Publisher’s version / Version de l'éditeur:
Electrochimica Acta, 61, pp. 198-206, 2011-12-09
READ THESE TERMS AND CONDITIONS CAREFULLY BEFORE USING THIS WEBSITE.
https://nrc-publications.canada.ca/eng/copyright
Vous avez des questions? Nous pouvons vous aider. Pour communiquer directement avec un auteur, consultez la première page de la revue dans laquelle son article a été publié afin de trouver ses coordonnées. Si vous n’arrivez pas à les repérer, communiquez avec nous à [email protected].
Questions? Contact the NRC Publications Archive team at
[email protected]. If you wish to email the authors directly, please see the first page of the publication for their contact information.
NRC Publications Archive
Archives des publications du CNRC
This publication could be one of several versions: author’s original, accepted manuscript or the publisher’s version. / La version de cette publication peut être l’une des suivantes : la version prépublication de l’auteur, la version acceptée du manuscrit ou la version de l’éditeur.
For the publisher’s version, please access the DOI link below./ Pour consulter la version de l’éditeur, utilisez le lien DOI ci-dessous.
https://doi.org/10.1016/j.electacta.2011.12.005
Access and use of this website and the material on it are subject to the Terms and Conditions set forth at
Nanocrystalline tungsten carbide (WC) synthesis/characterization and
its possible application as a PEM fuel cell catalyst support
Zhu, Weimin; Ignaszak, Anna; Song, Chaojie; Baker, Ryan; Hui, Rob; Zhang,
Jiujun; Nan, Feihong; Botton, Gianluigi; Ye, Siyu; Campbell, Stephen
https://publications-cnrc.canada.ca/fra/droits
L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE WEB.
NRC Publications Record / Notice d'Archives des publications de CNRC:
https://nrc-publications.canada.ca/eng/view/object/?id=161331f2-e814-4824-8629-e3cdd0fe3677
https://publications-cnrc.canada.ca/fra/voir/objet/?id=161331f2-e814-4824-8629-e3cdd0fe3677
ContentslistsavailableatSciVerseScienceDirect
Electrochimica
Acta
jo u r n al h om ep a ge :w w w . e l s e v i e r . c o m / l o c a t e / e l e c t a c t a
Nanocrystalline
tungsten
carbide
(WC)
synthesis/characterization
and
its
possible
application
as
a
PEM
fuel
cell
catalyst
support
Weimin
Zhu
a,
Anna
Ignaszak
a,
Chaojie
Song
a,∗,
Ryan
Baker
a,
Rob
Hui
a,
Jiujun
Zhang
a,
Feihong
Nan
b,
Gianluigi
Botton
b,
Siyu
Ye
c,
Stephen
Campbell
daInstituteforFuelCellInnovation,NationalResearchCouncilofCanada,4250WesbrookMall,Vancouver,BC,V6T1W5Canada
bDepartmentofMaterialsScienceandEngineering,McMasterUniversity,1280MainStreetWest,Hamilton,ON,L8S4L8Canada
cBallardPowerSystemsInc.,9000GlenlyonParkway,Burnaby,BC,V5J5J8Canada
dAFCCAutomotiveFuelCellCooperation,9000GlenlyonParkway,Burnaby,BC,V5J5J8Canada
a
r
t
i
c
l
e
i
n
f
o
Articlehistory:
Received5October2011
Receivedinrevisedform1December2011
Accepted2December2011
Available online 9 December 2011 Keywords:
Tungstencarbide
Catalystsupport
Oxygenreductionreaction
PEMfuelcells
a
b
s
t
r
a
c
t
Nanocrystallinetungstencarbide(WC)withahighsurfaceareaandcontainingminimalfreecarbonwas
synthesizedviaapolymerroute.Itsphysicalproperties,includingsolubilityinacidsolution,electronic
conductivity,andthermalstability,werethoroughlystudiedattwoelevatedtemperatures:95◦Cand
200◦C.ComparedtocommerciallyavailableWC,thisin-housesynthesizedWCshowedlowersolubility
inacidicmediaat200◦C,higherelectronicconductivity(comparabletothatofcarbonblack),aswellas
higherthermalstability.However,thismaterialexhibitedlowelectrochemicalstabilityinacidicmedia
whensubjectedtopotentialcyclingatpotentialslargerthan0.7Vvs.RHE,duetotheelectrooxidation
ofWC.ThemajorproductofWCelectrooxidationisWO3,whichwasconfirmedbyX-rayphoton
spec-troscopymeasurements.PtwasuniformlydepositedonthehighsurfaceareaWCtoforma20wt%ofPt
supportedcatalystfortheoxygenreductionreaction(ORR).TheORRmassactivitywasthenobtained
usingtherotatingdiskelectrodetechnique.
Crown Copyright © 2011 Published by Elsevier Ltd. All rights reserved.
1. Introduction
Inthelastseveraldecades,protonexchangemembrane(PEM) fuel cells, which have several advantages such as high power density,highenergydensity,andlow/zeroemission,have demon-stratedtheirfeasibilityaspracticalenergyconversiondevicesfor manypower-demandingfieldsincludingportable,automotive,and stationary applications. One of the major technical challenges, insufficientdurability,is identifiedasa barrier hindering wide-spreadcommercializationofPEMfuelcells.Ithasbeenrecognized thatthedegradationoffuelcellcatalystsisthemajorcausefor insufficientdurability.
Atthecurrentstateoftechnology,carbon-supportedplatinum (Pt)-basedcatalystsarethemostpracticalforPEMfuelcell opera-tionintermsofbothcatalystactivityanddurability.Unfortunately, duringfuelcelloperation,Ptdissolutionandcarbonsupport oxida-tion/corrosionwilloccur,degradingthecatalystperformance,and leadingtolowdurabilityofthefuelcell.Toaddressthischallenge, oneapproachistoreducecarbonsupportcorrosionbyhybridizing
∗ Correspondingauthor.Tel.:+16042213000x5577;fax:+16042213001.
E-mailaddress:[email protected](C.Song).
thesupportmaterialsorreplacingsomeofthecarboncontentusing non-carbonmaterials.Withrespecttothis,intensiveresearchhas beendonewitha focusonthecorrosion mechanism ofcarbon supportanditspreventioninthemostrecentyears[1–3].
