Core epithelial-to-mesenchymal transition interactome
gene-expression signature is associated with
claudin-low and metaplastic breast cancer subtypes
The MIT Faculty has made this article openly available.
Please share
how this access benefits you. Your story matters.
Citation
Taube, J. H., J. I. Herschkowitz, K. Komurov, A. Y. Zhou, S. Gupta, J.
Yang, K. Hartwell, et al. “Core epithelial-to-mesenchymal transition
interactome gene-expression signature is associated with
claudin-low and metaplastic breast cancer subtypes.” Proceedings of
the National Academy of Sciences 107, no. 35 (August 31, 2010):
15449-15454.
As Published
http://dx.doi.org/10.1073/pnas.1004900107
Publisher
National Academy of Sciences (U.S.)
Version
Final published version
Citable link
http://hdl.handle.net/1721.1/84510
Terms of Use
Article is made available in accordance with the publisher's
policy and may be subject to US copyright law. Please refer to the
publisher's site for terms of use.
Corrections
CELL BIOLOGY
Correction for “Core epithelial-to-mesenchymal transition in-teractome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes,” by Joseph H. Taube, Jason I. Herschkowitz, Kakajan Komurov, Alicia Y. Zhou, Supriya Gupta, Jing Yang, Kimberly Hartwell, Tamer T. Onder, Piyush B. Gupta, Kurt W. Evans, Brett G. Hollier, Prahlad T. Ram, Eric S. Lander, Jeffrey M. Rosen, Robert A. Weinberg, and Sendurai A. Mani, which appeared in issue 35, August 31, 2010 of Proc Natl Acad Sci USA (107:15449–15454; first published August 16, 2010; 10.1073/pnas.1004900107).
The authors note that on page 15453, right column,fifth full paragraph, sentence 2,“Microarray data for HMLE Gsc, Snail, Twist, TGF-β1, and vector control has been deposited in GEO under accession number GSE9691” should instead appear as “Microarray data for HMLE Gsc, Snail, Twist, TGF-β1, and vector control has been deposited in GEO under accession number GSE24202.”
www.pnas.org/cgi/doi/10.1073/pnas.1015095107
MEDICAL SCIENCES
Correction for“Detection of MLV-related virus gene sequences in blood of patients with chronic fatigue syndrome and healthy blood donors,” by Shyh-Ching Lo, Natalia Pripuzova, Bingjie Li, Anthony L. Komaroff, Guo-Chiuan Hung, Richard Wang, and Harvey J. Alter, which appeared in issue 36, September 7, 2010 , of Proc Natl Acad Sci USA (107:15874–15879; first published August 23, 2010; 10.1073/pnas.1006901107).
The authors note that the GenBank Accession Numbers for the gag gene are HM630557–HM630562, and GenBank Acces-sion Numbers for the env gene are HQ157342–HQ157343.
Core epithelial-to-mesenchymal transition interactome
gene-expression signature is associated with
claudin-low and metaplastic breast cancer subtypes
Joseph H. Taubea,1, Jason I. Herschkowitzb,1, Kakajan Komurovc,1, Alicia Y. Zhoud,e, Supriya Guptaf, Jing Yangg,
Kimberly Hartwellh, Tamer T. Onderd,e, Piyush B. Guptae,f, Kurt W. Evansa, Brett G. Holliera, Prahlad T. Ramc,
Eric S. Landerd,e,f,i, Jeffrey M. Rosenb, Robert A. Weinbergd,e,j,2, and Sendurai A. Mania,2
Departments ofaMolecular Pathology andcSystems Biology, University of Texas M.D. Anderson Cancer Center, Houston, TX 77054;bDepartment of Molecular
and Cellular Biology, Baylor College of Medicine, Houston, TX 77030;dWhitehead Institute for Biomedical Research, Cambridge, MA 02142;eDepartment of
Biology, Massachusetts Institute of Technology, Cambridge, MA 02142;gDepartment of Pharmacology, University of California, La Jolla, CA 92093-0636; hDepartment of Medicine, Brigham and Womens Hospital, Boston, MA 02115;fBroad Institute, Cambridge, MA 02142;iDepartment of Systems Biology,
Harvard Medical School, Boston, MA 02115; andjMassachusetts Institute of Technology, Ludwig Center for Molecular Oncology, Cambridge, MA 02139
Contributed by Robert A. Weinberg, June 24, 2010 (sent for review January 26, 2010)
The epithelial-to-mesenchymal transition (EMT) produces cancer cells that are invasive, migratory, and exhibit stem cell character-istics, hallmarks of cells that have the potential to generate meta-stases. Inducers of the EMT include several transcription factors (TFs), such as Goosecoid, Snail, and Twist, as well as the secreted TGF-β1. Each of these factors is capable, on its own, of inducing an EMT in the human mammary epithelial (HMLE) cell line. However, the interac-tions between these regulators are poorly understood. Overexpres-sion of each of the above EMT inducers up-regulates a subset of other EMT-inducing TFs, with Twist, Zeb1, Zeb2, TGF-β1, and FOXC2 being commonly induced. Up-regulation of Slug and FOXC2 by either Snail or Twist does not depend on TGF-β1 signaling. Gene expression sig-natures (GESs) derived by overexpressing EMT-inducing TFs reveal that the Twist GES and Snail GES are the most similar, although the Goosecoid GES is the least similar to the others. An EMT core signa-ture was derived from the changes in gene expression shared by up-regulation of Gsc, Snail, Twist, and TGF-β1 and by down-up-regulation of E-cadherin, loss of which can also trigger an EMT in certain cell types. The EMT core signature associates closely with the claudin-low and metaplastic breast cancer subtypes and correlates negatively with pathological complete response. Additionally, the expression level of FOXC1, another EMT inducer, correlates strongly with poor survival of breast cancer patients.
cancer stem cells
|
Twist|
Snail|
FOXC1T
he epithelial-to-mesenchymal transition (EMT) is a process in which adherent epithelial cells shed their epithelial charac-teristics and acquire, in their stead, mesenchymal properties, in-cludingfibroblastoid morphology, characteristic gene-expression changes, increased potential for motility, and in the case of cancer cells, increased invasion, metastasis, and resistance to chemo-therapy (1, 2). Recent studies have linked EMTs with both meta-static progression of cancer (3–5) and acquisition of stem-cell characteristics (6, 7), leading to the hypothesis that cancer cells that undergo an EMT are capable of metastasizing through their acquired invasiveness and, following dissemination, through their acquired self-renewal potential, which enables them to spawn the large cell populations that constitute macroscopic metastases.EMTs can be induced in vitro by exposing certain normal and neoplastic epithelial cells to various growth factors, including TGF-β1, hepatocyte growth factor, and PDGF (1, 8). Downstream of each of these growth factors and their cognate receptors lies an array of transcription factors (TFs), each of which is capable, on its own, of inducing an EMT. These TFs include the homeobox protein Goosecoid (Gsc) (9), the zinc-finger proteins Snai1 (Snail) and Snai2 (Slug) (10–12), the basic helix-loop-helix protein Twist1 (Twist) (3), the forkhead box proteins FOXC1 (8, 13) and FOXC2
(14), and the zinc-finger, E-box-binding proteins Zeb1 and Sip1 (Zeb2) (8, 15).
