Characterizing and engineering antibodies against
the epidermal growth factor receptor
by Ginger Chao
B.S. Chemical Engineering Rice University, 2002
Submitted to the Department of Chemical Engineering in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY IN CHEMICAL ENGINEERING
at the
Massachusetts Institute of Technology February 2008
© 2007 Massachusetts Institute of Technology All rights reserved
Signature of Author:_____________________________________________________________ Department of Chemical Engineering October 15, 2007 Certified by: ___________________________________________________________________ K. Dane Wittrup C.P. Dubbs Professor of Chemical Engineering and Bioengineering Thesis Supervisor Accepted by:___________________________________________________________________ William M. Deen Professor of Chemical Engineering
Characterizing and engineering antibodies against the epidermal
growth factor receptor
by Ginger Chao
Submitted to the Department of Chemical Engineering on October 15, 2007 in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemical Engineering
ABSTRACT
Epidermal growth factor receptor (EGFR) signaling leads to cellular proliferation and migration, and thus EGFR dysregulation can significantly contribute to the survival of tumor cells. Aberrant EGFR signaling due to receptor overexpression, mutation, or autocrine ligand has been observed in a wide variety of malignancies, and antibody drugs which inhibit EGFR signaling have been developed. However, the epitopes of most EGFR antibodies have not been characterized, and the marginal efficacies of current antibodies underscore the need for improved therapeutics.
In this thesis work, we have created a novel method of epitope mapping, which is the determination of antigen residues responsible for mediating an antibody-antigen interaction. In our technique, a library of random mutants of the EGFR antigen is displayed on the surface of yeast, and the library is combinatorially selected for loss of binding to the antibody being mapped. If a mutant shows loss of binding to an antibody, then that residue is a potential contact residue. In addition, we found that many mutants caused a global misfolding of the antigen, requiring the use of high-throughput sorting to remove the misfolded mutants. The development of our epitope mapping method using random mutagenesis and yeast surface display enabled the successful mapping of four different antibodies and three designed ankyrin repeat proteins binding to EGFR.
In addition, we continued work on engineering novel antibodies against EGFR domains II and IV. Antibodies against these domains are hypothesized to directly inhibit receptor dimerization and subsequent activation, as opposed to traditional anti-EGFR antibodies which block ligand binding. To accomplish this, peptide mimics of EGFR loops were used as antigens; however, antibodies generated using both yeast surface display and rabbit monoclonal technology were peptide-specific, but did not bind to EGFR protein. Finally, we developed a mathematical model to describe equilibrium EGFR ligand binding and dimerization. Based on observations that polyclonal antibodies against EGFR domain II or IV eliminate high affinity EGF binding normally observed on the surface of cells, we hypothesized that preformed inactive dimers were the high affinity component. Our model incorporating this hypothesis successfully reproduced experimental data, resulting in characteristic concave-up Scatchard plots.
Thesis Supervisor: K. Dane Wittrup
Dedicated to my parents, Jim and Grace, and my brother Eric. Thanks for your love and support.
Acknowledgements
I would like to thank my research advisor Dane Wittrup, who has been an ideal thesis advisor these four years. He was always there with great advice and guidance while allowing me the perfect amount of independence and autonomy. I am grateful for the way which he helped my project evolve into several successful side projects and for his contributions to the development of my abilities as a researcher. I also want to thank my thesis committee, Doug Lauffenburger and Harvey Lodish, who were always willing to take time out of their extremely busy schedules to offer great ideas and their unique perspectives.
Dane has created a great research environment, and I would like to thank everyone I’ve worked with in the Wittrup lab. In particular, Jennifer Cochran originally trained me during my first summer, getting me started with my project and providing invaluable assistance and ideas. Other people who had been working on the EGFR project before I joined were extremely helpful: Yong-Sung Kim, Mark Olsen, and Alejandro Wolf-Yadlin. I learned a lot from the people who were in the lab before me: Katarina Midelfort, Jeffrey Swers, Bala Rao, Andy Yeung, Dave Colby, Shaun Lippow, and Stefan Zajic. Of course, I also want to thank Andy Rakestraw for his assistance in the lab and moral support outside the lab. Thanks to Steve Sazinsky, for his insightful discussions and very detailed protocols; Ben Hackel, who has helped me learn about library design and the intricacies of Microsoft Word; and Shanshan Howland, a great benchmate and inventor of many ingenious lab tricks. Thank you to Dasa Lipovsek, who provided me with excellent advice and got me a job! I also want to thank everyone else who has joined the lab over the years: Wai Lau, Andrea Piatesi, Greg Thurber, Annie Gai, Mike Schmidt, Margie Ackerman, Pankaj Karande, Eileen Higham, Kelly Davis, David Liu, and the new kids Jamie Spangler, Jordi Mata-Fink, and Chris Pirie. Thanks everyone for letting me borrow reagents, answering questions, and providing insight at group meetings!
I would like to thank my UROPs Heather Pressler and Nick Nguyen. Also thanks to everyone at the flow cytometry core facility: Glen Paradis, Mike Doire, Mike Jennings, Yong Xie, and Michelle Perry. I am grateful to the various people with whom I have had the opportunity to collaborate: Arvind Sivasubramanian and Jeff Gray at Johns Hopkins, Jeff Way at EMD Lexigen, and Daniel Steiner and Andreas Plueckthun at the University of Zurich.
Thank you Amy Lewis, Mark Styczynski, and Jared Johnson for being great friends and helping me make it through my first semester at MIT. Thanks to my parents, Jim and Grace Chao, and my brother Eric for being supportive through all these years.
My research was supported by a National Defense Science and Engineering Graduate Fellowship, NIH grant CA96504, and a David Koch Graduate Fellowship from the MIT Center for Cancer Research.
Table of Contents
Chapter 1: Introduction and background ... 7
1.1 EGFR signaling and trafficking ... 7
