HAL Id: tel-03163893
https://tel.archives-ouvertes.fr/tel-03163893
Submitted on 9 Mar 2021HAL is a multi-disciplinary open access
archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Décryptage des mécanismes moléculaires menant au
Spectre Oculo-Auriculo-Vertébral : de la recherche de
gène aux modèles animaux
Aurélien Trimouille
To cite this version:
Aurélien Trimouille. Décryptage des mécanismes moléculaires menant au Spectre Oculo-Auriculo-Vertébral : de la recherche de gène aux modèles animaux. Médecine humaine et pathologie. Université de Bordeaux, 2020. Français. �NNT : 2020BORD0336�. �tel-03163893�
1 THÈSE PRÉSENTÉE
POUR OBTENIR LE GRADE DE
DOCTEUR DE
L’UNIVERSITÉ DE BORDEAUX
ÉCOLE DOCTORALE Sciences de la Vie et de la Santé SPÉCIALITÉ : Génétique
Par Aurélien TRIMOUILLE
Décryptage des mécanismes moléculaires menant au Spectre Oculo-Auriculo-Vertébral : De la recherche de gènes aux modèles animaux
Sous la direction du Pr Caroline ROORYCK-THAMBO Soutenue le 16/12/2020
Membres du jury :
M. DUBUS Pierre, PU-PH, Université de Bordeaux Président Mme ODENT Sylvie, PU-PH, Université de Rennes 1 Rapporteur M. VERLOES Alain, PU-PH, Université de Paris Rapporteur M. ARVEILER Benoit, PU-PH, Université de Bordeaux Examinateur Mme ROORYCK-THAMBO, PU-PH, Université de Bordeaux Invité
2
Titre : Décryptage des mécanismes moléculaires menant
au Spectre Oculo-Auriculo-Vertébral : De la recherche de
gènes aux modèles animaux
Résumé :
Le syndrome de Goldenhar, ou spectre Oculo-Auriculo-Vertébral (OAVS [MIM: 164210]), est une anomalie du développement embryonnaire associant des malformations des structures dérivées des premier et second arcs branchiaux, notamment l'oreille, l'œil, la mandibule et les vertèbres. Il s’agit du deuxième spectre malformatif embryonnaire le plus fréquent de la tête et du cou. Le phénotype clinique est hétérogène et caractérisé par une microsomie hémifaciale, des anomalies auriculaires asymétriques, des dermoïdes épibulbaires, et des malformations vertébrales.
Différentes causes non génétiques ont été rapportées dans l’OAVS, dont de nombreuses expositions toxiques (acide rétinoïque, mycophénolate mofetil, etc…). Par ailleurs, une origine génétique de l’OAVS a été rapportée chez des patients porteurs de variants de structure (locus 22q11.2, HMX1, OTX2, etc…), ou de variants ponctuels, touchant notamment MYT1, seul gène rapporté dans plusieurs études. L’objectif de ce travail a été, via l’application du séquençage de l’exome et du génome, d’identifier la ou les causes moléculaires de la survenue de l’OAVS chez des patients issus d’une cohorte de plus de 350 individus, et de confirmer cette identification de gènes par des études fonctionnelles. Celles-ci ont notamment utilisées des analyses en protéomique, à partir de modèles cellulaire et zebrafish, à même de permettre une étude de la morphologie cranio-faciale. Ce travail nous a permis d’identifier 3 gènes responsables d’OAVS : ZYG11B, ZIC3 et EYA3. Le séquençage de l’exome en trio chez un patient nous a permis d’identifier un variant non-sens de novo du gène ZYG11B. L’effet délétère de cette mutation sur la fonction de la protéine a pu être montré par surexpression de la forme mutée sur modèle cellulaire Hela, et par inactivation transitoire de zyg11 par morpholinos sur modèle zebrafish. Dans une famille où ségrège sur un mode récessif lié à l’X un phénotype OAVS modéré, la réalisation du séquençage d’exomes et d’un génome a pu montrer une association de ces atteintes avec la présence d’une expansion de polyalanines du gène ZIC3. Enfin, nous avons identifié chez deux familles différentes la présence d’un même variant faux-sens du gène
EYA3 ségrègeant avec des atteintes de l’OAVS. L’effet délétère de cette mutation a été confirmé par la
réalisation d’études fonctionnelles sur modèle cellulaire et modèle zebrafish, qui présente des malformations cranio-faciales. Par ailleurs, 3 diagnostics différentiels ont pu être fait chez des patients initialement considérés comme atteints d’OAVS, et concernant les gènes SOX5, EFTUD2, et EYA1. Enfin, deux autres gènes candidats ont pu être identifiés, et feront l’objet d’études fonctionnelles : SIX5 et SNRNP70. Si ce travail nous a permis de mieux comprendre les causes moléculaires impliquées dans l’OAVS, ses limites nous imposent de trouver de nouveaux outils pour poursuivre ces investigations. Parmi ceux-ci, l’intégration de technologies OMICs, l’identification de cibles après exposition toxique sur modèle animal, et l’utilisation de méthodes statistiques innovantes appliquées aux analyses génomiques semblent les plus pertinents.
3
Title : Deciphering molecular mechanisms of
Oculo-Auriculo-Vertebral Spectrum : from genes research to
animal models.
Abstract :
[4000 caractères maximum]
Goldenhar syndrom, or Oculo-Auriculo-Vertebral Sepctrum (OAVS [MIM: 164210]), is an embryonic developmental defect involving the first and second branchial arches. Defects associated with this syndrome are malformations of ears, eyes, mandibles and vertebrae. OAVS is the second most frequent embryonic malformative spectrum of head and neck. The phenotype is highly heterogeneous, and includes hemifacial microsomia, asymmetric auricular dysplasia, epibulbar dermoid and vertebral malformations.
Non genetic causes have been reported for OAVS, with several toxic expositions (retinoic acid, mycophenolate mofetil, etc…). Furthermore, genetic causes of OAVS have been described in patients harbouring structural variants (22q11.2, HMX1, OTX2, etc…), or single nucleotide variants, notably of MYT1 gene. The aim of this work is to identify molecular bases of OAVS with exome or genome sequencing, and to confirm the deleterious effect of variants with functional studies. Notably, we use proteomic analysis on cellular models and zebrafish models, allowing study of craniofacial development. This work identified 3 responsible genes of OAVS: ZYG11B, ZIC3 and EYA3. Trio-based exome sequencing in patient identified a de novo nonsense variant in ZYG11B. The deleterious effect of this mutation on protein function has been shown by mutant surexpression in Hela cells, and transient knockdown by morpholinos in zebrafish model. Exome and genome sequencing in a family affected by X-linked mild OAVS phenotype found an expansion of polyalanines in ZIC3 gene. We also found in two different families affected with OAVS the same missense variant of EYA3 gene. We confirmed the deleterious effect of this variant with functional studies on Hela cells and zebrafish model, which harbour craniofacial defects. Furthermore, differential diagnosis have been found in 3 patients, with mutations in SOX5, EFTUD2, and EYA1 genes. Finally, two other candidate genes have been identified, and will be investigated by functional studies: SIX5 and SNRNP70. This work allowed us to better understand molecular causes involved in OAVS. However, its limits force us to find new tools to further investigate this spectrum. Integration of OMICs technologies, toxicological studies on animal models, and innovative biostatistics tools applied to genomic analyses could be relevant.
Keywords :
OAVS, ZYG11B, ZIC3, EYA3, SNRNP70, SIX5Inserm UMR1211 – Maladies Rares, Génétique et
Métabolisme
4
Remerciements
Je remercie tous les membres du jury d’avoir accepté de consacrer de leur temps à la lecture de ce travail.
Je remercie le Professeur Pierre Dubus d’avoir accepté la présidence de ce jury. Je le remercie également pour son soutien constant et son accueil chaleureux dans la discipline d’histologie, embryologie et cytogénétique au sein de l’Université de Bordeaux.
Je remercie le Professeur Alain Verloes pour sa bienveillance et son soutien, depuis un passage marquant en tant qu’externe dans le service de génétique médicale de l’hôpital Robert Debré.
