Development of a Continuous-Flow Synthesis of Neostigmine Methylsulfate and Studies Toward a Continuous-Flow Synthesis of Lisinopril
By Liam P. Kelly
B.S. Chemistry, summa cum laude with honors Florida State University, 2014
Submitted to the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of
DOCTOR OF PHILOSOPHY IN CHEMISTRY at the
Massachusetts Institute of Technology June 2019
C 2019 Massachusetts Institute of Technology All rights reserved.
Signature redacted
Signature of Author: Department of Chemistry May 16, 2019Signature redacted
Certified by: Timothy F. Jamison Robert R. Taylor Professor of Chemistry Thesis SupervisorSignature redacted
Accepted by:
Robert W. Field
MASSACHUSETTS INSTITUTE Haslam and Dewey Professor of Chemistry OF TECHNOLOGY
This doctoral thesis has been examined by a committee in the Department of Chemistry as follows:
Signature redacted
Professor Stephen L . B uchw ald _Th s C m t e a
S Thesis Committee Chair Camille Dreyfus Professor of Chemistry
Signature redacted
Professor Timothy F. Jamison
Thesis Supervisor QR bert R. Taylor Professor of Chemistry
Signature redacted
Professor Klavs F. JensenThesis Committee Member Warren K Lewis Professor of Chemical Engineering, and Professor of Materials Science and Engineering
Development of a Continuous-Flow Synthesis of
Neostigmine Methylsulfate and Studies Toward a
Continuous-Flow Synthesis of Lisinopril
by Liam P. Kelly
Submitted to the Department of Chemistry on May 17, 2019
in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Chemistry ABSTRACT OH N 1.2 " -Solid handling '-- 0 S1.4 * Purification * Water content In-line evaporation 0 N N+ MeSO4
-neostigmine methyl sulfate
93,600 doses/day
46.8 g/day
15 min. average total residence time
Herein, we describe the development of a continuous flow synthesis of neostigmine methyl sulfate, an acetylcholinesterase inhibitor on the WHO list of essential medicines, and the transfer of the synthesis into a next-generation reconfigurable frame developed by our collaborators. Starting from 3-dimethylaminophenol, the synthesis provides a throughput of approximately 46.8 g/day (or 93,600 doses/day) of crude neostigmine methyl sulfate. The synthesis also showcases a prototype in-line evaporation unit that operates without any added carrier gas.
Dr. Christina Dai performed early screening of lithium bases. Dr. Yuqing Cui and Dr. Naomi Briggs developed the downstream purification sequence. Dr. Nopphon Weeranoppanant developed the in-line evaporator and, along with Dr. Dale Thomas, assisted with performing the synthesis within their developed frame. Liam P. Kelly developed the continuous synthesis
ABSTRACT + O + -CN H 0 + 0 OR OR 2 0 0 0 PGN O + CT OH 0 NH2 NH 2 Peptide coupling
HO 00 OH via activated ester or
N N-carboxyanhyd ride N N N3 I H N 0HO 0 0 0 OH Lisinopril0
2.1 Key C-N bond formation
via Strecker, aza-Michael, alkylation, or reductive amination
Lisinopril is a member of a large family of ACE inhibitors generally known as N-carboxyethyl dipeptides. Of this family, lisinopril is the most commonly prescribed. All known routes to lisinopril require isolation of several synthetic intermediates and protecting group manipulations, thus, development of an efficient continuous synthesis would provide great benefit. Herein we describe our investigation of several routes to generate intermediates of lisinopril with the end goal of a fully continuous synthesis, high material throughput, and minimal protecting group manipulations.
Liam P. Kelly performed all work described within this chapter.
Thesis Supervisor: Timothy F. Jamison
Title: Robert R. Taylor Professor of Chemistry
Portions of this thesis (Chapter 1) have been published in the following article and have been reprinted and/or adapted with permission from their respective publishers.