1.2 Biochemical and structural characterization of EGFR ... 11
1.3 EGFR in cancer... 15
1.4 Current EGFR-targeted therapeutics ... 18
1.5 Overview of thesis work... 25
1.6 Works cited... 27
Chapter 2: Epitope mapping of proteins binding to EGFR ... 36
2.1 Background... 36
2.2 Materials and methods ... 39
2.3 Results... 42
2.3.1 Creation of epitope mapping library... 42
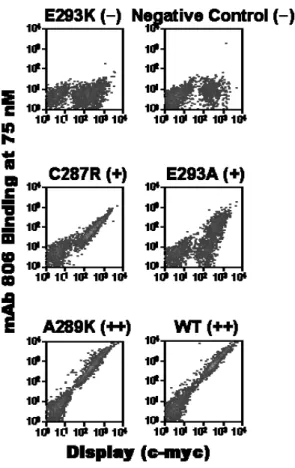
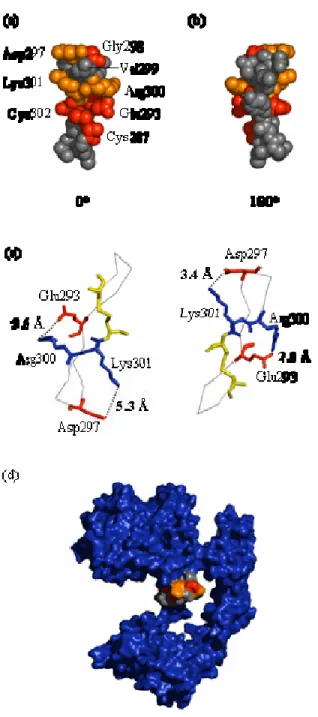
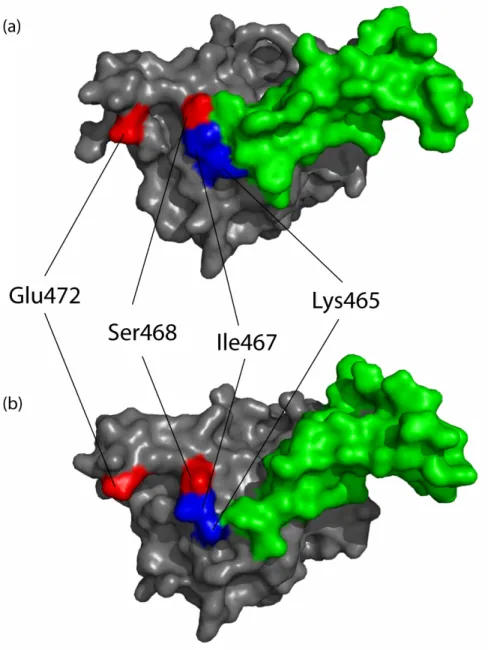
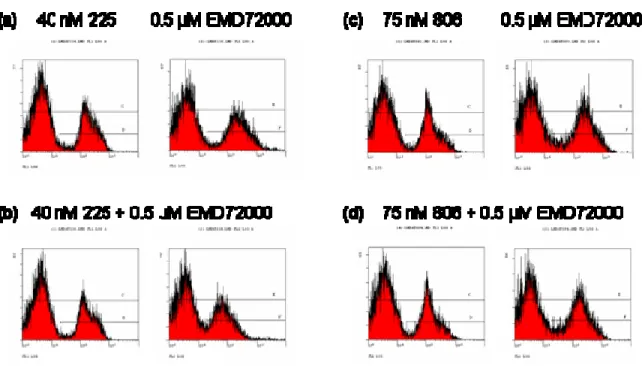
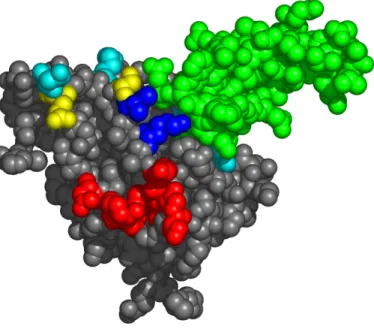
2.3.2 Epitope mapping of mAb 806... 44
2.3.3 Effects of EGFR mutations on binding of mAbs 806 and 175... 52
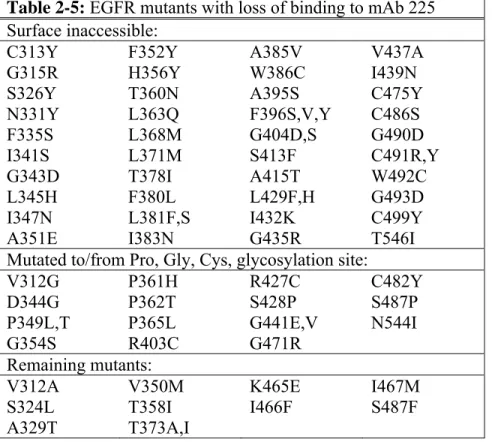
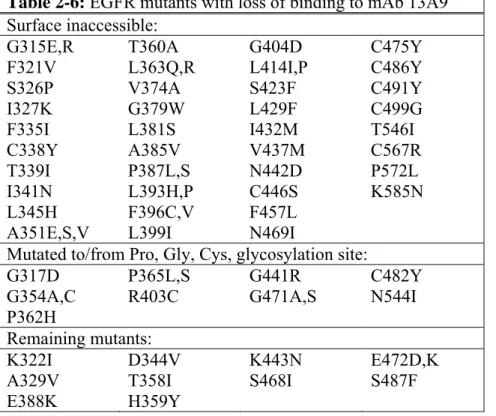
2.3.4 Epitope mapping of mAbs 225 and 13A9 ... 54
2.3.5 Epitope mapping of mAb EMD72000... 63
2.3.6 Epitope mapping of DARPins ... 68
2.4 Discussion ... 75
2.5 Works cited ... 80
Chapter 3: Engineering antibodies against EGFR ... 84
3.1 Background... 84
3.2 Materials and methods ... 94
3.3 Results ... 100
3.3.1 Characterization of antibody clones against EGFR peptides ... 100
3.3.2 Initial round of selection for scFvs against peptides ... 103
3.3.3 Second round of affinity maturation ... 107
3.3.4 Third round of affinity maturation... 110
3.3.5 Fab display libraries... 112
3.3.6 Rabbit monoclonal antibodies... 115
3.4 Discussion . ... 118 .
3.5 Works cited ... 120
Chapter 4: Mathematical modeling of high affinity EGF binding... 123
4.1 Background ... 123
4.2 Results... 126
4.2.1 Model formulation and output... 126
4.2.2 Model characterization ... 134 4.3 Discussion... 137 4.4 Works cited... 142 Appendix 1: Sequences ... 145 Appendix 2: Protocol... 153 Curriculum vitae... 154
Abbreviations Used
ADCC Antibody-dependent cellular cytotoxicity
APC Allophycocyanin
CDR Complementarity determining region DARPin Designed ankyrin repeat protein EGF Epidermal growth factor
EGFR Epidermal growth factor receptor
Fab Fragment antigen binding portion of an antibody FACS Fluorescence-activated cell sorting
FITC Fluorescein isothiocyanate
IgG Immunoglobulin G
Kd Apparent dissociation constant
koff Kinetic off-rate
kon Kinetic on-rate
mAb Monoclonal antibody MACS Magnetic cell sorting pAb Polyclonal antibody PBS Phosphate-buffered saline PCR Polymerase chain reaction
PE Phycoerythrin
scFv Single-chain variable fragment of an antibody TGF-α Transforming growth factor α
TKI Tyrosine kinase inhibitor
VH Variable heavy chain of an antibody
Chapter 1: Introduction and background
1.1 EGFR signaling and trafficking
The epidermal growth factor receptor (EGFR/Her1/ErbB1) is a type I transmembrane receptor tyrosine kinase and a member of the ErbB receptor family. This family includes three other members: Her2 (ErbB2/Neu), Her3 (ErbB3), and Her4 (ErbB4). The receptors are activated by a number of different ligands, such as epidermal growth factor (EGF) and transforming growth factor alpha (TGF-α) which bind to EGFR, and epiregulin and betacellulin which bind to both EGFR and Her4 (1). The exception to this is Her2, for which no natural ligand has been discovered. The binding of ligand drives receptor dimerization, and the various receptors in the EGFR family are capable of both homo- and heterodimerization with each other. However, it does not appear that Her2 homodimerizes at physiological expression levels, and homodimerization of Her3 receptors would not lead to receptor phosphorylation since Her3 is kinase dead and thus must be trans-activated by heterodimerization with other ErbB receptors.
The receptor dimerization event allows for kinase activation and trans-phosphorylation of residues in the intracellular domain. A number of different EGFR tyrosine residues can become phosphorylated, depending on the stimulating ligand and dimerization partner. Following stimulation of EGFR by EGF, the major sites of phosphorylation include tyrosines 1086, 1148, and 1173 (2). Phosphotyrosine-binding proteins then associate with these residues in the ErbB receptor tail and become activated, initiating various signaling cascades. Many of these interactions have been characterized in detail, such as Grb2 binding to EGFR tyrosine 1068 and SHC binding to EGFR tyrosines 1148 and 1173 (3,4). EGFR has been shown to activate a
phosphatidylinositol 3-kinase (PI3K), and protein kinase C (PKC) pathways. In addition, EGFR can be trans-activated by other proteins, such as G-protein-coupled receptors (GPCRs) (5). The various signaling cascades associated with EGFR activation are summarized in Figure 1-1. It has also been observed that activated EGFR may undergo nuclear translocalization and subsequently regulate gene expression, although the contribution of this pathway compared to more traditional signal transduction is unclear (6,7). The activation of EGFR thus leads to multiple cell responses, including cellular growth, differentiation, and migration.
Figure 1-1: EGFR signaling network. (a) EGFR family ligands are shown with their binding specificities. For
simplicity, arrows are only drawn for EGF and neuregulin 4 (NRG4), although other ligand specificities are shown in parentheses. (b) Signaling to the adaptor protein/kinase layer is only shown for two receptor dimers. (c) Possible cellular outcomes based on EGFR signaling. Figure has been reproduced from (5) with permission from Elsevier.
In the absence of ligand, EGFR is constitutively internalized with a half life of ~30 minutes and quickly recycled back to the cell surface, leading to a total receptor distribution of ~80-90% on the cell surface at any given time (Figure 1-2) (8). The metabolic half-life of EGFR
in a tumor cell line is 20 hours (9); thus, a single receptor will cycle through the endocytic pathway many times during its lifetime. Ligand activation increases the rate of EGFR endocytosis 5-10 fold, but does not increase the internalization rates of other ErbB receptors (10). In addition, activated EGFR dimers bound to EGF are preferentially retained in the endosomes and not recycled (11), and these endosomes mature into multivesicular bodies and are targeted to lysosomes (12). This leads to the specific degradation of activated receptors and overall receptor downregulation. Earlier reports indicated that ligand-accelerated internalization required EGFR kinase activity (13,14), but a recent study found that receptor dimerization was sufficient to induce rapid internalization and that kinase activity was unnecessary (15). Although receptor ubiquitination does not affect EGFR internalization rates (16,17), ubiquitination by c-Cbl targets receptors to the lysosome and is dependent on kinase activity and an intact C-terminal region (18).
