Université de Lille
Année Universitaire 2018 / 2019
Faculté de Pharmacie de Lille
THESE
POUR LE DIPLOME D'ETAT DE DOCTEUR EN PHARMACIE
Soutenue publiquement le 4 Octobre 2019 Par Mlle Rachel BOLDRON
_____________________________
LES CAR-T CELLS DANS LA PRISE EN CHARGE DES HEMOPATHIES MALIGNES : APPLICATION AU MYELOME MULTIPLE
_____________________________
Membres du jury :
Président : Monsieur Thierry DINE, Professeur de Pharmacie Clinique, Faculté de Pharmacie de Lille.
Directeur, conseiller de thèse : Monsieur Madjid TAGZIRT, Maitre de conférences Hématologie, Faculté de Pharmacie de Lille.
2
Université de Lille
Président : Jean-Christophe CAMART
Premier Vice-président : Damien CUNY
Vice-présidente Formation : Lynne FRANJIÉ
Vice-président Recherche : Lionel MONTAGNE
Vice-président Relations Internationales : François-Olivier SEYS Directeur Général des Services : Pierre-Marie ROBERT Directrice Générale des Services Adjointe : Marie-Dominique SAVINA
Faculté de Pharmacie
Doyen : Bertrand DÉCAUDIN
Vice-Doyen et Assesseur à la Recherche : Patricia MELNYK Assesseur aux Relations Internationales : : Philippe CHAVATTE Assesseur à la Vie de la Faculté et aux
Relations avec le Monde Professionnel : Thomas MORGENROTH
Assesseur à la Pédagogie : Benjamin BERTIN
Assesseur à la Scolarité : Christophe BOCHU
Responsable des Services : Cyrille PORTA
Liste des Professeurs des Universités - Praticiens Hospitaliers
Civ. NOM Prénom Laboratoire
Mme ALLORGE Delphine Toxicologie
M. BROUSSEAU Thierry Biochimie
M. DÉCAUDIN Bertrand Pharmacie Galénique
M. DEPREUX Patrick ICPAL
M. DINE Thierry Pharmacie clinique
Mme DUPONT-PRADO Annabelle Hématologie
M. GRESSIER Bernard Pharmacologie
M. LUYCKX Michel Pharmacie clinique
M. ODOU Pascal Pharmacie Galénique
M. STAELS Bart Biologie Cellulaire
Faculté de Pharmacie de Lille
3, RUE DU PROFESSEUR LAGUESSE - B.P. 83 - 59006 LILLE CEDEX 03.20.96.40.40 - : 03.20.96.43.64
3
Liste des Professeurs des Universités
Civ. NOM Prénom Laboratoire
M. ALIOUAT El Moukhtar Parasitologie
Mme AZAROUAL Nathalie Physique
M. BERTHELOT Pascal Onco et Neurochimie
M. CAZIN Jean-Louis Pharmacologie – Pharmacie clinique
M. CHAVATTE Philippe ICPAL
M. COURTECUISSE Régis Sciences végétales et fongiques
M. CUNY Damien Sciences végétales et fongiques
Mme DELBAERE Stéphanie Physique
M. DEPREZ Benoît Lab. de Médicaments et Molécules
Mme DEPREZ Rebecca Lab. de Médicaments et Molécules
M. DUPONT Frédéric Sciences végétales et fongiques
M. DURIEZ Patrick Physiologie
M. FOLIGNE Benoît Bactériologie
M. GARÇON Guillaume Toxicologie
Mme GAYOT Anne Pharmacotechnie Industrielle
M. GOOSSENS Jean François Chimie Analytique
M. HENNEBELLE Thierry Pharmacognosie
M. LEMDANI Mohamed Biomathématiques
Mme LESTAVEL Sophie Biologie Cellulaire
M. LUC Gerald Physiologie
Mme MELNYK Patricia Onco et Neurochimie
M. MILLET Régis ICPAL
Mme MUHR – TAILLEUX Anne Biochimie
Mme PAUMELLE-LESTRELIN Réjane Biologie Cellulaire
Mme PERROY Anne Catherine Législation
Mme ROMOND Marie Bénédicte Bactériologie
Mme SAHPAZ Sevser Pharmacognosie
M. SERGHERAERT Eric Législation
Mme SIEPMANN Florence Pharmacotechnie Industrielle
M. SIEPMANN Juergen Pharmacotechnie Industrielle
M. WILLAND Nicolas Lab. de Médicaments et Molécules
Liste des Maîtres de Conférences - Praticiens Hospitaliers
Civ. NOM Prénom Laboratoire
Mme BALDUYCK Malika Biochimie
Mme GARAT Anne Toxicologie
Mme GOFFARD Anne Bactériologie
M. LANNOY Damien Pharmacie Galénique
Mme ODOU Marie Françoise Bactériologie
4
Liste des Maîtres de Conférences
Civ. NOM Prénom Laboratoire
Mme ALIOUAT Cécile Marie Parasitologie
M. ANTHERIEU Sébastien Toxicologie
Mme AUMERCIER Pierrette Biochimie
Mme BANTUBUNGI Kadiombo Biologie cellulaire
Mme BARTHELEMY Christine Pharmacie Galénique
Mme BEHRA Josette Bactériologie
M BELARBI Karim Pharmacologie
M. BERTHET Jérôme Physique
M. BERTIN Benjamin Immunologie
M. BLANCHEMAIN Nicolas Pharmacotechnie industrielle
M. BOCHU Christophe Physique
M. BORDAGE Simon Pharmacognosie
M. BOSC Damien Lab. de Médicaments et Molécules
M. BRIAND Olivier Biochimie
M. CARNOY Christophe Immunologie
Mme CARON Sandrine Biologie cellulaire
Mme CHABÉ Magali Parasitologie
Mme CHARTON Julie Lab. de Médicaments et Molécules
M CHEVALIER Dany Toxicologie
M. COCHELARD Dominique Biomathématiques
Mme DANEL Cécile Chimie Analytique
Mme DEMANCHE Christine Parasitologie
Mme DEMARQUILLY Catherine Biomathématiques
M. DHIFLI Wajdi Biomathématiques
Mme DUMONT Julie Biologie cellulaire
Mme DUTOUT-AGOURIDAS Laurence Onco et Neurochimie
M. EL BAKALI Jamal Onco et Neurochimie
M. FARCE Amaury ICPAL
Mme FLIPO Marion Lab. de Médicaments et Molécules
Mme FOULON Catherine Chimie Analytique
M. FURMAN Christophe ICPAL
Mme GENAY Stéphanie Pharmacie Galénique
M. GERVOIS Philippe Biochimie
Mme GOOSSENS Laurence ICPAL
Mme GRAVE Béatrice Toxicologie
Mme GROSS Barbara Biochimie
M. HAMONIER Julien Biomathématiques
Mme HAMOUDI Chérifa Mounira Pharmacotechnie industrielle
Mme HANNOTHIAUX Marie-Hélène Toxicologie
Mme HELLEBOID Audrey Physiologie
M. HERMANN Emmanuel Immunologie
M. KAMBIA Kpakpaga Nicolas Pharmacologie
M. KARROUT Youness Pharmacotechnie Industrielle
Mme LALLOYER Fanny Biochimie
M. LEBEGUE Nicolas Onco et Neurochimie
Mme LECOEUR Marie Chimie Analytique
Mme LEHMANN Hélène Législation
Mme LELEU-CHAVAIN Natascha ICPAL
5
Mme MARTIN Françoise Physiologie
M. MOREAU Pierre Arthur Sciences végétales et fongiques
M. MORGENROTH Thomas Législation
Mme MUSCHERT Susanne Pharmacotechnie industrielle
Mme NIKASINOVIC Lydia Toxicologie
Mme PINÇON Claire Biomathématiques
M. PIVA Frank Biochimie
Mme PLATEL Anne Toxicologie
M. POURCET Benoît Biochimie
M. RAVAUX Pierre Biomathématiques
Mme RAVEZ Séverine Onco et Neurochimie
Mme RIVIERE Céline Pharmacognosie
Mme ROGER Nadine Immunologie
M. ROUMY Vincent Pharmacognosie
Mme SEBTI Yasmine Biochimie
Mme SINGER Elisabeth Bactériologie
Mme STANDAERT Annie Parasitologie
M. TAGZIRT Madjid Hématologie
M. VILLEMAGNE Baptiste Lab. de Médicaments et Molécules
M. WELTI Stéphane Sciences végétales et fongiques
M. YOUS Saïd Onco et Neurochimie
M. ZITOUNI Djamel Biomathématiques
Professeurs Certifiés
Civ. NOM Prénom Laboratoire
M. HUGES Dominique Anglais
Mlle FAUQUANT Soline Anglais
M. OSTYN Gaël Anglais
Professeur Associé - mi-temps
Civ. NOM Prénom Laboratoire
M. DAO PHAN Hai Pascal Lab. Médicaments et Molécules
M. DHANANI Alban Droit et Economie Pharmaceutique
Maîtres de Conférences ASSOCIES - mi-temps
Civ. NOM Prénom Laboratoire
M. BRICOTEAU Didier Biomathématiques
Mme CUCCHI Malgorzata Biomathématiques
M. FRIMAT Bruno Pharmacie Clinique
M. GILLOT François Droit et Economie pharmaceutique
M. MASCAUT Daniel Pharmacie Clinique
M. ZANETTI Sébastien Biomathématiques
6
AHU
Civ. NOM Prénom Laboratoire
Mme DEMARET Julie Immunologie
Mme HENRY Héloïse Biopharmacie
7
Faculté de Pharmacie de Lille
3, rue du Professeur Laguesse - B.P. 83 - 59006 LILLE CEDEXTel. : 03.20.96.40.40 - Télécopie : 03.20.96.43.64 http://pharmacie.univ-lille2.fr
L’Université n’entend donner aucune approbation aux opinions
émises dans les thèses ; celles-ci sont propres à leurs auteurs.