Advanced Continuous Flow Platform for On-Demand Pharmaceutical Manufacturing Zhang, P.; Weeranoppanant, N.; Thomas, D. A.; Tahara, K.; Stelzer, T.; Russell, M. G.; O'Mahony, M.; Myerson, A. S.; Lin, H.; Kelly, L. P.; Jensen, K. F.; Jamison, T. F.; Dai, C.; Cui, Y.; Briggs, N.; Beingessner, R. L.; Adamo, A. Chem Eur. J. 2018, 24, 2776-2784. DOI:
10.1002/chem.201706004. Copyright C 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. 0 PGN OH NH 2 PGN
Acknowledgements
My time at MIT has been the most challenging experience in my life. It has also been incredibly rewarding, both in terms of growth as a scientist and the personal connections I have made since being here. I would like to take a moment to thank the many people who have supported me in my journey leading me to MIT and through my graduate studies.
My realization that I was interested in chemistry as a career occurred during my AP chemistry class. My teacher, Mrs. Brenda Collias, provided an engaging environment to learn chemistry, and her passion for the subject was impossible to miss. Her encouragement, chemical demonstrations, and teaching shaped my desire to pursue a degree in chemistry.
At Florida State University, the chemistry department was filled with scientists passionate about chemical research. Dr. Mark Kearley and Dr. Ken Goldsby were my first contact with chemistry faculty at FSU, and were both incredible lecturers that I was fortunate to learn from early on in my career. I later took organic chemistry with Professor D. Tyler McQuade, whose instruction sparked my curiosity in the area for further study. Tyler later encouraged me to apply to MIT, and I am forever grateful for his recommendation and assistance in getting here. Thank you Tyler, for encouraging me as a young chemist, Dr. Paul Fleming, for recruiting me to the lab and being a supportive colleague, and Dr. Suzanne Opalka and Dr. Ashley Longstreet for your guidance and for introducing me to flow chemistry.
I owe my deepest gratitude to Professor Tim Jamison for accepting me into his lab and serving as a fantastic mentor during my time here at MIT. Tim's ability to generate ideas, store and recall extensive chemical knowledge, guide our research, and perform all other duties as both advisor and department head speaks to a level of efficiency and scientific prowess that I will forever strive to achieve. Tim, thank you for the incredible guidance and insight you have given me on all of the hurdles encountered during my graduate studies, and further for your trust in me to carry out the research. The lab environment that you foster has been invaluable in terms of scientific creativity, problem solving, and support.
I would also like to thank Professor Jeff Van Humbeck, and later, Professor Steve Buchwald, for serving as the chair of my thesis committee. The discussions we had regarding my chemistry throughout my PhD were helpful and insightful, and I am extremely thankful for Steve's willingness to step in as my thesis committee chair. I must also thank Professor Klavs Jensen for his collaboration, our discussions regarding the engineering aspects of flow chemistry, and for serving on my thesis committee. To Steve and Klavs, I am indebted to you for your extreme graciousness, patience, and flexibility as I wrote and edited this thesis.
I have had the pleasure of working with many talented and ambitious scientists throughout my time in the Jamison group, all of whom I felt I could call friends as well as colleagues. Dr. Jie Wu and Dr. Christina Dai were instrumental in my training when I first joined the Jamison lab, and Christina further mentored me through the completion of my synthesis of neostigmine. Thank you, Jie and Christina, for your guidance. Thank you to Dr. Rob Hicklin, for your mentorship during the development of routes toward lisinopril, for your practical view on chemistry, and for leading our lab volleyball team to back-to-back championships. I had the pleasure of working with several other postdoctoral researchers who gave great advice and shaped my development as a chemist. Thank you to Dr. Evan Styduhar,
work in and discuss science. Thank you to Dr. Victor Schultz, for your advice along the way and for bonding over hot sauce and peppers. To Grace Russell, thank you for your hard work, ideas, and flow chemistry expertise as we made it through the PoD project. To Chris Breen, thank you for all of the discussions on ACE inhibitors and flow chemistry. To my classmates, Dr. Jessica Weber, Dr. Tho Tran, Hyowon Seo, Dr. Kelley Danahy, and Amanda Wicker, thank you for your friendship and support both in and outside of the lab. I really enjoyed the times that we could all take a moment to grab brunch, even if you never forgive me for not wanting a matching track suit. Thank you to the graduate students in the lab that I looked up to, including Dr. Eric Standley, Dr. Sarah Tasker, Dr. Toma Halkina, Dr. Elizabeth Kelley, Dr. Kurt Armbrust, Dr. Andy McTeague, Dr. Laurel Heckman, and Dr. Charles Ocampo. Thank you to the other graduate students who have made the lab an enjoyable workplace, including Katie McGeough, Sarah Jane Mear, Grace Ahlqvist, and Corshai Williams. Finally, thank you to Dr. Rachel Beingessner. Your feedback has been an incredible resource for the entire lab. Thank you for all of your efforts to make us better scientists, and for sharing your writing expertise with us. I will miss sitting right around the corner from you.
