HAL Id: tel-01446638
https://tel.archives-ouvertes.fr/tel-01446638
Submitted on 26 Jan 2017HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
catalysées par des acides de Lewis : applications dans le
domaine des arômes et parfums.
Pierrick Ondet
To cite this version:
Pierrick Ondet. Synthèse d’éthers polycycliques par cycloisomérisations catalysées par des acides de Lewis : applications dans le domaine des arômes et parfums.. Autre. COMUE Université Côte d’Azur (2015 - 2019), 2016. Français. �NNT : 2016AZUR4075�. �tel-01446638�
Ecole Doctorale des Sciences Fondamentales et Appliquées
T H E S E
Docteur en Sciences
de l’UNIVERSITE de Nice-Sophia Antipolis
Discipline : Chimie
présentée et soutenue par
Pierrick ONDET
SYNTHESE D’ETHERS POLYCYCLIQUES PAR
CYCLOISOMERISATIONS CATALYSEES PAR DES ACIDES DE
LEWIS : APPLICATIONS DANS LE DOMAINE DES AROMES
ET PARFUMS
Thèse dirigée par le Dr
. Elisabet DUÑACH
et co-dirigée par le Dr.
Gilles LEMIERE
Soutenue le
20 octobre 2016Jury :
M. Jean-Marc CAMPAGNE, Professeur, Ecole Nationale Supérieure de Chimie de
Montpellier, Rapporteur
Mme Véronique MICHELET, Directrice de Recherche CNRS, Ecole Nationale Supérieure de
Chimie de Paris, Rapporteur
M. Christian CHAPUIS, Docteur, Firmenich SA, Examinateur
Mme Elisabet DU
Ñ
ACH, Directrice de Recherche CNRS, Université de Nice-Sophia
Antipolis, Directrice de thèse
M. Gilles LEMIERE, Chargé de Recherche CNRS, Université de Nice-Sophia Antipolis,
Co-directeur de thèse
« The best way to have a good idea is to have a lot of ideas » – Linus Pauling
« The harder you work, the luckier you get » – Gary Player
Sommaire
Sommaire ... 5
Remerciements ... 9
Abréviations, acronymes et symboles ... 11
Introduction générale ... 13
Chapitre 1 : Réactions de cyclisation catalysées par le triflate de bismuth(III) ... 17
I. Introduction ... 19
II. Superacides de Lewis ... 19
II. 1. Généralités ... 19
II. 2. Préparation des triflates métalliques ... 20
II. 3. Le triflate de bismuth(III) ... 22
III. Réactions de cyclisation catalysées par Bi(OTf)3 ... 25
III. 1. A ti atio d ol fi es ... 25
III. 2. A ti atio d al es ... 33
III. 3. A ti atio d all es ... 36
III. 4. Activation de fonctions alcools et dérivés ... 37
III. 5. Activation de fonctions carbonyles ... 46
III. 6. A ti atio d po des ... 52
III. 7. Activation de dérivés azotés... 53
IV. Bilan et objectifs de recherche ... 57
Chapitre 2 : C cloiso isatio s d’ the s d’ ol all i ues ... 59
I. Introduction ... 61
II. Généralités sur les allènes ... 61
II. 1. La fonction allène ... 62
II. 2. C lisatio d all es ... 63
III. R activit des the s d’ ol ... 66
IV. Etude préliminaire ... 70
IV. 1. Préparation des substrats de départ ... 71
IV. 2. Screening de catalyseurs ... 71
IV. 3. M a is e p opos pou l a ti atio de l the d ol ... 72
V. Réactivité en présence de triflate de bismuth(III) ... 73
V. 1. Ethe s d ols a li ues ... 73
V. 2. Thio the s d ol ... 75
V. 3. Ethe s d ol a li ues « inversés » ... 76
V. 4. Ethe s d ol li ues ... 76
V. 5. Bilan ... 84
VI. 1. Observations préalables ... 85
VI. 2. Additio d u u l ophile ... 87
VI. 3. Influence du catalyseur ... 88
VI. 4. Aspects mécanistiques ... 89
VII. Conclusion et perspectives ... 91
Chapitre 3 : S th se d’ the s pol c cli ues ... 93
I. Introduction ... 95
II. Etude préliminaire ... 95
II. 1. Préparation du substrat modèle ... 95
II. 2. Réactivités attendues ... 96
II. 3. Screening de catalyseurs ... 98
III. Exemplification ... 100
III. 1. Synthèse des substrats ... 100
III. 2. Cyclisation de dérivés dihydropyraniques ... 101
III. 3. Cyclisation de dérivés dihydrofuraniques ... 103
III. 4. C lisatio d aut es su st ats... 105
III. 5. C lisatio d al es te i au ... 110
IV. Etudes mécanistiques ... 113
IV. 1. Oléfine terminale vs trisubstituée ... 113
IV. 2. Diastéréospécificité ... 116
IV. 3. Calculs DFT ... 117
IV. 4. Mécanisme proposé ... 120
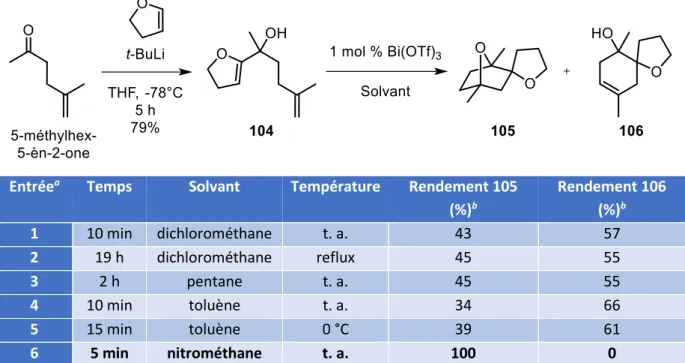
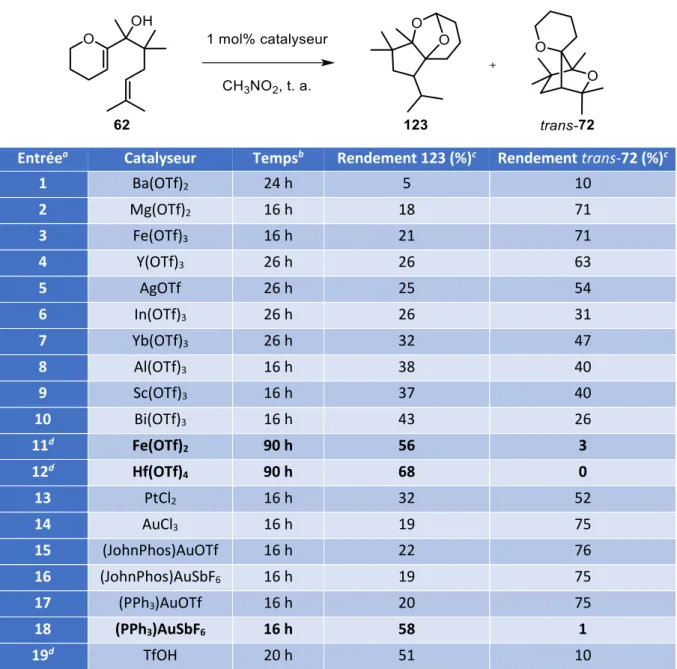
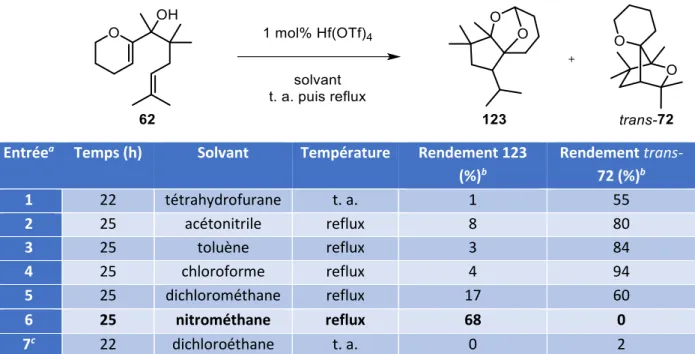
V. Nouvelle c clisatio ta de i pli ua t u t a sfe t d’h d u e ... 120
V. 1. Influence du solvant pour la cyclisation de 62 ... 121
V. 2. Les t a sfe ts d h d u es atal s s pa des a ides ... 122
V. 3. Etudes de l i flue e des pa a t es de la a tio ... 124
V. 4. Implications mécanistiques ... 127
V. 5. Fo atio d a tals pol li ues à pa ti d aut es p u seu s ... 128
V. 6. Nouvelle réactivité tandem ... 129
VI. Conclusion et perspectives ... 131
Chapitre 4 : Double cyclisation de dérivés du campholénal ... 133
I. Introduction ... 135
II. Le campholénal ... 135
II. 1. Production ... 135
II. 2. Utilisation pour la synthèse de molécules odorantes ... 137
III. Etude de la réaction de double cyclisation... 139
III. 1. Synthèse du substrat modèle ... 139
III. 2. Réactivités attendues ... 139
III. 3. Etude de la cyclisation du diasteréoisomère (R,R)-133 ... 140
III. 4. Etude de la cyclisation du diasteréoisomère (R,S)-133 ... 143
IV. 1. Préparation des précurseurs de cyclisation ... 146
IV. 2. Séparation énantiomérique des précurseurs de cyclisation ... 147
IV. 3. Cyclisation et analyses des alcools (R,R) et (S,S) ... 151
IV. 4. Cyclisation et analyses des alcools (R,S) et (S,R) ... 154
IV. 5. Post-fonctionnalisation ... 157
V. Conclusion ... 158
Chapitre 5 : Evaluation olfactive des éthers polycycliques ... 161
I. Introduction ... 163
II. Généralités ... 163
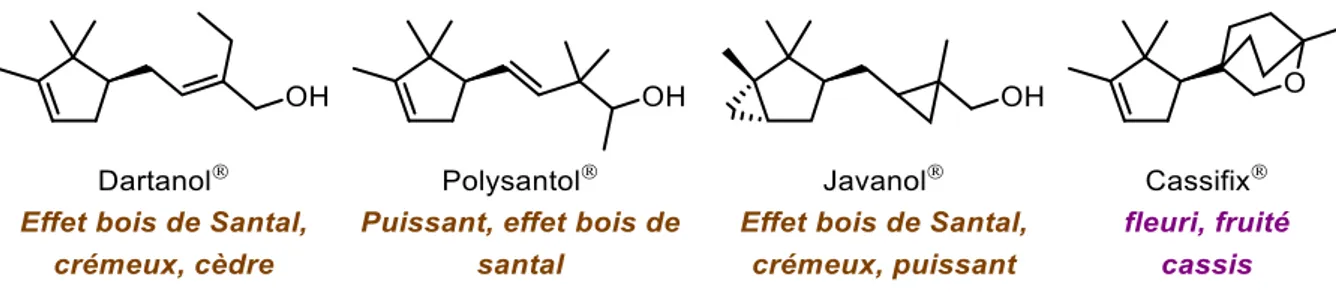
III. Les molécules (poly)cycliques en parfumerie ... 167
III. 1. Les molécules aux notes boisées ... 167
III. 2. Intérêt des éthers poly- et spirocycliques ... 169
IV. M thodes d’ valuatio olfactive... 172
IV. 1. Ch o atog aphie e phase gazeuse oupl e à l olfa to t ie ... 172
