Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées
Institut de chimie des milieux et matériaux de Poitiers - IC2MP (Diplôme National - Arrêté du 25 mai 2016)
École doctorale : Sciences pour l'environnement - Gay Lussac (La Rochelle) Secteur de recherche : Chimie appliquée
Présentée par :
Maïté Audemar
Hydrogénation catalytique de molécules biosourcées
Directeur(s) de Thèse :
Karine de Oliveira Vigier, Sébastien Royer Soutenue le 14 décembre 2016 devant le jury Jury :
Président Christophe Darcel Professeur des Universités, Université de Rennes 1 Rapporteur Nadine Essayem Directrice de recherche CNRS, IRCELyon
Rapporteur Mickaël Capron Maître de conférences, Université de Lille 1 Membre Karine de Oliveira Vigier Maître de conférences, Université de Poitiers Membre Sébastien Royer Professeur des Universités, Université de Lille 1 Membre Mario de Bruyn Research associate, University of York, Great Britain
Pour citer cette thèse :
Maïté Audemar. Hydrogénation catalytique de molécules biosourcées [En ligne]. Thèse Chimie appliquée. Poitiers : Université de Poitiers, 2016. Disponible sur l'Intranet de l'Université de Poitiers <http://theses.univ-poitiers.fr>
Pour l’obtention du grade de
DOCTEUR DE δ’UNIVERSITÉ DE POITIERS Faculté des Sciences Fondamentales et Appliquées Institut de Chimie des Milieux et Matériaux de Poitiers (IC2MP)
(Diplôme National - Arrêté du 7 août 2006)
École doctorale : Sciences pour l’Environnement GAY LUSSAC Secteur de Recherche : Chimie appliquée
Présentée par :
Maïté AUDEMAR
HYDROGÉNATION CATALYTIQUE DE MOLÉCULES
BIOSOURCÉES
Directrice de thèse : Karine DE OLIVEIRA VIGIER Co-directeur de thèse : Sébastien ROYER
************************* Soutenue le 14 Décembre 2016
Devant la commission d’examen
************************* JURY
Rapporteurs : Nadine ESSAYEM Mickaël CAPRON
Directrice de Recherches, IRCELYON, Villeurbanne Maître de Conférences - HDR, UCCS, Lille
Examinateurs : Christophe DARCEL Mario DE BRUYN Sébastien ROYER
Karine DE OLIVEIRA VIGIER
Professeur, ISCR, Rennes
Research Associate, GCCE, York (GB) Professeur, UCCS, Lille
REMERCIEMENTS
Ce travail de thèse a été effectué au sein de l’Institut de Chimie des Milieux et des Matériaux de Poitiers (IC2MP). Je tiens à remercier François Jérôme, Directeur de recherche au CNRS, et Christophe Coutanceau, Professeur à l’Université de Poitiers, de m’avoir accueilli au sein de leur équipe de recherche : Catalyse et Carbone Renouvelable. Je remercie la Région Poitou-Charentes pour le financement de ces trois années de thèse.
Je suis reconnaissante envers l’ensemble des membres du jury qui m’ont fait l’honneur d’accepter de juger ce travail.
En particulier j’adresse mes plus vifs remerciements à Nadine Essayem, Directrice de recherche au CNRS à l’IRCEδYON, et à εickaël Capron, εaître de Conférences à l’UCCS de Lille, pour avoir bien voulu accepter de juger mon travail et d’en être les rapporteurs.
εes remerciements vont également à l’égard de Christophe Darcel, Professeur à l’ISCR de Rennes et à Mario De bruyn, Chercheur au GCCE de York (GB) d’avoir accepté de d’examiner ce travail et de faire partie de mon jury de thèse. Je remercie également Mario pour son accueil au GCCE ainsi que pour ses conseils.
J’adresse des remerciements particuliers à ma directrice de thèse, Karine De Oliveira Vigier, à qui je tiens à exprimer toute ma gratitude pour son encadrement, sa présence et sa disponibilité au cours de mes trois années de thèse, ainsi que pour ses conseils et sa patience. Je tiens également à remercier Sébastien Royer, mon co-directeur, pour son aide et ses conseils. Merci à eux de m’avoir permis de concrétiser ce projet doctoral.
Je souhaite également remercier l’organisation : COST (European Cooperation in Science and Technology) ainsi que l’école doctorale Sciences pour l’Environnement Gay δussac qui m’ont permis de financer mon stage doctoral au Green Chemistry Centre of Excellence à l’Université de York (GB).
Ensuite, j’exprime ma reconnaissance à Dimitri et Michel pour leur aide sur les réacteurs. Je tiens à remercier également les personnes des plates-formes techniques pour leur aide : Sandrine, Mehrad, Lilian. Je tiens à remercier également Karine pour sa patience avec moi et mes colis.
Je remercie sincèrement les stagiaires qui ont contribué à une partie de ce travail :
Je souhaite spécialement remercier tous ceux que j’ai côtoyés au cours de ces trois années et qui ont contribué à tous ces souvenirs que je garderai de cette thèse.
Ainsi je veux remercier les permanents de l’équipe pour leur accueil, et bien sur mes collègue : Guillaume (désolé pour la quasi-amputation !), Alice (pour les sushis et les flamby !), Florent (pour ses allusions !), Wahiba, Clément, Nathalie, Jialu, Caroline, Joakim,
Houcine, Mohcin, Alban, Raluca, Eloi, Nassim, Ayman, Shi, Claudia. Ou encore les gars du
B30 : Soizic, Olivier, Julien et David.
Je termine ces remerciement par une note personnelle. Merci à Tica pour tout ce qu’elle me fait vivre (n’est-ce pas Lucie ?). Je remercie également mon « petit » frère, et bravo à toi ! Je tiens aussi à remercier du fond du cœur mes parents et grands-parents qui m’ont toujours soutenue et encouragée. Enfin, un immense merci à Lucie qui a réussi à me supporter pendant cette aventure !
TABLE DES MATIÈRES
LISTEDESABRÉVIATIONS 11
INTRODUCTION GÉNÉRALE 13
CHAPITRE I : ETUDE BIBLIOGRAPHIQUE 17
I. LA BIOMASSE LIGNOCELLULOSIQUE 19
I.1. COMPOSITION DE LA LIGNOCELLULOSE 19
I.1.1. La lignine 19
I.1.2. La cellulose 21
I.1.3. Les hémicelluloses 22
II. HYDROGENATION DU XYLOSE EN XYLITOL 23
II.1. GENERALITE SUR LE XYLITOL 23
II.2. PRODUCTION DE XYLITOL 26
II.2.1. Voie biotechnologique 26
II.2.2. Voie chimique 27
II.2.2.a. Catalyseur à base de nickel 27
II.2.2.b. Catalyseur à base de métaux nobles 29
II.3. Conclusion 33
III. HYDROGENATION DU FURFURAL 34
III.1. PRODUCTION DE L’ALCOOL FURFURYLIQUE 36
III.1.1. δ’alcool furfurylique 36
III.1.2. Hydrogénation en phase gazeuse 36
III.1.2.a. Catalyseurs chromite de cuivre 37
III.1.2.b. Catalyseurs à base de cuivre 39
III.1.2.c. Catalyseurs à base de métaux du groupe 10 42
III.1.β.d. εécanismes proposés pour l’hydrogénation catalytique du furfural en alcool
furfurylique 44
III.1.3. Hydrogénation en phase liquide 48
II.1.3.a. Catalyseurs monométalliques 50
II.1.γ.b. Effet de l’ajout d’un co-métal 51
• Catalyseurs à base de métaux nobles 51
• Catalyseurs à base de métaux non nobles 54
III.1.4. Hydrogénation du furfural par transfert catalytique d’hydrogène 62
II.1.5. Conclusion 62
III.2. HYDROGENOLYSE DU FURFURAL EN 2-METHYLFURANE 63
III.2.1. Le 2-méthylfurane 63
III.2.2. Hydrogénolyse en phase gazeuse 64
III.2.3. Hydrogénolyse en phase liquide 72
III.2.4. Hydrogénolyse par transfert catalytique d’hydrogène 75
III.2.5. Conclusion 78
IV. CONCLUSION ET OBJECTIF DE LA THESE 79
CHAPITRE II : PARTIE EXPÉRIMENTALE 81
I. SOLVANTS ET PRODUITS CHIMIQUES 83
I.1. SOLVANTS 83
I.2. PRODUITS CHIMIQUES 83
II. PREPARATION DES CATALYSEURS METALLIQUES 84
II.1. PREPARATION DES CATALYSEURS CO/SBA-15 84