Currently, carbon blacks,suchas Vulcan XC-72Rand Ketjen black,havebeenusedasthecatalystsupportsforPEMfuelcell catalystsbecauseof theirunique propertiesincludingsufficient electronic conductivity, high surface area, and suitable porous structure.However,underfuelcelloperatingconditions,carbon ispronetooxidationthroughthefollowingreaction:
C+H2O→CO2+4H++4e−, Eo= 0.207Vvs.SHE (1) At the fuel cell cathode, the normal operation potential (0.5–0.9V vs. RHE)is more positive than the carbon oxidation potential.Duringthestart-up/shutdownprocess,thepotentialcan reachas high as1.5V vs. RHE (reversible hydrogenelectrode), significantlyhigherthanthecarbonoxidationpotential.These elec-trodepotentialsfacilitatecarbonoxidation throughreaction(1). Furthermore,thepresenceofPtand/orhightemperature opera-tionofthePEMfuelcellcanacceleratecarbonoxidation/corrosion. Obviously,thiscarbon oxidation/corrosioncanresultinthe iso-lation of Pt catalyst particles and Pt catalyst agglomeration, degradingthefuelcellperformancesignificantly[4].Therefore,the
0013-4686/$–seefrontmatter.Crown Copyright © 2011 Published by Elsevier Ltd. All rights reserved.
developmentofhighperformancenon-carbonsupportmaterial,to replacecarbonisimperative.This,however,turnsouttobenot aneasytask,becauseahighperformancecatalystsupportshould meetallofthefollowingrequirements:highthermalstability,high electronicconductivity,lowsolubilityinacidicmedia,high electro-chemicalstability,highsurfacearea,aswellasfavouriteinteraction withthecatalystparticles.
Amongthenon-carbonsupportcandidates,tungstencarbide (WC)appearstobeattractive.Asarefractorymetalcarbide tra-ditionallyforerosionresistantcoatings,WC hasa highthermal stabilityandischemicallyinert.Theexperimentalresultsshowed that, under ambient conditions, the oxidation of WC starts at around600◦C [5],muchhigherthantheoperatingtemperature ofPEMfuelcells.WCalsoexhibitsgoodelectronicconductivity, whichmakesitanexcellentmaterialfordiffusionbarrierlayers inthesemi-conductorindustry.Inaddition,thePtlikecatalytic behaviourofWCcouldresultinasynergeticeffectwiththecatalyst, leadingtoanincreaseincatalyticactivity[6].
Tungsten carbide supported Pt catalysts have been synthe-sizedand usedfortheoxygenreduction reaction(ORR)aswell asmethanoloxidation, and havedemonstrated theirsynergetic catalytic effect. Meng and Shen [7] prepared a carbon-based nanocrystalline W2C using the intermittentmicrowave heating (IMH)method,thenmixed withPt/CandIMHtreated againto formacatalyst.Theirresultsshowedthatthecompositecatalyst obtainedcouldexhibitahigherORRonsetpotential,higher spe-cificactivity,aswellasbetteroxygenreductionselectivityinthe presenceofmethanolthanthatofthePt/Ccatalyst, demonstrat-ingasynergeticeffectbetweenthetungstencarbideandPt.Nie etal.[8]synthesizedaPt–WC–W2C/Ccatalystusinganimproved one-stepIMH.ThePt–WC–W2C/Ccatalyst showedaneven bet-terORRactivitythanthosePt–W2C/Ccompositecatalysts.Wang etal.[9]preparedhighsurfaceareatungstencarbidemicrospheres (withmixedWCandW2Cphases)usingahydrothermalpolymer method.ThePt/tungstencarbidemicro-spherescatalystwith uni-formlydistributedPtparticlesexhibitedabetterORRactivitythan thePt/Ccatalyst,whichwasattributedtothelarge electrochemi-calsurfaceareaandthePt–tungstencarbidesynergeticeffect.Zhu etal.[10]observedanenhancedORRcatalyticactivityforPton tungstencarbide-modified-carbonsupport.Modificationofcarbon withtungstencarbidecouldalsoimprovethethermalstabilityof thesupport.GanesanandHam[11–13]synthesizedhighsurface areatungstencarbide(W2C)microspheresandWCsupportsusing acarbonspheretemplate.ThecatalystwithPt(7.5%)loadingon theW2C microsphereshowedhigheractivitytowardsmethanol electro-oxidationthantheconventional20%Pt–Ru/Vulcancatalyst [11–13].Thisispossiblyduetoaco-catalyticeffectbetweenPtand tungstencarbide:tungstencarbideactivatesmethanoltoforma methoxyintermediate,andPtpromotesthedecompositionofthe methoxyspecies[12].
Thehighthermalstability,highelectronicconductivity,aswell asthesynergeticcatalyticeffectwiththecatalyst,maketungsten carbidepromisingasaPEMfuelcellcatalystsupportespeciallyfor hightemperatureoperation.However,theelectrochemical stabil-ityofWCmightbeanissuethatcouldpreventWCfromapplications inPEMfuelcells. Controversialliteratureresultswerereported regardingtheelectrochemicalstabilityofWCandWCsupported catalysts.MazzaandTrassatti[14]usedWCasaninertelectrode, whichdemonstratedgoodchemicalandelectrochemicalstability belowtheoxygenevolutionpotential.Chhina[15]foundthatWC supportedPtcatalystwaselectrochemicallymorestablethanthe carbonsupportedcommercialcatalystHiSpec4000TM(Pt/Vulcan XC-72R)whensubjectedtocontinuousCVscansatelevated tem-perature(80◦C).Voorhies[16]foundthatWCcouldbechemically oxidizedin1MH2SO4atroomtemperature,andelectrochemically oxidizedatmediumtohighanodicpotential.Weigertetal.[17]
observeda largeanodicpeak at0.6–0.8Vonthecyclic voltam-mograms(CVs)ofWCthinfilm,whichwasattributedtotheWC oxidation,andWO3 wasindeeddetectedbyXPSonthesurface aftertheCVscans.Lee[18]alsofoundthatWCwasnot electro-chemicallystable.However,somemetalsadditionscouldprevent orslowdowntheelectrochemicaloxidationofWC,possiblydueto asynergeticeffect.Forexample,theelectrochemicaloxidationof WCcouldbesloweddownbyalloyingwithTa,andtheWCthinfilm, onwhichamono-layerofPtwasdeposited,remainedunchanged afterCVmeasurements,whiletheonewithoutPtdepositionwas completelyoxidizedintoWO3.
Theimprovedelectrochemicalperformanceofthetungsten car-bidebasedPtcatalyst[7–13],andthecontroversialresults[14–18] regardingtheelectrochemicalstabilityoftungstencarbide moti-vatedtheauthorstomakeathoroughinvestigationofthephysical, chemical,andelectrochemicalpropertiesofWCtoevaluateits pos-sibilityasaPEMfuelcellcatalystsupport.
Itisworthwhiletonotethatintheliterature,aconsiderable amountof workwasfocused ontungstencarbide/carbon com-positesupports.Sincecarbonwillbeelectrochemicallyoxidized, leadingtofuelcellperformancedegradation,theexistenceof car-boninthesupportmaynotbedesirable.Inaddition,thetungsten carbidereportedinsomepapersisamixedphase(W2CandWC), whichisnot suitableforPEMfuelcellcatalystsupport applica-tionbecauseW2Ciselectrochemicallyandthermodynamicallyless stablethanWC[19].Fromthepointviewoffundamental under-standing,acarbon-free,single-phaseWCsupportisdesiredandis thefocusofthispaper.