IV. 2. Panel de parfumeurs ... 173
V. Propriétés organoleptiques des polycycles synthétisés ... 173
V. 1. E aluatio des p oduits de lisatio des the s d ol alléniques ... 173
V. 2. Evaluation olfactives de produits de double cyclisation ... 174
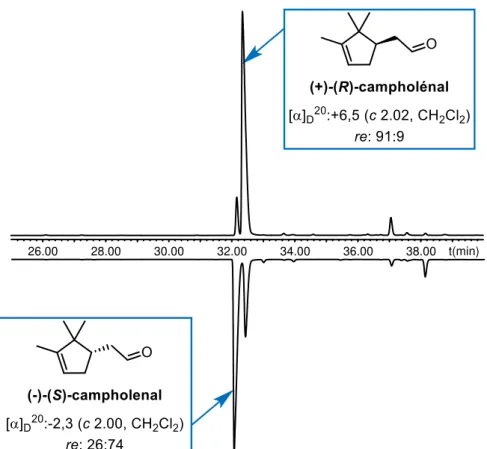
V. 3. Evaluation des dérivés cycliques du campholénal ... 177
VI. Conclusion et perspectives ... 183
Conclusion générale ... 185
Partie expérimentale ... 191
I. General information... 193
II. Experimental procedures ... 194
II. 1. Chapter 2 : Cycloisomerisation of allene enol ethers ... 194
II. 2. Chapter 3 : Synthesis of polycyclic ethers by double cyclisation ... 218
II. 3. Chapter 4 : Cyclisation of campholenic aldehyde derivatives ... 253
Références bibliographiques ... 265
Tout d a o d, je souhaite e e ie Dr. Elisa et Duña h pou a oi a o d sa o fia e pou di ige a th se et t a aille au sei de so g oupe, da s l uipe A ô es, Pa fu s, “ th se et Mod lisatio à l I stitut de Chi ie de Ni e. Ces t ois années remplies de conseils et de questions toujours plus pointues o t pe is de pa oui plei e e t e ta t ue hi iste o ga i ie . J ai eçu le soutien et la motivation nécessaires dans mes différents projets entrepris et lorsque les résultats se faisaient plus difficiles à obtenir.
For e t, j ad esse des co-remerciements au Dr. Gilles Lemière qui a accepté de me co-diriger pendant trois ans. Merci pour tes lumières quasiment quotidiennes, tes nombreux conseils en synthèse organique et tes e ou age e ts jus u au out. J ai d ou e t ue ta sou e d i spi atio était Linus Pauling ! Je me souviendrai aussi de tes connaissances linguistiques sur divers domaines et tes jeux de mots alambiqués.
Le travail de thèse ne va pas sans une évaluatio pa des s ie tifi ues du do ai e. Ai si j e p i e es profonds remerciements à mes rapporteurs Dr. Véronique Michelet et Prof. Jean-Marc Campagne pour a oi a ept de juge es t a au de th se e pa ti ipa t à o ju , est u ho eu pou oi. U e e ie e t pa ti ulie s adresse également au Dr. Christian Chapuis qui a a ept d examiner mon travail de recherche. Il me tenait à œu d a oi l oppo tu it de p se te es t a au à u industriel du domaine des arômes et parfums.
Forcément la p o hai e s ie de e e ie e t s ad esse au pe a e ts de l ICN, et plus particulièrement, au Dr. Sandra Olivero pour les conversations importantes sur le fonctionnement du laboratoire et les questions pertinentes lors des réunions ALEA. Dans le même couloir, je tiens à remercier le facétieux Dr. Sylvain Antoniotti pour les discussions prenantes et les analyses éclairées sur le monde de la chimie, ou le monde en général ainsi que le Dr. Sophie Martini-Poulain pour sa sympathie quotidienne. Merci aussi au Dr. Benoît Michel pour tes conseils sur les postdocs.
Faute de synonymes, je ais l utilise à ou eau : Merci ! Au Dr. Vincent Morizur pou a oi a ueilli à bras ouverts avec cette fameuse chaleur bretonne enivrante qui le caractérise si bien. A Antoine, mon Paulo et acolyte de pause-café ou aquaponey, entre autres, toujours suivi de près par son frère le Dr. Anthony Martin, dont la répartie a ga u peu plus ue je e l tais d jà. Je remercie également le stagiaire Guillaume Mossand que pour ses innombrables groupons et abricots. Dans le même registre, comment ne pas remercier les modélisateurs guidés par le rire regrett de l a ie e mascotte Julien et ses collègues Dr. Claire De March et Jean-Baptiste sans qui la conférence à la médiathèque de Co tes au ait ja ais atti auta t de o de.
Encore une fois, merci aux anciens, aux thésards et aux stagiaires en particulier Estelle, Hélène, Arnaud, Dr. Johanna Revel, Sophia, Loïc, Pascal, Florian, Laurane, Dr. Ilhem Diaf, Dr. Mélodie Maloubier, Marie-Aude, Daphné, Hella. Et plus généralement, les « «non-permanents » de tout l ICN pou es t ois a es e plies d ha ges s ie tifi ues et de a e ues, d ap o et de pauses af . Un grand merci au Dr. Guillaume Compain qui a apporté une certaine fraîcheur, de nombreux conseils et une bonne dose d hu ou lo s de son passage succinct dans notre équipe. De même, Lorenzo, merci pour ton implication en tant que stagiaire et pour ta sympathie quotidienne, ce fut un plaisir de travailler avec toi.
Rajoutons un merci particulier à mes portugais préférés (après CR7), Isa et Daniel toujours prêts à proposer de nouvelles activités pour a e l esp it ie ha g du th sa d la da.
Un grand merci au Dr. Jean-Jacques Filippi pour son intérêt dans notre collaboration et sa pédagogie ui a pe is d e i hi es o p te es e a al ses appli u es au odo a ts.
De plus, je souhaite exprimer ma gratitude au Dr. Caroline Plessis de la société V. Mane & Fils pour sa disponibilité dans notre collaboration et son intérêt à faire partager ses connaissances en chimie du parfum.
Je tiens à remercier le Dr. Jean-Claude Clinet, le Dr. Marc Gayzynski et Dominique Bonhomme pour leur aide précieuse dans la vie quotidienne du la o atoi e et de l i stitut.
Merci à Paul Bismuth de a oi pe is de pa le u peu plus au non-chimistes de e tal ue j ai utilisé sous forme de sel pendant trois ans.
Mes amis de longue date, Arnaud et son tractopelle, Medhi, Thomas, Guy ou Aurélien, je tiens à vous remercier chaleureusement pour les eaks ue ous a ez procurés quand le besoin de vacances se faisait sentir.
Me i a a et papa, d a oi essa de li e es pu lications pou o t e l i t êt que vous avez porté à mon travail et surtout votre soutien incessant depuis ue j ai e ta mes études. Enfin, dans la famille Ondet, je nomme la team Khalats du Mordor, Marjolaine, Eloïse et grand frère Cyril, qui
ont bien encouragé chaque année et notamment durant l été 2016.