In addition to TFs, members of the miR-200 family of micro-RNAs are regulated during an EMT (16). This down-regulation results, in turn, in the up-regulated expression of several critical target genes, notably Zeb1 and Zeb2. Expression of any one of these TFs or down-regulation of the miR-200 family in an appropriate epithelial cell suffices to induce an EMT (17, 18). Moreover, many of these TFs are expressed concomitantly in the mesenchymal cells that have passed through an EMT. The over-lapping and unique contributions of each inducer to the EMT program have not been adequately explored.
Recent microarray analyses have allowed stratification of clin-ical breast cancers into a large number of distinct subtypes, such as luminal, basal-like, and HER2+(19–21). Yet, other subtypes of tumors have recently been delineated by us and others (22, 23). These distinctions have proven to be useful in predicting responses to therapy, time to metastasis, and survival.
In the present study, we assayed gene expression signatures (GESs) in human mammary epithelial cells (HMLE) induced to undergo an EMT by expressing Gsc, Snail, Twist, or TGF-β1 or by knocking down expression of E-cadherin (24), and found that EMTs induced by these methods induce an overlapping set of changes in gene expression, which we term the“EMT core sig-nature.” We compared the EMT core signature with signatures that define breast cancer subtypes and found a close association with the claudin-low and metaplastic breast cancer subtypes. Results
Interaction of EMT-Related TFs as a Regulatory Network.To
eluci-date the network of interactions between EMT regulatory factors, wefirst assessed the expression of known EMT inducers and the genes known to be regulated during EMT in various breast cancer cell lines. To do so, we assayed four breast cancer cell lines for expression of the following TFs known to promote EMTs: Gsc (9), FOXC1 (8, 13), FOXC2 (14), Zeb1, Zeb2 (25, 26), Slug (10, 27), Snail (11, 28), and Twist (3), as well as other genes associated with
Author contributions: J.H.T., J.I.H., K.K., R.A.W., and S.A.M. designed research; J.H.T., J.I.H., K.K., A.Y.Z., S.G., J.Y., P.B.G., K.W.E., B.G.H., and S.A.M. performed research; J.Y., K.H., T.T.O., P.B.G., P.T.R., E.S.L., J.M.R., R.A.W., and S.A.M. contributed new reagents/ analytic tools; J.H.T., J.I.H., K.K., P.B.G., R.A.W., and S.A.M. analyzed data; and J.H.T., J.I.H., K.K., R.A.W., and S.A.M. wrote the paper.
The authors declare no conflict of interest.
1J.H.T., J.I.H., and K.K. contributed equally to this work.
2To whom correspondence may be addressed. E-mail: weinberg@wi.mit.edu or smani@
mdanderson.org.
This article contains supporting information online atwww.pnas.org/lookup/suppl/doi:10.
1073/pnas.1004900107/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1004900107 PNAS | August 31, 2010 | vol. 107 | no. 35 | 15449–15454
CELL
either the mesenchymal state [N-cadherin (29) and fibronectin (30)] or the epithelial state [E-cadherin (31)]. Relative to the lu-minal epithelial cell line, MCF-7, the MDA-MB 231, MDA-MB 435, and SUM 1315 basal B cell lines consistently expressed higher levels of Snail, Slug, and Zeb2, but not Gsc or FOXC1 (Fig. 1A). To further understand the interactions among EMT-inducing TFs, we tested how overexpression of a single EMT-inducing TF affects the expression of other TFs in this network, thereby estab-lishing an“EMT interactome.” To do so, we used immortalized HMLE cells that express high levels of E-cadherin and low levels of vimentin, similar to the MCF-7 breast cancer cell line and con-trasting with the more mesenchymal MDA-MB 231 and SUM1315 breast cancer cell lines (Fig. S1). HMLE cells were induced to undergo EMTs through overexpression of Gsc, Snail, Twist, or TGF-β1. We then used RT-PCR to confirm the expression levels of these genes, as well as various genes known to be associated with EMT programs (Fig. 1B andFig. S2).
Among the EMT-inducing TFs, expression of FOXC2, Slug, Zeb1, and Zeb2 was consistently elevated in response to over-expression of Gsc, Snail, Twist, or TGF-β1, suggesting that these four genes operate downstream of Gsc, Snail, Twist, and TGF-β1 in the EMT interactome (Fig. 1 B and C). In contrast, Gsc was not up-regulated by overexpression of the other EMT inducers, suggesting an independent mode of transcriptional regulation (Fig. 1B).
Recentfindings have demonstrated that EMT programs can also be induced by inhibiting members of the miR-200 family of microRNAs (17, 18, 32). We wished to determine the location of these miRNAs in the EMT interactome. The miR-200 family and Zeb transcription factors form a mutually inhibitory, double negative-feedback loop (16–18, 33). Consistent with a role for Zeb1 and Zeb2 in miR-200 repression, all miR-200 family mem-bers were repressed by the forced expression of either Gsc, Snail, Twist, or TGF-β1 (Fig. 1D). These results reinforce the findings that in addition to interactions of the EMT-inducing TFs,
re-pression of the miR-200 family of microRNAs accompanies and participates in execution of the EMT program.