Thank you to the PoD crew, including Dr. Nopphon Weeranoppanant, Dr. Dale Thomas, Dr. Luke Rogers, and Dr. Naomi Briggs, for all the support and ideas throughout our time on the PoD project. It was a pleasure getting to work with you all.
Thank you to those in the DCIF that have provided guidance and friendship throughout my time as a student instrument steward. It was a pleasure to work with Dr. Jeff Simpson, Anne Rachupka, and Li Li while they were at MIT. Thank you, Li Li, for your trust and training on the various MS instruments. It was wonderful to get to work so closely with you. Thanks also to Dr. Gang Liu for your invaluable knowledge of archaic instrumentation. To the current facility staff, Dr. Walt Massefski, Dr. Bruce Adams, John Grimes, and Dr. Mohanraja Kumar, thanks for all your efforts in improving the facilities. Walt and Bruce, thank you for all of your insightful conversations, practical advice, NMR wizardry, and for all of the donuts and bagels on those Thursday mornings. John, thank you for the great conversation and the books you lent me, which helped me rekindle my love of reading. Mohan, thank you for your help and guidance as we worked with the MS instrumentation. It's been great to get to work with you. To all of the student instrument stewards, I enjoyed all of the time we have spent in the DCIF. Best of luck to all of you.
To my friends outside of the lab, including Julian Cooper, Chase Olsson, Dr. Vera Schr6der, Dr. Phil Calabretta, Lindsey Orgren, Dr. Ken Kawamoto, and Gihan Hewage, your friendship has been invaluable. Thank you for all of your kindness and support.
Last, but certainly not least, I want to thank my family for their love and continued support. Thank you to my parents, Bruce and Amy Kelly. From a young age, you fostered my scientific interests by encouraging critical thinking, reading, and providing space for creativity, and for that I am grateful. I would also like to thank my late grandfather, Dr. Porter F. Crawford, who provided me with his old books of science experiments and sparked my interest in the scientific world. Thank you, Erin, for being a caring sister. To my aunt and uncle, Craig and Tom, thank you for being my home away from home when I moved to Boston. The time we've spent together has done wonders for my mental health. Finally, thank you to my incredible wife, Danielle, for her patience, love, and support during these five years. You have been my inspiration to keep improving as I have watched you grow and improve as a teacher. Thank you for sticking with me through all of the stress, the late nights, and the happy times too.