Luisa, la ate a d “o e s hei , e i d a oi suppo t et encouragé ma méticulosité quotidienne au laboratoire et mon french accent. To e o ti ued…
Ac : acétyle AL : acide de Lewis Atm : atmosphère Bn : benzyle
Boc : tert-butoxycarbonyle (groupement) Bu : butyle
CAN : it ate de iu et d a o iu CCM : chromatographie sur couche mince CH2Cl2 : dichlorométhane
CH3NO2 : nitrométhane
CPCM : conductor-like polarizable continuum model DBU : 1,8-diazabicyclo[5.4.0]undéc-7-ène
DCE : 1,2-dichloroéthane
DFT : density functional theory / théorie de la fonctionnelle de la densité DHP : dihydropyrane
DIBAL : hydrure de diisobutylaluminium DIEA : N,N-diisopropyléthylamine dig : digonal DMAP : 4-diméthylaminopyridine DMF : N,N-diméthylformamide DMSO : diméthylsulfoxyde DPG : dipropylène glycol équiv. : équivalent Et : éthyle Et2O : diéthyle éther g : gramme
GC : gas chromatography / chromatographie en phase gazeuse h : heure
HRMS : high resolution mass spectrometry Hz : Hertz
IBQ : isobutyle quinoline IBX : acide 2-iodoxybenzoïque
i-Pr : isopropyle
IR : infrarouge
J : constante de couplage (RMN)
JohnPhos : , ′-biphenyl-2-yl)di-tert-butylphosphine LBA : Lewis acid-assisted Brønsted acid
M : molarité Me : méthyle min : minute
MIP : istate d isop op le mL : millilitre
MS : mass spectrometrie / spectrométrie de masse Ms : mésyle
N : normalité
NOESY : nuclear Overhauser enhancement epectroscopy OTf- : trifluorométhylsulfonate ou triflate (CF
3SO3-)
PCC : chlorochromate de pyridinium PE : éther de pétrole
Ph : phényle
pKa : logarithme de la constante d a idit
PPh3: triphénylphosphine
ppm : partie par million
PPTS : pyridinium p-toluenesulfonate
p-TsOH : acide paratoluènesulfonique R : rectus
rd : ratio diastéréoisomérique re : ratio énantiomérique
RMN : résonance magnétique nucléaire
S : sinister
SbF6- : ion héxafluoroantimonate
SN1 : substitution nucléophile de type 1
SN : su stitutio u l ophile de t pe
T : température
t. a. : température ambiante t: temps
TBAF : fluorure de tétra-n-butylammonium
t-BuLi : tert-butylithium
TEA : triéthylamine Tf2NH : acide triflimidique
TFA : acide trifluoroacétique
TfOH : acide triflique ou trifluorométhanesulfonique TGDV : Triéthylène Glycol Divinyle Ether
THF : tétrahydrofurane TIPS : Triisopropylsilyle
TLC : thin layer chromatography TMS : triméthylsilyle
trig : trigonal Ts : tosyle
La plupart des molécules organiques biologiquement actives, et notamment, un grand nombre de produits naturels, présentent au sein de leur structure un ou plusieurs cycles. Dans ce contexte, les réactions de cyclisation et de cycloisomérisation sont devenues un outil incontournable en chimie et plus particulièrement en chimie fine pour trouver des applications dans différents domaines tels que les at iau , l ag o hi ie, la pha a euti ue ou ie la pa fu e ie.
Par conséquent, depuis les débuts de la chimie organique, le développement de nouvelles méthodes de s th se de st u tu es li ues a ess de sti ule l i t t de la o u aut des hi istes. Ainsi de nombreuses méthodes de cyclisation,1 intra- et intermoléculaires, ont été décrites pour
accéder à des squelettes retrouvés dans la nature2 ais aussi pou t ou e d aut es st u tu es et
a alogues afi d e plo e de ou elles a ti it s et appli atio s.
Malgré toutes les méthodes déjà disponibles, une possible amélioration de ces procédés reste toujours d a tualit , ota e t pou l i dust ie, aussi ie d u poi t de ue p u iai e ue du poi t de ue de l i pa t e i o e e tal. “a s pa le fo e t d u effet de ode, u i t t g a dissa t de la o u aut pou le espe t de l e i o e e t se le s i stalle du a le e t da s les œu s.3
Effe ti e e t, e plus des halle ges o solus pou l a s à e tai s s uelettes o ple es, des alternatives plus efficaces comparées aux méthodes classiques sont développées en accord avec les principes énoncés pour une chimie plus verte.4 Les récentes avancées visent à être économes en
tapes de s th se, e ato es et e e gie, e ui i pli ue e g al l e ploi de atal seu pou promouvoir les transformations chimiques et, spécialement, les cyclisations.5 De nombreuses
méthodes utilisent des catalyseurs basés sur des complexes de métaux de transitions, des métaux lourds, des métaux nobles ou encore des terres-rares, pour une partie toxiques. Il est donc intéressant de proposer des alternatives limitant leur utilisation.
Les molécules polycycliques et, plus particulièrement, les éthers polycycliques sont très utilisés dans le domaine des arômes et parfums. De nombreux odorants naturels possèdent ce type de structure et la recherche en chimie des fragrances a encouragé les chimistes à synthétiser de nouveaux composés cycliques possédant ce type de motifs dans le but de découvrir et de créer des odeurs nouvelles, parfois par sérendipité.6 Un autre objectif est de pouvoir mimer celles déjà existantes, difficiles à extraire ou
1 S. Ma, Handbook of Cyclization Reactions, John Wiley & Sons, 2009.
2 K. C. Nicolaou, D. J. Edmonds, P. G. Bulger, Angew. Chem. Int. Ed. 2006, 45, 7134-7186.
3 J.-M. Campagne, F. Agbossou, T. Ayad, O. Baudoin, G. Buono, I. Chataigner, C. Crévisy, I. Dez, M. Donnard, F.
Fache, L'Actualité Chimique 2010, 14-27.
4 P. T. Anastas, J. C. Warner, Principles of Green Chemistry, 1998. 5 B. Trost, Science 1991, 254, 1471-1477.
6 A. Christoforo, Peregrinaggio di tre giovani figliuoli del re di Serendippo, dall a Persiana nell'Italiana lingua,
retrouvées dans des huiles essentielles contenant parfois des composés allergènes. En effet, de nouveaux critères rigoureux concernant la réglementation, la biodégradabilité des odorants ou bien l adaptatio au ha ge e t de ode du a h justifie t l i t t de d ou i de ou elles molécules parfumantes.7
E plus de l i t t de es ol ules e pa fu e ie, le d eloppe e t de lisatio s pe etta t la s th se di e te de ol ules o igi ales pe et d ou i le ha p de a tio s et de st u tu es chimiques inexplorées.
Mon travail de thèse a porté sur le développement de nouvelles méthodologies catalytiques de s th se pou la fo atio d the s pol li ues pote tielle e t i t essa ts, e t e aut es, da s le domaine des arômes et parfums.
Dans un premier chapitre bibliographi ue, l tat de l a t de l utilisatio de supe a ides de Le is da s des réactions de cyclisation, et plus particulièrement, du triflate de bismuth(III), sera présenté. Ensuite, la loiso isatio atal ti ue de su st ats the s d ol all i ues, o tudiée auparavant, sera décrite pour la formation de molécules (poly)cycliques. Dans un troisième chapitre, une extension de la méthode développée avec les substrats alléniques fonctionnalisés sera extrapolée à des dérivés ol fi i ues ui o t pe is d o te ir efficacement de nouvelles structures complexes via une réaction de double cyclisation. Un cas plus particulier, à partir de dérivés du campholénal, sera décrit dans un quatrième chapitre pour la synthèse de nouvelles structures complexes. Enfin, le cinquième chapitre se a o sa à l aluatio et à la des iptio des p op i t s o ga olepti ues de uel ues ol ules polycycliques obtenues par les voies de synthèse décrites dans ce travail de thèse.
Chapitre 1 : Réactions de cyclisation
catalysées par le triflate de bismuth(III)
I.
Introduction
Les réactions de cycloisomérisation catalysées par des complexes métalliques ont émergé en tant u outils s th ti ues de choix pour générer efficacement de la complexité moléculaire via la formation de liaisons carbone-carbone ou carbone-hétéroatome. Lors des 30 dernières années, les lisatio s i t a ol ulai es se so t p i ipale e t fo alis es su l utilisatio de atal seu s d i s de métaux nobles (Au, Pd, Pt, Rh).8 Ainsi, le développement de nouvelles réactions de carbocyclisation
en utilisant des catalyseurs peu onéreuxs, non-toxiques et facilement réutilisables est d u i t t certain.
Un des principaux axes de recherche de notre groupe est le développement de nouvelles méthodologies catalytiques pour la synthèse de molécules cycliques, en utilisant en particulier des « superacides de Lewis » et plus précisément des triflates métalliques. Leur préparation par diverses voies, ainsi que leur utilisation en synthèse sera décrite.
Parmi les catalyseurs utilisés et étudiés, le triflate de bismuth(III) s est a d u e effi a it notoire pou l a ti atio sélective de certaines fonctions sur des molécules polyfonctionnelles permettant l a s di e t et sélectif à des structures complexes. Ce chapitre se concentrera donc principalement sur les réactions de cyclisation catalysées par le triflate de bismuth(III).
II.
Superacides de Lewis
II. 1.
Généralités
Un superacide de Brønsted est un composé protique dont l a idit est sup ieu e à elle de l a ide sulfurique pur.9 Les superacides de Lewis peuvent être définis comme les sels de cations métalliques
dérivant de superacides de Brønsted. Pa e e ple, l a ide t ifluo o tha esulfo i ue10 (ou acide
triflique) est un superacide de Brønsted qui possède un pKa de -14,7.11 Ainsi, les sels métalliques
constitués de sa base conjuguée trifluorométhylsulfonate (ou triflate) sont considérés comme des
8 Y. Yamamoto, Chem. Rev. 2012, 112, 4736; A. Fürstner, Chem. Soc. Rev. 2009, 38, 3208; V. Michelet, P. Y. Toullec,
J. P. Genêt, Angew. Chem., Int. Ed. 2008, 47, 4268; E. Jiménez-Núñez, A. M. Echavarren, Chem. Rev. 2008, 108, 3326; G. C. Lloyd-Jones, Org. Biomol. Chem. 2003, 1, 215; C. Aubert, O. Buisine, M. Malacria, Chem. Rev. 2002,
102, 813; B. M. Trost, M. J. Krische, Synlett 1998, 1; I. Ojima, M. Tzamarioudaki, Z. Li, R. J. Donovan, Chem. Rev. 1996, 96, 635; M. Lautens, W. Klute, W. Tam, Chem. Rev. 1996, 96, 49.