8
Table des matières
Remerciements 12
Liste des figures 14
Liste des tableaux 15
Liste des abréviations 16
Introduction 18
Partie 1 : Les hémopathies malignes 20
I. Définition 20
II. Des maladies qui se chronicisent en France 21
III. Classification 22
1. Les hémopathies myéloïdes 22
A. Leucémies aigues myéloïdes (LAM) 22
B. Syndromes myéloprolifératifs (SMP) 23
C. Syndromes myélodysplasiques (SMD) 23
2. Les hémopathies lymphoïdes 24
A. Les leucémies 24
1. Leucémie aigüe lymphoblastique (LAL) 24
2. Leucémie lymphoïde chronique (LLC) 24
B. Les lymphomes 25
1. Le lymphome Hodgkinien ou maladie de Hodgkin 25
2. Les lymphomes Non Hodgkiniens (LNH) 25
C. Le Myélome multiple 26
Partie 2 : Le myélome multiple 27
I. Qu’est-ce que le myélome multiple ? 27
1. Définition et historique du myélome multiple 27
2. Epidémiologie : Une maladie rare dont l’incidence progresse 28
3. Les signes cliniques de la maladie 29
A. Physiopathologie du myélome multiple 29
B. Des symptômes bien identifiés liés à la physiopathologie 30
1. Les atteintes osseuses 30
2. L’hypercalcémie 32
3. L’insuffisance rénale 32
4. L’anémie 33
5. Les infections 34
9
D. Comorbidités et facteurs de risques 37
E. Evolution de la maladie 37
II. Le diagnostic et les critères pronostic du MM 38
1. Les critères de diagnostic « CRAB » 39
2. Critères de l’IMWG 2014 (31) 39
3. Les examens permettant le bilan au diagnostic 42
A. Bilan sanguin (analyse via une prise de sang) (27,31) 42 B. Détection et évaluation d’une immunoglobuline monoclonale (34) 42
C. Myélogramme 44
D. Examens d’imagerie (31) 45
4. Les critères d’évaluation du pronostic du MM et les classifications qui en découlent 46
A. Classification de Durie et Salmon (38) 46
B. Classification R-ISS (Revised International Staging System) 47
III. La prise en charge du myélome multiple 49
1. Objectifs thérapeutiques 49
2. Principales classes thérapeutiques dans la prise en charge du MM 52
3. Recommandations de traitements 54
A. Traitements de 1ère ligne 55
1. Pour les patients < 65 ans et éligible à une autogreffe de CSH 56 2. Pour les patients > 65 ans et < 65 ans non éligibles à une autogreffe de CSH 58
B. Traitements en 1ère rechute ou en 2ème ligne 59
C. Traitements à partir de la deuxième rechute ou 3ème ligne 61
4. Les nouvelles thérapies à venir 62
Partie 3 : Les CAR-T-cells (Récepteurs antigéniques chimériques des Lymphocytes T) : Une innovation
de rupture 66
I. Biologie, concept et principes des CAR-T cells 66
1. Définition & mécanisme d’action 66
2. Structure et Optimisation des CAR : Les différentes générations 68
A. Première génération des CAR 69
B. Deuxième génération 69
C. Troisième génération 70
D. Quatrième génération 70
3. Le processus de fabrication des CAR-T 71
A. Leucaphérèse 71
B. Création des CAR-T cells 74
10
2. Modification génétique des Lc T : Transduction ex-vivo 74
C. Fabrication des récepteurs CAR 78
D. Multiplication & expansion des CAR-T cells ex-vivo 78
E. Réinjection au patient et suivi 79
II. Enjeu de cette immunothérapie innovante : Une efficacité clinique démontrée 79 associée à un processus complexe de fabrication nécessitant une organisation et une prise en
charge multidisciplinaire. 79
1. Une efficacité clinique démontrée et reconnue : Une révolution dans la prise en charge des
hémopathies malignes 79
2. La transition d’une production de petite à grande échelle 81 3. La nécessité d’une coordination exemplaire entre les différents établissements de santé 81
III. Différents challenges à l’emploi des CAR-T cells 84
1. Effets indésirables dû à l’emploi des CAR-T cells 84
A. Le syndrome de relargage cytokinique – SRC 84
B. Neurotoxicités 88
C. Cytopénies 89
D. Autres complications 89
2. Une réglementation en France entrainant des difficultés d’accès au marché 89 A. Un statut réglementaire établi mais susceptible d’évoluer 89 B. Une reconnaissance en termes d’innovation qui ne semble pourtant pas comblée les
problématiques d’accès au marché en France 93
3. Un impact économique important 95
Partie 4 : Les CAR-T cells : Une immunothérapie en plein essor dans le myélome multiple 97 I. Les essais actuels en cours dans le myélome multiple des CAR-T cells 97 II. Les différentes cibles antigéniques des CAR-T cells dans le MM 97
1. CAR-T cells dirigés contre l’antigène anti CD19 97
2. CAR-T cells dirigés contre l’antigène anti BCMA 99
A. Une cible intéressante 99
B. Bb2121 : Le CAR-T cells anti-BCMA du laboratoire Celgène 101
1. Structure 101
2. Essai clinique CRB-401 (94) 102
a. Design de l’étude 102
b. Caractéristiques des patients ayant intégré l’étude 103
c. Résultats de l’étude 104
C. LCAR-B38M : Le CAR-T cells anti-BCMA du laboratoire Janssen 107
1. Structure 108
11
a. Design de l’étude 109
b. Caractéristiques des patients ayant intégré l’étude 110
c. Principaux résultats de l’étude de phase 1 110
D. Autres CAR-T cell anti-BCMA avec des données dans le MM 113 3. CAR-T cells en cours d’essais sur d’autres antigènes cibles 115
Conclusion 117
Annexes 118
12
Remerciements
Aux membres du jury,
Tout d’abord, j’exprime toute ma reconnaissance et gratitude à Monsieur Thierry DINE, merci de me faire honneur en acceptant de présider cette thèse. Merci pour tout l’intérêt que vous portez à ce sujet.
A mon directeur de thèse, Monsieur Madjid TAGZIRT, pour avoir accepté de me soutenir et d’investir de votre temps dans l’élaboration de ce travail. Merci pour votre confiance et la disponibilité que vous m’avez accordée mais également pour vos précieux conseils qui ont permis d’enrichir mes connaissances sur le sujet. J’ai eu beaucoup de plaisir à travailler avec quelqu’un comme vous.
A Karima, qui est en plus d’être ma fidèle partenaire durant ces années d’études en pharmacie, clôture cette page jusqu’au bout en acceptant de faire partie de mon jury. Je ne te remercierai vraiment jamais assez pour toujours avoir été là pour moi, dans les meilleurs moments comme les pires. Tu es une amie en or et je souhaite que les années ne nous éloignent jamais.
A ma famille adorée,
A Maman, Papa, Maxime je ne sais pas par où commencer mais simplement merci de m’avoir soutenue durant toutes ses années car sans vous, tout cela n’aurait pas été possible. Merci de m’avoir transmis vos valeurs et votre volonté inouïe de réussir. Vous êtes et serez toujours un modèle pour moi. J’espère vous rendre fière.
A toute ma famille pour leur soutien et tous ces bons moments passés qui resteront inoubliables. Merci pour votre bienveillance et d’avoir toujours cru en moi.
A mes amis,
A mes amis de Pharmacie : Sarah, Karima, Tarik, Samir, Anthime et Youssef. Vous êtes des personnes en or et je suis très heureuse de vous avoir dans ma vie aujourd’hui. Merci pour ces merveilleux moments partagés durant ces années. Votre
13 amitié m’est si précieuse.
A mes amis de toujours, Marion, Marie et Benoit. Inséparables depuis la maternelle, j’espère que notre amitié durera au minimum toutes nos vies réunis. Merci de toujours me soutenir et d’être là pour moi quoi qu’il arrive.
A mes pinecos, sans qui ma vie ne serait définitivement pas la même. Merci pour tous ces moments de complicité, d’amitié, de fous rires, de joies mais surtout pour énormément d’amour. J’espère que nos amitiés seront éternelles.
A mes pintacassas, sans qui l’aventure ESSEC n’aurait définitivement pas été la même. Merci pour tous ces bons moments partagés qui resteront gravés dans ma mémoire. Des amitiés que je souhaite conserver aussi longtemps que possible
Et enfin, à mon didil, ma moitié, mon amour, mon sourire. Je ne serai comment te remercier d’être toujours là pour moi. Personne n’a ta gentillesse et ta tendresse. J’espère rester à tes cotés pour le reste de ma vie.
