1
I
2
L’hypertension artérielle (HTA) est la plus fréquente des maladies cardiovasculaires, elle est également la principale cause des accidents vasculaires cérebraux, d’insuffisance cardiaque et d’insuffisance rénale.
La prise en charge thérapeutique d’une HTA vise à réduire le risque de survenue de ses complications. Cet objectif nécessite à la fois un abaissement suffisant des chiffres de la pression artérielle et la prise en charge des autres facteurs de risques cardiovasculaires (tabagisme, dyslipidémie, diabète…).
Le système rénine-angiotensine-aldostérone (SRAA) joue un rôle clé dans la physiologie et la physiopathologie cardiovasculaire et rénale. En physiologie, le SRAA intervient de façon remarquable, dans la régulation de la pression artérielle et du bilan hydrosodé.
L’effecteur principal du SRAA est l'angiotensine II impliquée, en particulier, dans la genèse de l'hypertension artérielle par ses actions sur les vaisseaux (vasoconstriction) et les glandes surrénales (sécrétion d’aldostérone et de catécholamines). Il était donc logique que la recherche d’une inhibition de ces effets devienne un objectif majeur dans la mise au point de nouveaux traitements pharmacologiques antihypertenseurs.
Depuis une quarantaine d’années, l’exploration approfondie du SRAA, de la cascade enzymatique catalysée par la rénine et l’enzyme de conversion et du mode d’action de l’angiotensine II a permis de développer successivement plusieurs outils pharmacologiques puis thérapeutiques.
3
Le blocage du SRAA a des effets hémodynamiques, antihypertenseurs et anti-inflammatoires puissants et ralentit la progression de la maladie rénale au-delà des résultats obtenus par la baisse de la tension artérielle.
Après les décennies 70 et 80 qui ont vu successivement la mise sur le marché des bêta-bloquants, puis des inhibiteurs de l’enzyme de conversion (IEC), la décennie 90 a vu celle des antagonistes de l’angiotensine II ou sartans. Alors que les premiers inhibiteurs du SRA développés ont été les IEC, il aurait été plus intéressant d’un point de vue physiopathologique, de développer des médicaments qui ciblent plus spécifiquement le SRA tels qu’un inhibiteur direct de la rénine ou un ARA.
Ces médicaments agissant sur des éléments clés de ce système ont constitué des avancées majeures dans le traitement pharmacologique de l’hypertension artérielle et des maladies cardiovasculaires comme l’insuffisance cardiaque et l’infarctus du myocarde.
Des recherches sont en cours visant à développer de nouvelles stratégies thérapeutiques pour bloquer plus efficacement encore le SRAA de façon à mieux protéger les patients à haut risque cardio-rénal.
Cette thèse s’articulera donc selon 2 axes principaux : Une première partie consacrée aux généralités sur la physiopathologie de l’hypertension artérielle et la physiologie du système rénine-angiotensine et une seconde partie présentant les propriétés pharmacologiques des inhibiteurs de ce système ainsi que leur intérêt dans le traitement de l’hypertension artérielle et des maladies cardiovasculaires.
4
P
P
A
A
R
R
T
T
I
I
E
E
I
I
:
:
G
5
CHAPITRE I
RAPPELS SUR LA PRESSION
ARTERIELLE ET
6
I. PHYSIOLOGIE DE LA PRESSION ARTERIELLE
SANGUINE
I.1. Définition de la pression artérielle
La Pression Artérielle (PA) est la pression qui règne sur la paroi interne des vaisseaux pendant la circulation sanguine. On la dénomme également Tension artérielle. Elle est physiologiquement définie par la formule :
P = Q x R
P = pression artérielle,Q = débit cardiaque,
R= les résistances périphériques, essentiellement artériolaires [171].
Il faut aussi tenir compte de la masse sanguine circulante (MS), car ce sont ces trois facteurs (Q, R, MS) qui seront modifiés par les agents antihypertenseurs [147].
La pression artérielle est une variable entre deux extrêmes, la PA systolique (PAS) et la PA diastolique (PAD). La PAS résulte de l'activité du cœur pendant l'éjection sanguine systolique contre les résistances périphériques, on l'appelle encore en langage courant «maxima». La PAD, appelée en langage courant «minima», dépend seulement des résistances périphériques et de la volémie. Entre ces deux valeurs de PAS et de PAD, on a la pression différentielle, appelée encore la pression pulsée et c'est elle qui donne la sensation du pouls [171].
7
La pression artérielle (PA) normale chez l’adulte se définit comme une pression inférieure ou égale à 140/90 mm Hg.
I.2. Facteurs déterminants de la PA
I.2.1. Le débit cardiaque
Le débit cardiaque est le produit du volume d'éjection systolique par la fréquence cardiaque.
Q = Fc × VES.
Fc = Fréquence cardiaque,VES = Volume d'éjection systolique
Ces deux facteurs varient dans le même sens et sont sous contrôle du système neurovégétatif [56].
I.2.2. Les résistances artérielles
Selon Poiseuille, les résistances artérielles sont proportionnelles à la viscosité et à la longueur du vaisseau et inversement à son rayon.
R = 8.Ω.L / π.r
4* La longueur du vaisseau (L) est constante.
* La viscosité (Ω) ne varie que dans certaines circonstances pathologiques (les polyglobulies).
8
* Seul le calibre (r) peut faire varier la pression. Sa variation est assurée essentiellement par les artérioles (car elles ont une importante musculature lisse). [56]
I.2.3. Autres facteurs
La distensibilité de la paroi artérielle amortit les variations instantanées de la pression. Elle assure l’écoulement sanguin au cours de la diastole en restituant l’énergie emmagasinée lors de la systole [56].
I.3. Régulation de la PA
I.3.1. Régulation nerveuse
C’est une régulation rapide qui agit sur la vasomotricité et le débit cardiaque. Elle met en jeu les barorécepteurs de façon réflexe [56,136].
I.3.2. Régulation hormonale à moyen et à long terme [56,136]
Système rénine–angiotensine–aldostérone (SRAA)
L'appareil juxta-glomérulaire sécrète la rénine sous l’influence des variations de pression régnant dans l’artériole afférente. La rénine attaque l’angiotensinogène hépatique donnant l’angiotensine I transformée en angiotensine II sous l’effet d’une enzyme de conversion. L’angiotensine II est hypertensive par une vasoconstriction intense et en stimulant la sécrétion de l'aldostérone [150,271].
L’aldostérone
Elle intervient sur la PA en contrôlant la volémie lié au mouvement du sodium (Na+). Sa sécrétion est mise en jeu par le SRA et par la diminution du
9
réabsorption du Na+ dans le tubule distal et donc elle entraîne une augmentation de la PAS.
Les catécholamines
Ce sont des hormones hypertensives libérées par la médullosurrénale qui renforce la réaction de «fuite ou de lutte» amorcée par le sympathique en cas de stress.
Peptide atrial natriurétique (PAN)
Il est sécrété par les myocytes auriculaires en cas d’augmentation de la volémie ou d’hypertension auriculaire, il entraîne une vasodilatation et une excrétion hydrosodée ce qui concourt à la normalisation de la PAS.
Hormone anti-diurétique (ADH)
Elle est sécrétée par la neurohypophyse en cas d’hypovolémie ou d’hyperosmolarité d’eau (>280 mol/kg).
Les prostaglandines rénales
Facteurs endothéliaux (l’endothéline et l’oxyde nitrique (NO)) [56,136].
I.3.3. Régulation intrinsèque de la PAS
Autorégulation myogénique
C’est la réponse propre des fibres musculaires lisses à l’étirement, quand la PA augmente ces fibres se contractent et inversement, quand la PA diminue elles se relâchent.
10
Autorégulation chimique Elle est assurée par:
Des facteurs métaboliques vasodilatateurs tels que l’anoxie, l’hypercapnie, la diminution du pH, les ions potassiques (K+), l’acide lactique. L’effet de ces facteurs nécessite un temps de latence. [136].
Des facteurs humoraux vasodilatateurs (la Bradykinine)
Des facteurs humoraux constricteurs (sérotonine des plaquettes). [136]
II. PHYSIOLOGIE DE L’HYPERTENSION ARTERIELLE
L’hypertension artérielle (HTA) est définie par l’Organisation Mondiale de la Santé (OMS), comme une pression artérielle systolique (PAS) égale ou supérieure à 140 mmHg et/ou une pression artérielle diastolique (PAD) égale ou supérieure à 90 mmHg, il ne s’agit évidement que d’une valeur limite arbitraire fixée dans un but d’orienter la décision de traiter un hypertendu. En fait, ces chiffres seront modulés selon l’âge et le terrain (femme enceinte, diabète…)
Elle représente la principale cause de morbi-mortalité cardiovasculaire. Le traitement a réduit notablement l’incidence de ses complications. Ainsi, l’HTA pose un problème de santé publique par le nombre de sujets concernés [199, 19,79].
