α). (B) Proportion of short antiparallel (s-AP) structures in the total AP population under different experimental conditions. LT and RT label experiments where the adsorption of TFAP was performed below 220 K and at room temperature, respectively. The @ sign is followed by the temperature or tem-perature range at which the images were acquired. (C) Histogram of populations of structures as a function of α. (D) dphenyl-dhead scatter-plots for high angle (150-180°) structures. The latter group are all labeled as antiparallel (AP) structures. Each filled colored circle in the bottom panel represents an individual imaged structure. The α value for each structure is color-coded following the side panel. The black squares and the letters a, b refer to the specific structures A and B shown in Figure 5.2, and are included here as reference values. The pentagons refer to bimolecular structures pre-dicted by DFT calculations (Figure 5.4). The corresponding calculated complexation
energy (ΔE) is shown beside each pentagon. . . 91
5.4 Left panel: optB88-vdW DFT adsorption potential energies (ΔE) and stabilities of bimolecular structures formed by two TFAP and by TFAP and the hydroxyl species formed by adding one hydrogen atom to TFAP. The adsorption potential energy is referenced to the potential energy of two isolated adsorbed species. Right panel: Ex-ample STM images of (A) short and (B) long antiparallel bimolecular structures and
best calculated complex for (C) TFAP/TFAP and (D) TFAP/OH-TFAP structures. . . 92
6.1 Schematic illustration of a 2D potential energy landscape. In the search for the pathway from the initial, 𝑹𝐼, to the final state, 𝑹𝐹, a linear interpolation between the two points
(red dashed line in both panels) may be used as the starting guess for a NEB calculation. By diverting the pathway towards nearby local minima (using the yellow and blue vectors, 𝛽(𝑹𝐿,𝑖 − 𝑹𝑀), 𝑖 = 1, 2, 𝛽 = 0.25) better initial guesses (yellow and blue
dashed lines in lower panel) may be obtained. . . 99
6.2 Adsorption modes of TFAP on Pt(111). Upper left: Schematic drawings of possible Pt-bridge and Pt-hollow adsorption sites for TFAP. The letters A-G around the aromatic rings indicate the adsorption modes that would result if the side group was shifted to that location. Lower left: Ball models of a selection of most stable adsorption config-urations. Inserted numbers are the adsorption potential energy in eV. Right: Illustra-tion of possible elementary TFAP diffusion steps (short arrows: rotaIllustra-tions; long arrows: slides). The arrows are color-coded so that the darker the blue color the smaller the energy barrier for the step as calculated on a two-layer slab. The most favorable path-way is highlighted and each intermediate is related to its ball model by the color of the
border. . . 101
6.3 The lowest energy barrier diffusion pathway identified for TFAP. TS1, TS2 and TS3 represent 3 transistion states with 1-4 representing stable structures between the
6.4 DFT calculated adsorption geometries for (R)-NEA on Pt(111). The geometries are classified according to the naphthalene face pointing up from the surface (Re, Si), the number (6 or 7 - indicated by light grey shading) of Pt atoms coordinated to the naphthyl group, the orientation of amine group with respect to the naphthyl group (exo, endo), and the 2D-chirality of the naphthyl group footprint (-,+) as indicated by the curved arrows. The naphthyl groups in the middle row are all achiral. Nitrogen atoms are colored blue. All other atoms follow the coloring conventions in Figure 6.2 unless
otherwise indicated. . . 104
6.5 Translational diffusion pathways proposed for (R)-NEA-1 and (R)-NEA-2. For both
conformations, the translation is shown to occur from left to right in a stationary cell. 106
6.6 Rotational diffusion pathway proposed for (R)-NEA-1. Six local energy minima are found, with the overall energy barrier being defined by the transition state between 4 and 5. The molecule rotates by 60° while maintaining the nitrogen atom atop the same
underlying Pt atom. . . 107
6.7 (A) Large scale STM image after adsorption of (R)-NEA on Pt(111) at 261 K.The lines in the bottom left represent the orientation of the crystal axes. (B)-(G) Representative pairs of images taken immediately after each other showing a variety of movements of (R)-NEA. Molecular orientation is indicated by the black rulers drawn along the long axis of the dim oval protrusion that is believed to be the naphthyl group. The gray ruler in the second image of each pair recalls the position of the black ruler in the previous image. Data for (R)-NEA-1 are shown in (B)-(F) and data for (R)-NEA-2 are shown in
(G). . . 108
6.8 Time-lapsed STM images of (R)-NEA on Pt(111) taken at 268 K showing representa-tive diffusion behavior. Black rulers are defined as in Figure 6.7. In (A), a (R)-NEA-1 molecule is followed and is seen to display both translational and rotational behavior. In (B), a (R)-NEA-2 molecule is shown to display exclusively translational motion. The numbers indicate how many consecutive STM images were taken without observation of any additional motion. The images were extracted from a STM film and corrected for drift using the WsXM software package. The presence of many stationary (R)-NEA
molecules between any two subsequent images ensures a good correction of the drift. 109
6.9 (A) Histogram of (R)-NEA-2 translation amplitudes on Pt(111) at room temperature. (B) STM image of (R)-NEA-2 and definition of its axis. The black arrow represents the long axis of the naphthyl-related protrusion of the imaged molecule and is used as the reference for angles (0∘and 180∘). (C) Histogram of the orientational distribution
of (R)-NEA-2 translation events (> 0.15nm) relative to the defined axis. . . . 110