14
Liste des figures
FIGURE 1 : PHYSIOPATHOLOGIE DU MYELOME MULTIPLE (12,19,20) ... 30
FIGURE 2 : PHYSIOPATHOLOGIE DE L’ATTEINTE OSSEUSE LORS D’UN MYELOME MULTIPLE (22,23) ... 31
FIGURE 3 : L’INSUFFISANCE RENALE LORS DU MYELOME MULTIPLE (12,19) ... 33
FIGURE 4 : L’ANEMIE DANS LE CADRE DU MYELOME MULTIPLE (11,19) ... 34
FIGURE 5 : DEREGULATION DU SYSTEME IMMUNITAIRE INDUITE PAR LES PLASMOCYTES ANORMAUX (19,25) ... 35
FIGURE 6 : PATHOGENESE DU MYELOME MULTIPLE : UN PROCESSUS MULTI-ETAPES (12,31) ... 36
FIGURE 7 : UNE EVOLUTION DE LA MALADIE CARACTERISEES PAR DE MULTIPLES RECHUTES (12) ... 38
FIGURE 8 : CRITERES DE DIAGNOSTIC DU MM DE L’IMWG 2014 (31) ... 40
FIGURE 9 : PRESENCE D’UN PIC MONOCLONAL AU NIVEAU DES GAMMAGLOBULINES DETECTE PAR EPS CHEZ UN PATIENT ATTEINT DE MM (34) ... 43
FIGURE 10 : IDENTIFICATION D’UNE IMMUNOGLOBULINE G KAPPA A L’IFE (34) ... 44
FIGURE 11 : EXAMEN DU MYELOGRAMME DANS LE CADRE D’UN DIAGNOSTIC DU MM (31) ... 44
FIGURE 12 : LES ANOMALIES CHROMOSOMIQUES DANS LE MM ET LEURS CONSEQUENCES (37) ... 48
FIGURE 13 : CORRELATION ENTRE L’OS / PFS ET LES STADES R-ISS (37) ... 49
FIGURE 14 : RECOMMANDATIONS DE L’IMWG CONCERNANT L’EVALUATION DE LA REPONSE AU TRAITEMENT (41) ... 51
FIGURE 15 : LES STANDARDS DE TRAITEMENTS SELON LES RECOMMANDATIONS DE L’ESMO POUR LES PATIENTS ELIGIBLES A L’AUTOGREFFE DE CSH EN PREMIERE LIGNE DE TRAITEMENT. (49) ... 58
FIGURE 16 : UN PAYSAGE EN TERMES DE SOLUTIONS THERAPEUTIQUE QUI EVOLUE TRES VITE (52–54) ... 64
FIGURE 17 : QU’EST-CE QU’UN CAR ? (55) ... 66
FIGURE 18 : MODE D’ACTIVATION DES CAR-T CELLS (55) ... 67
FIGURE 19 : LES DIFFERENTES GENERATIONS DES CAR-T (55) ... 71
FIGURE 20 : GENERATIONS DE CAR-T ET CARACTERISTIQUES (57) ... 71
FIGURE 21 : LA LEUCAPHERESE ET SELECTION DES LC T (65) ... 73
FIGURE 22 : DEUX TECHNIQUES DE SEPARATIONS CELLULAIRES UTILISEES POUR ISOLER LES LC T (64) ... 74
FIGURE 23 : TRANSDUCTION VIA DES VECTEURS VIRAUX (64) ... 75
FIGURE 24 : METHODE DE TRANSFECTION PAR ELECTROPORATION.(64) ... 77
FIGURE 25 : LES PRINCIPAUX ACTEURS LORS DE LA MISE EN PLACE D’UN PROGRAMME DE THERAPIE CELLULAIRE PAR MEDICAMENTS DE THERAPIES INNOVANTES. (79) ... 84
FIGURE 26 : LES DIVERS SYMPTOMES DU SYNDROME DE RELARGAGE CYTOKINIQUE (80) ... 86
FIGURE 27 : CADRE REGLEMENTAIRE ET OPERATIONNEL POUR LA PRODUCTION, LA DISTRIBUTION ET L’ADMINISTRATION DE PRODUITS THERAPEUTIQUES CONSTITUES DE CELLULES VIABLES. (84) ... 91
FIGURE 28 : PREMIERS RESULTATS DES CAR-T CELLS ANTI CD-19 DANS LE MM EN RECHUTE (89,90) ... 98
FIGURE 29 : STRUCTURE DU CAR-T CELL DE 2EME GENERATION BB2121 (93) ... 101
FIGURE 30 : PROTOCOLES ET SCHEMA DE L’ETUDE CRB-401 (94) ... 103
FIGURE 31 : COURBE DE DE SURVIE SANS PROGRESSION (PFS) DANS L’ESSAI CRB-401 (94) ... 107
FIGURE 32 : PARTICULARITE DU CAR-T CELL LCAR-B38M (95) ... 108
FIGURE 33 : METHODOLOGIE DE L’ETUDE LEGEND-2 (95) ... 110
FIGURE 34 : COURBE DE DE SURVIE SANS PROGRESSION (PFS) DANS L’ESSAI LEGEND-2 (95) ... 112
15
Liste des tableaux
TABLEAU 1 : CLASSIFICATION DE SALMON ET DURIE (2) ... 47
TABLEAU 2 : LES DIFFERENTS STADES ISS (37) ... 47
TABLEAU 3 : MISE A JOUR DES CRITERES DE PRONOSTICS ISS : LA CLASSIFICATION R-ISS (37) ... 49
TABLEAU 4 : SCORE DE FRAGILITE DE L’IMWG (41) ... 56
TABLEAU 6 : LES DIFFERENTS GRADES DU SYNDROME DE RELARGAGE CYTOKINIQUE (81) ... 87
TABLEAU 7 : LES 4 CAR-T CELLS ANTI BCMA EN COURS D’EVALUATION CLINIQUE (92) ... 100
TABLEAU 8 : CARACTERISTIQUES DES PATIENTS INCLUS (94) ... 104
16
Liste des abréviations
AC Anticorps
ADN Acide désoxyribonucléique
AMM Autorisation de mise sur le marché ARN Acide ribonucléique
ARS Agence Régionale de Santé ASH American Society of Hematology
B/R Balance bénéfices/risques BCMA B cell Maturation Antigen
BCR Récepteur pour l’antigène des lymphocytes B
CAR Chimeric Antigen Receptor - Récepteur antigénique chimérique
CAR-T Cells Cellules (Lc) T génétiquement manipulées pour exprimer un récepteur chimérique à l’antigène.
CFU-L Colony Forming Unit – Lymphocytes
CFU-GEMM Colony Forming Unit Granulocytes, Erythrocytes, Monocytes,Mégacaryocytes CHMP Comité des médicaments à usage humain
CLL Chaines Légères Libres
CMH Complexe majeur d’histocompatibilité CMO Central Manufacturing Organization
CRAB Calcium elevation, Renal insufficiency, Anemia, Bone disease CRP protéine C réactive
CRS Cytokine Release Syndrome
CSH Cellules souches hématopoïétiques DKK Facteur Dickkopf
EI Effet indésirable
EMA European Medicines Agency
EFS Etablissement Français du Sang EPS Electrophorèse des protéines
ESMO European Society for Medical Oncology
EBMT European Society for Blood and Marrow Transplantation
FDA Food and Drug Administration
G-CSF Facteur de stimulation des colonies de granulocytes
GMSU Gammapathie monoclonale de signification indéterminée (=MGUS) HAS Haute Autorité de Santé
Hb Hémoglobine
HR Hazard ratio
IC Intervalle de confiance
IFM Intergroupe Francophone du Myélome
Ig Immunoglobuline
17 IMiD Agent immunomodulateur
IMWG International Myeloma Working Group
IP Inhibiteur du protéasome
IRM Imagerie par résonance magnétique
ISS International Staging System
ITT Intention de traitement L1G 1ère ligne greffe
L1NG 1ère ligne non greffe
Lc Lymphocyte
LLC Leucémie lymphoïde chronique LNH Lymphome non hodgkiniens
MM Myélome multiple
MOA Mécanisme d’action MRD Minimal Residual disease
MTI Médicament de Thérapie Innovante mPFS Médiane de survie sans progression MS Maladie stable
NA Non atteint
OPD Ostéoprotégérine
ORR Taux de réponses globale (Overall response rate) OS Survie globale (Overall survival)
PBMC Cellules mononucléées du sang périphérique PFS Survie sans progression (Progression Free Survival) RANK Receptor Activator of Nuclear factor Kappa-B
RC Réponse complète
RCs Réponse complète stringente
R-ISS Revised-International Staging System
RIPH Recherche impliquant la personne humaine RP Réponse partielle
SFGM-TC Société Francophone de Greffe de Moelle et de Thérapie Cellulaire
scFv : Single chaine variable fragment (Fragment variable d’AC à domaine unique)
SRC Syndrome de relargage cytokinique TBRP Très bonne réponse partielle
TCR Récepteur pour l’antigène des lymphocytes T
TNF Facteur de nécrose tumorale (Tumor Necrosis Factor) VIH Virus de l’immunodéficience humaine
18
Introduction
En France, les hémopathies malignes représentent près de 10% des nouveaux cancers diagnostiqués chaque année en France, soit 35 000 nouveaux patients. D’importantes avancées thérapeutiques ont été soulignées dans la prise en charge de ces maladies. Malgré de nombreux progrès réalisés, ces maladies restent à ce jour incurables pour la plupart d’entre elles et se caractérise comme des maladies devenues chroniques. (1,2)
La plus fréquente des hémopathies malignes est le myélome multiple (environ 5000 nouveaux cas par an), une maladie rare mais dont l’incidence augmente d’année en année. Il s’agit d’une maladie grave et invalidante, ayant de nombreuses répercussions sur la vie et le quotidien des patients. Malgré l’arrivée de nouvelles thérapies, la plupart des patients finissent par rechuter et peuvent développer des résistances aux traitements précédemment utilisés. L’essor de l’immunothérapie au cours de ces vingt dernières années a révolutionné la prise en charge de certaines hémopathies malignes, et notamment du myélome multiple.