9 N. F. Hall, J. B. Conant, J. Am. Chem. Soc. 1927, 49, 3047-3061; R. J. Gillespie, Acc. Chem. Res. 1968, 1, 202-209. 10 R. D. Howells, J. D. Mc Cown, Chem. Rev. 1977, 77, 69-92; L. R. Subramanian, A. G. Martínez, M. Hanack, G. K.
Surya Prakash, J. Hu, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2001.
superacides de Lewis (Figure 1). De même, les triflimidures métalliques sont des superacides de Lewis utilisés en synthèse organique.
Figure 1. Formules de triflate et triflimidure métallique.
La très forte électronégativité des groupements triflate et triflimidure augmente le caractère acide de Lewis du cation métallique.
II. 2.
Préparation des triflates métalliques
Bien que la plupart des triflates métalliques soient, à présent, commerciaux, différentes méthodes de préparation efficaces ont été mises au point. La voie de synthèse peut avoir une incidence sur le sel métallique synthétisé. Il peut être obtenu sous forme quasiment anhydre ou alors sous forme solvatée.
II. 2. a. Voie classique
La préparation classique des triflates métalliques procède, en général, par traitement à l acide triflique du carbonate (équation 1),12de l o de (équation 2),13 du chlorure,14 du sulfonate métalliques dans
l eau (Schéma 1 . Cette p o du e i pli ue la fo atio d u sel sous sa fo e h d at e. Le séchage à température trop élevée peut engendrer la dégradation du motif triflate.15
Schéma 1. Synthèse de triflates métalliques en milieu aqueux.
12 T. Imamoto, Y. Koide, S. Hiyama, Chem. Lett. 1990, 19, 1445-1446.
13 S. Kobayashi, Synlett 1994, 689-701; M. Nishizawa, E. Morikuni, K. Asoh, Y. Kan, K. Uenoyama, H. Imagawa,
Synlett 1995, 169-170; S. Répichet, A. Zwick, L. Vendier, C. Le Roux, J. Dubac, Tetrahedron Lett. 2002, 43,
993-995; M. Peyronneau, C. Arrondo, L. Vendier, N. Roques, C. Le Roux, J. Mol. Catal. A: Chem. 2004, 211, 89-91.
14 G. A. Olah, O. Farooq, S. M. F. Farnia, J. A. Olah, J. Am. Chem. Soc. 1988, 110, 2560-2565; T. Mukaiyama, N.
Iwasawa, R. W. Stevens, T. Haga, Tetrahedron 1984, 40, 1381-1390; I. Hachiya, M. Moriwaki, S. Kobayashi,
Tetrahedron Lett. 1995, 36, 409-412.
Des alte ati es pe etta t d o te i le sel a h d e de Bi(OTf)3 impliquent la réaction entre le
triphényle ou le triacétate de bismuth(III) et l a ide t ifli ue ou l a h d ide t ifli ue.16
II. 2. b. Voie oxydative
La voie de synthèse dite « oxydative » est u e oie alte ati e pe etta t d o te i des sels métalliques solvatés par le DMSO a e d e elle ts e de e ts à partir du métal sous forme de poudre, de la ide t iflique en quantité stœ hio t i ue et d o g e ol ulai e dans le DMSO à 100 °C (Schéma 2).17
Schéma 2. Préparation de triflate de bismuth(III) par dissolution oxydante de poudre métallique de bismuth. Cette méthode, qui peut aussi êt e appli u e à la p pa atio d aut es t iflates et triflimidures métalliques, a pour avantage de fournir les sels métalliques non hydratés bien que solvatés.
II. 2. c. Voie électrochimique
Une autre méthode pour obtenir le sel métallique anhydre passe par la voie électrochimique.18 Cette
méthode efficace est plus douce que les précédentes car elle est opérée à température ambiante, et pe et d o te i le sel talli ue a e des rendements quasiment quantitatifs, avec le dihydrogène comme seul sous-produit. L le t os th se s op e da s u e ellule mono-compartimentée avec une anode du métal à oxyder, u e athode i e te, et l a ide triflique ou triflimidique dans un solvant tel uel le it o tha e, le DMF ou l a to it ile. U ou a t o ti u pe et d effe tue la du tio de l a ide au i eau de la athode, l o datio à l a ode du tal ui est o so e fo a t le sel métallique correspondant en solution.
II. 2. d. Voie sonochimique
L o datio du tal sous forme de poudre en présence d u e esp e protique, telle que l a ide triflique, sous a ti atio ult aso pe et d o te i le sel a h d e avec du dihydrogène comme seul sous-produit de la réaction (Schéma 3).19
16 Y. Torisawa, T. Nishi, J.-i. Minamikawa, Organic Process Research & Development 2001, 5, 84-88; M.
Labrouillère, C. Le Roux, H. Gaspard, A. Laporterie, J. Dubac, J. R. Desmurs, Tetrahedron Lett. 1999, 40, 285-286.
17 S. Antoniotti, E. Duñach, Chem. Commun. 2008, 993-995.
18 I. Favier, E. Duñach, Tetrahedron Lett. 2003, 44, 2031-2032; E. Dunach, I. Favier, D. Hebrault, J. R. Desmurs,
FR2818994A1, 2002.
19 N. Legrave, A. Couhert, S. Olivero, J.-R. Desmurs, E. Duñach, Eur. J. Org. Chem. 2012, 901-904; J. R. Desmurs, E.
a tio el sui ie d u e évaporation. Ce recyclage a, par exemple, été mis en pratique pour des a tio s d h d oa latio .25
Le triflate de bismuth(III) possède une activité e ta t u a ide de Le is à la limite entre des catalyseurs oxophiles comme Al(OTf)3 ou Mg(OTf)2 et ceux plus carbophiles o e des d i s de l or.
Cela fait de lui un catalyseur à part qui peut réagir avec une grande sélectivité en fonction des groupements fonctionnels des substrats.
II. 3. c. Aspect mécanistique
L a ti atio de g oupements fonctionnels par Bi(OTf)3 mène généralement à la formation
d i te diai es carbocationiques ui agisse t e suite e p se e d u u l ophile par voie intra- ou intermoléculaire.
La atu e de l esp ce catalytique et du mode d a tio des t iflates métalliques, et plus particulièrement du triflate de bismuth(III), reste un sujet de débat dans la communauté scientifique malgré les quelques tudes a isti ues et th o i ues ises e œu e pa diff e ts groupes. En effet, la présence de traces d eau da s le ilieu a tio el i pli ue la possi ilit d u e h d ol se ou d u e h d atatio du triflate de bismuth(III).26 L h d ol se d u triflate métallique engendrerait la
fo atio d a ide t ifli ue in situ ui pou ait t e l esp e active en tant que superacide de Brønsted. Par ailleurs, la simple hydratation du métal est aussi à considérer (Figure 4). Ces deux phénomènes ont été évalués par calcul de la théorie de la fonctionnelle de la densité.27L h d atatio est haute e t
fa o is e o pa e à l h d ol se ui s a e t e e dothe i ue. Ce résultat corrobore une étude similaire avec le t iflate d tai .28
25 B. Cacciuttolo, S. Poulain-Martini, F. Fontaine-Vive, M. A. H. Abdo, H. El-Kashef, E. Duñach, Eur. J. Org. Chem. 2014, 7458-7468.
26 R. F. Lambert, R. J. Hinkle, S. E. Ammann, Y. Lian, J. Liu, S. E. Lewis, R. D. Pike, J. Org. Chem. 2011, 76,
9269-9277; D. B. G. Williams, M. Lawton, J. Mol. Catal. A: Chem. 2010, 317, 68-71; A. Dzudza, T. J. Marks, Chem. Eur. J.
2010, 16, 3403-3422; D. C. Rosenfeld, S. Shekhar, A. Takemiya, M. Utsunomiya, J. F. Hartwig, Org. Lett. 2006, 8,
4179-4182; T. C. Wabnitz, J.-Q. Yu, J. B. Spencer, Chem. Eur. J. 2004, 10, 484-493; S. Kobayashi, S. Nagayama, T. Busujima, J. Am. Chem. Soc. 1998, 120, 8287-8288.
27 J. Godeau, F. Fontaine-Vive, S. Antoniotti, E. Duñach, Chem. Eur. J. 2012, 18, 16815-16822.
28 P. Nava, Y. Carissan, J. Drujon, F. Grau, J. Godeau, S. Antoniotti, E. Duñach, S. Humbel, ChemCatChem 2014, 6,
Figure 4. Hydrolyse vs hydratation de Bi(OTf)3.
Da s le as d u e h d atation, la ol ule d eau o ple e au triflate de bismuth(III) possède une certaine charge positive, avec une forte acidité, comme cela a été calculé dans le cas de la cycloisomérisation d al ools i satu s catalysée par le t iflate d alu i iu .29 Par conséquent, dans le
cas du triflate de bismuth(III), le o ple e talli ue h d at agi ait e ta t u a ide de B ø sted assisté par un acide de Lewis (type LBA).30
Cepe da t, la o p he sio du a is e d a tio de es atal seu s est toujou s à l tude et elui-ci peut être différent selon la fonction activée.