II.1. Etiologies et classification
Dans 95 % des cas, le bilan ne permet pas de trouver une cause précise, on parle d’HTA «essentielle». Dans les 5 % restant, une cause précise peut être identifiée, l’HTA est dite «secondaire». En plus de causes organiques (rénales
11
ou surrénales), des circonstances comportementales peuvent contribuer à l’élévation de la pression artérielle, comme l’excès d’alcool ou l’obésité. En revanche, le stress, improprement appelé «tension nerveuse», est peu responsable d’HTA car les élévations tensionnelles imputables au stress ne sont souvent que temporaires. Enfin il existe des HTA génétiques (notamment de rares causes monogéniques) [208].
II.1.1. HTA essentielle
95 % des HTA n’ont pas de cause retrouvée. On parle alors d’HTA essentielle. Plusieurs facteurs peuvent favoriser l’apparition d’une HTA comme l’hérédité, les médicaments ou les toxiques (réglisse, vasoconstricteurs), l’excès pondéral (25 % des sujets en surpoids sont hypertendus), les facteurs nutritionnels ou environnementaux (consommation sodée, alcool, sédentarité, stress) [199, 19,79].
II.1.2. HTA secondaires
Les HTA secondaires sont relativement rares et leur fréquence au sein d’une population d'hypertendus tout venant est d'environ 5 à 10 %. Elles sont rattachées à une étiologie et une physiopathologie plus ou moins connue :
HTA secondaire aux maladies rénales telles que l’insuffisance rénale aigue ou chronique, la néphropathie unilatérale et la sténose des artères rénales.
HTA secondaire aux maladies de la surrénale telles que l’hyperaldostéronisme primaire, le phéochromocytome et le syndrome de Cushing.
12
HTA iatrogène: elle peut être secondaire à l’administration des vasoconstricteurs par voie nasale, des contraceptifs oestro-progestatifs, des corticoïdes, des anti-inflammatoires non stéroïdiens...
HTA toxique HTA gravidique
HTA secondaire au syndrome d’apnée du sommeil [199, 65].
II.2. Complications de l’HTA
Une pression diastolique et/ou systolique chroniquement élevée est associée à des changements structuraux et fonctionnels au niveau rénal, vasculaire et cardiaque qui favorisent l’apparition d’accidents vasculaires cérébraux et cardiaques [97].
Quelque soit l’origine de l’HTA (essentielle ou secondaire à une pathologie) elle provoque des effets délétères sur de nombreux organes:
Le cœur: les complications cardiaques de l’HTA peuvent se manifester par une hypertrophie ventriculaire gauche, une dilatation des cavités, une altération des fonctions systolique et diastolique, une insuffisance cardiaque et une insuffisance coronaire (angor, infarctus).
Les reins: les complications rénales de l’HTA peuvent se manifester par :
Une néphroangiosclérose secondaire à des lésions artériolaires, elle s’exprime d’abord par une microalbuminurie puis une macroalbuminurie, parallèlement à une baisse de la clairance glomérulaire. L’insuffisance rénale est souvent tardive.
13
Une ischémie rénale secondaire à une atteinte des artères rénales
Le cerveau: les complications cérébrales de l’HTA peuvent conduire à:
Des accidents vasculaires cérébraux: l’HTA demeure la principale cause des AVC qui peuvent être soit hémorragiques, soit ischémiques.
Une encéphalopathie hypertensive à l’occasion d’une HTA maligne avec un tableau d’hypertension intracrânienne.
Une démence artériopathique par atteinte diffuse des artères cérébrales par de l'athérome
Les vaisseaux: L’hypertension artérielle est à l’origine de nombreuses complications vasculaires qui peuvent conduire à:
Une artériopathie oblitérante et anévrisme par atteinte des grosses artères. Atteinte des organes sensoriels (rétinopathie, atteinte
14
CHAPITRE II
TRAITEMENT DE
15
I. TRAITEMENT NON MEDICAMENTEUX
La prise en charge de l’HTA commence par la mise en place de mesures non pharmacologiques. Ces règles hygiéno-diététiques sont poursuivies pendant 3 mois avant d’envisager un traitement médicamenteux. Chez certains patients ayant une HTA limite, elles peuvent suffire à normaliser les chiffres de la pression artérielle (PA). Chez d’autres, la mise en route d’un traitement antihypertenseur est nécessaire pour normaliser la PA, mais dans tous les cas, l’observance des mesures non pharmacologiques est fondamentale [183].
L’objectif des règles hygiéno-diététiques est de faire baisser le niveau tensionnel de l’individu, de diminuer au maximum le recours au traitement pharmacologique et le cas échéant d’en tirer l’efficacité maximale, de traiter les facteurs de risque associés et de s’intégrer dans la prévention primaire de l’HTA et des facteurs de risque cardiovasculaires à l’échelle d’une population.
Ces mesures hygiéno-diététiques comprennent [183]:
I.1. Arrêt du tabagisme
Le tabac entraîne une augmentation de la PA dans les 15 à 30 minutes qui suivent la consommation d’une cigarette. Il aggrave le pronostic cardiovasculaire par un rôle indépendant de la PA. Il faut largement inciter le patient hypertendu à arrêter de fumer.
I.2
.Réduction pondérale
L’obésité est associée à une augmentation des chiffres de PA. La réduction pondérale permet d’abaisser les chiffres de PA. Le régime constitue donc une étape primordiale dans la prise en charge de l’hypertendu obèse. Devant une
16
HTA légère chez un patient obèse, il faut commencer par une cure d’amaigrissement pendant 3 à 6 mois qui peut suffire à normaliser la PA avant d’envisager un traitement médicamenteux. Chez l’hypertendu traité qui présente un excès pondéral, le régime peut permettre d’alléger le traitement antihypertenseur, voire de l’arrêter.
I.3. Activité physique
L’exercice physique est un adjuvant précieux des traitements antihypertenseurs. Il procure en lui-même un bénéfice, au moins immédiat, sur le contrôle des PA systoliques et diastoliques. Il améliore la qualité de l’observance, aide à la stabilité du poids et contribue positivement à la sensation de «mieux-être» des sujets [2].
Le patient sédentaire a un risque de développer une HTA de 20 à 50% plus important que le patient pratiquant une activité physique régulière. La pratique régulière (15 à 20min, trois fois par semaine) d’une activité physique participe à la diminution des chiffres de PA (un exercice d’intensité modérée permet de diminuer la PA systolique d’environ 5 à 10mmHg).
I.4. Réduction de la consommation d’alcool
Bien que n’étant pas un facteur de risque d’athérosclérose dans la majorité des enquêtes épidémiologiques, la consommation excessive d’alcool est associée
à différentes pathologies cardiovasculaires: hypertension artérielle,
cardiomyopathie dilatée primitive, accident vasculaire cérébral, notamment hémorragique [25].
17
L’alcool élève le niveau de PA, il peut interférer avec le traitement médicamenteux et induire une résistance aux antihypertenseurs [183].
I.5. Réduction des apports en sodium
La relation positive entre la consommation duils sel et le niveau de pression artérielle laisse présager qu’une diminution des chiffres de pression artérielle pourrait être obtenue par réduction des apports en sel [25].
Une réduction modérée des apports quotidiens en chlorure de sodium aux environs de 100 mmol/j fait baisser les chiffres de PA systolique d’environ 5 mmHg.
Les patients hypertendus doivent suivre un régime modérément salé apportant environ 100 mmol/j de sel. Il ne faut pas prescrire de régime sans sel strict pour une HTA non compliquée.
I.6. Optimisation des apports en potassium
Un régime alimentaire supplémenté en potassium permet d’abaisser les chiffres de PA. Une alimentation riche en potassium est donc souhaitable chez les patients hypertendus.
Les sels potassiques doivent être utilisés avec prudence, notamment chez les sujets âgés ou chez les patients traités par des diurétiques épargneurs de potassium ou des IEC afin d’éviter tout risque d’hyperkaliémie [183].