II.2. PREPARATION DES CATALYSEURS CO/SIO2 ET CU/SIO2 85
III. TECHNIQUES DE CARACTERISATION PHYSICO-CHIMIQUES DES CATALYSEURS 85
III.1. ANALYSE ELEMENTAIRE PAR SPECTROMETRIE D’EMISSION OPTIQUE A PLASMA INDUCTIF
(ICP-OES) 85
III.1.a. Principe 85
III.1.b. Conditions d’analyse 85
III.2. SPECTROSCOPIE D’ABSORPTION ATOMIQUE 86
III.2.a. Principe 86
III.β.b. Conditions d’analyse 86
III.3. ADSORPTION-DESORPTION D’AZOTE 86
III.3.a. Principe 86
III.γ.b. Conditions d’analyse 87 III.4. MICROSCOPIE ELECTRONIQUE EN TRANSMISSION (MET) 87
III.4.a. Principe 87
III.4.b. Conditions d’analyse 87
III.5. DIFFRACTION DES RAYONS X(DRX) 88
III.5.a. Principe 88
III.η.b. Conditions d’analyse 88
III.5.c. Diffraction des rayons X in situ 88
III.6.a. Principe 89
III.θ.b. Conditions d’analyse 89
IV. REACTIONS D’HYDROGENATION 89
IV.1. HYDROGENATION DU XYLOSE EN XYLITOL 90
IV.2. HYDROGENATION DU FURFURAL EN ALCOOL FURFURYLIQUE 90 IV.3. REACTION D’HYDROGENOLYSE DU FURFURAL EN 2-METHYLFURANE 91 IV.4. HYDROGENATION DE L’ACIDE LEVULINIQUE EN -VALEROLACTONE 91
IV.4.a. Hydrogénation de l’acide lévulinique sous pression d’hydrogène 91
IV.4.b. Hydrogénation de l’acide lévulinique par transfert d’hydrogène assisté par
micro-ondes 92
V. QUANTIFICATION ET IDENTIFICATIONS DES PRODUITS OBTENUS 93
V.1. CHROMATOGRAPHIE EN PHASE LIQUIDE 93
V.2. CHROMATOGRAPHIE EN PHASE GAZEUSE 94
V.β.a. Produits d’hydrogénation du furfural et de l’acide lévulinique 94
V.β.b. Produits d’hydrogénolyse du furfural 97
V.3. SPECTROMETRE DE MASSE EN INFUSION DIRECTE 99
V.3.a. Principe 99
V.γ.b. Conditions d’analyse 99 V.4. CHROMATOGRAPHIE EN PHASE GAZEUSE COUPLEE A UN SPECTROMETRE DE MASSE (GC-MS)
99
V.4.a. Principe 99
V.4.b. Conditions d’analyse 100 V.5. SPECTROSCOPIE DE RESONNANCE MAGNETIQUE NUCLEAIRE DU PROTON :RMN1H 100
CHAPITRE III : HYDROGÉNATION DU XYLOSE 101
I. INTRODUCTION 103
II. EFFETS DES PARAMETRES REACTIONNELS 104
II.1. INFLUENCE DU TEMPS DE REACTION 104
II.2. INFLUENCE DE LA TEMPERATURE 105
II.3. INFLUENCE DE LA PRESSION EN DIHYDROGENE H2 106
II.4. INFLUENCE DE LA CONCENTRATION INITIALE EN XYLOSE 107
III. RECYCLAGE DU CATALYSEUR CO/SIO2 108
IV. INFLUENCE DE LA NATURE DU SUPPORT 111
CHAPITRE IV : HYDROGÉNATION/HYDROGÉNOLYSE DU FURFURAL 115
I. HYDROGENATION DU FURFURAL EN ALCOOL FURFURYLIQUE 117
I.1. HYDROGENATION DU FURFURAL EN PRESENCE DU CATALYSEUR CO/SIO2 118
I.2. HYDROGENATION DU FURFURAL EN PRESENCE DU CATALYSEUR CO/SBA-15 119
I.2.1. Caractérisation du catalyseur Co/SBA-15 120
I.2.2. Influence de la température de réduction du catalyseur sur son activité 124 I.2.3. Effets de différents paramètres réactionnels 128
I.2.3.a. Effets du temps de réaction 128
I.2.3.b. Effets de la pression en dihydrogène H2 129
I.2.3.c. Effets de la température de réaction 130
I.2.3.d. Effets des quantités en catalyseur Co/SBA-15 et en furfural 131
I.β.4. Stabilité du catalyseur pour la réaction d’hydrogénation du furfural 132
I.β.η. Influence de la nature du solvant sur la réaction d’hydrogénation 134 I.2.6. Influence de la pureté du furfural utilisé 135
I.3. CONCLUSION 139
II. HYDROGENOLYSE DU FURFURAL EN 2-METHYLFURANE 140
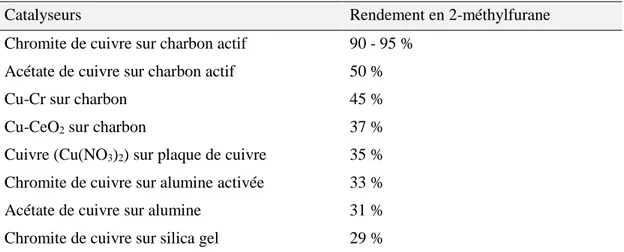
II.1. ÉTUDE DE CATALYSEURS A BASE DE CUIVRE 140
II.2. INFLUENCE DU SOLVANT 141
II.3. INFLUENCE DES PARAMETRES REACTIONNELS 143
II.3.1. Influence de la température de réaction 143
II.3.2. Influence du temps de réaction 143
II.3.3. Influence de la pression en dihydrogène H2 144
II.4. CONCLUSION 145
CHAPITRE V : HYDROGÉNATION DE L’ACIDE LÉVULINIQUE 147
I. HYDROGENATION EN PRESENCE DU CATALYSEUR CO/SBA-15 149 II. HYDROGENATION PAR TRANSFERT CATALYTIQUE D’HYDROGENE 152
II.1. INFLUENCE DE LA TEMPERATURE DE REACTION 153
II.2. INFLUENCE DE LA QUANTITE DE CATALYSEUR 153
II.3. INFLUENCE DE LA MASSE DE DEPART D’ACIDE LEVULINIQUE 154 II.4. INFLUENCE DE LA TENEUR EN PLATINE SUR LE CATALYSEUR 155
III. CONCLUSION 157
CONCLUSION GENERALE 159
LISTE DES ABRÉVIATIONS
LISTE DES ABRÉVIATIONS
ATG : Analyse thermo-gravimétrique ATD : Analyse thermo-différentielle
BET μ Surface spécifique d’un catalyseur mesurée par la méthode Brunauer Emmet et Teller DMSO : Diméthylsulfoxyde
DRX : Diffraction des rayons X FF : Furfural
FFA : Alcool furfurylique
GC : Chromatographie en phase gazeuse
GC-MS : Chromatographie en phase gazeuse couplée à un spectromètre de masse GVL μ -Valérolactone
HPLC : Chromatographie liquide à haute performance
ICP-OES : Spectroscopie par torche à plasma-spectrométrie d’émission optique LA : Acide lévulinique
MET : Microscopie électronique en transmission MF : 2-Méthylfurane
MIBK : Méthylisobutylcétone MTHF : 2-Méthyltétrahydrofurane RMN : Résonance magnétique nucléaire RTP : Réduction en température programmée THFA : Alcool tétrahydrofurfurylique TOF : Turnover Frequency
Aujourd’hui, en plus des préoccupations environnementales causées par la transformation, l’utilisation et la gestion de la fin de vie des produits issus des ressources fossiles, la fluctuation du prix du pétrole entraîne un changement dans la conception de la chimie.
Une chimie « alternative » s’est développée, une chimie qui se veut durable, qui a pour défis de satisfaire les besoins en énergie et produits chimiques, de réduire l'impact environnemental des procédés et de ces produits ainsi que de la gestion des déchets. Ce concept qui entraîne également le développement de nouveaux procédés de synthèse ou l’amélioration de ceux déjà existants ainsi que la diminution de leurs consommations énergétiques, est qualifié par le terme « chimie verte » depuis les années 70. Cependant, ce n’est en 1998 que ce concept de « chimie verte » a été codifié par Paul Anastas et John C. Warner, avec la mise en place de douze principes fondateurs.
δ’un des douze principes de la chimie verte repose sur l'utilisation de matières premières renouvelables à la place des ressources fossiles. Ainsi, la valorisation de la biomasse lignocellulosique pour l’énergie et la chimie fine est souhaitable. En effet cette biomasse lignocellulosique qui est disponible et abondante permet après transformation d’accéder à des molécules dites plateformes à partir desquelles des molécules à haute valeur ajoutée peuvent par la suite être obtenues. Cette biomasse lignocellulosique est constituée majoritairement de trois biopolymères que sont : la lignine, la cellulose et les hémicelluloses.
Cette thèse se consacre à l’étude de réactions d’hydrogénation catalytiques de trois molécules biosourcées. δa première est la réaction d’hydrogénation du xylose, obtenu par hydrolyse de la fraction hémicellulosique de la lignocellulose. Le xylose peut également être déshydraté en furfural qui est aujourd’hui une molécule plateforme ouvrant l’accès à de nombreuses molécules d’intérêt. Aussi, ce sont les réactions d’hydrogénation et d’hydrogénolyse du furfural pour la production d’alcool furfurylique et de β-méthylfurane, respectivement, qui ont été étudiées. De plus, les catalyseurs utilisés ont été testés pour l’hydrogénation de l’acide lévulinique en -valérolactone. Cette réaction a également été étudiée par transfert catalytique d’hydrogène.