III.
Réactions de cyclisation catalysées par Bi(OTf)
3Ces réactions seront classées selon la fonction assumée comme activée en premier dans le processus catalytique, afin de souligner la polyvalence du triflate de bismuth(III).
III. 1.
Activatio d’ol fi es
III. 1. a. Oléfines non-activées
Lh d oalko latio intramoléculaire d ol fi es représente un processus économique en atomes pour l accès à des éthers cycliques fonctionnalisés.
Ce type de réaction a été étudié en utilisant des oléfines non-activées. Après comparaison avec d aut es triflates métalliques, Bi(OTf)3 a montré une meilleure réactivité pour promouvoir
l h d oalko latio . Des éthers cycliques comportant diverses fonctions ont été obtenus avec de bons e de e ts et u e diast os le ti it e fa eu de l iso e anti dans la plupart des cas (Schéma 4).31 Une approche similaire a été décrite précédemment par notre groupe en utilisant le triflate
d alu i iu . 29, 32
29 L. Coulombel, M. Rajzmann, J.-M. Pons, S. Olivero, E. Duñach, Chem. Eur. J. 2006, 12, 6356-6365.
30 O. Kanno, W. Kuriyama, Z. J. Wang, F. D. Toste, Angew. Chem. Int. Ed. 2011, 50, 9919-9922; C. H. Cheon, O.
Kanno, F. D. Toste, J. Am. Chem. Soc. 2011, 133, 13248-13251; H. Yamamoto, K. Futatsugi, Angew. Chem. Int. Ed.
2005, 44, 1924-1942.
31 B. D. Kelly, J. M. Allen, R. E. Tundel, T. H. Lambert, Org. Lett. 2009, 11, 1381-1383. 32 L. Coulombel, I. Favier, E. Duñach, Chem. Commun. 2005, 2286-2288.
Schéma 4. S th se d’ the s cycliques catalysée par le triflate de bismuth(III).
Une hydroamination intramoléculaire de sulfonamides insaturées a été mise en œu e en utilisant des conditions douces pour accéder à des hétérocycles azotés saturés.33 Différents catalyseurs métalliques
ont aussi mené aux dérivés pyrrolidines protégées attendus avec, cependant, un temps de réaction plus long comparé au triflate de bismuth(III) (Schéma 5). Puisque la cyclisation ne se fait pas en p se e d u e ase, les auteu s o t sugg ue l a ide t ifli ue, g in situ par hydrolyse de
Bi(OTf)3, se ait l esp e atal ti ue a ti e.
Schéma 5. Hydroamination intramoléculaire de sulfonamides comportant une fonction alcène non-activée. Une hydroxyalkylation intermoléculaire de composés 1,3-dicarbonés et de dérivés styréniques a été développée en utilisant Bi(OTf)3 comme catalyseur.34 De façon intéressante, les auteurs ont observé
ue les p oduits d si s e pou aie t pas t e fo s e p se e d a ide t ifli ue e ua tit atal ti ue, etta t e a a t l a ti it atal ti ue diff e te du Bi OTf3. Bie ue les β-cétoesters ne
soient pas des substrats adéquats pour le processus intermoléculaire à cause de leur faible réactivité, ils ont pu être utilisés dans la version intramoléculaire. Le p o essus d h d oalk latio a t effe tu a e su s su u β-cétoester possédant une partie styrénique (Schéma 6). La cyclohexanone correspondante a été obtenue via une réaction de type Conia-ène avec un rendement de 72%.
Schéma 6. Hydroalkylation intramoléculaire catalysée par Bi(OTf)3.
33 F. Mathia, P. Szolcsanyi, Org. Biomol. Chem. 2012, 10, 2830-2839. 34 M. Rueping, B. J. Nachtsheim, A. Kuenkel, Synlett 2007, 1391-1394.
Lh d oa latio i t a ol ulai e atal ti ue de d i s ph les poss da t u e haî e a e u e double liaison a été développée (Schéma 7).35 Des tests avec différents systèmes catalytiques ont
indiqué que Bi(OTf)3, utilisé en quantité catalytique (1 mol%), était le catalyseur le plus actif pour la
cyclisation de ces dérivés aromatiques insaturés.
Schéma 7. H d oa latio i t a ol ulai e d’al es catalysée par Bi(OTf)3.
Lorsque des oléfines terminales 2,2-disubstitutées sont engagées dans les mêmes conditions de réaction, une lactonisation déalkylante a été observée, résultant de la réaction entre la fonction alcène et un des groupements esters (Schéma 8). 35 Des calculs DFT et les résultats expérimentaux ont indiqué
que la formation de la lactone avec un cycle à 5 chaînons est favorisée par rapport à la carbocyclisation menant à un cycle à 5 chaînons. Ce type de réactivité avait déjà été observé auparavant en utilisant le t ifli idu e d tai o e atal seu .36
Schéma 8. Lactonisation déalkylative catalysée par Bi(OTf)3.
Une extension du réarrangement de Wagner-Meerwein a été étudiée sur des dérivés terpéniques par catalyse au triflate de bismuth(III) e ta t u alte ati e plus ologi ue o pa e au thodes décrites auparavant. Le réarrangement du cycle E de dérivés lupanes a donné accès à des composés allobétuliniques avec de très bons rendements (Schéma 9).37
35 B. Cacciuttolo, S. Poulain-Martini, E. Duñach, Eur. J. Org. Chem. 2011, 3710-3714. 36 F. Grau, A. Heumann, E. Duñach, Angew. Chem. Int. Ed. 2006, 45, 7285-7289.
37 J. A. R. Salvador, R. M. A. Pinto, R. C. Santos, C. Le Roux, A. M. Beja, J. A. Paixao, Org. Biomol. Chem. 2009, 7,
Schéma 9. Double réarrangement de Wagner-Meerwein de la bétuline catalysé par Bi(OTf)3.
Il est intéressant de noter que le Sc(OTf)3 ainsi que le Bi(NO3)3 et le BiBr3 peuvent aussi catalyser la
réaction. Un deuxième réarrangement de Wagner-Meerwein est survenu sur le cycle A lorsque les conditions de réaction étaient plus vigoureuses. En effet, en passant de 5 à 20 mol%, un double réarrangement de Wagner-Mee ei s est p oduit a e d e elle ts e de e ts. Quel ues e p ie es o t pe is de sugg e ue l esp e elle e t a ti e se ait l a ide t ifli ue g in situ par hydrolyse du triflate de bismuth(III).
III. 1. b. Polyènes non activés
Le premier exemple de cyclisation de triènes non conjugués catalysée par le triflate de bismuth(III) a été décrit pour la formation de polycycles différemment fonctionnalisés. 27,38 Le mécanisme de la
a tio ai si ue l esp e atal ti ue a ti e et le ôle du triflate de bismuth(III) ont été étudiés pour cette transformation. La première fonction activée est la partie géranyle terminale, ensuite une double cyclisation sui ie d u arrangement pe et d a de à des o pos s polycycliques de manière « biomimétique » (Schéma 10). Des calculs DFT ont indiqué que Bi(OTf)3 subirait préférentiellement
une hydratation par rapport à une hydrolyse. La forme hydratée de Bi(OTf)3 serait un hybride acide de
Le is/B ø sted aug e ta t l a idit de la ol ule d eau oo di e ui pou ait p o ou oi la p oto atio de l al e t isu stitu .
Schéma 10. Cyclisation en cascade de polyènes.
E ta t u e te sio à la transformation des polyènes, la lisatio de di es a l s a pe is d off i l a s di e t à des d i s t i li ues e u e seule étape, en présence de 5 mol% de Bi(OTf)3 (Schéma
11). 25, 35 Une jonction de cycle trans est obtenue dans tous les cas à partir de dérivés du malonate de
géranyle.
Schéma 11. Cyclisation en tandem avec une réaction de Friedel-Crafts.
Les triènes aromatiques ont aussi permis la cyclisation dans les mêmes conditions réactionnelles. Dans ce cas spécifique, un produit tétracyclique est obtenu presque quantitativement avec une excellente diastéréosélectivité en une seule étape (Schéma 12). Une séquence de polycyclisation/réarrangement sui ie d u e iso isatio et d u e lisatio peut t e e isag e pou e pli ue la formation du produit final.
Schéma 12. Polycyclisation catalysée par Bi(OTf)3a e a a ge e t pa ig atio d’u g oupe e t th le.
Le même type de polycyclisation a été appliqué comme étape clé dans une stratégie de synthèse di e ge te pou l a s au Taiwaniaquinones A/F et Taiwaniaquinols B/D (Schéma 13).39 Parmi tous
les acides de Lewis testés, Bi(OTf)3 a été le meilleur catalyseur. Diverses transformations à partir de
l i te diai e o u via différentes étapes critiques a permis la synthèse des quatre produits naturels.
Schéma 13. S th se de l’i te diai e t i li ue l pour la formation de la taiwaniaquinone A et de la taiwaniaquinol B.
Il a été montré que Bi(OTf)3 pouvait être utilisé de manière efficace pour la cyclisation transannulaire
du produit naturel germacrone. Le noyau eudesmane est formé avec de bons rendements. (Schéma 14).40
Schéma 14. Cyclisation intramoléculaire de germacrone catalysée par le triflate de bismuth(III).