7.1 (A, B) Images d’un même TFAP qui change de prochiralité. (C, D) Image d’un substrat avec une protusion supplémentaire qui change de prochiralité. Au milieu un schéma du
mouvement montre la rotation de la liaison C-C. . . 114
7.2 Graphique représentant les principales familles de complexes entre le (R)-NEA et le TFAP entre 254 K et 262 K et la structure calculée par DFT la plus proche. 1a, 1b, 2b, 3c et 3d présentent un complexe (R)-NEA-1/TFAP et 2c présente un complexe
(R)-NEA-2/TFAP. . . . 115
7.3 Différents sites autour de l’amine du (R)-NEA. Les différents sites pour le phényle sont numérotés de 1 à 3 et les sites pour le COCF3sont classés de a à c. Par exemple, les sites 1a et 1b sont reliés par une rotation autour du phényle (rph) et les sites 1b et 2b
7.4 Paramétrisation de la géométrie des complexes entre le (R)-NEA et le TFAP. En dé-finissant deux angles θ et φ il est possible de quantifier les différentes géométries [1,
138]. Cette approche permet un classement plus facile et précise des géométries.. . . 116
7.5 Graphique représentant tous les complexes sur le (R)-NEA-1 en interaction avec l’amine entre 254 K et 262 K selon la paramétrisation définie à la Figure 7.4. Chaque point sur le graphique représente un complexe sur une image. Un complexe avec une durée de vie de N images y est donc compté N fois. On y distingue 7 groupes de complexes. Les
groupes sont identifiés avec la nomenclature présentée à la figure 7.3 . . . 117
7.6 Complexe 3d comparé avec la structure DFT stable non observée à température ambiante. 118
7.7 En haut : TFAP en complexe avec (R)-NEA-1 qui subit un changement de prochiralité pour passer de 1a (gauche) à 1b (droite). En bas : structure DFT correspondante. On voit que le phényle reste sur le même site et que l’oxygène du TFAP reste sur le même
atome de Pt. Cet atome est entouré en rouge. . . 119
7.8 Graphique représentant tous les complexes sur le (R)-NEA-1 entre 254 K et 262 K selon la paramétrisation définie à la Figure 7.4. Chaque complexe est représenté par un point indépendamment de sa durée de vie, contrairement à la figure 7.5 où chaque point représente un complexe dans une image. La taille des points et leur couleur re-présentent la durée de vie de chaque complexe, la charte de couleur sur la droite donne
la correspondance entre couleur et durée de vie en nombre d’images de présence. . . 120
7.9 Exemples d’événements de diffusion représentés sur la Figure 7.3 . . . 121
7.10 Rotation de TFAP autour du phényle lorsqu’il est en complexe. . . 121
7.11 Illustration des barrières énergétiques dans le cadre du formalisme de Curtin-Hammett
Table des schémas
3.1 Synthesis and Transformation of PNEA to lactones (+)-AF and (+)-Boc-AF. . . 53
3.2 (A) Proposed C−N Bond Scission in a Small Fraction of PNEA Adsorbed on Pt(111) in Ultrahigh Vacuum at Room Temperature To Form Aminolactone- And Ethylnaphthyl-Related Chemisorbed Fragments. (B) Proposed Dehydrogenation of the Ethylnaphthyl
Fragment To Yield Adsorbed Vinylnaphthalene. M Represents a Pt(111) Surface Site. 57
3.3 Two proposed adsorption geometries for (S)-AF. The hydrogen on the chiral carbon is
indicated in green. In structure (A) it is hidden behind the chiral carbon. . . 58
4.1 Schematic illustration of MBF adsorption. Formation of pro-R and pro-S adsorbed
states, and Pt catalysed hydrogenation to yield (R) and (S)-alcohol desorption products 75
4.2 Schematic drawing of plausible dimer structures corresponding to the dimer image
Remerciements
Je n’aurai pu accomplir autant de choses au cours de mon doctorat sans le soutien de tous les membres du laboratoire McBreen. Je tiens tout d’abord à remercier Peter pour sa confiance et son écoute atten-tive ainsi que pour l’attention qu’il porte à ses étudiants et à leur réussite. Je lui suis très reconnaissant de m’avoir permis de travailler dans des conditions exceptionnelles et de m’avoir envoyé à toutes ces conférences qui furent autant d’expériences enrichissantes. Ensuite je dois remercier Vincent qui m’a accueilli dans le laboratoire et m’a appris les ficelles du métier. Sans lui, les longues soirées d’expé-riences auraient été à la fois moins productives et moins agréables. Je remercie également ceux avec qui j’ai eu le plaisir de travailler sur le STM : Yi et Jean-Christian ainsi que tous ceux qui ont participé aux expériences d’une manière ou d’une autre, Federico, Yang, Gautier, Stéphane et Marc-André. Mon doctorat n’aurait pas été aussi enrichissant sans la participation de membres d’autres laboratoires. En premier lieu les membres du laboratoire Boukouvalas au département de chimie, pour leur colla-boration lors de la synthèse de molécules et les tests catalytiques, en particulier Raphaël, notre corres-pondant local en chimie organique mais également Richard, Vincent et Charles ainsi que John pour toutes les discussions que nous avons eues. Je tiens également à remercier les membres du laboratoire Hammer à l’université d’Aarhus pour leur participation cruciale à nos projets de recherche et égale-ment pour leur accueil chaleureux lors de ma visite, merci en particulier à Michael, Bjørk et à Anton qui m’a pris en charge lors de ma visite.
Je tiens également à remercier le département de chimie pour son soutien financier à travers les bourses Paul-Antoine Giguère et Arthur Labrie ainsi que tout le personnel de soutien du département, en parti-culier Mélanie, Sébastien, Jean et Magali pour leur aide précieuse. Je remercie également le FQRNT pour son soutien financier à travers une bourse de doctorat.
Je garde mes remerciements les plus sincères pour Pascale car son soutien et son amour qui m’ont donné une force et une confiance en moi que je n’aurai su trouver seul. Enfin je remercie ma petite Mathilde, l’amour réellement inconditionnel d’un enfant pour son parent est, je crois, la plus belle chose qu’il m’ait été donné de recevoir.