Actuellement qualifié par les experts comme ayant une place « incontournable » dans les hémopathies malignes, le développement des CAR-T cells, définies comme médicaments de thérapie innovante, constitue une avancée majeure et une véritable révolution dans le traitement et la prise en charge des patients souffrant de ces maladies. Cette thérapie génique fait l’objet de plus de 350 essais cliniques dans le monde entier et peut peut-être tendre vers un espoir de guérison et ainsi bouleverser les standards de traitements actuels. Cette thérapie innovante consiste à induire l’expression de récepteurs antigéniques chimériques (CAR), dirigés contre une cible bien spécifique, dans les cellules T. Lors de plusieurs essais pré cliniques et cliniques, certains CAR-T cells ont d’ores et déjà démontré des résultats de tolérance et d’efficacité très prometteurs.
Après avoir rappelé en première partie la classification des hémopathies malignes et leurs caractéristiques, nous nous focaliserons sur la plus fréquente d’entre elles, le myélome multiple, en faisant le point sur la physiopathologie jusqu’aux recommandations des différentes stratégies thérapeutiques actuelles dans la prise en charge du myélome.
19
Par la suite dans une troisième partie, nous développerons l’essor et le développement des CAR-T cells de leur production en laboratoire jusqu’à la réinjection au patient à l’hôpital. Il sera question de faire ressortir les enjeux de cette immunothérapie innovante ainsi que les différents challenges pouvant apparaitre lors de l’utilisation et la mise en place de cette thérapie.
Enfin, dans une dernière partie, nous parlerons du développement des CAR-T cells dans le myélome multiple à travers différentes actualisations et premiers résultats de plusieurs essais cliniques.
20
Partie 1 : Les hémopathies malignes
I. Définition
Les hémopathies malignes surviennent à partir de cellules d’origine hématopoïétique. Elles peuvent donc toucher l’ensemble de ces cellules sanguines à savoir les globules rouges ou érythrocytes, les globules blancs ou leucocytes, les plaquettes ou thrombocytes ainsi que leurs précurseurs.
Elles peuvent être définies comme des « cancers du sang » et résultent d’anomalie(s) au niveau de la balance physiologique de ces cellules hématopoïétiques : cela peut concerner à la fois la prolifération de ces cellules, leur apoptose ou leur différenciation.
Ces différentes anomalies entrainent toutes à une accumulation de cellules anormales et donc tumorales ou à la baisse de productions des cellules saines. Les manifestations cliniques dépendront de la lignée cellulaire concernée, du stade de maturation et du foyer de localisation des cellules atteintes.
Si les globules blancs sont concernés, il peut s’agir des plasmocytes, comme dans le cas du myélome multiple, ou des lymphocytes, comme pour les lymphomes et la leucémie lymphoïde chronique. En fonction de l’hémopathie maligne en question, la maladie peut se manifester à différents endroits : la moelle osseuse, les ganglions lymphatiques ou la rate.
Ces maladies restent souvent méconnues et mal comprises par les patients et leur entourage en raison de leur complexité, leur diversité, leur différent mécanisme de développement et leur évolution différente. L’effet de ces maladies ainsi que de leurs traitements associés entrainent une altération de la qualité de vie des patients, déclenchant en général une remise en cause et une incertitude sur leur avenir.
Les hémopathies malignes les plus connues et les plus fréquentes sont celles issues de la lignée lymphoïde : les lymphomes, les leucémies dont la leucémie lymphoïde chronique (LLC), le myélome multiple (MM) et la maladie de Waldenström.
21 II. Des maladies qui se chronicisent en France
Les hémopathies malignes représentent près de 10% des nouveaux cas de cancer diagnostiqué chaque année en France (sur 385 000 nouveaux cas de cancer en 2015).
Ces hémopathies touchent donc environ plus de 35 000 personnes en France chaque année. Environ 193 000 personnes (soit 6% des personnes atteintes de cancer) souffrent d’une hémopathie maligne en France. (1,3)
La plus fréquente de ces hémopathies malignes est le myélome multiple, même si cette maladie est considérée comme une maladie rare, puisqu’on diagnostique environ 5 000 nouveaux cas par an en France. La LLC compte environ 3 200 nouveaux cas diagnostiqués chaque année en France. Le lymphome constitue un groupe de pathologies complexes et multiples puisqu’on y compte près de 100 sous-types différents. On distingue les lymphomes Hodgkiniens (environ 20% des lymphomes) versus les lymphomes Non Hodgkiniens (LNH), majoritairement représentés. On distingue les LNH agressifs et les indolents. L’ensemble de ces lymphomes constitue environ 15 000 nouveaux patients diagnostiqué en France par an.
Concernant l’ensemble de ces pathologies malignes, plus de la moitié du diagnostic est posé chez les personnes âgées de plus de 60 ans. (4)
Ces hémopathies malignes ont un véritable impact sur le quotidien des patients et également sur leur entourage. En effet, ces maladies, de part des symptômes visibles ou non, peuvent avoir des conséquences très importantes sur la vie quotidienne des patients : cela peut se traduire par une altération de la qualité de vie physique (diminution de l’autonomie, de la mobilité …), morale ou socio-professionnel (sensation de dépendance, angoisse ect).
Ces dernières années, de nombreux progrès ont été réalisés concernant les traitements et la prise en charge des hémopathies malignes avec l’arrivée de nouvelles options thérapeutiques très prometteuses et ayant un profil balance bénéfice/risque amélioré. Ces progrès amènent cependant à une « chronicisation » de ces pathologies qui restent, à ce jour, incurables pour certaines. (2)
22 III. Classification
Avant de rappeler la classification des hémopathies malignes, il est essentiel de comprendre le processus de l’hématopoïèse. Toutes les cellules sanguines sont issues
d’une seule et même cellule, les cellules souches hématopoïétiques (CSH) localisées dans la moelle osseuse. Cette cellule subira une division cellulaire asymétrique, autrement appelée mitose pour donner naissance à deux types de cellules (qui donneront les deux lignées principales des hémopathies malignes) :
- Les cellules souches lymphoïdes CFU-L, faisant partie de la lignée lymphoïde et qui sera en charge du développement des lymphocytes.
- Les cellules souches myéloïde CFU-GEMM, faisant partie de la lignée myéloïde et qui sera en charge du développement des granulocytes, des érythrocytes, des monocytes et des mégacaryocytes.
En annexe 1, on retrouve un schéma résumant l’ensemble du processus de formation de l’ensemble de ces cellules sanguines.
De ce fait, on distingue les hémopathies malignes originaires de la lignée myéloïde et celles de la lignée lymphoïde.
1. Les hémopathies myéloïdes
Ces cancers de sang se développent à partir des progéniteurs myéloïde, issues des CSH ayant subies la différenciation myéloïde. Elles sont classées par 3 catégories selon l’organisation mondiale de la santé (OMS).
A. Leucémies aigues myéloïdes (LAM)
Sa physiopathologie résulte dans la prolifération de cellules immatures issues de la lignée myéloïde. Ces cellules rencontrent un blocage au niveau de leur maturation, provoquant ainsi une prolifération anormale et excessive de myéloblastes (cellules immatures) dans la moelle osseuse ainsi que dans la circulation sanguine (20%).
Les conséquences se caractérisent par une anémie (diminution des globules rouges et de l’hémoglobine) entrainant fatigue, essoufflement, palpitations. On note également une
23
neutropénie engendrant une sensibilité plus accrue aux infections. On parle de leucémie aigüe puisque les symptômes de la maladie s’installent de manière très rapide (quelques jours-semaines). L’origine reste encore inconnue. Cette pathologie peut survenir à tout âge même si la fréquence augmente avec l’âge : en effet, l’âge médian du diagnostic est de 65 ans. Selon la classification internationale Franco-Américano Britannique (FAB), on distingue huit types notés de 0 à 7 selon les caractéristiques des cellules (immatures) anormales retrouvées. (5)
B. Syndromes myéloprolifératifs (SMP)
Il s’agit d’hémopathies malignes caractérisées par la prolifération de cellules anormales provenant d’une ou plusieurs lignées myéloïdes ne présentant pas de blocage de différentiation. Elles entrainent une production excessive de cellules matures/ différenciées issues de la lignée myéloïde. Ces cellules peuvent donc être des globules rouges, des globules blancs ou des plaquettes (5). Selon la lignée concernée, on en distingue 4 :
- La maladie de Vaquez ou polyglobulie : cette hémopathie est caractérisée par une accumulation de globules rouges à la périphérie. On retrouvera comme principaux signes cliniques évocateurs une hyperviscosité sanguine ainsi qu’une érythrose faciale.