Je remercie le Professeur Sylvie Odent de nous offrir une opportunité de bénéficier de son expertise sur l’holoprosencéphalie, dont les voies génétiques s’avèrent recouper celles de l’OAVS. Je la remercie également pour ces échanges sur les projets qui font le futur de la prise en charge des maladies rares, du PNMR aux réseaux européens.
Je remercie le Professeur Benoit Arveiler pour son amitié et la confiance dont j’ai fait l’objet durant mes 3 années d’assistanat au sein du laboratoire de Génétique moléculaire, et je n’en doute pas pour les projets à venir.
5 Je remercie très chaleureusement le Professeur Caroline Rooryck-Thambo d’avoir accepté la direction de cette thèse. Je la remercie particulièrement pour sa ténacité tout au long de ces 3 dernières années, nous permettant d’aboutir finalement à ces beaux résultats.
Je remercie le Professeur Didier Lacombe pour toute ma formation en Génétique, de l’internat, à l’assistanat, à la thèse de sciences.
Un grand merci au Docteur Angela Tingaud-Sequeira pour son implication dans mon travail, pour tout ce temps qu’elle m’aura consacré, et pour son expertise. Ces remerciements viennent en écho à ceux déjà formulés lors de ma thèse d’exercice, et je l’espère le seront encore pour nos projets futurs. Merci également à Sara, décidemment aussi douée au bloc qu’à la paillasse, pour son aide très précieuse.
Bien évidemment, je remercie toute l’équipe du MRGM, Christophe, Johan, Christelle, Giovanni, Saharnaz, Gaia, Rodrigue, Isabelle, Nadège, Sophie, Jean-Paul, … et tous ceux que j’oublie.
Et bien sûr, je dédie ce travail à Chloé et Émile, soutiens indéfectibles à tout ce que j’entreprends.
6
Table des Matières
Introduction ... 14
Le syndrome de Goldenhar, ou OAVS : considérations cliniques et embryologiques ... 14
Embryologie des structures cranio-faciales ... 14
Les cellules des crêtes neurales ... 15
Développement des arcs branchiaux ... 18
Bourgeons faciaux ... 22
Historique du syndrome de Goldenhar et spectre Oculo-Auriculo-Vertébral ... 26
Microsomie hémifaciale ... 27
Atteintes ophtalmologiques ... 27
Atteintes auriculaires ... 28
Atteintes vertébrales ... 28
Système nerveux central ... 29
7
Etiologie de l’OAVS ... 35
Hypothèses étiologiques non génétiques ... 35
Fluoxetine ... 35 Cocaïne ... 36 Tamoxifène ... 36 Primidone ... 37 Thalidomide ... 37 Acide rétinoïque ... 38 Mycophénolate mofétil ... 39 Disruption vasculaire ... 40 Gémellité ... 41 Diabète maternel ... 44
Les causes moléculaires identifiées ... 44
8
Délétion 22q11.2 ... 44
Duplication de OTX2 ... 47
Duplication d’éléments régulateurs du gène HMX1 (NKX5-3) ... 49
Autres variants de structure et aneuploïdies... 51
Variants ponctuels ... 53 MYT1 ... 53 AMIGO2 ... 56 EFTUD2 ... 57 VWA1 ... 57 NFATC1 et TBX1 ... 58 CRKL, YPEL1 et OTX2 ... 60 GSC ... 61
Etude de Genome-Wide Association Study (GWAS) appliquée à la microsomie hémifaciale ... 62
9
Souris HOXA2 ... 63
Souris ZIC3 ... 63
Modèles animaux lié au gène HMX1 ... 65
Les diagnostics différentiels de l’OAVS ... 66
Syndrome de Treacher Collins [MIM : 154500] ... 66
Syndrome de Nager [MIM : 154400] ... 67
Syndrome de Miller [MIM : 263750] ... 67
Dysostose mandibulo faciale avec alopécie [MIM : 616367]... 68
Dysostose mandibulo-faciale de type Guion Almeida [MIM : 603892] ... 68
Syndrome Branchio-Oto-Rénal [MIM : 113650] ... 69
Objectif ... 70
Résultats... 70
Article 1 : ... 74
10
Article 2 : ... 96
Description d’une famille atteinte d’OAVS lié à l’X associé avec une expansion polyalanine de ZIC3 ... 96
Article 3 : ... 112
Un variant faux-sens récurrent d’EYA3 est associé au spectre Oculo-Auriculo Vertébral 112 Données non publiées : Analyses d’exomes de 23 cas d’OAVS et études fonctionnelles des gènes candidats... 148
Introduction ... 148
Matériels et Méthodes... 149
Séquençage de l’exome... 149
Inactivation de l’expression de SIX5 sur cellules HeLa par CRISPR/Cas9 ... 151
Inactivation transitoire du gène six5 de poisson zèbre par Morpholinos ... 152
Résultats ... 154
Diagnostics différentiels réalisés : SOX5, EFTUD2 et EYA1 ... 154
11
EFTUD2 ... 155
EYA1 ... 156
Nouveaux gènes candidats ... 158
SIX5 ... 158
SNRNP70 ... 161
Discussion générale ... 167
Limites et évolution des approches génomiques ... 167
ZYG11B et EYA3 : pertinence des analyses fonctionnelles en aval ... 170
Hypothèses concernant les mécanismes moléculaires de l’OAVS ... 172
Une multiplicité de voies physiopathologiques ... 172
Hypothèses : épigénétique, mosaïque, oligogénisme, environnement ? ... 173
Méthodes statistiques innovantes en génomique ... 175
Conclusion et Perspectives ... 176
12
Table des Figures et Tableaux
Figure 1 Formation de la crête neurale……….15
Figure 2 Formation du tube neural, et migration des cellules de la crête neurale ... 16
Figure 3 Gradients d’expression impliqués dans la différenciation antéro-postérieure du rhombencéphale ... 19
Figure 4 Développement des bourgeons faciaux ... 23
Figure 5 Résumé de l’évolution des éléments issus des bourgeons faciaux ... 25
Figure 6 Représentation graphique de la prévalence des atteintes en fonction des études. ... 32
Figure 7 Classification OMENS+ ... 34
Figure 8 Impact de l’acide mycophénolique sur la voie de synthèse de novo de GTP. ... 40
Figure 9 Jumelles monozygotes présentant un OAVS, avec atteintes “en miroir”. ... 43
Figure 10 Jumeaux monozygotes discordants ... 43
13
Figure 12 Visualisation de type Circos plot des variants de structure décrits dans l’OAVS. .. 52
Figure 13 Modèle de souris Zic3 PGK-NEO. ... 64
Figure 14 Composants du spliceosome impliqués en pathologie humaine ... 163
Figure 15 Mécanisme d’auto-régulation de l’expression de SNRNP70 ... 164
Figure 16 Impact prédit du variant sur SNRNP70... 166
Tableau 1 Prévalence des atteintes de l’OAVS dans les cohortes de patients publiées. MHF : Microsomie Hémifaciale ... 33
14
Introduction
Le syndrome de Goldenhar, ou OAVS : considérations cliniques et embryologiques Embryologie des structures cranio-faciales
La compréhension des mécanismes aboutissant à la survenue du syndrome de Goldenhar, ou spectre Oculo-Auriculo-Vertébral, est en grande partie liée aux évènements embryologiques aboutissant à la mise en place des structures anatomiques affectées par ce syndrome, et notamment les structures cranio-faciales.
Le développement embryonnaire est décrit comme une succession d’étapes, correspondant aux stades embryonnaires ou stade de Carnegie, dont la définition prend en compte trois critères : l’âge gestationnel, la taille de l’embryon, et ses caractéristiques morphologiques. La période embryonnaire, correspondant à 8 semaines, soit 56 jours, a ainsi été divisée en 23 stades de Carnegie, à la suite desquels débute la période fœtale.
Le développement cranio-facial a lieu durant deux périodes distinctes : au moment de l’organogénèse, entre la 2ème et la 8ème semaine, et durant la morphogénèse, après la 8ème
semaine. Trois composants vont notamment concourir au développement de la majorité de ces structures : les cellules des crêtes neurales, les arcs branchiaux, et les bourgeons faciaux.