Table of Contents
A b b rev iatio n s ... 11
Preface. Continuous API Synthesis Using Flow Chemistry... 13
Chapter 1. Development of a Continuous Synthesis of Neostigmine Methyl Sulfate in a Reconfigurable Frame ... 19
A . In tro d u ction ... 2 0 B. Synthetic Route Investigation ... 22
C. Route Development for Continuous Synthesis of Neostigmine... 23
D . In-L ine E vaporator ... 35
E. Continuous Synthesis of Neostigmine in the Compact Frame... 41
C hapter 1 Experim ental Section ... 43
A . M aterials and M ethods... 44
B. Batch Synthesis of Materials... 45
C. Setup and Construction of Tube-in-Tube Reactor ... 49
D. Continuous Synthesis of Neostigmine Methyl Sulfate ... 51
E . 'H and 13C N M R Spectra ... 55
Chapter 2. Studies Directed Toward the Development of a Continuous Synthesis of L isinopril... 65
A . In tro d u ction ... 6 6 B. Development of an Imine-Mediated Amino Acid Coupling... 71
C. Further Elaboration of the Imine-Mediated Coupling Route ... 76
D. Development of an aza-Michael Route for C-N Bond Formation ... 80
E. Route Based on N,N"-Protected Lysine... 82
I. Conclusions ... 100
Chapter 2 Experim ental Section ... 103
A . M aterials, M ethods, and General Considerations ... 104
B. Synthesis of Starting M aterials ... 105
C. Im ine-M ediated Coupling in Flow ... 115
D . Optim ization of aza-M ichael Reaction ... 119
E. Synthesis U sing Na,Nc-Protected Lysine ... 123
F. N-Carboxyanhydride Coupling Optim ization... 127
Abbreviations
API Active pharmaceutical ingredient
ASHP American Society of Health-System Pharmacists Boc tert-butoxycarbonyl Bn Benzyl BPR Backpressure regulator Bu Butyl Cbz Carboxybenzyl CDI 1,1 '-Carbonyldiimidazole
CSTR Continuously stirred tank reactor
d days
DABCO 1.,4-Diazabicyclo[2.2.2] octane DART Direct analysis in real time
DBU 1.,8-Diazabicyclo[5.4.0]undec-7-ene
DCM Dichloromethane
DIPEA N,N-Diisopropylethylamine
DMF Dimethylformamide
DMSO Dimethyl sulfoxide d.r. Diastereomeric ratio
ee Enantiomeric excess
ESI Electrospray ionization
Et Ethyl
FDA Food and Drug Administration Fmoc Fluorenylmethoxycarbonyl
FT Fourier transform
g Grams
GCMS Gas chromatography mass spectrometry
HATU 1-[Bis(dimethylamino)methylene]-1 H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
HoPhe Homophenylalanine
hr Hour
HMDS Hexamethyldisilazide
HRMS High resolution mass spectrometry ICR Ion cyclotron resonance
i.d. Inner diameter
IR Infrared
LCMS Liquid chromatography mass spectrometry
Lys Lysine
Me Methyl
MeCN Acetonitrile
mg Milligrams
PoD Pharmacy on demand PEEK Polyether ether ketone PFA Perfluoroalkoxy alkane
Ph Phenyl
Phth Phthaloyl
Pro Proline
psi Pounds per square inch p-TsOH para-Toluenesulfonic acid
T3P Propylphosphonic anhydride Tf Trifluoromethanesulfonyl THF Tetrahydrofuran TLC Thin-layer chromatography TMG N,N,N',N'-Tetramethylguanidine tR Residence time Ts p-Toluenesulfonyl
Preface
-
Continuous API Synthesis Using Flow Chemistry
Active pharmaceutical ingredient (API) manufacturing takes place on a massive scale, driving an industry that continues to increase in value. As life expectancy increases, and the major disease burden of the world continues to shift toward chronic diseases over acute diseases, the medicinal needs of the world are expected to continue growing.' Due to the large scale of the industry, minor improvements to efficiency in producing a drug substance can provide significant overall cost, material, and energy savings. One way that we look to improve the efficiency of the synthesis of APIs is through the adoption of continuous manufacturing over traditional batch processes. Logistically, batch processing can be a root cause of drug shortages, due to supply chain interruptions or variations in the quality control of an API. In fact, the U.S. FDA has reported well over 200 cases of such shortages per year during 2011-2014.2 Furthermore, a roundtable meeting held by the ASHP in 2017 reaffirmed that although newly reported shortages are fewer than at the height of the shortage crisis in 2012, the causes do not appear to have changed. At this roundtable, the members note that "drug shortages are largely the result of quality problems during the manufacturing process, which give rise to a halt in production in order to address the problem."3 This is compounded by the evidence that, in many countries, the storage capacity and conditions are inadequate for full access to the medicines required by their populations.4 To address this issue, one might envision that a continuous manufacturing process may alleviate these logistical burdens, where a fully-integrated system would remove the need for storage of intermediates and allow for continuous monitoring for quality control.5 With further development, a system capable of
World Health Organization. The World Medicines Situation 2011 - Global Health Trends: Global Burden of Disease and Pharmaceutical Needs. http://apps.who.int/medicinedocs/documents/s20036en/s20036en.pdf
(accessed Apr 24, 2019).