- La leucémie myéloïde chronique : Il s’agit d’une amplification de la lignée granuleuse.
- La thrombocytopénie essentielle : accumulation de plaquettes
- Une myélofibrose primitive : entrainant une fibrose de la moelle osseuse.
C. Syndromes myélodysplasiques (SMD)
Ces syndromes sont caractérisés par une prolifération anormale des précurseurs des cellules souches hématopoïétiques entrainant des cellules porteuses d’anomalies fonctionnelles et morphologiques ainsi qu’une insuffisance au niveau de la production de cellules matures saines (dû à un excès d’apoptose). L’hématopoïèse étant inefficace, ces syndromes entrainent une cytopénie en périphérie et peuvent donc toucher les trois
24
lignées : insuffisance de production de globules rouges (anémie), de plaquettes (thrombopénie) et globules blancs (neutropénie). (6)
2. Les hémopathies lymphoïdes
Opposées à celles de la lignée myéloïde, ces hémopathies se développent à partir d’anomalies survenant sur les cellules de la lignée lymphoïde. Environ 80% d’entre elles proviennent de la lignée des lymphocytes B. Selon la classification de l’OMS, elles se classent de manière suivante.
A. Les leucémies
1. Leucémie aigüe lymphoblastique (LAL)
Sa physiopathologie résulte d’une prolifération anormale des précurseurs lymphoïdes (B ou T) entrainant une multiplication excessive de lymphoblastes dans la moelle osseuse et dans le sang, empêchant ainsi la production de cellules sanguines saines et normales. On retrouve une insuffisance médullaire. Ces leucémies sont surtout observées chez les enfants mais également chez l’adulte après 50 ans, en effet 75% des LAL sont rapportés chez les moins de 18 ans. Elles peuvent être de deux types : constituée de lymphoblastes de type T ou B. Les LAL B sont celles qui sont les plus fréquentes chez les enfants. Actuellement, un traitement à base d’un CAR-T cell anti CD19 (récepteur antigénique chimérique dirigé contre l’Ag CD19), Kymriah®, a montré un intérêt clinique important dans le LAL de type B en rechute et réfractaire suite à une greffe ou après au moins la 2ème rechute chez les enfants et jeunes adultes (<25 ans). D’autres CAR-T cells sont également en cours de développement dans ce type de leucémie. (7,8)
2. Leucémie lymphoïde chronique (LLC)
Contrairement aux leucémies aiguës, la LLC est définie comme une maladie chronique du fait de son évolution très lente. Il s’agit d’une hémopathie touchant les lymphocytes B : on retrouve une accumulation de Lc B dans le sang ainsi que dans les ganglions. Il s’agit de la forme la plus fréquence des leucémies (3200 nouveaux cas par an en France) et touche plus souvent les hommes d’un facteur deux par rapport aux femmes. La LLC survient en général après 50 ans. Ces principaux symptômes sont reliés à sa physiopathologie, notamment un nombre important de Lc B accumulées. On distingue trois stades du A au C : simple
25
augmentation de Lc B, avec ou sans autre anomalies sanguines et présence ou non de ganglions. (7,9)
B. Les lymphomes
1. Le lymphome Hodgkinien ou maladie de Hodgkin
Il ‘agit d’un type de lymphome présentant de grandes cellules atypiques issues de la lignée lymphoïdes B. Elles sont appelées les cellules de Reed-Stemberg et peuvent infiltrer les ganglions. Plus la maladie est avancée, plus l’augmentation de ces cellules sont importantes. La prolifération de cellules anormales provenant de la lignée B entraine une augmentation du volume des ganglions lymphatiques et explique la présence de ces ganglions. Il représente environ 10% des lymphomes et le nombre de nouveaux cas par an est estimé à 1 900. Ce type de lymphome survient principalement chez les jeunes adultes entre 20 et 30 ans et chez les personnes âgées de plus de 60 ans. Selon la classification d’Ann Arbor, on distingue 4 stades : les stades I (atteinte des ganglions lymphatiques) et II (atteinte des ganglions lymphatiques et du médiastin) sont localisés alors que les stades III (atteintes du stade II + atteintes de la rate) et IV (atteinte de la rate, du foie et de la moelle osseuse) sont
les plus étendues. (7,10)
2. Les lymphomes Non Hodgkiniens (LNH)
Ces lymphomes peuvent concerner l’ensemble des trois lignées lymphoïdes : B, T et NK (Natural Killer). Dans ces différents types de lymphomes, les lymphocytes sont produits en quantité trop importante et leur durée de vie est anormalement plus longue.
Ces Lc malins seront à l’origine des différents LNH et se multiplieront dans les ganglions lymphatiques (dans ce cas, on les qualifiera de lymphome ganglionnaire) ou dans le tissu lymphoïde retrouvée dans la majorité des organes (lymphomes extra-ganglionnaires). Ces LNH touchent souvent les ganglions lymphatiques mais plus de la moitié ont tout de même une localisation initiale extra-ganglionnaire. On distingue deux grands groupes de LNH :
- Les lymphomes à cellules ou Lc B anormaux.
Il s’agit des plus fréquents et des plus nombreux : ils représentent 85% des LNH.
Dans ce groupe, on retrouve les lymphomes folliculaire, le lymphome à cellules du manteau, le lymphome diffus à grandes cellules B, le lymphome de Burkitt, le lymphome de la zone marginale et la Maladie de Waldenström (lymphome lympho-plasmocytaire)
26 - Les lymphomes à cellules ou Lc T anormaux.
On en distingue également quelques formes mais sont moins nombreux et moins communs que les lymphomes à cellules B. (7)
C. Le Myélome multiple
Le myélome multiple est une hémopathie maligne caractérisée par la sécrétion anormale de plasmocytes en quantité plus importante. Ces plasmocytes sont issus de la lignée B des lymphocytes. De plus, il s’agit d’une prolifération médullaire d’un clone de lymphoïde sécrétant une immunoglobuline monoclonale. Nous développerons plus précisément cette hémopathie maligne dans la seconde partie de cet exercice.
Au vu de l’amélioration de l’espérance de vie, le nombre de nouveaux cas d’hémopathies malignes progresse et augmente chaque année. Ces pathologies occupent alors de plus en plus une place centrale dans la lutte contre le cancer. Il est donc essentiel de développer et d’optimiser les différentes options thérapeutiques qui peuvent être mis à disposition pour ces patients souffrants d’hémopathies malignes.
27
Partie 2 : Le myélome multiple
I. Qu’est-ce que le myélome multiple ?
1. Définition et historique du myélome multiple
Le myélome multiple ou autrement appelé « Maladie de Kahler » fait partie des hémopathies malignes de la lignée lymphocytaire B et se caractérise comme un cancer du sang. Cette maladie se définit par la prolifération d’un clone de plasmocytes anormaux ou tumoraux dans la moelle osseuse hématopoïétique.(11)
Pour la plupart des patients, ces plasmocytes anormaux ou malins produisent et sécrètent une immunoglobuline (Ig) monoclonale anormale (ou protéine M). Ces plasmocytes peuvent sécrétés de manière complète ou uniquement l’un des fragments de cette immunoglobuline.
L’Ig ou protéine M est retrouvée dans le sang ou les urines des patients.
• Plus de 50% des patients atteints d’un myélome multiple sécrètent une protéine M de type IgG : Il s’agit de la forme la plus courante.
• Environ 20% des patients atteints de myélome multiple sécrètent une protéine M de type IgA
• 16% des patients présentent des plasmocytes anormaux sécrétant un fragment de l’Ig : On parle alors de myélome à chaines légères.(12)
Dans le cas contraire chez certains patients, les plasmocytes malins ne sécrètent pas de protéine M : On désigne donc le myélome multiple comme étant non sécrétant et concerne entre 1 et 3% des patients atteints.(12)
Les datés clés concernant l’historique de cette maladie sont les suivantes :
• 1889 : Publication de la 1ère description clinique détaillée du myélome multiple par le Dr Otto Kahler, sous le nom de la « maladie de Kahler ».
• 1890 : Description des plasmocytes(13)
• 1939 : identification d’un pic de protéines sériques dans le cas d’un myélome multiple (14)
28
2. Epidémiologie : Une maladie rare dont l’incidence progresse
Même si le myélome multiple représente moins de 2% de l’ensemble des cancers, il est la deuxième hémopathie maligne la plus fréquente en France (entre 10 et 15% des hémopathies malignes), derrière les lymphomes non hodgkiniens.(15)
De nos jours, environ 16 000 patients sont atteints de myélome multiple en France.