15
Les cellules des crêtes neurales
Les cellules des crêtes neurales sont des cellules embryonnaires transitoires, à caractère multipotent, présentes chez tous les vertébrés, y compris ceux appartenant aux taxons les plus anciens, telle la lamproie1. L’embryologiste suisse Wilhelm His les découvre en 1868, et les
nomme “Zwischenstrang”, ou bord intermédiaire, en raison de leur localisation entre l’ectoderme dorsal et le tube neural (Figure 1).
Figure 1 Formation de la crête neurale2, A) Plaque neurale B) Gouttière neurale 1) Epiblaste 2)
16 Figure 2 Formation du tube neural, et migration des cellules de la crête neurale. 1) épiblaste 2) bourrelets neuraux 3) cellules des crêtes neurales en migration 4) neuroépithélium 5) canal épendymaire 6) tube neural
Elles proviennent en effet de l’ectoderme situé à la bordure du tube neural, et apparaissent en fin de 3ème semaine de développement3. Intégrées au sein du neuroépithélium, elles y sont
initialement indistinguables histologiquement des autres cellules. Si la bordure du tube neural donne naissance aux progéniteurs des cellules des crêtes neurales, d’autres types cellulaires en dérivent également. En effet, le marquage de cellules des replis neuraux avant la fermeture du tube neural montre que celles-ci pourront être retrouvées dans les crêtes neurales, le tube neural, ou l’épiderme4. Par ailleurs, l’injection de cellules des crêtes neurales dans le tube neural ventral
montre que celles-ci peuvent se différencier comme les cellules environnantes et former la plaque du plancher, et les motoneurones5. Ces données montrent bien que si l’induction des
17 bords de la plaque neurale est bien à l’origine des cellules des crêtes neurales, leur différenciation est un processus remarquablement flexible1.
A la suite de signaux d’induction faisant suite à des interactions de contact entre la plaque neurale et l’ectoderme de surface, la délamination des cellules des crêtes neurales débute par une transition épithélio-mésenchymateuse, avant de migrer dans de nombreuses régions de l’embryon (Figure 2). Les cellules des crêtes neurales vont ainsi participer aux développement de nombreux tissus, tels que le système nerveux périphérique, le tissu squelettique, ou encore les cellules pigmentaires6.
Le long du tube neural, il est possible de distinguer anatomiquement une partie crâniale, à destinée encéphalique, et une partie caudale, à destinée médullaire. La partie crâniale peut elle-même être segmentée en trois territoires : les crêtes neurales prosencéphalique, mésencéphalique et rhombencéphalique, qui vont ensuite migrer dans des territoires spécifiques. Les crêtes neurales rhombencéphaliques vont notamment migrer en direction des arcs branchiaux, avant de donner naissance à différents dérivés mésenchymateux.
18
Développement des arcs branchiaux
Les arcs branchiaux, constituant l'appareil pharyngé, sont les structures anatomiques précurseures de la plupart des éléments de la face et du cou. Ceux-ci se forment entre la 4ème et la 7ème semaine de gestation. Chacun des cinq arcs branchiaux, numérotés 1, 2, 3, 4 et 6, le 5ème arc ne formant pas de structure chez l'humain, vont contribuer au développement de structures spécifiques. Le développement de ces structures, aboutissant à la formation des différents éléments de la face et du crâne, dont l’ensemble des nerfs crâniens, est étroitement lié à l’établissement de gradients d’expression de nombreux gènes, permettant une différenciation des tissus selon les axes antéro-posterieur et cranio-caudal (Figure 3). Par ailleurs, le rhombencéphale est régionalisé grâce à l’existence d’un gradient d’acide rétinoïque, dérivé biologique actif de la vitamine A, dont la concentration augmente selon l’axe cranio-caudal du rhombencéphale. Ce gradient est constitué grâce à l’expression différentielle de des enzymes de synthèse RALDH 1 à 4, et d’enzymes du cytochrome P450. L’acide rétinoïque se lie aux facteurs de transcription RAR et RXR, qui vont à leur tour activer différents gènes cibles, dont HOXA1 et HOXB1.
19 Figure 3 D’après Méndez-Maldonado7. La différenciation antéro-postérieure et cranio-caudale
lors du développement cranio-facial, la migration des cellules des crêtes neurales, et leur contribution aux différents tissus nécessitent une régulation fine impliquant l’établissement de gradients d’expression de nombreux gènes. (A) Schéma d’un embryon humain au stade de Carnegie 13 (30 jours, 32 somites), indiquant les principaux facteurs de transcription impliqués dans le développement des progéniteurs des cellules des crêtes neurales (BMP, FGF8, HIPPO,
20
NOTCH, SHH, WNT1). (B) Gradients d’expression impliqués dans la différenciation
antéro-postérieure du rhombencéphale. (C) Schéma du rhombencéphale d’un embryon humain au stade 13 de Carnegie (33 jours), indiquant à gauche le niveau d’origine des cellules des crêtes neurales (NCCs) contribuant à la formation des différents nerfs crâniens (CN) et des vésicules otiques (OV), le niveau d’expression des différents gènes de la famille HOX, et à droite, les différents facteurs de transcription impliqués dans la formation des nerfs crâniens.
Ces arcs branchiaux sont constitués d’un bourrelet mésenchymateux, provenant à la fois du mésoblaste para axial et de la lame latérale, et des cellules des crêtes neurales. Ces bourrelets sont recouverts d’ectoblaste en dehors, et d’entoblaste en dedans8. Par ailleurs, entre chaque arc
branchial, on peut définir deux poches à la face interne et externe de l'embryon : les poches ectobranchiales et endobranchiales.
Enfin, chaque arc branchial sera constitué 1) d'un noyau cartilagineux, 2) d'un noyau musculaire, 3) d’une artère issue de l'arc aortique, et 4) d’un nerf crânien. Nous détaillerons ici ces quatre éléments pour chacun des 5 arcs branchiaux.
Le premier arc branchial, ou arc mandibulaire, apparaît au 22ème jour de développement, et est associé à deux paires de bourgeons, contenant chacun un élément cartilagineux central transitoire : les bourgeons maxillaires et mandibulaires. On notera que si les bourgeons maxillaires ont initialement été considérés comme directement dépendants du 1er arc branchial, le mésenchyme dont ils dérivent est en réalité situé crânialement par rapport au 1er arc branchial. L’élément cartilagineux central des bourgeons maxillaires est le cartilage
palato-21 ptérygo-carré, qui donnera naissance à l’enclume et à la grande aile du sphénoïde, tandis que celui des bourgeons mandibulaires est le cartilage de Meckel, à l’origine du marteau. Par ailleurs, le premier arc branchial donnera naissance à d’autres éléments squelettiques par ossification dermique : les os maxillaire et zygomatique, l’écaille de l’os temporal, et la mandibule. Le premier arc branchial est à l'origine de différents muscles de la mandibule, notamment les muscles permettant la mastication, le vaste antérieur du muscle digastrique, le muscle tenseur du tympan, le muscle tenseur du voile du palais. Il est par ailleurs vascularisé par l'artère maxillaire, et innervé par le nerf trijumeau (V).
La première poche endobranchiale deviendra le récessus tubo-tympanique, à l'origine de la trompe auditive et de la caisse du tympan. La première poche ectobranchiale deviendra le méat acoustique externe.
Le 2ème arc branchial a pour élément cartilagineux le cartilage de Reichert, à l’origine du 3ème os de l’oreille moyenne, de l’étrier, ainsi que du processus styloïde de l’os temporal, et des petites cornes et du bord supérieur de l’os hyoïde. Le 2ème arc est également à l’origine des muscles permettant la mimique (orbiculaires de l’œil et de la bouche, risorius, platysma, auriculaire, frontal et buccinateur), du ventre postérieur du digastrique, du stylo-hyoïdien, et du stapédien. Il est vascularisé par l’artère stapédienne chez l’embryon, et l’artère cortico-tympanique chez l’adulte, et est innervé par le nerf facial (VII).
Le 3ème arc n’est à l’origine que d’un seul os, l’os hyoïde, avec la formation de ses grandes cornes et de la partie inférieure de son corps, et d’un seul muscle, le stylo-pharyngien. Il est vascularisé et innervé par le nerf glosso-pharyngien (IX).
