T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul SabatierDiscipline ou spécialité : Cancérologie
JURY
Rodriguez-Lafrasse Claire - Rapporteur Bettaieb Ali - Rapporteur
Iovanna Juan - Rapporteur Levade Thierry - Examinateur Le Stunff Hervé - Examinateur
Ecole doctorale : Biologie Santé et Biotechnologies Unité de recherche : I2MR- U858- Equipe 16
Directeur(s) de Thèse : Christiane Susini/Corinne Bousquet Rapporteurs :
Présentée et soutenue par LISE DAVENNE Le 15 décembre 2008
Titre : Implication des métabolites sphingolipidiques dans la prolifération, la survie et la
UNIVERSITE PAUL SABATIER – TOULOUSE III Ecole Doctorale Biologie-Santé-Biotechnologies
THÈSE
Présentée en vue de l’obtention du DOCTORAT de l’Université Paul Sabatier Spécialité CANCEROLOGIE
par
Lise DAVENNE
IMPLICATION DES METABOLITES SPHINGOLIPIDIQUES DANS LA
PROLIFERATION, LA SURVIE ET LA REPONSE DES CELLULES
CANCEREUSES PANCREATIQUES AUX MOLECULES
CHIMIOTHERAPEUTIQUES
Soutenue le 15 Décembre 2008 devant un jury composé de :
- Pr. Claire Rodriguez-Lafrasse, Professeur des Universités, Lyon Rapporteur - Pr. Ali Bettaïeb, Professeur des Universités, Dijon Rapporteur - Dr. Juan Iovanna, Directeur de Recherche, U624, Marseille Rapporteur - Pr. Thierry Levade, Professeur des universités, UPS, Toulouse Examinateur - Dr. Hervé Le Stunff, MCU, UMR 8619, Pais 11 Examinateur - Dr. Christiane Susini, Directeur de Recherche, U858, Toulouse Directrice de thèse - Dr. Corinne Bousquet, Chargée de Recherche, U858, Toulouse Co-responsable de thèse
Institut de Médecine Moléculaire de Rangueil (I2MR), INSERM U858, Equipe 16 CHU Rangueil, 1 avenue Jean Poulhès, 31432 TOULOUSE CEDEX 4
REMERCIEMENTS
Je voudrais tout d’abord remercier le Pr Thierry Levade, Président du jury, les Pr Rodriguez-Lafrasse et Bettaïeb ainsi que les Dr Iovanna et le Stunff, membres du jury, d’avoir accepté d’évaluer mon travail et d’assister à sa défense.
Je tiens à remercier le Dr Christiane Susini de m’avoir accueillie au sein de son équipe et d’avoir suivi, avec le Dr Corinne Bousquet, la progression de mon travail.
Merci également aux autres membres de l’équipe 16 : au Dr Stéphane Pyronnet pour ses conseils scientifiques avisés, à Nathalie Saint-Laurent pour son aide précieuse à tous points de vue et à Audrey Sicard qui a contribué à l’avancée de ce travail
Je voudrais aussi exprimer toute ma reconnaissance et mon affection aux « Susinettes », Amandine, Hanane, Rania, Séverine et Souad. Merci pour votre bonne humeur, votre soutien et nos fous-rires. Je n’oublierai jamais avoir fait partie de ce clan !
Je suis également reconnaissante à tous nos voisins de pallier, membres des équipes 12 et 13, pour leurs conseils techniques, les dépannages de produits ou encore de matériel (n’est-ce pas Pierre ?).
Une mention spéciale aux indéfectibles de la brigade de la cantine (Nathalie, Hanane, Véronique, Pascal, Amandine…) grâce à qui j’ai appris beaucoup de choses au cours de discussions plus ou moins animées. Merci pour ces grands moments de détente et de rire (et de sérieux un peu parfois aussi !).
Enfin, je souhaite dire ici toute ma gratitude et ma reconnaissance à ma famille et mes amis proches sans qui rien de tout cela n’aurait été possible. Merci du fond du cœur !
Résumé
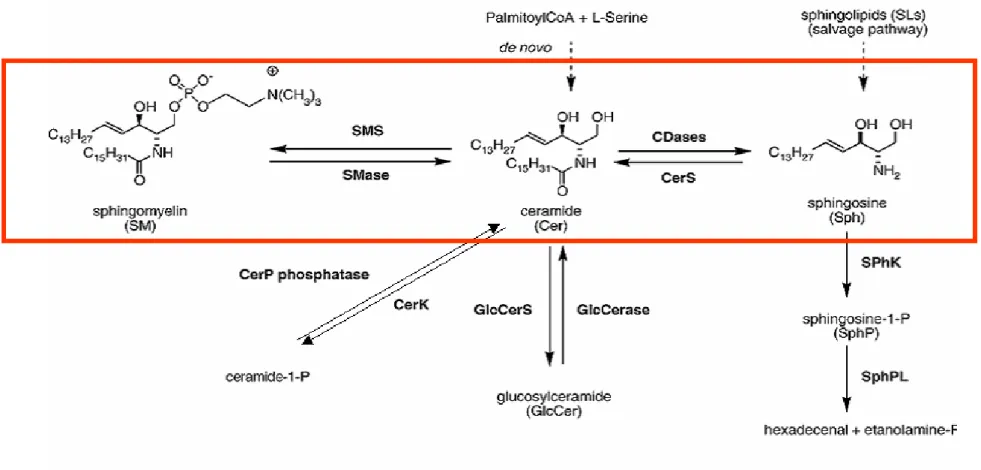
Les métabolites sphingolipidiques représentent une nouvelle classe de messagers lipidiques impliqués dans la régulation de la prolifération et de la mort cellulaire. Parmi ces messagers, le céramide, la sphingosine 1-phosphate (S1P) et les enzymes sphingosine kinases 1 et 2 (SphK1 et SphK2), phosphorylant la sphingosine, métabolite du céramide, en S1P, sont particulièrement impliqués dans différents aspects du cancer (son développement, sa progression et la réponse aux traitements). L’existence d’un rhéostat sphingolipidique entre le céramide (apoptotique) et la S1P (pro-survie) décidant de la survie ou de la mort cellulaire fait de ces lipides des cibles potentielles pour l’amélioration des stratégies thérapeutiques anti-tumorales actuelles.
Le premier objectif de ce travail a été de rechercher le rôle du rapport céramide/S1P dans la résistance des cellules cancéreuses pancréatiques à la gemcitabine, agent chimiothérapeutique qui dans le cas du cancer pancréatique améliore la qualité de vie des patients mais n’allonge pas leur durée de vie. Nos résultats ont tout d’abord révélé une forte expression de la SphK1 dans les tumeurs pancréatiques alors qu’elle est absente du pancréas sain. De plus, nous avons mis en évidence une corrélation inverse entre la sensibilité à la gemcitabine et l’activité de la SphK1 dans quatre lignées cellulaires cancéreuses pancréatiques. Nous avons ensuite montré qu’un rapport céramide/S1P élevé associé à une faible activité SphK1 corrèle avec une plus grande sensibilité à la gemcitabine dans ces cellules. L’augmentation de ce ratio par ajout de céramide exogène ou par inhibition de la SphK1 (par inhibition pharmacologique ou siRNA spécifique) diminue la survie des cellules cancéreuses pancréatiques et sensibilise les cellules les plus résistantes à la gemcitabine. A l’inverse, une diminution de ce ratio par augmentation de S1P grâce à la surexpression de la SphK1 rend ces cellules encore plus résistantes à la gemcitabine et augmente leur prolifération. Enfin, nos résultats suggèrent que l’effet sensibilisant du céramide exogène ou de l’inhibition de SphK1 serait la conséquence d’une diminution de l’activité du facteur de transcription NF-κB, déjà connu pour son rôle dans les phénomènes de résistance des cellules cancéreuses pancréatiques à la gemcitabine.
La seconde partie de mes travaux a consisté à rechercher dans plusieurs lignées pancréatiques humaines l’expression et les effets biologiques de la SphK2, qui contrairement à la SphK1, aurait des effets anti-prolifératif et pro-apoptotique. Les résultats obtenus ont montré que seule la lignée pancréatique ductale « saine » HPDE exprime la SphK2 alors que quatre lignées cancéreuses ne l’expriment pas. L’inhibition d’expression de la SphK2 par siRNA dans les cellules HPDE entraîne une augmentation de la prolifération cellulaire, suggérant un rôle anti-prolifératif de cette protéine. De même, la réexpression conditionnelle de cette kinase dans la lignée cancéreuse pancréatique Panc-1 diminue la prolifération et la survie cellulaires et augmente l’activité des caspases effectrices, enzymes clés du processus apoptotique. Enfin, l’induction de la SphK2 dans ces cellules xénogreffées chez la souris immuno-déprimée entraîne une perte de leur potentiel tumoral par comparaison aux cellules n’exprimant pas la SphK2.