Inourhightemperature(HT)PEMfuelcellapproach,inorderto overcomethechallengeofcarbonsupportcorrosioninHTPEMfuel cells,wesynthesizedasingle-phaseWCsupportwithhighsurface area,andinvestigateditselectronicconductivity,chemicalstability inacidicsolution,aswellastheelectrochemicalstability.ForORR activity,thisWCmaterialwasusedasthePtcatalystsupportto formPt/WCcatalyst,andthecorrespondingORRmassactivityas wellastheelectrochemicalstabilitywereinvestigated.
2. Experimental
2.1. SynthesisofWCsupportandPt/WCcatalyst
Tungstencarbide(WC)supportsweresynthesizedusinga poly-merprecursorroutemodifiedbasedonGanesan’sprocedure[7,8]. Typically, 4.73gof ammoniummetatungstate(AMT, Fluka) and 1.21g of resorcinol (Sigma–Aldrich) were dissolved in 1.63mL of formaldehyde (37% in H2O, Sigma–Aldrich) and 19.8mL of deionizedwater.Thesolutionwasthenrefluxedat95◦Cfor24h forminganorange precipitate.Theprecipitate waswashed and driedatroomtemperature,andthenheat-treatedinatubefurnace underamixtureofAr(flowrate:160mL/min)andH2(flowrate: 60mL/min)gasflowat1000◦Cfor2htoformWC.Afterwards,the furnacewascooleddownto700◦Candkeptfor2hunderaH
2flow (flowrate:115mL/min)toremovetheextracarbon.The synthe-sizedWCisdenotedasWC-SYNinthispaper.ForPtdepositionto formWC-supportedcatalyst(Pt/WC),anintermittentmicrowave heating(IMH)assistedpolyolmethodwasemployedtodepositPt ontotheWCsupport.Typically,90mgWCsupportwasdispersed in20mLethyleneglycol(EG),andthepHwasadjustedto8.0with 0.2MNaOHEGsolution.ThisWCsuspensionwasthensonicated for1h,followedbyadding46.6mgofH2PtCl6 (dissolvedin3mL EG)dropwise intotheWC suspension.Afterwards,thecatalyst precursorsuspensionwasheatedinamicrowaveoven(LBP111RS, LaddResearch,USA)at600Wfor60s,andthenintermittentlyfor 6timeswith10sofmicrowavingeachtime.Theproductwasthen filtered,washed,anddried.
2.2. Structureandmorphologycharacterizations
ThestructureoftheseWCsampleswascharacterizedbyX-ray diffraction(XRD)andthelatticepatternoftheWCsupportwas observedusinghighresolutiontransmissionelectronmicroscopy (HRTEM)togetherwithelectrondiffraction.Theinter-planar dis-tance of the lattice was measuredbased on the latticefringe. Scanning electron microscopy (SEM) and transmission electron microscopy(TEM)wereemployedtostudythemorphologiesof theWCprecursors,WCproducts,andPt/WCcatalysts.
TheXRD measurementswere carried outusing a Bruker D-8diffractometerwithastepsizeof0.02◦ and anexposuretime of 0.1s for each step. SEM observations were carried out on a Hitachi S3500N scanning electron microscope with an oper-ating voltage of 20kV. Samples for TEM characterization were directlysupportedona coppermeshwithacarbon micro-grid. TEMinvestigationsatrelativelowmagnificationwereperformed using a Philips CM12 operated at 120kV. HRTEMobservations were carried out using a JEOL-2010F electron microscope at an accelerating voltage of 200kV with a Field-Emission Gun (FEG). This instrument is equipped with a Gatan Imaging Fil-ter (GIF) for energy-filtered imaging and an energy dispersive X-ray(EDX)detectorforelementalanalysisandmapping. Exper-imentallyobtainedelectron diffractionpatternswerecompared to electron diffraction patterns simulated with the software programJEMS.
2.3. Solubilitytest
Thesolubilityofcommerciallyavailable(AlfaAesar)and synthe-sizedWCpowdersweretestedin1MH2SO4 at95◦Cand200◦C, respectively,accordingtothefollowingprocedures:forthe solubil-itytestat95◦C,aflaskcontaining200mgofWCpowderand50mL of1MH2SO4wasputinanoilbathwithconstantstirring,keptat thistemperaturefor24h.Afterwards,thesuspensionwasfiltered, andthefilteredsolutionwassentforICP/MS(inductivelycoupled plasmamassspectroscopy)toanalyzeforW,fromwhichthe sol-ubilityofWC wascalculated.Theleftoverpowderwaswashed, dried,andsubjectedtoXRDcharacterizationtolookforanyphase changes;forthesolubilitytestat200◦C,anin-housemade chemi-callyinertvesselwithapressuresealingsystemwasused.200mg ofWCpowdersampleand50mLof1MH2SO4wereputintothis vessel,whichwasthenfilledwithN2gasatapressureof200psi.The highpressure(200psi)keptinsidethevesselcanpreventH2Ofrom evaporatingat200◦C,sothattheconcentrationofH
2SO4doesnot changeduringthetest.Thevesselwasthenheatedinanoilbath at200◦Cfor2h.Aftercoolinganddepressurization,the suspen-sionwasfiltered,andthesolutionanalyzedusingICP/MSwhere theleftoverpowderwasthenwashed,dried,andsubjectedtoXRD characterization.
2.4. Thermalstabilitytest
Thethermal stabilitytest wascarried outat200◦C for96h. Typically,certainamountofsamplewasputinaMuffleFurnace, heldat200◦Cinairfor96h.Thenthesamplewascooleddown toroomtemperatureandsentforXRDcharacterizationtoobserve anyphasechanges.
2.5. Conductivitymeasurement
Anin-housemadeconductivitycell,whichconsistsofanUltem cylinder(innerdiameter=0.5cm)toholdthesampleandtwoCu pistonsaselectrodes,wasusedforconductivitymeasurement.A smallamountofWCpowderwasplacedintheconductivitycell, andaloadwasappliedontheupperpistontokeepapressureof
80kPa.ThewholesetupwasputinaMufflefurnacetocontrolthe temperature.ATsuruga3566ACm-Ohmtesterwithafour-point setupwasusedtomeasuretheconductivityataconstantfrequency of1kHz.TheelectronicconductivityofWCwasmeasuredat95and 200◦C.