22 Les 4ème et 6ème arcs branchiaux forment ensemble le larynx, composé des cartilages thyroïdes, cunéiformes, corniculés, aryténoïdes et cricoïdes. Ils sont également à l’origine des muscles constricteurs du pharynx, crico-thyroïdien et élévateur du voile du palais. Le 4ème arc branchial est vascularisé du côté gauche par l’arc de l’aorte, du côté droit par l’artère subclavière droite, ainsi que par l’origine des artères pulmonaires. Le 6ème arc est lui vascularisé par le canal artériel et les racines des artères pulmonaires. Ces 2 arcs innervés respectivement par les branches laryngées supérieures et récurrentes du nerf vague (X)
Bourgeons faciaux
La formation de la face a lieu entre la 4ème et 10ème semaine de développement, grâce à la croissance et à la fusion des 5 bourgeons faciaux : le bourgeon fronto-nasal, et les paires de bourgeons maxillaires et mandibulaires, associées au 1er arc branchial (Figure 4). Une partie de ces différents bourgeons va être constituée de la migration de cellules des crêtes neurales, migrant du mésencéphale et du prosencéphale pour constituer le mésenchyme du bourgeon fronto-nasal, et du mésencéphale et du rhombencéphale pour contribuer à la formation des bourgeons maxillaires et mandibulaires.
23 Figure 4 D’après Dixon et al9. a) Développement de la proéminence fronto-nasale, des
processus maxillaires et mandibulaires à la 4ème semaine de développement. b) Formation des placodes olfactives (Nasal pits) à la 5ème semaine de développement. c) Fusion des bourgeons
nasaux médiaux et du bourgeon maxillaire, formant le philtrum et le palais primaire à la 7ème
semaine de développement.
A la 4ème semaine de développement, ces 5 bourgeons entourent la cavité orale, le stomodéum. Celle-ci est séparée du tractus digestif par la membrane oro-pharyngienne. Au cours de la 5ème semaine, les bourgeons se développent dans leurs parties ventrales et médianes, et les placodes olfactives apparaissent sur le bourgeon fronto-nasal sous la forme d’un épaississement ectodermique (Figure 5). Celles-ci s’invagineront au cours de la 6ème semaine, formant les cupules nasales, divisant le bourgeon fronto-nasal en bourgeons nasaux latéral et médial. On définit le sillon séparant les bourgeons nasaux latéraux des bourgeons maxillaires comme le sillon naso-lacrymal, dont l’ectoderme s’invaginera au cours de la 7ème semaine pour former le conduit lacrymo-nasal et le sac lacrymal.
24 A partir de la fin de la 7ème semaine, les extrémités inférieures des bourgeons nasaux médiaux fusionnent pour donner naissance au processus intermaxillaire, qui fusionnera à son tour avec le bourgeon maxillaire, pour donner le philtrum et le palais primaire.
Concernant les bourgeons mandibulaires, ceux-ci sont initialement séparés par une fissure médio-ventrale, qui se comblera du fait d’une prolifération mésenchymateuse, au cours des 4 et 5ème semaines de développement. Par ailleurs, la membrane oro-pharyngienne se rompt, ouvrant ainsi la bouche embryonnaire.
25 Figure 5 D’après “Reference Module in Biomedical Sciences : Development of the Face”10.
Résumé de l’évolution des éléments issus des bourgeons faciaux et intervenant dans la formation de la face, de la 4ème à la 8ème semaine de développement. A droite, images correspondantes, obtenues par microscopie électronique en balayage.
26
Historique du syndrome de Goldenhar et spectre Oculo-Auriculo-Vertébral
La première description du spectre Oculo-Auriculo-Vertébral a été faite par Maurice Goldenhar, ophtalmologue belge-américain, en 195211. Il rapporte dans cette première étude
l’association d’une constellation de malformations congénitales impliquant des anomalies oculaires, des oreilles et de la face. En 1960, puis en 1963, Gorlin précise ce spectre malformatif, en insistant sur la variabilité des symptômes, ainsi que sur leur potentiel caractère unilatéral12. Différentes études ont par la suite pu établir que le syndrome de Goldenhar, ou
spectre Oculo-auriculo-vertébral, constitue le 2ème spectre malformatif d’origine embryonnaire impliquant la sphère tête et cou le plus fréquent, avec une prévalence estimée à 1/2650013.
En fonction des études considérées, les critères cliniques permettant de considérer un patient comme atteint d’un syndrome de Goldenhar peuvent être plus ou moins stringents14,15.
Cependant, on notera que quels que soient les critères diagnostiques choisis, l’une des caractéristiques majeures de l’OAVS est le caractère asymétrique des malformations. Nous décrirons ici les atteintes rapportées, en nous appuyant notamment sur les cohortes de patients les plus récentes, telles que Rooryck et al.16, Beleza-Meireles et al.17 et Barisic et al.13, cette
27
Microsomie hémifaciale
La microsomie hémifaciale, décrite comme microsomie cranio-faciale, dysostose oto-mandibulaire unilatérale, ou encore dysplasie faciale unilatérale, peut être définie comme une hypoplasie de l’oreille, du squelette facial, et des tissus mous. Il s’agit de la 3ème malformation
congénitale cranio-faciale la plus fréquente, après les fentes labiales et palatines, et la craniosténose, avec une incidence évaluée entre 1/3500 et 1/5600 naissances18,19. Dans 89% à
100% des cas, la microsomie est caractérisée par une hypoplasie de la mandibule, conduisant à l’aspect d’asymétrie faciale20. Cette atteinte a été considérée à elle seule par certains auteurs
comme suffisante, même isolée, pour considérer un patient comme atteint d’OAVS. Cette définition peut expliquer la prévalence plus élevée de l’OAVS dans différentes études. Deux études portant respectivement sur 755 et 911 patients atteints de microsomie hémifaciale font apparaître qu’entre 35% et 47% d’entre eux présentent d’autres atteintes, appartenant en majorité au spectre OAV21,22. Par ailleurs, la microsomie hémifaciale apparaît comme un critère
minimum pour considérer qu’un patient est bien atteint d’une forme d’OAVS, ou syndrome de Goldenhar dans plusieurs études1. Devant l’existence de 17 à 18% de patients présentant des
atteintes caractéristiques de l’OAVS, sans microsomie hémifaciale franche, il a été considéré dans l’étude de notre cohorte de patients qu’un aspect d’asymétrie faciale pouvait constituer le critère minimum à retenir1.
Atteintes ophtalmologiques
Les atteintes ophtalmologiques du syndrome de l’OAVS peuvent être multiples, uni ou bilatérales. De nombreuses atteintes ont été décrites : anophtalmie, microphtalmie, ou
28 colobome, pouvant concerner la paupière, l’iris, ou la rétine. Par ailleurs, l’un des symptômes les plus fréquemment décrits est la présence d’un dermoïde épibulbaire. Cette tumeur bénigne congénitale est constituée d’annexes cutanées, de tissu conjonctif, adipeux ou neurologique, localisée au contact de la cornée et d’une partie de la sclère. Si l’atteinte est principalement esthétique, une atteinte de la vision est également possible, avec astigmatisme, obstruction du champ visuel, ou infiltration graisseuse de la cornée23. Le pronostic est cependant très bon après
prise en charge chirurgicale.24
Atteintes auriculaires
De très nombreuses anomalies auriculaires, uni ou bilatérales avec un caractère asymétrique, ont été décrites dans l’OAVS. Parmi les plus fréquentes, nous pouvons citer l’anotie, la microtie, l’atrésie du canal auditif externe, et la présence d’enchondromes pré-auriculaires. Par ailleurs, ont été décrites des dysplasies touchant l’hélix, l’anthélix, ou le lobule. Du fait de la morphologie complexe de l’oreille externe, différentes classifications ont été établies, afin d’objectiver la sévérité de ces anomalies25–28. Celles-ci sont souvent associées à une surdité, de
conduction dans la plupart des cas, mais également de perception ou mixte.
Atteintes vertébrales
Il existe une abondante littérature scientifique concernant la survenue d’atteintes vertébrales chez les patients présentant une microsomie hémifaciale, pouvant être la seule atteinte cranio-faciale, et retrouvant une prévalence allant de 12 à 79%29–35, cette variabilité pouvant être
29 patients. L’étude rétrospective de Renkeman et al.35, portant sur 991 patients présentant une
microsomie hémifaciale, soit le plus grand nombre de patients rapportés, retrouve, quant à elle, une prévalence de 28%. Les atteintes les plus fréquentes sont la scoliose, le bloc vertébral, et l’hémivertèbre.