Avant-propos
Les chapitre2à5sont constitués d’articles scientifiques. Les chapitre2à6ont déjà fait l’objet d’une publication dans une revue scientifique alors que le chapitre5présente un manuscript en préparation en collaboration avec Michael Groves et le Prof. Bjørk Hammer de l’Université d’Aarhus en vue d’une soumission prochaine dans une revue scientifique. Les résultats présentés au chapitre7n’ont pas encore fait l’objet de publication.
Les travaux présentés dans cette thèse sont le résultats de travaux en collaboration avec le groupe de recherche du Prof. Bjørk Hammer en calculs de la théorie de la fonctionnelle de la densité (DFT) à l’Université d’Aarhus au Danemark pour les chapitres 5 et 6et en collaboration avec le groupe de recherche du Prof. John Boukouvalas en chimie de synthèse totale de produits naturels à l’Université Laval pour le chapitre3.
Je suis premier auteur de tous les articles présentés dans cette thèse. J’ai participé à leur écriture en collaboration avec le Prof. Peter McBreen, avec le Prof. Bjørk Hammer et Michael Groves pour les chapitres5et6et avec le Prof. John Boukouvalas pour le chapitre3. Pour tous ces articles j’ai réalisé les mesures de microscopie à effet tunnel (STM) en collaboration avec Vincent Demers-Carpentier pour les chapitres 2et3, avec Jean-Christian Lemay pour les chapitres3et5et avec Yi Dong pour le chapitre 5, 6 et 7. J’ai développé la méthodologie d’analyse semi-automatisée des images STM et effectué l’analyse, en collaboration avec Yi Dong, Yang Zheng et Jean-Christian Lemay pour les chapitre5et7. Les mesures spectroscopiques infrarouge pour le chapitre2ont été réalisés par Federico Masini et Yi Dong.
J’ai planifié les expériences et coordonné le travail de nos collaborateurs en chimie organique et en calculs théoriques. Tous les calculs DFT ont été réalisés par le Prof. Hammer, Anton Rasmussen et Michael Groves de l’Université d’Aarhus. Les synthèses de molécules organiques, tests catalytiques et les mesures de diffraction par rayons X, RMN et dichroïsme circulaire ont été réalisées par Richard P. Loach et Raphaël Lafleur-Lambert du groupe du Prof. Boukouvalas.
Introduction
0.1 Chiralité
La chiralité est la propriété des objets qui ne peuvent être superposés à leur image miroir. Le mot vient du grec χειρ qui signifie “main”. Les mains sont en effet un exemple de chiralité, la main gauche étant l’image miroir de la main droite (figure0.1). Les deux formes miroir d’un objet chiral sont appelées ses énantiomères et un mélange à 50% de chaque énantiomère est appelé un mélange racémique. La chiralité se retrouve en fait dans tout le monde vivant, aussi bien au niveau de la structure des orga-nismes (fleurs de tournesol, coquilles des escargots...) que des acides aminés, les blocs de base des protéines. Excepté la glycine, tous les acides aminés sont chiraux. En fait, ces derniers sont presque ex-clusivement rencontrés sous une seule forme énantiomérique à l’état naturel, on parle d’homochiralité du vivant.
F. 0.1 : Illustration de la chiralité, deux mains sont images miroir l’une de l’autre et les deux énan-tiomère d’un acide aminé également.
F. 0.2 : Règle de Cahn, Ingold et Prelog pour la détermination de la configuration absolue des énan-tiomères.
0.1.1 Définition
De façon générale on peut dire qu’un objet est chiral s’il ne possède pas de centre de symétrie, de plan de symétrie ou de rotation impropre (composition d’une rotation d’un angle 𝜃 et d’une réflexion par rapport à un plan 𝑃) qui conserve sa géométrie. En chimie organique, la chiralité est généralement issue de la présence d’un centre asymétrique, la plupart du temps un carbone tétravalent (on peut aussi définir des énantiomères pour les centres métalliques hexavalents). Dans ce cas on utilise les règles de Cahn, Ingold et Prelog (nomenclature R/S) pour assigner à chaque énantiomère un nom (R ou S) de façon non ambiguë en donnant un ordre de priorité aux substituant du carbone asymétrique. On place le substituant le moins prioritaire derrière le carbone et on énumère les substituants restants du plus prioritaire au moins prioritaire. Si on énumère les substituants dans le sens antihoraire, l’énantiomère est S, sinon il est R (figure0.2).
Étant donné leur proximité structurelle, les seules façons de différencier deux énantiomères sont :
• L’absorption de la lumière polarisée circulairement qui est différente pour la polarisation circu-laire gauche et circucircu-laire droite (le dichroïsme circucircu-laire).
• L’interaction avec une autre molécule chirale (utilisé en chromatographie).
• La diffraction par les rayons X pour déterminer la structure exacte du cristal de chaque énantio-mère si les énantioénantio-mères cristallisent séparément.
Parmi ces techniques, seule la troisième permet de déterminer de façon absolue quel énantiomère est le R et quel énantiomère est le S.
0.1.2 Découverte
L’existence de la chiralité dans les molécules organiques a été découverte par Pasteur en 1848 [3]. Il a en effet isolé deux formes de cristaux d’acide tartrique qui réagissent différemment à la lumière
F. 0.3 : Les deux formes miroir des cristaux d’acide tartrique.
polarisée circulairement. Les deux formes de l’acide tartrique forment des cristaux différents qui sont symétriques l’un de l’autre (figure0.3).