While receptor internalization has often been viewed as a means of signaling attenuation, it has been shown that activated EGFR continues to signal while in the endosome through certain signaling pathways (19,20). Although Her2 has often been described as the preferred heterodimerization partner for ErbB receptors (21), mathematical modeling and experiments have shown that EGFR homodimerization and EGFR/Her2 heterodimerization occur with comparable affinities (22), and EGFR/Her2 heterodimers appear to predominate because they have a reduced rate of internalization and increased fraction of recycling (23). EGFR trafficking has been the subject of much mathematical modeling, and the system serves as a paradigm for other ligand-binding receptors (24). The constitutive trafficking patterns of EGFR can even affect the global pharmacokinetics of antibody drugs which bind to the receptor due to antibody
internalization and subsequent degradation (25). Additional experiments on EGFR trafficking are reviewed in (8) and (26).
Figure 1-2: Trafficking of EGFR. Activated EGFR homo- and heterodimers are internalized through clathrin-coated
pits and preferentially degraded. Times represent approximate mean time of the processes indicated. Figure has been reproduced from (8) with permission from Elsevier.
1.2 Biochemical and structural characterization of EGFR
EGFR is a 170 kDa protein that is heavily N-glycosylated but not O-glycosylated (27), with carbohydrate accounting for ~20% of the molecular mass. Mass spectrometry characterization has determined that eight of the eleven canonical N-linked sites are glycosylated in the A431 cell line, with two sites unglycosylated and one site glycosylated in a fraction of the receptors (28). The EGFR extracellular region is divided into four domains, numbered I-IV (Figure 1-3). Domains I and III (DI and DIII), also designated L1 and L2, are members of the leucine rich repeat family and are similar to domains found in the insulin receptor family (29). Prior to receptor crystallization, it was known that both DI and DIII contribute to ligand binding (30). However, domain III appears to contribute most of the binding energy, since the affinity of DIII alone for EGF is ~400 nM as measured solubly by isothermal titration calorimetry (31), while soluble EGFR ectodomain binds immobilized EGF with an affinity of ~200 nM (32). Domains II and IV (DII and IV), or the cysteine-rich (CR) domains, contain multiple small disulfide-bonded modules similar to those in laminin (33).
Figure 1-3: Domains of EGFR. The extracellular region consists of domains I-IV, followed by a transmembrane
region (TM) and the intracellular portion. The regions inside the cell include the juxtamembrane portion (JM), tyrosine kinase (TK), and C-terminal tail containing tyrosine residues which become phosphorylated upon receptor activation.
The EGFR ectodomain has been crystallized in both monomer and dimer forms, and a review of these structures can be found in (34). The monomer exists in a tethered, closed, or autoinhibited state (35), with DII and DIV forming a tether contact (Figure 1-4, left). However, in the dimer structures of EGFR bound to EGF (36) or TGF-α (37) ligand, the receptor is in an untethered, open, or extended conformation, with ligand binding between DI and DIII (Figure 1-4, right). Although most cytokine receptors dimerize through ligand bridging the two receptor monomers (38), all dimerization contacts in the EGFR dimer are fully receptor-mediated. The dimer exists in a 2:2 receptor to ligand ratio, with multiple contacts through the DII dimerization arm (residues 242-259). An additional DII loop, residues 271-283, also contributes important dimer contacts (32). The dimerization interface may include DIV; however, in the dimer structures, DIV is either unresolved (36) or was not present in the crystallized protein (37). Peptides mimicking portions of DIV can inhibit EGFR heterodimerization with Her3 (39), and DIV mutations can impair the ability of ligand to bind and induce EGFR phosphorylation (40). However, these potential DIV contacts contribute <9% of the free energy of dimerization as demonstrated by studies on soluble EGFR dimer formation (32). For illustrative purposes, DIV has been added to the dimer structure in Figure 1-4 in the same relative orientation to DIII as seen in the tethered monomer. A low-resolution molecular envelope structure of the full EGFR dimer in solution has been obtained from small-angle X-ray scattering (SAXS), and it is consistent with this dimer structure (41).
Figure 1-4: Crystal structures of monomeric and dimeric EGFR. Left, autoinhibited EGFR monomer (1NQL), with
domain I (DI), red; DII, orange; DIII, blue; DIV, green. A 130o rotation of DI and DII allows DI and DIII to come together to bind EGF ligand. Right, dimeric EGFR bound to EGF ligand in yellow (1IVO). DIV is not resolved in this structure, but has been added in the same relative orientation to DIII as seen in the monomer. This figure was adapted from (34).
From the crystal structures of the monomer and dimer, a model for EGFR activation has been proposed (34). In this model, the monomer mostly exists in a tethered conformation, in which the DII dimerization arm and other residues are obscured, thus preventing receptor dimerization. The monomer equilibrates between the tethered conformation and an extended conformation similar to that seen in the dimer. The binding of ligand between DI and DIII stabilizes the extended conformation, which exposes the DII dimerization arm and other residues, thus allowing the formation of the dimer. However, the breaking of the tether is necessary but not sufficient for dimer formation, as demonstrated through mutations and deletions of DIV (37,42,43). It also appears that certain types of receptor glycosylation can shift the equilibrium toward the extended form (44,45). It has subsequently been found that the DII-DIV tether interaction is not necessarily responsible for maintaining the receptor in the autoinhibited conformation; in fact, mutating every tether interaction in soluble EGFR still leads
to an autoinhibited conformation (41). In addition, it appears that ligand binding is necessary to stabilize the conformation of the DII loop 271-283, which is an important dimerization contact (32).
Ectodomain crystal structures are also available for the EGFR family members Her2 (46), Her3 (47), and Her4 (48). The Her2 monomer, in contrast to EGFR, adopts an extended conformation similar to that of EGFR in the dimer structure. Since Her2 has no activating ligand, it is extended and poised to interact with other ErbB receptors. However, since ligand binding is necessary to activate Her3 and Her4, these monomers adopt a tethered conformation.
EGFR has a single transmembrane domain, and the intracellular portion contains a short juxtamembrane region, the tyrosine kinase, and a C-terminal tail containing tyrosine residues which become phosphorylated upon receptor activation (Figure 1-3). The EGFR kinase domain has been independently crystallized, revealing an intrinsically autoinhibited domain resembling Src and cyclin-dependent kinases (CDKs) (49). In contrast to most kinases, phosphorylation of the EGFR activation loop is not necessary for its activation (50). Instead, the EGFR kinase is activated by an asymmetric dimer in which the C-terminal lobe of one kinase domain binds to the second kinase domain in a manner analogous to cyclin in activated CDK/cyclin complexes. Thus, ligand binding brings two receptor monomers together and allows for the dimerization and subsequent activation of the kinase domains. In this model, the extent of EGFR kinase activation depends on the effective local concentration of the receptor, since the probability of dimerization will increase with increasing kinase domain density. This phenomenon has been demonstrated experimentally using increasing concentrations of EGFR kinase domains tethered to the surface of vesicles (49).
1.3 EGFR in cancer
EGFR was the first receptor to be directly linked to human cancer (51), and because EGFR activation often leads to cellular growth, its signaling can provide tumor cells with substantial survival advantages. In addition, EGFR signaling has been implicated in tumor cell production of pro-angiogenic factors and cellular migration and invasion (52). EGFR dysregulation has been observed in a wide variety of carcinomas, including head and neck, breast, bladder, and non-small-cell lung cancer (5). Excessive EGFR signaling can arise from receptor overexpression, autocrine signaling, or mutation. Normal cells usually express 4x104 to 1x105 EGF receptors per cell, but tumor cells can express as many as 2x106 (53). Receptor overexpression commonly develops due to gene amplification, although it was recently reported that the hypoxic microenvironment of tumors can also induce overexpression of EGFR by increasing EGFR mRNA translation (54). In head and neck cancer, at least 80% of tumors overexpress EGFR (55), and the observed percentages of tumors overexpressing EGFR in various types of cancer are listed in Table 1-1. Furthermore, elevated levels of EGFR serve as a prognostic indicator for poor survival rates (56).
Tumor type Percentage of tumors overexpressing EGFR
Colon 22-75%
Head and neck 80-100%
Pancreatic 30-50% Non-small-cell lung 40-80% Breast 14-91% Renal 50-90% Ovarian 35-70% Glioma 40-63% Bladder 31-48% Table 1-1: EGFR overexpression in different tumor types. Table has been adapted from (53).
