Caractérisation des mécanismes moléculaires à
potentiel thérapeutique dans la progression
métastatique du mélanome uvéal
Thèse
Cindy Weidmann
Doctorat en biologie cellulaire et moléculaire
Philosophiæ doctor (Ph. D.)
Caractérisation des mécanismes moléculaires à
potentiel thérapeutique dans la progression
métastatique du mélanome uvéal
Thèse
Cindy Weidmann
Sous la direction de :
Résumé
Le mélanome uvéal, qui origine de la transformation anormale des mélanocytes de l’uvée, et représente la tumeur intraoculaire la plus fréquente chez l’adulte. Plus de 40% des patients ne ressentent ni douleur, ni troubles visuels lors du développement de la tumeur oculaire, ce qui explique le dépistage tardif de ce cancer et la présence de métastases au foie lors du diagnostic initial dans près de la moitié des cas. Une méta-analyse a souligné l’inefficacité des thérapies disponibles étant donné le peu d’études cliniques de phase III complétées dans les 30 dernières années pour traiter le mélanome uvéal métastatique. L’objectif général de mon projet de doctorat consistait à caractériser des mécanismes moléculaires à potentiel thérapeutique dans la progression des métastases du mélanome uvéal qui pourraient être ciblés pour traiter les patients ayant reçu un mauvais pronostic. Mon premier objectif consistait à caractériser les effets de la répression pharmacologique du récepteur 2B de la sérotonine (HTR2B) dans les cellules cancéreuses métastatiques du mélanome uvéal. Nous avons démontré que l’antagoniste sélectif PRX-08066 réduit la viabilité des cellules du mélanome uvéal et la population en mitose et altère leur potentiel d’auto-renouvellement et de migration avec une sensibilité variable d’une lignée à l’autre. De plus, nous avons observé une diminution de la phosphorylation de kinases des voies de signalisation classiquement activées par HTR2B, ainsi que des kinases β-caténine, PYK2 et STAT5 dans les cellules traitées. Mon second objectif consistait à caractériser le profil d’hydroxyméthylation (5-hmC) de l’ADN durant la progression du mélanome uvéal. Nous avons montré une réduction du niveau global de 5-hmC dans les lignées cancéreuses du mélanome uvéal comparativement à la choroïde et aux mélanocytes. De plus, nos analyses ont révélé une diminution de l’expression de l’enzyme IDH1 dans les cellules cancéreuses et une seule lignée comportait une mutation dans ce gène. Mon troisième objectif consistait à tester différents taux d’oxygène (3% vs 21% O2) pour la culture in vitro des lignées
mélanocytaires. Nous avons démontré que la morphologie des mélanocytes et leur pigmentation sont légèrement changées lorsqu’exposés à 3% O2 comparativement aux
cellules cancéreuses. De plus, leur temps de doublage était significativement plus rapide dans ces conditions. Le profilage génique des mélanocytes choroïdiens n’a pas révélé une longue liste de transcrits considérablement dérégulés entre les deux concentrations d’oxygène: seul le transporteur de lactate MCT4 était dérégulé de manière significative à
3% O2 chez tous les donneurs. Mes travaux de doctorat ont donc permis l’avancement des
connaissances sur le promoteur de métastases HTR2B de la signature moléculaire pronostique, l’hydroxyméthylation de l’ADN et l’hypoxie dans le mélanome uvéal, ce qui contribuera au développement d’une thérapie adjuvante ou épigénétique pour améliorer la survie des patients.
Abstract
Uveal melanoma (UM) originates from the abnormal transformation of uveal melanocytes and is the most common intraocular tumor in adults. More than 40% of patients do not experience pain or visual disturbances during the development of their ocular tumor, which explains the late detection of this cancer in nearly half of the cases, and the presence of liver metastases at the initial diagnosis. A meta-analysis highlighted the ineffectiveness of available therapies given the limited number of phase III clinical studies completed in the past 30 years to treat metastatic UM. The general objective of my PhD project was to characterize molecular mechanisms with therapeutic potential in the metastatic progression of UM that could be targeted to treat patients with poor prognosis. My first objective was to characterize the effects of the pharmacological repression of the serotonin 2B receptor (HTR2B) in metastatic UM cells. We demonstrated that the selective antagonist PRX-08066 reduces both the viability of UM cells and the population in mitosis, and alters their potential for self-renewal and migration, with an interindividual variability in sensitivity. In addition, we observed a decrease in the phosphorylation of kinases of the signaling pathways classically activated by HTR2B, as well as new targets such as β-catenin, PYK2 and STAT5 in the treated cells. My second objective was to characterize the DNA hydroxymethylation profile (5-hmC) during UM progression. We have shown a reduction in the overall level of 5-hmC in UM cell lines compared to choroid and uveal melanocytes. Furthermore, our analyzes revealed a decrease in the expression of the IDH1 enzyme in UM cells and only one UM cell line showed a mutation in this gene. My third objective was to test different levels of oxygen (3% vs 21% O2) for the in vitro culture of
melanocytic lines. We showed that the morphology of melanocytes and their pigmentation are slightly changed when exposed to 3% O2 compared to UM cells. Moreover, their
doubling time was significantly faster under these conditions. The gene profiling of choroidal melanocytes did not reveal a long list of significantly deregulated transcripts between both oxygen concentrations: only the lactate transporter MCT4 was significantly deregulated in all donors at 3% O2. My doctoral work enabled the advancement of
knowledge on the HTR2B metastasis promoter from the prognostic molecular signature, DNA hydroxymethylation and hypoxia in UM, which will contribute to the development of an adjuvant or epigenetic therapy to improve patient survival.
Table des matières
Résumé ... iii
Abstract ... v
Table des matières ... v
Liste des tableaux ... ix
Liste des figures ... x
Liste des abréviations et des sigles ... xii
Remerciements ... xvii
Avant-Propos ... xix
1 Chapitre 1: Introduction ... 1
1.1 Anatomie du globe oculaire ... 1
1.2 La choroïde ... 5
1.2.1 Anatomie ... 5
1.2.2 Les fonctions de la choroïde... 6
1.2.3 Les mélanocytes oculaires ... 6
1.3 Le mélanome uvéal ... 9
1.3.1 Étiologie et facteurs prédisposant au mélanome uvéal ... 9
1.3.2 Les facteurs pronostiques du mélanome uvéal ... 11
1.3.2.1 Les facteurs cliniques ... 11
1.3.2.2 Les facteurs histologiques ... 12
1.3.2.3 Les facteurs cytogénétiques et génétiques ... 12
1.3.3 Le tropisme du mélanome uvéal ... 20
1.3.4 Diagnostic et traitement du mélanome uvéal ... 21
1.3.4.1 Le traitement de la tumeur primaire ... 22
1.3.4.2 Le traitement des métastases ... 24
1.4 Les mécanismes moléculaires du cancer ... 28
1.4.1 Les propriétés caractéristiques des cellules cancéreuses... 28
1.4.2 L’hétérogénéité des cellules cancéreuses ... 29
1.4.3 La progression tumorale ... 29
1.4.4 Le récepteur 2B de la sérotonine (HTR2B) dans la signature moléculaire pronostique du mélanome uvéal ... 32
1.4.4.1 Les récepteurs de la sérotonine ... 33
1.4.4.2 La sérotonine ... 35
1.4.4.3 Le récepteur 2B de la sérotonine (HTR2B) ... 36
1.4.5 Les modifications épigénétiques ... 37
1.4.5.1 La méthylation/déméthylation de l’ADN ... 38
1.4.5.2 La méthylation de l’ADN dans le cancer ... 40
1.4.5.3 La méthylation de l’ADN dans le mélanome ... 41
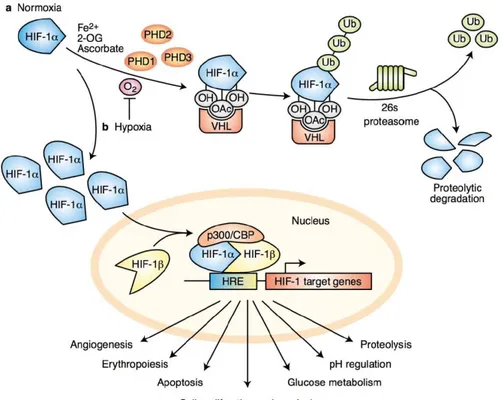
1.4.6.1 Les facteurs de transcription HIFs ... 42
1.4.6.2 L’hypoxie et le métabolisme énergétique ... 44
1.4.6.3 L’hypoxie et l’angiogenèse ... 44
1.4.6.4 L’hypoxie et la résistance aux traitements ... 46
1.4.6.5 L’hypoxie et les stratégies thérapeutiques ... 46
1.4.6.6 L’hypoxie dans le mélanome ... 46
1.5 Enjeux, hypothèses et objectifs des travaux de recherche ... 47
Bibliographie ... 49
2 Chapitre 2: Expression of serotonin receptor 2B and effects of an antagonist on uveal melanoma cell lines ... 65