2 Food and Drug Administration. Strategic Plan for Preventing and Mitigating Drug Shortages (October 2013). www.fda.gov/downloads/Drugs/DrugSafety/DrugShortages/UCM372566.pdf (accessed Apr 24, 2019).
' ASHP. Drug Shortages Roundtable: Minimizing Impact on Patient Care (November 6, 2017). https://www.ashp.org/-/media/assets/drug-shortages/docs/drug-shortages-nov-20 1
7-shortage-meeting-providing on-demand synthesis could also offer relief in cases of epidemic where sudden changes in demand or need for treatment occurs.
In addition to the aforementioned logistical advantages, continuous manufacturing can offer significant improvements to efficiency or selectivity over batch generation of APIs6. For
many batch processes, significant challenges in reactor and reaction engineering must be overcome during scale-up.7 When appropriate considerations are taken, the advantages of continuous processing can help develop a robust, scalable route.8 In brief, some of the
significant advantages are as follows: efficient thermal or mass transfer,9 the ability to use
hazardous and/or exothermic reagents,10 access to extreme temperature or operation windows,"' and improved irradiation of the reaction mixture. The rapid thermal equilibrium attained within the small flow channel suppresses the formation of hot spots and temperature gradients which can result in decreased selectivity. Additionally, this fine thermal control allows for the use of significantly exothermic reagents with minimal concern for runaway reactions.'2 Mass transfer is also enhanced, particularly so with biphasic plug flow due to
internal circulation and high surface area:volume ratios associated with each plug.'3 Mixing can further be enhanced in these systems through the use of static mixers within the reaction channel. 4 The small active volume of a reactor (as compared to the total volume of material
processed) allows for the safe use of hazardous reagents." Furthermore, direct telescoping of a reaction means that a hazardous intermediate can be generated and consumed fully, preventing accumulation of a dangerous quantity. The compact size and contained nature of these systems also allow for safer access to extreme temperatures and pressures with minimal concern regarding reactor breaches. With high pressures tolerated, the use of gaseous reagents
6 Morse, P. D.; Beingessner, R. L.; Jamison, T. F. Isr. J. Chem. 2017, 57 (3-4), 218-227.
7 Caygill, G.; Zanfir, M.; Gavriilidis, A. Org. Process Res. Dev. 2006, 10 (3), 539-552.
' (a) Hartman, R. L.; McMullen, J. P.; Jensen, K. F. Angew. Chem. Int. Ed Engl. 2011, 50 (33), 7502-7519; (b) Plutschack, M. B.; Pieber, B.; Gilmore, K.; Seeberger, P. H. Chem. Rev. 2017, 117 (18), 11796-11893.
9 Wang, K.; Luo, G. Chem. Eng. Sci. 2017, 169, 18-33.
1 Gutmann, B.; Cantillo, D.; Kappe, C. 0. Angew. Chemie Int. Ed. 2015, 54 (23), 6688-6728.
"Hessel, V.; Kralisch, D.; Kockmann, N.; No61, T.; Wang, Q. ChemSusChem 2013, 6 (5), 746-789.
12 Gemoets, H. P. L.; Su, Y.; Shang, M.; Hessel, V.; Luque, R.; No61, T. Chem. Soc. Rev. 2016, 45 (1), 83-117. 13 Wang, K.; Li, L.; Xie, P.; Luo, G. React. Chem. Eng. 2017, 2 (5), 611-627.
" Lvesque, F.; Rogus, N. J.; Spencer, G.; Grigorov, P.; McMullen, J. P.; Thaisrivongs, D. A.; Davies, 1. W.; Naber, J. R. Org. Process Res. Dev. 2018, 22 (8), 1015-1021.