In addition, EGFR ligands can be overexpressed, resulting in high levels of autocrine signaling when the receptor is present (57). Autocrine production of TGF-α or EGF is also associated with reduced cancer survival (21). Finally, various activating EGFR mutations have been observed in tumor samples. Gene rearrangements in glioblastoma often lead to truncation mutants, the most common of which is EGFRvIII (EGFRΔ2-7), where amino acids 6-273 (encoded by exons 2-7) are deleted from the gene (58-60). This leads to a mutant which is missing DI and most of DII, with EGFR amino acids 1-5 fused to the rest of the receptor through a novel glycine residue resulting from the gene fusion. Although most frequently seen in glioblastoma, EGFRvIII has also been observed in other types of tumors, including non-small-cell lung, breast, and ovarian cancers (61-63). EGFRvIII is not activated by ligand, but signals constitutively at low levels (64) and thus confers enhanced tumorigenicity to cancer cells (65). There is also evidence that EGFRvIII is downregulation defective, leading to prolonged signaling (66). In addition, point mutants of the EGFR ectodomain have recently been observed in glioblastoma, and select mutants showed high basal phosphorylation and conferred tumorigenicity to NIH-3T3 cells (67).
In non-small-cell lung cancer (NSCLC), mutations in the EGFR kinase domain, which are clustered around the ATP-binding pocket of the enzyme, have been observed (68). These mutations appear to be restricted to a subset of NSCLC patients, particularly non-smokers, women, and those of Asian ethnicity (69). Exon 19 in-frame deletions of some or all of residues 746-750 account for 45% of reported mutations, exon 21 substitutions (especially L858R) account for 45%, and the remaining 10% involve exons 18 and 20, such as G719S and V765A (68). Most of these mutations have been shown to hyperactivate the kinase and have oncogenic
activity. Although predominately seen in NSCLC, very rare kinase domain mutations have also been observed in other types of tumors, including ovarian, colorectal, and head and neck (68).
Other EGFR family members have been shown to be dysregulated in cancer. Her2 overexpression due to gene amplification has been observed in various tumors, including breast, ovarian (70), and bladder cancer (71). In particular, Her2 overexpression is an indicator of poor prognosis in breast cancer (72). Her3 and Her4 signaling can also contribute to a malignant phenotype (5).
1.4 Current EGFR-targeted therapeutics
Because of the importance of the EGFR family in cancer, the receptors are promising targets for rational oncology therapies designed to inhibit receptor signaling. The two main classes of such therapeutics are tyrosine kinase inhibitors (TKIs) and monoclonal antibodies. TKIs are small molecule drugs which are designed to bind the ATP pocket of the kinase domain and inhibit EGFR phosphorylation (73). Two EGFR-targeted TKIs have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of non-small-cell lung cancer: gefitinib (Iressa, ZD1839; AstraZeneca) (74) and erlotinib (Tarceva, OSI-774; OSI/Genentech/Roche) (75), which has been co-crystallized with the EGFR kinase domain (76). Although these two TKIs are targeted to the EGFR tyrosine kinase, with IC50 values
(concentration required for 50% inhibition of activity) in the nanomolar range, they also will inhibit the Her2 tyrosine kinase with IC50 values in the micromolar range (73). A small subset of
NSCLC patients dramatically respond to treatment with gefitinib (73) or erlotinib, and the characterization of these patient tumors led to the discovery of activating EGFR kinase domain mutations (described in Section 1.3). In fact, 77% of NSCLC patients who respond to gefitinib or erlotinib harbor certain kinase domain mutations, as opposed to a 7% incidence of mutation in non-responders. It has been hypothesized that these activating EGFR kinase domain mutations lead to a state of oncogene addiction for tumor cells, thus explaining the extreme sensitivity of mutation-bearing tumors to EGFR TKIs (68). Unexpectedly, in cells expressing the ectodomain truncation mutant EGFRvIII, gefitinib induces signaling at low concentrations (77). Gefitinib and erlotinib are reversible kinase inhibitors; however, irreversible TKIs which bind the kinase domain covalently, such as HKI-357 (78), have also been developed. These have been shown to
be effective against reversible-TKI resistant cells expressing a secondary kinase mutation T790M. Additional TKI drugs in development are reviewed in (79) and (68).
An alternative to TKI therapeutics is treatment with anti-EGFR monoclonal antibodies (mAbs; for a general discussion on antibody structure, see Section 3.1). While TKIs can be conveniently delivered orally and have a reduced chance of inducing allergic reactions, monoclonal antibodies have superior stability in vivo and lower gastrointestinal toxicity (79). Cetuximab (Erbitux, IMC-C225; ImClone Systems) was the first anti-EGFR monoclonal antibody to be approved by the FDA. Originally approved for the treatment of EGFR-expressing metastatic colorectal cancer, it has also recently been approved for head and neck cancer. The antibody was initially isolated by immunization of mice with EGFR, and the murine monoclonal antibody was designated mAb 225 (80). In vitro experiments revealed that this antibody inhibited the binding of ligand to EGFR, resulting in decreased tyrosine phosphorylation and reduced cell proliferation (80,81). The crystal structure of the cetuximab Fab bound to the EGFR ectodomain reveals that the antibody directly competes with EGF ligand by binding to DIII on an epitope which overlaps the EGF binding site (82). It was also found that mAb 225 can induce EGFR internalization (83), which may reduce the number of receptors available to bind ligand. The bivalency of the mAb 225 molecule was shown to be important for its ability to internalize and downregulate EGFR and inhibit subsequent growth (84). In addition, cetuximab has been shown to exhibit anti-angiogenic properties, leading to even more effective treatment of tumor xenografts in vivo (85). Cetuximab may also induce antibody-dependent cellular toxicity (ADCC) in vivo (86), and the antibody’s various mechanisms of action are further discussed in (87). To decrease its potential immunogenicity, the mouse mAb 225 was chimerized with human IgG1 by adding the mouse antibody variable regions to the human constant IgG regions. This
chimeric antibody, designated C225, was shown to have a higher affinity for EGFR and was effective against human tumor xenografts (88). Additionally, cetuximab is able to inhibit the growth of lung cancer xenografts expressing the L858R/T790M EGFR kinase mutants, which are resistant to reversible TKI treatment (89). In clinical trials, cetuximab was well-tolerated with mild toxicities, which included acneiform rash, fatigue, and hypersensitivity (90). The effects to normal tissue appear to minimal, since many healthy cells expressing EGFR are not rapidly turned over, with the intestinal epithelium as an exception (53). In a Phase III clinical trial with 329 irinotecan-refractory metastatic colorectal cancer patients, 10.8% responded to cetuximab therapy alone while 22.9% responded to cetuximab plus irinotecan (90), leading to the FDA approval of this drug. Interestingly, cetuximab treatment was able to reverse tumor cell resistance to the chemotherapeutic irinotecan.
Other antibodies have been developed which similarly inhibit ligand binding to EGFR as cetuximab does. Panitumumab (Vectibix, ABX-EGF; Amgen/Abgenix) has also been FDA-approved for the treatment of metastatic colorectal cancer and blocks the binding of ligands to EGFR (91). Panitumumab has one potential advantage over cetuximab; it is a fully human antibody developed from the XenoMouse technology, in which human antibody genes were cloned into mice (91). However, panitumumab is a class IgG2 antibody and thus incapable of activating ADCC, although it is unclear how important this is for tumor inhibition (92). Data from clinical trials shows that panitumumab produces an 8% overall response rate in metastatic colorectal cancer, with progression-free survival extended from 60 days to 96 days (92). Another cetuximab-like antibody is matuzumab (EMD72000; EMD Pharmaceuticals/Merck KgaA), which is currently in various phase II clinical trials. The antibody was originally isolated from mice (mAb 425/EMD55900) and inhibits the binding of ligands to EGFR (93). The murine
version of the antibody was humanized by grafting the murine complementarity-determining regions (CDRs) onto a human IgG1 framework (94). A phase I study of matuzumab treatment of patients with advanced pancreatic adenocarcinoma showed partial responses and stable disease in eight of twelve patients (95). However, in a recent phase II trial, matuzumab showed no evidence of significant clinical activity in ovarian tumors (96). In addition, a bispecific version of the humanized mAb 425, named MDX-447 (Medarex/Merck KgaA), has been developed, with the other arm of the antibody binding to the high affinity Fc receptor (FcγRI) (97). MDX-447 is currently undergoing further clinical trials in combination with activated monocytes in an effort to direct these immune cells to tumor sites. Finally, nimotuzumab (hR-3, TheraCIM; YM Biosciences/Center of Molecular Immunology, Cuba) is a humanized antibody which also blocks ligand binding to EGFR (98). Phase I/II trials of glioblastoma and anaplastic astrocytoma patients showed a 38% objective response rate (99), and the antibody is currently undergoing further trials in the U.S.