La rédaction de ce manuscrit s’organise selon la structure suivante :
● δe premier chapitre de ce manuscrit présente l’état de l’art des différents systèmes
avantages et inconvénients de divers systèmes catalytiques proposés seront présentés, ainsi que les problèmes rencontrés pour ces réactions d’hydrogénation.
● δe second chapitre présente l’ensemble des procédures expérimentales utilisées
durant le Doctorat : modes opératoires adoptées pour les différentes réactions d’hydrogénation ainsi que les méthodes analytiques permettant d’identifier et/ou de quantifier les produits de ces réactions ; techniques de caractérisations physico-chimiques et conditions d’analyse des catalyseurs.
● Le chapitre trois est consacré à la présentation des résultats obtenus lors de l’étude
de la réaction d’hydrogénation du xylose en xylitol, un édulcorant.
● δe chapitre quatre concerne l’étude de catalyseurs actifs et sélectifs en alcool
furfurylique lors de la réaction d’hydrogénation du furfural. Les caractérisations du catalyseur développé, et les performances de ce catalyseur seront présentées dans ce chapitre. Ce chapitre présente également les résultats préliminaires obtenus pour l’hydrogénolyse du furfural en 2-méthylfurane.
● Le chapitre cinq est consacré quant à lui à la présentation des résultats obtenus lors de l’hydrogénation de l’acide lévulinique en -valérolactone, ainsi que ceux obtenu par transfert catalytique d’hydrogène.
● Finalement une conclusion, résumant l’ensemble des résultats obtenus pour ces
réactions d’hydrogénation et décrivant les avantages et inconvénients des divers systèmes catalytiques développés, clôture cet ouvrage. Enfin, des perspectives de poursuite de ces travaux sont proposées.
CHAPITRE I
I. La biomasse lignocellulosique
Face à une demande sans cesse croissante en matières premières pétrolières pour l’énergie ou la chimie fine, un intérêt croissant se porte sur le développement de l’utilisation de la biomasse lignocellulosique comme source alternative de molécules. En effet, cette dernière représente une source de carbone renouvelable accessible et abondante pouvant provenir, par exemple, des résidus agricoles et forestiers, des déchets municipaux ou encore des déchets de la transformation du bois. La lignocellulose est composée de trois composés majeurs : la lignine, les hémicellulose et la cellulose.
I.1. Composition de la lignocellulose
La biomasse lignocellulosique est majoritairement composée de trois biopolymères : les hémicelluloses (25 - 35 %), la cellulose (40 - 50 %), et le lignine (15 - 20 %).1 Sa structure est illustrée sur la Figure 1. Les autres constituants de la lignocellulose retrouvés dans de faibles proportions sont : les protéines et les pectines (1 à 2 %), les huiles essentielles (< 2 %) ainsi que certains minéraux (2 à 4 %).2
Figure 1 : Structure de la lignocellulose.
Les trois paragraphes suivants présentent les caractéristiques des trois biopolymères majoritaires de la lignocellulose.
I.1.1. La lignine
La fraction lignine de la biomasse lignocellulosique est un polymère branché qui forme une structure tridimensionnelle amorphe. C’est cette lignine qui fournit leur rigidité structurelle aux plantes.
1 C.E. Wyman, B.E. Dale, R.T. Elander, M. Holtzapple, M.R. Ladisch, Y.Y. Lee, Bioresour. Technol., 2005, 96, 1959-1966.
Figure 2 : Structure de lignine avec diverses liaisons C-O et C-C typiquement présentes dans la lignine native. 3
La lignine est composée majoritairement de trois monomères que sont : l'alcool coumarylique sans groupe méthoxy, l’alcool coniférylique à un groupe méthoxy et l'alcool sinapylique à deux groupes méthoxy.4
HO OH HO OH H3CO HO OH OCH3 H3CO
Alcool coumarylique Alcool coniférylique Alcool sinapylique
Figure 3 : Monomères composant la lignine.
Aujourd’hui, la plus grande partie de la lignine est brulée pour une production d’énergie et/ou de chaleur. En effet, la valorisation de la lignine pour la chimie fine reste difficile en raison de sa structure qui diffère en fonction à la fois de la source de la lignine et de la méthode utilisée pour isoler cette lignine de la lignocellulose.
Cependant, des solutions permettant la production d'intermédiaires fonctionnels à partir de la lignine sont tout de même proposées dans le but de produire des produits chimiques
3 F. Boissou, Université de Poitiers 2015.
d’intérêts. Par exemple, étant donné que la lignine est riche en espèces aromatiques oxygénées, elle peut être utilisées comme matières premières dans la production de résines phénoliques.5,6 Une autre stratégie repose sur la pyrolyse de la lignine pour la production de bio-huiles composées majoritairement de composés phénoliques.7
I.1.2. La cellulose
La cellulose est un homopolymère linéaire constitué de monomères d’unités D-anhydroglucose (AGU) liées par des liaisons -1→4 glycosidiques. Le motif de répétition de ce polymère est le dimère cellobiose qui est constitué de deux unités AGU.
Figure 4 : Structure de la cellulose.
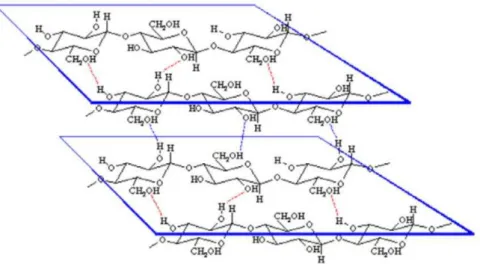
Le nombre de répétition des unités AGU défini le degré de polymérisation (DP) de la cellulose. Ce DP dépend largement de l’origine de la cellulose ainsi que des prétraitements qu’elle a subit. δa valeur de ce degré de polymérisation peut alors varier d’une centaine à plusieurs milliers unités AGU. Chaque unité AGU possède trois groupements hydroxyles, deux alcools secondaires sur les atomes de carbone Cβ et Cγ, ainsi qu’un alcool primaire sur le carbone C6 (Figure 4). Ces groupements hydroxyles sont à même de former entre eux des liaisons hydrogène intra- et intermoléculaires (Figure 5). La présence de ces nombreuses liaisons hydrogène créent une forte cohésion entre les chaines de cellulose ce qui lui confère une structure semi-cristalline et qui a pour effet de rendre la cellulose insoluble dans l’eau et dans la plupart des solvants organiques.
5 J.H. Lora, W.G. Glasser, Journal of Polymers and the Environment, 2002, 10, 39-48. 6 S. Sarkar, B. Adhikari, J. Adhes. Sci. Technol., 2000, 14, 1179-1193.
7 J. Jae, G.A. Tompsett, Y.-C. Lin, T.R. Carlson, J. Shen, T. Zhang, B. Yang, C.E. Wyman, W.C. Conner, G.W. Huber, Energy Environ. Sci., 2010, 3, 358-365.
Figure 5 : Réseau de liaisons hydrogène engendrant un agencement parallèle des chaines de cellulose. 8 δa cellulose peut être utilisée sous forme de fibres brutes pour l’industrie de la pâte à papier. La présence des trois groupements hydroxyles situés sur les carbones C2, C3 et C6 permettent également diverses transformations chimiques à la surface de la cellulose. Une autre valorisation possible est la dépolymérisation de la cellulose afin d’obtenir du glucose.
I.1.3. Les hémicelluloses
Les hémicelluloses sont liées à la lignine et entrelacées avec les fibres de cellulose.9 Ce sont des hétéropolysaccharides constitués d’hexoses (glucose, mannose et galactose) et de pentoses (xylose et arabinose). Selon l’origine de la plante, la composition chimique des hémicelluloses diffère, mais le xylose est dans la plupart des cas le monomère le plus abondant,10 formant chez les feuillus des polymères nommés xylane. Toutefois, dans le cas d’un bois résineux, le mannose est le monomère majoritaire.
δes xylanes sont des polymères constitués d’une chaine d’unités -D-xylopyranose liées entre elles par des liaisons -1,4 glycosidiques et ramifiés (en partie) par du L-arabinose, de l’acide glucuronique ou sous sa forme éthérifiée et des acides acétique, ferulique et p-coumarique dans le cas d’un xylane de graminée (Figure 6).
Ces hémicelluloses peuvent être hydrolysées en présence de solutions acides ou basiques. Cette dépolymérisation permet d’obtenir les monomères de sucre. D’autres molécules peuvent également être obtenues, tel que des furanes : le 5-hydroxyméthylfurfural provenant de la déshydratation des hexoses, le furfural provenant de la déshydratation des
8 P. Harmsen, W. Huijgen, L. Bermudez, R. Bakker, Energy Research Centre of the Netherlands, 2010, 10-13. 9 D.M. Alonso, J.Q. Bond, J.A. Dumesic, Green Chem., 2010, 12, 1493-1513.
pentoses, des acides carboxyliques (acides acétique, lévulinique et formique) provenant de la déshydratation et réhydratation des sucres.
O O O O O O O O O O OH O O O HO HO O HO O O CH3 O RO HO OH HOOC R1= H, CH3 O HO OH OH O O OH OH O CH3 OCH3 OH R2 O
R2= acide ferulique ou acide p-coumarique
H3CO HO OH O HO OH O Partie glycosidique O n HO
Figure 6 : Exemple de structure de xylanes d'une plante graminée.3
Le xylose alors obtenu à partir des hémicelluloses peut être hydrogéné afin de produire du xylitol. Il peut également être déshydraté pour conduire à la molécule plateforme qu’est le furfural.