On compte près de 5450 nouveaux cas diagnostiqués de myélome multiple chaque année en France avec une augmentation estimée de 1 à 2% par an.(15) (16)
Il s’agit d’une hémopathie maligne dont la fréquence augmente avec l’âge. L’âge médian au diagnostic est de 70 ans chez l’homme et 74 ans chez la femme. 52% des patients touchés sont de sexe masculin. On peut donc conclure que le myélome multiple est un cancer du sang du sujet âgé puisqu’environ ¾ des patients ont 65 ans ou plus au moment de leur diagnostic. Cependant il peut également toucher les sujets plus jeunes puisque près d’1/4 des patients sont diagnostiqués à moins de 65 ans. (Environ 3% des patients sont diagnostiqués avant 40 ans).(16)
Compte tenu de l’accroissement de la population des personnes ayant plus de 65 ans, l’incidence du myélome multiple pourrait augmenter en 2030. En effet, une étude américaine récente a montré une augmentation du nombre de patients âgés atteint de myélome multiple puisque les nouveaux diagnostics de myélome multiple concerneront environ trois quarts des patients de plus de 65 ans. Actuellement, deux tiers des nouveaux patients diagnostiqués correspondent à des patients âgés de plus de 65 ans.(17) Le myélome multiple se classerait donc en 3ème position des cancers les plus fréquents sur cette population de patients.(18)
Il faut donc garder en tête que l’incidence de cette hémopathie maligne ne cesse d’augmenter.
Cette pathologie reste encore, au jour d’aujourd’hui incurable et son évolution reste encore imprévisible. Le myélome multiple est une maladie hétérogène dont les causes restent encore inconnues.
29
3. Les signes cliniques de la maladie
A. Physiopathologie du myélome multiple
Le myélome multiple est une hémopathie maligne touchant la lignée lymphocytaire B au stade du plasmocyte. Il va donc être responsable de la prolifération excessive de plasmocytes anormaux ou malins. Ces plasmocytes anormaux et en quantité excessive migreront progressivement dans la moelle osseuse, ce qui amènera à son dysfonctionnement. On parle donc de « myélome multiple » dû à une atteinte de plusieurs os.(11)
Ces plasmocytes malins vont produire en quantité anormalement plus élevée :
• Une immunoglobine monoclonale dans le sang et/ou dans les urines et dont la nature varie selon les patients.
Cette immunoglobuline anormale se traduira par un pic étroit et fin appelé « pic monoclonal » détectée à l’aide de l’examen d’électrophorèse des protéines (EPS).
Cependant, parfois ce pic monoclonal n’apparait pas sur les résultats de l’examen d’EPS : dans ce cas, il s’agit d’un type de myélome dit à chaines légères, c’est-à-dire que les plasmocytes anomaux ne produisent alors qu’un fragment de l’immunoglobuline monoclonale.
La quantité anormale et importante de protéines monoclonales entrainera l’augmentation de la viscosité plasmatique, qui expliquera un syndrome d’hyperviscosité retrouvé généralement chez des patients souffrants de myélome.
La sécrétion d’Ig anormale entraine également une synthèse anormale de chaines légères. Ces chaines légères (de type - kappa ou - lambda) s’accumuleront dans les urines et provoqueront des néphropathies conduisant à une insuffisance rénale.
• Des cytokines inflammatoires comme des facteurs de nécroses tumorales (TNFa) ou bien encore des interleukines. Cela entraine :
o Une immunodéficience due à un déficit de production d’Ig normal entrainant donc une susceptibilité augmentée d’être sujet à des infections pour un patient atteint de myélome multiple.
o L’activation d’ostéoclastes responsable des lésions ostéolytiques, d’où l’apparition de douleurs osseuses et l’augmentation de la concentration de calcium CA2+ dans le sang.
30
o Un dérèglement de l’hématopoïèse provoquant l’infiltration de certaines cellules sanguines dans la moelle osseuse et générant une anémie.(12) (19) (20)
Figure 1 : Physiopathologie du myélome multiple (12,19,20)
B. Des symptômes bien identifiés liés à la physiopathologie
Les symptômes associés à la maladie, lorsqu’ils se manifestent sont peu spécifiques (anémie, douleur, fractures) et peuvent en premier lieu être attribués à d’autres troubles ou maladies. (21)
1. Les atteintes osseuses
L’accumulation des plasmocytes malins dans la moelle osseuse sera responsable du déséquilibre osseux, responsable des atteintes.
En effet, ces atteintes osseuses représentent la cause majeure de mortalité et morbidité chez les patients souffrant de myélome multiple. Plus de 80% des patients seront sujets à des atteintes osseuses.
31
❖ De l’augmentation anormale de l’activation des ostéoclastes (cellules responsables de la destruction du tissu osseux).
Les plasmocytes malins seront responsables de la surproduction des chimiokines activatrices des ostéoclastes comme le Tumor Necrosis Factor (TNF-α), l’interleukine 6 (IL-6) et 7 (IL-7) et le Macrophage Inflammatory Protein-one alpha (MIP-1α).
Une dérégulation du système récepteur RANK, du ligand RANK-L et de l’ostéoprotégérine (OPD) contribue également à l’activation et la différenciation de ces ostéoclastes. De plus, ce processus de résorption osseuse induit le relargage de facteurs de croissance, augmentant alors la masse tumorale responsable du sur-emballement du processus ostéolytique.(22,23)
❖ Du blocage de la production et de la différenciation des ostéoblastes (cellules responsables de formation continue du tissu osseux.) essentiellement dû à la surproduction anormale d’IL-6, IL-7 et du facteur DKK-1.
Figure 2 : Physiopathologie de l’atteinte osseuse lors d’un myélome multiple (22,23)
Ces atteintes associées au myélome multiple sont le résultat donc d’une augmentation de la résorption osseuse couplée à une réduction de la formation osseuse. Il se produit donc un déséquilibre : le tissu osseux est davantage détruit que reconstruit. Ce déséquilibre induit
32
donc des lésions ostéolytiques qui seront irréparables et seront à l’origine de plusieurs atteintes osseuses comme :
❖ Des douleurs osseuses (situées la plupart du temps au niveau du dos ou des côtes) ; ❖ Une augmentation du risque de fractures pathologiques au vue de l’augmentation de
la fragilité des os : 20% des patients présenteront des fractures pathologiques ; ❖ Un risque de tassement vertébral pouvant entrainer une compression de la moelle
épinière ;
❖ Une hypercalcémie (augmentation anormale de la concentration de calcium dans le sang). (10) (22) (23)
Ces complications osseuses sont à ne pas prendre à la légère et sont responsable d’une altération de la qualité de vie importante, associées à une augmentation de la mortalité.(24)
2. L’hypercalcémie
Comme vu précédemment, les atteintes osseuses conduisent à l’augmentation du taux de calcium dans le sang. Quand le calcium est présent en trop grande quantité dans le sang, on pourra alors retrouver différents troubles caractéristiques tels que des maux de tête, des urines très abondantes, une bouche sèche, des nausées ou encore des vomissements.
L’hypercalcémie est également responsable de l’augmentation de la résorption tubulaire du calcium et de la baisse du débit de filtration glomérulaire, pouvant conduire dans certains cas à une insuffisance rénale aigue. (11,12)
3. L’insuffisance rénale
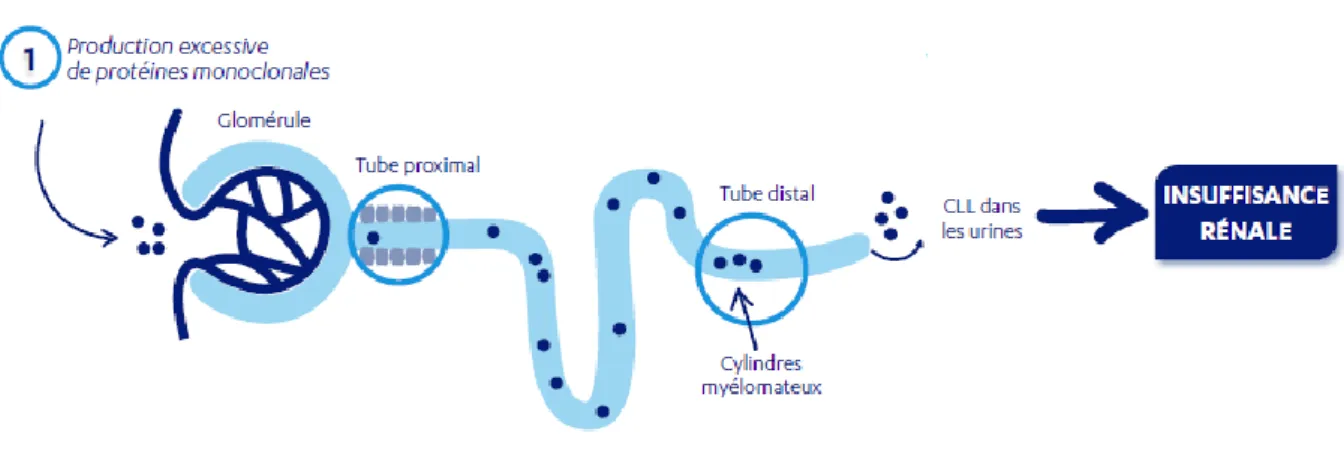
L’hypercalcémie, à l’origine de la destruction osseuse, peut provoquer un dysfonctionnement au niveau des reins puisque cela entraine également une accumulation du calcium au niveau rénale. Parallèlement dans le cas d’un myélome multiple, on retrouve une production excessive de protéines monoclonales, donc une augmentation de chaines légères libres (CLL) au niveau du glomérule qui provoquera donc un dépassement de la capacité d’absorption au niveau du tube proximal. Cela aura pour conséquence :
o Une accumulation des CLL au niveau du segment distal du néphron o Une association des CLL avec la glycoprotéine urinaire Tamm-Horsfall o Une obstruction du tube distale conduisant à une insuffisance rénale.