Antibodies which target the truncation mutant EGFRvIII have also been developed. Since EGFRvIII contains a novel glycine residue at the junction between two EGFR fragments, this junctional peptide is a potential target for antibody binding and represents an epitope specific to tumor cells. One antibody targeting this peptide is MR1-1, which is an affinity-matured single chain variable fragment (scFv) specific for EGFRvIII (100). However, antibodies against the junctional peptide have no intrinsic anti-EGFRvIII activity, and thus must be conjugated to cytotoxic agents such as immunotoxins in order to kill tumor cells (101). Another antibody, mAb 806, was isolated from immunization of mice with EGFRvIII, and it was shown to bind and inhibit the growth of cells expressing the mutant protein. However, it was discovered that mAb 806 also bound to a small portion of wild-type EGFR (~5-10%) on cells overexpressing the
receptor and inhibited cell growth, but it did not bind to or inhibit cells expressing normal EGFR levels (102-104). Therefore, mAb 806 has the unique ability to recognize EGFR expressed by tumor cells but not on normal tissues. Characterization of mAb 806 revealed that it binds a peptide epitope shared by both EGFRvIII and EGFR (105). However, this epitope is only exposed in certain situations: in EGFRvIII due to the deletion of DI and DII, when wild-type EGFR transitions from the autoinhibited to open conformation, or by high-mannose forms of the receptor in cells overexpressing EGFR (44). The mAb 806 epitope is discussed in further detail in Chapter 2. mAb 806 has also been chimerized to reduce murine sequences, generating antibody ch806 (106). In mouse models of cancer, 806 was effective in inducing regression of lung tumors expressing either EGFR harboring an activating kinase domain mutation or EGFRvIII (107). However, 806 is ineffective against kinase-dead EGFRvIII, suggesting that it functions by blocking receptor trans-phosphorylation (108). Phase I clinical trial data showed that ch806 was well-tolerated by patients, with highly specific targeting to tumors versus normal tissues (109), and further trials are currently ongoing.
The only FDA-approved antibody which targets Her2, trastuzumab (Herceptin, 4D5; Genentech), was isolated by mouse immunization (110) and humanized (111). Trastuzumab inhibited the proliferation of breast tumor-derived cell lines (110) and also supported ADCC killing against the SK-Br-3 tumor line (111). Trastuzumab was shown to be effective against human gastric carcinoma xenografts (112), as well as breast cancer BT-474 xenografts (113). In phase III trials, the addition of trastuzumab treatment to chemotherapy was associated with a longer time to disease progression (7.4 vs. 4.6 months) and a higher objective response rate (50% vs. 32%) (114), leading to the FDA approval of this drug for the treatment of Her2-positive metastatic breast cancer. The main toxicities were cardiac adverse events, since Her2 signaling
appears to be important for adult heart function (115). Recent trials have shown that adjuvant trastuzumab therapy reduces the 3-year risk of recurrence by half in early breast cancer patients (116). Despite the success of trastuzumab, its mechanism of action is not immediately apparent. A crystal structure of trastuzumab bound to Her2 reveals that it binds the C-terminal portion of domain IV (117); however, it does not block Her2 heterodimerization with Her3 (118). Although initial reports suggested that trastuzumab could inhibit tumors through downregulation of the Her2 receptor (119,120), a more recent study demonstrated that the antibody is internalized and recycled back to the cell surface without mediating receptor degradation (121). The Her2 ectodomain undergoes cleavage, and the resulting membrane-bound kinase fragment shows increased tyrosine kinase activity (122). Trastuzumab blocks this Her2 shedding (123), presumably by restricting access to the protease cleavage site, thus preventing the formation of the activated kinase fragment (119). Finally, trastuzumab can induce ADCC killing of tumor cells (124), but this cannot be its sole mechanism since ADCC cannot explain the antibody’s efficacy in vitro. Additional experiments with trastuzumab are reviewed in (125). Another anti-Her2 antibody named pertuzumab (Omnitarg, 2C4; Genentech) binds to domain II of anti-Her2 (demonstrated by co-crystallization (126)), and thus it inhibits Her2/Her3 heterodimerization and breast and prostate xenograft tumor growth (118). The murine version, mAb 2C4, was humanized (127), and initial results from a phase II trial treating prostate cancer showed no objective responses (128). However, a clinical trial evaluating pertuzumab treatment plus trastuzumab is currently ongoing, motivated by preclinical studies where the two antibodies synergistically inhibited the survival of breast cancer cells (129).
Although therapeutics directed at the EGFR receptor family have been somewhat effective in inhibiting the growth of tumor cells, optimal cancer treatments will likely include
some form of combination therapy. Since multiple genetic alterations contributing to a malignant phenotype normally arise during the course of tumor development (130), it may be difficult to inhibit these tumors by blocking only one receptor. Synergistic inhibition effects of combining anti-EGFR antibodies with chemotherapeutic agents in vitro were initially reported by (131). Treatments of trastuzumab plus chemotherapy (114) or cetuximab plus irinotecan (90) both improved patient outcome in clinical trials. Additional trials examining combinations of anti-EGFR antibodies with other chemotherapeutics have been and are currently being investigated (3,53,132). However, not all combinations are synergistic or even additive, as demonstrated by failed clinical trials combining gefitinib or erlotinib with chemotherapy (133). Using two different classes of anti-EGFR inhibitors may be effective, since tumor xenograft experiments have shown synergy between mAb 806 and the TKI AG1478 (134). In addition, the combination of two anti-EGFR antibodies directed against different epitopes has been shown to stimulate receptor downregulation and inhibit signaling (135,136). Other successful combinations include simultaneously inhibiting EGFRvIII and c-Met (137), and rationales for treatment using ErbB plus mTOR inhibitors or ErbB plus estrogen inhibitors are discussed in (132).
1.5 Overview of thesis work
In this thesis work, we have sought to characterize and engineer antibodies against the epidermal growth factor receptor. Multiple anti-EGFR antibodies have been developed, but their binding epitopes had not been determined due to a lack of convenient epitope mapping methods. To alleviate this, we developed a novel method of epitope mapping using random mutagenesis and yeast surface display. In our approach, we disrupted the antibody-antigen interaction through point mutations. However, since we did not know the epitopes of the antibodies, we made a library of random mutations, and then combinatorially screened this library using yeast surface display. EGFR mutants which displayed a loss of binding to the antibody being mapped were thus potential epitope residues. It was also necessary to screen for retention of binding to a conformation-specific antibody to remove misfolded EGFR mutants. The development of our novel method enabled the epitope mapping of a total of 4 antibodies: mAbs 806, 225, 13A9, and EMD72000. The determination of the antibody epitopes enabled further understanding of their mechanisms of action. Since mAbs 806, 225, and EMD72000 have high therapeutic relevance, this information may have clinical impact as well. In addition, the epitope mapping method was extended to mapping the binding of three designed ankyrin repeat proteins (DARPins) against EGFR.
Instead of using the common inhibition strategy of using antibodies to block ligand binding, we opted to engineer antibodies to directly inhibit receptor dimerization. Such antibodies could have advantages in tumors driven by autocrine signaling or the EGFRvIII mutant. We selected epitopes in EGFR domain II and domain IV to target with antibodies, and synthesized peptide mimics of these loops to use as antigen. Starting from a nonimmune human scFv library in yeast, candidate binders to the peptide epitopes were isolated and characterized.
The majority of binders were heavy chain only constructs, many of which were likely mediating binding through the VH/VL interface instead of the antibody CDRs. In addition, rabbit
monoclonal antibodies against a domain IV peptide were developed, but appeared to only bind the peptide and not the EGFR ectodomain.
Finally, previous experiments had shown that polyclonal antibodies (pAbs) against either EGFR domain II or domain IV could eliminate the high affinity facet of EGF binding normally observed on the surface of cells. Based on these results, we hypothesized that the pAbs inhibited receptor dimerization, and thus inactive preformed EGFR dimers could represent the high affinity binding component. We developed a mathematical model incorporating these hypotheses to determine whether such a model could be consistent with experimental observations. The equilibrium ligand binding and receptor dimerization model was fit to the experimental data using three parameters: the high affinity binding constant, the dimerization constant, and the maximum binding signal. The model was able to successfully reproduce the experimental data, and it could produce characteristic concave-up Scatchard plots for a wide range of values. Overall, we have increased understanding of the mechanisms of action of a number of anti-EGFR antibodies.
1.6 Works cited
1. Harris, R. C., Chung, E., and Coffey, R. J. (2003) EGF receptor ligands, Exp Cell Res
284, 2-13.
2. Erba, E. B., Matthiesen, R., Bunkenborg, J., Schulze, W. X., Di Stefano, P., et al. (2007) Quantitation of multisite EGF receptor phosphorylation using mass spectrometry and a novel normalization approach, J Proteome Res 6, 2768-2785.
3. Sebastian, S., Settleman, J., Reshkin, S. J., Azzariti, A., Bellizzi, A., et al. (2006) The complexity of targeting EGFR signalling in cancer: from expression to turnover, Biochim
Biophys Acta 1766, 120-139.