Ces travaux ont donc permis de montrer que les métabolites sphingolipidiques jouent un rôle essentiel dans la réponse à la gemcitabine des cellules cancéreuses pancréatiques, mais également dans leur prolifération et leur survie. Ils représentent donc une cible potentielle pour l’amélioration de la thérapie de l’adénocarcinome
Abstract
Sphingolipids are a new class of lipid messengers involved in many cellular processes such as cell survival, proliferation, migration and angiogenesis. Ceramide and S1P, the major sphingolipids, exert opposite functions on cell survival ; whereas ceramide is a pro-apoptotic agent, sphingosine 1-phosphate (S1P) promotes cell growth. It has been hypothesized that the balance between these two lipids determines cell fate. This sphingolipid rheostat is in part regulated by the sphingosine kinases (SphKs), which phosphorylate sphingosine, a ceramide metabolite, to generate S1P.
Our first work aimed to investigate the implication of this sphingolipid rheostat and of SphK1 in the resistance of pancreatic cancer cell lines to gemcitabine. We first demonstrated a strong SphK1 expression in pancreatic cancer tissues while it is not expressed in normal pancreatic ducts. We also showed an interesting correlation between SphK1 activity and gemcitabine resistance in four pancreatic cancer cell lines. We then established that an elevated ceramide/S1P ratio associated to a weak SphK1 activity correlated with a better sensitivity to gemcitabine. Stickingly, inhibiting SphK1 activity (using pharmacological inhibitor or small interference RNA) decreased pancreatic cancer cell survival and sensitized the most resistant cells to gemcitabine. A similar result was obtained by addition of exogenous ceramide. Conversely, overexpressing SphK1 in these cells rendered them more resistant to gemcitabine and increased their proliferation. Taken together, these results show that the cellular ceramide/S1P ratio is a critical biosensor for predicting pancreatic cancer cell sensitivity to gemcitabine.
In a second time, we investigated in pancreatic normal and cancer cells the expression and the role of the second isoform of SphK, SphK2, which as opposed to SphK1 is described as an anti-proliferative and pro-apoptotic protein. We showed that SphK2 is expressed in human normal, but not cancerous, pancreatic cells and tissues. Inhibiting SphK2 expression (using small interference RNA) in pancreatic normal cells increased their proliferation. Conversely, re-expressing SphK2, in a tetracyclin-inducible manner, in a pancreatic cancer cell line decreased cell proliferation and survival in part by a mechanism involving the activation of executioner caspases. Interestingly, this inducible re-expression of SphK2 also impaired pancreatic cancer cells to form tumor, when sub-cutaneously xenografted in immuno-deficient mice.
All these results suggest that the sphingolipid metabolism plays a critical role in pancreatic tumorogenesis and response to chemotherapeutic treatments of pancreatic cancer.
SOMMAIRE
PARTIE I- Introduction bibliographique
Introduction
générale
1Chapitre I : Le cancer du pancréas
3A- Généralités
31-Epidémiologie et facteurs de risque 4
a- Facteurs environnementaux 4 b- Facteurs pathologiques 4 c-Facteurs génétiques 5 2- Bases moléculaires de la carcinogenèse pancréatique 5
a- Les oncogènes 6 b- Les gènes suppresseurs de tumeur 8
c- Les autres altérations dans le cancer pancréatique 9
1- Activité télomérase 10
2- La mort cellulaire programmée 10
3- L'angiogenèse 11 4- La prolifération cellulaire 11 5- Invasion tissulaire 12 6- Le microenvironnement tumoral 12 d- Modifications épigénétiques 12 e- Les microARN 13 3- Modélisation histologique de la cancérogenèse pancréatique 14
B – L’adénocarcinome pancréatique en pratique clinique
151- Diagnostic et explorations pré-thérapeutiques 15
2- Les traitements actuels du cancer du pancréas 16
a- Les traitements chirurgicaux 16 b- Les traitements palliatifs 17
3- Les thérapeutiques émergentes 19
a- Les inhibiteurs de la topoisomérase I 19 b- Les inhibiteurs de la transduction du signal 19
C- La gemcitabine
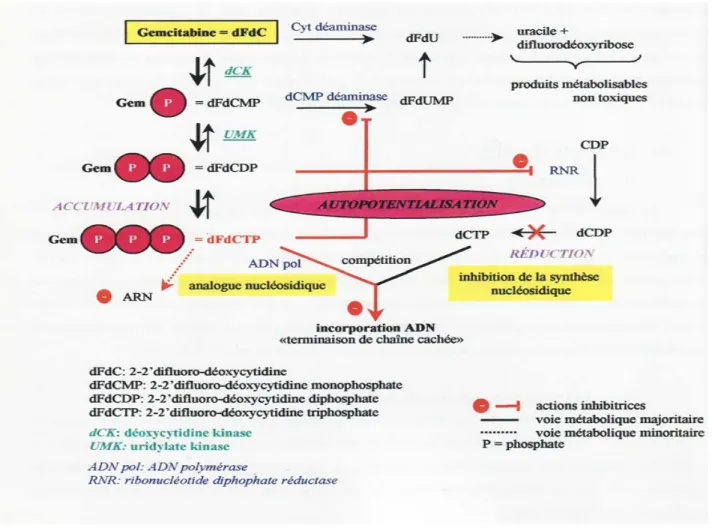
23 1- Généralités 23 2- Voies métaboliques 24 a- Transporteurs 24 b- Métabolisation intra-cellulaire 24 3- Mode d’action 25a- La gemcitabine : un analogue nucléosidique 25 b- Inhibition de la synthèse nucléosidique 25
4- La gemcitabine en clinique humaine 26
5- Mécanismes moléculaires de résistance et sensibilité à la chimiothérapie 26
Chapitre II : Les sphingolipides ou les nouveaux messagers cellulaires
30A- Le céramide : premier messager sphingolipidique
331- Métabolisme du céramide 34
a- Synthèse 34 b- Génération de céramide par les sphingomyélinases 34
c- Transport et métabolisme du céramide 37
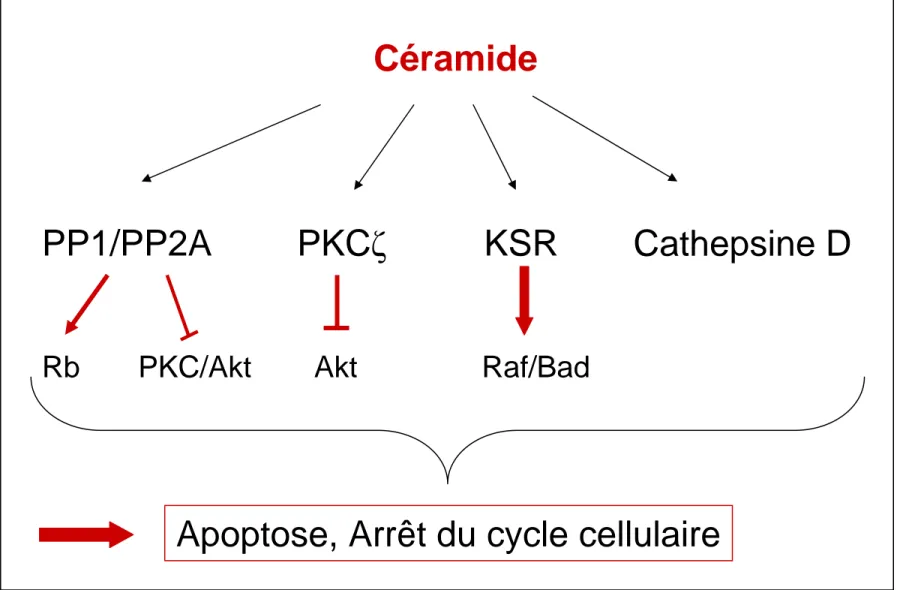
d- Interactions protéiques 38 2- Rôles physiologiques 38 a- Apoptose 38 1- Récepteurs de mort 39 2- Stress oxydatif 41 3 -Radiations ionisantes 41 4- Rayonnements UV 41 5- Apoptose mitochondriale 42 6-Molécules chimiothérapeutiques 42
b- Arrêt du cycle cellulaire 43 c- Réarrangement du cytosquelette 43
d- Autres fonctions 44
B- La sphingosine : l’ombre du céramide ?