"5 Kockmann, N.; Thende, P.; Fleischer-Trebes, C.; Laudadio, G.; No61, T. React. Chem. Eng. 2017, 2 (3), 258-280.
is also operationally simple in flow, through plug flow or other means.'6 Finally, the short path length through the reaction channel also benefits photochemical17 or microwave-heated 8 reactions due to more effective irradiation of the reaction stream.
Figure P-1. Schematic representations of a selection of components commonly used in continuous synthesis.
Coiled PFA Tubing Reactor In-line phase separator
7
[7
-*- - -- Organic or gas (permeate) Aqueous (retained phase)T-mixer Y-mixer Cross mixer
T- *Y
Syringe Pump BPR Packed Bed
Although there are many logistical and chemical advantages accessible by the adoption of flow chemistry, appropriate considerations should be taken prior to implementation. One
such consideration, the solubility of reagents, products, and byproducts, can be critical for route selection and development. Unchecked solid formation within a reactor can lead to clogging or system instability. However, if alternative soluble reagents are not feasible, or if the goal is to generate a solid product, principles and solutions for solid handling in flow have been discussed at length.19 Specific examples include crystal seeding21 to induce controllable
16 (a) Brzozowski, M.; O'Brien, M.; Ley, S. V; Polyzos, A. Acc. Chem. Res. 2015, 48 (2), 349-362; (b) Yang, L.; Jensen, K. F. Org. Process Res. Dev. 2013, 17 (6), 927-933; (c) Mallia, C. J.; Baxendale, I. R. Org. Process Res. Dev. 2016, 20 (2), 327-360; (d) Mo, Y.; Imbrogno, J.; Zhang, H.; Jensen, K. F. Green Chem. 2018, 20 (16), 3867-3874.
10276-precipitation or the use of ultrasonic agitation2 1 to evenly suspend the precipitate. If the
addition of solid reagents or starting materials is desired, CSTRs have been demonstrated on multiple occasions for such reactions on pilot- to production-scale.2 2 Further considerations
are required when performing multistep synthesis in flow. In a telescoped flow system, all material added to a reaction stream and any generated products or byproducts will come in contact with the material infused further downstream.23 Thus, analysis of the chemical incompatibilities that could arise within the system is required. These challenges have led to the development of many technologies to enable further increases in synthetic complexity, including solid-supported reagents2 4 and continuous liquid-liquid2 5 or gas-liquid26 separators
which can effectively compartmentalize a specific step. Cost and design of reactors is also a consideration for the adoption of a continuous process. Recently, several methods for 3D-printing reactors from stainless steel27 and thermoplastics 28 have been developed to address these issues. For reactions that are extremely sensitive to scale, strategies for splitting flow in a "numbering up" approach have been developed29 to access increased throughput without substantial equipment requirements. Although many specialized reactor designs have been developed, it should be noted that the laboratory-scale experiments described herein utilize
2 1
(a) Hartman, R. L.; Naber, J. R.; Zaborenko, N.; Buchwald, S. L.; Jensen, K. F. Org. Process Res. Dev. 2010,
14 (12), 1347-1357; (b) Bddard, A.-C.; Longstreet, A. R.; Britton, J.; Wang, Y.; Moriguchi, H.; Hicklin, R. W.; Green, W. H.; Jamison, T. F. Bioorg. Med. Chem. 2017, 25 (23), 6233-6241.
22 (a) Klokov, B. A. Org. Process Res. Dev. 2001, 5 (3), 234-240; (b) Kopach, M. E.; Roberts, D. J.; Johnson,
M. D.; McClary Groh, J.; Adler, J. J.; Schafer, J. P.; Kobierski, M. E.; Trankle, W. G. Green Chem. 2012, 14
(5), 1524-1536; (c) Wong, S.-W.; Changi, S. M.; Shields, R.; Bell, W.; McGarvey, B.; Johnson, M. D.; Sun,
W.-M.; Braden, T. M.; Kopach, M. E.; Spencer, R. D.; Flanagan, G.; Murray, M. Org. Process Res. Dev. 2016,
20 (2), 540-550.