4. Schulze, W. X., Deng, L., and Mann, M. (2005) Phosphotyrosine interactome of the ErbB-receptor kinase family, Mol Syst Biol 1, 2005 0008.
5. Yarden, Y., and Sliwkowski, M. X. (2001) Untangling the ErbB signalling network, Nat
Rev Mol Cell Biol 2, 127-137.
6. Wells, A., and Marti, U. (2002) Signalling shortcuts: cell-surface receptors in the nucleus?, Nat Rev Mol Cell Biol 3, 697-702.
7. Lo, H. W., and Hung, M. C. (2006) Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival, Br J
Cancer 94, 184-188.
8. Wiley, H. S. (2003) Trafficking of the ErbB receptors and its influence on signaling, Exp
Cell Res 284, 78-88.
9. Stoscheck, C. M., and Carpenter, G. (1984) Characterization of the metabolic turnover of epidermal growth factor receptor protein in A-431 cells, J Cell Physiol 120, 296-302. 10. Waterman, H., Sabanai, I., Geiger, B., and Yarden, Y. (1998) Alternative intracellular
routing of ErbB receptors may determine signaling potency, J Biol Chem 273, 13819-13827.
11. Herbst, J. J., Opresko, L. K., Walsh, B. J., Lauffenburger, D. A., and Wiley, H. S. (1994) Regulation of postendocytic trafficking of the epidermal growth factor receptor through endosomal retention, J Biol Chem 269, 12865-12873.
12. Futter, C. E., Pearse, A., Hewlett, L. J., and Hopkins, C. R. (1996) Multivesicular endosomes containing internalized EGF-EGF receptor complexes mature and then fuse directly with lysosomes, J Cell Biol 132, 1011-1023.
13. Chen, W. S., Lazar, C. S., Poenie, M., Tsien, R. Y., Gill, G. N., et al. (1987) Requirement for intrinsic protein tyrosine kinase in the immediate and late actions of the EGF receptor,
Nature 328, 820-823.
14. Schmidt, M. H., Furnari, F. B., Cavenee, W. K., and Bogler, O. (2003) Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization, Proc Natl Acad Sci U S A 100, 6505-6510.
15. Wang, Q., Villeneuve, G., and Wang, Z. (2005) Control of epidermal growth factor receptor endocytosis by receptor dimerization, rather than receptor kinase activation,
EMBO Rep 6, 942-948.
16. Duan, L., Miura, Y., Dimri, M., Majumder, B., Dodge, I. L., et al. (2003) Cbl-mediated ubiquitinylation is required for lysosomal sorting of epidermal growth factor receptor but is dispensable for endocytosis, J Biol Chem 278, 28950-28960.
17. Huang, F., Kirkpatrick, D., Jiang, X., Gygi, S., and Sorkin, A. (2006) Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain, Mol Cell 21, 737-748.
18. Levkowitz, G., Waterman, H., Zamir, E., Kam, Z., Oved, S., et al. (1998) c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor,
Genes Dev 12, 3663-3674.
19. Haugh, J. M., Huang, A. C., Wiley, H. S., Wells, A., and Lauffenburger, D. A. (1999) Internalized epidermal growth factor receptors participate in the activation of p21(ras) in fibroblasts, J Biol Chem 274, 34350-34360.
20. Wang, Y., Pennock, S., Chen, X., and Wang, Z. (2002) Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival, Mol Cell Biol 22, 7279-7290.
21. Citri, A., and Yarden, Y. (2006) EGF-ERBB signalling: towards the systems level, Nat
Rev Mol Cell Biol 7, 505-516.
22. Hendriks, B. S., Opresko, L. K., Wiley, H. S., and Lauffenburger, D. (2003) Quantitative analysis of HER2-mediated effects on HER2 and epidermal growth factor receptor endocytosis: distribution of homo- and heterodimers depends on relative HER2 levels, J
Biol Chem 278, 23343-23351.
23. Hendriks, B. S., Opresko, L. K., Wiley, H. S., and Lauffenburger, D. (2003) Coregulation of epidermal growth factor receptor/human epidermal growth factor receptor 2 (HER2) levels and locations: quantitative analysis of HER2 overexpression effects, Cancer Res
63, 1130-1137.
24. Wiley, H. S., Shvartsman, S. Y., and Lauffenburger, D. A. (2003) Computational modeling of the EGF-receptor system: a paradigm for systems biology, Trends Cell Biol
13, 43-50.
25. Lammerts van Bueren, J. J., Bleeker, W. K., Bogh, H. O., Houtkamp, M., Schuurman, J., et al. (2006) Effect of target dynamics on pharmacokinetics of a novel therapeutic antibody against the epidermal growth factor receptor: implications for the mechanisms of action, Cancer Res 66, 7630-7638.
26. Carpenter, G. (2000) The EGF receptor: a nexus for trafficking and signaling, Bioessays
22, 697-707.
27. Cummings, R. D., Soderquist, A. M., and Carpenter, G. (1985) The oligosaccharide moieties of the epidermal growth factor receptor in A-431 cells. Presence of complex-type N-linked chains that contain terminal N-acetylgalactosamine residues, J Biol Chem
260, 11944-11952.
28. Zhen, Y., Caprioli, R. M., and Staros, J. V. (2003) Characterization of glycosylation sites of the epidermal growth factor receptor, Biochemistry 42, 5478-5492.
29. Ward, C. W., and Garrett, T. P. (2001) The relationship between the L1 and L2 domains of the insulin and epidermal growth factor receptors and leucine-rich repeat modules,
BMC Bioinformatics 2, 4.
30. Lax, I., Fischer, R., Ng, C., Segre, J., Ullrich, A., et al. (1991) Noncontiguous regions in the extracellular domain of EGF receptor define ligand-binding specificity, Cell Regul 2, 337-345.
31. Lemmon, M. A., Bu, Z., Ladbury, J. E., Zhou, M., Pinchasi, D., et al. (1997) Two EGF molecules contribute additively to stabilization of the EGFR dimer, Embo J 16, 281-294.
32. Dawson, J. P., Berger, M. B., Lin, C. C., Schlessinger, J., Lemmon, M. A., et al. (2005) Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface, Mol Cell Biol 25, 7734-7742.
33. Ward, C. W., Hoyne, P. A., and Flegg, R. H. (1995) Insulin and epidermal growth factor receptors contain the cysteine repeat motif found in the tumor necrosis factor receptor,
Proteins 22, 141-153.
34. Burgess, A. W., Cho, H. S., Eigenbrot, C., Ferguson, K. M., Garrett, T. P., et al. (2003) An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors, Mol
Cell 12, 541-552.
35. Ferguson, K. M., Berger, M. B., Mendrola, J. M., Cho, H. S., Leahy, D. J., et al. (2003) EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization, Mol Cell 11, 507-517.
36. Ogiso, H., Ishitani, R., Nureki, O., Fukai, S., Yamanaka, M., et al. (2002) Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains, Cell 110, 775-787.
37. Garrett, T. P., McKern, N. M., Lou, M., Elleman, T. C., Adams, T. E., et al. (2002) Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha, Cell 110, 763-773.
38. Stroud, R. M., and Wells, J. A. (2004) Mechanistic diversity of cytokine receptor signaling across cell membranes, Sci STKE 2004, re7.
39. Berezov, A., Chen, J., Liu, Q., Zhang, H. T., Greene, M. I., et al. (2002) Disabling receptor ensembles with rationally designed interface peptidomimetics, J Biol Chem 277, 28330-28339.
40. Saxon, M. L., and Lee, D. C. (1999) Mutagenesis reveals a role for epidermal growth factor receptor extracellular subdomain IV in ligand binding, J Biol Chem 274, 28356-28362.
41. Dawson, J. P., Bu, Z., and Lemmon, M. A. (2007) Ligand-Induced Structural Transitions in ErbB Receptor Extracellular Domains, Structure 15, 942-954.
42. Mattoon, D., Klein, P., Lemmon, M. A., Lax, I., and Schlessinger, J. (2004) The tethered configuration of the EGF receptor extracellular domain exerts only a limited control of receptor function, Proc Natl Acad Sci U S A 101, 923-928.
43. Walker, F., Orchard, S. G., Jorissen, R. N., Hall, N. E., Zhang, H. H., et al. (2004) CR1/CR2 interactions modulate the functions of the cell surface epidermal growth factor receptor, J Biol Chem 279, 22387-22398.