441- Métabolisme 44
a- Synthèse 44 b- Catabolisme 45
2- La sphingosine : un messager apoptotique 45 a- La génération de sphingosine induite par le stress 45 b- Mécanismes de régulation de l’apoptose par la sphingosine 46
C- Les sphingosines kinases : des enzymes clés
481- Caractérisation des différentes sphingosine kinases 48
a- Clonage et identification 48
b- Expression 50
c- Caractéristiques structurelles 50
d- Caractéristiques enzymatiques 51
e- Invalidation génétique 52
2- Régulation de l’activité des sphingosine kinases 53
a- Mécanismes de régulation spécifiques de la SphK1 54 b- Mécanismes de régulation spécifiques de la SphK2 56
c- Régulation par la localisation subcellulaire 56 d- Signalisation «inside-out » des sphingosine kinases 58
3- Rôles biologiques 58
a-L’inflammation 58 b- Régulation de l’homéostasie calcique 59
c- Prolifération et survie cellulaires 59
d- Migration cellulaire 63 e- Régulation du tonus vasculaire 63
f- Métabolisme de la sphingosine et de la sphingosine 1-phosphate 64
g- Parallèle avec les levures 64
D- La sphingosine 1-phosphate : « l’anti-céramide »
651- Métabolisme de la sphingosine 1-phosphate 65
a- Génération de la sphingosine 1-phosphate 66 b- Catabolisme de la sphingosine 1-phosphate 66 2- S1P intra- et extracellulaires : « inside-out signaling 66
3- Rôles physiologiques de la sphingosine 1-phosphate 69
a- Prolifération et survie cellulaires 69
b- Angiogenèse 70 c- Migration cellulaire 71
d- Immunité 71 e- Inflammation 72 4- La balance sphingosine/sphingosine 1-phosphate : 73
E Conclusion 74
Chapitre III : Sphingolipides et Cancer
76A- Implication du métabolisme sphingolipidique dans la pathologie
cancéreuse
761- Modifications du métabolisme du céramide 76
a‐ Analogues du céramide 77
b‐ Inhibition des sphingomyélinases 78
1 La sphingomyélinase acide 78
2- La sphingomyélinase neutre 78
c- La céramidase acide 79 2- Modifications du métabolisme de la sphingosine 1-phosphate 79
a- Inhibition de la SphK1 80 b- Inhibition des S1PRs 83
1- Le FTY720 83
2- Le VPC23019 84
3- Le JTE-013 84
c- Anticorps bloquant dirigé contre la sphingosine 1-phosphate 84
3- Autres sphingolipides - autres rôles 85
a- Rôle de protection cellulaire 85 b- Régulation de la P-glycoprotéine 85
c- Les gangliosides 86
B- Sphingolipides et cancer du pancréas
861- Effet chimio-sensibilisant de la sphingomyéline dans les cellules Panc-1 87
2- Rôle du céramide dans la mort de cellules cancéreuses pancréatiques
induite par des cannabinoïdes 87
3- Rôle du céramide dans la mort de cellules cancéreuses pancréatiques
induite par le paclitaxel 88
PARTIE II- Résultats expérimentaux
Article 1 : La modulation du métabolisme sphingolipidique améliore la
réponse à la gemcitabine des cellules cancéreuses pancréatiques
90Introduction de l’article
91Discussion de l’article
92Article 2 : La sphingosine kinase 2 induit une diminution de la
prolifération et de la survie des cellules cancéreuses pancréatiques
95Introduction de l’article
96Discussion de l’article
97PARTIE III- Conclusion générale et perspectives
100
PARTIE IV- Références bibliographiques
103
Annexes
Table des illustrations
Figure 1 : Evolution histologique des lésions cancéreuses pancréatiques et évènements moléculaires associés 15 Figure 2 : Voies métaboliques et schéma représentatif du mode d’action de la gemcitabine. 24
Figure 3 : Représentation schématique du métabolisme sphingolipidique. 31
Figure 4 : Cibles intracelllulaires du céramide 38
Figure 5 : Représentation schématique des voies de sigalisation apoptotique. 39
Figure 6 : Structure de la SphK1. 51
Figure 7 : Structure chimique d’inhibiteurs de sphingosine kinases 51 Figure 8 : Modélisation intégrée des effets opposés des deux isoformes de sphingosine kinase sur le métabolisme sphingolipidique et la prolifération et la survie cellulaires 64 Figure 9 : Inside-out signaling » de la sphingosine 1-phosphate 68 Figure 10 : Mécanismes d’action de la S1P via ses récepteurs membranaires. 68 Figure 11 : Représentation schématique de la balance entre les effets du céramide et de la sphingosine 1-phosphate 73 Tableau 1 : Tableau récapitulatif des tumeurs surexprimant la SphK1. 80
Liste des abbréviations
BH3 : Bcl-2 Homology domain Case : céramidase
CEACAM : CarcinoEmbryonic Antigen-related Cell Adhesion Molecule CerK : céramide kinase
DISC : Death-Inducing Signaling Complex DR5 : Death Receptor 5
EGF : Epidermal Growth Factor
FADD : Fas-Associated protein with Death Domain GluCer : glucosylcéramide
GlyCer : glycosylcéramide HGF : Hepatocyte Growth Factor
MAPK : Mitogen Activated Protein Kinase NF-κB : Nuclear Factor-kappa B
NGF : Neuronal Growth Factor PDGF : Platelet D Growth Factor Pi3K : Phosphoinositide 3-kinase PKB/Akt : Protéine Kinase B PKC : Protéine Kinase C S (Ser) : sérine
SAPK : Stress Activated Protein Kinase Smase : sphingomyélinase
SphK1 : sphingosine kinase 1 SphK2 : sphingosine kinase 2 T (Thr) : thréonine
TNFα : Tumor Necrosis Factor alpha TRAF2 : TNF Receptor Associated Factor VEGF : Vascular Endothelial Growth Factor
Introduction
Introduction
générale
Le cancer du pancréas, dont l’incidence est en forte progression, est situé au cinquième rang des causes de décès par cancer dans les pays occidentaux et se place au quatrième rang de fréquence des cancers digestifs. Les causes de son mauvais pronostic (3,5% de survie à 5 ans) sont multiples au rang desquelles l'on retrouve : une agressivité responsable d'une invasion précoce par voies lymphatique et nerveuse, un diagnostic tardif en raison de signes cliniques de début de la maladie absents ou non spécifiques et l'absence de marqueurs biologiques précoces disponibles en pratique clinique quotidienne, n'autorisant pas un diagnostic précoce. Le seul traitement curatif est la résection chirurgicale, instituée à visée curative dans seulement 10 à 15% des cas. La faible sensibilité de ces tumeurs à la chimiothérapie ou à la radiothérapie ne permet pas un traitement adjuvant efficace.
La plupart des lignées cancéreuses pancréatiques sont en effet résistantes à l’action cytotoxique de la gemcitabine (un analogue difluoré de la désoxycytidine), qui pourtant constitue le traitement chimiothérapeutique de première ligne de l’adénocarcinome pancréatique chez l’homme, bien qu’améliorant faiblement la survie des patients. Les mécanismes de cette résistance impliquent entre autres l’activation excessive de voies de survie (Pi3K, Nuclear Factor-κB), ou la surexpression de protéines régulant négativement le métabolisme et le mécanisme d’action proapoptique (Bcl-xL) de cet agent chimiothérapeutique. En outre, des travaux originaux ont montré que la modulation du métabolisme des sphingolipides affecte profondément la viabilité et la résistance des cellules cancéreuses aux agents chimiothérapeutiques.
En l’espace d’une décennie, les métabolites sphingolipidiques ont émergé comme les représentants d’une nouvelle classe de messagers lipidiques régulant de nombreux processus biologiques fondamentaux : réponse inflammatoire, prolifération et migration cellulaires, apoptose ou encore sénescence. Le céramide et la sphingosine 1-phosphate ont plus particulièrement attiré l’attention de par leur implication dans différents aspects du cancer : son développement ou sa progression et la réponse aux traitements. Le céramide - molécule centrale du métabolisme sphingolipidique - fonctionne comme un “suppresseur” de tumeur, induisant des réponses antiprolifératives et proapoptotiques dans une grande variété de cellules cancéreuses. A l’opposé, la sphingosine 1-phosphate (S1P) favorise la survie cellulaire sous l’effet de stimuli positifs (facteurs de croissance) ou en réponse aux stress et
2 ceci in vitro, ex vivo et in vivo. Les effets opposés de ces deux sphingolipides ont conduit à introduire le concept selon lequel la balance dynamique entre les taux de céramide et de sphingosine 1-phosphate - ainsi que leurs effets antagonistes sur diverses voies de signalisation régulant la survie cellulaire telles que les MAPKinases - représente un facteur déterminant pour le devenir de la cellule. Un des acteurs majeurs contrôlant cette balance est la sphingosine kinase qui phosphoryle la sphingosine (le catabolite immédiat du céramide) en sphingosine 1-phosphate (S1P). Deux isoformes de sphingosine kinase ont été clonés, nommés SphK1 et SphK2, qui contribuent tous deux aux niveaux cytosolique et sérique de S1P. Le rôle pro-oncogénique de la SphK1 est bien documenté et il a été montré que sa surexpression dans de nombreux cancers favorise la croissance tumorale ainsi que la résistance aux agents cytotoxiques. A l’inverse, la SphK2, beaucoup moins étudiée pour l’heure, pourrait contrôler négativement la prolifération cellulaire et induire l’apoptose des cellules. La divergence du rôle de ces deux enzymes pourrait être attribuée à des différences au sein de leur de séquence protéique ainsi que dans leur localisation subcellulaire.
Il n'existe pas à l’heure actuelle de données sur l'implication de la voie sphingolipidique dans le contrôle la prolifération et de l'apoptose des cellules cancéreuses pancréatiques et de leur résistance aux agents chimiothérapeutiques.
Mes premiers travaux de thèse ont eu pour but d’étudier l’implication des métabolites sphingolipidiques en général et de la SphK1 en particulier dans la résistance des cellules cancéreuses pancréatiques à la gemcitabine.
Le second objectif de ma thèse a été de rechercher le rôle de la SphK2 dans le contrôle de la prolifération et de la survie des cellules cancéreuses pancréatiques.