Ces atteintes semblent être le plus souvent sous-diagnostiquées, car n’ayant pas toujours de retentissement fonctionnel. Par ailleurs, les radiographies standards ne permettent pas toujours d’évaluer l’ensemble de ces anomalies, contrairement à la réalisation d’un scanner avec reconstruction 3D36, qui apparaît également comme pouvant avoir un intérêt dans l’évaluation
des anomalies mandibulaires et orthodontiques de l’OAVS37.
Système nerveux central
La description de troubles du neurodéveloppement associés au spectre OAV doit être faite avec prudence, du fait d’une prévalence de la déficience intellectuelle estimée à 1% dans la population générale38, et de l’existence de diagnostics différentiels correspondant à des retards
de développement syndromiques. Le retard des acquisitions concerne entre 5 et 20%16,13,17 des
patients dans les dernières séries rapportées, associé ou non à des malformations cérébrales ou à une microcéphalie. Il est également rapporté dans la littérature la survenue d’anomalies touchant les différentes paires crâniennes, associées aux malformations cranio-faciales39. Une
revue complète de la littérature décrivant des patients atteints de microsomie hémifaciale s’intégrant ou non dans le cadre de l’OAVS rapporte des estimations de la prévalence
30 d’anomalies du système nerveux central allant de 2% à 69%, et de troubles du neurodéveloppement allant de 8% à 73%40.
Autres malformations
De nombreuses autres malformations (arbre urinaire, génitales, viscérales, etc…) ont pu être décrites chez les patients présentant par ailleurs des atteintes de l’OAVS. Parmi celles-ci, les malformations cardiaques sont les plus fréquentes, avec une prévalence estimée entre 5 et 47% en fonction des cohortes. Celles-ci sont le plus souvent de type conotroncal, comme la tétralogie de Fallot, ou les communications inter-auriculaires ou inter-ventriculaires41,42.
Les cohortes de patients décrites démontrent l’hétérogénéité clinique de l’OAVS
Dans la littérature, nous retrouvons de nombreuses études portant sur des cohortes de patients atteints d’OAVS13,15,17,22,29–35,39–41,43–60. Cependant, celles-ci présentent une grande
hétérogénéité de nombre de patients inclus, de critères d’inclusion, et de catégories d’atteintes considérées. Deux catégories peuvent néanmoins être définies : les études portant sur des patients présentant un OAVS ou Goldenhar, avec par définition une association de plusieurs atteintes, et les études incluant des patients présentant une microsomie hémifaciale isolée. Il faut noter que le nombre de patients inclus est généralement bien plus important pour ces dernières, du fait de la prévalence de la microsomie hémifaciale. Concernant les atteintes, certaines études se basent sur la classification OMENS+31,33, permettant d’évaluer
31 Tissue, Tessier Cleft). Cette classification peut également s'associer à l’utilisation de scanner 3D34 pour évaluer l’atteinte mandibulaire, par la classification de Pruzansky61 et Kaban62. Les
explorations menées pour définir les atteintes vertébrales (radiographies ou scanner 3D) sont également très différentes entre les études. Les troubles du neurodéveloppement sont également définis de façons très diverses, selon la considération du degré d’atteinte fonctionnelle (autisme, troubles des apprentissages, déficience intellectuelle, etc...), et des anomalies cérébrales sans retentissement clinique décelables à l’IRM cérébrale (hypoplasie du corps calleux, ventriculomégalie, etc…). Enfin, la prévalence estimée des atteintes oculaires dépend de l’inclusion ou non de la présence de dermoïde épibulbaire dans cette catégorie. L’ensemble de ces éléments incitent à considérer les prévalences rapportées par ces études avec une grande prudence, leur comparaison étant très difficile du fait de ces divers critères d’inclusion.
32 Figure 6 Représentation graphique de la prévalence des atteintes en fonction des études.
33 Tableau 1 Prévalence des atteintes de l’OAVS dans les cohortes de patients publiées.
Publication Année Nb de patients Types d’atteintes
Microsomie Hémifaciale Oculaire Auriculaire Vertébrale Cardiaque Neuro.
Feingold and Baum44 1978 16 81 100 100 56 19 25
Rollnick et al.45 1987 202 69 18 100 19 5 13 Morrison et al.41 1992 24 72 88 100 64 32 36 Tasse et al.15 2005 44 83 25 100 19 15 15 Touliatou et al.53 2006 14 59 65 100 23 18 29 Strömland et al.56 2007 13 89 72 100 67 33 39 Johansson et al.55 2007 20 80 75 100 50 40 50 Engiz et al.54 2007 31 77 39 90 70 39 52 Barisic et al.13 2014 259 49 24 100 24 28 10 Tuin et al.34 2015 44 100 41 94 61 19 NA Beleza-Meireles et al.17 2015 51 90 29 92 20 16 10 Tuin et al.34 2015 94 100 5 85 21 27 NA Pegler et al.58 2016 41 90 54 100 66 37 17
Berenguer et al. (Cohorte Colovati et al.)59 2017 57 98 38 98 40 28 5
Berenguer et al. (Cohorte Rooryck et al.)59 2017 169 82 74 100 89 47 21
34 Figure 7 adapté de Tuin et al34. Classification OMENS+, permettant de coter les différentes
atteintes orbitales, mandibulaires, auriculaires, des nerfs faciaux, des tissus mous, ainsi que les fentes faciales retrouvées dans l’OAVS
35
Etiologie de l’OAVS
Hypothèses étiologiques non génétiques
Différents travaux ont rapporté la survenue d’associations malformatives congénitales pouvant être considérées comme appartenant à l’OAVS, à la suite d’expositions toxiques ou iatrogènes durant la période anténatale. Ces cas soulignent l’impact que peuvent avoir les facteurs environnementaux dans la survenue de l’OAVS. On peut noter cependant que dans de nombreux cas, les liens de causalité suggérés par ces études n’ont pas été confirmés par la suite.
Fluoxetine
Une étude décrit un patient présentant un syndrome de Goldenhar, caractérisé par la présence d’une asymétrie faciale avec une microtie droite, une hypoplasie mandibulaire, une hypoplasie bilatérale de la macula, une scoliose et une communication interventriculaire63. Dans les antécédents anténataux, les auteurs rapportent que durant la grossesse, la mère du patient était sous traitement par Fluoxetine à la dose de 20mg/jour. Cet inhibiteur sélectif de la recapture de la sérotonine étant très largement prescrit dans le cadre du traitement de la dépression, différentes études de tératogénicité ont été menées, avec des résultats parfois en contradiction, ne démontrant pas de façon formelle l’existence d’un sur-risque de malformations congénitales64,65.
36 Cocaïne
En 1991, Lessik et al.66 décrivent le cas d’un enfant atteint d’OAVS, présentant une asymétrie
faciale, une microphtalmie droite, une anotie droite, un enchondrome préauriculaire à gauche, une scoliose avec absence de 4 côtes à droite, 2 hémivertèbres et une hypoplasie de la paroi thoracique à droite. Par ailleurs, ce patient présentait une polydactylie pré-axiale de la main droite. Durant la grossesse, une consommation de cocaïne est rapportée par la mère, à l’exception d’autres toxiques. Les auteurs de cette étude font un lien entre cette consommation et la survenue de l’OAVS chez ce patient, via un mécanisme de disruption vasculaire décrit pour d’autres malformations associées à une exposition anténatale à la cocaïne en 1990 par Hoyme et al.67. Il faut noter cependant que si l’hypothèse d’un risque accru de malformations
congénitales par disruption vasculaire est reprise par plusieurs auteurs par la suite, les études menées ultérieurement n’ont pas permis de confirmer ce risque avec certitude68,69.