2.1 Résumé ... 66
2.2 Abstract ... 67
2.3 Introduction ... 68
2.4 Material and Methods ... 70
2.4.1 Primary tumor samples and tissue culture... 70
2.4.2 Anchorage-independent colony formation assay ... 73
2.4.3 Western blotting ... 73
2.4.4 Viability assay ... 74
2.4.5 Cell proliferation assay... 74
2.4.6 Migration assay ... 74
2.4.7 Human phospho-kinase proteome ... 75
2.4.8 Statistical analysis ... 75
2.5 Results ... 76
2.6 Discussion ... 82
2.7 Acknowledgements ... 85
2.8 References ... 86
3 Chapitre 3: Analysis of DNA hydroxymethylation in melanocytes and uveal melanoma cell lines ... 92
3.1 Résumé ... 93
3.2 Abstract ... 94
3.3 Introduction ... 95
3.4 Material and Methods ... 97
3.4.1 Primary tumor samples and tissue culture... 97
3.4.2 PCR amplification and DNA sequencing... 100
3.4.3 DNA slot blot ... 100
3.4.4 LC-MS/MS ... 101 3.4.5 Gene profiling ... 101 3.4.6 Western blotting ... 102 3.4.7 Statistical analysis ... 103 3.5 Results ... 104 3.6 Discussion ... 110
3.7 Acknowledgements ... 112
3.8 References ... 113
4 Chapitre 4: Differential responses of choroidal melanocytes and uveal melanoma cells to low oxygen conditions ... 117
4.1 Résumé ... 118
4.2 Abstract ... 119
4.3 Introduction ... 120
4.4 Material and Methods ... 122
4.1.1 Human choroidal melanocyte cell culture ... 122
4.1.2 Cell count and doubling time ... 124
4.4.3 Anchorage-independent colony formation assay ... 124
4.4.4 Cell proliferation assay and cell nucleus characteristics ... 124
4.4.5 Gene expression profiling ... 125
4.4.6 Western blotting ... 126 4.4.7 Statistical analysis ... 126 4.5 Results ... 128 4.6 Discussion ... 141 4.7 Acknowledgements ... 145 4.8 References ... 146
5 Chapitre 5 : Discussion, conclusions et perspectives ... 151
5.1 Étude #1: Expression of serotonin receptor 2B and effects of an antagonist on uveal melanoma cell lines ... 151
5.2 Étude #2: Analysis of DNA hydroxymethylation in melanocytes and uveal melanoma cell lines ... 155
5.3 Étude #3: Differential responses of choroidal melanocytes and uveal melanoma cells to low oxygen conditions ... 157
5.4 Forces et limites de nos études ... 159
5.5 Conclusion générale ... 160
Bibliographie ... 161
Liste des tableaux
Tableau 1.1: Éléments de la classification TNM. ... 11
Tableau 1.2: Facteurs pronostiques du mélanome uvéal.. ... 14
Tableau 1.3: Mutations somatiques du mélanome uvéal. ... 15
Tableau 1.4: Traitements recommandés selon la taille de la tumeur... 23
Table 2.1: Summary of clinicopathological features, survival data, and mutation status of uveal melanoma patients. ... 71
Table 2.2: Sequence of forward and reverse primers used for PCR amplification and DNA sequencing. ... 72
Table 3.1: Summary of clinicopathlogical features, survival data, and mutation status of uveal melanoma patients. ... 98
Table 3.2: Sequence of forward and reverse primers used for PCR amplification and DNA sequencing. ... 98
Table 3.3: LC-MS/MS analysis of 5-hmC and 5-mC percentages in DNA from UM cell lines (T97-T131), normal uveal melanocytes (NUM) and choroids (C). ... 106
Table 3.4: Mutation status in exon 4 of IDH1 and IDH2 genes in UM cell lines. ... 109
Table 4.1: Clinicopathological features and survival data of uveal melanoma cases. ... 123
Table 4.2: Number of deregulated transcripts between both O2 conditions for each donor. ... 134
Liste des figures
Figure 1.1: Coupe sagittale de l'œil humain. ... 2
Figure 1.2: Organisation laminaire des cellules de la rétine et schématisation de la structure des photorécepteurs. ... 4
Figure 1.3: Schématisation des différentes couches de la choroïde.. ... 5
Figure 1.4: Schématisation des principales structures dérivées des crêtes neurales lors de la morphogénèse embryonnaire. ... 7
Figure 1.5: Schématisation de la structure des mélanosomes. ... 8
Figure 1.6: Apparences cliniques du mélanome uvéal.. ... 10
Figure 1.7: Structure des domaines protéiques et profil des mutations dans les gènes fréquemment mutés dans le mélanome uvéal. ... 18
Figure 1.8: Schématisation des principales fonctions des protéines traduites des gènes fréquemment mutés dans le mélanome uvéal. ... 19
Figure 1.9: Anomalies génétiques impliquées dans la progression du mélanome uvéal.. ... 20
Figure 1.10: Schématisation du principe de la curiethérapie par plaque. ... 23
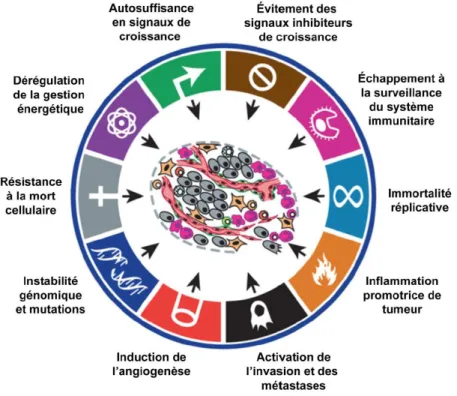
Figure 1.11: Propriétés caractéristiques des cellules cancéreuses selon le modèle de Hanahan et Weinberg. ... 28
Figure 1.12: La cascade métastatique. ... 31
Figure 1.13: Signature moléculaire pronostique du mélanome uvéal et survie selon la classe. ... 33
Figure 1.14: Structure typique des récepteurs couplés aux protéines G. . ... 34
Figure 1.15: Voies de signalisation sous le contrôle du récepteur HTR2B. ... 37
Figure 1.16: Mécanismes épigénétiques. ... 38
Figure 1.17: Cycle de déméthylation de la cytosine.. ... 40
Figure 1.18: Régulation de l'hypoxie par HIF1A. ... 43
Figure 1.19: Hypoxie et angiogenèse.. ... 45
Figure 2.1: Characteristics of UM cells lines.. ... 72
Figure 2.2: Expression of HTR2B in UM cell lines. ... 76
Figure 2.3: The HTR2B antagonist PRX-08066 reduced the viability of UM metastatic cell lines.. ... 77
Figure 2.4: The HTR2B antagonist PRX-08066 decreased the proliferation of UM metastatic cell lines.. ... 78
Figure 2.5: The HTR2B antagonist PRX-08066 reduced the self-renewal of UM metastatic cell lines... 79
Figure 2.6: The HTR2B antagonist PRX-08066 decreased the migration of UM metastatic cell lines... 79
Figure 2.7: The HTR2B antagonist PRX-08066 reduced the phosphorylation of various signaling kinases. ... 80
Figure 3.1: Characteristics of UM cells lines.. ... 99
Figure 3.3: Expression of IDH transcripts. ... 107 Figure 3.4: Expression of IDH proteins in UM and NUM.. ... 108 Figure 4.1: Phase contrast micrographs of monolayer cultures of UM cells T111, T131 and
T132 and their expression of MILANA. ... 123 Figure 4.2: Low physiologic oxygen conditions favor the expansion of choroidal
melanocytes.. ... 128 Figure 4.3: Low physiologic oxygen conditions improve the viability of choroidal
melanocytes... ... 129 Figure 4.4: Low physiologic oxygen conditions do not increase cell senescence.. ... 130 Figure 4.5: Low physiologic oxygen conditions shorten the doubling time of choroidal
melanocytes and increased proliferative capacity.. ... 132 Figure 4.6: Low physiologic oxygen conditions are asociated with the greater siez and
circularity of choroidal melanocyte nuclei.. ... 133 Figure 4.7: Low physiologic oxygen conditions change slightly the transcriptome of
choroidal melanocytes. ... 135 Figure 4.8: Low physiologic conditions increased the mRNA and protein expression of
lactate transporter MCT4 in choroidal melanocytes. ... 136 Figure 5.1: Édition du génome avec CRISPR/Cas9. ... 153 Figure 5.2: Cellules HEK293T transfectées à la lipofectamine avec l’un des plasmides
pSpCas(BB)-2A-GFP-ARNg HTR2B. ... 154 Figure 5.3: Test enzymatique de surveyor.. ... 154 Figure 5.4: Prolifération cellulaire suite à des traitements avec l’oncométabolite 2-HG... 157
Liste des abréviations et des sigles
2-HG 2-hydroxyglutarate 5-caC 5-carboxycytosine 5-fC 5-formylcytosine 5-hmC 5-hydroxyméthylcytosine 5-mC 5-méthylcytosineADN Acide désoxyribonucléique
ANGPTL4 Angiopoietin-like 4
ARN Acide ribonucléique
ATP Adénosine triphosphate
B2M Beta-2-microglobulin
BAP1 BRCA1 associated protein 1
BCA Bicinchoninic acid
BCG Bacille Calmette-Guérin
bFGF Basic fibroblast growth factor
BRCA1 Breast Cancer 1
BER Base excision repair
cAMP Adénosine monophosphate cyclique
CAV1 Caveolin-1
CDH11 Cadhérine 11
CpG Cytosine-phosphate-guanine
CRISPR Clustered Regularly Interspaced short Palindromic Repeats CSCs Cellules souches cancéreuses
CTBP1 C-terminal binding protein 1
CXCR4 C-X-C motif chemokine receptor 4
CYSLTR2 Cystein leukotriene Receptor 2
DMEM Dulbecco’s Modification of Eagle’s Medium
DNMT DNA methyltransferase
DAG 1,2-diaglycérol
DCT Dopachrome tautomérase
ECM1 Extracellular matrix protein 1
EIF1B Eukaryotic translation initiation factor 1