2.6. Otherphysicaltests
Forsurfaceareaanalysis,aBeckmanCoulterSA-3100Surface AreaAnalyzerwasemployedtoobtainN2adsorption/desorption isotherm,fromwhichtheBETsurfaceareaoftheWCsamplewas calculated.Forthedeterminationoffreecarboncontentinthe syn-thesizedWCsamples,thermalgravityanalysis(TGA)usingaPerkin ElmerParis6ThermalGravityAnalyzerwascarriedoutontheWC samplesfromroomtemperatureupto730◦Cinair.TheX-ray pho-toelectronspectroscopy(XPS)oftheas-synthesizedWCpowder andtheoneafter700cyclicvoltammogramscans(from0.05Vto 1.2V,seebelow),takenbyaLeyboldMAX-200X-rayPhotoelectron Spectrometer,wasusedtoanalyzethesurfacecompositionsofthe samples.
2.7. Electrodepreparationandelectrochemicalmeasurements Toprepareacatalyst(Pt/WC)orsupport(WC)ink,19mg cat-alystorsupportwasdispersedin9.5mLisopropanoland0.5mL de-ionizedwater,sonicatedfor1htoformacatalystorsupport ink.24Lofcatalystorsupportinkwasuniformlydepositedona glassycarbonelectrodewithageometricareaof0.20cm2(Glassy carbon electrode diameter: 5mm, AFE7M050GC, Pine Research Instrument)toformacatalystorsupportlayer.Afterwards,7.1L Nafion®solution(50:1inmethanol)wasdepositedontoptoform theelectrodecoatinglayer.
Theelectrochemicalmeasurements werecarriedout usinga conventionalthree-electrodecellcontaining0.1MHClO4aqueous solution.ForORRmeasurement,thiselectrolytesolutionwas sat-uratedwithpureoxygen(bubbledbypureoxygengas).For CV, thissolutionwasbubbledwithpurenitrogengastoremoveall dis-solvedoxygen.AnRHE(reversiblehydrogenelectrode)wasusedas thereferenceelectrodeandaPtwireasthecounterelectrode.The measurementsweretakenonaSolartron1480multi-potentiostat, controlledbyCorrwaresoftware.Fortheelectrochemicalstability test, the electrode wasimmersed in 0.1M deoxygenated (bub-blingwithN2)HClO4 solutionat30◦C, andtheCVcurves were recorded from 0.05V to 1.2V with a scan rate of 20mV/s up to1000scans.From theCV curves,theelectrochemicalsurface areawascalculatedfromhydrogenadsorption/desorptionpeaks between0.05Vand0.4V.FortheORRmeasurements,the voltam-mogramwasrecordedwithaRDE(rotatingdiscelectrode)ina 0.1MO2-saturatedHClO4 solutionwitha scanspeedof 5mV/s. AnASRrotator(PineInstrument)wasusedtocontroltherotation rate.
Forelectrochemicalactivesurfacearea(ECA)measurement,CO strippingmethodwasused.Thepotentialofthecatalystloaded electrodewillbeheldat0.05Vvs.RHEin0.1MHClO4 withCO purginguntilsaturationofCOadsorptionisreached.Theelectrolyte solutionispurgedwithN2for30mintoremoveun-adsorbedCO. Thenthepotentialoftheelectrodewillbescannedfrom0to1.0V vs.RHEatascanrateof20mV/s.TheECAwillbecalculatedfrom thechargeoftheCOoxidationpeakaccordingtoEq.(1′):
ECA = QCOadsorption
0.42 ×1000 (1
′) whereQCOadsorptionisthechargeforCOoxidationinC,and0.42mC isthechargerequiredtooxidizeamonolayerofadsorbedCOon 1cm2ofPtsurface.
Table1
PropertiesofWCsamples.
BETsurfacearea(m2/g) Solubility(wt%) Conductivity(S/cm) Thermalstability
95◦ C 200◦ C 95◦ C 200◦ C WC-Alfa 1.6 0.15 0.63 0.75 0.87 Stableupto200◦C WC-SYN 89 0.16 0.27 2.4 3.0 Stableupto200◦C
3. Resultsanddiscussion
3.1. PropertiesofcommerciallyavailableWC
As a preliminary assessment of WC as a catalyst support, thesolubility,electronicconductivity,thermalstability,and elec-trochemicalstability of commerciallyavailable WC (denotedas WC-Alfahereafter)wereinvestigated.Thesurfaceareaofthis cat-alystisonly1.6m2/g.
Asshown inTable1,thesolubilityofWC-Alfain1MH2SO4 at95and200◦Cis0.15wt%and0.63wt%,respectively.Nophase changewasobservedontheXRDpatternsaftersolubilitytests(not shownhere),indicatingthatthismaterialhasalowsolubilityand highchemicalstabilityin1MH2SO4.TheWC-Alfawasalsofound tobethermallystableat200◦C.NophasechangeontheXRD pat-ternwasobserved(notshownhere)after96hofheattreatmentat 200◦Cinair.TheelectronicconductivityoftheWC-Alfawasfound toincreasewithtemperaturefrom0.75S/cmatroomtemperature to0.87S/cmat200◦C,whichisinthesameorderasthatofcarbon black.
ThecyclicvoltammetryscanswereconductedonWC-Alfato evaluateitselectrochemicalstability.Nooxidation/reductionpeaks wereobservedwithinthepotentialrange(0.05V–1.2V)upto1000 CVscans(notshownhere).Thisindicatesthegoodelectrochemical stabilityofWC-Alfa.
BasedontheabovepreliminaryassessmentresultsonWC-Alfa, itseemsthatWCcouldbeasuitablecandidateasaPEMfuelcell catalystsupport.However,thesurfaceareaofthisWC-Alfaisfairly low(only1.6m2/g).InordertoobtainhighORRperformance,a highsurfaceareaWCisdesirable.Wehavethussynthesizedahigh puritynano-crystallineWCwithhighsurfacearea.
3.2. Synthesisandpurificationofnano-crystallineWC
Fig.1showstheXRDpatternofthesynthesizedtungsten car-bide(WC-SYN).Itcanbeseenthat apureWC phasewithonly
90 80 70 60 50 40 30 47 48 49 50 2θ (Degree) Inte nsity (a.u. ) WC (2 0 1 ) WC (1 0 2 ) WC (2 0 0 ) WC (1 1 1 ) WC (0 0 2 ) WC (1 1 0 ) W(110) WC (1 0 1 ) WC (1 0 0 ) WC (0 0 1 ) Intensity (a.u.) 2θ (Degree) WC (101)
Fig.1.XRDpatternofin-housesynthesizedWCsample(WC-SYN).
smallamountofWmetal(0.9wt%basedontheXRDpeakarea) wasobtained, indicating thatWC wassuccessfully synthesized. Usually,WCsynthesis involveshightemperature,which results inlowsurfaceareaproducts.However,apolymersynthesis pro-cedurecanreducethesynthesistemperaturesignificantly.Inthis polymersynthesisprocedure,aBakelitetypeW–Ccomplex precur-sorwasformedasanintermediatephase[20].Thisintermediate W–Ccomplexphase couldresult ina W–Chomogeneityatthe molecularlevel,leadingtosignificantdecreaseoftheW–Calloying temperaturecomparedtothatforsolid-statesynthesis.