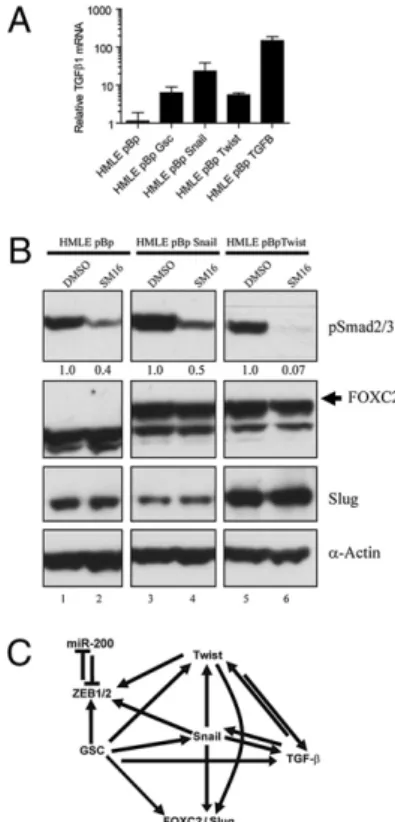
A widely accepted model of the EMT during tumorigenesis proposes that TGF-β1, produced by the tumor microenvironment, promotes tumor progression by inducing expression of the EMT-inducing TFs (34). Indeed, treatment of HMLE cells with TGF-β1 for a period of 12 d induced an EMT (14) and the expression of a number of EMT-inducing TFs (Fig. 1B). We undertook to de-termine whether another type of feedback loop operated here, specifically whether induction of an EMT achieved by overex-pressing EMT-inducing TFs yields, in turn, the expression of TGF-β1. For this experiment, we measured the expression of TGF-β1 mRNA by quantitative RT-PCR (qRT-PCR) in HMLE cells expressing either an empty vector control, Gsc, Snail, Twist, or TGF-β1. In all cases, expression of TGF-β1 mRNA increased by more than 5-fold compared with control cells (Fig. 2A).
Because TGF-β1 sufficed to induce an EMT, we investigated whether the TGF-β1 expressed in response to an EMT operates in an autocrine manner to induce and maintain expression of down-stream EMT effectors, specifically FOXC2 and Slug. Because Snail and Twist are known to induce a complete EMT, including ex-pression of FOXC2, Slug, and TGF-β1, we blocked TGF-β1 sig-naling in these cells using SM16, a compound developed to inhibit the TGF-β1 pathway by interfering with the type I receptor sig-naling (35). As anticipated, treating HMLE cells expressing either an empty control vector, Snail, or Twist with SM16 significantly reduced the levels of phosphorylated SMAD2/3 by 50 to 90%, compared with DMSO (Fig. 2B). Under these conditions, neither
Fig. 1. Expression of EMT marker genes in breast cancer cell lines and nontransformed EMT-induced human mammary epithelial cells. (A) Expres-sion of EMT marker genes was measured by semiquantitative RT-PCR per-formed on RNA extracted from MCF-7, MDA-MB 231, MDA-MB 435, and SUM1315 cells. (B–D) HMLE cells were transduced with a retrovirus over-expressing the indicated gene and expression of the indicated gene was measured by semiquantitative RT-PCR (B) or quantitative PCR (C and D). GAPDH (C) or U6 snoRNA (D) was amplified for normalization. Error bars represent the SD from three independent experiments.
Fig. 2. TGF-β1 is regulated by EMT inducers, but is not required for up-regulation of FOXC2 or Slug. (A) HMLE cells were transduced with a retro-virus overexpressing the indicated genes and expression of TGF-β1 mRNA was measured by quantitative RT-PCR. GAPDH was amplified for normali-zation. (B) HMLE cells transduced with a retrovirus overexpressing the in-dicated gene were treated with DMSO or SM16, and a TGF-β signaling inhibitor and gene expression was assayed by Western blot for the indicated proteins. Relative levels of pSmad2/3 were calculated by densitometry and listed beneath the bands.α-Actin was used as a loading control.
the Snail- nor Twist-expressing cells altered expression of FOXC2 or Slug protein (Fig. 2B). Thisfinding indicated that induction of an EMT by Snail or Twist does not depend on ongoing TGF-β1 autocrine signaling and suggests, instead, that Snail and Twist can induce FOXC2 and Slug through alternative mechanisms, quite possibly involving only intracellular signaling.
Hierarchical Clustering of GESs from EMTs Induced by Gsc, Snail,
Twist, TGF-β1, and by Down-Regulating E-Cadherin.Because various
inducers of EMT seemed to be capable of transactivating a com-mon set of downstream effectors, we extended these comparisons by determining the larger effects of these TFs on overall gene ex-pression within cells. We began by deriving individual GESs of the cells induced to undergo EMT by forced expression of Gsc, Snail, Twist, or TGF-β1, or by knocking down E-cadherin, and cata-loguing genes exhibiting at least a 2-fold up- or down-regulation under any of the conditions of EMT induction. Cells in which E-cadherin was experimentally down-regulated were also included in these analyses, as we previously demonstrated that this alteration could also serve to induce an EMT in these cells (24). We then performed hierarchical clustering of these GESs to measure their degree of similarity (Fig. 3A).
Of thefive methods used to induce EMTs in this analysis, Snail and Twist generated the most similar GESs, consistent with the fact that Twist is a direct target gene of the Snail transcription factor (36), and Gsc showed the most distinct GES (Fig. 3A), consistent with the observation that Gsc was not induced by ex-pression of either Snail, Twist, or TGF-β1 (Fig. 1B). Not surpris-ingly, expression of TGF-β1 or knockdown of E-cadherin pro-duced GESs that diverged slightly from both Snail and Twist, likely because of the fact that, even though either method induces an EMT, neither involves ectopic expression of a TF and, therefore, may affect its downstream targets less directly.
EMT Core Signature. Localized paracrine signals arising in the
tu-mor microenvironment appear to be important in inducing an EMT in nearby cancer cells; accordingly, only a minority of cells within a tumor may display characteristics of having entered into or passed through an EMT. These observations complicate attempts to id-entify groups of cells within a tumor that have undergone an EMT. For this reason, we sought to identify a GES common to several known EMT-inducing signals. Establishment of such a core signa-ture should prove useful in the fusigna-ture for identifying the small subset of cells that have undergone an EMT within a tumor, even if such an EMT is induced by a currently unknown inducer of this program.
To identify such a signature, we reanalyzed the microarray-based gene-expression changes from HMLE cells expressing Gsc, Snail, Twist, or TGF-β1 or an siRNA targeting E-cadherin. We identified an EMT core signature consisting of 159 genes that were down-regulated and 87 genes that were up-regulated at least 2-fold by all of these EMT-inducing signals (Table S1). Several of these gene-expression changes were validated by qRT-PCR (Fig. S3). As expected, the epithelial adhesion molecule E-cadherin (CDH1) was down-regulated in all samples and the mesenchymal markers N-cadherin (CDH2), vimentin, and the invasion-associated pro-tease, matrix metalloproteinase (MMP2), were commonly up-regulated (Table S1). In addition, Zeb1, one of the TFs capable of orchestrating an EMT, was commonly up-regulated.
Overexpression of Snail has been shown to down-regulate the expression of the cell-cycle protein cyclin D2 (CCND2) (37), and indeed we found that CCND2 was down-regulated in the EMT core signature. Cells undergoing an EMT are known to be resistant to apoptosis (38–41). Consistent with this finding, the proapo-ptosis gene BIK was down-regulated in all samples. In addition, genes with a Zeb1 binding site present in their promoters were enriched in the set of genes down-regulated by EMT, including the gene discoidin domain receptor 1 (DDR1), which encodes an RTK involved in E-cadherin localization and distinguishes basal A from basal B cell lines (42–45), and the gene follistatin (FST), which is a TGF-β antagonist (Tables S2andS3) (46–48).