Une cyclisation intramoléculaire catalysée par Bi(OTf)3d a l tosulfo es pe etta t d a de à des
dérivés benzéniques substitués a été décrite (Schéma 15).41
Schéma 15. Annulation, aromatisation desulfonative et alkylation intramoléculaire de Friedel-Crafts en tandem. Du poi t de ue a isti ue, à la fois la o ple atio du tal et la fo atio d a ide t ifli ue in situ catalyserait la cyclisation et une aromatisation désulfonative. Qua d u d i α-géranyle a été engagé dans ces conditions, une seconde réaction de Friedel-C afts a pe is d a de au o pos i li ue correspondant.
III. 1. c. Activation de diènes conjugués
Une synthèse de dérivés chromanes a été étudiée par prénylation directe de phénols pauvres en électrons en présence de Bi(OTf)3, via une réaction de type Friedel-Crafts avec lisop e sui ie d u e
cyclisation (Schéma 16).42 Le mécanisme proposé impliquerait en premier lieu, une C-alkylation en
position ortho du noyau aromatique par réaction avec le carbocation généré après l a ti atio de
40 M. C. Pérez Morales, J. V. Catalán, V. Domingo, M. Jaraíz, M. M. Herrador, J. F. Quílez del Moral, J.-L.
López-Pérez, A. F. Barrero, Chem. Eur. J. 2013, 19, 6598-6612.
41 M.-Y. Chang, Y.-C. Cheng, Y.-J. Lu, Org. Lett. 2015, 17, 3142-3145. 42 K. E. Judd, L. Caggiano, Org. Biomol. Chem. 2011, 9, 5201-5210.
l isop e pa le t iflate de is uth III . E suite la cyclisation du phénol o-prénylé permettrait d obtenir le chromane.
Schéma 16. Prénylation et cyclisation catalysée par Bi(OTf)3.
Notre groupe a reporté la bis-hydroarylation tandem régiosélective intermoléculaire à partir de diènes- , et d a es (Schéma 17).43Cette thode pe et d o te i des d i s i da es su stitu s
en une seule étape à partir de produits de départ disponibles commercialement.
Schéma 17. S th se d’i da es par hydroarylation tandem de diènes-1,3.
Le mécanisme proposé implique une première activation du diène par Bi(OTf)3 et le cation allylique
formé serait piégé par le noyau aromatique pour fournir un arène alkylé qui pourrait subir une deuxième hydroarylation intramoléculaire et former le produit de cyclisation.
III. 1. d.
Activatio d’ the s d’ ol
Une cyclisation régiosélective catalysée par Bi(OTf)3 d the s i li ues a o ati ues fa ile e t
accessibles a été développée pour obtenir des polycycles aromatiques a e d e elle ts e de e ts (Schéma 18).44
Schéma 18. Synthèse de 2-méthylephénantrène par catalyse au bismuth(III).
43 B. Cacciuttolo, P. Ondet, S. Poulain-Martini, G. Lemiere, E. Duñach, Org. Chem. Front. 2014, 1, 765-769. 44 M. Murai, N. Hosokawa, D. Roy, K. Takai, Org. Lett. 2014, 16, 4134-4137.
La a tio est aussi possi le e p se e d I OTf 3ai si u a e les a ides de Le is tels ue AuCl3 ou
BiCl3. Le a is e p opos i pli ue ait u e p e i e a ti atio de l the d ol pou fo e u
intermédiaire oxocarbénium qui pourrait être attaqué par le noyau aromatique selon un mécanisme de type Friedel-C afts. E suite, u e a o atisatio sui ie d u e li i atio d u e ol ule de méthanol mènerait au produit cyclisé correspondant.
III. 2.
Activatio d’alc es
Des composés bicycliques azotés ont été obtenus à partir de précurseurs trifonctionnels comportant une amine protégée, un alcyne et un alcène terminal en présence de Bi(OTf)3 (Schéma 19).45
Schéma 19. C lisatio d’a i o-1,6-énynes catalysée par le Bi(OTf)3.
Bie ue peu d e e ples aie t t d its et ue les e de e ts e p oduits isol s e soient pas disponibles, uel ues e p ie es a isti ues o t t ises e œu e. Des e p ie es de o t ôle pa ‘MN du fluo o t i di u ue l esp e a ti e pou ette t a sfo atio est Bi OTf 3 et non pas
l a ide t ifli ue. Du poi t de ue a isti ue, la fo tio a ti e préférentiellement par Bi(III) serait la triple liaison permettant une addition nucléophile de la double liaison menant à un intermédiaire carbocationique qui pourrait être piégé dans une seconde addition nucléophile intramoléculaire de l ato e d azote. E fin, la protodémétallation mènerait au produit bicyclique correspondant.
Une cyclisation intramoléculaire catalysée par Bi(OTf)3a t d ite à pa ti d a ides a o li ues
possédant une fonction alcyne pour la synthèse de lactones insaturées à 5 ou 6 chaînons. (Schéma 20).46 Le mécanisme proposé pour cette transformation impliquerait une complexation à la fois de
l al e et de l a ide a o li ue. La t iple liaison subirait une attaque nucléophile de la fonction acide pour former majoritairement la lactone à 6 chaînons. La formation de lactones fonctionnalisées,
45 K. Komeyama, M. Miyagi, K. Takaki, Heteroat. Chem 2008, 19, 644-648. 46 K. Komeyama, K. Takahashi, K. Takaki, Chem. Lett. 2008, 37, 602-603.
g ale e t à haî o s, a t d ite à pa ti d a ides a t l i ues pa atal se à l o I), notamment.47
Schéma 20. Hydro-oxycarbonylation intramoléculaire catalysée par Bi(OTf)3.
La méthode a été étendue aux esters acétyléniques pour accéder à de nouvelles lactones polysubstituées (Schéma 21).48 La réaction doit être opérée en présence de tamis moléculaire pour
pi ge l eau et ite l h d ol se des este s de d pa t. D aut es t iflates métalliques, tels que Fe(OTf)2,
Fe(OTf)3, Cu(OTf)2, Zn(OTf)2 and Ni(OTf)2, catalysent aussi la réaction, cependant, avec des rendements
se si le e t plus fai les. Il est i t essa t de ote ue l a ide t ifli ue a seulement donné accès à l acide carboxylique correspondant. Des expériences croisées ont indiqué que la réaction procède de manière intramoléculaire. Le mécanisme proposé correspondrait à la double activation de la triple liaiso et de l este pa le tal e a t à u i te diai e zwitterionique. Ensuite, une migration-1,5 du groupement benzyle sur le carbone métallé mènerait au produit final.
Schéma 21. Carbo-o a o latio i t a ol ulai e d’al e este .
Une carboarylation d al e-arènes via la fo atio d io s N-acyliminiums générés par des N,O-acétals a été décrite. Fe(OTf)3 et Bi(OTf)3o t pe is d o te i le p oduit de lisatio a e des e de e ts
47 E. Tomás-Mendivil, P. Y. Toullec, J. Borge, S. Conejero, V. Michelet, V. Cadierno, ACS Catalysis 2013, 3,
3086-3098; E. Genin, P. Y. Toullec, S. Antoniotti, C. Brancour, J.-P. Genêt, V. Michelet, J. Am. Chem. Soc. 2006, 128, 3112-3113.
similaires.49 Le mécanisme proposé procéderait par une double chélation de l ato e d o g e et de
la t iple liaiso ui fa o ise ait l additio u l ophile du o au a o ati ue pou e e au p oduit de carboarylation (Schéma 22).
Schéma 22. Ca oa latio d’a e al e ia u io a li i iu atal s e par Bi(OTf)3.
La synthèse de furanes sulfonyles différemment fonctionnalisés a été décrite en conditions douces à partir de précurseurs acétyléniques (Schéma 23).50 Un mécanisme par cycloisomérisation
intramoléculaire 5-exo-dig a t p opos . Tout d a o d, le atal seu activerait la fonction alcyne qui serait ensuite attaquée par la fonction carbonyle pour mener à une intermédiaire oxonium. Une suite de protonation, déprotonation, débismuthation et isomérisation génèrerait le produit de cyclisation.
Schéma 23. C loiso isatio d’une a l to e γ-alcyne catalysée par Bi(OTf)3.
Une voie simple pour la formation de pipérazine-2-o e a t tudi e à pa ti d al e alk la ide e présence de Bi(OTf)3 comme catalyseur de cycloisomérisation. De façon intéressante, lorsque PtCl2 est
utilisé sur le même substrat, une hydroamination endo est observée donnant lieu à la formation sélective de dérivés diazépanone-1,4 (Schéma 24).51 Avec Bi(OTf)
3, le mécanisme proposé procéderait
par une première activation π de l al e pa le atio talli ue. E suite, u e lisatio -exo-dig au ait lieu suite à l additio u l ophile de l a ide su le a o e i te e de la t iple liaiso . E fi , u e tape de p otod talatio et d iso isatio de la dou le liaiso fo e ait le p oduit de lisatio .
49 K. Komeyama, T. Yamada, R. Igawa, K. Takaki, Chem. Commun. 2012, 48, 6372-6374. 50 M.-Y. Chang, Y.-C. Cheng, Y.-J. Lu, Org. Lett. 2015, 17, 1264-1267.
Schéma 24. Hydroamination 6-exo-dig d’alkynylamide catalysée par le triflate de bismuth(III) et comparaison avec la catalyse par PtCl2.
III. 3.
Activatio d’all es
Récemment, notre groupe s est i t ess à la a ti it de la fo tio all e e p se e de superacides de Lewis avec des substrats comportant un groupement aryle ou carbonyle.