Chapitre I : Le cancer du pancréas
A- Généralités
Le cancer du pancréas représente environ 10 % des cancers de l’appareil digestif et se place au troisième rang des tumeurs digestives après les cancers colorectal et gastrique. On estime qu'en 2005 en France, 7218 nouveaux cas de cancers du pancréas ont été diagnostiqués et qu'il a été responsable de 7787 décès. Cette pathologie est rare avant 50 ans et affecte plus souvent l'homme que la femme avec une incidence augmentant avec l'âge (pic de fréquence à 69 ans pour l'homme, 74 ans pour la femme). Dans le monde, on en diagnostique plus de 216 000 nouveaux cas chaque année.
Ce cancer est considéré comme l’une des maladies les plus meurtrières puisqu’il est responsable de 40000 décès par an en Europe et près de 28000 aux Etats-Unis où il est la cinquième cause de mortalité par cancer. Son pronostic reste très sombre avec une espérance de vie à 5 ans généralement inférieure à 3,5% et une médiane de survie de 4 mois chez les patients présentant une évolution métastatique de la maladie. 65% des patients meurent six mois après le diagnostic et environ 90% sont morts après un an. La chirurgie d’exérèse, quand elle peut être instituée à temps, est la seule forme de thérapie curative mais moins de 20% des patients peuvent en bénéficier et parmi ceux-ci seuls 5% sont encore en vie après 5 ans. Ce pronostic péjoratif s’explique par l’absence ou la non-spécificité des symptômes retardant le diagnostic (on parle de phase clinique latente), par une agressivité tumorale marquée (invasion métastatique rapide) et enfin, par le manque de moyens thérapeutiques efficaces. De ce fait, au moment du diagnostic, 85% des patients présentent déjà un envahissement loco-régional avec souvent une extension métastatique rendant impossible la chirurgie à visée curative (Marshaw et al., 1992).
L'adénocarcinome pancréatique canalaire représente 95 % de l'ensemble des cas et 70% des cancers de la tête du pancréas. Il se développe à partir de cellules exocrines canalaires et peut présenter plusieurs formes histologiques :
- le cystadénome mucineux, de meilleur pronostic mais pouvant dégénérer ;
- le carcinome intra-ductulaire mucineux, tumeur des canaux excréteurs du pancréas (ou TIPMP pour Tumeurs Intra-canalaires Papillaires Mucineuses du Pancréas) également de pronostic plus favorable ;
4 - finalement, l’adénocarcinome ductulaire pancréatique, dans sa forme la plus agressive.
L’adénocarcinome acinaire quand à lui se développe à partir des cellules exocrines acinaires.
1-Epidémiologie et facteurs de risque
Connaître l’épidémiologie d’un cancer et ses facteurs de risques est essentiel pour développer une approche thérapeutique rationnelle et efficace. Le cancer du pancréas se développe principalement dans les pays industrialisés et son incidence, par ailleurs en légère augmentation, approche celle de la mortalité (ratio mortalité/incidence = 0,99) selon les données de l’Organisation Mondiale de la Santé.
Bien que les causes de ce cancer ne soient pas réellement identifiées, différents facteurs de risques ont été avancés. On les classe en facteurs environnementaux, pathologiques et génétiques.
a- Facteurs environnementaux
Le tabagisme, facteur de risque générique dans la maladie cancéreuse, augmente le risque de développer un cancer du pancréas et des données pré-cliniques suggèrent que les nitrosamines et le tabac sont carcinogènes pour le pancréas. En revanche, la relation de cause à effet entre l’alimentation et le cancer du pancréas reste très floue et souvent contradictoire ; il semblerait cependant que le développement d’un cancer du pancréas soit favorisé par une prise trop importante de calories (régimes riches en graisse, sel et pauvres en fibres). Enfin, aucune étude n’a pu réellement mettre en évidence de lien éventuel entre la consommation de café ou d’alcool et l’incidence du cancer pancréatique.
Parmi les risques liés à l’environnement, on peut aussi compter l’exposition longue et répétée à des agents chimiques, au charbon et à certains métaux (aluminium). En effet, bien que le risque soit limité et les mécanismes inconnus, les personnes exposées à ces sources toxiques ou encore travaillant dans l’industrie textile (tanneries) constitueraient une population plus prédisposée au développement d’un cancer du pancréas.
b- Facteurs pathologiques
Il n’existe pas de véritable population à risque pour le cancer pancréatique. Cependant certaines maladies comme le diabète ou la pancréatite chronique pourraient jouer un rôle favorisant (Silverman et al., 1999).
Le diabète non insulino-dépendant est en effet connu pour être une manifestation précoce de la maladie et un facteur de prédisposition puisque près de 20% des patients diagnostiqués pour un cancer pancréatique sont diabétiques.
Enfin, les personnes souffrant d’une pancréatite chronique calcifiante présentent un risque accru de développer un cancer du pancréas, bien que les mécanismes physiopathologiques de ce phénomène ne soient pas encore connus.
c-Facteurs génétiques
Des formes héréditaires d’adénocarcinome pancréatique ont été décrites et pourraient représenter jusqu’à 3% des cas pour ce cancer.
Ainsi, une prédisposition génétique au cancer du pancréas exocrine a été évoquée pour quelques maladies ou syndromes rares (pour la plupart des maladies autosomales dominantes) telles que les néoplasies endocrines multiples de type 1 (NEM-1), les pancréatites héréditaires, les syndromes de Von Hippel-Lindau et de Peutz-Jeghers, l’ataxie-télangiectasie ou certains mélanomes familiaux (Efthimiou et al., 2001). Certains, telle la pancréatite héréditaire, sont organe-spécifiques. D’autres, dont l’ataxie-télangiectasie ou le syndrome de Von Hippel-Lindau, sont associés à l’apparition de multiples tumeurs à la fois dans le pancréas et dans différents autres organes (rétine, système nerveux central, glandes surrénales et foie). Il existe également quelques formes familiales dont les gènes mis en cause sont encore inconnus.
En résumé, bien que de nombreuses études s’attachent à déterminer l’étiologie de ce type de cancer, le cancer du pancréas se caractérise par l’absence de facteurs de risques très spécifiques. Il est alors difficile de définir des groupes à risque en vue d’un dépistage précoce ou de tout acte de prévention, éléments qui permettraient d’améliorer quelque peu le sombre pronostic de cette maladie.
2- Bases moléculaires de la carcinogenèse pancréatique
Connaître les mécanismes moléculaires à la base de la cancérogenèse pancréatique est primordial car cela permettrait l’identification de marqueurs moléculaires caractéristiques de cette maladie, rendant ainsi son diagnostic incontestable, voire plus précoce, et offrirait de nouvelles cibles thérapeutiques spécifiques pour ce cancer.
6 Le cancer du pancréas est un processus complexe résultant de l’accumulation de modifications génétiques et d’une cascade d’événements dans le patrimoine génétique des cellules pancréatiques normales.
En effet, le cancer se développe à partir d’une certaine population cellulaire qui, après avoir subi une série de changements structuraux et fonctionnels, acquiert un avantage sélectif de croissance et une prolifération clonale.
Les gènes impliqués dans la carcinogenèse se définissent globalement en oncogènes et gènes suppresseurs de tumeurs, encore appelés anti-oncogènes. Un oncogène est au départ un gène courant, que nos cellules possèdent toutes (proto-oncogène = du grec « protos » premier, précurseur et « onkos », masse) et dont la principale fonction est le contrôle de la prolifération et de la différenciation cellulaires. Lorsque ce gène échappe aux systèmes de régulation cellulaires (activation aberrante par surexpression ou mutations), il acquiert un pouvoir transformant et immortalisant, contribuant à l’établissement d’un phénotype cancéreux. Contrairement aux oncogènes (qui acquièrent un gain de fonction), les gènes suppresseurs de tumeur contribuent à la cancérisation du fait de la perte de leur fonction. En effet ces gènes exercent ordinairement le rôle d’inhibiteur de la progression dans le cycle cellulaire. De ce fait, leur inactivation contribue au phénomène de cancérisation.
Enfin, la susceptibilité d’un individu à développer un phénotype tumoral dépend également d’autres déterminants génétiques tels une dérégulation des gènes impliqués dans la réparation de l’ADN (on parle alors d’instabilité génétique).
En résumé, la cancérogenèse résulte de l’activation constitutive ou excessive d’oncogènes et de l’inactivation ou répression de gènes suppresseurs de tumeurs.
a- Les oncogènes
• Le gène Ras est l’oncogène le plus fréquemment détecté dans les tumeurs malignes humaines. Il appartient à une famille constituée de trois membres (K-ras, H-ras et N-ras). Le proto-oncogène K-ras (Kirsten-ras) code pour une petite protéine cytoplasmique de 21 kDa qui a des effets pléïotropes tels que la prolifération, la différenciation et la survie cellulaires. Dans les adénocarcinomes pancréatiques, ce proto-oncogène est converti en oncogène activé par des mutations ponctuelles dans sa séquence génomique. Les mutations de K-ras ont été retrouvées dans 80 à 95% des cancers pancréatiques ; cela représente l’incidence la plus forte parmi les tumeurs malignes humaines connues pour porter la mutation. De même, des tumeurs à potentiel dégénératif comme les TIPMP présentent elles aussi une mutation de K-ras dans 92% des cas. Des mutations de cet oncogène pourraient donc représenter un
marqueur précoce de l’adénocarcinome pancréatique et leur recherche présente donc un intérêt diagnostique certain dans cette pathologie (Berthelemy et al., 1996).