" Webb, D.; Jamison, T. F. Chem. Sci. 2010, 1 (6), 675-680.
24 (a) Ley, S. V; Baxendale, I. R.; Bream, R. N.; Jackson, P. S.; Leach, A. G.; Longbottom, D. A.; Nesi, M.;
Scott, J. S.; Storer, R. I.; Taylor, S. J. J Chem. Soc. Perkin Trans. 1 2000, No. 23, 3815-4195; (b) Hodge, P. Curr. Opin. Chem. Biol. 2003, 7 (3), 362-373; (c) Maier, M.; Radtke, C. P.; Hubbuch, J.; Niemeyer, C. M.;
Rabe, K. S. Angew. Chemie Int. Ed. 2018, 57 (19), 5539-5543.
21 (a) Adamo, A.; Heider, P. L.; Weeranoppanant, N.; Jensen, K. F. Ind. Eng. Chem. Res. 2013, 52 (31),
10802-10808; (b) O'Brien, M.; Koos, P.; Browne, D. L.; Ley, S. V. Org. Biomol. Chem. 2012, 10 (35), 7031; (c) Vural
Giirsel, I.; NoEl, T.; Wang, Q.; Hessel, V. Green Chem. 2015, 17 (4), 2012-2026.
26 Wu, J.; Yang, X.; He, Z.; Mao, X.; Hatton, T. A.; Jamison, T. F. Angew. Chemie -Int. Ed. 2014, 53 (32),
8416-8420.
27 Gutmann, B.; K6ckinger, M.; Glotz, G.; Ciaglia, T.; Slama, E.; Zadravec, M.; Pfanner, S.; Maier, M. C.;
Gruber-W6lfler, H.; Oliver Kappe, C. React. Chem. Eng. 2017, 2 (6), 919-927.
21 (a) Neumaier, J. M.; Madani, A.; Klein, T.; Ziegler, T. Beilstein J Org. Chem. 2019, 15, 558-566;(b) Rao, Z.
X.; Patel, B.; Monaco, A.; Cao, Z. J.; Barniol-Xicota, M.; Pichon, F.; Ladlow, M.; Hilton, S. T. European J
Org. Chem. 2017, 2017 (44), 6499-6504.
2
1(a) Iwasaki, T.; Kawano, N.; Yoshida, J. Org. Process Res. Dev. 2006, 10 (6), 1126-1131; (b) Nagaki, A.;
Hirose, K.; Tonomura, 0.; Taniguchi, S.; Taga, T.; Hasebe, S.; Ishizuka, N.; Yoshida, J. Org. Process Res. Dev.
commercially available PFA tubing with an i.d. of 0.02 - 0.063 in and an o.d. of 1/16 or 1/8 in. These reactors represent a cheap, simple, and customizable method for construction of reactors. Though simple, this style of reactor has even been used to screen reaction conditions
in parallel through the use of segmented flow,30 providing further complementarity to traditional batch chemistry.
The research herein describes collaborative efforts to further advance the field of multistep flow synthesis. Where the commodity chemical and petrochemical industries perform their operations in a continuous manner, pharmaceutical manufacturing traditionally operates using batch processes. In part, this is due to the complexity of the APIs that are synthesized. As advances are made through development of new enabling technologies3' and adoption of modular32 or automated5 33 processes, we expect the adoption of continuous synthesis to continue growing. Advances to the field thus far have led to a slow, but steadily growing, adoption of continuous processes for API synthesis.34
Previous research with colleagues at MIT led to the development of a continuous automated system for manufacturing of aliskiren hemifumarate.35 This shipping container-sized unit was capable of performing all of the steps needed to continuously produce tablets of the API-multistep chemical reactions, separations, crystallizations, drying, formulation, and tablet production. This work allowed for the identification of areas for further advances in continuous API manufacturing, which culminated in the design of a reconfigurable system the size of a refrigerator capable of synthesizing multiple APIs. 36 This first-generation system was able to produce hundreds to thousands of liquid doses of diphenhydramine hydrochloride, diazcpam, lidocaine hydrochloride, and fluoxetine hydrochloride, each from simple starting
3 Weiberth, F. J.; Powers, M. R.; Gallin, C.; McDonald, D. Org. Process Res. Dev. 2018, 22 (4), 512-519.
Ley, S. V; Fitzpatrick, D. E.; Ingham, R. J.; Myers, R. M. Angew. Chemie Ini. Ed. 2015, 54 (11), 3449-3464.