EIF1AX Eukaryotic translation initiation factor 1A, X-linked
EPR Épithélium pigmentaire rétinien
EPO Érythropoïétine
ERK1/2 Extracellular signal-regulated kinases 1 and 2
FAK Focal adhesion kinase
FXR1 Fragile X mental retardation gene 1
GDP Guanosine diphosphate
GEO Gene Expression Omnibus
GTP Guanosine triphosphate
GNAQ G protein subunit alpha q
GNA11 G protein subunit alpha 11
GIPs GPCR-interacting proteins
GPCRs G protein-coupled receptors
GOLGA1 Golgin A1
GSK3β Glycogen synthase kinase 3 beta
HATs Histone acetyltransferases
HCF1 Host cell factor 1
HDACs Histone deacetylases
H&E Hematoxylin and eosin
HGF Hepatocyte growth factor
HK Housekeeping
HIFs Hypoxia inducible factors
HPLC-MS/MS High performance liquid chromatography tandem-mass spectrometry
HREs Hypoxia-response elements
HRP Horseradish peroxidase
HTR1D 5-hydroxytryptamine receptor 1D
HTR2A 5-hydroxytryptamine receptor 2A
HTR2B 5-hydroxytryptamine receptor 2B
HTR2C 5-hydroxytryptamine receptor 2C
HTR7 5-hydroxytryptamine receptor 7
IDHs Isocitrate deshydrogenases
ID2 Inhibitor of DNA binding 2
IGF-1 Insulin-like growth factor 1
IP3 Inositol triphosphate
JAK Janus kinase
LINEs Long interspersed nuclear elements
LTRs Long terminal repeat sequences
LTA4H Leukotriene A4 hydrolase
LMCD1 LIM and cystein rich-domains 1
MAPK Mitogen activated kinase-like protein
Met-tRNAi Methionyl initiator tRNA
MGMT O-6-methylguanine-DNA methyltransferase
MLANA Melan-A
MLPH Melanophilin
MITF Micropthalmia-associated transcription factor
MTS 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
MTT 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
MTUS1 Microtubule associated tumor suppressor 1
MYO5A Myosine 5A
NUM Normal uveal melanocytes
PDGFRs Platelet-derived growth factor receptors
PD-1 Programmed cell death 1
PD-L1 Programmed cell death ligand 1
PH3 Phospho-histone H3 PHDs Prolyl hydroxylases PIP2 Phosphatidylinositol-4,5-bisphosphate PKC Protein kinase C PLC Phospholipase C PLCB4 Phospholipase C bêta 4
PMEL Premelanosome protein
pO2 Pression partielle d’oxygène
PR-DUB Polycomb repressive deubiquitinase complex
PYK2 Proline-rich tyrosine kinase 2
PVDF Polyvinylidene fluoride
RASSF1 RAS association domain family member 1
ROBO1 Roundabout guidance receptor 1
ROS Reactive oxygen species
RUNX3 Runt related transcription factor 3
SAM S-adénosyl-méthionine
SATB1 SATB homeobox 1
SEM Standard error of the mean
SRC SRC proto-oncogene non-receptor tyrosine kinase STAT5 Signal transducer and activator of transcription 5
SF3B1 Splicing factor 3b subunit 1
snRNP Small nuclear ribonucleoprotein
SINEs Short interspersed nuclear elements
TDG Thymine DNA glycosylase
TERT Telomerase reverse transcriptase
TM Transmembranaire
TNM Tumor-Nodes-Metastasis
TRAIL Tumor necrosis factor-related apoptosis inducing ligand-like
TETs Tet methylcytosine dioxygenases
TYR Tyrosinase
TYRP1 Tyrosinase-related protein 1
UICC Union internationale contre le cancer
UM Uveal melanoma
UV Ultraviolet
VEGFs Vascular endothelial growth factors
Remerciements
Je tiens tout d’abord à remercier ma directrice de recherche Pre Solange Landreville, de m’avoir accueillie dans son laboratoire et de m’avoir laissé la chance de réaliser ces travaux sous sa direction. Merci pour ta patience et ton soutien. Merci également pout toutes les opportunités de communications dans des congrès, de formations et de collaborations. Je te souhaite une belle carrière en recherche.
Merci à tous les membres de l’équipe de recherche que j’ai pu côtoyer durant mes études doctorales: Laurence, Ariane, Constance, Renée, Jade, Julie, Ioana, Aïcha, Christine, Cully, Marie-Pier, Léo et Emilie. Un gros merci à Jade pour ton soutien lors de nos deux années passées ensemble. J’ai apprécié ta gentillesse, ta douceur, ton professionnalisme et toutes nos activités en dehors du laboratoire. Merci à ta famille pour leur accueil très chaleureux et sincère. Un merci particulier à Julie pour ta disponibilité, ton soutien et ton professionnalisme. Merci à Mathieu pour ton accueil, mon intégration à mon arrivée au laboratoire et ton amitié. Un gros merci à mes deux « choupinettes », mes voisines de bureau Manel et Aïcha. Les filles, vos jolis sourires et vos joies de vivre ont égayé mes heures au laboratoire et mes repas du midi. Merci à ma belle Hélèna pour cette amitié née au laboratoire, pour ton soutien, tes appels et tes visites à nos retours dans l’Hexagone. Merci à Anissa pour ton amitié, ton soutien et nos escapades à travers le monde! Je n’oublierai jamais nos galères traversées ensemble à mon arrivée sur le continent.
Merci à tous les membres du CUO-Recherche pour leur expertise, leurs conseils et le prêt de matériel. Un merci particulier à Dre Karine Zaniolo pour tes conseils très précieux et ton soutien. Merci aux équipes de la clinique des tumeurs oculaires et de la recherche clinique du CUO, ainsi qu’au Dr Mohib Morcos et Mme Michèle Orain du service de pathologie de l’Hôpital du Saint-Sacrement. Merci à Carolyne M. Lowry et au Pr J. Richard Wagner de l’Université de Sherbrooke pour leur collaboration sur le projet hydroxyméthylation. Enfin, je remercie l’équipe du Pr Jacques P. Tremblay pour son accueil en laboratoire pour l’apprentissage de la technique CRISPR/Cas9, particulièrement Jean-Paul Iyombe-Engembe et Joël Rousseau.
Je remercie les donneurs volontaires de globes oculaires et les patients de la Clinique des tumeurs oculaires pour leurs précieux dons de tissus ou sang. Merci au FRQS pour le financement partiel des banques de tissus oculaires sains et cancéreux.
Je tiens également à remercier la Faculté de médecine de l’Université Laval, la Fondation du CHU de Québec et le Centre LOEX de l’Université Laval pour le soutien financier qui a permis la réalisation de cette thèse de doctorat, ainsi que les IRSC et le Réseau ThéCell du FRQS pour les bourses de congrès.
Enfin, merci à ma famille pour leur amour et leur soutien. Malgré les 6 000 km qui nous séparent et le décalage horaire, vous avez toujours été présents, surtout toi maman que ce soit par les nombreux courriels, les visites ou encore les appels journaliers matinaux. Mes nombreux aller-retours transatlantiques ont rythmé ma vie d’expatriée et comblé le manque du pays. Mes visites dans l’Hexagone m’ont toujours permis de me ressourcer, de m’évader et de me rappeler quelle Française de cœur je suis. Merci à Bruno pour ton amour et ton soutien durant toutes ces années. Ta présence ici a rendu mon séjour tellement plus agréable surtout avec toutes les galères traversées durant ces quatre années. Nos weekends dans les parcs nationaux et nos escapades sur le continent ont rendu mes années nord-américaines inoubliables. Merci pour tout, je vous aime !
Avant-Propos
La présente thèse de doctorat est divisée en 5 chapitres.
Le premier chapitre est une introduction qui décrit le globe oculaire, les principes généraux du cancer et l’avancée des connaissances sur le mélanome uvéal.
Le deuxième chapitre concerne le manuscrit « Expression of serotonin receptor 2B and effects of an antagonist on uveal melanoma cell lines » qui sera soumis au journal Clinical
and Experimental Metastasis. Ma contribution est la suivante: j’ai effectué 90% des
expériences, analysé les résultats et rédigé la première version du manuscrit sous la supervision de la Pre Solange Landreville.
Le troisième chapitre concerne le manuscrit en préparation « Analysis of DNA hydroxymethylation in melanocytes and uveal melanoma cell lines » qui sera soumis au journal Pigment Cell & Melanoma Research. La contribution des auteurs est la suivante: j’ai effectué 70% des expériences assistée de Julie Bérubé. Jade Pomerleau et Marie-Pier Veilleux ont participé à la caractérisation des lignées cancéreuses utilisées et ont également réalisé l’immunobuvardage Western pour les IDHs. Carolyne M. Lowry a réalisé les analyses HPLC-MS\MS. Ces dernières analyses ont été supervisées par le Pr Richard J. Wagner (Université de Sherbrooke). J’ai analysé les résultats et rédigé la première version du manuscrit sous la supervision de la Pre Solange Landreville.