44. Johns, T. G., Mellman, I., Cartwright, G. A., Ritter, G., Old, L. J., et al. (2005) The antitumor monoclonal antibody 806 recognizes a high-mannose form of the EGF receptor that reaches the cell surface when cells over-express the receptor, Faseb J 19, 780-782. 45. Whitson, K. B., Whitson, S. R., Red-Brewer, M. L., McCoy, A. J., Vitali, A. A., et al.
(2005) Functional effects of glycosylation at Asn-579 of the epidermal growth factor receptor, Biochemistry 44, 14920-14931.
46. Garrett, T. P., McKern, N. M., Lou, M., Elleman, T. C., Adams, T. E., et al. (2003) The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors, Mol Cell 11, 495-505.
47. Cho, H. S., and Leahy, D. J. (2002) Structure of the extracellular region of HER3 reveals an interdomain tether, Science 297, 1330-1333.
48. Bouyain, S., Longo, P. A., Li, S., Ferguson, K. M., and Leahy, D. J. (2005) The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand,
Proc Natl Acad Sci U S A 102, 15024-15029.
49. Zhang, X., Gureasko, J., Shen, K., Cole, P. A., and Kuriyan, J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor, Cell
125, 1137-1149.
50. Gotoh, N., Tojo, A., Hino, M., Yazaki, Y., and Shibuya, M. (1992) A highly conserved tyrosine residue at codon 845 within the kinase domain is not required for the transforming activity of human epidermal growth factor receptor, Biochem Biophys Res
Commun 186, 768-774.
51. de Larco, J. E., and Todaro, G. J. (1978) Epithelioid and fibroblastic rat kidney cell clones: epidermal growth factor (EGF) receptors and the effect of mouse sarcoma virus transformation, J Cell Physiol 94, 335-342.
52. Holbro, T., Civenni, G., and Hynes, N. E. (2003) The ErbB receptors and their role in cancer progression, Exp Cell Res 284, 99-110.
53. Herbst, R. S., and Shin, D. M. (2002) Monoclonal antibodies to target epidermal growth factor receptor-positive tumors: a new paradigm for cancer therapy, Cancer 94, 1593-1611.
54. Franovic, A., Gunaratnam, L., Smith, K., Robert, I., Patten, D., et al. (2007) Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer, Proc Natl Acad Sci U S A 104, 13092-13097.
55. Ford, A. C., and Grandis, J. R. (2003) Targeting epidermal growth factor receptor in head and neck cancer, Head Neck 25, 67-73.
56. Nicholson, R. I., Gee, J. M., and Harper, M. E. (2001) EGFR and cancer prognosis, Eur J
Cancer 37 Suppl 4, S9-15.
57. Sizeland, A. M., and Burgess, A. W. (1992) Anti-sense transforming growth factor alpha oligonucleotides inhibit autocrine stimulated proliferation of a colon carcinoma cell line,
Mol Biol Cell 3, 1235-1243.
58. Sugawa, N., Ekstrand, A. J., James, C. D., and Collins, V. P. (1990) Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas, Proc Natl Acad Sci U S A 87, 8602-8606.
59. Wong, A. J., Ruppert, J. M., Bigner, S. H., Grzeschik, C. H., Humphrey, P. A., et al. (1992) Structural alterations of the epidermal growth factor receptor gene in human gliomas, Proc Natl Acad Sci U S A 89, 2965-2969.
60. Frederick, L., Wang, X. Y., Eley, G., and James, C. D. (2000) Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas, Cancer Res 60, 1383-1387.
61. Garcia de Palazzo, I. E., Adams, G. P., Sundareshan, P., Wong, A. J., Testa, J. R., et al. (1993) Expression of mutated epidermal growth factor receptor by non-small cell lung carcinomas, Cancer Res 53, 3217-3220.
62. Wikstrand, C. J., Hale, L. P., Batra, S. K., Hill, M. L., Humphrey, P. A., et al. (1995) Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas, Cancer Res 55, 3140-3148.
63. Moscatello, D. K., Holgado-Madruga, M., Godwin, A. K., Ramirez, G., Gunn, G., et al. (1995) Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors, Cancer Res 55, 5536-5539.
64. Huang, H. S., Nagane, M., Klingbeil, C. K., Lin, H., Nishikawa, R., et al. (1997) The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling, J Biol Chem 272, 2927-2935.
65. Nishikawa, R., Ji, X. D., Harmon, R. C., Lazar, C. S., Gill, G. N., et al. (1994) A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity, Proc Natl Acad Sci U S A 91, 7727-7731.
66. Han, W., Zhang, T., Yu, H., Foulke, J. G., and Tang, C. K. (2006) Hypophosphorylation of residue Y1045 leads to defective downregulation of EGFRvIII, Cancer Biol Ther 5, 1361-1368.
67. Lee, J. C., Vivanco, I., Beroukhim, R., Huang, J. H., Feng, W. L., et al. (2006) Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain, PLoS Med 3, e485.
68. Sharma, S. V., Bell, D. W., Settleman, J., and Haber, D. A. (2007) Epidermal growth factor receptor mutations in lung cancer, Nat Rev Cancer 7, 169-181.
69. Sequist, L. V., Joshi, V. A., Janne, P. A., Bell, D. W., Fidias, P., et al. (2006) Epidermal growth factor receptor mutation testing in the care of lung cancer patients, Clin Cancer
Res 12, 4403s-4408s.
70. Slamon, D. J., Godolphin, W., Jones, L. A., Holt, J. A., Wong, S. G., et al. (1989) Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer, Science 244, 707-712.
71. Sauter, G., Moch, H., Moore, D., Carroll, P., Kerschmann, R., et al. (1993) Heterogeneity of erbB-2 gene amplification in bladder cancer, Cancer Res 53, 2199-2203.
72. Slamon, D. J., Clark, G. M., Wong, S. G., Levin, W. J., Ullrich, A., et al. (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene, Science 235, 177-182.
73. Fry, D. W. (2003) Mechanism of action of erbB tyrosine kinase inhibitors, Exp Cell Res
284, 131-139.
74. Barker, A. J., Gibson, K. H., Grundy, W., Godfrey, A. A., Barlow, J. J., et al. (2001) Studies leading to the identification of ZD1839 (IRESSA): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer, Bioorg Med Chem Lett 11, 1911-1914.
75. Moyer, J. D., Barbacci, E. G., Iwata, K. K., Arnold, L., Boman, B., et al. (1997) Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase, Cancer Res 57, 4838-4848.
76. Stamos, J., Sliwkowski, M. X., and Eigenbrot, C. (2002) Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor, J Biol Chem 277, 46265-46272.
77. Pedersen, M. W., Pedersen, N., Ottesen, L. H., and Poulsen, H. S. (2005) Differential response to gefitinib of cells expressing normal EGFR and the mutant EGFRvIII, Br J
Cancer 93, 915-923.
78. Kwak, E. L., Sordella, R., Bell, D. W., Godin-Heymann, N., Okimoto, R. A., et al. (2005) Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib, Proc Natl Acad Sci U S A 102, 7665-7670.
80. Sato, J. D., Kawamoto, T., Le, A. D., Mendelsohn, J., Polikoff, J., et al. (1983) Biological effects in vitro of monoclonal antibodies to human epidermal growth factor receptors,
Mol Biol Med 1, 511-529.
81. Gill, G. N., Kawamoto, T., Cochet, C., Le, A., Sato, J. D., et al. (1984) Monoclonal anti-epidermal growth factor receptor antibodies which are inhibitors of anti-epidermal growth factor binding and antagonists of epidermal growth factor binding and antagonists of epidermal growth factor-stimulated tyrosine protein kinase activity, J Biol Chem 259, 7755-7760.
82. Li, S., Schmitz, K. R., Jeffrey, P. D., Wiltzius, J. J., Kussie, P., et al. (2005) Structural basis for inhibition of the epidermal growth factor receptor by cetuximab, Cancer Cell 7, 301-311.
83. Sunada, H., Magun, B. E., Mendelsohn, J., and MacLeod, C. L. (1986) Monoclonal antibody against epidermal growth factor receptor is internalized without stimulating receptor phosphorylation, Proc Natl Acad Sci U S A 83, 3825-3829.
84. Fan, Z., Lu, Y., Wu, X., and Mendelsohn, J. (1994) Antibody-induced epidermal growth factor receptor dimerization mediates inhibition of autocrine proliferation of A431 squamous carcinoma cells, J Biol Chem 269, 27595-27602.
85. Petit, A. M., Rak, J., Hung, M. C., Rockwell, P., Goldstein, N., et al. (1997) Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of solid tumors, Am J
Pathol 151, 1523-1530.
86. Zhang, W., Gordon, M., Schultheis, A. M., Yang, D. Y., Nagashima, F., et al. (2007) FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab, J Clin Oncol 25, 3712-3718.
