Etude de l'hydroaminométhylation asymétrique des alcènes et identification d'espèces impliquées dans la catalyse
Texte intégral
Figure
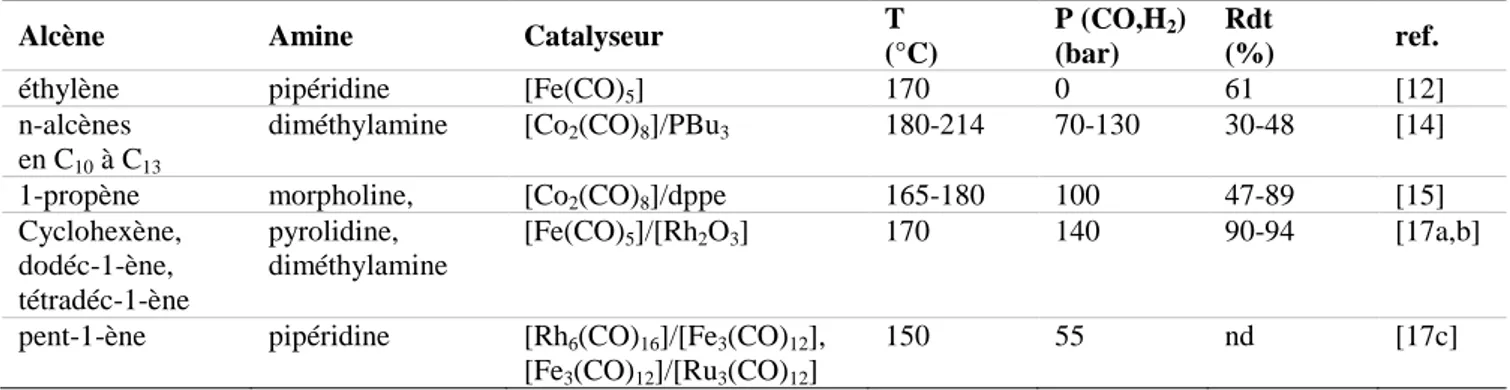
![Tableau II. 5. Influence du solvant dans l’HAM du pent-1-ène avec la pipéridine, adapté de la référence [45]](https://thumb-eu.123doks.com/thumbv2/123doknet/3655102.107922/78.892.86.779.821.983/tableau-influence-solvant-ham-pent-pipéridine-adapté-référence.webp)
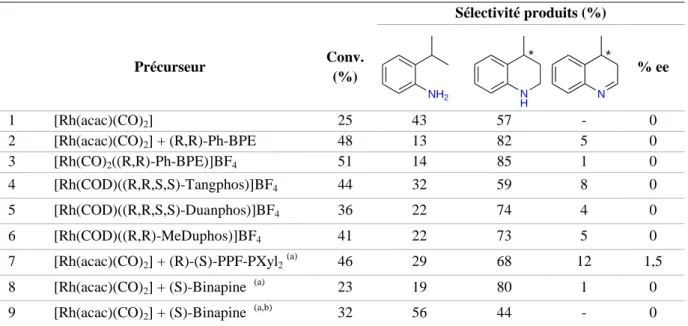
Outline
Documents relatifs
Lors de cette étude en une dimension, nous nous sommes concentrées sur des caractéristiques physiques d’une éruption volcanique, tels que le rayon des aérosols et la
Cette étude a été réalisée sur 12 ovins abattus dans un abattoir de la région d’Alger et ayant présenté des abcès .Les prélèvements de pus ont été analysés au niveau
La présente étude est dès lors centrée sur les problèmes de mammites au sein de la station sahélienne expérimentale de Toukounous afin d’identifier : (i) les
Visualiser un modèle MIMOSA consistera dès lors en la mise place de certaines relations entre composants, composé, relations du modèle et ceux de visualisation (exemple :
La présente étude réalisée sur le thème « Elevage des bovins dans la Commune de Kalalé et changement climatique : perception des éleveurs et approches de solutions »
La difficulté de mise en œuvre de l’obligation d’information lors de la procédure de data-room ŕ Dès lors quřelle nřa pas fait lřobjet dřune contractualisation dans
Une étude clinique a été réalisée au Sénégal et au Soudan dans le but de comparer une solution de la pulpe du fruit du baobab avec une solution de
Aucun effet sur la sécrétion n’a été observé avec la déplétion de Arf4 et Arf5 validant ainsi l’hypothèse que GBF1 possède 2 fonctions indépendantes régulées par