The WC phase obtained in this work is different from that reported in the literature. For example, Ganesen et al. [11,12] showedthatthetungstencarbideproductwasdominatedbyW2C. Normally,inWCsynthesis,thereduction/carburizationundergoes thefollowingsequence:WO3–WO2–W–W2C–WC[21,22]. Forma-tionoftungstencarbiderequiresmetallicWandactivecarbonthat isproducedontheWsurface.Thephaseofthetungstencarbide productcanbedeterminedbytwocriticalsteps:theformationof activecarbonC*ontheWsurface,andthediffusionofC*intothe metallicW[22].IfthereductionoftungstenoxideintoWisslow, theamountofC*formedwillbelowduetothelackoffreshW surface,andthediffusionofCintoWwillbefast,leadingtothe formationof W2C.On theotherhand,ifthereductionof tung-stenoxideintoWisfast,thereisenoughWfortheformationof C*,and thediffusionofCintoWwillbecomethelimitingstep. Asaresult, WCphase willbeformed.Inthis work,inorderto obtainpureWCphase,weincreasedtheWprecursor(ammonium metatungstate)contenttocreatemoreWavailableforthe forma-tionofC*,andchangedthegasenvironmentfrompureArtomixed gasflowofArandH2tospeedupthereductionoftungstenoxide intoW.Thesechanges successfullyshowtheformationof pure WCphase.
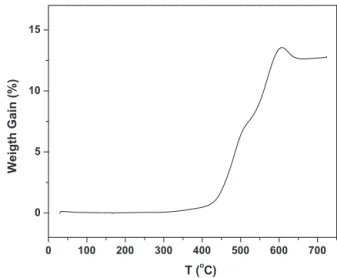
Fig.2showstheTGAresult(inair)ofthesynthesizedWC-SYN after2hofH2treatment.Aweightgainstartingfrom400◦Ccanbe observed,whichisattributedtotheoxidationofWCintoWO3.This weightgainreachesitsmaximumataround600◦C,andthenstarts todecrease,whichisduetoburningofexcesscarbon.After650◦C,
700 600 500 400 300 200 100 0 0 5 10 15 Weigth Gain (%) T (oC)
theweightgainremainsconstant,indicatingthattheproducthas beencompletelyoxidized.Itisworthwhiletonotethatinthis WC-SYNsample,somefreecarbonexists,whichisnotdesirable.
Inordertoremovecarbon(especiallyonthesurface),themost commonwayisflushingthesamplewithH2atanelevated tem-peratureviaamethanizationreaction.However,H2canalsoreduce WCintoWdependingontemperature,heattreatmenttime,aswell asH2concentration.Sincethemethanizationreactionhasalower GibbsfreeenergythantheWCreductionreaction,itmightbe pos-sible,byoptimizingtheseexperimentalparameters,toremovethe excesscarbonatareasonableratewithoutsignificantformationof metallicW.Ribeiroetal.[23]reportedthatWCsurfaceabsorbed polymericcarbon,whichcouldberemovedafterheattreatmentin H2at700◦Cfor1.5h.Howeverafter2hofheattreatment, metal-lictungstencouldbedetectedbyXRD.Theoptimalconditionof carbonremovalbyH2treatmentdependsontheparticlesizeand surfacepropertiesoftheWCproductaswell.
Inthiswork,wetreatedtheas-synthesizedWCproductat700◦C for2hinaH2atmosphere.AsshowninFig.2,thecompositionof theWCproductcanbefoundtobe94.2wt%ofWC,0.9wt%ofWand 4.9wt%ofC,assumingthatWC,W,andCaretheonlycomponents. WealsoobservedthatextendingH2treatmenttimedidnotlead tofurtherreductioninCcontent,ratheritincreasedWcontent. However,whiledecreasingH2treatmenttime,ahigherCcontent wasobserved.Therefore,2hofH2treatmentat700◦Cseemedtobe theoptimaltimetoremovethesurfacecarbonontheWCproduct. Itisbelievedthattheremaining4.9wt%ofcarbonisinsidetheWC particles.
3.3. Propertiesofthein-housesynthesizedWC-SYN
Thesurfacearea,solubility,electronicconductivity,aswellas thethermalstabilityofWC-SYNweretestedinthesamewayas thoseforcommerciallyavailableWC-Alfa.Table1summarizesthe results.Forcomparison,thepropertiesofWC-Alfawerealso pre-sented.
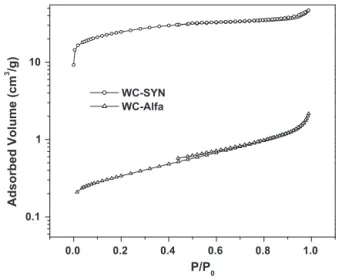
ThesurfaceareaofWC-SYNwasmeasuredusingN2 adsorp-tion/desorption isothermand theobtained datawas compared withthatofWC-Alfa.Fromtheisothermcurve(Fig.3),aBETsurface areaof89m2/gwasobtainedforWC-SYN,whichissignificantly higherthanWC-Alfathatisonly1.6m2/g.
ThesolubilityofWC-SYNat200◦Cis0.27wt%,lowerthanthatof WC-Alfa.Aftersolubilitytesting,astrongWO3peakcouldbeseen ontherecoveredWC-SYN,whileontherecoveredWC-Alfasample, therewasnosuchWO3peak.Thisobservationmaysuggestthatthe formationofinsolubleWO3ontheWC-SYNshouldberesponsible foritslowersolubility.
TheconductivityofWC-SYNmeasuredat95and200◦Cislisted inTable1togetherwiththoseforWC-Alfaforcomparison.Itcan beseenthattheWC-SYNhasahigherconductivitythanWC-Alfa.
1.0 0.8 0.6 0.4 0.2 0.0 0.1 1 10 WC-SYN WC-Alfa Adsorbed Volume (cm 3 /g) P/P0
Fig.3.N2adsorption/desorptionisothermcurvesforWC-AlfaandWC-SYN.
ThishigherconductivityofWC-SYNmightbepartiallyduetothe existenceofWmetal,causedbypostH2treatment.
Regardingthethermalstability ofWC-SYN,noimpurephase couldbeidentifiedontheXRDpatternafterthesamplewasheated inairat200◦Cfor96h,indicatinghighthermalstabilityofWC-SYN. 3.4. WC-SYNmorphologiesandstructure
Fig. 4 shows the SEM images of the WC-SYN precursor (a) andproductaftercarbonremoval(b).TheWC-SYNprecursorhas asphericalmorphologywithsizesaround0.5–1.0m.However, afterthesyntheticprocessandheattreatment,thesemicrospheres collapsedasseeninFig.4bandFig.5.Thisresultisdifferentfrom theliterature[11],wheremicrospheresmorphologywasretained inthefinalproduct.Thiscollapseofthemicrosphereinthefinal WCproductmightbecausedbylowercarboncontentinthe pre-cursor.Normally,carboncanactasskeletonforthemicrosphere.If thecarboncontentintheprecursorisbelowacriticalextent,the microspherestructuremightcollapse.Thebeneficialaspectisthat thiscollapsecanexposemoresurfacesforcarbonremoval,leading toalowCcontent.