18
I.7. Thérapeutiques comportementales
Le stress émotionnel peut provoquer une élévation aiguë de la PA. Issues des pratiques orientales de relaxation psychique et physique (yoga, zen, méditation transcendantale...), les techniques comportementales se proposent d’éduquer l’individu à contrôler volontairement les réponses du système nerveux sympathique aux différents stress quotidiens. La permanence de cet effet à distance des moments de relaxation, l’applicabilité de ces méthodes au-delà du champ d’action de leurs initiateurs restent toutefois à démontrer. S’il est peu
probable qu’elles pourraient être généralisées, ces méthodes non
médicamenteuses s’inscrivent dans un comportement d’ensemble qui, orienté et contrôlé, permet un meilleur équilibre physique et psychique, et secondairement une meilleure observance des traitements [2].
II. TRAITEMENT MEDICAMENTEUX
Les mesures non médicamenteuses améliorent le pronostic cardiovasculaire non seulement en diminuant la pression artérielle mais également en agissant favorablement sur les paramètres du bilan métabolique dans le dessein d’une moindre évolutivité de l’athérosclérose, en particulier coronaire. En cas d’échec des mesures non médicamenteuses en termes d’objectif tensionnel, un traitement médicamenteux est donc instauré [25].
II.1. Classification des antihypertenseurs
Les antihypertenseurs appartiennent à sept grandes classes thérapeutiques qui sont les diurétiques, les bêtabloquants, les inhibiteurs de l’enzyme de conversion (IEC), les inhibiteurs calciques, les antagonistes des récepteurs de
19
l’angiotensine II, les vasodilatateurs et les antihypertenseurs centraux sans oublier de citer les inhibiteurs directs de la rénine comme nouveaux antihypertenseurs [2].
II.1.1
Les diurétiquesLes diurétiques sont des agents pharmacologiques qui ont pour but
d’augmenter l’excrétion rénale du sodium (Na+) et d’eau.
Il existe trois types principaux de diurétiques dont le site d’action se place différemment au niveau du néphron:
Les thiazidiques et apparentés: l’action de ces diurétiques se situe entre la branche ascendante de Henlé et le segment de dilution distal. Ils n’ont pas d’action sur la concentration urinaire proximale, mais ils inhibent la réabsorption du chlorure de sodium.
Ce type de diurétiques n’est pleinement efficace que lorsque la fonction rénale est normale. Ils sont contre-indiqués car inefficaces en cas d’insuffisance rénale sévère.
Les diurétiques de l’anse: ils bloquent les mouvements de chlore et de sodium au niveau de la branche ascendante de Henlé, empêchant ainsi
la réabsorption du Na+. Ces diurétiques sont puissants par leur action rapide
et brève et ils ne sont actifs en cas d’insuffisance rénale.
les diurétiques distaux: leur action se fait au niveau du tube contourné distal ; ils diminuent l’excrétion du potassium et d’ion H+ et augmentent la fraction de sodium excrétée d’environ 2%. Ce sont des
20
épargneurs potassiques, faiblement natriurétiques et contre-indiqués en cas d’insuffisance rénale.
Les diurétiques provoquent une natriurèse, entraînant une contraction du volume plasmatique et extracellulaire, une diminution initiale du débit cardiaque et une baisse de la PA. On observe ensuite un retour vers les valeurs initiales du volume plasmatique et du débit cardiaque et l’apparition d’une diminution des résistances périphériques.
II.1.2. Les bêtabloquants
Les bêtabloquants sont des antagonistes spécifiques et compétitifs des récepteurs bêta-adrénergiques postganglionnaires du système nerveux sympathique. Le mécanisme d’action des bêtabloquants n’est pas encore éclairci ; trois hypothèses principales sont invoquées pour expliquer l’action des bêtabloquants: la diminution de la contractilité cardiaque, la diminution de la
libération de rénine et une action sur le système nerveux.
Les antagonistes bêta-1-adrénergiques ou «cardiosélectifs» ont peu d’affinité pour les récepteurs bronchiques et vasculaires, respectant ainsi les effets vasodilatateurs périphériques des catécholamines circulantes et l’équilibre entre récepteurs alpha et bêta-adrénergiques. Ils limitent le risque d’aggravation d’un trouble de la microcirculation ou d’une hypoglycémie.
Certains bêtabloquants sont doués d’une activité agoniste partielle ou «activité sympathomimétique intrinsèque» (ASI). Ils ont un effet inhibiteur moins marqué sur la fréquence et le débit cardiaques.
21
Le labétalol se situe à part, car il exerce des propriétés alpha et bêtabloquantes. Il peut être utilisé en cas de trouble de la circulation périphérique.
Le céliprolol possède une action bêta-1-inhibitrice et une action bêta-2-agoniste. Son profil pharmacologique permettrait son utilisation en cas de broncho-pneumopathie chronique obstructive.
II.1.3. Les inhibiteurs de l’enzyme de conversion de l’angiotensine
L’effet antihypertenseur des IEC résulte principalement de l’inhibition de la synthèse d’angiotensine II. L’enzyme de conversion est l’enzyme responsable de la transformation d’angiotensine I en angiotensine II, hormone hémodynamiquement active. Les IEC entrent en compétition avec l’angiotensine I au niveau des sites catalytiques de l’enzyme. L’inhibition enzymatique est spécifique, les IEC ne bloquent pas d’autres protéases.
Les effets de l’inhibition de l’enzyme de conversion portent [215]:
sur le système rénine-angiotensine-aldostérone les IEC entraînent une augmentation de l’activité rénine plasmatique, une augmentation du taux d’angiotensine I, une réduction du taux d’angiotensinogène et surtout une diminution des concentrations plasmatiques d’angiotensine II et d’aldostérone.
sur le système kallicréine-kinine, par un blocage de la dégradation de la bradykinine.
22
La résultante en est une suppression des effets vasopresseurs et antinatriurétiques de l’angiotensine II et de l’aldostérone, elle peut-être potentialisée par l’action vasodilatatrice et natriurétique de la bradykinine.
sur les prostaglandines: Les IEC pourraient également exercer leurs effets en stimulant la production de prostaglandines, vasodilatatrices et natriurétiques.
II.1.4. Les inhibiteurs calciques
À l’intérieur de cette classe hétérogène, on distingue habituellement quatre familles ou groupes chimiques dont les chefs de file possèdent des profils pharmacologiques différents, liés à leur sélectivité tissulaire ou au type de canal calcique bloqué préférentiellement.
la nifédipine , représentant du groupe des dihydropyridines (DHP), possède une sélectivité vasculaire marquée qui l’expose à une moindre réduction de la contractilité et de la conduction cardiaque que le vérapamil, chef de file des phénylalkylamines, dont la sélectivité cardiaque est plus importante. Le diltiazem, chef de fil des benzothiazépines, présente un profil intermédiaire. A l’inverse des molécules précédentes, bloqueurs préférentiels des canaux de type L, le mibéfradil, dérivé tétralol non commercialisé, agit préférentiellement sur les canaux de type T [149].
Les inhibiteurs calciques semblent agir en inhibant le transfert transmembranaire du calcium. Ils diminuent ainsi le taux du calcium libre intracellulaire, ce qui a pour conséquence de réduire le tonus du muscle lisse vasculaire. Cet effet s’exerce essentiellement au niveau précapillaire,
c’est-à-23
dire sur les vaisseaux de petit calibre dont le tonus vasoconstricteur est anormalement élevé chez la plupart des hypertendus. Il s’agit du principal mécanisme d’action des antagonistes calciques sur la PA [2].
II.1.5. Les antagonistes des récepteurs de l’angiotensine II
Après le succès des IEC dans les années 1980, notamment dans le traitement de l’insuffisance cardiaque et de la néphropathie diabétique, il est apparu que le blocage du système rénine-angiotensine n’était pas maximal, en raison notamment du rétrocontrôle exercé par la rénine et l’angiotensine I. Aussi, le blocage compétitif des récepteurs AT1 de l’angiotensine II par des
inhibiteurs peptidiques, puis non peptidiques, est devenu un mode thérapeutique de l’HTA [105].
Tous les antagonistes des récepteurs de l’angiotensine II sont des
antagonistes non peptidiques et sont tous spécifiques des récepteurs AT1. En
grande majorité, les antagonistes AT1 sont non compétitifs [2].
L’administration d’un antagoniste des récepteurs AT1 s’accompagne d’une
augmentation dose-dépendante de la rénine dans le plasma, liée à l’interruption du rétrocontrôle négatif de l’angiotensine II sur la sécrétion de rénine par
24
II.1.6. Les vasodilatateurs
Les vasodilatateurs artériolaires directs sont capables de réduire le tonus artériolaire et devraient abaisser la PA de façon non spécifique dans tous les types d’hypertension.