Récemment, un modèle transgénique chez la souris ciblant l’expression de l’oncogène K-ras dans les cellules acinaires et centro-acinaires a permis de reproduire les lésions précancéreuses (PanIN) et cancéreuses de type adénocarcinome ductal tel que l’on observe chez l’humain (Guerra et al., 2003).
• La protéine c-erbB-2 est elle aussi impliquée dans les phénomènes de prolifération et de différenciation cellulaires. Sa surexpression intervient dans de nombreux adénocarcinomes humains (dont à 20 à 40% des cancers pancréatiques) et est souvent associée à une forte agressivité tumorale, un pronostic pessimiste et un taux de survie limité. Cette surexpression est uniforme dans la tumeur primaire (affectant toutes les cellules malignes) et se retrouve également au sein de ses métastases (Pryczynicz et al., 2008).
• NF-κB est un facteur de transcription impliqué dans de nombreux processus physiologiques tels que l’embryogenèse, la prolifération, l’apoptose, l’invasion, l’angiogenèse, l’inflammation et la réponse immunitaire. Il semble exister une activation constitutive du facteur de transcription NF-κB (Nuclear Factor-kappa B) dans les cellules cancereuses pancréatiques entraînant leur résistance au ligand de mort TNF-α et favorisant plutôt la survie des cellules au détriment de leur mort (Wang et al., 1999). De plus, l’inhibition de NF-κB dans des modèles de cancer pancréatique réduit l’invasion métastatique et l’angiogenèse tumorale via l’inhibition respective de l’expression du VEGF et de l’IL8 (Fujioka et al., 2003). De même, l’activation de facteurs d’invasion comme l’ « urokinase plasminogene Activator » via NF-κB pourrait jouer un rôle dans l’invasion tumorale des adénocarcinomes pancréatiques (Sirivatanauksorn et al., 1998). Le rôle de NF-κB dans les processus de chimiorésistance à la gemcitabine a également été évoqué compte-tenu de son implication dans les processus anti-apoptotiques. La modulation de son activité est donc une voie de recherche dans le traitement du cancer pancréatique (Wang et al., 1999).
• D’autres protéines issues d’oncogènes ont également été étudiées dans le cadre de la cancérogénèse pancréatique. Ainsi, la surexpression des protéines oncogéniques myc et c-fos, dans les adénocarcinomes pancréatiques, est très faible et retrouvée dans peu de cas (environ 10%). L’oncogène Akt2 (codant pour une sérine-thréonine kinase impliquée dans la signalisation de la phosphatidyl-inositol-3 kinase ou Pi3K) est quant à lui surexprimé dans
8 20% des tumeurs pancréatiques humaines ainsi que dans certaines lignées pancréatiques immortalisées comme la lignée Panc-1 (Ruggeri et al., 1998). Le degré de phosphorylation d’Akt dans les cellules cancéreuses pancréatiques est d’ailleurs inversement corrélé avec la survie des patients (Giovannetti et al., 2006).
• Enfin, l’étude récente de matériel tissulaire microdisséqué à partir de lésions prénéoplasiques de type PanIN (Pancreatic IntraNeoplasic lesions) a montré, à des étapes précoces de la carcinogenèse pancréatique, la surexpression de marqueurs tels que S100P (une protéine liant le Calcium et régulant son transport intracellulaire) (Lin et al., 2008), RAB1B (membre de la famille Ras), cathepsine E ou CEACAM5 (Carcinoembryonic Antigen-related Cell Adhesion Molecule 5) (pour revue Fukushima et al., 2004).
b- Les gènes suppresseurs de tumeur
• Le gène p53 code pour une phosphoprotéine nucléaire de 53 kDa jouant un rôle primordial dans la régulation du cycle cellulaire. Cette protéine est un facteur de transcription capable de réguler l’expression de nombreux gènes impliqués dans la prolifération cellulaire, la réparation et la surveillance du patrimoine génétique ainsi que dans le déclenchement des phénomènes apoptotiques. La survenue de mutations avec en parallèle une perte d’hétérozygotie -LOH- (propre aux gènes suppresseurs de tumeur) de ce gène sont responsables de son inactivation. Le gène p53 est muté dans plus de 50% des adénocarcinomes pancréatiques humains ; la plupart de ces mutations sont responsables d’un allongement significatif de sa demi-vie conduisant à l’accumulation dans la cellule de la protéine mutée inactive ne pouvant plus se lier à l’ADN (Suwa et al., 1997)
• Les INK4 appartiennent à la famille des inhibiteurs de kinases dépendantes des cyclines (ou CDKI) et entraînent, en inhibant la formation des complexes cyclines/CDK, un arrêt du cycle cellulaire en phase G1. La perte de ces protéines favorise donc la transition G1/S et la progression dans le cycle cellulaire. Ainsi, La protéine p16, premier membre identifié de la famille des INK4 bloque la division cellulaire en inhibant l’interaction de la cycline D avec la CDK4. La perte d’activité de p16 lève alors tous les effets inhibiteurs et régulateurs de la transition G1/S et conduit à une dérégulation de la croissance cellulaire. Des travaux ont mis en évidence une inactivation de ce gène dans près de 80% des lignées cancéreuses pancréatiques et dans 40% des tumeurs pancréatiques primaires (Kawesha et al., 2000). Divers mécanismes d’inactivation de cette protéine ont été décris : mutations, perte
allélique (LOH dans 85% des cas), promoteur rendu silencieux par hyperméthylation d’ilôts CpG (Schutte et al., 1997).
• La protéine Smad4/DPC4 (pour Deleted in Pancreatic Cancer) intervient en tant qu’effecteur central de la signalisation du TGF-β (Transforming Growth Factor-bêta), facteur antiprolifératif des cellules épithéliales et inducteur d’apoptose (Hu et al., 1998). L’adénocarcinome pancréatique se caractérise par une délétion homozygote du gène codant pour Smad4 dans près de 30% des cas. De plus, la seule perte d’hétérozygotie s’accompagne de mutations sur l’autre allèle du gène Smad4 dans 20% des tumeurs. Ce gène présente donc une inactivation biallélique dans 50% des cas, suggérant son rôle clé et précoce dans la carcinogenèse pancréatique (Hahn et al., 1996).
• Dans près de 90% des adénocarcinomes pancréatiques et des lignées cellulaires qui en dérivent, une perte sélective d’expression du sous-type de récepteur de somatostatine sst2 (hormone inhibitrice de nombreux processus cellulaires dont les sécrétions endocrines et exocrines et la prolifération cellulaire) a été observée, ce qui en fait l’altération génique la plus fréquente. Les travaux de l’équipe ont par ailleurs monté que la réintroduction du gène codant pour sst2 dans des cellules cancéreuses pancréatiques diminuait leur prolifération et leur survie, augmentait les phénomènes d’apoptose ainsi que leur sensibilité au ligand de mort FasL et enfin inhibait la progression tumorale et métastatique in vivo (effet « «bystander » local et à distance) dans des modèles de xénogreffes chez la souris Nude et le hamster doré (Guillermet et al., 2003 ; Vernejoul et al., 2002 ; Delesque et al., 1997 ; Rochaix et al., 1999).
• Enfin, la mutation du gène APC et les mutations du gène BRCA2 sont respectivement responsables d’une pathologie appelée la polypose adénomateuse familiale et de cancers familiaux du sein et de l’ovaire. La présence de ces mutations augmenterait le risque chez ces familles de développer des cancers colorectaux et pancréatiques, mais ces données sont cependant contradictoires (Giardiello et al., 2000 ; Naderi et al., 2002).
c- Les autres altérations dans le cancer pancréatique
Environ 10% des tumeurs pancréatiques réséquées s’avèrent être des pancréatites chroniques d’un point de vue histologique et, inversement, 5 à 10% des spécimens de pancréatite chronique sont en fait des adénocarcinomes pancréatiques.
10 Devant cette incertitude, proposer une chirurgie est une décision délicate quand on connaît le taux de morbidité relative (30-40%) et la mortalité (5%) associées à la duodéno-pancréatectomie céphalique. Il est donc nécessaire, à côté des facteurs plus ou moins pronostiques classiques (taille de la tumeur, détection de métastases ganglionnaires, grade de différenciation), de trouver d’autres facteurs déterminants sur la base des altérations moléculaires rencontrées dans cette pathologie cancéreuse.
1- Activité télomérase
Récemment, de hauts niveaux d’activité télomérase ont été retrouvés sur du tissu et du matériel de cytoponction d’adénocarcinomes pancréatiques alors que cette activité est basse voire nulle dans le tissu normal ou inflammatoire (Hiyama et al., 1997). La télomérase est une ribonucléoprotéine qui assure l’élongation des télomères chromosomiques par l’ajout d’hexamères TTAGGG. L’activation de la télomérase associée à une stabilisation des télomères est une des caractéristiques classiques des cellules immortalisées. Une élévation de l’activité télomérase est en effet observée dans de nombreux cancers et participerait au développement mais surtout au maintien de la croissance tumorale (Bollmann et al., 2008). La chronologie de l’élévation de cette activité au cours de la cancérogenèse pancréatique est toutefois encore peu connue.