Le quatrième chapitre concerne le manuscrit « Differential responses of choroidal melanocytes and uveal melanoma cells to low oxygen conditions » publié dans le journal
Molecular Vision. La contribution des auteurs est la suivante: j’ai effectué 50% des
expériences et Jade Pomerleau ainsi que Laurence Trudel-Vandal ont réalisé les autres. J’ai analysé les résultats et rédigé la première version du manuscrit avec l’aide de Jade Pomerleau sous la supervision de la Pre Solange Landreville. Tous les auteurs ont lu et approuvé le manuscrit avant sa soumission.
Enfin, j’ai également collaboré à un autre projet durant mon doctorat dont les résultats ne sont pas mentionnés dans cette thèse. Ainsi, je suis co-auteure d’un article publié dans le journal Investigative Ophthalmology & Visual Science1 en 2016.
1Mouriaux F, Zaniolo K, Bergeron MA, Weidmann C, De La Fouchardiere A, Fournier F, et al. Effects of
long-term serial passaging on the characteristics and properties of cell lines derived from uveal melanoma primary tumors. Invest Ophthalmol Vis Sci. 2016;57(13):5288-301.
1
Chapitre 1: Introduction
1.1 Anatomie du globe oculaire
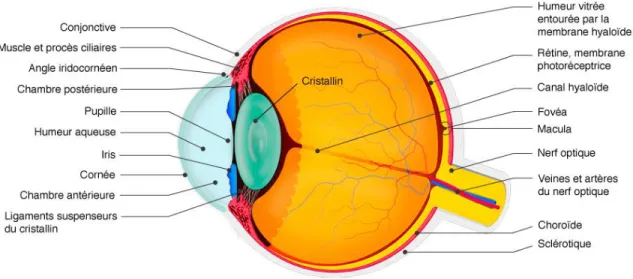
L’œil est l’organe de la vision. La vue est précieuse, elle est essentielle pour intégrer le monde qui nous entoure. L’œil adulte est une structure creuse de forme sphérique d’une longueur axiale de 2,3 cm ayant une masse d’environ 7 g (Figure 1.1) [1, 2]. Inséré dans l’orbite et protégé par une couche de graisse, il est entouré de plusieurs structures annexes qui ont pour principal objectif de le préserver.
Le globe oculaire est composé de trois chambres : la chambre antérieure (espace entre la cornée et l’avant de l’iris), la chambre postérieure (espace entre l’iris et le cristallin) et la chambre vitrée (derrière le cristallin) [1, 2]. Les chambres antérieures et postérieures sont remplies d’humeur aqueuse, un liquide transparent. Le corps vitré, une substance gélatineuse transparente qui occupe 90% du volume de l’œil est contenu dans la chambre vitrée. Sécrétées par le corps ciliaire et essentiellement composées d’eau, ces substances permettent le maintien de la forme du globe et la régulation de la pression intraoculaire. Le globe oculaire est formé de trois tuniques: fibreuse, vasculaire et nerveuse [1, 2]. La tunique fibreuse est constituée de la sclère (aussi appelée sclérotique) en position postérieure et de la cornée en position antérieure. La sclère est la couche protectrice externe qui enveloppe le globe oculaire en postérieur. C’est un tissu fibreux blanchâtre et opaque très résistant. La surface postérieure de cette couche protectrice est traversée par le nerf optique. Sa surface antérieure est recouverte d’une fine muqueuse transparente appelée la conjonctive [1]. Ensuite, la cornée est une couche transparente non vascularisée de forme incurvée, qui permet la focalisation de la lumière sur la rétine. La cornée est en continuité avec la sclére et la conjonctive au niveau du limbe cornéen.
Figure 1.1: Coupe sagittale de l'œil humain. Tirée de http://chenieux-ophtalmologie.com.
La tunique vasculaire, également appelée uvée, est la couche nutritive du globe [1, 2]. L’uvée se divise en trois parties: l’iris, le corps ciliaire et la choroïde. L’iris est la structure la plus antérieure de l’uvée située entre la cornée et le cristallin. C’est une membrane circulaire et contractile; elle constitue la partie colorée de l’œil grâce aux lamelles pigmentaires contenant de la mélanine qui confèrent la couleur des yeux. Au centre de l’iris, la pupille permet de réguler la quantité de lumière pénétrant dans le globe oculaire via des muscles radiaux et circulaires. Le corps ciliaire, la couche la plus épaissie de l’uvée, constitue la portion antérieure de la choroïde sur laquelle est attaché le cristallin par l’intermédiaire des ligaments suspenseurs. Le corps ciliaire sécrète l’humeur aqueuse et influence la courbure du cristallin. La choroïde sera décrite à la section 1.2.
La tunique nerveuse également appelée rétine, est une membrane mince pluristratifiée qui tapisse les trois quarts postérieurs de l’œil [1, 2]. La rétine est un tissu neurosensoriel dont la fonction est de capter la lumière et de la convertir en stimuli nerveux (i.e. phototransduction) qui seront transmis par l’intermédiaire du nerf optique au cortex visuel qui est le siège du traitement de l’information visuelle [2]. Elle se compose de l’épithélium pigmentaire rétinien (EPR) et de la rétine neurale [3]. Cette dernière est divisée en trois couches de neurones: les photorécepteurs, les cellules bipolaires et les cellules ganglionnaires (Figure 1.2) [1, 2]. Il existe deux grands types de photorécepteurs en
fonction de leur morphologie : les bâtonnets et les cônes. Les photorécepteurs sont composés de quatre parties distinctes : un segment externe, un segment interne, un corps cellulaire et une terminaison synaptique. Les bâtonnets présentent un long segment externe cylindrique alors que les cônes ont un segment externe très court (Figure 1.2). Les bâtonnets sont beaucoup plus sensibles à la lumière et sont plus nombreux que les cônes. Ils permettent la vision nocturne et l’appréciation des formes et des mouvements. Les cônes interviennent dans la vision des couleurs et l’acuité visuelle. Les bâtonnets sont localisés en périphérie de la rétine alors que les cônes sont concentrés dans la fovéa de la macula [1, 2]. Lorsque les cônes sont excités par la lumière, ils la transforment en influx nerveux qui vont transiter vers les cellules bipolaires et ganglionnaires, dont les axones forment le nerf optique. Ces signaux sont alors acheminés vers le cerveau en passant par la couche plexiforme externe qui contient les cellules horizontales et la couche plexiforme interne composée des cellules amacrines. La rétine neurale est également composée de cellules gliales (i.e. cellules de Müller, astrocytes, cellules microgliales), ainsi que de cellules vasculaires (i.e. cellules endothéliales et péricytes) localisées dans la couche des fibres nerveuses et dans les couches plexiformes interne et externe [1, 2]. Enfin, l’EPR est une monocouche de cellules épithéliales dont les rôles principaux sont le transport de nutriments, d’ions et d’eau, l’absorption de la lumière, la réisomérisation du tout-trans rétinal en 11-cis-rétinal (un élément clé du cycle visuel), ainsi que le renouvellement des segments externes des photorécepteurs par phagocytose [4].
Figure 1.2: Organisation laminaire des cellules de la rétine et schématisation de la structure des photorécepteurs. Tirées de https://www.annabac.com (A) et http://lecerveau.mcgill.ca (B). La vascularisation de l’œil est assurée par l’artère ophtalmique, une branche de l’artère carotide interne. L’artère ophtalmique se ramifie pour donner naissance à d’autres artères qui vont irriguer l’uvée [1]. Ainsi, la majorité de l’irrigation du globe oculaire provient des vaisseaux uvéaux (e.g. longue et courte artères ciliaires postérieures); seuls les vaisseaux rétiniens (e.g. artère centrale rétinienne) sont responsables de l’apport sanguin de la rétine interne [1]. Ce sont les longue et courte artères ciliaires postérieures qui sont responsables de l’irrigation de l’uvée. Des vaisseaux lymphatiques sont présents dans certaines structures de l’œil, notamment le corps ciliaire et le limbe cornéen [5] et permettent le maintien de l’homéostasie du globe oculaire. L’absence de vaisseaux lymphatiques fonctionnels dans la choroïde explique la dissémination des métastases exclusivement via la circulation sanguine [6].
1.2 La choroïde
1.2.1 Anatomie
Située entre la sclère et la rétine, la choroïde constitue la membrane nourricière de l’œil. Selon la zone considérée, son épaisseur est variable [7]. La choroïde se divise en quatre couches principales : la membrane de Bruch, la couche de choriocapillaires, la couche des gros vaisseaux et la suprachoroïde (Figure 1.3). Riche en fibres élastiques et de collagènes, la membrane de Bruch est située entre l’EPR et la couche de choriocapillaires [7]. Cette dernière représente un important réseau de capillaires fenêtrés qui permet le passage des nutriments et de l’oxygène vers les couches externes de la rétine. La couche de gros vaisseaux est composée de nombreux mélanocytes, fibroblastes et vaisseaux sanguins enrobés dans du tissu conjonctif (i.e. stroma) [7]. Elle est subdivisée en deux couches vasculaires: les couches de Sattler et de Haller. La couche de Sattler contient des vaisseaux sanguins de diamètre moyen à petit (i.e. artérioles et veinules), alors que la couche de Haller renferme des vaisseaux de gros diamètre (i.e. artères ciliaires postérieures). Enfin, la suprachoroide est constituée essentiellement de fibroblastes, de mélanocytes, de fibres élastiques et de collagènes.