0.1.3 Importance technologique
À cause de l’homochiralité du vivant, les deux énantiomères d’un produit chiral peuvent avoir une activité biologique très différente. Par exemple un énantiomère peut être actif et l’autre non, l’un être un médicament efficace et l’autre un poison dangereux [4–6]. Pour ces raisons, les corps réglementaires en agroalimentaire et en pharmacologie (FDA au États-Unis, ACIA/Santé Canada au Canada) édictent des règles strictes. Il n’est pas possible de mettre sur le marché un médicament dont le principe actif est chiral sans avoir évalué l’activité des deux énantiomères et sans fournir une molécule active énantiopure si nécessaire [7].
Les deux possibilités pour arriver à une synthèse énantiopure sont les suivantes :
• Une synthèse racémique suivie d’une séparation des deux énantiomères.
• Une synthèse directement énantiosélective.
Étant donné la stabilité thermodynamique égale des deux énantiomères, si aucune source préexistante de chiralité n’est introduite dans la réaction, le produit est nécessairement un mélange racémique. La séparation des deux énantiomères est souvent difficile et toujours coûteuse puisqu’elle nécessite des
étapes de synthèse supplémentaires. De plus, sans stratégie de racémisation de l’énantiomère non désiré après synthèse, le rendement est mécaniquement divisé par 2.
Ainsi, obtenir des synthèses très énantiosélectives est un objectif très important en chimie pharmaceu-tique [6].
0.2 Catalyse
0.2.1 Principe
Si au cours d’une réaction une substance participante n’est pas consommée et si sa présence augmente la vitesse de réaction sans changer la nature des produits et des réactifs, on appelle cette substance additionnelle le catalyseur. Il est utilisé en petite quantité par rapport aux réactifs et sa présence altère la vitesse des différentes réactions concurrentes. Le catalyseur ne modifie pas la thermodynamique de la réaction, il ne modifie pas la stabilité des réactifs ou des produits, mais il permet d’abaisser, sélectivement, la barrière de réaction (figure 0.4) en changeant la nature de l’état de transition. Le catalyseur peut être sélectif envers le groupe fonctionnel qui réagit, la position d’un groupe fonctionnel donnée sur la molécule ou bien l’énantiomère produit dans le cas d’une réaction asymétrique. En supposant un processus chimique thermique, la dépendance du taux de ce processus 𝛤 avec la température absolue 𝑇 fait apparaître la barrière d’activation 𝐸𝑎𝑐𝑡 selon la loi suivante :
𝛤(𝑇 ) = 𝜈𝑒−𝐸𝑎𝑐𝑡𝑘𝐵𝑇 avec 𝑘
𝐵 la constante de Boltzmann (1)
De manière classique, on interprète le préfacteur 𝜈 comme une fréquence d’essai, c’est-à-dire la fré-quence du processus en question en l’absence de barrière d’activation. On voit que diminuer la barrière d’activation augmente exponentiellement le taux de la réaction.
Pour qu’un catalyseur soit considéré comme utile en pratique il faut qu’il présente une forte activité, ce qu’on peut mesurer par la fréquence de rotation catalytique (Turn-Over Frequency, TOF), qu’il soit stable, c’est-à-dire qu’il permette d’effectuer un grand nombre de réaction avant de devenir inefficace, ce qu’on mesure par le nombre de rotation catalytique (Turn-Over Number, TON) et également qu’il soit sélectif pour la réaction désirée. Souvent, un compromis doit être trouvé entre stabilité et activité.
0.2.2 Différentes méthodes de catalyse
Catalyse homogène
En catalyse homogène, le catalyseur se situe dans la même phase que les réactifs, généralement la phase liquide. Cette forme de catalyse se rencontre principalement avec des catalyseurs organométalliques [8], organiques, on parle d’organocatalyse [9,10] ou avec des enzymes [11]. L’avantage de cette forme de catalyse est la simplicité relative des sites catalytiques et leur bonne compréhension. La suppression du catalyseur après la réaction demande cependant des étapes supplémentaires puisqu’il est en solution
F. 0.4 : Profil énergétique d’une réaction sans catalyseur (ligne pleine) et avec un catalyseur (ligne pointillée). La barrière d’activation est réduite de 𝐸∗à 𝐸∗
𝑐
avec les produits. Cette méthode est rencontrée en particulier en chimie pharmaceutique et en synthèse organique [12–14].
Catalyse hétérogène
En catalyse hétérogène, le catalyseur ne se trouve pas dans la même phase que les réactifs, en géné-ral le catalyseur est solide et les réactifs en phase liquide ou gazeuse. Les formes de catalyseurs les plus courantes sont les oxydes poreux [15] et les nanoparticules métalliques supportées sur un oxyde [16]. Dans le domaine de la photocatalyse on trouve également beaucoup de réactions catalysées par des oxydes comme le TiO2[17]. Les catalyseurs hétérogènes sont généralement complexes car ils pré-sentent une variété de sites catalytiques différents dans une concentration généralement inconnue qui dépend de la géométrie exacte des particules de catalyseur [18,19]. L’avantage de la catalyse hétéro-gène est la facilité de récupération du produit puisqu’il suffit de le filtrer et de le prétraiter au besoin avant réutilisation. La catalyse hétérogène permet également la mise en place de procédés en continu avec l’utilisation de réacteurs à écoulement (“flow chemistry”). Pour ces raisons, la catalyse hétérogène se prête très bien à la mise à grande échelle et représente de gigantesques volumes de production en industrie pétrochimique, pour la production d’engrais ou la dégradation des polluants [20–22]. Pour le reste de cette section, on se concentrera sur la catalyse hétérogène puisque c’est le domaine de recherche dans le cadre duquel se place mes travaux de thèse.