33
Figure 3 : L’insuffisance rénale lors du myélome multiple (12,19)
L’insuffisance rénale est retrouvée chez près de la moitié des patients lors d’un diagnostic du myélome.(12,19)
4. L’anémie
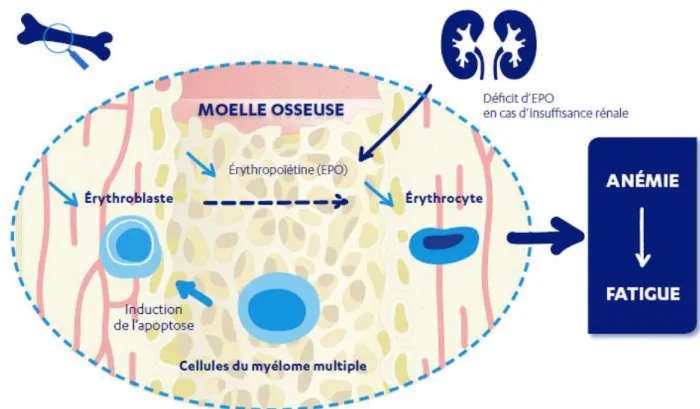
Elle est rencontrée chez environ 73% des patients atteints de myélome multiple.
Plusieurs facteurs contribuent à son développement. Elle est tout d’abord la conséquence de l’infiltration dans la moelle osseuse des plasmocytes malins qui provoquera une diminution de la production des globules rouges. La principale raison est liée à l’induction de l’apoptose des érythroblastes (précurseurs des globules rouges) par les cellules atteintes du myélome, responsable donc de la diminution de production d’érythroblaste.
La diminution du taux de globule rouge dans le sang provoque naturellement une baisse du taux d’hémoglobine entrainant donc une anémie. Elle se traduit donc par une fatigue importante et un essoufflement.
L’insuffisance rénale peut engendrer une diminution d’érythropoïétine (EPO), cytokine précurseur des érythrocytes dans la moelle osseuse, pouvant donc renforcer l’anémie.(11,19)
34
Figure 4 : L’anémie dans le cadre du myélome multiple (11,19) 5. Les infections
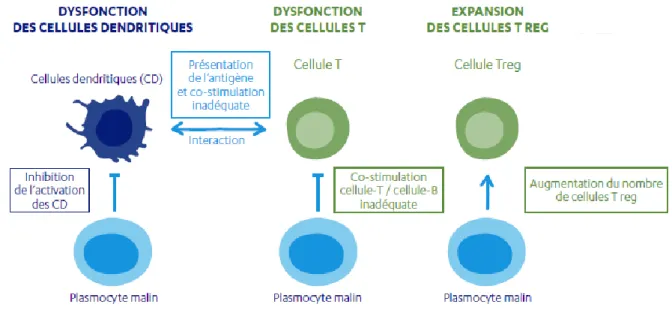
Le risque accrue d’infections du à la production anormale de plasmocytes malins est la principale cause de décès chez les patients atteints de myélome multiple.
Les infections chez les patients souffrant de myélome multiple sont le résultat d’une dérégulation du système immunitaire induite par les plasmocytes anormaux (resultant d’une surproduction d’immunoglobulines anormales). Les infections les plus fréquentes retrouvées chez ces patients sont :
o Harmophilus influenzae o Streptococcus pneumoniae o Bactéries Gram –
o Grippe et zona
De plus, les traitements utilisés pour traiter le myélome multiple peuvent amplifier le risque d’infections pour les patients. Certaines vaccinations pourront être envisagées ou recommandées afin de diminuer l’incidence de ces événements infectieux. (19,25)
35
Figure 5 : Dérégulation du système immunitaire induite par les plasmocytes anormaux (19,25)
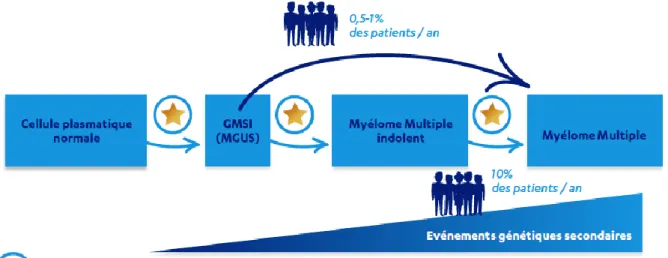
C. Pathogénèse moléculaire
Le myélome multiple est une hémopathie maligne qui se développe en plusieurs étapes. Généralement, Il est précédé de deux phases asymptomatiques diagnostiquées de manière fortuite, notamment dans le cadre d’examens médicaux de routine (26) :
❖ La gammapathie monoclonale de signification indéterminée (GMSI) ou MGUS. Présente chez environ 4% des personnes âgées de plus de 50 ans, cette phase se caractérise par l’apparition d’un faible pic d’immunoglobuline monoclonale détecté lors d’une électrophorèse des protéines sériques (EPS). Cela traduit la présence d’Ig monoclonale en faible quantité dans le sang, toutefois inférieur à 30g/L. Le taux de plasmocytes de la moelle osseuse est également inférieur à 10% et le patient ne présente aucun signe clinique ou biologique caractéristique du myélome. Chaque année, près de 1% des patients présentant une GMSI développeront un risque d’évolution vers un myélome multiple symptomatique. (22) La GMSI peut également évoluer vers un myélome multiple asymptomatique.
❖ Le myélome indolent, connu également sous le nom de myélome multiple asymptomatique.
Les patients diagnostiqués de myélome indolent ne présentent aucun symptôme clinique ou biologique. Le pic monoclonal détecté par EPS est en général supérieur 30 g/L et le taux de plasmocytes dans la moelle osseuse se situe entre 10 à 60%. Il représente environ 20 % des
36
cas de myélome.(27) La durée de cet état est variable. Les patients diagnostiqués d’un myélome indolent représentent un groupe hétérogène vis-à-vis du risque de développement et de progression vers un myélome actif :
- Certains d’entre eux présenteront un faible risque de progression (environ 5 à 8% à 5 ans), proche du risque de progression d’une MGUS à un myélome actif ;
- d’autres présenteront un risque bien plus important, de lors de 50% à 5 ans. (28) Afin de surveiller ces patients présentant un myélome indolent à haut risque de progression, un bilan biologique doit être réalisé tous les 4 à 6 mois ainsi d’une surveillance de l’imagerie osseuse. (29,30)
Ces deux phases asymptomatiques ne nécessitent habituellement pas de traitements mais une surveillance de certains paramètres cliniques plus ou moins régulière.
Enfin, la troisième phase est celle du myélome multiple symptomatique ou forme active de la maladie. Elle nécessite une prise en charge par un traitement dans les plus brefs délais. Cette phase est caractérisée par de nombreux symptômes vus précédemment mais également des critères diagnostics précis que nous verrons par la suite.
37 D. Comorbidités et facteurs de risques
Les comorbidités les plus fréquentes chez les patients atteints de myélome multiple sont l’hypertension (environ 47% des patients) et d’autres troubles cardiovasculaires (pour 19% des patients).(32)
A l’heure actuelle, la cause du développement du myélome reste encore inconnue. Cependant, certains facteurs de risques possibles ont été étudiés et sont plus ou moins établis. L’origine du myélome serait donc, probablement, multifactorielle :
✓ L’âge avancé du patient : En effet, le risque de tumeur maligne augmente avec l’âge. ✓ Les antécédents familiaux : les formes familiales du myélome (c’est-à-dire retrouvée
chez plusieurs membres d’une même famille) sont très rares. (12,33) ✓ L’origine afro-américaine
✓ Les facteurs génétiques : ces facteurs sont toujours en cours d’investigation. ✓ Les facteurs environnementaux.
Aujourd’hui, seules les radiations ionisantes, liées à des expositions accidentelles, sont reconnues comme facteur de risque avéré. Certaines expositions professionnelles à des substances comme des solvants organiques ou pesticides sont également évoqués et sont en cours d’investigation. (33)
E. Evolution de la maladie
L’évolution du myélome multiple se caractérise par une alternance entre des phases de rémission et de rechutes. Ces multiples rechutes sont bien souvent inévitables. En effet, cette maladie doit être suivi à vie comme une maladie chronique. Les phases de rémission se distinguent lorsque le pic d’immunoglobuline monoclonal anormal diminue fortement et n’est plus détectable lors des examens. La durée de ces phases est variable d’un patient à un autre. Les phases de rechutes quant à elles surviennent après des phases de rémission, et se caractérisent par une reprise de l’évolution de la maladie après une première ligne de traitement. (13)
38
Figure 7 : Une évolution de la maladie caractérisées par de multiples rechutes (12)
En conclusion, le myélome multiple est une maladie dont la progression reste une majorité pour la plupart des patients et s’accompagner de symptômes cliniques nécessitant des traitements spécifiques et en général assez lourd pour les patients.
Le myélome multiple est encore de nos jours une maladie invalidante et incurable.