Contributions of EMT-Inducing TFs to Breast Cancer. We next
at-tempted to use existing GESs of various types of breast cancer to uncover possible connections between the various TFs and breast cancer pathogenesis. We sought to understand the relatedness of individual EMT-inducers and their respective GESs to the gene-expression profiles with individual breast carcinomas in a large (244 patient) cohort of these tumors (49). The gene-expression profiles derived from many tumors displayed a high correlation to GESs derived from Gsc, Snail, Twist, and TGF-β1, but correlated less strongly with the signature derived from knocking down E-cadherin (Fig. 3B). As predicted from previous observations, tumors with a high correlation to the Snail GES also displayed a high correlation to the Twist GES. In addition, the gene-expression profiles derived from each tumor in this dataset tend to correlate with the GESs derived from more than one EMT in-ducer. Moreover, the expression signature of individual tumors did not make it possible to resolve between alternative mechanisms of EMT induction (i.e., Twist-, Snail-, TGF-β1-, or Gsc-induced EMT) in these breast cancers. Nonetheless, the GESs of individual EMT-inducers could be used to assay for the occurrence of an EMT in a breast tumor, even if the EMT was induced by a still-unknown inducer and activation of this transdifferentiation pro-gram occurred in only a minority of the cells within each tumor. Importantly, the possibility that stromal elements within the tumor samples rather than the carcinoma cells themselves resulted in the detection of a mesenchymal GES could not be discounted. Correlation of EMT Core Signature with the Basal B, Claudin-Low, and
Metaplastic Signatures.We also wished to determine how the EMT
core signature relates to the GESs of various subtypes of breast cancer. We compared the mean expression values of genes up- or
Fig. 3. Clustering of the individual EMT-inducer gene expression profiles based on their similarity to each other and to a large cohort of breast cancer patient gene-expression samples. (A) Heatmap of gene expression profiles in each sample. Values represent the log2 ratio over control. The diagram above each heatmap shows the similarities of EMT-inducer profiles to each other. (B) Heat map of correlations of each EMT-inducer profile with the gene expression profiles of patient tumors in a large cohort of breast cancer patients (49). Pearson correlation coefficients were calculated for all of the EMT-inducer–patient pairs and plotted as a heat map, in which red indicates a highly significant positive correlation, green indicates a highly significant negative correlation and black indicates a weak or absent correlation.
Taube et al. PNAS | August 31, 2010 | vol. 107 | no. 35 | 15451
CELL
down-regulated by an EMT against the GES of various breast cancer cell line subtypes (50) and found that the EMT core sig-nature most strongly correlated with the sigsig-nature derived from basal B cell lines (Fig. S4). This subtype is characterized by high vimentin expression and a stem cell-like expression profile (45, 50), similar to cells that have undergone an EMT; in contrast, the signatures derived from the basal A and luminal subtypes (45, 50) (Fig. S4) were not closely related to the EMT core signature.
We also measured the association of the EMT core signature to GESs derived from breast cancer subtypes defined by Hennessy et al. (23) and against the GES of a cohort of metaplastic tumors that had been previously analyzed using the same microarray platform (22). The up- and down-regulated genes from the EMT core signature were also significantly up- and down-regulated in both the metaplastic and claudin-low breast cancers, but not in other breast cancer subtypes (Fig. 4 A and B). This finding is consistent with recent reports that have linked low E-cadherin expression, indicative of passage through an EMT, to clinically encountered breast tumors of either the claudin-low or meta-plastic subtype (22, 23). Because passage through an EMT has also been linked with acquisition of stem cell characteristics (6), this suggests that the neoplastic cells in these tumors contain relatively high proportions of cancer stem cells.
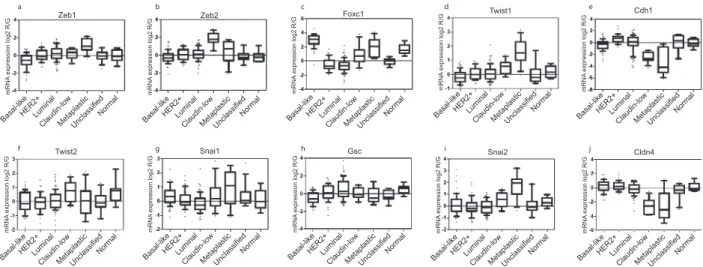
We also assayed the expression of the mRNAs encoding EMT-inducing TFs in breast cancers that had been classified into basal-like, HER2+, luminal, claudin-low, and metaplastic subtypes (22, 23). We found that the Snail, Slug, Zeb1, Twist1, and FOXC1 TFs were up-regulated in metaplastic tumors, and Zeb2 was most commonly up-regulated in claudin-low tumors (Fig. 5A). Both metaplastic and claudin-low tumor subtypes showed significant down-regulation of E-cadherin mRNA expression (CDH1) as well as claudin 4 (Fig. 5A). Although nearly all claudin-low tu-mors express high levels of Zeb2, only half of these tutu-mors con-comitantly expressed high levels of other EMT-inducing genes (Snail, Twist, FOXC1) (Fig. 5A). Strikingly, nearly all basal-like breast tumors, but not luminal or HER2+tumors, display consis-tently high expression of the EMT-inducer FOXC1, which is known to be associated with increased cell motility and invasion and decreased expression of E-cadherin (13) (Fig. 5A andFig. S5). Correlation of Expression of an EMT-Inducer with Patient Survival. We anticipated that tumors expressing the EMT-associated genes would exhibit a poorer survival than tumors not expressing the EMT-associated genes. For this reason, we performed clinical prediction analyses on the breast tumors in the Netherlands Cancer Institute (NKI) and University of North Carolina (UNC) databases (49, 51), using the EMT core signature. We found,
A
B
MetaplasticClaudin-low
LuminalBasal-likeHER2+Normal
s e n e G n w o D T M E s e n e G p U T M E Mean Expression Mean Expression
MetaplasticClaudin-lowLuminalBasal-like HER2+Normal 0 . 0 0 . 0 -0.2 -0.4 0.2 0.4 -0.2 -0.4 0.2 0.4 0.6 -0.6
Fig. 4. The core EMT signature correlates with metaplastic and claudin-low breast cancers. (A and B) Gene-expression data were plotted as box plots for the mean expression of the EMT-up genes (A) and the EMT-down genes (B) by subtype using the dataset from Hennessey et al. (23) with the addition of 12 metaplastic tumors. Subtypes were called as in Herschkowitz et al. (22). The list was derived using Significance Analysis of Microarrays and cut off at the top∼1,155 probes, 544 up and 611 down. Next, the genes were ex-tracted in the dataset and averaged in each tumor (up and down sepa-rately). The one-way ANOVA significance for each plot was P < 0.0001.