La classe thérapeutique des vasodilatateurs antihypertenseurs est très hétérogène par la chimie, mais trouve son unité dans son site d’action ; ces vasodilatateurs se fixent électivement et agissent directement sur l’artériole, en entraînant la relaxation du muscle lisse vasculaire.
Vasodilatateurs artériolaires directs
Les vasodilatateurs directs ont pour effet quasi exclusif une relaxation du muscle lisse vasculaire artériolaire, leurs autres effets étant pour l’essentiel des adaptations secondaires à la vasodilatation. On cite comme molécules appartenant à cette classe pharmacologique:
L’hydralazine et la dihydralazine
Le minoxidil dont l’emploi est réservé aux hypertensions réfractaires, en cas d’échec des polythérapies habituelles
Alphabloquants
La prazosine est un vasodilatateur indirect et fut le premier alphabloquant disponible par voie orale [110]. C’est un alpha-1-bloquant remarquablement sélectif qui antagonise la noradrénaline par compétition au niveau de ses récepteurs alpha-1 postsynaptiques périphériques [92].
25
La vasodilatation induite par la prazosine est liée à l’inhibition de la vasoconstriction noradrénergique, sans effet sur la vasoconstriction induite par l’angiotensine II.
Urapidil: Comme la prazosine, l’urapidil abaisse la PA via un blocage des récepteurs alpha-1-postsynaptiques, avec réduction des résistances périphériques totales sans modification de la fréquence ou du débit cardiaque et via un effet sur la régulation centrale de la PA.
II.1.7. Les antihypertenseurs centraux
Les antihypertenseurs centraux inhibent le système nerveux sympathique au niveau central. Il s’agit principalement de l’alphaméthyldopa, de la clonidine, de la guanfacine, de la rilmenidine, du guanoxabenz et de la moxonidine. L’importance des effets secondaires de cette classe thérapeutique, notamment l’hypotension orthostatique chez le sujet âgé, l’absence d’effet cardio ou néphroprotecteur démontré et le choix thérapeutique offert par les autres classes thérapeutiques en ont limité les indications.
II.2. Choix des médicaments antihypertenseurs
Le choix de l’antihypertenseur se fait en fonction des recommandations internationales, du terrain et d’éventuelles pathologies associées et des habitudes du prescripteur [183].
Dans l’HTA essentielle non compliquée, les cinq classes
d’antihypertenseurs majeurs (les diurétiques thiazidiques, les bêtabloquants, les inhibiteurs calciques, les inhibiteurs de l’enzyme de conversion (IEC) et les
26
antagonistes des récepteurs de l’angiotensine II (ARA-II) ont montré un bénéfice sur la morbi-mortalité cardiovasculaire dans les essais cliniques.
Ces cinq classes d’antihypertenseurs peuvent donc être proposées en première intention dans la prise en charge d’un hypertendu essentiel non compliqué.
Le choix d’un traitement médicamenteux sera adapté à chaque patient en fonction:
des indications préférentielles de certaines classes dans des situations cliniques particulières (en accord avec les études cliniques) ;
de l’efficacité et de la tolérance des médicaments déjà pris par le patient;
des interactions médicamenteuses possibles [199];
de l’existence de comorbidités pouvant justifier ou contre-indiquer certains antihypertenseurs ;
du coût du traitement et de sa surveillance, en sachant que le diurétique thiazidique fait partie des classes dont le coût journalier est le plus faible.
27
II.3. Stratégie d’adaptation du traitement antihypertenseur
II.3.1. Monothérapie initiale
On débute en général un traitement antihypertenseur par une monothérapie ou par une plurithérapie faiblement dosée ayant l’indication en première intention. Pour les monothérapies, l’une des cinq classes pharmacologiques suivantes est utilisée: les bêtabloquants, les diurétiques, les antagonistes calciques, les IEC et les antagonistes des récepteurs à l’angiotensine II [25].
La monothérapie permet habituellement une normalisation des chiffres tensionnels chez 50 à 70%des hypertendus.
En cas d’inefficacité totale de la monothérapie initiale, il est recommandé de l’arrêter et de lui substituer un produit appartenant à une autre classe. C’est la stratégie de «la monothérapie séquentielle». On débute par un premier traitement antihypertenseur dont on adapte la posologie. Si il n’y a pas de normalisation tensionnelle ou d’effet tensionnel satisfaisant après 4 à 6 semaines de traitement maximal, on passe à un autre médicament appartenant à une autre classe d’antihypertenseur (figure 1).
On peut essayer ainsi différentes classes pharmacologiques en monothérapie avant d’avoir recours à une association. Cette stratégie permet de n’utiliser qu’une seule molécule et donc de limiter les effets secondaires et d’accroître l’observance du traitement.
La monothérapie séquentielle est cependant difficile à appliquer chez l’hypertendu en pratique quotidienne. Le succès de cette approche pour un
28
patient donné dépend en effet largement du hasard puisqu’il faut trouver le produit qui soit à la fois efficace et bien toléré.
Figure 1: Stratégie d’adaptation du traitement antihypertenseur et
29
II.3.2. Plurithérapies fixes
Le fait que l’HTA présente de multiples mécanismes a motivé l’apparition
de plurithérapies fixes microdosées. En cas d’efficacité partielle de la
monothérapie initiale, il convient d’adjoindre une molécule d’une autre classe plutôt que d’augmenter les doses du médicament initial (figure 2). Cette association d’antihypertenseurs permet de contrôler les chiffres tensionnels de 70 à 90% des patients. Par ailleurs, cette stratégie diminue la survenue de certains effets secondaires dose-dépendants (trouble de la kaliémie sous diurétiques, flush et oedème sous antagonistes calciques) [25].
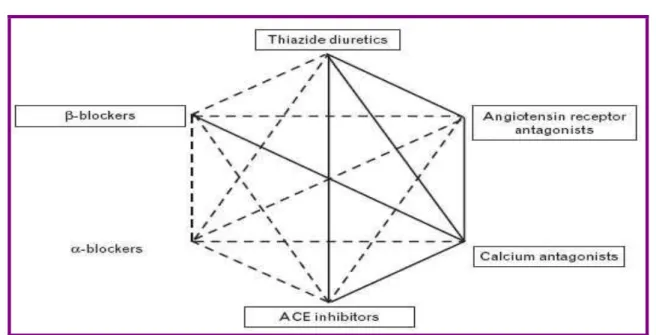
Les associations à privilégier pour leur synergie d’action sont les suivantes (figure 3):
bêtabloquant + diurétique ;
bêtabloquant + antagoniste calcique ; IEC + diurétique ;
IEC + antagoniste calcique ; antagoniste calcique + ARA II; ARA II + diurétique
Il existe des associations d’antihypertenseurs à doses fixées qui permettent
une prise médicamenteuse unique contenant plusieurs principes
30
Figure 2: les associations possibles entre les différentes classes
d’antihypertenseurs (les associations les plus rationnelles sont figurées par des lignes pleines) [267]
Figure 3: Schéma résumant les associations des classes thérapeutiques
31
A l’inverse, certaines associations d’antihypertenseurs sont déconseillées car elles augmentent le risque d’effets secondaires :
IEC+diurétique épargneur de potassium en raison du risque accru d’hyperkaliémie
bêtabloquant+vérapamil et bêtabloquant + diltiazem : les deux associations doivent être évitées en raison du risque accru de troubles conductifs
alphabloquant+antagoniste calcique : car cette association présente risque accru d’hypotension orthostatique.
En cas d’échec de la bithérapie antihypertensive, il convient de rechercher une cause de la résistance au traitement antihypertenseur (mauvaise observance, fausse résistance, HTA secondaire, interaction médicamenteuse). Ce n’est qu’après avoir éliminé ces différentes étiologies qu’une trithérapie peut être initiée [85, 243].
32
CHAPITRE III
LE SYSTEME
33
I. SYSTEME RENINE-ANGIOTENSINE
I.1. Introduction
Le système rénine-angiotensine, l’un des principaux complexes de régulation de la pression sanguine, est distribué entre le sang circulant et l’espace péricellulaire de l’interstium tissulaire. Il participe en physiologie et en pathologie de la régulation de la vasomotricité et du remodelage tissulaire dans le système cardiovasculaire. Dans le cadre de ces effets, le système rénine-angiotensine tissulaire agit sur les cellules musculaires lisses vasculaires et les fibroblastes, tandis que le système rénine-angiotensine plasmatique a pour cibles les cellules endothéliales et les leucocytes circulants [32].