II. Le diagnostic et les critères pronostic du myélome multiple
En effet, il existe plusieurs critères diagnostic qui se sont affinés dans le temps et sont aujourd’hui utilisés pour permettre un diagnostic précis de la maladie. Ils permettent de classer les différents stades d’évolution et sont essentiels pour adapter la solution thérapeutique qui sera alors choisie pour le patient.
En effet, établis en 2003 et clarifiés en 2009 par l’International Myeloma Working Group (IMWG), le diagnostic du myélome reposait sur trois critères biologiques qui étaient les suivants :
✓ Une plasmocytose médullaire ;
✓ La présence d’un pic d’immunoglobuline monoclonale ; ✓ L’identification d’au moins l’un des 4 critères CRAB.
39
1. Les critères de diagnostic « CRAB »
Jusqu’à la révision des critères de diagnostic du myélome de l’IMWG en 2014, le diagnostic du myélome multiple passait essentiellement par l’identification d’au moins un des 4 symptômes caractéristiques de la pathologie, regroupés sous le l’acronyme de « CRAB » :
• C pour « Calcium elevation » correspondant à une hypercalcémie.
Caractéristique d’une concentration de calcium dans le sang ou calcémie au-dessus de la limite normale supérieure (c’est-à-dire supérieur à 2,75 mmol/L ou à 0,25 mmol/L).
• R pour « Renal insufficiency » représentant l’insuffisance rénale (12)
Une clairance de créatinine inférieure à 40 mL/ min ou un taux de créatinine sérique supérieur à 2mg/dL ou 177 μmol/L.
• A pour « Anémia » : Anémie, correspondant à un taux d’hémoglobine inférieur à 10g/dL
(valeur normale).
• B pour « Bone lesions » : Atteinte osseuse reflétant la présence d’au moins une lésion osseuse par différents moyens : radiographie, scanner ou PET-scan.
2. Critères de l’IMWG 2014 (31)
En 2014, ce groupe de travail a revu ces critères afin de permettre également l’identification de patients souffrant de myélome multiple asymptomatique à haut risque de développer un myélome symptomatique. Des marqueurs biologiques ou également appelés marqueurs de malignités (permettant d’identifier des patients à haute probabilité de progression vers un myélome actif) ont été ajoutés.
Le diagnostic actuel du myélome multiple symptomatique se base donc sur l’arborescence ci-dessous :
40
Figure 8 : Critères de diagnostic du myélome multiple de l’IMWG 2014 (31)
Ce diagnostic se base donc sur la présence d’une plasmocytose médullaire supérieure ou égale à 10% ou plasmocytose extra médullaire confirmé par biopsie associé à l’un des critères diagnostic suivants (31) :
✓ ET la présence d’au moins un critère CRAB pouvant être attribué à une prolifération plasmocytaire sous-jacente ;
✓ ET/OU la présence d’au moins des marqueurs biologiques de malignité :
o La présence d’une plasmocytose médullaire supérieure ou égale à 60% (valeurs utilisées par Freelite assay).
o La présence du ratio Kappa/Lambda des chaines légères libres sériques affectées/non affectées supérieur ou égale à 100. Le ratio normal pour ces chaines légères kappa/lambda est compris entre 0,26 et 1,65. De ce fait, si ce ratio est inférieur à 0,26, le patient a une chaine légère libre monoclonale lambda impliqué et si au contraire, ce ratio est supérieur à 1,65, la chaine légère libre kappa est impliqué.
o La présence de plus d’une lésion focale à l’IRM et chacune de ces lésions doit être supérieure à 5 mm.
41
Cette revue a également permis d’établir les critères diagnostic du myélome multiple indolent et ceux du MGUS.
Le myélome multiple indolent se base donc sur :
✓ La présence d’au moins l’un des trois items suivants
o Une prolifération plasmocytaire ou plasmocytose médullaire clonale compris entre 10% et 60% ET/OU
o La présence d’une immunoglobuline monoclonale sérique supérieure ou égale à 30 g/L.
o La présence d’une immunoglobuline ou protéine monoclonale urinaire supérieur ou égale à 500 mg/24h.
✓ L’absence de critères CRAB.
Quant au MGUS, les critères demeurent les suivants :
✓ La présence d’une plasmocytose médullaire inférieur à 10% ; ✓ La présence d’une protéine monoclonale inférieure à 30 g/L ; ✓ L’absence de critères CRAB.
L’IMWG a également inclus de nouveaux critères CRAB, spécifiques à l’insuffisance rénale et l’atteinte osseuse, fondés sur l’utilisation d’examens plus sensibles.
Comme exposé précédemment, le myélome multiple est une maladie incurable dont la majorité des patients diagnostiqués présenteront de multiples rechutes. La rechute peut être évoquée à partir du pic monoclonal et du tableau clinique avec la réapparition de symptômes cliniques ou biologiques. Elle peut également être significative d’un point de vue biochimique : elle sera donc marquée par le doublement de l’immunoglobuline monoclonale lors de deux mesures consécutives espacées de 2 mois minimum avec une valeur de référence de 5 g/L ou l’une des augmentations suivantes présentes dans les deux mesures consécutives :
o Ig monoclonale ≥ 10 g/L ;
o Augmentation Ig urinaire de ≥ 500 mg par 24 heures ;
o Augmentation chaines légères libres d’au moins 20 mg/dL avec un ratio anormal ou une augmentation de 25%.
42
3. Les examens permettant le bilan au diagnostic
La réalisation de plusieurs examens médicaux est donc nécessaire pour établir le diagnostic du myélome et confirmer les critères instaurés par l’IMWG, vus précédemment.
A. Bilan sanguin (analyse via une prise de sang) (27,31)
Un bilan sanguin avec numérotation de la formule sanguine (NFS) sera nécessaire car il permettra de mettre en évidence la présence ou non de signes cliniques spécifiques du myélome : l’anémie, l’insuffisance rénale et l’hypercalcémie. Les techniques sont donc les suivantes :
✓ Un hémogramme avec frottis sanguin permettant la recherche d’une anémie (Hémoglobine < 10g/dL) et d’une éventuelle leucopénie et/ou thrombocytopénie. ✓ Une créatininémie pour rechercher une éventuelle insuffisance rénale (créatinine
sérique > 2 mg/dL ou > 177 μmol/L).
✓ Une calcémie (par ionogramme sanguin et urinaire) permettant la recherche d’une hypercalcémie (calcium sérique > 11mg/dL ou > 2,75 mmol/L).
✓ Dosage de la protéine C réactive (CRP), vitesse de sédimentation et lactates déshydrogénases (LDH). Ces marqueurs sont le reflet de la masse tumorale et sont des facteurs pronostiques de la maladie. Une VS élevée et un taux de CRP normale peuvent conduire à la conclusion d’une éventuelle anomalie des IgG. En revanche, si le taux de CRP était également élevé, cela correspondrait plutôt à un syndrome inflammatoire.
B. Détection et évaluation d’une immunoglobuline monoclonale (34)
A partir des analyses sanguines et urinaires recueillies, une recherche de protéines sera réalisée permettant ainsi la détection de l’immunoglobuline monoclonale et ses caractéristiques. De ce fait, deux principaux examens seront réalisés afin de caractériser ce composant monoclonal :
❖ Electrophorèse des protéines sériques et/ou urinaires (EPS) (34)
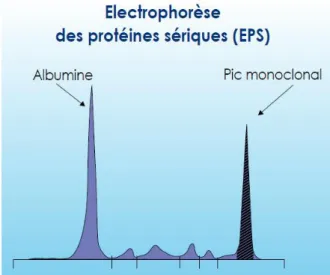
Cette technique permet de doser les protéines présentes dans le sang/ urine, de les séparer et ainsi de voir lesquelles se trouvent en grande quantité. Dans le cadre du myélome, elle permet de mettre en évidence la présence d’un pic étroit de l’immunoglobuline
43
monoclonale ou protéine M. Dans la majorité des cas, Ce pic est situé au niveau des gamma-globulines ou béta-gamma-globulines, évoquant plutôt une Immunoglobuline de type G ou A. Cette technique permet une évaluation correcte et suffisante de l’identification et de la quantification du taux de l’immunoglobuline monoclonale. Lorsqu’un pic étroit est mis en évidence, il conviendra, par la suite, de faire des analyses supplémentaires afin de déterminer les caractéristiques d’une immunoglobuline monoclonale comme un dosage des immunoglobulines monoclonales par densitométrie.
Figure 9 : Présence d’un pic monoclonal au niveau des gammaglobulines détecté par EPS chez un patient atteint de myélome multiple (34)
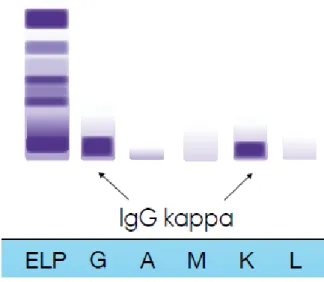
❖ Immunofixation des protéines sériques et/ ou urinaires ou immunoélectrophorèse (IFE)
Cette technique se fait à la suite d’une détection du pic monoclonal par EPS. Elle permet de confirmer les caractéristiques monoclonales du pic et donc de caractériser l’isotype du composant monoclonal (G, A ou M pour les chaines lourdes et Kappa ou Lambda pour les chaines légères libres.)