Fig. 5. EMT-inducing genes are up-regulated in metaplastic and claudin-low tumors and FOXC1 expression marks basal-like tumors and is a predictor of poor clinical outcome. (A) Data were extracted for EMT-related genes and samples were ordered by intrinsic subtype as in Herschkowitz et al. (22). The twelve metaplastic tumors from Hennessey et al. (23) were also included. (B) Patients from the NKI and UNC datasets were divided into high- and low-FOXC1 expressers and their survival was compared. The P value was generated using theχ2test of equality.
however, that expression by a tumor of the EMT core signature failed to predict patient survival.
We therefore pursued an alternative hypothesis: that expression of individual genes capable of inducing an EMT, rather than multigene signatures, could predict patient survival. Although the expression of Gsc, Snail, Twist, or TGF-β1 genes did not predict clinical outcome, high expression of the FOXC1 TF was indeed a powerful predictor of poor clinical outcome. Thus, those patients whose tumors exhibited high levels of FOXC1 gene expression showed a significantly poorer survival outcome (Fig. 5B). Al-though, FOXC1 was not induced by Gsc, Snail, Twist, or TGF-β1, expression of FOXC1 in immortalized, but nontumorigenic, hu-man mammary epithelial MCF12A cells has been shown to induce an EMT (8, 13).
Recent reports have linked expression of specific sets of genes to resistance to chemotherapy in breast cancer patients (52, 53). Using an M.D. Anderson Cancer Center study of response to neoadjuvant chemotherapy with paclitaxel, 5-fluorouracil, doxo-rubicin, and cyclophosphamide (54), we asked if tumor gene-expression profiles from patients that had a pathological complete response (pCR) correlated with the EMT core signature. We found that gene-expression profiles from patient tumors with pCR showed a negative correlation to the EMT core signature, whereas GESs from tumors from patients without pCR showed a slightly positive correlation to the EMT Core Signature (Fig. S6). Discussion
The EMT core signature that we have identified was generated by comparing the gene-expression changes that occurred by overexpressing either Gsc, Snail, Twist, or TGF-β1, or by re-ducing levels of E-cadherin. As we found, this signature is en-riched for genes containing the Zeb1 transcription factor-binding site near their transcription start sites (Table S2), and is most similar to GES from claudin-low and metaplastic breast cancers, as well as cancers without pCR.
Of note, a recent study demonstrated that the GES obtained from normal mammary epithelial stem cells is most similar to the claudin-low signature (55). Thisfinding is consistent with the presentfindings that the expression changes associated with the EMT-inducing TFs correlate most closely with the claudin-low and metaplastic breast cancers and with our earlier demonstration that passage through an EMT results in the acquisition of stem-cell characteristics (6).
Analysis of mRNA expression levels in cells overexpressing EMT-inducers reveals that Twist, Snail, and TGF-β1 each up-regulate expression of Foxc2, Zeb1, and Zeb2 (Fig. 2C). Fur-thermore, we demonstrate that Snail and Twist generate the most closely related GESs, and expression of TGF-β1, Gsc, and knock-down of E-cadherin generate quite distinct, less closely related signatures. Further exploration of the differences between these various GESs will likely yield insights into the mechanisms re-quired to activate the EMT program and those involved in main-taining the resulting mesenchymal/stem cell state.
Surprisingly, we were unable to observe any correlation between expression of genes in the EMT core signature and a poorer sur-vival outcome among breast cancer patients, despite the observa-tion that the EMT core signature correlates with metaplastic tumors, which are themselves linked with poorer patient survival (56). Interestingly, a recent report by Creighton et al. found that the GES derived from breast tumor-initiating cells was also enriched in claudin-low tumors, but likewise did not serve as a useful prog-nostic marker for clinical progression (53). However, they found that cell populations that survive conventional chemotherapeutic treatment are enriched for cells with EMT-associated mesenchy-mal and stem cell-associated tumor-initiating features (53). Addi-tionally, an association between EMT and chemotherapy resistance, rather than survival, is suggested by Farmer et al., who show that a stroma-related GES predicts shorter relapse-free
sur-vival among patients who received chemotherapy, but not among untreated breast cancer patients (52). Thesefindings are consistent with our data indicating that gene expression profiles of tumors from patients that responded to chemotherapy correlate negatively with the EMT core signature.
The use of total mRNA isolated from entire tumors for an-notation of the expression datasets may preclude detection of cells that have undergone EMT, as only a small proportion of the neoplastic cells in each tumor may exhibit an EMT phenotype. Poor-prognosis tumors may therefore harbor an insufficient num-ber of cells with a mesenchymal phenotype, so that there is no detectable effect on the gene-expression profile of the tumor as a whole. Accordingly, it may be necessary to collect tumor cells at the invasive edges of tumor cell islands and analyze these for the expression of the EMT-associated genes to gauge the true ma-lignant potential of the tumor as a whole.
Among the group of EMT-inducing genes studied here, FOXC1 expression most closely correlates with a poorer survival of the breast cancer patients in the NKI and UNC datasets (Fig. 5B). FOXC1 was highly expressed in metaplastic and basal-like breast cancer subtypes (Fig. 5A), for which highly effective treatments are not currently available. The closely related FOXC2 TF is known to play a key role in inducing an EMT, promoting metastasis and to be associated with aggressive basal-like breast cancers as studied using immunohistochemistry methods (14). However, expression of FOXC2 could not be correlated with survival in the cited microarray-based stratification of breast cancer patients, because the arrays used did not contain suitable FOXC2 probes. Given the consistently high levels of FOXC1 in basal-like and metaplastic breast cancer subtypes, the high levels of Zeb2 in claudin-low breast cancers, and the contribution of FOXC2 to basal-like breast cancer (14), these genes or the pathways that regulate these genes would seem to represent potential targets for the development of novel anticancer therapeutics.
Methods
RT-PCR. RNA was prepared from cultured cells by TRIzol extraction (Invi-trogen). Complementary DNA was synthesized using Moloney Murine Leu-kemia virus reverse transcriptase (Invitrogen). Relative quantification values were calculated using the ddCt method (57) and values were plotted with SD using GraphPad Prism v5.0 (GraphPad Software, Inc.).