Le système rénine-angiotensine-aldostérone gouverne l’homéostasie hydrosodée dans l’organisme. Il s’agit d’une cascade enzymatique dont le seul précurseur connu est l’angiotensinogène et qui aboutit à la synthèse d’un octapeptide vasoactif, l’angiotensine II [153].
Le système rénine-angiotensine-aldostérone (SRAA) est un superbe exemple de l’application en clinique humaine des données de la recherche fondamentale. La découverte et l’utilisation des inhibiteurs du système rénine-angiotensine, qu’il s’agisse d’inhibiteurs de l’enzyme de conversion de l’angiotensine (ECA), de bloqueurs des récepteurs de l’angiotensine II ou actuellement d’inhibiteurs de la rénine illustrent bien ce que l’on appelle aujourd’hui «la recherche translationnelle» [53].
34
I.2. Les composants du système rénine-angiotensine
I.2.1. Angiotensinogène [153]
L’angiotensinogène est le substrat unique et spécifique de la rénine [246]. Il s’agit d’une glycoprotéine de 60 kDa constituée de 452 acides aminés dont les dix acides aminés de l’Ang I comportant le site d’hydrolyse par la rénine à l’extrémité N-terminale et les 442 acides aminés du des-Ang I-angiotensinogène porteur d’un site de clivage C-terminal [48].
L’angiotensinogène est un peptide sécrété par les hépatocytes de façon constitutive et n’est donc pas, contrairement à la rénine, stocké dans des granules de sécrétion. Une hépatectomie totale entraîne une chute du taux d’angiotensinogène plasmatique [242]. Les sujets porteurs d’une cirrhose hépatique ou d’une anastomose portocave ont un taux d’angiotensinogène plasmatique bas et une pression artérielle diminuée.
La sécrétion hépatique de l’angiotensinogène est finement régulée de façon transcriptionnelle et/ou post-transcriptionnelle par les stéroïdes, l’Ang II et les cytokines.
La concentration d’angiotensinogène plasmatique est élevée dans les hypercorticismes, pendant la grossesse, sous contraception orale œstrogénique et lors de l’inflammation. Elle est abaissée lors d’une surrénalectomie, d’une thyroïdectomie et d’un traitement inhibiteur de l’ECA [69, 68, 61]. Pendant la grossesse, l’angiotensinogène plasmatique augmente d’un facteur 4 à 6 parallèlement à l’élévation croissante du taux d’estrogènes [130] et une forme particulière d’angiotensinogène dite de «haut poids moléculaire» est sécrétée.
35
I.2.2. Rénine
Propriétés biochimiques et régulation
La rénine est une aspartylprotéase sécrétée par les cellules myoépithéliales de l’appareil juxta-glomérulaire rénal [151]. Ce sont des cellules musculaires lisses ayant subi une métaplasie endocrine et situées à l’extrémité de la paroi de l’artériole efférente.
La rénine est une glycoprotéine dont le poids moléculaire est de 40 kDa environ et qui comprend deux sous unités de 20 et 25 kDa [96]. Elle est principalement sécrétée sous forme de prorénine inactive (90 %) et peu sous forme de rénine active (10 %). L’enzyme de maturation n’est pas encore clairement connue bien que plusieurs candidats soient discutés dans la littérature [176,228]. L’activation se fait exclusivement dans les granules de sécrétion et la rénine est libérée dans la circulation après stimulation [80].
La concentration de rénine est le facteur limitant de la cascade enzymatique du système rénine-angiotensine et sa sécrétion est régulée de façon très fine et rapide, la régulation du taux d’angiotensinogène étant un mécanisme beaucoup plus lent [126]. La concentration physiologique de rénine est de l’ordre de la picomole, mais peut varier de 10 à 1 000 fois.
Quatre mécanismes principaux sont impliqués dans la régulation de la synthèse et de la sécrétion de rénine :
La stimulation des barorécepteurs situés dans la paroi de l’artériole afférente du glomérule ; la sécrétion de rénine est stimulée par une baisse de la pression de perfusion et vice-versa;
36
L’innervation sympathique avec une stimulation par les agonistes adrénergiques et une diminution de la sécrétion lors de l’utilisation de bêta-bloquants ;
Le rétrocontrôle négatif exercé par l’Ang II, par action directe, sur les récepteurs AT1 et AT2 situés au niveau des cellules de l’appareil
juxta-glomérulaire ;
Enfin, le signal paracrine généré par le débit urinaire du chlorure de sodium dans le tube distal en regard de la macula densa: la baisse de ce débit aboutit à une diminution de la réabsorption du chlore par les cellules
juxta-glomérulaires stimulant la sécrétion de rénine et inversement.
Rôles de la rénine
La rénine a longtemps été considérée comme une enzyme ayant pour seul rôle la génération d’Ang I avec une action du SRA essentiellement médiée par la fixation de l’Ang II à ses récepteurs. Plus récemment, on a montré que la rénine
avait des effets cellulaires indépendants de la génération d’Ang II [188].
C’est en partant de ce constat qu’on est parvenue à mettre en évidence un récepteur de la rénine sur des cellules mésangiales humaines en culture [188] puis à le cloner [189]. Ce récepteur est une protéine de 350 acides aminés, avec un unique domaine transmembranaire, et n’a pas d’homologie de structure avec d’autres familles de récepteur connues ; c’est le premier récepteur d’une aspartylprotéase décrit. Il lie de façon spécifique la rénine et la prorénine et cette liaison entraîne:
37
L’augmentation d’un facteur quatre de l’efficacité de la conversion de l’angiotensinogène en Ang I lorsque la rénine est liée à la membrane (par rapport à la rénine non liée) ;
L’activation intracellulaire de la voie des MAP-kinases mitogènes, indépendamment de la génération et de l’action de l’Ang II.
Ce récepteur a une expression élevée au niveau cardiaque, cérébral, oculaire et placentaire, et faible au niveau rénal et hépatique. Des techniques d’immunofluorescence ont permis de le localiser plus précisément au niveau du mésangium glomérulaire, de l’endothélium de l’artère rénale et des artères coronaires [190].
Ce récepteur pourrait avoir une implication physiologique importante : un rôle fonctionnel de la prorénine indépendant de l’Ang II pourrait expliquer l’efficacité des inhibiteurs du système rénine-angiotensine dans la prévention de la néovascularisation rétinienne chez l’animal [182], et chez l’homme dans la correction de l’hypertrophie myocardique et aortique et la prévention de la néphroangiosclérose indépendamment du niveau tensionnel [190].L’inhibition de ce récepteur pourrait représenter une nouvelle perspective thérapeutique [182].
I.2.3. Angiotensine I
L’Ang I est un décapeptide dépourvu d’activité biologique propre mais il est clivé par l’enzyme de conversion pour former un octapeptide actif qui est l’Ang II. Il est également dégradé en desaspartyl-angiotensine I par une aminopeptidase et en angiotensine par une endopeptidase [178].
38
I.2.4. Enzyme de conversion
Définition et propriétésL’enzyme de conversion, ou kininase II (ECA) est une ectoenzyme membranaire de 180 kDa qui existe sous deux formes [21]: l’une soluble dans le plasma, le liquide céphalorachidien (LCR) et de nombreux fluides physiologiques et l’autre ancrée à la membrane plasmique de différents types de
cellules (cellules endothéliales, cellules épithéliales et cellules
neuroépithéliales). C’est la forme tissulaire qui joue un rôle prépondérant dans la conversion d’Ang I en Ang II
L’ECA possède deux sites catalytiques distincts appelés site N-terminal et C-terminal. Ces deux sites ont certaines propriétés communes et quelques propriétés distinctes. Les deux sites catalysent le clivage de l’angiotensine I et de la bradykinine avec une efficacité identique. Le domaine N-terminal clive de manière physiologique l’angiotensine 1-7 [58] et le peptide hémorégulateur AC-SDKP [17, 218]. Le substrat physiologique du site C-terminal n’est pas connu.
Rôles de l’enzyme de conversion
L’enzyme de conversion (ECA) est une métalloprotéase à zinc qui possède deux domaines amino et carboxyterminaux avec chacun son site actif (dipeptidyl-carboxypeptidase) [274].
Elle a de multiples substrats, en dehors de l’Ang I, elle est capable de dégrader la bradykinine (BK), peptide vasodilatateur, en un métabolite inactif (BK1-7) mais aussi le tétrapeptide N-Acétyl-Séryl-Aspartyl-Lysyl-Proline
(AC-39
SDKP) impliqué dans le contrôle de la prolifération des cellules souches
hématopoïétiques [17].