Fig.5shows theTEMimageofWC-SYN.Nano-particleswith morphologyofrice-likenanocrystalsandwhiskerscanbeclearly observed.Theaverageparticlesizeisabout35nmaccordingtothe measurementover200particles(Fig.6).
In orderto furtherinvestigatethestructure ofWC-SYN, the HRTEMimagingtogetherwiththeelectrondiffractionwascarried out.AsshowninFig.7,theWC-SYNexhibitsaclearlatticefringes withwell-definedsinglecrystal-likeelectrondiffractionpattern (Fig.7c), indicating high crystallinity of WC-SYN particles. The
Fig.5. TEMimageofWC-SYNparticles.
primaryinterplanarspacingsona lowindexzone axisare2.57 and2.92 ˚A,respectively(Fig.7c),whichareattributedtothe(010) and(001)reflectionsandconsistentwiththehexagonalWClattice (spacegroupofP-6m2).
3.5. Pt/WC-SYN
Tomakeacatalystfortheoxygenreduction reaction,Ptwas depositedontheWC-SYNsupportbyapolyolmethod.Fig.8shows theTEMimageof20wt%Pt/WCcatalyst.Itcanbeseenthatthe PtdistributionontheWCsupportisfairlyuniform,demonstrating thesuccessinsynthesizingPt/WCcatalyst.TheaveragePtparticle sizewasfoundtobearound5nm.
3.6. ElectrochemicalstabilityofWC-SYNandPt/WC-SYN
InordertotesttheelectrochemicalstabilityofWC-SYNsupport anditssupportedPtcatalyst(Pt/WC-SYN),thesematerialswere coatedontheelectrodeindividually,thentestedinN2-saturated 0.1MHClO4solutionat30◦Cusingcyclicvoltammetry.Fig.9shows thecyclicvoltammograms(CVs)ofWC-SYN.Inthefirstcycle,a largeanodiccurrentwasobservedatpotentials>0.7Vvs.RHE.This
Fig.6. SizedistributionofWC-SYNparticles.
Fig.7.(a)EnlargedHRTEMimageofWC-SYNparticle;(b)lowermagnificationviewofaparticle;and(c)thecorrespondingindexedselectedareaelectrondiffraction(SAED)
Fig.8. TEMimageof20wt%Pt/WC-SYNcatalyst(averagePtparticlesize:5nm).
peakdecreasesastheCVscanproceeds,andfinallydisappearsat the100thCVscan.Atthesametime,redoxpeaksbetween0.1and 0.4Vvs.RHEonbothanodicandcathodicscansbecomemoreand moreprominentwithincreasingCVscan,andthenslightlydecrease after100CVcycles,especiallyforthecathodicpeaks(Fig.9b).
Thelargeanodiccurrentat>0.7V vs.RHEshouldarisefrom theelectrochemicaloxidationofWC,asreportedintheliterature [17–19].WiththeproceedingofCVscan,moreandmoreWCwas oxidizedintoWO3, which coveredtheWC surface, resultingin graduallydiminishingofthiscurrent.
0.0 0.2 0.4 0.6 0.8 1.0 1.2 -0.2 0.0 0.2 0.4 0.6 0.8 1.0 1.2 I (mA/cm 2 ) E (V) vs RHE 1st CV scan 10th CV scan 30th CV scan 100th CV scan CV of WC support
(a)
0.0 0.2 0.4 0.6 0.8 1.0 1.2 -0.20 -0.15 -0.10 -0.05 0.00 0.05 0.10 200th CV scan 600th CV scan 1000th CV scan CV of WC support I (mA/cm 2 ) E (V) vs RHE(b)
Fig.9.CyclicvoltammogramsofWC-SYNinaN2-saturated0.1MHClO4solution.
(a)First100scans,and(b)remaining900scan.Scanrate:20mV/s,temperature:
30◦
C,andWCloadingontheelectrode:48g/cm2.
42 40 38 36 34 32 30 28 (a) WC as-synthesized
Binding Energy (ev) (b)
WO 3
WC WC after 700 CV scans
Intensity (A.U.)
Fig.10. XPSspectraofWC-SYN(a)beforeCVscanand(b)after700CVscans.
Thepeaksbetween0.1and0.4Vvs.RHEcouldbeattributed tothereversibleredoxprocess ofWO3/HxWO3 inacidic media [24–26]:
WO3+xH++xe−↔HxWO3 (2)
At thecathodicscan, proton intercalatesinto WO3, forming hydrogen tungsten bronze, resulting in cathodic peaks. At the anodicscans,whenthepotentialincreasestoacertainvalue,H+will de-intercalatefromthehydrogentungstenbronzelattice, oxidiz-ingHxWO3backtoWO3,leadingtoanodicpeaks.Theintensitiesof theseanodicandcathodicpeakswereexpectedtoincreasewithCV cyclingbecausemoreWO3wasproduced.However,adecreaseof thepeakintensitieswasobservedafter100CVscans.Thisislikely duetothedecreaseinsurfaceareacausedbytheWO3coverage [26].
InordertoconfirmtheformationofWO3 ontheWCsurface, XPSspectraweretakenonWC-SYNbeforeandaftertheCVscan, asshowninFig.10.Fortheas-synthesizedWC,aW4fdoubletwith bindingenergiesof31.5eVand33.6eV,canbeobserved,which cor-respondstotheWCphase.Inaddition,twopeakscanalsobefound athigherbindingenergy(35.2eVand37.4eV),correspondingto theW4f7/2andW4f5/2 peaksofWO3phase(Fig.11a).The pres-enceofsmallamountofWO3onthesurfaceoftheas-synthesized
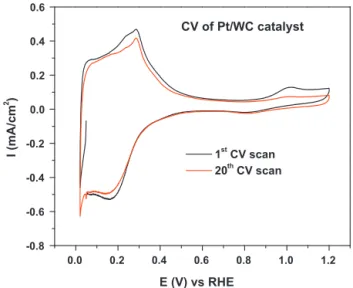
1.2 1.0 0.8 0.6 0.4 0.2 0.0 -0.8 -0.6 -0.4 -0.2 0.0 0.2 0.4 0.6 CV of Pt/WC catalyst I (mA/cm 2 ) E (V) vs RHE 1st CV scan 20th CV scan
Fig.11.Cyclicvoltammogramsof20wt%Pt/WC-SYNinN2-saturated0.1MHClO4
solutionat30◦
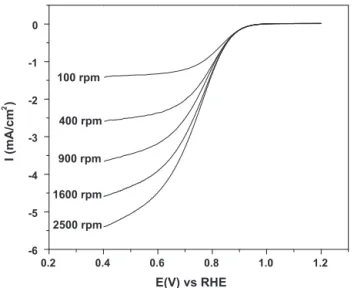
1.2 1.0 0.8 0.6 0.4 0.2 -6 -5 -4 -3 -2 -1 0 2500 rpm 1600 rpm 900 rpm 400 rpm 100 rpm I (mA/cm 2 ) E(V) vs RHE
Fig.12.Voltammetriccurvesatdifferentrotationrates,recordedonarotating
diskelectrodecoatedwith20wt%Pt/WC-SYNcatalystinO2-saturated0.1MHClO4
solutionat30◦C.Scanrate:5mV/s.Ptloading:47.8
g/cm2.