2- La mort cellulaire programmée
La croissance des tumeurs n’est pas uniquement déterminée par la capacité des cellules à proliférer mais également par le taux de mort cellulaire. Un des deux mécanismes par lequel une cellule meurt est la « mort cellulaire programmée » connue sous le nom d’apoptose (l’autre étant le processus de nécrose). L’apoptose fait partie intégrante de la physiologie normale d’un organisme. Au cours des nombreuses mitoses et différenciations cellulaires, il est en permanence nécessaire d’éliminer les cellules « superflues » ou potentiellement dangereuses. Ce processus de suicide se traduit par de nombreux changements morphologiques : la membrane plasmique se désorganise (formation de vésicules apoptotiques) et la chromatine se condense avant d’être dégradée en un profil caractéristique (échelle de fragmentation de l’ADN). Ce phénomène relativement complexe résulte d’une cascade d’activation faisant intervenir entre autres la voie apoptotique mitochondriale activant des protéases spécifiques apoptogènes appelées caspases (initiatrices et effectrices) (Solary et al., 2003). Le cancer du pancréas est connu pour être résistant au phénomène d’apoptose. Les mécanismes exacts ne sont pas bien décortiqués mais impliquent la perte de fonction de p53
et l’altération de protéines pro- et anti-apoptotiques telles que respectivement Bax et Bcl-xL (membres de la famille de Bcl-2) (Friess et al, 1998). Une augmentation de la sécrétion de Fas-ligand (FasL/CD95/APO-1), prototype du ligand de mort, par les cellules cancéreuses pancréatiques a également été décrite mais il semble que ces cellules soient apparemment insensibles à ce facteur. Il est possible que ce FasL soit muté et empêche l’interaction entre le récepteur Fas et FasL normal (Ungefroren et al., 1998).
3- L'angiogenèse
Le phénomène d’angiogenèse (formation de nouveaux capillaires sanguins à partir de vaisseaux pré-existants) est également essentiel pour amener les facteurs, les nutriments et l’oxygène indispensables à la croissance d’une tumeur solide. Cependant, la néo-vascularisation s’établissant de façon anarchique dans les tumeurs, les apports en oxygène et nutriments sont insuffisants. La conséquence de cette hypoxie est l’expression de facteurs de croissance inductibles par l’hypoxie (Hypoxia Inducible Factors - HIF) et la stimulation de facteurs pro-angiogéniques comme le VEGF, le PDGF, l’HGF ainsi que les FGF et leurs récepteurs (Reinmuth et al., 2003). Des travaux ont d’ailleurs mis en évidence une surexpression du VEGF et de son récepteur dans les lignées cancéreuses pancréatiques humaines (von Marschall et al., 2000).
4- La prolifération cellulaire
Différentes études réalisées sur culture cellulaire, modèles animaux et pièces d’exérèse pancréatiques font également état d’une surexpression de facteurs de croissance. Les plus importants sont l’EGF et son récepteur ainsi que les peptides de la même famille (TGFα, c-erB, amphiréguline) (pour revue Garcea et al., 2005). La surexpression de ces facteurs participe à la croissance tumorale. Ces événements sont par ailleurs associés à une perte de l’effet de facteurs inhibiteurs de la croissance cellulaire comme la somatostatine et le TGF-β (Wagner et al., 1998). De même que la perte du récepteur sst2 empèche toute action de la somatostatine, la perte de l’expression de Smad 4 prive le récepteur du TGF-β de son système de transduction et donc de l’effet inhibiteur de son ligand sur la prolifération cellulaire (Schutte, 1999). D’autres facteurs de croissance tels que le NGF, la gastrine et la bombésine participent également à l’auto-entretien de la prolifération des cellules cancéreuses pancréatiques (pour revue Yamaoka et al., 1999).
12
5- Invasion tissulaire
La croissance et la progression tumorale impliquent une forte capacité des cellules cancéreuses à l’invasion locale et métastatique. Dans le cancer pancréatique, le caractère invasif des cellules tumorales semble être en rapport avec l’action d’enzymes protéolytiques (Matrix MetalloProteinases ou MMP) et de protéases (cathepsines et E-cadhérine). En effet on observe dans ce cancer une augmentation de l’expression de métalloprotéinases (MMP-2, MMP-3 et MMP-9) et une diminution de l’expression de leurs inhibiteurs (TIMP-1) qui participent au modelage de la matrice extracellulaire (Bramhall et al., 1997). La surexpression de TIMP-1 dans les cellules cancéreuses pancréatiques xénogreffées chez la souris athymique est d’ailleurs responsable d’une diminution de la croissance tumorale, de son angiogenèse et de sa progression métastatique (Bloomstom et al., 2002). Enfin, la perte de facteurs réduisant la mobilité et l’adhésion cellulaire (comme la E-cadhérine ou la laminine) est corrélée au mauvais pronostic et/ou au caractère métastatique de ce cancer (pour revue Garcea et al., 2005). L’ensemble de ces anomalies est responsable de l’acquisition d’une mobilité cellulaire et d’une activité collagénolytique favorisant la migration cellulaire. Participe également au phénotype invasif une augmentation de la conversion du plasminogène en plasmine (via « l’urokinase plasminogen activator » par exemple) permettant une dégradation de la matrice extracellulaire (Sirivatanauksorn et al., 1998). De même, les protéines de type S100A4, S100A6, S100P et l’intégrine β4 participent à la migration cellulaire des cellules cancéreuses (Cruz-Monserrate Z, 2007 ; Mahon et al., 2007). Toutes ces anomalies contribuent à la progression et au caractère invasif par voies vasculaire, lymphatique et nerveuse de ce type de tumeur.
6- Le microenvironnement tumoral
Il est maintenant reconnu que le microenvironnement tumoral joue un rôle critique dans la progression tumorale. Ceci est particulièrement vrai pour l’adénocarcinome ductal pancréatique qui présente une forte réaction desmoplasique fibreuse. Cette réaction provient d’une communication étroite et réciproque entre cellules épithéliales cancéreuses et cellules du stroma, les cellules épithéliales altérant leur microenvironnement de manière à le rendre plus propice à leurs propres prolifération, survie, migration et invasion (Koninger et al., 2004).
d- Modifications épigénétiques
L’ADN, lorsqu’il est méthylé sur des motifs spécifiques appelés ilôts CpG, est reconnu par des « methyl binding proteins » qui recrutent des histones déacétylases qui vont agir en
compactant l’ADN, rendant ainsi sa transcription impossible (Szyf et al., 2006). Une hyperméthylation aberrante est responsable de l’inactivation de nombreux gènes suppresseurs de tumeurs dans les cas de cancers pancréatiques, le cas le plus étudié étant celui du gène p16INK4. A l’inverse, suite à une hypométhylation, certains gènes impliqués dans la progression tumorale se retrouvent fortement exprimés au sein de tumeurs pancréatiques tels que les protéines Maspin et S100P (pour revue Feldmann et al., 2008).
Les processus de méthylation/déméthylation de l’ADN étant réversibles, des espoirs de traitement reposent sur une meilleure compréhension des mécanismes de maintien de l’information épigénétique. Des inhibiteurs de l’ADN méthyltransférases (Zebularin®) sont actuellement en cours d’essai cliniques et montrent déjà des résultats prometteurs sur le cancer de la vésicule. Seules quelques études se sont intéressées aux effets de ces drogues modifiant les modifications épigénétiques sur des cas de cancer pancréatique (Neureiter et al., 2007).
e- Les microARN
La voie de l’interférence à l’ARN est un processus cellulaire présent chez les eucaryotes qui régule l’expression génique de façon transcriptionnelle et post-traductionnelle. La spécificité de la répression génique par interférence à ARN provient de petits ARNs guides d’environ 21 à 23 nucléotides (appelés miRNA ou miARN). La synthèse des miRNA est induite par la production de longs transcrits endogènes appelés miARN primaires (pri-miRNA). Le pri-miARN est ensuite clivé dans le noyau pour donner une structure en tête d’épingle plus courte appelée miARN précurseur (pré-miARN). L’exportation du pré-miARN au cytoplasme est suivie du clivage et de la maturation du pré-miARN en miARN mature. Le miARN mature est ensuite incorporé dans le complexe inhibiteur RISC et le guide vers une séquence sur l’ARNm lui étant partiellement complémentaire. Cette association conduit à la répression de la traduction de l’ARNm cible. Ainsi, les miARN se positionnent comme des molécules essentielles dans le contrôle du développement, de la prolifération cellulair et, plus récemment, de l’oncogenèse. L’altération de l’expression de différents miARN, notamment impliqués dans le contrôle de la prolifération, de l’apoptose ou de la progression tumorale a été récemment rapportée. Certains miRNA exercent des propriétés oncogéniques ou anti-oncogéniques. Leur profil d’expression signe le lignage d’un tissu tumoral, ce qui peut être utile pour le diagnostic. L’existence de miARN dans les tumeurs solides et les hémopathies ouvre des perspectives inédites pour la compréhension de l’oncogenèse et potentiellement dans la prise en charge des maladies cancéreuses. Plus particulièrement, l’expression de Let7
14 est retrouvée altérée dans le cancer du poumon et plus récemment dans le cancer colorectal. Les premiers résultats des études concernant le cancer du pancréas font état d’une surexpression de miR-100 et miR-196a-2 dans les adénocarcinomes, mir-196 étant corrélé à une survie plus courte (Szafranska et al., 2007 ; Lee et al., 2007).