87. Baselga, J. (2001) The EGFR as a target for anticancer therapy--focus on cetuximab, Eur
J Cancer 37 Suppl 4, S16-22.
88. Goldstein, N. I., Prewett, M., Zuklys, K., Rockwell, P., and Mendelsohn, J. (1995) Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model, Clin Cancer Res 1, 1311-1318.
89. Perez-Torres, M., Guix, M., Gonzalez, A., and Arteaga, C. L. (2006) Epidermal growth factor receptor (EGFR) antibody down-regulates mutant receptors and inhibits tumors expressing EGFR mutations, J Biol Chem 281, 40183-40192.
90. Cunningham, D., Humblet, Y., Siena, S., Khayat, D., Bleiberg, H., et al. (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer, N Engl J Med 351, 337-345.
91. Yang, X. D., Jia, X. C., Corvalan, J. R., Wang, P., and Davis, C. G. (2001) Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy,
Crit Rev Oncol Hematol 38, 17-23.
92. Messersmith, W. A., and Hidalgo, M. (2007) Panitumumab, a monoclonal anti epidermal growth factor receptor antibody in colorectal cancer: another one or the one?, Clin
Cancer Res 13, 4664-4666.
93. Murthy, U., Basu, A., Rodeck, U., Herlyn, M., Ross, A. H., et al. (1987) Binding of an antagonistic monoclonal antibody to an intact and fragmented EGF-receptor polypeptide,
94. Kettleborough, C. A., Saldanha, J., Heath, V. J., Morrison, C. J., and Bendig, M. M. (1991) Humanization of a mouse monoclonal antibody by CDR-grafting: the importance of framework residues on loop conformation, Protein Eng 4, 773-783.
95. Graeven, U., Kremer, B., Sudhoff, T., Killing, B., Rojo, F., et al. (2006) Phase I study of the humanised anti-EGFR monoclonal antibody matuzumab (EMD 72000) combined with gemcitabine in advanced pancreatic cancer, Br J Cancer 94, 1293-1299.
96. Seiden, M. V., Burris, H. A., Matulonis, U., Hall, J. B., Armstrong, D. K., et al. (2007) A phase II trial of EMD72000 (matuzumab), a humanized anti-EGFR monoclonal antibody, in patients with platinum-resistant ovarian and primary peritoneal malignancies, Gynecol
Oncol 104, 727-731.
97. Fury, M. G., Lipton, A., Smith, K. M., Winston, C. B., and Pfister, D. G. (2007) A phase-I trial of the epidermal growth factor receptor directed bispecific antibody MDX-447 without and with recombinant human granulocyte-colony stimulating factor in patients with advanced solid tumors, Cancer Immunol Immunother.
98. Mateo, C., Moreno, E., Amour, K., Lombardero, J., Harris, W., et al. (1997) Humanization of a mouse monoclonal antibody that blocks the epidermal growth factor receptor: recovery of antagonistic activity, Immunotechnology 3, 71-81.
99. Ramos, T. C., Figueredo, J., Catala, M., Gonzalez, S., Selva, J. C., et al. (2006) Treatment of high-grade glioma patients with the humanized anti-epidermal growth factor receptor (EGFR) antibody h-R3: report from a phase I/II trial, Cancer Biol Ther 5, 375-379.
100. Kuan, C. T., Wikstrand, C. J., Archer, G., Beers, R., Pastan, I., et al. (2000) Increased binding affinity enhances targeting of glioma xenografts by EGFRvIII-specific scFv, Int J
Cancer 88, 962-969.
101. Ochiai, H., Archer, G. E., Herndon, J. E., 2nd, Kuan, C. T., Mitchell, D. A., et al. (2007) EGFRvIII-targeted immunotoxin induces antitumor immunity that is inhibited in the absence of CD4+ and CD8+ T cells, Cancer Immunol Immunother.
102. Luwor, R. B., Johns, T. G., Murone, C., Huang, H. J., Cavenee, W. K., et al. (2001) Monoclonal antibody 806 inhibits the growth of tumor xenografts expressing either the de2-7 or amplified epidermal growth factor receptor (EGFR) but not wild-type EGFR,
Cancer Res 61, 5355-5361.
103. Johns, T. G., Stockert, E., Ritter, G., Jungbluth, A. A., Huang, H. J., et al. (2002) Novel monoclonal antibody specific for the de2-7 epidermal growth factor receptor (EGFR) that also recognizes the EGFR expressed in cells containing amplification of the EGFR gene,
Int J Cancer 98, 398-408.
104. Jungbluth, A. A., Stockert, E., Huang, H. J., Collins, V. P., Coplan, K., et al. (2003) A monoclonal antibody recognizing human cancers with amplification/overexpression of the human epidermal growth factor receptor, Proc Natl Acad Sci U S A 100, 639-644. 105. Johns, T. G., Adams, T. E., Cochran, J. R., Hall, N. E., Hoyne, P. A., et al. (2004)
Identification of the epitope for the epidermal growth factor receptor-specific monoclonal antibody 806 reveals that it preferentially recognizes an untethered form of the receptor, J
Biol Chem 279, 30375-30384.
106. Panousis, C., Rayzman, V. M., Johns, T. G., Renner, C., Liu, Z., et al. (2005) Engineering and characterisation of chimeric monoclonal antibody 806 (ch806) for targeted immunotherapy of tumours expressing de2-7 EGFR or amplified EGFR, Br J
107. Li, D., Ji, H., Zaghlul, S., McNamara, K., Liang, M. C., et al. (2007) Therapeutic anti-EGFR antibody 806 generates responses in murine de novo anti-EGFR mutant-dependent lung carcinomas, J Clin Invest 117, 346-352.
108. Johns, T. G., Perera, R. M., Vernes, S. C., Vitali, A. A., Cao, D. X., et al. (2007) The efficacy of epidermal growth factor receptor-specific antibodies against glioma xenografts is influenced by receptor levels, activation status, and heterodimerization, Clin
Cancer Res 13, 1911-1925.
109. Scott, A. M., Lee, F. T., Tebbutt, N., Herbertson, R., Gill, S. S., et al. (2007) A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors, Proc Natl Acad Sci U S A 104, 4071-4076.
110. Hudziak, R. M., Lewis, G. D., Winget, M., Fendly, B. M., Shepard, H. M., et al. (1989) p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor, Mol Cell Biol 9, 1165-1172.
111. Carter, P., Presta, L., Gorman, C. M., Ridgway, J. B., Henner, D., et al. (1992) Humanization of an anti-p185HER2 antibody for human cancer therapy, Proc Natl Acad
Sci U S A 89, 4285-4289.
112. Tokuda, Y., Ohnishi, Y., Shimamura, K., Iwasawa, M., Yoshimura, M., et al. (1996) In vitro and in vivo anti-tumour effects of a humanised monoclonal antibody against c-erbB-2 product, Br J Cancer 73, 136c-erbB-2-1365.
113. Baselga, J., Norton, L., Albanell, J., Kim, Y. M., and Mendelsohn, J. (1998) Recombinant humanized anti-HER2 antibody (Herceptin) enhances the antitumor activity of paclitaxel and doxorubicin against HER2/neu overexpressing human breast cancer xenografts,
Cancer Res 58, 2825-2831.
114. Slamon, D. J., Leyland-Jones, B., Shak, S., Fuchs, H., Paton, V., et al. (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2, N Engl J Med 344, 783-792.
115. Citri, A., Skaria, K. B., and Yarden, Y. (2003) The deaf and the dumb: the biology of ErbB-2 and ErbB-3, Exp Cell Res 284, 54-65.
116. Baselga, J., Perez, E. A., Pienkowski, T., and Bell, R. (2006) Adjuvant trastuzumab: a milestone in the treatment of HER-2-positive early breast cancer, Oncologist 11 Suppl 1, 4-12.
117. Cho, H. S., Mason, K., Ramyar, K. X., Stanley, A. M., Gabelli, S. B., et al. (2003) Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab, Nature 421, 756-760.
118. Agus, D. B., Akita, R. W., Fox, W. D., Lewis, G. D., Higgins, B., et al. (2002) Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth, Cancer Cell
2, 127-137.
119. Baselga, J., Albanell, J., Molina, M. A., and Arribas, J. (2001) Mechanism of action of trastuzumab and scientific update, Semin Oncol 28, 4-11.
120. Cuello, M., Ettenberg, S. A., Clark, A. S., Keane, M. M., Posner, R. H., et al. (2001) Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2, Cancer Res 61, 4892-4900.
121. Austin, C. D., De Maziere, A. M., Pisacane, P. I., van Dijk, S. M., Eigenbrot, C., et al. (2004) Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin, Mol Biol Cell 15, 5268-5282.