II. Hydrogénation du xylose en xylitol
II.1. Généralité sur le xylitolCes dernières années, l’augmentation des problèmes liés à l’alimentation (obésité, diabète de type II) a entrainé un changement de l’industrie agro-alimentaire. En effet, le sucre (saccharose) ajouté dans de nombreux produits alimentaires manufacturés est aujourd’hui remplacé par des édulcorants. Ces derniers peuvent être artificiels tel que l’aspartame ou l’acésulfame K, ou naturels tel que les polyols.
Les édulcorants présentent l’avantage d’avoir un faible apport calorique ainsi qu’une faible activité cariogène. Toutefois, la méfiance envers les édulcorants artificiels suscite de plus en plus une remise en question de leur innocuité de la part des consommateurs, que ce
soit de la molécule en elle-même ou des produits issus de sa dégradation et/ou métabolisation. Les consommateurs souhaitent donc se tourner vers les édulcorants dits naturels.
δe xylitol est l’un des polyols qui est considéré comme étant un substitut potentiel du saccharose du fait de ses propriétés physiques et chimiques. Un grand intérêt lui est porté dans les industries alimentaires, pharmaceutiques et cosmétiques. Le xylitol possède la même saveur que celle du saccharose ainsi qu’un pouvoir sucrant équivalent. De plus, son apport calorique n’est que de β,4 kcal/g contre 4,0 kcal/g pour celui du saccharose11 et son index glycémique est de 13 alors que celui du saccharose (référence) est de 100.12 Le xylitol présente également l’avantage d’être non-cariogène.11 En plus de ces propriétés, il présente une enthalpie de solution négative (-36 cal/g) ce qui lui confère, lorsqu’il entre en contact avec de la salive, une sensation de fraicheur en bouche.11 Cette dernière propriété accentue encore l’intérêt que lui porte l’industrie des confiseries, des chewing-gums et du dentifrice. De plus, son emploi dans l’industrie alimentaire dédiée à l’Homme a largement été approuvé. En effet, les organismes que sont le Joint FAO/WHO Expert Committee on Food Additives ainsi que la European Food Safety Authority ont attribué au xylitol une dose journalière admissible « non spécifiée », ce qui représente la meilleure évaluation en terme de sécurité pouvant être donnée à tout additif alimentaire.
La Figure 7 présente la structure du xylitol (C5H12O5). Le xylitol est un polyol non cyclique à 5 atomes de carbone et 5 groupes hydroxyles. Il a été isolé presque simultanément pour la première fois en 1891 par deux groupes de chercheurs allemands et français.13,14
Figure 7 : molécule de xylitol.
Le xylitol est naturellement présent en faible quantité dans des lichens, des algues ou encore dans certains fruits ou légumes, le Tableau 1 présente la teneur en xylitol présent dans certains aliments.
11 L.O.B. Nabors, "Alternative Sweeteners", 2012, 4th ed., CRC Press. 12 G. Livesey, Nutr. Res. Rev., 2003, 16, 163-191.
13 E. Fischer, R. Stahel, Ber. Dtsch. Chem. Ges., 1891, 24, 528-539. 14 M.G. Bertrand, Bull. Soc. Chim. Paris, 1891, 5, 554-557.
Tableau 1 : Teneur en xylitol de quelques fruits ou légumes. 15
Produits Xylitol (mg/100 g matière sèche)
Prune 935 Fraise 362 Chou-fleur 300 Framboise 268 Endive 256 Myrtille 213 Aubergine 180 Laitue 131 Champignon blanc 128 Épinards 107
Les fruits et légumes ne sont pas les seules sources « naturelles » qui contiennent du xylitol, certains déchets agro-industriels en contiennent également à des teneurs faibles (< 50 mg/100 g). Le Tableau 2 indique la teneur en xylitol de certains déchets agro-industriels.
Tableau 2 : Teneur en xylitol de quelques déchets agro-industriels. 16
Matière Xylitol (mg/100 g) Coquilles de pistaches 33-50 Son d'orge 29.2 Épis de maïs 28-35.3 Chêne 21.7 Paille de sorgho 21.5
Bagasses de cannes à sucre 20.5-25.6
Paille de blé 19.2-21
Bouleau 18.5-24.9
Tremble 18-27.3
Paille de riz 14.8-23
Sarment de vigne 12.8
La production de xylitol, basée sur l’extraction à partir de déchets agro-industriels, de fruits ou de légumes, n’est pas économiquement viable dû aux faibles teneurs qu’ils
15 J. Washuettl, P. Riederer, E. Bancher, J. Food Sci., 1974, 38, 1262-1263.
contiennent (moins de 100 mg/100 g de matière sèche). Il est donc produit industriellement, soit par un procédé chimique d’hydrogénation catalytique du xylose, soit par voie biotechnologique reposant sur l’utilisation de microorganismes.
II.2. Production de xylitol
II.2.1. Voie biotechnologique
La voie biotechnologique pour la production de xylitol a pour avantage qu’elle ne nécessite pas d’étapes de purification du xylose. De nombreux microorganismes, capables de produire du xylitol à partir de matières premières contenant du xylose, ont été identifiés ces deux dernières décennies.17 Ces microorganismes sont de trois types : bactéries, champignons et levures. Le Tableau 3 liste quelques un de ces microorganismes utilisés pour la production de xylitol.
Tableau 3 : Microorganismes permettant la production de xylitol. 17
Levures Champignons Bactéries
Petromyces albertensis Aspergillus niger Corynebacterium sp. Candida boidinii Penicillium brevicompactum Enterobacter liquifaciens C. maltosa Xu316 P. citrinum Mycobacterium smegmatis
C. mogii P. expansum
C. tropicalis HXP2 P. griseoroseum Debaromyces hansenii UFV-170 P. roqueforti Hansenula polymorpha P. purpurogenum Pachysolen tannophilus P. janthinellum Pichia caribica P. chrysogenum C. guilliermondii FTI-20037 P. italicum
C. intermedia P. crustosum
Toutefois, un des inconvénients majeurs de la production de xylitol par fermentation est la sensibilité des souches des microorganismes aux conditions expérimentales. Il en résulte ainsi un coût plus élevé par rapport à une production par voie chimique.
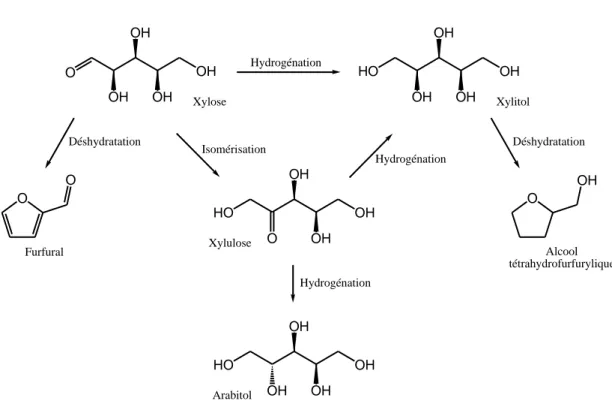
II.2.2. Voie chimique HO OH OH OH OH O OH OH OH OH Hydrogénation O O O OH HO OH O OH OH HO OH OH OH OH Xylose Xylitol Furfural Alcool tétrahydrofurfurylique Xylulose Arabitol Hydrogénation Hydrogénation Déshydratation Déshydratation Isomérisation
Figure 8 : Produits de réaction possibles lors de l'hydrogénation du xylose.
δe xylitol peut être obtenu par l’hydrogénation catalytique du xylose. Au cours de cette réaction, il est possible d’observer également la formation de l’arabitol provenant de l’hydrogénation du xylulose ou encore les formations du furfural ou de l’alcool furfurylique provenant respectivement de la déshydratation du xylose et du xylitol (Figure 8).
Dans la littérature, la réaction d’hydrogénation du xylose en xylitol a été étudiée en milieux aqueux en présence de divers catalyseurs métalliques supportés. Ces derniers sont généralement à base de nickel, de ruthénium ou de platine.
II.2.2.a. Catalyseur à base de nickel
Actuellement, la production industrielle du xylitol par hydrogénation du xylitol est réalisée en présence d’un catalyseur nickel de Raney avec un rendement de η0 à 60 %.18 De plus, de nombreuses études sont basées sur l’utilisation de ce catalyseur nickel de Raney.19,20,21
18 A. Melaja, L. Hämäläinen, H. O. Heikkilä, patent FI 589388. 1981
19 J. Wisniak, M. Hershkowitz, R. Leibowitz, S. Stein, Ind. Eng. Chem. Prod. Res. Dev., 1974, 13, 75-79. 20 J.-P. Mikkola, T. Salmi, Chem. Eng. Sci., 1999, 54, 1583-1588.
δ’utilisation du nickel de Raney présente des avantages qui sont : son faible coût ainsi qu’une activité et sélectivité élevées (> 90 %). Cependant, le nickel de Raney possède également des inconvénients qui sont sa désactivation rapide ainsi que la lixiviation du métal. Par conséquent, il est important de purifier la solution de xylitol après la réaction afin d’éliminer le nickel ayant pu passer en solution pour en permettre l’utilisation dans l’industrie alimentaire, pharmaceutique ou cosmétique, ce qui induit un coût supplémentaire.