Il est cependant encore trop tôt pour dire si l’un ou plusieurs d’entre eux vont constituer de nouvelles cibles diagnostiques ou thérapeutiques, mais des molécules si importantes dans la vie cellulaire doivent forcément jouer un rôle ou être dérégulées au cours de la cancérogenèse pancréatique.
3- Modélisation histologique de la cancérogenèse pancréatique
Les cellules d’adénocarcinome pancréatique présentent un phénotype de cellules pancréatiques ductales avec une organisation cuboïdale, la présence d’antigènes ductaux (cytokératines, mucines, antigène carcinoembryonnaire ACE, synaptophysine) et une croissance en structures tubulaires.
Cependant, différentes hypothèses ont été avancées quand au contingent cellulaire d’origine impliqué dans le processus de carcinogenèse pancréatique : cellules souches multipotentes dérivant de la portion endocrine du pancréas (transdifférenciation ilôt/canal) (Pour et al., 1999) ou encore cellules acineuses transdifférenciées. En effet, des résultats obtenus dans le modèle de souris transgéniques ElasCCK2 (surexprimant le gène du récepteur de la CholeCystoKinine 2 humain dans les cellules exocrines) montrent des altérations histologiques du pancréas (structures ductulaires d’origine acinaire) et le développement de lésions pré-néoplasiques (transdifférenciation acino-canalaire) (Clerc et al., 2002).
Malgré ces données encore peu documentées, un modèle de cancérogenèse pancréatique à partir de cellules ductales a été établi sur la base de l’observation des modifications histo-morphologiques et des altérations génétiques. Ainsi, par analogie avec le modèle de progression tumorale décrit pour les carcinomes colo-rectaux (séquence adénome-cancer invasif), des auteurs ont établi dans le pancréas un modèle évolutif de progression histologique dérivant des ductules exocrines normaux vers un adénocarcinome invasif (Hruban et al., 2000). Dans les canaux adjacents à la tumeur infiltrante, la modification la plus précoce est une hyperplasie ductale ; ensuite, cette progression tumorale résulte à la fois d’une augmentation du degré d’atypie cytologique et architecturale (lésions prolifératives ductales ou PanIN pour Pancreatic Intraepithelial Neoplasia) et d’une accumulation des altérations gèniques associées à ce cancer. On distingue alors trois sous-types de lésions ductales : - PanIN-1 (regroupant PanIN-1A et PanIN-1B), lésions papillaires avec cellules allongées ;
- PanIN-2, lésions ductales avec des anormalités nucléaires ;
- PanIN-3, lésions pancréatiques de haut grade avec noyaux atypiques et mitoses anormales. L’activation de l’oncogène K-ras et la surexpression de la protéine c-erbB-2 apparaissent dans les étapes précoces de la modification morphologique. L’inactivation du gène p16INK4a se produit généralement dans des stades intermédiaires de la néoplasie pancréatique (PanIN-2). Enfin, les mutations de p53 et Smad/DPC4 semblent intervenir dans des événements beaucoup plus tardifs, correspondant à des états de dysplasie sévère (PanIN-3 de haut grade) ou d’adénocarcinome. Du fait de cette chronologie des altérations géniques, il a été évoqué le concept de processus carcinologique multi-étapes (Torrisani et al., 2002) (cf figure 1).
Connaître l’origine et le schéma séquentiel de la pathogenèse de l’adénocarcinome pancréatique apparaît comme un élément essentiel dans un but préventif, diagnostique et surtout thérapeutique (détection des lésions précoces potentiellement curables).
B – L’adénocarcinome pancréatique en pratique clinique
1- Diagnostic et explorations pré-thérapeutiques
L’adénocarcinome pancréatique siège le plus souvent dans la tête du pancréas (75% des cas). La symptomatologie habituelle est alors représentée par un ictère nu progressif lié à la compression extrinsèque du bas cholédoque accompagné d’une altération de l’état général (asthénie, anorexie, perte de poids) et est souvent associée à des douleurs abdominales hautes modérées.
Dans les 25% restants, les tumeurs sont de siège corporéo-caudal. Les douleurs inaugurent la symptomatologie : elles sont vagues, peu intenses au début, ce qui explique le diagnostic à un stade tardif. La présence de douleurs épigastriques intenses transfixiantes correspond à un envahissement nerveux (plexus coeliaque) caractéristique de l’agressivité loco-régionale de ce type de tumeur. Elle est de mauvais pronostic car elle signe le plus souvent une inextirpabilité chirurgicale.
Le bilan d’extension pré-opératoire et le diagnostic de cancer du pancréas se sont considérablement améliorés grâce aux progrès de l’imagerie médicale. Une échographie abdominale est souvent réalisée en première intention. En visualisant une dilatation des voies biliaires et du bas cholédoque, cet examen confirme un obstacle bas situé sur le cholédoque.
Figure 1- Evolution histologique des lésions cancéreuses pancréatiques et évènements moléculaires associés
PDF processed with CutePDF evaluation edition
Compte-tenu de la situation profonde du pancréas dans l’abdomen (recouvert par l’estomac et barré par le colon transverse), celui-ci ne peut être correctement examiné dans sa totalité du fait d’artefacts digestifs. Le scanner abdominal est alors l’examen non-invasif de référence pour confirmer l’origine tumorale de l’obstacle biliaire et pour apprécier son extension loco-régionale. Cet examen est souvent préféré à l’échographie qui dans 20% des cas ne montre pas bien le pancréas ou ne détecte pas la tumeur. En effet, le scanner spiralé montre une masse pancréatique hypodense faite d’une tumeur associée à une forte réaction de fibrose périphérique, caractéristique de l’adénocarcinome canalaire du pancréas. De plus, il permet d’estimer la résécabilité chirurgicale de la tumeur en appréciant ses supports vis-à-vis du pédicule mésentérique supérieur (artère et veine), en recherchant une infiltration du rétropéritoine (plexus nerveux coeliaque), du pédicule hépatique et la présence d’adénopathies à distance du pancréas. Enfin, en raison de ses performances satisfaisantes, il permet également de réaliser un bilan d’extension métastatique en recherchant des métastases hépatiques et une carcinose péritonéale.
Afin de pouvoir mieux juger des traitements néo- et/ou adjuvants mis en œuvre lors des protocoles (qui sont fonction du statut métastatique et de la résécabilité tumorale), la détermination pré-thérapeutique du grade tumoral doit être la plus précise possible. Ainsi, en cas de doute par rapport à l’extirpabilité de la tumeur, un angio-IRM (Imagerie par Résonnance Magnétique) ou un examen échoendoscopique peuvent être envisagés. Ce dernier est plus invasif que les autres examens d’imagerie mais il permet dans le même temps la réalisation de biopsies tumorales transduodénales et d’un drainage par pose d’une endoprothèse biliaire. La ponction guidée sous échoendoscopie est dès lors utile si l’on s’oriente vers un traitement néoadjuvant pour lequel la preuve histologique de malignité est indispensable (Barish et al., 1999 ; Buscail et al., 1999). La place des examens d’imagerie ainsi que les traitements est alors resituée sur un arbre décisionnel.
2- Les traitements actuels du cancer du pancréas
D’après les considérations cliniques précédentes, il apparaît clairement que le traitement curatif du cancer du pancréas viendra de la possibilité d’élargir un diagnostic précoce de ce cancer.
a- Les traitements chirurgicaux
Le pancréas est un organe profond dont l’abord chirurgical est souvent très délicat et complexe.
17 La chirurgie du pancréas est une chirurgie lourde car elle est souvent réalisée chez des patients en mauvais état général, entraînant une morbi-mortalité élevée.
Les extensions loco-régionales, vasculaires et ganglionnaires ainsi qu’à distance sont les trois éléments principaux à considérer pour la résécabilité d’une tumeur. Ainsi, les tumeurs localement inextirpables sont des tumeurs avec une atteinte de l’artère mésentérique, de l’artère hépatique ou du tronc coeliaque. De même l’extension ganglionnaire mésentérique métastatique péri-pancréatique est habituellement considérée comme un critère de non-résécabilité.
Le traitement chirurgical à visée curative est réalisé quand l’exploration chirurgicale confirme la résécabilité de la tumeur et l’absence de métastases. Il consiste en l’exérèse de la tumeur et n’est praticable que chez 20% des patients. Si celle-ci siège dans la tête du pancréas, le chirurgien réalise une duodéno-pancréactectomie céphalique (DPC), sinon il s’agit d’une pancréatectomie caudale. De plus, cet acte chirurgical s’accompagne d’une morbi-mortalité non négligeable ; en effet, la mortalité post-opératoire après DPC est inférieure à 2% mais peut toutefois atteindre 8 à 12% dans des centres non spécialisés. Pour ces patients opérés, le taux de survie à 5 ans est de l’ordre de 10% en fonction de la taille de la tumeur.