1.2.2 Les fonctions de la choroïde
La structure de la choroïde lui assure plusieurs fonctions, mais la plus essentielle est d’assurer l’oxygénation et la nutrition de la rétine externe [7]. Elle est très vascularisée; son important réseau de vaisseaux sanguins lui permet donc une régulation mineure de la pression intraoculaire par la modification de son volume et une thermorégulation de la rétine en protégeant les photorécepteurs des températures extrêmes [7]. Une autre de ses fonctions est de former un écran qui va permettre de reproduire à l’intérieur de l’œil une chambre noire. En effet, les mélanocytes, présents en grand nombre dans la choroïde, produisent un pigment (i.e. la mélanine) qui absorbe la lumière diffuse et permet d’obtenir une image nette [8]. La choroïde assure un rôle mineur dans la modulation de la vascularisation en sécrétant des facteurs de croissance comme le VEGF (vascular
endothelial growth factor) et le bFGF (basic fibroblast growth factor) [7, 9]. Enfin, elle
peut également agir sur la position de la rétine en changeant son épaisseur [7].
1.2.3 Les mélanocytes oculaires
Les mélanocytes ont pour précurseurs les mélanoblastes, des cellules dérivées des crêtes neurales [10]. Lors de la morphogénèse embryonnaire, ces structures transitoires donnent naissance à plusieurs lignées cellulaires qui se différencient progressivement durant leur migration jusqu'à leur territoire cible: certains types de neurones et de cellules gliales, les cellules chromaffines, certaines cellules osseuses et mésenchymateuses du squelette, les cellules des leptoméninges et les cellules mélanocytaires (Figure 1.4). Dans la choroïde, les mélanocytes sont différenciés, pigmentés et caractérisés par une morphologie dendritique et fusiforme [11]. Les mélanocytes contiennent un organite intracellulaire, le mélanosome, dans lequel est synthétisé le pigment de la mélanine, qui confère sa couleur brunâtre à la choroïde [8]. Dépendamment du type de mélanine produite (i.e. eumélanine ou phéomélanine), on peut distinguer deux types de mélanosomes. Ainsi, les eumélanosomes sont larges, de forme elliptique et contiennent une matrice très riche en glycoprotéines
fibrillaires requises pour la synthèse de l’eumélanine de couleur brune à noire, alors que les phéomélanosomes sont plus petits, sphériques et produisent de la phéomélanine de couleur jaune à rouge [12].
Figure 1.4: Schématisation des principales structures dérivées des crêtes neurales lors de la morphogénèse embryonnaire. A, crêtes neurales; B, tube neural (Zone du manteau); C,
mésenchyme; D, tube neural (couche épendymaire); 1a, neuroblaste bipolaire; 1b, neuroblaste bipolaire en différenciation; 1c, neurone sensitif pseudo-unipolaire des ganglions rachidiens; 2a, neuroblaste unipolaire; 2b, neurone multipolaire des ganglions sympathiques; 2c, médulloblaste (cellules chromaffines) dans la médullaire de la glande surrénale; 3a, glioblaste; 3b, cellule de Schwann; 3c, cellule satellite; 4a, cellule mésenchymateuse; 4b, cellule des leptoméninges; 4c, cellule de l’ecto-mésenchyme; 5a, mélanoblaste; 5b, mélanocyte. Tirée de http://www.embryology.ch.
La maturation du mélanosome se divise en quatre stades (Figure 1.5). Au stade I, le mélanosome immature appelé prémélanosome, correspond à un organite non mélanisé. Il
est de forme sphérique et contient des filaments et des vésicules internes dérivés de l’invagination de la membrane [12]. Au stade II, le mélanosome se remplit de fibres intraluminales disposées de façon parallèle ce qui allonge sa structure. Deux protéines sont essentielles à la maturation du stade I au stade II : PMEL (aussi appelée gp100) et MILANA (Melan-A, aussi appelée MART-1). PMEL permet la production des fibres intraluminales, en effet plusieurs fragments de PMEL constituent la matrice fibrillaire de l’organite. MLANA est nécessaire à l’expression et à la stabilité de PMEL. Au stade III, le mélanosome est partiellement pigmenté, la mélanine est répartie de manière uniforme. La pigmentation devient homogène et uniforme au stade IV; les structures fibrillaires ne sont alors plus visibles sous le dépôt de la mélanine.
Figure 1.5: Schématisation de la structure des mélanosomes. Schéma d’un corps cellulaire de la
peau représentant les différents stades de maturation des mélanosomes (A). Le degré de pigmentation de la mélanine apparait en noir. Images en microscopie électronique à balayage représentant les différents stades de maturation des mélanosomes (B). Images adaptées de [13].
Plusieurs enzymes sont nécessaires à la mélanogénèse. Le facteur de transcription MITF (micropthalmia-associated transcription factor) joue un rôle essentiel dans la biogénèse des mélanocytes et leur pigmentation. Il contrôle l’expression de trois enzymes essentielles à la pigmentation : TYR (tyrosinase), TYRP1 (tyrosinase-related protein 1) et DCT
(dopachrome tautomérase, aussi appelée TYRP2). La TYRP1 est essentiellement synthétisée dans le réticulum endoplasmique, puis elle mature dans le réseau de l’appareil de Golgi avant d’être introduite dans l’endosome. La DCT suit relativement les mêmes étapes de maturation. Au cours de leur maturation, les mélanosomes migrent de la région périnucléaire, où ils sont produits, vers l’extrémité des dendrites. Ce déplacement intracellulaire implique les microtubules, la dynéine, la kinésine, les filaments d’actine, RAB27A, MLPH (melanophilin) et MYO5A (myosine 5A). La dynéine et la kinésine sont impliquées dans le transport antérograde/rétrograde des microtubules, alors que la MYO5A permet le transport des mélanosomes dans les filaments d’actine [12].
Les mélanocytes jouent également un rôle dans la chélation d’agents toxiques, certains processus immunitaires ainsi que la photoprotection en absorbant les rayons ultraviolets (UVs) et en neutralisant les radicaux libres et d’autres espèces chimiques réactives induites par les UVs [8]. Contrairement aux mélanocytes de la peau, les mélanocytes oculaires ne synthétisent pas de novo la mélanine et leurs mélanosomes ne sont pas transférés aux cellules voisines. Ils contiennent peu de mitochondries, ce qui suggère une faible activité métabolique [8]. Une transformation anormale des mélanocytes de l’uvée conduit au développement d’un naevus (tumeur bénigne) ou d’un mélanome uvéal (tumeur maligne) [14].
1.3 Le mélanome uvéal
1.3.1 Étiologie et facteurs prédisposant au mélanome uvéal
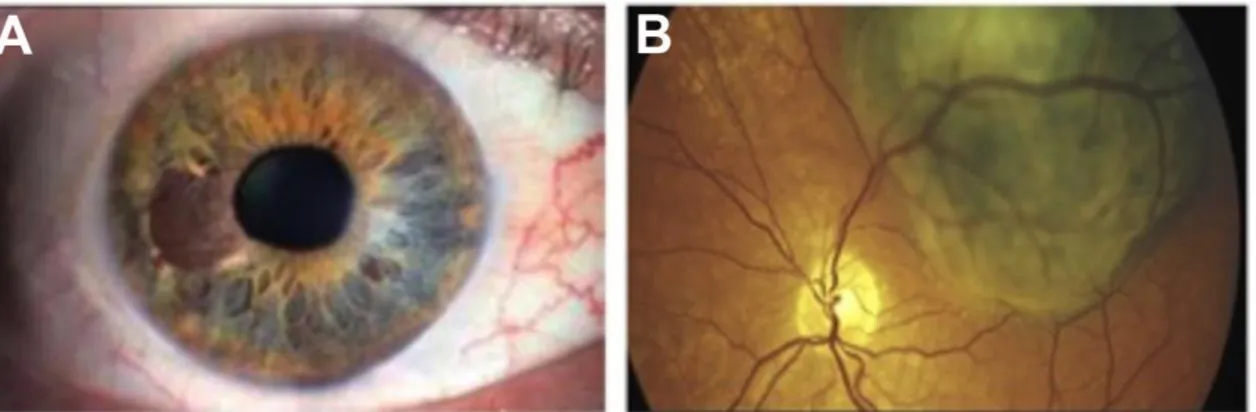
Le mélanome uvéal représente la tumeur intraoculaire primaire maligne la plus fréquente chez l’adulte [14]. La tumeur est le plus souvent localisée dans la choroïde (80% des cas), mais peut également se retrouver au niveau du corps ciliaire et de l’iris (Figure 1.6). Dans 10 à 30% des cas, le mélanome uvéal est asymptomatique [14]. Ce cancer est fortement métastatique: plus de 50% des patients développent des métastases mortelles au foie dans les 15 années suivant le diagnostic de la tumeur oculaire [14]. Il n’existe aucune évidence d’hérédité familiale; les cas familiaux représentent seulement 0,6% des patients [15].