0.2.3 Importance technologique
On estime que 85% des carburants et des produits de l’industrie chimique utilisent la catalyse hétéro-gène [18]. La pétrochimie, avec notamment le procédé Fischer-Tropsch qui permet de synthétiser des hydrocarbures utiles (principalement des alkanes) à partir de gaz de synthèse [21] et le procédé
Haber-Bosch qui permet la production d’ammoniac à partir d’azote et d’hydrogène gazeux pour la production d’engrais azotés [22] sont des exemples de production de masse qui sous tendent une bonne partie de l’industrie moderne. La catalyse hétérogène est également utilisée dans tous les véhicules récents dans le pot catalytique pour oxyder le monoxyde de carbone et les hydrocarbures non brûlés ainsi que pour réduire les oxydes d’azote [20].
0.2.4 Défi environnementaux qui nécessitent son amélioration
Entre autres applications de la catalyse, notamment hétérogène, se trouve la décontamination des pol-luants [20], la production de carburants issus de sources renouvelables [23,24] et la production de molécules organiques de base pour la synthèse chimique utilisant des sources renouvelables, comme la biomasse [25]. Aucun des défis reliés à la production renouvelable de ressources ne pourra être résolu sans faire appel à la catalyse.
La synthèse de molécules organiques et de biocarburants à partir de la biomasse comme la lignine [26] ou la cellulose [25, 27, 28], polymères d’alcools (monolignols) et de glucose respectivement, est une voie de synthèse intéressante. La catalyse, hétérogène ou homogène peut être utilisée pour transformer la lignine en composé aromatiques simples qui peuvent être intégrés aux techniques de synthèse usuelles [26]. De la même manière, la catalyse hétérogène est développée pour transformer la cellulose en sucres, alcools ou glycols en demandant moins d’énergie que la gasification complète en gas de synthèse [24,27,28].
0.3
Sélectivité et mécanismes en catalyse hétérogène
0.3.1 Complexité du catalyseur hétérogène et nature du site actif
Un catalyseur hétérogène est un matériau complexe qui présente en général plusieurs types de sites actifs en compétition. Ces sites peuvent être représentés par :
• Différents plans cristallins à la surface [29]
• Des atomes sous-conjugués au bord d’une terrasse [30–32]
• Des défauts comme un atome manquant, un atome en trop ou une impureté [33,34] • L’interface avec le support [35]
• Des joints de grain au sein d’un polycristal [23]
Le nombre de ces sites peut varier avec la taille des particules, le type de support ou les conditions expérimentales [36,37]. Il faut ajouter à cela la prise de conscience récente que les sites actifs possèdent un comportement dynamique et qu’ils ne présentent pas une activité constante au cours du temps, comme les protéines qui alternent des périodes de travail et de sommeil [18]. La compréhension du
F. 0.5 : Illustration de la structure macroscopique, microscopique et nanoscopique (A) d’un cataly-seur oxyde mésoporeux et (B) d’un catalycataly-seur métallique supporté [18]. Reproduit depuis Buurmans, I.L.C ; Weckhuysen, B.M. ; Nature Chemistry, 2012, 4, 873-886 ©Nature Publishing Group 2012.
F. 0.6 : Différents sites rencontrés à la surface d’un cristal outre les terrasses atomiques : (1) atome supplémentaire (2) et (3) atomes manquants à une marche et dans une terrasse atomique (4) Coin et (5) marche atomique.
site actif est au cœur de la recherche moderne en catalyse hétérogène. Dans les sections suivantes on présentera plusieurs approches fructueuses pour étudier le comportement des sites actifs et définir leur structure.
0.3.2 Spectroscopie in-situ et operando
Une des approches qui s’est développée le plus dans les dernières années est l’étude des catalyseurs sous des conditions réalistes, “in-situ”, ou même pendant le déroulement de la réaction, les études
“operan-do” [38,39]. Ces techniques permettent d’étudier l’effet des conditions extérieures et de l’avancement de la réaction sur le catalyseur. Les techniques operando consistent à combiner des techniques de spec-troscopie pour étudier le réacteur catalytique avec la spectromètrie de masse ou la chromatographie pour mesurer l’avancement de la réaction en temps réel. Cette étude nécessite la combinaison de plu-sieurs techniques complémentaires avec pour but une résolution temporelle et spatiale suffisante pour analyser la variation du comportement des sites en fonction de leur position dans le catalyseur. Les expériences operando combinent généralement spectroscopie UV-visible et spectroscopie vibra-tionnelle (infrarouge et Raman) [40] avec parfois des mesures d’absorption des rayons X, comme l’EXAFS [40,41].
Un des défis des méthodes de spectroscopie est leur faible résolution spatiale. Il est possible d’obtenir une résolution de l’ordre de 15-50 nm avec la micro-spectroscopie rayon X [42–44] ou bien d’utili-ser le rayonnement synchrotron pour obtenir une résolution entre 10 µm et 100 µm en spectroscopie vibrationnelle infrarouge [44]. Pour obtenir du signal qui vient uniquement de sites actifs il est pos-sible d’utiliser la spectroscopie par fluorescence [45], elle a été utilisée en conditions operando avec des marqueurs spécifiques pour différents sites pour l’analyse de l’acidité de Bronsted d’un catalyseur d’oligomérisation [46]. Ce type de techniques demande des marqueurs spécifiques et n’est pas forcé-ment généralisable. En utilisant les techniques de microscopie par fluorescence issues de la recherche en biologie il est possible d’obtenir de l’information sur des sites uniques et de mesurer leur activité [33]. Ceci n’est cependant possible que pour une réaction qui transforme une molécule non fluorescente en molécule fluorescente.
Une méthode qui permet l’étude de catalyseurs à l’échelle du site unique est la microscopie à transmis-sion électronique (TEM) “environnementale” c’est-à-dire sous atmosphère gazeuse pour se rapprocher des conditions réactionnelles [41, 47,48], ceci permet d’étudier la structure atomique du catalyseur en fonction de l’environnement. Cette technique est cependant difficile à mettre en place puisque le TEM demande normalement le vide pour permettre la transmission des électrons et ne permet pas de travailler dans les conditions réelles de pression.