Le traitement chirurgical palliatif est réalisé quand le bilan d’extension per-opératoire montre des métastases ou quand il s’agit d’une tumeur de la tête du pancréas inextirpable. Pour la majorité des patients opérés (80%), il est envisagé cette chirurgie palliative qui consiste en une double dérivation digestive faite d’une gastro-entéroanastomose et d’une anastomose cholédoco-jéjunale, permettant le drainage de l’ictère et parant à l’obstruction duodénale par la tumeur laissée en place.
b- Les traitements palliatifs
Actuellement, la chirurgie d’exérèse est le seul traitement curatif du cancer du pancréas. Il est donc nécessaire de développer de nouvelles thérapeutiques néo-adjuvantes ou adjuvantes à la chirurgie dont le but ultime est de permettre secondairement une résection des tumeurs.
Pour les patients avec un cancer pancréatique non-résécable métastatique ou non, la palliation des symptômes est le principal objectif thérapeutique.
En cas d’ictère obstructif, la pose d’endoprothèse biliaire permet un écoulement de la bile et représente une alternative à la chirurgie palliative (double dérivation) (van den Bosch et al., 1994).
La prise en charge de la douleur par traitement antalgique adapté (utilisation d’antalgiques per os et de dérivés morphiniques -patch-) et la prise en charge d’un symptôme dépressif souvent associé sont indispensables. Une alcoolisation du plexus coeliaque à visée antalgique peut également être réalisée.
La chimiothérapie et la radiothérapie sont des traitements palliatifs du cancer du pancréas (comme pour de nombreux cancers) dont le but est d’améliorer la qualité de vie. L’adénocarcinome du pancréas présente une chimiorésistance marquée et peu de drogues évaluées ces dernières années ont donné un taux de réponse supérieure à 15%, tant en mono-chimiothérapie qu’en association. Malgré ces données pré-cliniques et des études de phase II encourageantes, aucun traitement de chimiothérapie n’a montré son efficacité depuis les trente dernières années. La radiothérapie permet parfois d’améliorer la qualité de vie des patients en réduisant les symptômes, principalement la douleur. Cependant elle ne s’applique qu’à des patients avec un état général compatible avec la réalisation d’une irradiation.
La mono-chimiothérapie a fait l’objet de nombreuses études chez des patients présentant une tumeur non-résécable ou métastatique. L’inhibiteur de la thymidylate synthase, le 5-Fluoro-Uracile (5-FU), est l’agent chimiothérapeutique le mieux évalué pour le cancer du pancréas mais le taux de réponse objective et l’impact sur la survie sont faibles. Depuis 1990, de nombreuses molécules (iproplatine, trimétrexate, édatrexate, fazarabine, diaziquone, amonafide…) se sont révélées totalement inactives. Un nouvel analogue nucléosidique de la cytidine, la gemcitabine, semble toutefois un médicament prometteur avec un taux de réponse objective compris entre 5,4 et 11% (Casper et al., 1994). Les résultats des études actuelles sont cependant discutables car ils ne montrent aucune amélioration en terme de survie globale (5,6 mois vs 4,4 mois pour les patients traités par 5-Fluoro-Uracile) et de progression tumorale mais rapportent seulement l’existence d’un « bénéfice clinique » observé chez un grand nombre de patients (Burris et al., 1997). Dans les études de phase II, la gemcitabine semble plus efficace en association à l’oxaliplatine (GEMOX) (Alberts et al., 2003), au cisplatine et/ou au 5-FU (Oettle et al., 2000). Néanmoins, aucune de ces associations n’a pour l’instant montré d’amélioration de la survie globale versus la gemcitabine seule dans des investigations cliniques de phase III (Scheithauer et al., 2003).
La radiochimiothérapie concomitante (RT-CT) pourrait être une nouvelle approche thérapeutique prometteuse du cancer du pancréas. Des résultats encourageants ont été obtenus lorsque chimiothérapie et irradiation sont associées de façon concomitante. De ce fait, elle est proposée aux patients pour lesquels on espère secondairement une résection chirurgicale.
19 Cependant, ces essais en faveur d’une RT-CT méritent confirmation avant de recommander ce traitement (Mornex et al., 2000).
Etant donné sa faible toxicité et l’amélioration du bénéfice clinique, la gemcitabine (Gemzar®) est devenue le traitement chimiothérapeutique de référence dans le cancer du pancréas métastatique depuis l’obtention de l’AMM en Europe dans cette indication en 1998. Les cliniciens et oncologues restent cependant très critiques et vigilants quand à l’évaluation de cette drogue et à sa réelle efficacité (Hammel et al., 2002). Ses mécanismes d’action et son métabolisme feront l’objet d’un chapitre à part entière.
3- Les thérapeutiques émergentes
La connaissance de la biologie du cancer évolue rapidement et permet d’entrevoir de nouvelles approches et cibles thérapeutiques pour le cancer du pancréas.
A l’heure actuelle, les voies de recherche pour améliorer le pronostic du cancer pancréatique sont :
- le développement de traitements complémentaires ou plus efficaces que la gemcitabine pour ralentir la progression de la maladie ;
- l’identification de marqueurs moléculaires spécifiques et fiables permettant, si possible, un diagnostic plus précoce, voire d’apprécier le pronostic et/ou la réponse thérapeutique à la gemcitabine.
a- Les inhibiteurs de la topoisomérase I
Les topoisomérases interviennent dans la relaxation de la chaîne d’ADN au cours de la réplication ou de la transcription, ce qui entraîne des petites interruptions dans la chaîne d’ADN. Les inhibiteurs de topoisomérases freinent le processus de religation et bloquent ainsi la réplication et la transcription de l’ADN. Ces inhibiteurs ont pour avantage d’être très spécifiques de leur cible et sont des analogues structuraux de la camptothécine. Ces anti-topoisomérases tels l’irinotécan®, le topotécan® et plus récemment le rubitécan®, en cours d’investigation clinique, contribueront dans le futur au traitement du cancer pancréatique chez l’homme.
b- Les inhibiteurs de la transduction du signal
Un certain nombre de molécules ont été développées afin de stopper une signalisation aberrante rencontrée dans les cellules néoplasiques pancréatiques.
• Le PTZ/ZK (SU5416 ou vatalinib®) est un puissant inhibiteur sélectif du récepteur flk-1/KDR, récepteur au facteur de croissance vasculaire VEGF ; il inhibe la croissance cellulaire et empêche le développement d’une vascularisation tumorale dense dans des modèles expérimentaux de tumeurs humaines. Il a ainsi été montré que cette molécule inhibait la croissance tumorale de cellules cancéreuses pancréatiques dans un modèle in vivo de xénogreffe chez la souris Nude (Bocci et al., 2004). Des résultats de phase III d’étude clinique d’utilisation de cette molécule dans le traitement du cancer colorectal sont par ailleurs encourageants.
• Des petites molécules inhibitrices de l’activité tyrosine kinase telles que le OSI-774 (Erlotimib®) s’opposant à la transduction du signal induit par l’EGF ont été testées. L’Erlotimib permet effectivement d’apporter un gain de survie (bien que modeste) en association à la gemcitabine par rapport à la gemcitabine seule pour le cancer du pancréas (Senderowicz et al., 2007).
• Le TNP-470, analogue de la fumagilline, inhibe l’angiogenèse. Il a été rapporté une réponse complète chez certains patients présentant un cancer cervical métastatique ou de la prostate (Kudelka et al., 1997 ; Logothetis et al., 2001). Par ailleurs, deux autres molécules, l’endostatine (peptide de la collagénase XVIII) et l’angiostatine (peptide du plasminogène) présentent une activité anti-tumorale prouvée sur différents modèles. Elles font actuellement l’objet d’études cliniques pour évaluer leur potentialité anti-angiogénique dans le cancer du pancréas.
• L’utilisation d’inhibiteurs de l’activité du récepteur au TGF-β, TGFβRI, a montré leur efficacité pour inhiber in vitro et in vivo le pouvoir migratoire, invasif et métastatique de ces cellules cancéreuses pancréatiques dans des modèles de xénogreffes sous-cutanées et orthotopiques de cellules cancéreuses pancréatiques chez la souris Nude (Subramanian et al., 2004).
• L’expression de la cyclo-oxygénase 2 (Cox-2) dans les cellules cancéreuses leur confère un avantage de croissance par activation de la prolifération cellulaire, inhibition de l’apoptose et stimulation de l’angiogenèse. De plus, son expression se trouve augmentée dans les cancers pancréatiques humains (Tucker et al., 1999) ce qui laisse présager d’utiliser des inhibiteurs spécifiques de la Cox-2 (rofecoxib®, celecoxib®) dans le traitement du cancer du pancréas (Kokawa et al., 2001).
• Enfin, un nouvel antagoniste compétitif du facteur angiogénique HGF, le NK4, possède une action anti-tumorale in vitro et in vivo significative en inhibant l’angiogenèse et