Tamoxifène
Cullins et al.70 rapportent en 1994 le cas d’une patiente sous traitement par tamoxifène à la dose
de 20mg/j dans les suites d’un cancer du sein, chez qui une grossesse est découverte tardivement à 26 semaines d’aménorrhée, avec nécessité de réaliser une césarienne en urgence pour chorioamniotite. Il faut également noter que durant cette grossesse, une scintigraphie osseuse au Technetium-99m a été réalisée, et que la patiente rapporte une consommation de cocaïne et de cannabis hebdomadaire. Dans la littérature, l’association entre tamoxifen et anomalies cranio-faciales n’est rapportée que dans une seule autre étude portant sur un enfant présentant
37 une séquence Pierre Robin71. Des travaux récents chez la souris suggèrent qu’une exposition
anténatale peut être à l’origine de la survenue de fentes palatines72.
Primidone
Un seul cas d’OAVS attribué à la prise d’un anti-épileptique, le primidone, durant la grossesse, a été rapporté en 198573. Ce patient présentait plusieurs atteintes caractéristiques, notamment
une asymétrie faciale, un colobome de la paupière droite, des dermoïdes épibulbaires bilatéraux, des tags pré-auriculaires bilatéraux, une atrésie modérée des choanes à droite, ainsi qu’une tétralogie de Fallot. Ces symptômes étaient associés par ailleurs à un encéphalocèle antérieur et à une hydrocéphalie liée à une hémorragie intraventriculaire. Les auteurs de cette étude proposent une hypothèse selon laquelle l’exposition au primidone est responsable de la survenue de l’OAVS chez ce patient, via la survenue d’une diathèse hémorragique, et un effet antagoniste sur l’acide folique.
Thalidomide
Depuis les premières descriptions, en 196174,75, de l’embryopathie qui lui est associée, le
thalidomide est connue pour être responsable en cas d’exposition in utero de malformations congénitales touchant préférentiellement les membres, classiquement à type de phocomélie ou d’amélie76. Cependant, les atteintes rapportées sont nombreuses, et comprennent notamment
des malformations retrouvées dans l’OAVS, et notamment des anomalies auriculaires77,78, des
38 à partir de modèles animaux par Poswillo et al.19,81 retrouvent également ces atteintes
cranio-faciales. Si les mécanismes physiopathologiques de l’embryopathie au thalidomide sont encore débattus, l’une des principales hypothèses est que le défaut de formation des membres est dû à un effet anti-angiogénique du thalidomide, bloquant la maturation des vaisseaux en formation, et aboutissant à la perte de ceux-ci82.
Acide rétinoïque
Une étude visant à caractériser le spectre malformatif associé à l’exposition intra-utérine à l’acide rétinoïque, dérivé actif de la vitamine A (rétinol) a pu rapporter les cas de 154 grossesses marqués par une exposition à l’isotrétinoïne, prescrit dans le cadre du traitement de l’acné sévère83. Parmi les atteintes décrites, certaines correspondent à celles de l’OAVS, notamment
la microtie ou anotie (15 cas), la micrognathie (6 cas), les fentes palatines (3 cas), les malformations cardiaques (8 cas). La publication de différents autres cas confirme ce phénotype84–86, Par ailleurs, une étude87 rapporte le cas d’une grossesse marquée par une
exposition massive à la vitamine A, due à l’ingestion accidentelle par la mère d’une solution en laboratoire. L’enfant issu de cette grossesse présentait un dermoïde épibulbaire bilatéral, et des appendices prétragiens à gauche, faisant évoquer le diagnostic de syndrome de Goldenhar. Ces atteintes des arcs branchiaux dans l’embryopathie à l’acide rétinoïque sont à mettre en relation avec son rôle connu durant l’embryogénèse. Par ailleurs, il a été montré que MYT1, premier gène causal mis en évidence dans l’OAVS, est surexprimé après traitement à l’acide rétinoïque d’un modèle cellulaire88, et que sa surexpression induit une diminution de l’expression du
39 ces éléments tend à démontrer que l’acide rétinoïque est une voie physiopathologique importante dans la survenue de l’OAVS, et que son exploration pourrait permettre de mettre en évidence de nouveaux interacteurs également impliqués. Cette stratégie a été appliquée par notre équipe, dans une étude en protéomique de têtes d’embryons de souris soumises à une intoxication à l’acide rétinoïque in utero, permettant ainsi de rapporter de nouvelles cibles candidates89.
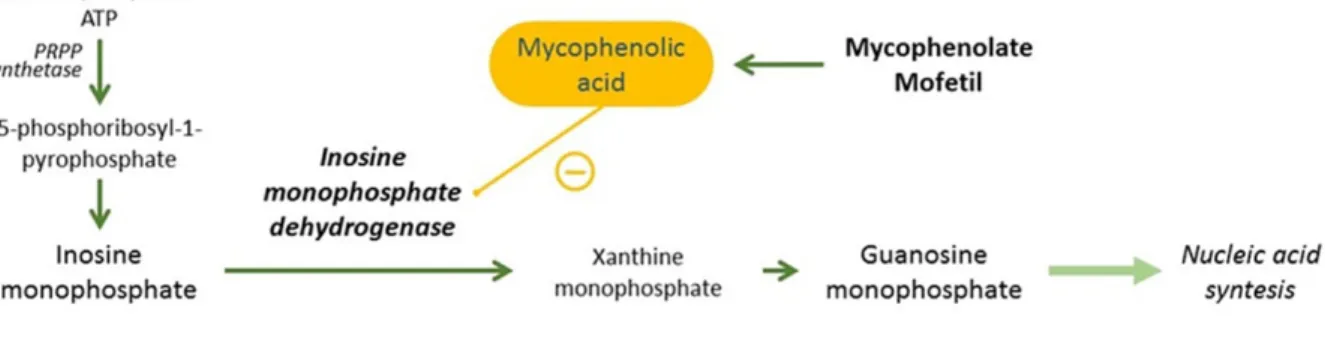
Mycophénolate mofétil
Le mycophénolate mofetil (CellCept®) est un immunosuppresseur largement prescrit dans le cadre du traitement post transplantation, et des maladies auto-immunes. Le mycophénolate mofetil a été désigné comme une prodrogue presque totalement hydrolysée en sa forme active, l’acide mycophénolique, dans l’estomac, l’intestin grêle, le sang, et le foie90–92. L’acide
mycophénolique est un inhibiteur non compétitif de l’inosine 5’-monophosphate déshydrogénase (IMPDH). L’IMPDH est impliquée dans la synthèse de novo de guanosine, en catalysant l'oxydation de l’inosine monophosphate en xanthine 5’-monophosphate, métabolite intermédiaire de la production de guanosine triphosphate (GTP)93 (Figure 8).
De nombreuses observations rapportent un effet tératogène chez au moins 25 patients, avec la survenue de phénotypes malformatifs pouvant se rapprocher de l’OAVS. Ont notamment été rapportées la survenue de microtie ou anotie, d’atrésie du canal auditif externe, de fentes orofaciales, de colobome, de micrognathie, et d’atrésie de l’oesophage92,94–105. Des expériences
40 sur modèles zebrafish ont également été menées, rapportant un phénotype caractérisé par des malformations de la queue et un œdème péricardiaque106,107.
Figure 8 D’après Allisson et al.93 Impact de l’acide mycophénolique sur la voie de synthèse de novo de GTP.
Disruption vasculaire
L’hypothèse de la survenue d’une disruption vasculaire, impliquant notamment l’artère stapédienne, a été évoquée dès 1940 comme cause de malformations cranio-faciales108–110, et
donc possiblement de l’OAVS111, dont le caractère unilatéral des atteintes pourraient
s’expliquer par ce phénomène local. La disruption vasculaire est également la cause supposée d’autres anomalies du développement, telles que le syndrome des brides amniotiques, les anomalies transverses de membres112, ou le syndrome de Poland113, avec pour hypothèse que
41 endothéliales, de pertes tissulaires et de phénomènes de réparation114. Enfin, une étude portant
sur 93 cas de fœtus exposés au misoprostol rapporte chez 17,2% d’entre eux la survenue d’une microsomie hémifaciale115. Cet analogue de la prostaglandine est utilisé dans le cadre
d’interruption de grossesse, et induit des contractions utérines. Il a été supposé que celles-ci pouvaient conduire à l’apparition de disruptions vasculaires chez le fœtus, à l’origine de différentes atteintes, comme le syndrome de Moebius ou des anomalies des membres115,116.