La cycloisomérisation catalysée par Bi(OTf)3d a les possédant une fonction allène a été étudiée en
conditions douces pour la synthèse de structures polycycliques. Cette allylation intramoléculaire catalytique de type Friedel-Crafts de noyaux aromatiques o a ti s a pe is d a de à di e s squelettes polycycliques comme des tétralines, des chromanes et des tétrahydroquinolines (Schéma 25).52 Cette méthodologie utilise le caractère électrophile de la fonction allène par son activation
fa o is e e p se e d u t iflate métallique.
Schéma 25. C loh d oa latio d’all es atal s e pa le t iflate de is uth III .
Afin de profiter de la présence de la nouvelle insaturation C–C formée sur le produit de cyclisation, une bis-hydroarylation a aussi été développée en modifiant les conditions. Cette nouvelle réaction tandem a donné lieu à divers structures tri- et t t a li ues d i t t Schéma 26). 52
Schéma 26. Cyclisation tandem intramoléculaire.
De plus, la e sio i te ol ulai e de la a tio ta de d all latio de F iedel-Crafts suivie de l alk latio a pu t e d elopp e de a i e gios le ti e afi d o te i des o pos s i da i ues fonctionnalisés (Schéma 27). 52
Schéma 27. Cyclisation tandem intermoléculaire.
III. 4.
Activation de fonctions alcools et dérivés
III. 4. a. Activation des fonctions alcools
La e z latio d a es et d h t oa es atal s e pa le triflate de bismuth(III) a été décrite.53
L al ool e z li ue est a ti pa le atal seu pou g e u a o atio sta ilis ui peut su i u e addition directe du noyau aromatique. Cette méthodologie a été appliquée en version i t a ol ulai e pou l a latio de F iedel-Crafts constituant une voie efficace pour la synthèse de dérivés fluorènes (Schéma 28).
Schéma 28. Arylation intramoléculaire catalysée par Bi(OTf)3.
Une réaction de Friedel-Crafts intramoléculaire similaire a été développée avec un plus grand champ d appli atio pour la formation du squelette thiochromane de la molécule SHetA2. (Schéma 29).54
Cette méthode a pu être étendue à la synthèse de dérivés tétrahydronaphtalènes, chromanes, thiochromanes et tétrahydroquinolines.
Schéma 29. Synthèse de tétrahydroisoquinolines et dérivés.
Une réaction tandem de benzylation sui ie d u e h d oa i atio i t a ol ulai e a t d elopp e pou la s th se d h t o les azot s à pa ti d a i o ol fi es et d al ools facilement accessibles (Schéma 30).55 Du point de vue mécanistique, la fonction alcool serait activée préférentiellement lors
de la p e i e tape et l atta ue de la dou le liaiso e ait à un intermédiaire carbocationique dans un processus de type ène. Le groupement amine piégerait ensuite le carbocation permettant d a de au p oduit de lisatio o espo da t.
Schéma 30. Réaction d’a i o-oléfine avec un alcool énophile.
La synthèse de furanes fonctionnalisés catalysée par le triflate de bismuth(III) a été décrite à partir d a loï es et de β-cétoesters ou de dicétones-1,3 (Schéma 31).56 Parmi les catalyseurs testés, il est
i t essa t de ote ue l a ide t ifli ue a pas pe is de atal se la a tio . Du poi t de ue a isti ue, la fo tio al ool de l a loï e se ait a ti e p f e tiellement par le triflate de
54 B. Nammalwar, R. A. Bunce, Tetrahedron Lett. 2013, 54, 4330-4332.
55 K. Komeyama, Y. Kouya, Y. Ohama, K. Takaki, Chem. Commun. 2011, 47, 5031-5033. 56 K. Komeyama, Y. Ohama, K. Takaki, Chem. Lett. 2011, 40, 1103-1104.
bismuth(III) permettant de la transformer en meilleur groupe partant qui pourrait ensuite subir une condensation avec la dicétone- , . E suite, l i te diai e lise ait puis l li i atio d u e
ol ule d eau pe ett ait d a der au dérivé furanique correspondant.
Schéma 31. S th se de d i s fu a i ues à pa ti d’a loï e et de dicétone catalysée par Bi(OTf)3.
Lo s du d eloppe e t d u e app o he a tios le ti e de la synthèse du squelette de la (–)-plastensimycine, une réaction diastéréosélective intramoléculaire de Friedel-Crafts sur une fonction lactol catalysée par Bi(OTf)3 a été étudiée (Schéma 32).57 La réactivité désirée a pu être observée en
utilisa t ui ale ts de hlo u e d tai . U e alte ati e a t t ou e e utilisa t le t iflate de bismuth(III) en quantité catalytique en présence de LiClO4 comme co-catalyseur. La fo atio d u e
esp e o o a iu pe hlo e fa o ise ait l atta ue u l ophile du g oupe e t a o ati ue pou former le cycle. De plus, le sel de lithium annihilerait le caractère base de Lewis du groupement sulfonate évitant la désactivation du catalyseur.
Schéma 32. Cyclisation de Friedel-C afts d’u la tol atal s e pa Bi OTf3.
III. 4. b. Activation des fonctions alcool allylique
La synthèse intramoléculaire stéréocontrôlée de tétrahydroisoquinolines à partir d al ools allyliques chiraux via un transfert de chiralité-1,3 a été développée (Schéma 33).58D aut es atal seu s, tels ue
SnCl4, FeCl3, AuCl3, SbCl3, GaCl3, In(OTf)3, o t pe is d effe tue la lisatio a e des e de e ts
moins bons et de moins bonnes sélectivités comparés au Bi(OTf)3. La réaction est opérée à -15 °C pour
éviter la perte de chiralité lors du transfert. Un mécanisme de type SN est p opos pou expliquer la
stéréochimie de nouveau centre stéréogène formé. Bi(OTf)3 activerait d a o d l al ool all li ue pa
o ple atio sui i d u e pe te de ol ule d a ide t ifli ue pour mener à la fo atio d u o plexe où le métal coordinerait la fonction carbonyle du groupement Boc. Cet intermédiaire clé déterminerait la sélectivité de la face, ensuite une cyclisation syn intramoléculaire de type SN mènerait au produit
correspondant en régénérant le catalyseur.
Schéma 33. Transfert de chiralité intramoléculaire-1,3 catalysé par le triflate de bismuth(III).
Plus récemment, une cyclisation catalysée par le triflate de bismuth(III) avec des dérivés possédant un the d ol a été développée par notre groupe. Plusieurs cyclopenténones ont été obtenues avec de très bons rendements à partir de produits de départ facilement accessibles, par catalyse avec du triflate de bismuth(III). Le mécanisme procèderait via une électrocyclisatio π d u i te diai e
58 N. Kawai, R. Abe, M. Matsuda, J. i. Uenishi, J. Org. Chem. 2011, 76, 2102-2114; N. Kawai, R. Abe, J. i. Uenishi,
a o atio i ue pe tadi le fo pa l a ti atio p f e tielle de l al ool all li ue pa le atal seu . E suite, l atta ue de la ol ule d eau li i e lo s de la p e i e tape fo e ait u la tole ui pou ait s ou i pou former la cyclopenténone correspondante, plus stable (Schéma 34).59
Schéma 34. Réarrangement de 2-alkoxy diallyle alcools catalysé par Bi(OTf)3.
Lors du développement de la synthèse totale de (+)-Taiwaniaquinole F et autres Taiwaniaquinoides, une nouvelle cyclisation catalytique de type Nazarov a été explorée à partir de dérivés aryldiallycarbinoles (Schéma 35).60 La réaction diastéréosélective catalysée par Bi(OTf)
3 a permis de
former une structure carbotétracyclique clé utile pour la synthèse des mérosesquiterpènes naturels o e pa e e ple l Alkaol A.
Schéma 35. C lisatio de t pe Naza o d’a l i l a i ol.
59 L. Lempenauer, E. Duñach, G. Lemière, Org. Lett. 2016, 18, 1326-1329. 60 B. N. Kakde, N. Kumar, P. K. Mondal, A. Bisai, Org. Lett. 2016, 18, 1752-1755.
III. 4. c. Activation de dérivés halogénés allyliques
Une cyclisation 5-exo-trig catalysée par Bi(OTf)3 a été communiquée pour la formation de produits
diéniques-1,4 (Schéma 36).61Le a is e p o de ait d a o d pa l a ti ation du bromure allylique
pour donner un carbocation allylique qui subirait u e atta ue u l ophile de l al e pou former un carbocation intermédiaire. Une élimination mènerait ensuite au produit diénique de cyclisation.
Schéma 36. Cyclisation 5-exo-trig catalysée par Bi(OTf)3.
Une alternative plus douce et régiosélective à la cyclisation de type Friedel-Crafts intramoléculaire décrite précédemment pour la synthèse de squelettes benzoazépine a été décrite (Schéma 37).62
Schéma 37. Réaction de Friedel-Crafts intramoléculaire catalysée par Bi(OTf)3.
61 R. Hayashi, G. R. Cook, Tetrahedron Lett. 2008, 49, 3888-3890.
La a tio essite l utilisatio d u ui ale t de hlo u e d i diu , le e e de e t est o te u e p se e d u e ua tit atal ti ue de Bi OTf 3. D u poi t de ue a isti ue, le o u e
allylique serait activé par le triflate de bismuth(III), soit par chélation π de l ol fi e et du ome, soit e d plaça t l halog e pou fo e u a o atio all li ue. E suite, l atta ue du o au a o ati ue suivie de la réaromatisation mènerait au bicycle aryle fusionné à un cycle à 7 chaînons.