0.3.3 Science des surface
Les processus de catalyse hétérogène sont intrinsèquement des processus de surface, il n’est donc pas étonnant que des techniques spécifiques à l’analyse de surface soient utilisée dans ce domaine. De
nombreuses techniques de surface nécessitent l’utilisation de conditions ultra-haut vide (UHV, cha-pitre1). Cependant certaines techniques se prêtent également à l’analyse in-situ. Par exemple l’étude par spectroscopie photo-électronique par rayons X (XPS) permet, après une modification complexe de l’instrument, l’étude sous une atmosphère gazeuse, ce qui a permis d’étudier l’état de surface de nanoparticules de Pt-Pd et Rh-Pd et de montrer que la concentration des deux composants en surface varie de façon très importante selon que l’atmosphère est oxydante ou non [36,37].
Les techniques de microscopie à sonde locale (SPM) permettent l’analyse dans l’air ou en phase li-quide. En particulier il est possible d’utiliser des montages de microscopie à force atomique (AFM) ou de microscopie à effet tunnel (STM) pour mesurer des spectres Raman en utilisant l’effet TERS (Tip Enhanced Raman Spectroscopy, spectroscopy raman exaltée par la pointe) [49,50]. Cet effet est une variante du SERS (spectroscopie Raman exaltée par une surface), qui permet d’augmenter le signal Raman de plusieurs ordres de grandeurs dans les “points chauds” sur des métaux plasmoniques (or et argent principalement) [51]. Les points chauds sont les endroits où un espaces de quelques nanomètres séparent deux régions du métal, ces endroits peuvent être une cavité dans le matériau ou bien l’espace en deux particules. En TERS, l’espace entre la pointe (AFM ou STM) et l’échantillon sert d’environ-nement propice (de “point chaud”) pour cet effet. Il est alors possible de détecter un signal issu d’un faible nombre de molécules voire de molécules uniques. Ceci peut être utilisé pour suivre une réduc-tion photocatalytique [52] et mesurer des changement dans une couche de réactif pendant la réaction. Le TERS est une technique qui ne nécessite pas de marqueur fluorescent ce qui la rend théoriquement plus facile à généraliser que les techniques de microscopie par fluorescence. Il est cependant néces-saire d’utiliser une pointe en argent ou or et une surface du même métal pour la détection de molécules uniques.
Les techniques de science des surfaces sont cependant utilisées la plupart du temps sous UHV et le plus souvent sur une face bien définie d’un monocristal. Ceci permet de contrôler parfaitement les conditions expérimentales et d’éliminer les contaminants non désirés. Ainsi le système étudié n’est pas nécessairement identique à celui présent en solution mais il permet d’élucider certains des mécanismes fondamentaux qui sous-tendent la catalyse et se transfèrent souvent à la compréhension de systèmes réels. On peut donner en exemple le travail de Gerhard Ertl [22,53] qui a reçu le prix Nobel de chimie pour ses études des réactions sur des surfaces solides en particulier l’oxydation du CO ou la synthèse de l’ammoniac (procédé Haber-Bosch), dans ce dernier cas, ses travaux ont permis de déterminer la nature d’intermédiaires en surface. Un autre exemple est l’étude des catalyseurs pour l’hydro-désulfuration par STM par l’équipe de Fleming Besenbacher. Ces études par microscopie à effet tunnel (STM) a permis d’isoler la structure des particule de MoS2et de catalyseurs CoMoS [54–56] ou de catalyseurs pour le vaporeformage d’hydrocarbures [57]. D’autres études ont permis la détermination par STM de sites catalytiques asymétriques et de leur sélectivité [1] ou bien l’étude du débordement entre le catalyseur et son support [58,59].
Avant de terminer cette section, on présentera une technique SPM qui permet d’obtenir une résolution atomique, y compris dans les molécules organiques, c’est l’AFM en mode sans contact (NC) avec une
molécule (généralement du CO) adsorbé à l’extrémité de la pointe. Ces expériences, en exploitant les interactions de très courte portée comme la répulsion de Pauli, fournissent une résolution inégalée. Cette technique a été utilisée pour déterminer la structure de molécules organiques [60] et récemment a permis d’observer des liaisons hydrogène [61] ce qui permet de penser qu’elle offre des possibilités pour l’étude de la catalyse hétérogène et d’assemblages sur une surface.
0.3.4 Travaux théoriques
Une grande partie des travaux présentés précédemment contiennent une partie théorique, en général sous la forme de calculs de théorie fonctionnelle de la densité (DFT). Les études DFT permettent de compléter les études de science de surface en assignant une structure atomique précise aux géométries observées [1]. Les études DFT se sont montrées également capables de déterminer des tendances dans la réactivité et de prédire la réactivité de divers catalyseurs [62,63]. Un avantage de la DFT est qu’il est possible d’essayer de nombreuses combinaisons de catalyseurs et d’analyser séparément plusieurs sites (terrasses, marches atomiques, défauts...) pour prédire leur réactivité. C’est donc un outil exploratoire puissant pour l’étude de nouveaux catalyseurs.