Cependant, plusieurs éléments ne permettent pas d’expliquer la survenue de l’OAVS par la seule existence d’une disruption vasculaire117. Si le caractère unilatéral des atteintes est l’une
des caractéristiques décrites de l’OAVS, celles-ci sont dans de nombreux cas bilatérales, bien qu’asymétriques, ce qui ne peut être expliqué par un phénomène latéralisé. Par ailleurs, l’OAVS comprend des malformations qui n’appartiennent pas à la sphère cranio-faciale, telles que les malformations vertébrales et cardiaques.
Gémellité
De nombreux cas d’OAVS chez des patients issus de grossesses gémellaires ont été rapportés, qu’il s’agisse de jumeaux avec phénotypes concordants ou discordants 118–123, notamment chez
des jumeaux monozygotes (Figure 9 et 10). De plus, les cohortes de patients OAVS rapportent un taux significativement supérieur de grossesses gémellaires par rapport à la population générale (2%), avec des estimations allant de 3,9 à 14%17,60,124. Parmi les patients inclus dans
la cohorte de patients étudiée au laboratoire MRGM Inserm U1211, on estime la prévalence de ces grossesses gémellaires à près de 14,5% (Données non publiées). On remarque cependant
42 que d’après l’étude de Wieczorek et al.124, le taux de grossesse obtenue après aide médicale à
la procréation (AMP), facteur de risque de grossesse gémellaire, est supérieur chez les patients OAVS par rapport à la population générale. Ainsi, il n’est pas encore établi que la gémellité soit en lien avec l’OAVS de façon directe. On peut cependant noter que la discordance entre jumeaux monozygotes a été étudiée dans le cadre des malformations cardiaques congénitales, avec différentes hypothèses : facteurs épigénétiques, anomalies des voies de signalisation de la latéralité, ou facteurs vasculaires125–127. Ces mêmes facteurs pourraient être en cause dans la
survenue de l’OAVS. Enfin, l’existence de jumeaux discordants pourrait être en faveur de l’implication de mutations somatiques. Cependant, une étude par séquençage d’exome de jumeaux OAVS n’a pas permis de mettre en évidence de tels évènements123.
43 Figure 9 Jumelles monozygotes présentant un OAVS, avec atteintes “en miroir”121.
Figure 10 D’après Chen et al.123. Jumeaux monozygotes discordants, dont seul l’un des deux
44 Diabète maternel
La présence d’un diabète maternel préexistant à la grossesse est l’un des facteurs fréquemment retrouvé dans la littérature comme associé à la survenue de l’OAVS13,128.
L’étude de Barisic et al.13, basée sur les données du réseau EUROCAT, rapporte un taux de
diabète préexistant à la grossesse de 3% chez les mères de patients OAVS, à comparer au taux estimé de 0,3% dans la population générale129. Une étude cas- contrôles portant sur une
cohorte de 108 enfants présentant une microsomie hémifaciale et 84 enfants sains a pu montrer une association significative avec le diabète maternel (OR 4.01, 95% CI 1.6-10.5). Différentes hypothèses ont été évoquées pour expliquer la tératogénicité du diabète maternel, notamment l’existence de modifications épigénétiques130, de même que la survenue de stress
oxydatif131, même si les mécanismes précis ne sont à jour pas connus.
Les causes moléculaires identifiées Variants de structure
Délétion 22q11.2
De nombreuses études ont rapporté l’existence de réarrangements chromosomiques intéressant le locus 22q11.2, avec la présence de délétions ou de duplications, chez des patients atteints d’OAVS132–142. Les délétions de ce locus sont bien connues, le syndrome de délétion 22q11.2,
45 prévalence estimée à 1/1000 foetus143. Cette pathologie associe une déficience intellectuelle
variable, des anomalies vélo-palatines, cardiaques, ainsi qu’une auto-immunité. Le remaniement chromosomique le plus récurrent, rapporté dans ce syndrome chez 90% des patients144, est la présence d’une délétion hétérozygote d’environ 3 Mb, dont les points de
cassure correspondent à deux des régions Low-copy repeat de ce locus, les LCR22A et LCR22D. Celle-ci aboutit à l'haploinsuffisance d’une centaine de gènes, dont l’influence de chacun dans la survenue du phénotype a été étudiée extensivement143,145–149. Parmi ceux-ci, les
gènes TBX1150 et DGCR8151, ont été identifiés comme jouant un rôle majeur dans cette
pathologie.
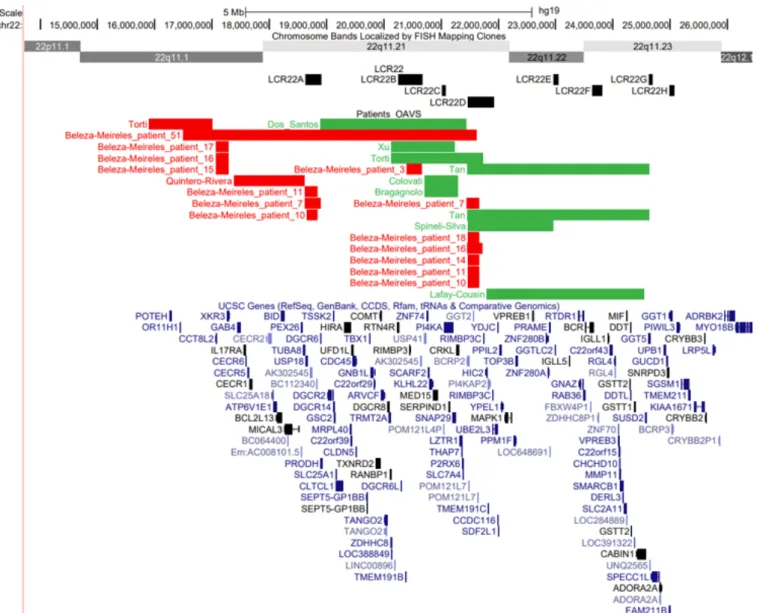
Concernant les patients atteints d’OAVS, l’analyse des différents remaniements rapportés dans la littérature à ce locus montre que ceux-ci ne sont pas pour la plupart superposables aux délétions retrouvées dans le syndrome de DiGeorge (Figure 11). On peut noter que plusieurs patients inclus dans l’étude de Beleza-Meireles et al.17 présenteraient chacun deux duplications
distinctes identifiées par CGH-array, et localisées au niveau des LCR22A et LCR22D. Cependant, la présence de remaniements complexes, incluant une inversion par exemple, n’a pas été exclue chez ces patients. L’objectif d’une étude réalisée par Glaeser et al.152 a été
d'identifier le ou les gènes les plus fréquemment impactés par ces remaniements chromosomiques, et d’en déduire ceux qui sont le plus susceptibles d’être responsables de l’OAVS. Ces travaux rapportent ainsi les gènes CLTCL1, GSC2, HIRA, MAPK1, TBX1, et
YPEL1 comme étant les meilleurs candidats pour ce locus, sans que leur implication ne puisse
46 divers remaniements chromosomiques chez les patients OAVS, les mécanismes physiopathologiques conduisant aux différentes atteintes ne sont pas connues.
Figure 11 Localisation des remaniements chromosomiques du locus 22q11.2 chez les patients OAVS (UCSC Genome Browser153). En noir : régions de Low-Copy Repeat LCR22A-H. Les
47 Duplication de OTX2
Au moins 3 études portant sur des patients présentant un phénotype OAVS ont rapporté la présence de duplications situées en 14q22.3 et comprenant le gène OTX2154–156.
Le cas index de la première famille décrite est une patiente présentant un phénotype sévère avec retard de développement, associant des enchondromes préauriculaires, des oreilles dysplasiques, une sténose des canaux lacrymaux, une hypoplasie de la mandibule, une micrognathie, associés à des malformations cardiaque, vertébrale, rénale et un méningocèle154.
Le père de cette patiente, ainsi que plusieurs autres membres de la branche paternelle sont rapportés comme présentant également des anomalies cranio-faciales quoique moins sévères. La réalisation d’une CGH-array a retrouvé chez le cas index ainsi que chez son père la présence de remaniements complexes entraînant une duplication de 11,79 Mb du chromosome 14 comprenant notamment les gènes OTX2, SIX1 et SIX6, et une délétion de 4,38 Mb du chromosome 13.