L’ECA augmente donc la production d’un vasoconstricteur puissant, l’Ang II, en dégradant parallèlement un vasodilatateur potentiel, la BK.
ECA-2
Un homologue humain de l’ECA a récemment été décrit [251]. L’ECA-2 est également une métalloprotéase à zinc de 805 acides aminés ayant une homologie de structure significative avec l’ECA [151]. Contrairement à l’ECA, elle a une fonction carboxypeptidase et dipeptidyl-carboxypeptidase, ce qui aboutit à l’hydrolyse de l’Ang I en Ang 1-9 et de l’Ang II en Ang 1-7 ainsi qu’à la dégradation de la BK. En revanche, l’ECA-2 est incapable de convertir l’Ang I en Ang II et son activité enzymatique n’est pas bloquée par les inhibiteurs de l’ECA.
L’ECA-2 est donc un inhibiteur de la formation de l’Ang II en stimulant des voies alternatives de dégradation de l’Ang I. Cette enzyme a été localisée au niveau de la membrane des cardiomyocytes, des cellules de l’endothélium et du tubule rénal ainsi qu’au niveau du testicule.
La mutation du gène de l’ECA-2 n’a pas de conséquence sur le plan tensionnel mais entraîne une augmentation de la concentration d’Ang II et une anomalie de la contractilité cardiaque [55]. Cette enzyme est donc en partie un contre-régulateur physiologique de l’ECA
40
I.2.4. Angiotensine II
Définition et effets biologiques
L’angiotensine II est un octapeptide généré par clivage de l’angiotensine I (décapeptide inactif) sous l’action de l’enzyme de conversion de l’angiotensine (figure 4).
Figure 4 : Schéma illustrant la cascade enzymatique du SRA qui aboutit à la
formation de l’octapeptide active (angiotensine II) [153] Les principaux effets biologiques de l’angiotensine II sont [153] :
Des effets vasculaires: L’Ang II exerce une action vasopressive et trophique sur les cellules musculaires lisses des parois du système cardiovasculaire. Sur le plan vasculaire, on peut distinguer les effets
41
immédiats de l’Ang II sur l’hémodynamique et les effets tardifs sur la synthèse des constituants de la paroi artérielle [81];
Des effets surrénaliens: l’Ang II stimule la production d’aldostérone par les cellules de la zone glomérulée du cortex surrénal ;
Des effets rénaux: l’Ang II intervient dans la régulation du débit sanguin rénal et de la filtration glomérulaire par une action vasoconstrictrice des artérioles efférente et afférente glomérulaires, dans la réabsorption tubulaire de sodium (soit directement dans le tube proximal en activant l’échangeur Na/H, soit indirectement dans le canal collecteur en stimulant la sécrétion surrénale de l’aldostérone) et dans la régulation de la sécrétion de rénine au niveau de l’appareil juxta-glomérulaire ;
Des effets cérébraux: à ce niveau, l’Ang II agit par la stimulation de la soif en activant la sécrétion de vasopressine et par la régulation centrale de la pression artérielle ;
Des effets sur le système nerveux sympathique: l’Ang II stimule la libération de noradrénaline ;
Des effets tissulaires: l’Ang II possède une action trophique et hyperplasique.
En effet, l’Ang II a une action rapide qui vise à contrebalancer une baisse de pression artérielle et/ou de volémie alors qu’à long terme elle a une action sur le remodelage vasculaire en favorisant l’hypertrophie des cellules musculaires lisses et la production de matrice extracellulaire. Ces effets trophiques associés à son action vasoconstrictrice aboutissent à une diminution de la compliance des artères élastiques et à une élévation des résistances périphériques.
42
L’Ang II a un effet endocrine sur sa propre sécrétion en exerçant un feed-back négatif rapide sur la sécrétion de rénine et un feed-feed-back positif sur la
synthèse hépatique d’angiotensinogène [126]. Alors que les demi-vies
plasmatiques de la rénine et de l’angiotensinogène sont assez longues, l’Ang II est dégradée en quelques secondes par des peptidases en plusieurs métabolites actifs: l’Ang III qui agit sur les mêmes récepteurs que l’Ang II, l’Ang IV et l’Ang 1-7 qui possèdent leurs propres récepteurs avec des effets distincts.
Voies de formation de l’angiotensine II
L’enzyme de conversion (ECA) n’est pas la seule enzyme capable de transformer l’angiotensine I en angiotensine II, et une partie de cette synthèse peut être conservée malgré l’inhibition totale de l’ECA.
Ainsi, un groupe d’enzymes tissulaires présent au niveau aortique et dénommé CAGE (Chymostatin sensitive Angiotensin II Generating Enzyme), et la chymase (une enzyme présente chez l’homme au niveau du cœur et des vaisseaux) peuvent réaliser cette transformation. L’affinité de la chymase pour l’angiotensine I est nettement supérieure à celle de l’ECA. Elle serait responsable de 75% de la production intracardiaque d’angiotensine II chez l’homme [259, 258].
D’autres enzymes tissulaires (t-PA, cathepsine G et tonine) transforment directement l’angiotensinogène en angiotensine II [82].
Récepteurs de l’angiotensine II:
Les effets de l’Ang II sont médiés par des récepteurs membranaires faisant partie de la famille des récepteurs à sept domaines transmembranaires couplés
43
aux protéines G. Deux gènes codant pour deux récepteurs à l’Ang II dont la pharmacologie est différente ont été clonés chez l’homme [153].
Les récepteurs AT1
Le récepteur AT1 est une chaîne polypeptidique de 359 acides aminés
contenant sept segments hydrophobes intramembranaires connectés par trois boucles intra et extracellulaires. Le gène codant pour ce récepteur est exprimé dans les vaisseaux (cellules musculaires lisses), les reins, les surrénales, le cœur, le cerveau et l’hypophyse.
L’activation du récepteur AT1 par l’Ang II est responsable de la plupart des
actions visant à augmenter la pression artérielle (vasoconstriction, prolifération cellulaire, croissance tissulaire, réabsorption rénale de sodium, stimulation du
système nerveux sympathique, sécrétion de l’aldostérone...) [45].
Les récepteurs AT2:
Le récepteur AT2 est une chaîne polypeptidique de 363 acides aminés à sept
domaines transmembranaires, couplé à une protéine G, qui a seulement 34 %
d’homologie avec le récepteur AT1. Il est fortement exprimé durant le
développement fœtal dans les tissus mésenchymateux. Chez l’adulte, son expression est réduite mais persiste à un faible taux au niveau de l’endothélium vasculaire, du cœur et du rein [47].
Le mécanisme d’action principal du récepteur AT2 est médié par la
libération de BK avec pour conséquence la génération de monoxyde d’azote (NO) et GMPc [231, 232].Cette action a pour conséquences une vasodilatation,
44
une inhibition de la prolifération cellulaire et possiblement une augmentation de
la natriurèse [44].Son action s’oppose donc à celle du récepteur AT1.
Les récepteurs non AT1 non AT2
Ils ont été décrits dans les tissus cancéreux et les fibroblastes cardiaques. Leur nature et leur rôle restent à préciser.
I.2.6. Aldostérone
L’aldostérone est une hormone minéralocorticoïde sécrétée par la zone glomérulée du cortex surrénal. Contrairement à la rénine et aux angiotensines (qui sont des protéines et des peptides), l’aldostérone est un stéroïde synthétisé à partir du cholestérol par une série de réactions comprenant plusieurs étapes faisant intervenir différentes enzymes [153].
La sécrétion d’aldostérone est essentiellement régulée par l’Ang II et le potassium extracellulaire. Physiologiquement, les concentrations d’aldostérone et de rénine sont étroitement corrélées. L’Ang II stimule la biosynthèse de l’aldostérone par un effet sur les enzymes stéroïdogènes lors de sa liaison à son
récepteur AT1 situé à la surface des cellules de la zone glomérulée.
L’aldostérone a un rôle central dans la régulation de la volémie et son implication est importante dans certaines formes d’HTA et d’insuffisance cardiaque [206]. Son action principale est la régulation du transport transépithélial du sodium (réabsorption de sodium, excrétion de potassium) dans le tubule collecteur cortical du rein mais aussi dans d’autres organes comme la parotide ou le côlon. Par ailleurs, l’aldostérone a également des effets rapides, non génomiques qui ont été mis en évidence dans plusieurs types cellulaires
45
notamment les cardiomyocytes, les cellules musculaires lisses vasculaires et les cellules du muscle strié squelettique [95].