Afin de pallier à ces problèmes de lixiviation, la stabilisation du catalyseur nickel de Raney a été proposée à partir de différentes approches. Ainsi, Mikkola et Salmi20,22 ont mené des études sur l’utilisation de la sonochimie afin de limiter la désactivation d’un catalyseur nickel de Raney lors de l’hydrogénation du xylose en xylitol. δa raison évoquée par ces auteurs pour expliquer une désactivation moins importante du catalyseur pour le 2nd cycle catalytique est que l'irradiation acoustique fragmente les particules du catalyseur, augmentant ainsi sa surface et exposant alors de nouveaux sites actifs pour la réaction d'hydrogénation.
Dans une autre étude, un catalyseur nickel de Raney a été dopé par du molybdène.23 Cependant, malgré ce dopage, il se produit tout de même une désactivation importante. Les auteurs ont constaté que la teneur en aluminium (que contiennent les catalyseurs nickel de Raney) ainsi que celle du promoteur, le molybdène, a diminué de moitié après réaction. De plus, ils ont constaté une diminution de la surface spécifique significative après 7 cycles catalytiques de 60 m²/g à 41 m²/g. L’utilisation d’un solvant éthanol-eau permet néanmoins de limiter la désactivation du catalyseur.23 Les auteurs émettent l’hypothèse que le taux de recouvrement de l’hydrogène à la surface du catalyseur est plus élevé, du fait d’une solubilité plus importante de l'hydrogène dans ce solvant mixte éthanol-eau.
Ceci a été confirmé ultérieurement,24 dans une étude démontrant que le transfert de masse d'hydrogène de la phase gazeuse à la phase liquide, et donc vers la surface du catalyseur, est nettement amélioré par l'utilisation d'un solvant alcoolique que dans l’eau seule.
Face au manque de stabilité des catalyseurs à base de nickel, des recherches ont été menées sur des catalyseurs à base de métaux nobles.
22 J.-P. Mikkola, T. Salmi, Catal. Today, 2001, 64, 271-277.
23 J.-P. Mikkola, H. Vainio, T. Salmi, R. Sjoholm, T. Ollonqvist, J. Vayrynen, Appl. Catal. A: Gen., 2000, 196, 143-155.
II.2.2.b. Catalyseur à base de métaux nobles
Parmi les catalyseurs à base de métaux nobles étudiés pour la réaction d’hydrogénation du xylose en xylitol, il y a les catalyseurs à base de platine. Pour ces catalyseurs, l’influence du support et de l’association avec l’étain a été étudiée.
Tathod et al.25 ont montré que l’association d’un support basique hydrotalcite (HT) à un catalyseur à base de platine déposé sur un support acide Al2O3 permettait d’avoir un effet bénéfique pour l’hydrogénation du xylose. Cette association permet :
a) la stabilisation des particules métalliques à des tailles inférieures (dispersion plus importante).
b) une immobilisation des ions hydrogène (H+ et H−) sur l'ensemble des sites actifs (paire O2- - cations métalliques) et que ces sites soient séparés les uns des autres, engendrant l’impossibilité de la recombinaison des ions hydrogène. Ce « réservoir » d'ions hydrogène pourrait ensuite être disponible pour la réaction d'hydrogénation, ce qui permet d’augmenter l'activité du catalyseur. Ainsi, un rendement de 79% en xylitol a été obtenu avec le système Pt/ -Al2O3 + HT (60 °C, 16 bar, 4 h). δ’étude sur le recyclage de ce système catalytique (Pt/ -Al2O3 + HT) montre que le catalyseur subit une désactivation sur 3 cycles catalytiques, avec une diminution du rendement après chaque cycle constatée (1er cycle : 82 % ; 2nd cycle : 80 % ; 3ème cycle : 74 %). Cette désactivation du catalyseur est due à la lixiviation d’une fraction du platine. Ce même groupe (Tathod et al.)26 a étudié l’effet promoteur de l’étain sur des catalyseurs à base de platine Pt/ -Al2O3 et Pt/C. Une comparaison des performances des différents systèmes étudiés est présentée dans le Tableau 4.
Tableau 4 : Conversion du xylose et rendement en xylitol sur divers catalyseurs à base de Platine. 26
Entrée Catalyseur Conversion
du xylose (%) Rendement en xylitol (%) 1 Pt(2)/ -Al2O3 90 24 2 Pt(2)Sn(0,25)/ -Al2O3 98 64 3 Pt(2)/C 71 15.5 4 Pt(2)Sn(0,25)/C 65 22 5 Pas de catalyseur 47 9.5
Conditions réactionnelles : xylose 0,15 g, catalyseur 0,075 g, eau 35 mL, 190 °C, 16 bar H2, 15 min
25 A. Tathod, T. Kane, E.S. Sanil, P.L. Dhepe, J. Mol. Catal. A: Chem., 2014, 388–389, 90-99. 26 A.P. Tathod, P.L. Dhepe, Bioresource Technol., 2015, 178, 36-44.
Les produits secondaires de cette réaction sont des glycols (éthylène glycol, glycérol et 1,2-propanediol). Dans ces conditions expérimentales, les catalyseurs supportés sur charbon (Pt(2)/C et Pt(2)Sn(0,25)/C) présentent des rendements en xylitol inférieurs à ceux obtenus par les catalyseurs supportés sur -Al2O3 (Tableau 4). Concernant les catalyseurs supportés sur -Al2O3, le rendement en xylitol est augmenté d’environ β,7 fois après l’ajout d’étain (Tableau 4, Entrées 1 et 2). Les auteurs proposent que l’étain qui est présent sous forme ionique forme alors des sites acides de Lewis qui seraient à même de polariser le groupe carbonyle du xylose, favorisant l’activité catalytique lors de la réaction d’hydrogénation. Le rendement en xylitol de 64 % avec le catalyseur Pt(β)Sn(0.βη)/ -Al2O3 peut être améliorée à 85 % en diminuant la température à 130 °C et en augmentant le temps de réaction à 120 minutes.
L’influence de la nature du métal du catalyseur sur la réaction d’hydrogénation du xylose en xylitol a été étudiée par Wisniak et al.27 qui indiquent que l’activité des catalyseurs pour cette réaction d’hydrogénation diminue dans l'ordre : Ru > Ni > Rh > Pd. Un ordre d’activité comparable a également été obtenu par Lee et al.28, qui annoncent que le nombre de moles de substrat converties par site actif du catalyseur et par unité de temps : le TOF (Turn Over Frequency) des catalyseurs décroit selon l’ordre : Ru > Ni ∼ Co > Pt > Rh ∼ Pd.
Par la suite, Lee et al.29 ont à nouveau étudié des catalyseurs à base de palladium, platine ou ruthénium, mais cette fois avec addition de Ni, de Co ou de Fe. Le Tableau 5 présente certains des résultats obtenus.
Dans le cas des catalyseurs à base de palladium et de platine (Tableau 5, Entrées 1 à 8), l’addition d’un co-métal améliore l’activité du catalyseur en comparaison de celle du monométallique. Les catalyseurs à base de ruthénium (Tableau 5, Entrées 9 à 12) présentent des comportements différents puisque les catalyseurs bimétalliques présentent une activité inférieure à celle du catalyseur monométallique. δe catalyseur qui présente l’activité la plus élevée est le catalyseur monométallique au ruthénium (Tableau 5, Entrée 12), avec un TOF de 2476,9 h-1. Ainsi l’utilisation de catalyseurs à base de ruthénium pour la réaction d’hydrogénation du xylose en xylitol a fait l’objet de nombreuses publications.
27 J. Wisniak, M. Hershkowitz, S. Stein, Ind. Eng. Chem. Prod. Res. Dev., 1974, 13, 232-236. 28 J. Lee, Y. Xu, G.W. Huber, Appl. Catal. B: Environ., 2013, 140-141, 98-107.
Tableau 5 : Vitesse de réaction pour l'hydrogénation (en phase aqueuse) du xylose en présence de catalyseurs mono- et bimétalliques supportés sur γ-Al2O3. 29
Entrée Catalyseur TOF (h-1)
1 3wt% Pd 13.8 2 Pd1Ni3 1552.0 3 Pd1Co3 724.7 4 Pd1Fe3 701.3 5 3wt% Pt 51.3 6 Pt1Ni3 872.7 7 Pt1Co3 390.1 8 Pt1Fe3 200.2 9 3wt% Ru 2476.9 10 Ru1Ni1 1255.4 11 Ru1Co1 1735.7 12 Ru1Fe1 899.7 13 20wt% Ni 333.0 14 20wt% Co 196.0 15 20wt% Fe 0
Conditions réactionnelles : 100 °C, 5,41 MPa H2, 5 wt% xylose solutions as the feed
Yadav et al.30 ont étudié l'activité de catalyseurs à base de ruthénium supporté sur divers supports (Tableau 6).