0.3.5 Catalyse asymétrique
Comme expliqué à la section0.1.3, la catalyse asymétrique est fondamentale en chimie pharmaceu-tique et agroalimentaire. Les catalyseurs homogènes sont le plus souvent utilisés puisqu’ils présentent une meilleure activité et sélectivité, en grande partie grâce à leur site catalytique généralement bien défini. La catalyse asymétrique homogène a été récompensée par le prix Nobel de chimie en 2001 [13,
64,65]. En catalyse asymétrique homogène on trouve des catalyseurs organométalliques [13,66], des catalyseurs organiques [9,10,67] et des catalyseurs enzymatiques [11]. Les désavantages de ces mé-thodes sont principalement la difficulté de séparation du catalyseur après la réaction, qui nécessite des étapes supplémentaires et des coûts importants et le problème de contamination par des métaux pour les catalyseurs organométalliques. En raison des avantages de la catalyse hétérogène décrit à la section
0.2.2, le développement de catalyseurs asymétrique hétérogènes reste un objectif important [68,69]. C’est le domaine de recherche dans lequel se placent mes travaux de thèse.
On peut dénombrer plusieurs types de catalyseurs hétérogènes asymétriques. On trouve en premier lieu les catalyseurs homogènes immobilisés sur un substrat, cette méthode consiste à utiliser un catalyseur homogène performant et à essayer transférer cette activité et sélectivité sur un support qui peut com-porter des nanopores ou mesopores, l’étude de catalyseurs supportés sur des particules de polymères est également explorée [70,71]. C’est une technique prometteuse mais les catalyseurs résultants sont souvent moins actifs que leur contrepartie homogène libre.
Une autre méthode est l’utilisation de matériaux mésoporeux chiraux, elle permet de créer des cata-lyseurs non supportés présentant une grande densité de sites bien définis [72–74]. La synthèse de ces matériaux est encore un sujet de recherche actif et le lien structure-sélectivité n’est pas encore assez
F. 0.7 : Le 2,2,2-trifluoroacétophénone (TFAP) peut être adsorbé de deux façons sur la surface selon le côté de la molécule (énantioface) en contact avec la surface. On défini alors deux prochiralités [77]. compris pour qu’on puisse prédire le comportement d’une structure donnée. Cette approche permet également d’immobiliser un catalyseur organique asymétrique dans le matériau [75].
La dernière méthode est la modification chirale d’une surface de catalyseur achirale. Cette méthode est particulièrement développée pour des réactions d’hydrogénation. Les catalyseurs métalliques utilisés sont le Pt, le Pd ou le Ni [66,76]. Elle consiste à adsorber à la surface du catalyseur un énantiomère d’une molécule organique appelée modificateur chiral. Ce faisant un site catalytique énantiosélectif est créé lorsque le réactif se lie au modificateur de manière réversible, en général par des liaisons hydrogène. Ces liaisons vont maintenir une face spécifique du réactif en contact avec la surface (figure
0.7). Étant donné que l’hydrogène est ajouté au réactif depuis la surface du métal, l’hydrogénation par une seule énantioface et donc la formation d’un seul énantiomère est favorisée. On peut définir alors la notion de prochiralité : un réactif prochiral est une molécule qui peut devenir chirale après une réaction (ici l’ajout de 2 atomes d’hydrogène). Un réactif prochiral proR va devenir un produit R après réaction et un réactif prochiral proS deviendra un produit S. Ce qui va différencier un réactif proR et proS est la face du réactif en contact avec la surface du catalyseur (figure0.8). On peut ainsi dire que le principe de la réaction asymétrique est de stabiliser une forme prochirale sur la surface ou bien d’accélérer la réaction d’une forme prochirale par rapport à l’autre.
Deux types de réactions sont connues pour donner des excès énantiomériques important avec cette méthode : l’hydrogénation énantiosélective de beta céto-esters sur le Ni [78] et l’hydrogénation énan-tiosélective de cétones activées (alpha céto-esters et trifluorocétones [79] notamment) sur le Pt ou le Pd [80], cette dernière réaction étant connue sous le nom de réaction d’Orito [81,82]. Les chapitres3
F. 0.8 : La prochiralité du réactif contrôle la chiralité du produit puisque l’hydrogène est ajouté au réactif depuis la surface. Un réactif proR produit un énantiomère R et un réactif proS produit un énantiomère S.
et4sont consacrés à son étude. C’est le sujet de la section suivante.
0.4
Catalyse hétérogène asymétrique : hydrogénation de cétones
activées sur le Pt supporté
0.4.1 Description et applicabilité
Dans cette réaction, la symétrie d’un catalyseur achiral comme le Pt est brisée par l’adsorption d’un énantiomère d’une molécule organique appelé modificateur chiral. Le modèle généralement accepté pour les interactions entre le modificateur est le complexe entre un modificateur et un réactif à la fois (complexes 1 :1). Des mesures catalytiques ont supporté cette hypothèse [68,83] et des mesures di-rectes par STM ont montré des complexes 1 :1 et 1 :2 [1]. Cette réaction fonctionne avec de nombreuses cétones (figure0.9) mais le nombre de réactif qui permettent d’atteindre des excès énantiomériques (ee) supérieurs à 90% est plus limité. Les réactifs qui offrent une grande sélectivité ont tous en com-mun un groupe électrophile en alpha de la cétone comme le 2,2,2-trifluoroacetophenone, TFAP (92 % ee, c’est une des seules trifluorocétones présentant un bon excès énantiomérique), ou les alpha céto-ester comme le méthyle pyruvate, MP (98 % ee), le cétopantolactone, KPL (92 % ee) et le méthyle benzoylformate, MBF (98 % ee) [68,76,83–90].
Les deux axes de recherche les plus actifs sont la compréhension des mécanismes pour les réactifs qui présentent une bonne sélectivité et l’augmentation du nombre de systèmes qui offrent une bonne énantiosélectivité. Pour atteindre ces buts, nous devons d’abord nous intéresser à ce qui fait un bon modificateur chiral.
F. 0.9 : Sélection de réactifs étudiés pour la réaction d’Orito avec le meilleur excès énantiomérique répertorié [68,76,83–90].