I.3.
Compartimentation du système rénine-angiotensine [179]
Le système rénine-angiotensine a été décrit comme un système endocrine par H. Goldblatt en 1934 [104]. La rénine active est synthétisée et stockée par les cellules myoépithélioïdes de l’artériole afférente du glomérule rénal. Sa sécrétion est contrôlée par divers stimuli diminuant la concentration du calcium libre dans la cellule myoépithélioïde (stimulation β-adrénergique, baisse de la tension pariétale dans l’artériole et diminution de la réabsorption du sodium et du chlore par le cotransport Na-K-2Cl de la macula densa). Inversement, l’augmentation du NaCl dans la macula densa et de l’angiotensine II freine la sécrétion de rénine en augmentant la concentration du calcium libre dans les cellules myoépithélioïdes.
La rénine active sécrétée diffuse dans les compartiments plasmatique, lymphatique et interstitiel, de même que son substrat, l’angiotensinogène, synthétisé et sécrété par le foie (il est également probable que de faibles quantités d’angiotensinogène soient directement synthétisées au niveau de la paroi artérielle). A l’opposé, les peptides dérivés de l’angiotensinogène (angiotensines I et II) n’agissent vraisemblablement que dans le compartiment dans lequel ils ont été produits.
Un point important est que si la rénine active diffuse librement dans le plasma, elle s’adsorbe dans les tissus, en particulier dans la paroi artérielle.
46
Dans le compartiment interstitiel, les résidus mannose-6 phosphate retiennent la rénine par des liaisons électrostatiques de faible affinité [263] conduisant à un enrichissement en rénine et en angiotensine du milieu interstitiel par rapport au plasma [60]. Un récepteur membranaire ayant une forte affinité pour la prorénine et la rénine active [189] a récemment été mis en évidence. Son implication physiologique et physiopathologique doit encore être précisée [54].
I.4. SRA et remodelage vasculaire
L’activation du système rénine-angiotensine in vivo peut être étudiée de deux façons: soit en analysant les effets de l’activation du système sur la structure vasculaire, soit en étudiant les conséquences de l’inhibition de l’angiotensine II dans des modèles ne dépendant pas directement de l’activation du système rénine-angiotensine. Dans tous les cas, il est toujours difficile de séparer complètement la réponse fonctionnelle sur la pression artérielle de la réponse structurale sur la paroi des vaisseaux [179].
Classiquement, la production d’angiotensine II conduit à une hypertension artérielle et à une hypertrophie de la paroi artérielle. Cette dernière s’accompagne d’une augmentation de la synthèse de matrice extracellulaire et de la sécrétion plasmatique d’antiprotéases [34].
L’hypertension, en augmentant la «tenségrité» (tension interne réglée par la conformation du cytosquelette) des cellules musculaires lisses liée à leur adhérence à la matrice extracellulaire, s’ajoute ici aux effets directs de l’angiotensine II sur la trophicité des cellules vasculaires. Inversement, lorsque l’hypertension artérielle n’est pas directement liée à l’activation du système
47
rénine-angiotensine, comme chez le rat spontanément hypertendu ou dans le modèle d’intoxication à la nitroarginine (suppression de la production endothéliale de monoxyde d’azote (NO), au potentiel vasodilatateur), le blocage du système rénine-angiotensine entraîne une baisse importante de la pression artérielle et de la trophicité de la paroi artérielle [124].
48
I.5.
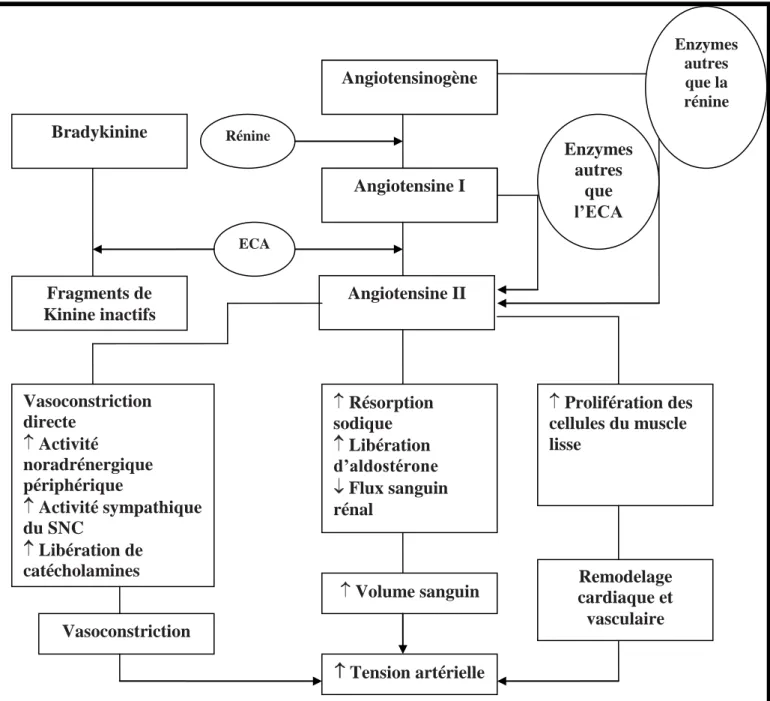
Rôle du SRA dans la pathogenèse de l’hypertension
[9]
Figure 5
: Schéma illustrant le rôle des composants du SRA dans la
pathogenèse de l’HTA Angiotensine II Enzymes autres que l’ECA Fragments de Kinine inactifs Bradykinine Rénine ECA Prolifération des cellules du muscle lisse Résorption sodique Libération d’aldostérone Flux sanguin rénal Vasoconstriction directe Activité noradrénergique périphérique Activité sympathique du SNC Libération de catécholamines surrénale Vasoconstriction Tension artérielleVolume sanguin cardiaque et Remodelage vasculaire Angiotensinogène Angiotensine I Enzymes autres que la rénine
49
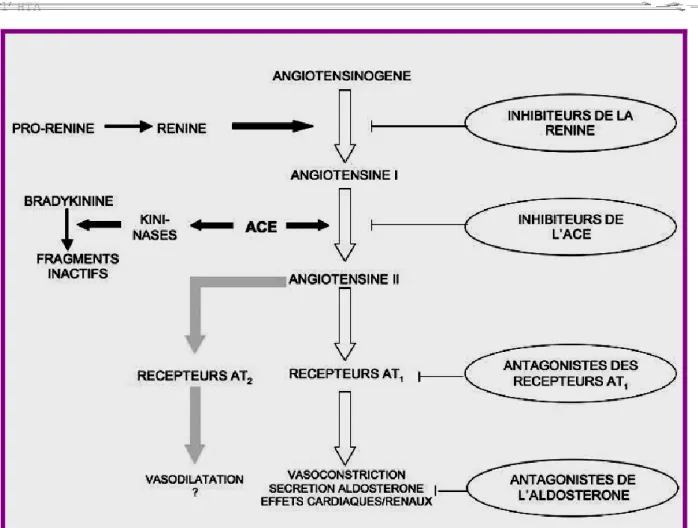
II. BLOCAGE DU SYSTEME RENINE-ANGIOTENSINE [223]
Le blocage du SRAA, au niveau systémique et local, joue un rôle protecteur dans certaines pathologies cardiaques ou rénales [240]. Le blocage du SRAA exercerait des effets pléiotropes dont certains sont encore méconnus mais pourraient contribuer aux effets thérapeutiques et ouvrir de nouvelles perspectives à l’avenir [46].L’inhibition du SRAA occupe désormais une place privilégiée dans le traitement de l’hypertension artérielle [276,144], de la décompensation cardiaque [52,135], de la protection post-infarctus [52,116] et de la néphropathie avec microalbuminurie ou protéinurie, notamment la néphropathie diabétique [273,115]. La présence d’un diabète sucré représente une condition clinique qui oriente volontiers le choix thérapeutique vers un bloqueur du SRAA de façon à tirer profit de ses effets bénéfiques à la fois vasculaires, cellulaires et métaboliques [248]. Une amélioration a été démontrée, non seulement en ce qui concerne les critères d’évaluation intermédiaire, mais aussi les événements cliniques, y compris la mortalité [223].
Il existe plusieurs possibilités pharmacologiques pour obtenir un blocage du SRAA (figure 6).