WC-SYNmightbeduetotheoxidationofWCunderambient con-ditions[18].After700CVscans,theintensityoftheW4f7/2 and W4f5/2peaksincreasedsignificantly(Fig.11b),indicatingan elec-trochemicaloxidationofWCintoWO3.TheseXPSresultsconfirm theconversionofWCintoWO3onthesurfaceoftheWC-SYNas aresultofCVcycling.Itisinagreementwiththosereportedby Weigertetal.[17]andLeeetal.[18].
CVcyclingwasalsoconductedonthePt/WC-SYNcatalystto test the electrochemical stability of WC supported Pt catalyst. Fig.11showstheCVcurveofthe20wt%ofPtdepositedon WC-SYN(20wt%Pt/WC-SYN). Largehydrogenadsorption/desorption (from0.05Vto0.4V)peaks,anddisproportionallysmallPt oxida-tion/reductionpeakswereobserved.TheshapeoftheCVcurvefor the20wt%Pt/WC-SYNcatalystisverysimilartothatofthe WC-SYNsupport(after100CVcycles),andthepeakpositionsforthe lowpotentialregion(0.1Vand0.3V)matchwellwiththatofH+ intercalation/de-intercalationprocessinWO3.Similarphenomena werealsoreportedintheliterature[27].Therefore,thislarge hydro-genadsorption/desorptioncurrentiscomposedoftwoprocesses:H adsorption/desorptiononPtandhydrogenadsorption/desorption onWO3.
Fig.11alsoshowsthattheintensityofthePtoxidationpeak decreasesas theCV scan proceeds. This is possibly due tothe electrochemical oxidation of WC-SYN. The formation of non-conductiveWO3couldisolatePtparticlesdepositedonit,leadingto electrochemicallyinaccessiblePtparticles.However,thelarge oxi-dationcurrentat>0.7VonbareWC-SYN(Fig.9)wasnotobserved on20wt%Pt/WC-SYNcatalyst,indicatingthattheoxidationofWC inPt/WC-SYNisnotassevereasbareWC. Thismeansthatthe presenceofPtonWCmightstabilizeWCorinhibitWCoxidation, consistentwiththeresultsreportedinliterature[17,18].
3.7. ElectrocatalyticactivityofPt/WC-SYNtowardsoxygen reductionreaction
InordertoevaluatetheelectrocatalyticactivitytowardORR,the in-housesynthesized20wt%Pt/WC-SYNcatalystwasdepositedon theelectrodesurfacetoformacatalystlayer,thenputintoan elec-trochemicalcellforORRmeasurements.Fig.12showstheCVcurves recordedatvariouselectroderotationratesat30◦CinO
2-saturated 0.1MHClO4 solution.AnORRmassactivityof3.3mA/mgPtwas obtainedfromthe1600rpmcurveat0.9Vvs.RHEafterthe lim-itingcurrentcorrection.Thisvalueissignificantlylowerthanthat
-0.3 -0.2 -0.1 0 0.1 0.2 0.3 0.4 0.5 1.2 1 0.8 0.6 0.4 0.2 0 -0.2 E (V) vs RHE I (mA/cm 2 )
Fig.13.TheCOadsstrippingvoltammogramon20wt%Pt/WC-SYNin0.1MHClO4
at30◦
C.Scanrate:20mV/s,Ptloading:48g/cm2.
obtainedwithcommercialPt/C(110mA/mgPt[28]).Inthe litera-ture,largerORRmassactivitieswerereportedforPt/WCcatalysts suchas12mA/mgPt[7],and24mA/mgPt[8](convertedfrom spe-cificactivityandPtloading).However,inthesecases,largeamount ofcarbonwaspresentinthecatalysteitherintheformofPt/Cand WCmixture[7]orPtdepositedontheWC+Ccompositesupport [8].Obviously,partoftheORRmassactivitycomesfromthePt depositedonC.Inaddition,thePtloadinginPt/WCisveryhigh (from50%to100%)[8].IfthehighdensityofWCisconsidered, suchahighPtloadingcouldleadtotheformationofaconducting Ptmultilayer.ElectronsinvolvedintheHOR(hydrogenoxidation reaction)andORRdonotneedtogothroughthesupportandthe catalystbehaveslikePtblack,resultinginhighORRperformance.
InordertobetterunderstandwhatcausesthelowORRmass activity,theECAwasmeasuredbyCOstripping,whichissupposed toavoidWCsupportinterference.Fig.13showstheCOstripping voltammogram.AclearCOoxidationpeakbetween0.6–0.8Vwas observed.FromEq.(1′),theECAwascalculatedtobe9m2/gPt.For Ptwithparticlesizeof5nm,thetheoreticalexternalsurfacearea shouldbe56m2/gPt.Thisindicatesthatonly16%oftheexternal surfaceiselectrochemicallyactive,whichmightbethemaincause ofthelowORRmassactivity.TheECAcanalsobecalculatedby subtractingtheHdesorptionpeakonWC(Fig.9)fromthatonPt/WC (Fig.11),whichgivesavalueof45m2/g.Thisvalueisclosetothe theoreticalvalueandsignificantlylargerthanthatobtainedwith COstripping.Mostpossibly,theinteractionbetweenPtandWC enhancedtheHadsorption/desorption.Howeveritdidnotaffect O2adsorptionandreductiononPt,whichisevidencedbythesmall PtoxidationpeakinFig.11.
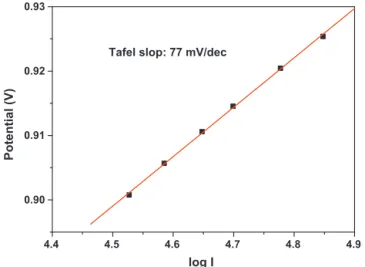
Tafeldiagramofthelinearscanwasplotted.The>0.9V poten-tialregionwasselectedfortheTafelplot,becauseinthatpotential region,theORRcurrentisindependentoftheRDErotationrate, asobservedinFig.12,indicatingapurekineticbehaviour.Fig.14 shows the Tafel plot of the Pt/WC-SYN catalyst. A linear rela-tionbetweenlogarithmcurrentdensityandpotentialisobserved, whichhasaTafelslopeof77mV/dec,largerthanthe60mV/dec usuallyreportedforPtcatalyzedORRreactionatlowoverpotentials [29].ThislargerTafelslopeindicatesthatthislowmassactivityis partlycausedbythesluggishreactionkinetics.