Plusieurs facteurs peuvent avoir un impact sur l’incidence de ce cancer: l’âge, le sexe, la race, le lieu géographique et les maladies congénitales oculaires. L’âge moyen au moment du diagnostic est de 50 à 60 ans [14]. Les cas chez les enfants et jeunes adultes sont très rares [16]. Il existe une certaine prédominance chez les hommes [17].
Figure 1.6: Apparences cliniques du mélanome uvéal. Mélanome de l’iris (A). Mélanome de la choroïde (B). Tirées de http://www.snof.org.
Ensuite, l’incidence annuelle est plus élevée chez la population caucasienne, soit de 4 à 7 nouveaux cas par million en Amérique du Nord [14] et de 6 à 8 cas par million en Australie [18]. En Europe, l’incidence annuelle montre un gradient du nord au sud, avec 8 nouveaux cas par million dans les pays du nord, comparativement à 2 nouveaux cas par million dans les pays du sud [19]. De plus, l’incidence est trois fois supérieure chez les personnes aux iris pâles et à la peau claire [16, 20]. Les Caucasiens avec une peau mate et des yeux bruns ont ainsi moins de chance de développer le mélanome uvéal [21]. Les mélanomes uvéaux nés de la transformation d’un naevus sont très rares (0,02% des cas) [22]. Cependant, les mélanocytoses congénitales oculaires et oculodermiques sont plus courantes chez les personnes ayant développé un mélanome uvéal [17]. Ces deux pathologies se caractérisent par une prolifération anormale des mélanocytes.
1.3.2 Les facteurs pronostiques du mélanome uvéal
En oncologie, le système de classification TNM est un langage commun utilisé par les médecins pour décrire les tumeurs solides selon l’Union internationale contre le cancer (UICC) [23]. La classification TNM se base sur trois éléments (Tableau 1.1).
Tableau 1.1: Éléments de la classification TNM.
Les facteurs pronostiques du mélanome uvéal peuvent être regroupés en quatre groupes: cliniques (avant énucléation), histologiques (post-énucléation), cytogénétiques (anomalies chromosomiques) et génétiques (mutations et signature génique) (Tableaux 1.2 et 1.3) [24].
1.3.2.1 Les facteurs cliniques
La localisation de la tumeur et sa taille sont disponibles avant une énucléation grâce à l’ultrasonographie [14]. Les mélanomes situés dans le corps ciliaire ont le plus mauvais pronostic avec 53% de mortalité sur 5 ans, tandis que les mélanomes de l’iris sont associés à un meilleur pronostic avec 6% de mortalité sur 10 ans [24]. Le diamètre de la tumeur fait également partie des facteurs déterminant le pronostic du patient; une grosse tumeur est ainsi davantage associée à un mauvais pronostic [24].
Lettre Signification Description
T Tumeurs Taille de la tumeur primaire/degré d’extension aux tissus voisins (T0-T4)
N
Ganglions lymphatiques (Nodes)
Classification des ganglions en fonction de leur nombre, de leur taille et de leur extension locale (N0-N3)
1.3.2.2 Les facteurs histologiques
La classification cytologique, l’activité mitotique, la présence de microvaisseaux, de lymphocytes et d’une extension extra-sclérale sont les facteurs histologiques qui permettent d’établir un pronostic. Ainsi, les mélanomes uvéaux sont classés selon trois types cellulaires: épithéloïde, fusiforme ou mixte (i.e. un mélange des deux types précédents). Les cellules fusiformes sont moins propices à la migration cellulaire vers le foie et donc associées à un meilleur pronostic, tandis que le type épithéloïde est corrélé à un plus grand risque de développer des métastases [16, 24]. Le pronostic des mélanomes mixtes est mauvais si la tumeur est composée de plus de 50% de cellules épithélioïdes. Ensuite, l’activité mitotique est utilisée pour le pronostic dans de nombreuses études [16, 24]: un grand nombre de mitoses cellulaires dans la tumeur est associé à un mauvais pronostic [25]. Les cellules agressives du mélanome uvéal ont la capacité d’exprimer un phénotype de cellules endothéliales et à former des pseudo-vaisseaux dépourvus de cellules endothéliales. Ce processus est nommé « mimétisme vasculogénique » [26-29]. Il existe neuf types de patrons microvasculaires et les types complexes « parallèle », « en boucle » et « en réseau » ont été associés aux tumeurs de patients décédés des suites du mélanome uvéal [29]. Également, plusieurs études ont démontré qu’une infiltration de lymphocytes dans la tumeur est corrélée à une diminution de la survie du patient (mortalité de 62% sur 15 ans) [24, 26]. Enfin, la présence d’une extension extra-sclérale est accompagnée d’un risque accru de mortalité (mortalité de 75% sur 10 ans) [16, 24].
1.3.2.3 Les facteurs cytogénétiques et génétiques
Des études cytogénétiques ont démontré des anomalies chromosomiques fréquentes sur les chromosomes 1, 3, 6 et 8 dans le mélanome uvéal [14, 30, 31]. Présente dans ¼ des mélanomes uvéaux, la perte du bras court du chromosome 1 (1p-) est souvent associée à la perte d’une copie du chromosome 3 (monosomie 3-) [31]. La perte de 1p est l’une des rares anomalies chromosomiques qui a un pouvoir pronostique indépendant du statut du chromosome 3 et sa présence est corrélée à une diminution de la survie [30]. La monosomie 3 est détectée dans 50-60% des mélanomes uvéaux énucléés et la majorité des patients
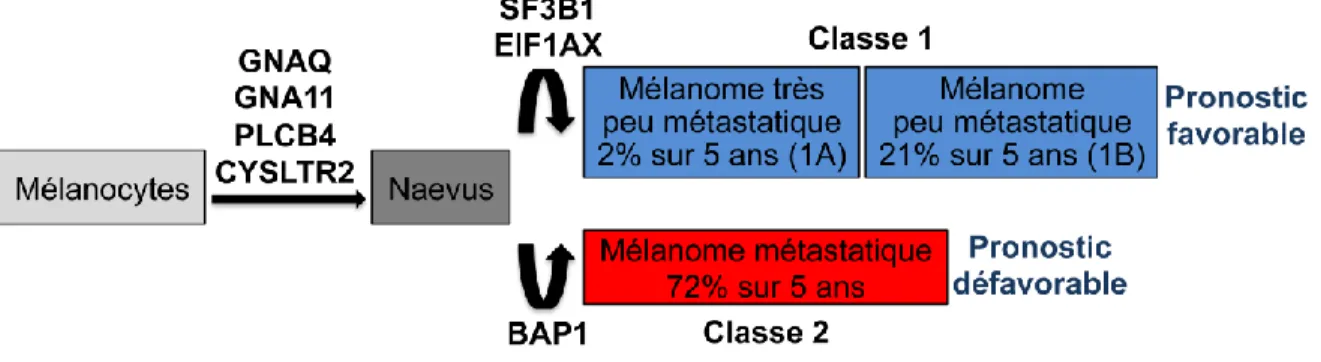
présentant cette anomalie décèdent de métastases au foie [32, 33]. Une perte du bras long ou un gain du bras court du chromosome 6 (6q-, 6p+) se retrouvent dans plus de ⅓ des mélanomes uvéaux [30]. Ces deux anomalies peuvent être présentes dans une même tumeur, suggérant la formation d’un isochromosome 6p [30, 34]. Ensuite, une perte du bras court du chromosome 8 (8p-) est présente dans ¼ des mélanomes uvéaux, comparativement au gain du bras long (8q+) qui représente 40% des cas [35]. Une perte d’une copie du chromosome 3 et une amplification du chromosome 8 (trisomie 8 ou amplification 8q) co-occurent fréquemment et sont associées à un très mauvais pronostic [24, 33, 35, 36]. Au contraire, un gain du bras court du chromosome 6 (6p) est associé à un très bon pronostic [14, 30, 37]. Ces deux évènements mutuellement exclusifs représentent une bifurcation dans la progression métastatique du mélanome uvéal (Figure 1.11) [14, 38].
Tableau 1.2: Facteurs pronostiques du mélanome uvéal. Adapté de [16, 24].
Catégorie Facteur Pronostic* Mortalité/Période
Clinique Localisation Iris Choroïde Corps ciliaire B I M 5-6% sur 10 ans 14% sur 5 ans 22-53% sur 5 ans Taille (diamètre) Petite (D<10 mm) Moyenne (D<10-15 mm) Grande (D>15 mm) B I M 1 à 3% sur 5 ans 14-16% sur 5 ans 32% sur 5 ans Histologique Type cellulaire Fusiforme Mixte Épithéloïde B I M 20% sur 15 ans 60% sur 15 ans 75% sur 15 ans Activité mitotique Faible Moyenne Forte B I M 13% sur 6 ans 43% sur 6 ans 56% sur 6 ans Patrons microvasculaires complexes Absence Présence B M 10% sur 10 ans 50% sur 10 ans Lymphocytes dans tumeur Absence Présence B M 30% sur 15 ans 60% sur 15 ans Extension extra-sclérale Absence Présence B M 37% sur 10 ans 75% sur 10 ans Cytogénétique Anomalies chromosomiques Absence 1p- 6p+ 6q- Monosomie 3 8q+ Monosomie 3/8q+ 8p- B M B B M M M M 6% sur 5 ans ND** ND ND 40% sur 5 ans 31% sur 5 ans 66% sur 5 ans ND
Plusieurs mutations somatiques ont été identifiées dans le mélanome uvéal (Tableau 1.3, Figures 1.7-1.9).