50
Figure 6 : Illustration du système rénine-angiotensine-aldostérone et des
sites d’action des différentes approches pharmacologiques pour bloquer le système [223]
Les bêta-bloquants inhibent certes partiellement la sécrétion de rénine [28], mais de façon insuffisante pour être considérés comme de vrais antagonistes du SRAA. Les antagonistes des récepteurs de l’aldostérone (spironolactone, éplérénone) ne peuvent pas, non plus, être considérés comme des inhibiteurs du SRAA à part entière puisqu’ils ne bloquent que les effets minéralocorticoïdes de l’aldostérone, mais n’interfèrent en rien avec les effets vasoconstricteurs et cellulaires de l’Ang II.
51
Les effets de l’Ang II peuvent être bloqués par un IEC [63], par un antagoniste des récepteurs AT1 (ARA) [63, 39] et/ou par un inhibiteur direct de la rénine comme l’aliskiren [213].
52
P
P
A
A
R
R
T
T
I
I
E
E
I
I
I
I
:
:
L
L
e
e
s
s
i
i
n
n
h
h
i
i
b
b
i
i
t
t
e
e
u
u
r
r
s
s
d
d
u
u
s
s
y
y
s
s
t
t
è
è
m
m
e
e
r
r
é
é
n
n
i
i
n
n
e
e
-
-
a
a
n
n
g
g
i
i
o
o
t
t
e
e
n
n
s
s
i
i
n
n
e
e
53
CHAPITRE I
LES INHIBITEURS DE L’ENZYME
DE CONVERSION DE
54
I. HISTORIQUE
La découverte des IEC trouve sa source dans les années soixante lorsqu’un pharmacologue brésilien nommé FIERRERA a montré qu’un extrait de venin de la vipère BOTHROPS JARARACA était capable de potentialiser les effets biologiques de la bradykinine qui est une substance vasodilatatrice, par inhibition de sa dégradation [223, 4].
En 1966, il a été montré que ce facteur potentialisateur de la bradykinine était capable d’inhiber une enzyme décrite comme une carboxypeptidase et appelée kininase II. En 1968, VANE remarqua que la kininase et l’enzyme de conversion avaient le même mécanisme d’action et constituaient le même enzyme.
Au début des années 70, deux types d’inhibiteurs du système rénine-angiotensine ont été utilisés dans le cadre d’études expérimentales puis en investigations physiologiques chez l’Homme ; il s’agit de la «saralazine» qui est une substance antagoniste compétitive de l’angiotensine II au niveau de ses récepteurs, et du teprotide qui est un inhibiteur peptidique de l’enzyme de conversion de l’angiotensine [178].
Le teprotide n’est malheureusement utilisable que par voie veineuse ; ce qui limitait son utilisation à long cours. Ainsi s’ouvrait de nouvelles perspectives de recherche pour aboutir à la naissance de la première molécule de cette classe thérapeutique en 1970.
55
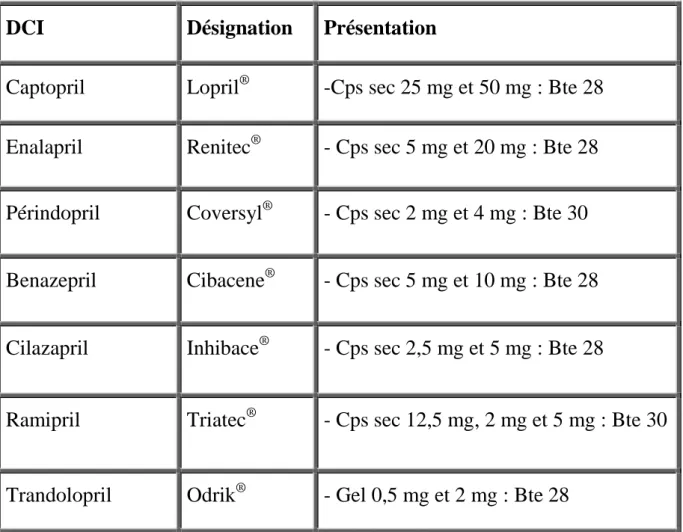
L’inhibition de l’enzyme de conversion n’a pu être utilisée au long cours qu’après l’avènement du captopril et de l’enalapril dont l’utilisation a considérablement augmenté vers la fin des années 70 ; ainsi et dans l’espace de 5 ans, plus d’un million de malades ont été traités par les inhibiteurs de l’enzyme de conversion dont environ 70% pour une hypertension artérielle et 30% pour une insuffisance cardiaque ; il est à signaler que cette utilisation a été limitée aux hypertensions artérielles et insuffisances cardiaques thérapeutico-résistantes. Actuellement, les inhibiteurs de l’enzyme de conversion sont à l’étude dans le traitement des stades précoces de l’insuffisance cardiaque et dans le traitement des cardiopathies ischémiques [83, 249].
II. CLASSIFICATION ET STRUCTURE CHILIQUE:
II.1. Classification:
La classification des différents inhibiteurs de l’enzyme de conversion peut se concevoir :
II.1.1. Selon leur structure chimique (tableau n°1): actuellement, on
56
Tableau I: Classification des IEC selon leur structure chimique [249]
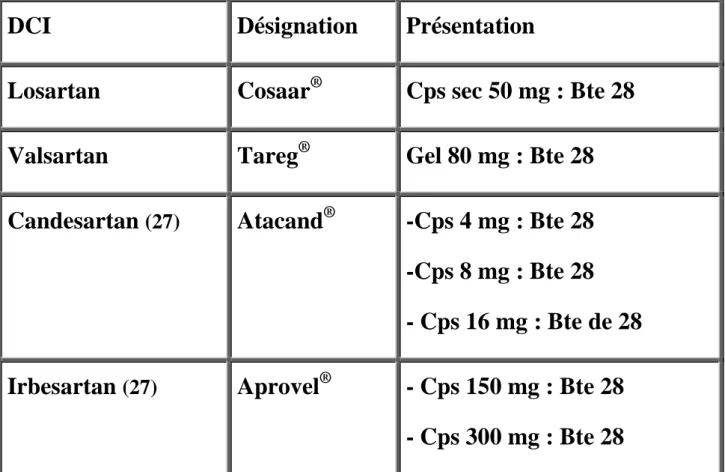
Classification par structure chimique DCI
IEC possédant une
fonction sulfhydryle
libre ou masquée
IEC possédant une
fonction sulfhydryle libre
Captopril Fentiapril IEC possédant une
fonction sulfhydryle masquée
Pivopril, Alacepril, Zofénopril Des IEC non soufrés
comportant un radical carboxyl
IEC non soufrés possédant une fonction acide et une fonction ester Enalapril, Périndopril, Ramipril, Cilazapril Spirapril, Quinapril Indolapril, Pentopril Trandolapril,
IEC non soufrés possédant deux fonctions acides
Lisinopril, Acide enalaprilique, Acide périndoprilique
57
II.1.2. Classification des IEC selon qu’ils soient directement actifs ou qu’ils soient des «prodrogues»:
Tableau II: Classification des IEC selon qu’ils se comportent comme des
prodrogues [4]
Classes des IEC DCI
IEC directement actifs et ayant un groupement thiol
Captopril
IEC «prodrogue» avec groupement thiol Alacepril
IEC directement actifs et sans groupement thiol Lisinopril
IEC «prodrogue» sans groupement thiol Enalapril
Périndopril Ramipril Cilazapril Quinapril Pentopril Fosinopril Delapril
![Figure 1: Stratégie d’adaptation du traitement antihypertenseur et fréquence du suivi [267]](https://thumb-eu.123doks.com/thumbv2/123doknet/15062197.699127/28.892.83.819.200.895/figure-stratégie-adaptation-traitement-antihypertenseur-fréquence-suivi.webp)
![Figure 4 : Schéma illustrant la cascade enzymatique du SRA qui aboutit à la formation de l’octapeptide active (angiotensine II) [153]](https://thumb-eu.123doks.com/thumbv2/123doknet/15062197.699127/40.892.122.785.332.754/figure-schéma-illustrant-cascade-enzymatique-formation-octapeptide-angiotensine.webp)
![Tableau III: Structure chimique des principaux inhibiteurs de l’enzyme de conversion [93]](https://thumb-eu.123doks.com/thumbv2/123doknet/15062197.699127/58.892.98.799.280.797/tableau-iii-structure-chimique-principaux-inhibiteurs-enzyme-conversion.webp)
![Tableau VI: Synopsis des antagonistes non peptidiques du récepteur AT 1 à l’angiotensine II [37,105]](https://thumb-eu.123doks.com/thumbv2/123doknet/15062197.699127/115.892.94.818.234.1039/tableau-vi-synopsis-antagonistes-peptidiques-récepteur-at-angiotensine.webp)