Cell Culture. Immortalized HMLE were grown as previously described (58). MCF-7, SUM1315, and MDA-MB 231 cells were maintained according to ATCC instructions. The SUM159 cell line used for the study was developed from pleural effusions of breast cancer patients (59). SUM159 cells were cultured in F-12 Hams (Gibco) supplemented with 5% FBS (Tissue Culture Biologicals), 5 μg/mL of insulin, and 1 μg/mL hydrocortisone at 37 °C in 5% CO2.
Microarray Data Analysis and Deposition. Microarray data for HMLE shCDH1 and vector control were extracted from the Gene Expression Omnibus (GEO) database under the accession GSE9691. Microarray data for HMLE Gsc, Snail, Twist, TGF-β1, and vector control has been deposited in GEO under accession number GSE9691.
To compare gene expression among different EMT inducers, a heat map was generated using genes with at least a 2-fold change in at least one condition included in the clustering. To compare individual EMT signatures to breast cancer patient gene-expression data, a heat map showing correla-tions between gene-expression profiles of the EMT inducers and the gene-expression profiles of tumors from breast cancer patients in the UNC cohort was created. Pearson correlation coefficients were calculated between the gene-expression profiles of all EMT inducer-patient pairs.
Significance Analysis of Microarrays. The list of up- and down-regulated EMT genes was derived using Significance Analysis of Microarrays and cut off at the top 1,155 probes, 544 up and 611 down. The genes in the dataset were then extracted and averaged in each sample (up and down separately). ANOVA was performed and boxplot graphs were plotted with gene expression using GraphPad Prism v5.0 (GraphPad Software, Inc.). The one-way ANOVA for each plot was P< 0.0001.
Taube et al. PNAS | August 31, 2010 | vol. 107 | no. 35 | 15453
CELL
Survival Analysis. Patients from the NKI and UNC datasets were divided into high- and low-FOXC1 expressers and their survival was compared. The P value was generated using theχ2test of equality using the survival package in R
(www.r-project.org).
ACKNOWLEDGMENTS. We thank the members of the Mani laboratory for helpful discussions and Biogen for the SM16 (TGF-β inhibitor). Additional help with data analysis was provided by Dr. Keith Beggarly and Nianxiang
Zhang. This work was supported in part by the Broad Institute, Komen post-doctoral Fellowship PDF0707744 (to. J.I.H.), KG091219 (to B.G.H), National Institutes of Health Grant R01CA125109 (to P.T.R.), a pilot grant from the Dan L. Duncan Cancer Center (to J.M.R. and S.A.M.), a Research Trust award from M. D. Anderson Cancer Center, the V Foundations V scholar award, and Cancer Center Support Grant CA016672 (to S.A.M.), and National Institutes of Health Grant RO1 CA78461, the Breast Cancer Research Foundation, and Ludwig Center for Molecular Oncology at the Koch Institute (to R.A.W.).
1. Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin
Invest 119:1420–1428.
2. Gupta PB, et al. (2009) Identification of selective inhibitors of cancer stem cells by
high-throughput screening. Cell 138:645–659.
3. Yang J, et al. (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117:927–939.
4. Frixen UH, et al. (1991) E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol 113(1):173–185.
5. Sabbah M, et al. (2008) Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist Updat 11(4-5): 123–151.
6. Mani SA, et al. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704–715.
7. Morel AP, et al. (2008) Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 3:e2888.
8. Polyak K, Weinberg RA (2009) Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat Rev Cancer 9:265–273. 9. Hartwell KA, et al. (2006) The Spemann organizer gene, Goosecoid, promotes tumor
metastasis. Proc Natl Acad Sci USA 103:18969–18974.
10. Nieto MA, Sargent MG, Wilkinson DG, Cooke J (1994) Control of cell behavior during
vertebrate development by Slug, a zincfinger gene. Science 264:835–839.
11. Batlle E, et al. (2000) The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2(2):84–89.
12. Cano A, et al. (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2(2):76–83.
13. Bloushtain-Qimron N, et al. (2008) Cell type-specific DNA methylation patterns in the human breast. Proc Natl Acad Sci USA 105:14076–14081.
14. Mani SA, et al. (2007) Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci USA 104: 10069–10074.
15. Aigner K, et al. (2007) The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26: 6979–6988.
16. Gregory PA, Bracken CP, Bert AG, Goodall GJ (2008) MicroRNAs as regulators of
epithelial-mesenchymal transition. Cell Cycle 7:3112–3118.
17. Gregory PA, et al. (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10:593–601. 18. Park SM, Gaur AB, Lengyel E, Peter ME (2008) The miR-200 family determines the
epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22:894–907.
19. Hu Z, et al. (2006) The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics 7:96.
20. Sørlie T, et al. (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98:10869–10874. 21. Sorlie T, et al. (2003) Repeated observation of breast tumor subtypes in independent
gene expression data sets. Proc Natl Acad Sci USA 100:8418–8423.
22. Herschkowitz JI, et al. (2007) Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol 8(5):R76.
23. Hennessy BT, et al. (2009) Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res 69:4116–4124.
24. Onder TT, et al. (2008) Loss of E-cadherin promotes metastasis via multiple down-stream transcriptional pathways. Cancer Res 68:3645–3654.
25. Comijn J, et al. (2001) The two-handed E box binding zincfinger protein SIP1
downregulates E-cadherin and induces invasion. Mol Cell 7:1267–1278.
26. Vandewalle C, et al. (2005) SIP1/ZEB2 induces EMT by repressing genes of different
epithelial cell-cell junctions. Nucleic Acids Res 33:6566–6578.
27. Savagner P, Yamada KM, Thiery JP (1997) The zinc-finger protein slug causes de-smosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol 137:1403–1419.
28. Carver EA, Jiang R, Lan Y, Oram KF, Gridley T (2001) The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol Cell Biol 21:8184–8188. 29. Derycke LD, Bracke ME (2004) N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signalling. Int J Dev Biol 48:463–476. 30. Burdsal CA, Damsky CH, Pedersen RA (1993) The role of E-cadherin and integrins in
mesoderm differentiation and migration at the mammalian primitive streak. Devel-opment 118:829–844.
31. Hay ED (1995) An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 154(1):8–20.
32. Yu M, et al. (2009) A developmentally regulated inducer of EMT, LBX1, contributes to breast cancer progression. Genes Dev 23:1737–1742.
33. Bracken CP, et al. (2008) A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res 68: 7846–7854.
34. Bierie B, Moses HL (2006) Tumour microenvironment: TGFbeta: The molecular Jekyll and Hyde of cancer. Nat Rev Cancer 6:506–520.