Tableau 1.3: Mutations somatiques du mélanome uvéal. Adapté de [39-46]. Mutations somatiques Localisation
chromosomique Pronostic* GNAQ Chromosome 9 B GNA11 Chromosome 7 B BAP1 Chromosome 3 M SF3B1 Chromosome 2 I EIF1AX Chromosome X I PLCB4 Chromosome 20 B CYSLTR2 Chromosome 13 B
*Pronostic: B, bon; I, intermédiaire; M, mauvais.
Les mutations les plus fréquentes sont observées dans la voie des MAPK
(mitogen-activated protein kinases), dans les exons 4 (R183) et 5 (Q209) des gènes GNAQ/GNA11
(Figure 1.7). Ces mutations oncogéniques sont mutuellement exclusives et très communes dans les cas de mélanomes uvéaux (83%) [39-41]. Les mutations qui affectent le codon Q209 du gène GNA11 sont présentes dans 32% des tumeurs primaires et 57% des cas avec métastases. Au contraire, les mutations dans le codon Q209 du gène GNAQ affectent 45% des tumeurs primaires et 22% des cas métastatiques. De plus, les mutations qui touchent le codon R183 sont similaires dans les deux gènes avec une prévalence de 6% dans le mélanome uvéal. Ces mutations conduisent à une activation constitutive des sous-unités Gαq et Gα11 et bloquent l’activité GTPase intrinsèque qui conduit à un retour à l’état inactif;
il en résulte alors une prolifération cellulaire incontrôlée (Figure 1.8). Le fait que ces mutations soient aussi détectées dans les naevi suggère que les gènes mutés GNAQ/GNA11 sont impliqués dans les évènements précoces de la tumorigenèse, mais qu’ils ne sont pas suffisants pour conduire au stade métastatique (Figure 1.9) [47].
Des mutations inactivatrices ont été retrouvées dans le gène BAP1 (BRCA1-associated
protein 1) dans 84% des cas métastatiques (Figures 1.7-1.8); ce serait donc un gène
BAP1 est localisé sur le chromosome 3, ce qui peut expliquer l’association entre la
monosomie du chromosome 3 et la présence de métastases (Figure 1.9) [47]. Les mutations dans BAP1 sont retrouvées tout le long du gène et les régions codantes sont les plus touchées (Figure 1.7). Le gène BAP1 code pour une ubiquitine carboxy-terminale hydrolase qui se lie à BRCA1 et potentialise sa fonction anti-proliférative [48]. BAP1 est un régulateur clé de la différenciation des mélanocytes. En effet, la déplétion du gène BAP1 induit un phénotype de cellules souches et favorise la progression tumorale [49].
Récemment, des mutations ont été découvertes dans les gènes SF3B1 (splicing factor 3
subunit beta 1), EIF1AX (eucaryotic translation initiation factor 1A, X–linked), PLCB4
(phospholipase C bêta 4) et CYSLTR2 (cysteinyl leukotriene receptor 2) (Figure 1.9, Tableau 1.3) [43-46]. SF3B1 est une composante des petites protéines nucléaires snRNP (small nuclear ribonucleoprotein) du spliceosome qui participe à l’épissage des ARN pré-messagers (Figure 1.8) [43, 44]. Les mutations dans le gène SF3B1 sont retrouvées dans les domaines non redondants (HEAT) (Figure 1.7) [43, 44]. La plus commune est positionnée dans l’exon 14 (R625) (Figure 1.7), mais d’autres mutations ont été rapportées (K666 et K700) [43, 44, 50]. Retrouvées dans 18% des tumeurs primaires, ces mutations sont hétérozygotes et mutuellement exclusives avec celles du gène BAP1; elles sont donc associées à un meilleur pronostic [43, 44, 47].
Figure 1.7: Structure des domaines protéiques et profil des mutations dans les gènes fréquemment mutés dans le mélanome uvéal. Les gènes GNAQ et GNA11 codent pour les
sous-unités Gαq et Gα11 qui sont composées d’un domaine hélicoïdal et d’un domaine GTPase
catalytique. Les mutations (épingles) sont localisées dans les exons 4 et 5 pour les deux acides aminés (Q209 et R183) (A). BAP1 est composé d’un domaine ubiquitine carboxyl-terminal hydrolase (UCH) catalytique en N-terminal, d’un domaine de liaison BARD, d’un motif de liaison HCF1 (host cell factor 1), d’un domaine de liaison à BRCA1 et de séquences de localisation nucléaire (NLS). Les mutations non tronquantes (barres) et tronquantes (épingles) sont indiquées (B). SF3B1 possède des motifs d’interaction (U) et (S), ainsi que 17 domaines non redondants HEAT (HD). La mutation la plus commune est sur le codon R625 de l’exon 14 (épingle). Les autres mutations moins communes sont également indiquées (barres) (C). EIF1AX présente une extrémité N-terminale possédant les mutations identifiées jusqu’à maintenant correspondant à des délétions de 1-2 acides aminés (barres), un site de liaison d’oligonucléotide (OB fold) et une extrémité C-terminale (D). Le gène PLCB4 code pour la phospholipase C composée de plusieurs domaines en gris (PH, EF, X, Y et C2 (catalytiques)). La mutation la plus commune est indiquée (épingle; D630) (E). Localisation des mutations dans le gène CYSLTR2 dans la cohorte TCGA. La mutation sur la leucine 129 (flèche rouge) est détectée dans les mélanomes uvéaux (F). Tirée de [45, 47, 51].
Le gène EIF1AX code pour un facteur de transcription qui stimule le transfert du Met-tRNAi (methionyl initiator tRNA) à la sous-unité 40S du ribosome (Figure 1.8) [52]. Les
mutations dans les deux premiers codons de ce gène altèrent l’épissage de certains transcrits d’ARNm, mais son rôle dans la tumorigenèse demeure inconnu (Figure 1.7) [44]. Les mutations dans EIF1AX sont non tronquantes et hétérozygotes [44, 47]. Elles sont également mutuellement exclusives avec celles du gène SF3B1 et associées à un bon pronostic (Figure 1.9) [44, 47].
Figure 1.8: Schématisation des principales fonctions des protéines traduites des gènes fréquemment mutés dans le mélanome uvéal. Les mutations dans GNAQ/GNA11 désactivent l’activité GTPase, résultant en une activation constitutive de la voie des MAPK, conduisant à une prolifération cellulaire incontrôlée. Le gène BAP1 code pour une enzyme qui enlève une mono-ubiquitine (« ub ») de substrats spécifiques, soient l’histone H2A, HCF1 et BRCA1. L’interaction de BAP1 avec AXSL1 affecte la régulation transcriptionelle. Des mutations dans le gène SF3B1 altèrent l’épissage des transcrits d’ARN spécifiques. Enfin, des mutations dans le gène EIF1AX affectent la régulation post-transcriptionelle en altérant la production protéique. Des mutations dans les gènes PLCB4ou CYSLTR2conduisent également à une activation constitutive de la voie des MAPK [46, 47].
Ensuite, une mutation récurrente (D630) a été découverte dans le gène PLCB4 qui joue un rôle dans la transduction intracellulaire des signaux extracellulaires [45]. Tout comme les mutations dans GNAQ/GNA11, cette dernière conduit à l’activation de la voie des MAPK (Figure 1.8) [45]. Cette mutation est mutuellement exclusive avec celles des gènes
Enfin, une dernière mutation récurrente a été identifiée dans le gène CYSLTR2. La mutation (L129) active GNAQ de manière constitutive [46]. La présence de cette mutation dans les mélanocytes favorise la tumorigenèse in vivo et indique donc que CYSLTR2 joue un rôle d’oncogène dans la pathogenèse du mélanome uvéal (Figure 1.9) [46].
Figure 1.9: Anomalies génétiques impliquées dans la progression du mélanome uvéal. Des
mutations dans les gènes GNAQ/GNA11, PLCB4 ou CYSLTR2 initient la transformation maligne, puis celles dans le gène BAP1 sont à l’origine de la cascade d’évènements moléculaires favorisant la dissémination métastatique de ce cancer (classe 2). Les mutations dans les gènes SF3B1/EIF1AX sont associées à un meilleur pronostic. Adaptée de [45-47].
Ensuite, un test basé sur une signature moléculaire permet maintenant de prédire le risque de développer des métastases à partir d’une biopsie de tumeur primaire. Le test
DecisionDx-UM, utilisé de routine dans plusieurs cliniques à travers le monde, divise les
mélanomes uvéaux en plusieurs classes, selon leur probabilité de développer des métastases sur 5 ans (Figure 1.9) [53]. Ce test sera décrit plus en détails à la section 1.4.4.