ThelowPtutilizationandlargeTafelslopeobtainedwiththe Pt/WC-SYNmightbeduetothecontactresistancebetweenthe catalystandthesupportifthesupportisoxidized.Aninsulating WO3layermayblocktheelectrontransferbetweenthecatalystand thesupport,isolatingthePtcatalystresultinginlowPtutilization. Inaddition,therearemanyfactorsthatcancausealowmass activity. For example, the non-uniform distribution and/or the agglomeration ofthePtcatalyst onthesupportcoulddecrease Pt utilization, unfavourable electronic interaction between the
4.9 4.8 4.7 4.6 4.5 4.4 0.90 0.91 0.92 0.93 Potential (V) log I
Tafel slop: 77 mV/dec
Fig.14. TafelplotoftheORRobtainedwith20%Pt/WC-SYNcatalystinO2-saturated,
0.1MHClO4,30◦C.Scanrateof5mV/s,andRDErotationrate:1600rpm.
catalyst and thesupportcouldalsoshift theonset potentialof ORRtonegativepotential,andsoon.Thesetopicsaredefinitely acontinuingresearchareaforfuturework.
4. Conclusions
Nano-crystallineWCwithasurfaceareaof89m2/gand min-imalcarboncontentwassynthesizedusingapolymerroute.The in-housesynthesizedWC(WC-SYN)hasahighelectronic conduc-tivity,highthermalstability,aswellaslowsolubilityinacidwhen comparedtocommerciallyavailableWC-Alfainthetemperature rangeof95◦C–200◦C.However,WC-SYNhasaslightlylower elec-trochemicalstabilityduetotheelectrooxidationintoWO3,which wasconfirmedbyXPSmeasurementsaftertheWCsamplewas potentialcyclinginthepotentialrangeof0.0–1.2Vvs.RHE.This instabilitywasattributedtoitshighsurfaceareabecausethe com-merciallyavailableWCsample,whichhasamuchlowersurface area,wasfoundtobeelectrochemicallystable.
WC-SYNwas testedas a Ptcatalyst support.Uniformly dis-tributedPtparticleswereobservedonWC-SYN.Electrochemical measurementsshowedthatthepresenceofPtdidnotcatalyzeWC oxidation.Incontrary,itcouldslightlystabilizetheWC-SYN sup-port.However,themassactivityofORRona20wt%ofPtsupported onWC-SYNwasonly3.3mA/mgPt,significantlylowerthan com-mercialPtsupportedoncarbon.Moreworkshouldbefocusedon
improvingthemassactivityandthesupportstabilityoftheWC supportedPtcatalysts.
Acknowledgements
Financial supportfrom NationalResearch Council of Canada (NRC),InstituteforFuelCellInnovation(IFCI)NRC,BallardPower Systems, andAFCC (Automotive FuelCell Cooperation)through theTechnologyDevelopmentProgram(TDP)isgreatlyappreciated. TheauthorswouldliketothankMr.KenTsayatNRC-IFCIfortheCO strippingmeasurement.Partoftheelectronmicroscopywas car-riedoutattheCanadianCentreforElectronMicroscopy,afacility supportedbyNSERCandMcMasterUniversity.GABisgratefulfor aNSERCstrategicgrantforpartiallysupportingFNandthiswork. References
[1]W.Li,A.M.Lane,Electrochem.Commun.11(2009)1187.
[2] A.A.Franco,M.Gerard,M.Guinard,B.Barthe,O.Lemaire,ECSTrans.13(2008)
35.
[3]H.Tang,Z.Qi,M.Ramani,J.F.Elter,J.PowerSources158(2006)1306.
[4]H.Chhina,S.Campbell,O.Kesler,J.PowerSources164(2007)431.
[5] C.A.Ribeiro,W.R.DeSouza,M.S.Crespi,J.A.GomezNeto,J.Therm.Anal.Calorim.
90(2007)801.
[6]R.Levy,M.Bourdat,Science181(1973)547.
[7]H.Meng,P.K.Shen,J.Phys.Chem.B109(2005)22705.
[8] M.Nie,P.K.Shen,M.Wu,Z.Wei,H.Meng,J.PowerSources162(2006)173.
[9]Y.Wang,S.Q.Song,V.Maragou,P.K.Shen,P.Tsiakaras,Appl.Catal.B:Environ.
89(2009)223.
[10]Q.Zhu,S.H.Zhou,X.Q.Wang,S.Dai,J.PowerSources193(2009)495.
[11] R.Ganesan,J.Lee,Angew.Chem.Int.Ed.44(2005)6557.
[12] R.Ganesan,J.Ham,J.Lee,Electrochem.Commun.9(2007)2576.
[13]J.Ham,K.Kim,H.Han,J.Lee,Catal.Today132(2008)117.
[14]F.Mazza,T.Trassatti,J.Electrochem.Soc.110(1963)847.
[15] H.Chhina,S.Campbell,O.Kesler,J.PowerSources179(2008)50.
[16] J.D.Voorhies,J.Electrochem.Soc.119(1972)219.
[17]E.Weigert,D.Esposito,J.G.Chen,J.PowerSources193(2009)501.
[18]K.Lee,A.Ishihara,S.Mitsushima,N.Kamiya,K.Ota,Electrochim.Acta49(2004)
3479.
[19] M.Zellner,J.G.Chen,Catal.Today99(2005)299.
[20]D.J.Ham,R.Ganesan,J.S.Lee,Int.J.HydrogenEnergy33(2008)6865.
[21]E.Lessner,W.-D.Schubert,Tungsten:Properties,Chemistry,Technologyofthe
Element,AlloysandChemicalCompounds,Springer,1999.
[22] R.Koc,S.K.Kodambaka,J.Eur.Ceram.Soc.20(2000)1859.
[23]F.H.Ribeiro,R.A.DallaBetta,G.J.Guskey,M.Boubart,Chem.Mater.3(1991)
805.
[24] H.Binder,A.Kohling,W.Kuhn,W.Lindner,G.Sandstede,Nature224(1969)
1299.
[25]B.S.Hobbs,A.C.C.Tseung,Nature222(1969)556.
[26]X.Cui,H.Zhang,X.Dong,H.Chen,L.Zhang,L.Guo,J.Shi,J.Mater.Chem.18
(2008)3575.
[27]D.Ham,Y.Kim,S.Han,J.Lee,Catal.Today132(2008)117.
[28]H.A.Gasteiger,S.S.Kocha,B.Sompalli,F.T.Wagner,Appl.Catal.B:Environ.56
(2005)9.