35. Suzuki E, et al. (2007) A novel small-molecule inhibitor of transforming growth factor beta type I receptor kinase (SM16) inhibits murine mesothelioma tumor growth in vivo and prevents tumor recurrence after surgical resection. Cancer Res 67:2351–2359. 36. Ip YT, Park RE, Kosman D, Yazdanbakhsh K, Levine M (1992) Dorsal-twist interactions establish snail expression in the presumptive mesoderm of the Drosophila embryo. Genes Dev 6:1518–1530.
37. Vega S, et al. (2004) Snail blocks the cell cycle and confers resistance to cell death.
Genes Dev 18:1131–1143.
38. Vitali R, et al. (2008) Slug (SNAI2) down-regulation by RNA interference facilitates apoptosis and inhibits invasive growth in neuroblastoma preclinical models. Clin
Cancer Res 14:4622–4630.
39. Inoue A, et al. (2002) Slug, a highly conserved zincfinger transcriptional repressor,
protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2:279–288.
40. Roy HK, et al. (2004) Down-regulation of SNAIL suppresses MIN mouse tumorigenesis: Modulation of apoptosis, proliferation, and fractal dimension. Mol Cancer Ther 3: 1159–1165.
41. Sayan AE, et al. (2009) SIP1 protein protects cells from DNA damage-induced apoptosis and has independent prognostic value in bladder cancer. Proc Natl Acad Sci USA 106:14884–14889.
42. Eswaramoorthy R, et al. (2010) DDR1 regulates the stabilization of cell surface E-cadherin and E-E-cadherin-mediated cell aggregation. J Cell Physiol 224:387–397. 43. Wang CZ, Yeh YC, Tang MJ (2009) DDR1/E-cadherin complex regulates the activation
of DDR1 and cell spreading. Am J Physiol Cell Physiol 297:C419–C429.
44. Maeyama M, et al. (2008) Switching in discoid domain receptor expressions in SLUG-induced epithelial-mesenchymal transition. Cancer 113:2823–2831.
45. Blick T, et al. (2010) Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia 15:235–252.
46. Subramanian A, et al. (2005) Gene set enrichment analysis: A knowledge-based
approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA
102:15545–15550.
47. Mootha VK, et al. (2003) PGC-1alpha-responsive genes involved in oxidative phos-phorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273.
48. Nogai H, et al. (2008) Follistatin antagonizes transforming growth factor-beta3-induced epithelial-mesenchymal transition in vitro: Implications for murine palatal development supported by microarray analysis. Differentiation 76:404–416. 49. Hoadley KA, et al. (2007) EGFR associated expression profiles vary with breast tumor
subtype. BMC Genomics 8:258.
50. Neve RM, et al. (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10:515–527.
51. van de Vijver M (2005) Gene-expression profiling and the future of adjuvant therapy. Oncologist 10 (Suppl 2):30–34.
52. Farmer P, et al. (2009) A stroma-related gene signature predicts resistance to neo-adjuvant chemotherapy in breast cancer. Nat Med 15(1):68–74.
53. Creighton CJ, et al. (2009) Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA 106:13820– 13825.
54. Hess KR, et al. (2006) Pharmacogenomic predictor of sensitivity to preoperative
chemotherapy with paclitaxel andfluorouracil, doxorubicin, and cyclophosphamide
in breast cancer. J Clin Oncol 24:4236–4244.
55. Lim E, et al. (2009) kConFab (2009) Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med
15:907–913.
56. Luini A, et al. (2007) Metaplastic carcinoma of the breast, an unusual disease with worse prognosis: The experience of the European Institute of Oncology and review of the literature. Breast Cancer Res Treat 101:349–353.
57. Heid CA, Stevens J, Livak KJ, Williams PM (1996) Real time quantitative PCR. Genome Res 6:986–994.
58. Elenbaas B, et al. (2001) Human breast cancer cells generated by oncogenic trans-formation of primary mammary epithelial cells. Genes Dev 15(1):50–65.
59. Ethier SP (1996) Human breast cancer cell lines as models of growth regulation and disease progression. J Mammary Gland Biol Neoplasia 1(1):111–121.
Supporting Information
Taube et al. 10.1073/pnas.1004900107
SI Methods
Microarray Data Collection.We used 1μg of total RNA to prepare
complementary DNA (cDNA) using the Genechip HT One-Cycle cDNA synthesis Kit (Affymetrix 900687) and the GeneChip HT IVT Labeling Kit (Affymetrix 900688). Total RNA wasfirst re-verse transcribed using a T7-Oligo (dT) promoter primer. Fol-lowing RNase H-mediated second strand cDNA synthesis, the double stranded cDNA was purified and served as a template for in an in vitro transcription reaction. The in vitro transcription re-action was carried out in the presence of T7 RNA polymerase and a biotinylated nucleotide analog/ribonucleotide mix for cRNA amplification and biotin labeling. The biotinylated cRNA targets were then cleaned up, fragmented and hybridized to Affymetrix HT-HG U133 A peg arrays (Affymetrix 900751). The hybridiza-tion and subsequent washing and staining was performed on the Affymetrix GeneChip Array Station automation platform. Arrays with signal intensity< 100 failed because of high noise and high background. Samples with percent presents within the range of 40 to 60% of genes present and GAPDH andβ-actin ratios less than 3 passed and were incorporated in the analysis.
Pathological Complete-Response Analysis.Genes with at least 2-fold
up- or down-regulation in allfive epithelial-to-mesenchymal trans-ition (EMT) condtrans-itions were collected and their fold-changes were averaged to generate an EMT profile. Patient microarray data (1) were row-normalized to their median, so that each row had a me-dian of 0. Pearson correlation coefficients were calculated between the EMT profile and each patient gene expression profile. Average correlation values of patients annotated as“pCR” were compared with those annotated as“non-pCR” with a Welch t test.
Gene Set Enrichment Analysis.Gene set enrichment analysis was
performed using Genepattern (http://broad.mit.edu/cancer/soft-ware/genepattern/) (2, 3). The rank order of genes from human mammary epithelial (HMLE) cell lines in two biological states, mesenchymal versus epithelial controls, was compared with gene sets within the molecular signatures database and significant en-richments reported. For statistical strength of these enen-richments, gene set enrichment analysis uses family wise error rate to correct for multiple testing and false-discovery rate to reduce false positive reporting.
1. Hess KR, et al. (2006) Pharmacogenomic predictor of sensitivity to preoperative
chemotherapy with paclitaxel andfluorouracil, doxorubicin, and cyclophosphamide in
breast cancer. J Clin Oncol 24:4236–4244.
2. Subramanian A, et al. (2005) Gene set enrichment analysis: A knowledge-based
ap-proach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102:
15545–15550.