Le cas index de la deuxième famille est une patiente présentant des tags et pits préauriculaires, une agénésie du conduit auditif externe à gauche, une oreille dysplasique à droite, une micrognathie et une hypoplasie mandibulaire155. Cinq autres membres de sa famille présentaient
également des enchondromes préauriculaires. La réalisation d’une CGH-array a retrouvé la présence d’une duplication hétérozygote interstitielle de 1,34 Mb comprenant le gène OTX2, et ségrégeant avec le phénotype dans cette famille.
48 Enfin, une troisième famille a été rapportée, dont cinq membres présentaient une asymétrie faciale avec fente faciale et une hypoplasie mandibulaire et maxillaire, associées en fonction des individus avec une rétrognathie, une microtie, une anotie et/ou des tags préauriculaires156.
La réalisation d’un séquençage d’exomes n’a pas retrouvé de variant ponctuel pertinent, tandis que la réalisation de SNP array a retrouvé la présence d’une duplication hétérozygote de 1,3 Mb comprenant OTX2 chez les individus atteints.
Enfin, une étude par caryotype et FISH chez un patient de 9 mois présentant une asymétrie faciale, une microtie à droite, une communication interventriculaire, ainsi qu’une hémivertèbre retrouve la présence d’une inversion péricentrique du chromosome 14, dont le point de cassure distal a été localisé dans un intervalle comprenant le gène OTX2. Bien que les analyses réalisées ne permettent pas de savoir si ce point de cassure interrompt le gène OTX2, où est situé à proximité, cette inversion pourrait avoir un impact sur son expression, et ainsi conduire à la survenue du phénotype.
OTX2 code pour un facteur de transcription à homeobox de type bicoïde, orthologue du gène orthodenticle chez la drosophile, jouant un rôle critique durant le développement du
prosencéphale et du mésencéphale, des glandes pituitaires et pinéales, de l’oreille interne et de la rétine157,158. Dans les modèles animaux étudiés, les orthologues d’OTX2 apparaissent comme
soumis à une régulation spatiale et temporelle complexe, aboutissant à des gradients d’expression nécessaires à la différenciation antéro-postérieure lors du développement cranio-facial156,159. Les souris KO pour Otx2 présentent un phénotype létal avec anomalies de la
49 comprendre une anencéphalie, une micrognathie, une anophtalmie ou microphtalmie160,161. Par
ailleurs, les variants délétères entraînant la perte de fonction d’OTX2 ont été rapportés comme étant responsables de microphtalmie, anophtalmie, et d’agnathie-otocéphalie162,163.
L’ensemble de ces éléments sont en faveur d’une implication des duplications d’OTX2 dans l’OAVS, du fait de son rôle dans le développement cranio-facial. Cependant, les mécanismes physiopathologiques par lesquels la perte de fonction ou surexpression d’OTX2 aboutissent à ces différentes atteintes cranio-faciales restent à élucider.
Duplication d’éléments régulateurs du gène HMX1 (NKX5-3)
L’existence de plusieurs modèles animaux avait suggéré une implication d’éléments régulateurs situés à distance de HMX1 dans la survenue d’anomalies auriculaires (Voir Modèles animaux). L’étude de Si et al.164 réalisée en 2020 rapporte chez cinq familles dans lesquelles ségrègent
des microties bilatérales, la présence de duplications en 4p16, avec un intervalle critique minimum de 21,8 kb, comprenant une région conservée d’enhancers d’HMX1. Ces résultats sont compatibles avec une étude antérieure par analyse de liaison165, qui avait déjà identifié le
locus comprenant HMX1 comme lié à la microtie bilatérale. Les analyses fonctionnelles réalisées par test sur gène rapporteur luciférase montrent que cet enhancer est actif chez l’humain, et est sensible au facteur de transcription HOXA2.
Dans la cohorte de patients rapportée par Bragagnolo et al.60, trois patients présentaient des
50 avec asymétrie faciale, atteinte mandibulaire, microtie, et anomalies vertébrales. L’analyse des coordonnées de ces duplications montrent que celles-ci comprennent HMX1, et excluent a priori la région de 600 paires de bases identifiée comme enhancer, sans que la résolution des puces utilisées ne permette de l’affirmer avec certitude.
HMX1 code pour un facteur de transcription à homeobox jouant un rôle majeur dans la
différenciation mésenchymateuse durant le développement craniofacial166. Par ailleurs, les
variants entraînant la perte de fonction biallélique de HMX1 sont responsables chez l’humain du syndrome oculo-auriculaire167,168, caractérisé par des anomalies oculaires (microcornée,
microphtalmie, dysgénésie du segment antérieur, cataracte, colobome, anomalies de l’épithélium pigmentaire, dystrophie rod-cone), ainsi qu’une anomalie auriculaire spécifique, avec incisure du lobe de l’oreille.
L’ensemble de ces éléments montre que les remaniements chromosomiques du locus du gène
HMX1 semblent bien être responsables d’atteintes du spectre OAV, les duplications limitées
aux éléments régulateurs étant associées à des microties bilatérales isolées, les duplications du gène HMX1 lui-même causant des atteintes plus sévères de l’OAVS. On pourra noter que les auteurs de l’étude de Si et al.164 soulignent que l’analyse de ces remaniements doit prendre en
compte l’effet de l’inclusion ou non des barrières des Topologically Associated Domains (TADs), pouvant expliquer que des sujets asymptomatiques soient porteurs de duplications de ce locus dans la base de données DGV.
51 Autres variants de structure et aneuploïdies
Dans la littérature, quatre études indépendantes ont rapporté de façon systématique les remaniements chromosomiques détectés dans des cohortes de patients OAVS16,17,60,169 (Figure
12). Par ailleurs, de très nombreux cas individuels ont été rapportés, présentant chacun un ou plusieurs remaniements chromosomiques, qu’il s’agisse de délétions, duplications, inversions, ou translocations, dont la présence a été associée à la survenue de l’OAVS chez les patients170– 185. Des études antérieures ont également rapporté la présence de trisomies en mosaïque
intéressant les chromosomes 7, 9 et 22186–189. Enfin, un cas de mosaïcisme 45, X / 46, XY a été
rapporté190. Néanmoins, en l’absence de réplication de ces différents résultats, et bien que des
gènes candidats soient décrits dans nombre de ces études du fait de leurs rôles biologiques connus, l’implication de ces différents variants de structure dans l’OAVS ne peut être affirmée.
52 Figure 12 Visualisation de type Circos plot résumant l’ensemble des variants de structure décrits dans l’OAVS. En vert : délétion, en rouge : duplication, arcs gris : translocation ou inversion.
53
Variants ponctuels
Plusieurs études décrites ci-dessous ont rapporté la présence de variants ponctuels dans différents gènes, associés à la survenue d’un OAVS. Ces études ont été singulièrement facilitées par le développement du séquençage de l’exome dans l’étude des causes des maladies rares, permettant d’obtenir en une seule expérience la totalité des variants situés dans les régions codantes du génome. Cependant, des variants d’un seul gène, MYT1, a pu être retrouvé dans 3 études de 2 équipes indépendantes. De plus, l’ensemble des variants ponctuels décrits n’explique la survenue d’un OAVS que chez un très faible pourcentage de patients. On notera également qu’une étude de type Genome Wide Association Study (GWAS) a pu retrouver différents gènes candidats associés à la présence d’une microsomie hémifaciale.
MYT1
Le gène MYT1 a été le premier gène identifié en 2016 comme responsable d’une forme autosomique dominante d’OAVS59,88. Ce gène code pour le facteur de transcription myelin
transcription factor 1, appartenant à une famille de protéines se liant à l’ADN via un domaine à doigt de zinc de type C2HC. Cette famille comprend également les protéines MYT1L et ST18. Toutes trois sont fortement exprimées dans le système nerveux en formation, et sont indispensables à la migration des précurseurs neuronaux dans la région subventriculaire et la plaque corticale191,192. MYT1 forme un complexe multiprotéique stable la Lysine-Specific
Demethylase 1 via une interaction directe193, complexe jouant un rôle majeur dans la régulation