Tableau 6 : Rendement en xylitol sur différents catalyseurs à base de ruthénium (+ Ni de Raney). 30
Entrée Catalyseurs Conversion
du xylose (%) Rendement en xylitol (%) Sélectivité en xylitol (%) 1 Ru(1%)/NiO(5%)-TiO2 > 99 > 99 > 99 2 Ru(1%)/TiO2 97 96 99 3 Ru(1%)/C 97 94 98 4 Raney Ni 97 94 97
Conditions réactionnelles : 120 °C, 5,5 MPa H2, 2 h, solution 20 wt% xylose
Un catalyseur Ru/C présente une activité très proche de celle d’un catalyseur nickel de Raney (Tableau 6, Entrées 3 et 4). Cette étude indique également qu’un catalyseur à base de ruthénium supporté sur TiO2 est un peu plus actif et sélectif que lorsqu’il est supporté sur charbon. La modification du support TiO2 par NiO (Tableau 6, Entrée 1) permet d’augmenter
l’activité du catalyseur ainsi que la sélectivité en xylitol. δ’ordre d’activité issu de cette étude est alors : Ru/NiO(5%)-TiO2 > Ru/TiO2 > Ru/C > Raney Ni. Les auteurs ayant réalisé cette étude indiquent que le catalyseur modifié Ru/NiO-TiO2 subit toutefois une lixiviation importante du nickel, avec près de 11,0 mg/L de nickel en solution en fin de cycle.
Hernandez-Mejia et al.31 ont également étudié des catalyseurs à base de ruthénium supporté sur oxyde de titane TiO2. Ces derniers ont mis en évidence une influence de la structure cristalline du support TiO2 sur l’activité du catalyseur lors de l’hydrogénation du xylose en xylitol comme le montre la Figure 9.
Structure cristalline du support TiO2 A Anatase B Anatase C Rutile D Anatase et rutile
Figure 9 : Rendement en xylitol en fonction de la structure cristalline du support et de la température. 31 Lorsque le catalyseur à base de ruthénium est supporté sur du TiO2 de structure anatase, les rendements en xylitol obtenus restent inférieurs à 75 %. Cependant, lorsque le TiO2 utilisé est de structure rutile, des rendements de 98 % sont obtenus, dès 15 minutes de réaction à 120 °C. Des résultats comparables ont été obtenus avec un support composé des deux polymorphes de TiO2. Les catalyseurs supportés sur TiO2 de structure anatase possèdent des particules de ruthénium d’une taille plus importante que ceux supportés sur rutile. Les auteurs expliquent cette observation par le fait que RuO2 possède également une structure de type rutile, ce qui empêcherait ou limiterait la mobilité des particules de ruthénium à la surface du support de structure rutile au cours des traitements thermiques. Le catalyseur déposé sur TiO2 rutile apparait assez stable, des résultats similaires à ceux obtenus lors du 1er cycle catalytique ont été obtenus après 4cycles de réaction, sans traitement intermédiaire de reconditionnement du catalyseur.
Mishra et al.32 ont étudié les propriétés du ruthénium lorsque supporté sur zéolite Y, et déterminé l’effet de l’acidité du support sur les propriétés du catalyseur. Une augmentation du
31 C. Hernandez-Mejia, E.S. Gnanakumar, A. Olivos-Suarez, J. Gascon, H.F. Greer, W. Zhou, G. Rothenberg, N. Raveendran Shiju, Catal. Sci. Technol., 2016, 6, 577-582.
rapport Si/Al de 5,1 à 80 engendre une diminution progressive du nombre et de la force des sites acides de Brønsted et des sites acides de δewis. δ’influence de l’acidité sur les activités et sélectivités en xylitol est présentée dans le Tableau 7. Une augmentation du rapport Si/Al favorise l'hydrogénation sélective du xylose en xylitol, la sélectivité en xylitol allant de 68 % pour un ratio Si/Al de 5,1 à 98 % pour un ratio de 80. δa formation d’arabitol est, elle, expliquée par une réaction d’isomérisation/hydrogénation du xylose, sur les catalyseurs présentant une acidité plus élevée (Figure 8).
Tableau 7 : Effet du rapport Si/Al de la zéolite Y sur l'hydrogénation du xylose catalysée par Ru/HYZ. 32
Entrée Catalyseur Ratio Si/Al Conversion du xylose (%) Sélectivité en xylitol (%) Sélectivité en arabitol (%) 1 Ru/HYZ-5,1 5,1 46 68 22 2 Ru/HYZ-30 30 52 86 11 3 Ru/HYZ-60 60 58 94 4 4 Ru/HYZ-80 80 62 98 2
Conditions : xylose = 20 g, catalyseur (Ru/HYZ) = 0,5 g, eau = 100 mL, 120 °C, 5,5 MPa H2, 1 h
La recyclabilité de ce catalyseur ruthénium supporté sur zéolite Y a été évaluée pour 5 cycles catalytiques successifs. Une légère baisse d’activité a été constatée au cours de ces 5 cycles, de 7 % pour la conversion du xylose et de 6 % pour la sélectivité en xylitol. Les auteurs ne donnent aucune information ou hypothèse sur l’origine de cette légère diminution d’activité.
II.3. Conclusion
En résumé, il a été montré que le métal le plus actif pour l’hydrogénation catalytique du xylose en xylitol est le ruthénium. Toutefois, le recyclage de ces catalyseurs à base de ruthénium reste problématique. De plus, il serait intéressant de proposer des alternatives à ce métal coûteux. Le nickel, qui est présenté comme le second métal le plus actif pour cette réaction d’hydrogénation du xylose, présente également l’inconvénient de se désactiver rapidement, du fait de la lixiviation d’une fraction du nickel. Lee et al.28 montrent néanmoins que l’activité du cobalt pour cette réaction est similaire à celle du nickel. Toutefois, les références traitant de l’hydrogénation du xylose en xylitol en présence de catalyseurs à base de métaux non noble autre que le nickel reste rares, et leur stabilité en réaction inconnue.
III. Hydrogénation du furfural
L'hydrolyse de polysaccharides en C5 présents dans la fraction hémicellulose de la biomasse lignocellulosique permet d’obtenir des sucres (pentoses et hexoses) dont le xylose est généralement majoritaire. Ce xylose peut ensuite être déshydraté en furfural (Figure 10). Dans les années 2000, plus de 280 kT/an de furfural étaient produits chaque année. Le plus gros producteur est la Chine (200 kT/an), suivie par la République Dominicaine (32 kT/an) et l'Afrique du Sud (20 kT/an).33
Figure 10 : Schéma simplifié de la conversion du xylan en furfural.
Le premier procédé commercial pour la production de furfural a été mis en service en 1921 par Quaker Oats.34 Ce procédé permet d’obtenir un rendement en furfural de 40 à 50 % calculé à partir des hémicelluloses, et repose sur l’utilisation de l'acide sulfurique aqueux (H2SO4) à 170-185 °C. La première étape de ce procédé consiste à hydrolyser les hémicelluloses afin d’obtenir le xylose, qui, en perdant ensuite trois molécules d’eau, forme le furfural. Les rendements relativement faibles en furfural résultent de réactions secondaires. Celles-ci comprennent la formation d’humines par condensation du furfural ou encore la résinification du furfural. De plus, l’utilisation d’acides minéraux pour cette production est problématique en raison des problèmes de corrosion et environnementaux (traitement des déchets). Dans l’optique d’améliorer les rendements en furfural, des systèmes biphasiques H2O-solvants organiques ont été étudiés afin de limiter la dégradation du furfural au cours de sa production.35 Ce principe repose sur l’affinité plus élevée du furfural pour la phase organique, où les réactions de dégradation sont le plus souvent inhibées en raison de l'absence du catalyseur. La conversion catalytique des pentoses en furfural sur des catalyseurs solides a été largement étudiée au cours des dernières décennies. Une grande variété de matériaux (zéolithes, résines échangeuses d'ions, silices mésoporeuses modifiés par des groupements
33 SRI Consulting, Furfural. Chemical Economics Handbook.2011
34 K.J. Zeitsch, "The chemistry and technology of furfural and its many by-products", Vol. 13, 2000, Elsevier. 35 J.N. Chheda, Y. Roman-Leshkov, J.A. Dumesic, Green Chem., 2007, 9, 342-350.
acide sulfonique, oxydes métalliques sulfonés, etc.)36 ont été proposés pour cette transformation.
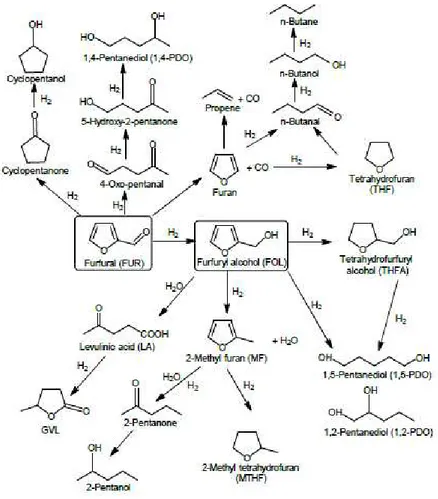
Récemment, Bozell et al.37 ont identifié le furfural comme l’un des composés chimiques les plus prometteurs pour une production durable de carburant et de produits chimiques. En effet, le nombre de transformations possibles du furfural en produits chimiques et en carburant est colossal. Une des réactions possibles est l’hydrogénation du furfural, la Figure 11 suivante donne un aperçu complet de tous les composés pouvant être obtenus à partir de l'hydrogénation du furfural.