0.4.2 Modificateurs chiraux
Le modificateur doit remplir deux rôles pour offrir un haut excès énantiomérique :
• Le modificateur doit interagir sélectivement avec une des deux formes prochirales du réactif. Ceci facilite alors la production d’un énantiomère du produit.
• Le modificateur doit également accélérer la production de l’énantiomère désiré. Augmenter le nombre de sites sélectifs n’est en général pas suffisant car la réaction racémique, c’est-à-dire sur des sites non modifiés du catalyseur, est toujours présente. En formant une liaison hydrogène avec le groupement à hydrogéner, le carbonyle, le modificateur l’active et favorise son hydrogénation en abaissant la barrière d’hydrogénation [68,91,92].
Le second effet, l’accélération de la réaction en présence du modificateur, est observé pour la grande majorité des couples modificateur/réactif sélectifs [93]. Un contre-exemple intéressant est le TFAP pour lequel l’accélération est faible (10-30%) [94].
Des études en science de surface par STM au sein de notre groupe ont montré que le TFAP forme des dimères stables à température ambiante. Ces dimères pourraient avoir un effet sur la cinétique de la réaction [95–97]. Ces assemblages de TFAP sont étudiés plus précisément dans le chapitre5de cette thèse.
Parmi les modificateurs connus, on se concentrera sur deux modificateurs bien étudiés, la cinchonidine (CD), le 1-(1-naphtyl)éthylamine (NEA) et leur dérivés. Dans tous les modificateurs efficaces connus, trois caractéristiques sont nécessaires :
F. 0.10 : Trois modificateurs chiraux, (A) la cinchonidine, (B) le NEA, (C) le pantoyl-naphtyléthylamine (PNEA). Le chiffre 1 indique le groupement aromatique, 2 indique l’amine et 3 indique le carbone chiral entre les deux groupes précédents.
• Un groupement polyaromatique, généralement formé de deux cycles, qui permet une forte chi-misorption du modificateur sur la surface. [76]
• Un groupement donneur de proton pour une liaison hydrogène, généralement une amine [68]. • Une centre stéréogénique à proximité des deux fonctionnalités décrites précédemment [76,83]. Le rôle exact du groupement aromatique n’est pas nécessairement limité à un rôle d’ancre, des études par STM ont montré que ces groupements sont capables de former des liaisons hydrogène qui par-ticipent à la sélectivité de la réaction [2]. La possibilité de former des liaisons avec des hydrogènes aromatiques et alkyls est explorée dans le chapitre2de cette thèse.
Le modificateur chiral découvert initialement [81] et le plus étudié par la suite est la cinchonidine, un alkaloïde naturel (figure0.10). C’est également le modificateur qui offre la meilleure sélectivité pour un grand nombre de réactifs [76,83]. La CD est un diastéréoisomère, elle possède une amine tertiaire, un groupement aromatique quinoline comme ancrage et un carbone asymétrique entre les deux. Le diastéréoisomère cinchonine (CN) donne l’énantiomère inverse de la CD, en général avec un excès énantiomérique plus faible [76]. Le solvant peut également avoir un effet important sur la sélectivité en particulier en modifiant la conformation de la CD. Dans de nombreuses conditions expérimentales, l’amine est protonée [68]. La plupart des études concluent que la quinoline est adsorbée parallèle à la surface [68].
Le NEA est le modificateur chiral synthétique le plus étudié. Il possède une structure plus simple que la CD, en particulier il ne présente qu’un seul centre stéréogénique et il est plus facile à modifier syn-thétiquement. Il contient une amine primaire et un groupement naphtyle. Plusieurs études ont montré que le groupement naphtyle est adsorbé parallèle à la surface [1,98] mais des résultats récents sur le platine polycristallin a remis en cause ce résultat au moins pour un haut recouvrement de modificateur [99]. Les travaux du groupe d’A. Baiker ont montré que le NEA peut condenser in-situ avec le methyl-pyruvate pour former un nouveau modificateur [83,100]. Par la suite de nombreuses condensations ont été testées et testées pour l’hydrogénation asymétrique du cétopantolactone (KPL) [101]. Parmi
F. 0.11 : Les deux conformères de NEA sur le Pt(111), (A) NEA-1, la forme exo et (B) NEA-2, la forme endo.
ces produits de condensation, le pantoyl-naphtyléthylamine (PNEA), sur la Figure0.10, montre la plus grande sélectivité. Ce modificateur et sa décomposition in-situ en aminolactone sur le catalyseur sont étudiés dans le chapitre3. Bien que le NEA soit plus simple que les alkaloïdes de la famille des cin-chona, les images STM ont montré qu’il existe deux conformères de NEA sur la surface (figure0.11) et que ces deux formes peuvent présenter une activité différente.
0.4.3 Étude de sites sélectifs
Les études sur la réaction d’Orito ont montré qu’il n’est pas possible de trouver un mécanisme de transfert de chiralité qui puisse expliquer le fonctionnement d’un modificateur pour tous les réactifs. Le mode d’interaction de chaque couple modificateur/réactif lui est potentiellement spécifique. Les études sur le NEA et deux réactifs, le TFAP et le methyle trifluoropyruvate, MTFP, (figure 0.12) en sont un exemple. Ainsi pour le MTFP, il existe une géométrie d’interaction dominante, ce qui n’est pas le cas pour le TFAP [1,2]. Dans ce site catalytique dominant, le réactif est lié au modificateur par des liaisons hydrogène à la fois avec l’amine et le cycle aromatique (figure0.12). Il en résulte qu’un des deux conformères du NEA est beaucoup plus actif que l’autre pour la formation de complexes.



![Figure 3.1: Originally reported [101, 167] and revised structures of PNEA and its epimer.](https://thumb-eu.123doks.com/thumbv2/123doknet/6591473.178862/82.918.268.603.112.537/figure-originally-reported-revised-structures-pnea-epimer.webp)