3. Mootha VK, et al. (2003) PGC-1alpha-responsive genes involved in oxidative phos-phorylation are coordinately downregulated in human diabetes. Nat Genet 34:267– 273. E-Cadherin Vimentin 0.00001 0.0001 0.001 0.01 0.1 1 10 100 1000 10000 100000 MCF-7 MDA MB 231 SUM 1315 HMLE Re la ti ve m RNA
Fig. S1. Expression of E-cadherin (CDH1) and Vimentin (VIM) in HMLE, MCF-7, MDA-MB 231, and SUM1315 cells. Quantitative RT-PCR was performed for indicated genes on RNA from the indicated cell lines and graphed relative to expression in empty vector control cells. Error bars represent the standard deviation from at least three separate measurements.
Goo secoi d Sna il Twis t TGF B1 0.1 1 10 100 1000 10000 HMLE-Gsc HMLE-Snail HMLE-Twist HMLE HMLE-TGFbeta1 Gene Re la ti v e m R NA
Fig. S2. Overexpression of retrovirally introduced transgenes. Quantitative RT-PCR was performed for indicated genes on RNA from the indicated cell lines and graphed relative to expression in empty vector control cells. Error bars represent the standard deviation from at least three separate measurements.
x x x x x x x x x x x
Relative Expression of EMT associated genes
CA9 COL 1A2 COL3 A1 DCN GRE M1 IGFB P2 KRT 5 SERP INB SPR R1A SPRR1 B 1.0 10- 6 1.0 10- 5 1.0 10- 4 1.0 10- 3 1.0 10- 2 1.0 10- 1 1.0 100 1.0 101 1.0 102 1.0 103 1.0 104 HMLE-Gsc HMLE-Snail HMLE-Twist HMLE HMLE-TGFb Re la tiv e m RNA
Fig. S3. Validation of gene-expression changes of select members of the EMT core signature. Quantitative RT-PCR was performed for indicated genes on RNA from the indicated cell lines and graphed relative to expression in empty vector control cells. Error bars represent the standard deviation from at least three separate measurements.
Fig. S4. The EMT core signature is enriched in basal B breast cancer cell lines. (A and B) Gene-expression data were plotted as box plots for the mean ex-pression of the EMT up genes and the EMT down genes by subtype using the dataset from Neve et al. (1). The list was derived using Significance Analysis of Microarrays and cut off at the top 1,155 genes, 544 up and 611 down. Next, the genes were extracted in the dataset and averaged in each tumor (up and down separately). The one-way ANOVA for the EMT up genes was P = 0.0001 and for the EMT down genes was P = 0.0075.
a b c d e f g h i j e k il-l a s a B G/ R 2 g ol n oi s s er p x e A N R m Cdh1 + 2 R E H l a n i m u L w o l-n i d u a l C c it s a l p a t e M d e ifi s s a l c n U l a m r o N e k il-l a s a B G/ R 2 g ol n oi s s er p x e A N R m Cldn4 + 2 R E H l a n i m u L w o l-n i d u a l C c it s a l p a t e M d e ifi s s a l c n U l a m r o N e k il-l a s a B G/ R 2 g ol n oi s s er p x e A N R m Snai2 + 2 R E H l a n i m u L w o l-n i d u a l C c it s a l p a t e M d e ifi s s a l c n U l a m r o N e k il-l a s a B G/ R 2 g ol n oi s s er p x e A N R m Twist1 + 2 R E H l a n i m u L w o l-n i d u a l C c it s a l p a t e M d e ifi s s a l c n U l a m r o N e k il-l a s a B G/ R 2 g ol n oi s s er p x e A N R m Foxc1 + 2 R E H l a n i m u L w o l-n i d u a l C c it s a l p a t e M d e ifi s s a l c n U l a m r o N e k il-l a s a B G/ R 2 g ol n oi s s er p x e A N R m Zeb2 + 2 R E H l a n i m u L w o l-n i d u a l C c it s a l p a t e M d e ifi s s a l c n U l a m r o N e k il-l a s a B G/ R 2 g ol n oi s s er p x e A N R m Zeb1 + 2 R E H l a n i m u L w o l-n i d u a l C c it s a l p a t e M d e ifi s s a l c n U l a m r o N e k il-l a s a B G/ R 2 g ol n oi s s er p x e A N R m Gsc + 2 R E H l a n i m u L w o l-n i d u a l C c it s a l p a t e M d e ifi s s a l c n U l a m r o N e k il-l a s a B G/ R 2 g ol n oi s s er p x e A N R m Twist2 + 2 R E H l a n i m u L w o l-n i d u a l C c it s a l p a t e M d e ifi s s a l c n U l a m r o N e k il-l a s a B G/ R 2 g ol n oi s s er p x e A N R m Snai1 + 2 R E H l a n i m u L w o l-n i d u a l C c it s a l p a t e M d e ifi s s a l c n U l a m r o N
Fig. S5. Gene-expression data were plotted as box plots for EMT-related genes and samples were classified by intrinsic subtype as in Herschkowitz et al. (1). The number of samples in each class was: basal-like = 58, HER2+= 44, luminal = 93, claudin-low = 13, unclassified = 15, and normal = 9. The 12 metaplastic
tumors from Hennessey et al. (2) were also included.
pCR * non-pCR -0 .4 -0 .2 0 .0 0 .2 0 .4 Co rre la ti o n w it h E M T si g na tur e
Fig. S6. The EMT core signature negatively correlates with gene expression profiles of patients with pathological complete response (pCR). Patients from Hess et al. (1) were stratified based on their pCR status and their gene-expression profiles were correlated to the EMT core signature. A significant difference between the two populations was determined by a Welch t test. *P = 0.005.
1. Herschkowitz JI, et al. (2007) Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol 8:R76.
2. Hennessy BT, et al. (2009) Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res 69:
4116–4124.
Table S1. Genes in the EMT core signature
Table S1 (DOCX)
Genes up- or down-regulated at least 2-fold by overexpression of Twist, Snail, Gsc, TGF-β1, and by down-regulation of E-cadherin are listed with fold-changes relative to control cells.
Table S2. Gene set enrichment analysis of the EMT core signature
Table S2 (DOCX)
Using gene set enrichment analysis, the rank order of genes from HMLE cell lines in two biological states, mesenchymal versus epithelial controls, was compared with gene sets within the molecular signatures database and significant enrichments reported. FDR, false discovery rate; FWER, family-wise error rate.
Table S3. Complete list of EMT-down-regulated genes containing putative, conserved Zeb1 binding elements in their promoters
Table S3 (DOCX)
Gene set enrichment analysis was performed comparing HMLE cells induced to undergo EMT with HMLE control cells. The gene set V$AREB6_01 (Zeb1 binding sites) was found to be significantly enriched in genes down-regulated when cells underwent EMT. ES, enrichment score.