Figure 11 : Produits pouvant être formés au cours de l'hydrogénation du furfural.
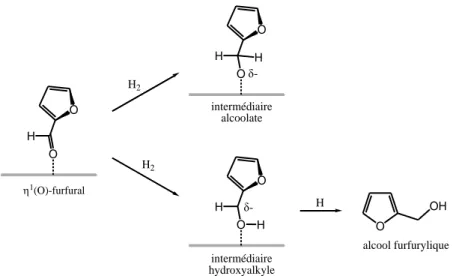
Ces composés peuvent être : l’alcool furfurylique, l’alcool tétrahydrofurfurylique (THFA) produit suite à l’hydrogénation totale du furfural, ou le 2-méthylfurane (2-MF) produit d’hydrogénolyse pouvant être ultérieurement hydrogéné en 2-méthyltétrahydrofurane, ou encore le furane à la suite de la décarboxylation du furfural.
36 R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sadaba, M. Lopez Granados, Energy Environ. Sci., 2016, 9, 1144-1189.
La sélectivité envers le produit désiré lors de cette réaction d’hydrogénation dépend très fortement des conditions réactionnelles appliquées ainsi que de la nature du catalyseur utilisé.
Dans ce travail, la littérature concernant la production de deux composés issus de l’hydrogénation du furfural est reportée. Ces derniers sont l’alcool furfurylique (obtenu par hydrogénation sélective de la liaison −C=O) et le β-méthylfurane (produit d’hydrogénolyse du furfural).
III.1. Production de l’alcool furfurylique III.1.1. δ’alcool furfurylique
La Figure 12 présente la molécule de l’alcool furfurylique de formule C5H6O2.
Figure 12 : Molécule d'alcool furfurylique.
δ’alcool furfurylique est un composé qui trouve de nombreuses applications en tant que solvant ou pour la production de résines, de fibres synthétiques, d’adhésifs, de produits de chimie fine (tel que la vitamine C, la lysine, …), ou de produits chimiques agricoles. Toutefois, il trouve sa principale application dans la fabrication des résines de fonderie.38
Pour cette raison, l’alcool furfurylique est le dérivé le plus important du furfural.34 Il a été estimé que ~ 62 % du furfural produit dans le monde chaque année sont convertis en alcool furfurylique.39
La production de l'alcool furfurylique est effectuée par hydrogénation catalytique du furfural, qui peut être réalisée en phase gazeuse ou en phase liquide. De nombreux catalyseurs ont donc été étudiés et/ou développés au cours de ces dernières décennies pour la production sélective d’alcool furfurylique.
III.1.2. Hydrogénation en phase gazeuse
δ’hydrogénation du furfural en alcool furfurylique en phase gazeuse avec un catalyseur à base de cuivre a été rapportée pour la première fois en 1927.40 En 1931, la société Du Pont
38 J.B. Barr, S.B. Wallon, J. Appl. Polym. Sci., 1971, 15, 1079-1090.
39 K. Yan, G. Wu, T. Lafleur, C. Jarvis, Renew. Sust. Energ. Rev., 2014, 38, 663-676. 40 R. Eloi, G.H. Martin, patent US1739919. 1929.
de Nemours a breveté un procédé d’hydrogénation du furfural pour la production d’alcool furfurylique en phase gazeuse avec l'utilisation d’un catalyseur de type chromite de cuivre.41
Aujourd’hui le procédé industriel d’hydrogénation du furfural en phase gazeuse est réalisé en vaporisant le furfural dans un flux d’hydrogène à 1β0 °C qui est ensuite surchauffé avant d’être introduit dans un réacteur multitubulaire chauffé par bain d’huile. Après passage à travers le réacteur, la conversion du furfural dépasse 99 % et l'alcool furfurylique brut est condensé avant d’être distillé dans une colonne sous vide afin d’obtenir une pureté de plus de 99 %.
III.1.2.a. Catalyseurs chromite de cuivre
δes premiers catalyseurs utilisés pour l’hydrogénation du furfural étaient des catalyseurs chromite de cuivre,57 cependant ces derniers montrent une désactivation significative.
Dans une étude menée afin d’expliquer la désactivation des catalyseurs chromite de cuivre,42 plusieurs phénomènes ont été proposées. Ces derniers sont :
- la formation de coke,
- l'empoisonnement du catalyseur par adsorption du furfural ou des autres produits de la réaction,
- le frittage des particules de cuivre au cours du procédé catalytique, - la lixiviation des espèces de cuivre,
- un changement de l'état d'oxydation du cuivre.
- le recouvrement des sites Cu par des espèces Cr provenant de la décomposition partielle du chromite de cuivre.
Une des causes principales de la désactivation des catalyseurs chromite de cuivre utilisés pour l’hydrogénation catalytique du furfural en phase gazeuse semble être un empoisonnement des sites actifs dû à une forte adsorption d’espèces formées par la polymérisation du réactif et/ou des produits à la surface du catalyseur, comme le montre les résultats du Tableau 8.
41 W.A. Lazier, patent US2077422. 1937.
42 D. Liu, D. Zemlyanov, T. Wu, R.J. Lobo-Lapidus, J.A. Dumesic, J.T. Miller, C.L. Marshall, J. Catal., 2013, 299, 336-345.
Tableau 8 : Analyses XPS de diverses espèces identifiées sur la surface des catalyseurs frais et utilisés. 42
Espèces Energie de
liaison (eV)
Composition atomique (%) Catalyseur frais Catalyseur
utilisé
C−C 284,8 90 55
C−OH ou C−O−C 286,1–286,3 4 24
C=O 287,8 – 14
O−C=O 288,6–288,7 6 7
Des analyses XPS ont mis en évidence la présence d’espèce carbonyle, alcool ou éther à la surface du catalyseur après réaction, espèces initialement absentes.
Les auteurs indiquent également qu’à plus haute température ( > 200 °C), le rapport Cr/Cu augmente de 50 %, ce qui suggère qu’un recouvrement des sites de cuivre par le chrome devient une cause supplémentaire de désactivation du catalyseur. Ces derniers indiquent également que la perte des sites Cu(I) au cours de l'hydrogénation n’est pas une cause de désactivation, par opposition à ce qui avait été rapporté précédemment dans la littérature.43,44 Toutefois, des études plus récentes contredisent cette interprétation.45,47
Il a été mis en évidence qu’à la surface d’un catalyseur à base de cuivre déposé sur SBA-15, la présence de dépôt carboné est moindre que dans le cas d’un catalyseur chromite de cuivre sans que toutefois les auteurs en donnent l’explication.45 Ce catalyseur est légèrement moins sélectif que les catalyseurs chromite de cuivre, avec un rendement maximal en alcool furfurylique de 85 % pour une conversion du furfural de 92 %.
Avec comme objectif de stabiliser les catalyseurs chromite de cuivre, il a été démontré que le dépôt d'une couche atomique d'alumine (ou d’oxyde de titane) s’avérait efficace.46,47 Le dépôt de ces couches atomiques d'alumine (ou d’oxyde de titane) entraîne l'encapsulation des particules de cuivre par une surcouche d’Al2O3 ou de TiO2. Une calcination à haute température, conduit alors à une microporosité dans cette surcouche, exposant ainsi le cuivre
43 R.S. Rao, R.T.K. Baker, M.A. Vannice, Catal. Lett., 1999, 60, 51-57.
44 B.M. Nagaraja, A.H. Padmasri, B.D. Raju, K.S.R. Rao, J. Mol. Catal. A: Chem., 2007, 265, 90-97.
45 D. Vargas-Hernandez, J.M. Rubio-Caballero, J. Santamaria-Gonzalez, R. Moreno-Tost, J.M. Merida-Robles, M.A. Perez-Cruz, A. Jimenez-Lopez, R. Hernandez-Huesca, P. Maireles-Torres, J. Mol. Catal. A: Chem., 2014, 383-384, 106-113.
46 H. Zhang, Y. Lei, A.J. Kropf, G. Zhang, J.W. Elam, J.T. Miller, F. Sollberger, F. Ribeiro, M.C. Akatay, E.A. Stach, J.A. Dumesic, C.L. Marshall, J. Catal., 2014, 317, 284-292.
47 H. Zhang, C. Canlas, A. Jeremy Kropf, J.W. Elam, J.A. Dumesic, C.L. Marshall, J. Catal., 2015, 326, 172-181.
en dessous, tout en empêchant le frittage des particules de cuivre ainsi que le recouvrement des particules de cuivre par des espèces chromite.
Depuis lors, de nombreux autres systèmes catalytiques ont été proposés pour cette transformation, notamment pour limiter les problèmes environnementaux liés à la présence du chrome dans les catalyseurs chromite de cuivre. Il en ressort qu’une très large majorité des catalyseurs utilisés dans la littérature pour l’hydrogénation du furfural en alcool furfurylique en phase gazeuse sont à base de cuivre (associé ou non à d’autres métaux). Il est à noter que quelques autres études ont étudiés des catalyseurs à base de palladium ou de nickel (seul ou en association avec un autre métal).