1.3.3 Le tropisme du mélanome uvéal
L’absence de vaisseaux lymphatiques fonctionnels dans la choroïde et son degré de vascularisation élevé permettent d’expliquer la dissémination hématogénique des cellules métastatiques du mélanome uvéal [6]. Plus de 50% des patients vont développer des métastases dans les 15 années suivant le diagnostic de la tumeur oculaire [14]. Les
métastases sont localisées dans plus de 90% des cas au foie [20]. Elles sont également retrouvées dans les poumons (24%), les os (16%) et la peau (11%) [30]. L’établissement des métastases dépendrait des propriétés des cellules tumorales et des facteurs de l’organe hôte, soit l’hypothèse du « soil and seed » [54]. Cette dernière sera discutée à la section 1.4. Il a été suggéré qu’il existe une symbiose particulière entre les cellules du mélanome uvéal et le microenvironnement hépatique [55]. Ainsi, le foie produit une grande quantité de HGF (hepatocyte growth factor), un puissant mitogène pour les cellules hépatiques qui accroit la dispersion et la mobilité des cellules endothéliales et épithéliales. De nombreuses cellules cancéreuses possèdent le récepteur c-Met spécifique au HGF et il joue entre autres un rôle dans la dissémination métastatique [55, 56]. Des études ont montré que les cas métastatiques de mélanome uvéal présentaient une forte expression de c-Met dans leur tumeur primaire comparativement aux cas sans métastases [56-58]. Par ailleurs, un second facteur de croissance sécrété par le foie, l’IGF-1 (insulin-like growth factor 1), est impliqué dans la prolifération, la migration et la survie cellulaire. Une forte expression de son récepteur (IGF1R) dans le mélanome uvéal corrèle avec la mortalité [57-59].
1.3.4 Diagnostic et traitement du mélanome uvéal
Dans la majorité des cas, le mélanome uvéal est asymptomatique; il est donc détecté tardivement lors d’un examen de routine par ophtalmoscopie indirecte chez l’ophtalmologiste ou l’optométriste [59]. Des symptômes tels qu’une baisse d’acuité visuelle (vision embrouillée), une photopsie (perception d’éclairs, de flashs) et de la douleur peuvent êtres des signes annonciateurs de la présence d’une tumeur oculaire [60]. Actuellement, il n’existe pas de traitement curatif pour le stade métastatique. De plus, des micrométastases seraient présentes avant le traitement de la tumeur oculaire [61]. La période de latence avant la détection clinique des macrométastases est longue et à ce stade, le traitement est inefficace [61].
Le choix du traitement est orienté selon plusieurs facteurs: l’âge du patient, le diamètre de la tumeur et sa localisation, son extension dans les tissus environnants et les analyses histopathologiques de la tumeur énucléée (Tableau 1.4).
1.3.4.1 Le traitement de la tumeur primaire
L’objectif principal du traitement de la tumeur primaire est de minimiser la dissémination des cellules cancéreuses. Le choix du traitement est généralement dicté par sa taille en absence de complications (Tableau 1.4). Lorsque le diamètre est supérieur à 15 mm, l’oncologiste oculaire a recours à l’énucléation, qui consiste à retirer le globe oculaire de l’orbite, tout en préservant les muscles oculaires pour la pose d’une prothèse. Lorsque la tumeur est de taille moyenne, des traitements plus conservateurs sont utilisés comme la thermothérapie et la radiothérapie.
La thermothérapie utilise un laser diode qui émet dans le rouge (810 nm) dont l’énergie est absorbée par les tissus pigmentés. Avec un taux de récidive élevée de 30%, cette technique nécessite encore des progrès [60].
Les principaux types de radiothérapie sont la curiethérapie, la radiothérapie stéréotaxique et la protonthérapie [62]. La radiothérapie utilise des rayons à haute énergie (faisceaux de photons, d’électrons ou de protons) pour affecter l’ADN des cellules cancéreuses. La radiation va affecter toutes les cellules, mais les cellules cancéreuses ont une capacité limitée à réparer leur ADN, alors que les cellules normales vont être capables de réparer les dommages entre les différents traitements. En effet, les points de contrôles importants du cycle cellulaire sont souvent dérégulés ou mutés dans les cancers, ce qui va limiter la réparation de l’ADN par les cellules cancéreuses suite à la radiation. Le dosage radiologique dépend du degré de radiosensibilité de la tumeur, du degré de tolérance du tissu normal et enfin de la quantité de tissus à traiter par radiation.
Tableau 1.4: Traitements recommandés selon la taille de la tumeur. Adapté de [63].
Taille de la tumeur Traitements recommandés
Petite
(diamètre <10 mm; épaisseur < 3 mm) Surveillance Moyenne
(diamètre 10-15 mm; épaisseur 3-8 mm) Curiethérapie, protonthérapie, thermothérapie Grosse
(diamètre >15 mm; épaisseur > 8 mm) Énucléation, protonthérapie
Figure 1.10: Schématisation du principe de la curiethérapie par plaque. Des grains radioactifs
sont contenus dans une plaque en or installée sur la sclère au niveau de la tumeur lors d’une chirurgie. La plaque est gardée de 4 à 7 jours selon le type de substance radioactive. Adaptée de http://www.cancer.ca.
La curiethérapie par plaque, aussi appelée brachythérapie, est une technique de radiothérapie très localisée qui consiste à placer une plaque en or radioactive sur la sclère au niveau de la tumeur (Figure 1.10). Les principales substances radioactives utilisées sont l’iode-125 et le palladium-103 (Canada et États-Unis) ou encore le ruthénium 106 (Europe) [64]. Le choix de la substance radioactive dépend de la durée du traitement en fonction de sa demi-vie. Lorsque la dose de radiation a été administrée, la plaque est retirée par chirurgie (après 4-7 jours d’irradiation). Les avantages de ce traitement sont de préserver la vision du patient et les tissus avoisinants étant donné que la plaque est placée le plus près possible de la tumeur. Cette technique s’utilise pour des tumeurs de taille petite à moyenne.
Des complications locales peuvent survenir telles qu’une rétinopathie radique, une cataracte et un glaucome secondaire.
La radiothérapie stéréotaxique est un type de radiothérapie externe qui dirige de façon très précise les faisceaux de radiation vers une région particulière comme par exemple le fond de l’œil. Elle s’utilise dans le cas d’une tumeur localisée près du nerf optique si la curiethérapie ne peut être utilisée. Une seule dose élevée de radiations vers la tumeur, appelée fraction unique, est administrée pendant 5 jours [65]. Elle nécessite une formation et des équipements spécialisés et n’est donc pas offerte dans tous les centres d’oncologie oculaire.
La protonthérapie utilise un faisceau de protons très précis qui irradie la tumeur en préservant les tissus sains proches de la tumeur. Son utilisation est pertinente pour des interventions de très grande précision. C’est une technique fiable et efficace puisqu’elle permet une conservation du globe oculaire dans 90% des cas et un contrôle local de la tumeur dans 96% des cas [64]. Elle requiert une formation et des équipements spécialisés (cyclotron) et n’est donc pas offerte dans tous les centres d’oncologie oculaire. Cependant, le choix entre les thérapies radicales et conservatrices n’a pas un grand impact sur la survie étant donné la dissémination précoce de ce cancer [61, 66].
1.3.4.2 Le traitement des métastases
Après le traitement de la tumeur primaire, il est important de surveiller l’apparition de métastases hépatiques. Un bilan sanguin comprenant des tests biochimiques sur la fonction hépatique et une imagerie du foie (ultrasonographie abdominale et tomographie à émission de positron) sont réalisés de 1-2 fois par année afin de diagnostiquer des lésions [67]. La survie des patients suite au diagnostic des métastases hépatiques varie de 4 à 30 mois [68]. Des études ont démontré que la survie des patients est indépendante de la méthode de traitement de la tumeur primaire [24, 69] et le taux de survie des patients demeure inchangé depuis les années 70s [69]. Plusieurs données supportent le concept de micrométastases
![Figure 1.3: Schématisation des différentes couches de la choroïde. Image adaptée de [1]](https://thumb-eu.123doks.com/thumbv2/123doknet/3448109.100709/25.918.190.729.662.1004/figure-schématisation-couches-choroïde-image-adaptée.webp)
![Tableau 1.3: Mutations somatiques du mélanome uvéal. Adapté de [39-46].](https://thumb-eu.123doks.com/thumbv2/123doknet/3448109.100709/35.918.128.639.220.481/tableau-mutations-somatiques-mélanome-uvéal-adapté.webp)
![Tableau 1.4: Traitements recommandés selon la taille de la tumeur. Adapté de [63].](https://thumb-eu.123doks.com/thumbv2/123doknet/3448109.100709/43.918.130.794.142.284/tableau-traitements-recommandés-taille-tumeur-adapté.webp)
![Figure 1.14: Structure typique des récepteurs couplés aux protéines G. Adaptée de [114]](https://thumb-eu.123doks.com/thumbv2/123doknet/3448109.100709/54.918.138.780.425.736/figure-structure-typique-récepteurs-couplés-protéines-adaptée.webp)
![Figure 1.15: Voies de signalisation sous le contrôle du récepteur HTR2B. Tirée de [131]](https://thumb-eu.123doks.com/thumbv2/123doknet/3448109.100709/57.918.206.717.162.356/figure-voies-signalisation-contrôle-récepteur-htr-b-tirée.webp)