III. 4. d.
Activatio d’ the s allyliques silylés
En recherchant une voie de synthèse plus efficace pour la formation de produits naturels, une nouvelle carbocyclisation a été développée pour accéder à la citridone A (Schéma 38).63
Schéma 38. Hétérocyclisation tandem sélective.
Cette lisatio est pas atal ti ue a u ui ale t de Bi(OTf)3 a été nécessaire pour effectuer
l h th o a o lisatio . La positio all li ue distale se ait a ti e pa l a ide de Le is ap s la fo atio d u e o ple e is uth-amide. Le carbocation obtenu serait piégé par le groupement alcool pour accéder à la molécule cible.
III. 4. e.
Activatio d’ac tates propargyliques
Une réaction domino catalysée par Bi(OTf)3 pour la formation de furanes 2-éthyléniques à partir
d a tates p opa g li ues et d the s d ol sil l s a t tudiée (Schéma 39).64 La cyclisation a aussi
lieu a e des e de e ts si ilai es e p se e d aut es a ides de Le is ou B ønsted tels que InCl3,
AgOTf, Sc(OTf)3 et TfOH. Une étude mécanistique a permis de montrer une suite de deux substitutions
de type SN. E effet, le atal seu a ti e ait e p e ie l a tate p opa g li ue pou fo e u
63 A. D. Fotiadou, A. L. Zografos, Org. Lett. 2011, 13, 4592-4595. 64 D. Nitsch, T. Bach, J. Org. Chem. 2014, 79, 6372-6379.
carbocation tertiaire qui pourrait subir une substitution nucléophile SN a e l the d ol sil l pou
former un intermédiaire allénique. Une deuxième substitution nucléophile SN fo e ait u e liaiso
oxygène- a o e sulta t de l atta ue de l o g e su le a o e e t al de l all e et la pe te d u e molécule de méthanol. Le noyau furane serait obtenu après déprotonatio de l i te diai e cationique.
Schéma 39. Synthèse de dérivés furaniques à pa ti d’u a tate p opa g li ue et d’u the d’ ol sil l .
III. 4. f.
Activatio d’alcools propargyliques
Une al l lop opa atio i t a ol ulai e d ol fi es atal s e pa Bi OTf3 a été développée
(Schéma 40).65 La s th se s est a e st os le ti e pe etta t l a s à des squelettes azotés
intéressants retrouvés dans des produits biologiquement actifs. Il est important de noter que la bipyridine a du être utilisée comme additif pour les oléfines pauvres en électrons. La réaction a permis d o te i les es st u tu es avec des rendements sensiblement plus faibles en utilisant le Fe(OTf)2
ou le Fe(OTf)3. D u poi t de ue a isti ue, les auteu s o t p opos u e p e i e a ti atio pa
u e dou le oo di atio du is uth a e l al e et l o g e du g oupe h d o le. E suite, l ol fi e atta ue ait l al ool p opa g li ue a ti pou e e à u a o atio i te diai e o te a t u e partie vinylidène qui pourrait subir une deuxième cyclisation sur le carbocation pour former un cation vinylique. Enfin, une déprotonation sui ie d u e pe te d u e ol ule d eau e ait à l al lp opa e de a i e st osp ifi ue e fo tio de la st o hi ie de la dou le liaiso .
Schéma 40. Alk l lop opa atio d’un azaényol catalysée par le triflate de bismuth(III).
Une version intramoléculaire de la réaction tandem du réarrangement de Meyer-Schuster sui ie d u e addition nucléophile-1,4 sur des alcools propargyliques a été étudiée pour la formation de dihydroquinolones (Schéma 41).66 Le mécanisme proposé passe ait, tout d a o d, pa u
réarrangement de Meyer-“ huste pa a ti atio de l al ool p opa g li ue pou fo e u intermédiai e all ol do a t la to e i li ue o epo da te. E suite, l i te diai e su i ait u e addition nucléophile- , de l ato e d azote du a a ate pou e e au p oduit de lisatio . De a i e su p e a te, l ajout d tha ol e e s est essai e pour effectuer la réaction et aucune additio de l tha ol sulta t de l additio de Mi hael a t o se e su la to e α,β-insaturée intermédiaire.
Schéma 41. Synthèse de dihydroquinolones catalysée par Bi(OTf)3.
III. 5.
Activation de fonctions carbonyles
III. 5. a.
Activatio d’ald h des et c to es
La cyclisation de dicétones-1,4 catalysée par Bi(OTf)3 dans le liquide ionique [bmim]BF4 a permis de
former des dérivés furaniques trisubstitués (Schéma 42).67 Quand une aniline ou le réactif de Lawesson
sont engagés dans le milieu réactionnel, des squelettes pyrrole ou thiophène sont respectivement obtenus.
Schéma 42. Synthèse de dérivés furaniques.
Une synthèse hautement diastéréosélective de diols syn-1,3 masqués a été développée à partir de diols monoallyliques-1,5 et d ald h des pa atal se à l o (I) ou au bismuth(III) (Schéma 43).68
Schéma 43. Hémiacétalisation et cyclisation diastéréosélective de diols-1,5 catalysées par Bi(III) et Au(I) en tandem. Quand les oléfines Z so t e gag es e p se e d u atal seu à l o (I), le produit désiré est obtenu
ais seul l a tal est fo a e Bi OTf3 . Au o t ai e pou l iso e E, la catalyse au triflate de
is uth III a pe is d a de au p oduit de façon diastéréosélective alors ue le atal seu à l o e
67 J. S. Yadav, B. V. S. Reddy, B. Eeshwaraiah, M. K. Gupta, Tetrahedron Lett. 2004, 45, 5873-5876. 68 J. A. Goodwin, C. F. Ballesteros, A. Aponick, Org. Lett. 2015, 17, 5574-5577.
o e ait pas. D u poi t de ue a isti ue, l ald h de se ait a ti e p e ie pa le tal pou subir une hémiacétalisation, ensuite, une cyclisatio et d sh d atatio de l al ool li e du g oupe e t allylique permettrait de do e l a tal o espo da t. Les dioxolanes-1,3 ont pu être déprotégés par la suite en diols-syn-1,3, ou directement transformés en tétrahydrofuranes fonctionnalisés.
La fo atio st os le ti e de p olidi es à pa ti d ald h des α-ca o l s et d a i oall lsila es a été décrite (Schéma 44).69 D u poi t de ue a isti ue, la a tio do i o o e e ait pa
l a ti atio de l ald h de pa le catalyseur pour former un ion imminium qui subirait une réaction intramoléculaire de t pe “aku ai pe etta t d a de à l h t o le o espo da t.
Schéma 44. Synthèse de pyrrolidi es à pa ti d’all lsila es -aminés.
La synthèse « one-pot » d iso thio h o a es a t d elopp e à pa ti d ald h des et de phényléthanethiol ou phényléthanol (Schéma 45).70 D u poi t de ue a isti ue, la a tio
procèderait via une cyclisation de type oxa(thia)-Pictet-“pe gle . Tout d a o d, l ald h de se ait a ti pa le t iflate de is uth III pou fo e u h ia tal ui su i ait la pe te d u e ol ule d al ool ou de thiol. L i te diai e o o iu ou sulfo iu sulta t ycliserait de façon intramoléculaire pour former le produit attendu.
Schéma 45. S th se d’isothio h o a es à partir de phényléthanethiol.
69 P. Mohr, M.-P. Imhoff, F. Reggiani, Tetrahedron Lett. 2015, 56, 2262-2264.
70 C. Lherbet, D. Soupaya, C. Baudoin-Dehoux, C. André, C. Blonski, P. Hoffmann, Tetrahedron Lett. 2008, 49,
Une réaction de type Prins catalysée par le triflate de bismuth(III) dans un solvant ionique a été décrite pou la fo atio de t t ah d op a oles su stitu s à pa ti d ald h des et d al ools ho oall li ues (Schéma 46).71 La a tio a o duit ajo itai e e t à l iso e cis de manière hautement
diast os le ti e. D u poi t de vue mécanistique, comme dans u e a tio de P i s, l ald h de se ait a ti e p e ie pa le atal seu et atta u pa l ol fi e. U e pe te d u e ol ule d eau et cyclisation mènerait à un carbocation qui subirait une attaque nucléophile du groupement hydroxyle.
Schéma 46. Synthèse de dérivé tétrahydropyranolique catalysé par le triflate de bismuth(III).
Des réactions multicatalytiques tandem de type Mukaiyama suivies d u e h d oalko latio et d u e addition de Sakurai puis d u e h d o alko latio o t t tudi es. La thode a pe is d a de à des tétrahydrofuranes fonctionnalisées avec de bons rendements et diastéréosélectivités (Schéma 47). 31 Dans les deu as, la p e i e tape se ait l a ti atio de l aldéhyde par le catalyseur puis
l additio du nucléophile silylé. Selon les auteurs et les études précédentes,72 la vraie espèce
catalytique serait le TMSOTf produit par Bi(OTf)3 et les dérivés silylés. La p se e d u u l ophile
externe, comme le méthanol, est nécessaire pour effectuer la deuxième étape du procédé « one-pot » constitant en la déprotection de l i te édiaire alcool sil l . E suite l h d o alko latio catalytique prendrait place pour fournir le tétrahydrofurane correspondant.
Schéma 47. Synthèse de tétrahydrofuranes fonctionnalisés.
71 M. S. R. Murty, K. Rajasekhar, V. Harikrishna, J. S. Yadav, Heteroat. Chem 2008, 19, 104-106. 72 E. M. Carreira, R. A. Singer, Tetrahedron Lett. 1994, 35, 4323-4326.