III.1.2.b. Catalyseurs à base de cuivre
Une étude menée sur des catalyseurs à base de cuivre monométalliques a montré que l’utilisation de la silice pour l’hydrogénation du furfural en phase gazeuse est préférable à l’utilisation d’autres supports comme l’alumine ou l’oxyde de zinc (Cu/SiO2 > Cu/Al2O3 > Cu/ZnO).48 Il semblerait que le support SiO2 puisse interagir avec les espèces de cuivre ce qui permettrait de disperser uniformément les particules de cuivre. En présence de ce catalyseur Cu/SiO2, l'alcool furfurylique est le principal produit lors de l’hydrogénation du furfural en phase gazeuse à basse température (73 % d’alcool furfurylique à 140 °C). Cependant, la sélectivité en 2-méthylfurane augmente fortement avec l'augmentation de la température de réaction (de 140 à 220 °C), la formation de pentanone et de pentanol étant également observée. La diminution de la sélectivité à haute température étant liée à la formation de produits secondaires, la température de réaction est une condition importante permettant d’améliorer la sélectivité en alcool furfurylique. Les auteurs ne donnent pas d’information sur la stabilité de ce catalyseur Cu/SiO2. Cependant ils indiquent que ce dernier ne montre pas de désactivation à une température de 220 °C (pour une application à la production de 2-méthylfurane).
Nagaraja et al.49 ont, quant à eux, étudié un catalyseur Cu/MgO. Ces derniers ont également constaté une baisse de la sélectivité en alcool furfurylique avec l'augmentation de la température au-dessus de 180 °C. La raison de cette baisse de sélectivité en alcool furfurylique s’explique par la formation de produits secondaires, à savoir : du 2-méthylfurane,
48 F. Dong, Y. Zhu, H. Zheng, Y. Zhu, X. Li, Y. Li, J. Mol. Catal. A: Chem., 2015, 398, 140-148. 49 B.M. Nagaraja, A.H. Padmasri, B.D. Raju, K.S.R. Rao, J. Mol. Catal. A: Chem., 2007, 265, 90-97.
de l’alcool tétrahydrofurfurylique, du tétrahydrofurane. Aucune indication sur la stabilité de ce catalyseur n’est donnée dans cette étude.
δ’effet bénéfique de l’addition d’un co-métal à des catalyseurs à base de cuivre a fait l’objet de quelques autres travaux.
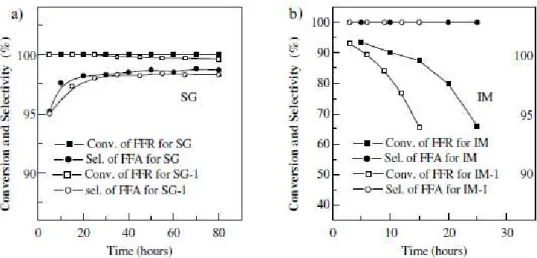
δa performance catalytique la plus élevée pour l’hydrogénation en phase gazeuse jusqu’à aujourd’hui a été obtenue avec un catalyseur CuCa/SiO2.50 La méthode de préparation du catalyseur semble avoir un effet important. En effet, un catalyseur préparé par voie sol-gel (SG) présente une plus grande stabilité au cours du temps qu’un catalyseur préparé par imprégnation (IM). Ainsi La Figure 13 montre que le catalyseur préparé par voie sol-gel (SG) ne subit pas ou peu de perte d’activité après 80 heures de réaction tandis que dans le cas du catalyseur préparé par imprégnation (Iε) l’activité chute d’environ γ0 % après 25 heures. Les auteurs indiquent que les particules sont plus petites pour le catalyseur préparé par voie sol-gel, menant à une plus grande dispersion du cuivre à la surface du catalyseur.
Figure 13 : Performance des catalyseurs pour l'hydrogénation du furfural en alcool furfurylique. 50 Le calcium est habituellement utilisé pour améliorer la stabilité des catalyseurs par son rôle de promoteur structurel.50 Ce dernier permet d’améliorer la dispersion du cuivre et limiterait le frittage des particules de cuivre, ce qui améliore la stabilité du catalyseur. Toutefois, comme le montre la Figure 13, l’effet de l’addition de calcium est moins marqué dans le cas du catalyseur préparé par la méthode sol-gel (SG : avec Ca ; SG-1 : sans Ca) que dans le cas du catalyseur préparé par imprégnation (IM : avec Ca ; IM-1 : sans Ca). En effet, lors de la réaction avec le catalyseur préparé par la méthode sol-gel, la conversion du furfural est totale même dans le cas du catalyseur sans calcium promoteur et la
sélectivité en alcool furfurylique de 98 % n’est pas significativement améliorée par le calcium promoteur.
Dans leur étude sur des catalyseurs bimétalliques à base de métaux de transition supportés sur silice, Reddy et al.51 indiquent que l'ordre d'activité des catalyseurs pour la réaction d’hydrogénation du furfural en alcool furfurylique en phase gazeuse est la suivante : Cu-Co/SiO2 > Ni-Cu/SiO2 > Co-Ni/SiO2 (Figure 14).
Figure 14 : Activité et sélectivité de divers catalyseurs bimétallique pour l'hydrogénation du furfural en alcool furfurylique. 51
Quelle que soit la température de réaction utilisée, la sélectivité du catalyseur Cu-Co/SiO2 est supérieure à celle des deux autres systèmes catalytiques. D’après eux, l’activité plus élevée observée pour le catalyseur Cu-Co/SiO2 (environ 85 % de rendement en alcool furfurylique à 225 °C) semble être due à une plus grande surface spécifique.
Un autre système bimétallique a été étudié pour l’hydrogénation du furfural en phase gazeuse, il s’agit d’un catalyseur Pd-Cu supporté sur zéolithe Y.52 Les auteurs expliquent le choix du palladium par la capacité de ce dernier à activer l’hydrogène. Toutefois, un rendement en alcool furfurylique de seulement 58 % a été obtenu avec ce catalyseur Pd-Cu/Y. Les auteurs indiquent que la désactivation de ce catalyseur semble être due à la réduction des espèces d’oxyde de cuivre en cuivre métallique à hautes températures ( > 300 °C), ainsi qu’à la formation de coke à la surface du catalyseur pour des températures plus faibles ( < 200 °C). Après un traitement thermique sous air, l’activité du catalyseur est revenue à 95 % de son activité initiale.
51 B.M. Reddy, G.K. Reddy, K.N. Rao, A. Khan, I. Ganesh, J. Mol. Catal. A: Chem., 2007, 265, 276-282. 52 G. Seo, H. Chon, J. Catal., 1981, 67, 424-429.
III.1.2.c. Catalyseurs à base de métaux du groupe 10
D’autres métaux sont connus pour leur activité dans les réactions d’hydrogénation. Ainsi dans le cas de l’hydrogénation du furfural en alcool furfurylique, des catalyseurs à base de platine, de palladium ou encore de nickel ont également été étudiés.
Divers catalyseurs à base de platine déposé sur des supports SiO2 préalablement recouverts d’une ou deux monocouches d’un oxyde de métal de transition ont été étudiés. Parmi ces derniers, le catalyseur Pt/TiO2-V2O5-SiO2 semble être le plus sélectif (avec un rendement de 79 % pour une conversion du furfural de 87 %).53 La raison de cette haute sélectivité serait qu’une polarisation plus importante de la surface du catalyseur se produit si la différence entre l'électronégativité du platine et celle du métal de transition atteint un maximum. En effet, les systèmes polaires adsorbent plus facilement et activent le groupe carbonyle polaire.
Pour les catalyseurs à base de platine, la taille des particules métalliques semble être un paramètre important à prendre en compte pour la sélectivité de la réaction. Les nanoparticules de platine possèdent deux sites actifs différents, dont la proportion varie en fonction de la taille et de la forme des nanoparticules, pour induire soit une décarbonylation en furane, soit une hydrogénation du groupe carboxyle.54,55
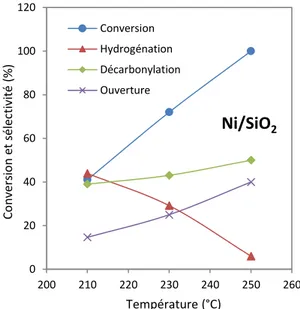
La Figure 15 montre que pour des nanoparticules de platine ayant une taille inférieure à 2 nm, le produit majoritaire de la réaction est du furane avec une sélectivité supérieure à 90 %. Pour des particules dont la taille est supérieure à 3,6 nm la production d’alcool furfurylique est favorisée par rapport à la production de furane. La sélectivité en alcool furfurylique augmente alors jusqu’à θ0-65 % pour des tailles de particules supérieures ou égales à 5 nm.
53 J. Kijenski, P. Winiarek, T. Paryjczak, A. Lewicki, A. Mikolajska, Appl. Catal. A: Gen., 2002, 233, 171-182. 54 V.V. Pushkarev, N. Musselwhite, K. An, S. Alayoglu, G.A. Somorjai, Nano Lett., 2012, 12, 5196-5201. 55 K. An, N. Musselwhite, G. Kennedy, V.V. Pushkarev, L. Robert Baker, G.A. Somorjai, J. Colloid Interface Sci., 2013, 392, 122-128.