32 Ghislieri, D.; Gilmore, K.; Seeberger, P. H. Angew. Chemie Int. Ed. 2014, 678-682.
131 B6dard, A.; Adamo, A.; Aroh, K. C.; Russell, M. G.; Bedermann, A. A.; Torosian, J.; Yue, B.; Jensen, K. F.; Jamison, T. F. Science 2018, 361 (6408), 1220-1225.
3 (a) McWilliams, J. C.; Allian, A. D.; Opalka, S. M.; May, S. A.; Journet, M.; Braden, T. M. Org. Process Res. Dev. 2018, 22 (9), 1143-1166; (b) May, S. A. J. Flow Chem. 2017, 7 (3-4), 137-145; (c) Malet-Sanz, L.;
Susanne, F. J Med. Chem. 2012, 55 (9), 4062-4098.
materials. It also served as a proof-of-principle for an on-demand API manufacturing platform that could be replicated and operated by an end user, potentially offering a solution to the critical drug shortages that can occur as a result of batch manufacturing. Herein referred to as the Pharmacy on Demand, or "PoD", the system underwent development to further generations as synthetic complexity and manufacturing capabilities were increased, while also striving for efficiency and a smaller footprint. During the course of the project, the PoD team consisted of the upstream synthesis team in the Jamison research group, the systems development team in the Jensen research group, and the downstream purification and formulation team in the Myerson research group. Through this collaboration, we have been able to further advance the capabilities of multistep flow synthesis of APIs.
Figure P-2. An overview of the collaborative structure of the PoD project.
Upstream Systems Downstream Purification
Synthesis Development and Formulation
(Jamison Group) (Jensen Group) (Myerson Group)
I
I
Design and development t Design and development of compact, of purification and reconfigurable system formulation system
1
ContinuousInnLarge-scale synthesis - purification and A
Mn-out formulation of APIs
New flow technology Development of development continuous purification Development of
synthetic route
Purified, Formulated APIs
Chapter
1
Development of a Continuous Synthesis of
Neostigmine Methyl Sulfate in a
Reconfigurable Frame
ABSTRACT
OH 0
N N,
1.2 hadln N 1.4
"eSO-Solid handling -Purfication In-line evaporation . 4
SWater content neostigmine methyl sulfate
93,600 doses/day 46.8 g/day
15 min. average total residence time
Herein, we describe the development of a continuous flow synthesis of neostigmine methyl sulfate, an acetylcholinesterase inhibitor on the WHO list of essential medicines, and the transfer of the synthesis into a next-generation reconfigurable frame developed by our collaborators. Starting from 3-dimethylaminophenol, the synthesis provides a throughput of approximately 46.8 g/day (or 93,600 doses/day) of crude neostigmine methyl sulfate. The synthesis also showcases a prototype in-line evaporation unit that operates without any added carrier gas.
Dr. Christina Dai performed early screening of lithium bases. Dr. Yuqing Cui and Dr. Naomi Briggs developed the downstream purification sequence. Dr. Nopphon Weeranoppanant developed the in-line evaporator and, along with Dr. Dale Thomas, assisted with the in-frame synthesis runs. Liam P. Kelly developed the continuous synthesis of neostigmine methyl sulfate.
A. Introduction
The development of a continuous-flow synthesis of neostigmine methyl sulfate was performed as a part of the continuation of the PoD research project introduced within the preface section. Overall, the complexity of the APIs targeted for synthesis in the second-generation system being developed concurrently was greater than that of the APIs targeted and synthesized in the first-generation system.
The synthesis of neostigmine methyl sulfate (1.1) was first disclosed in 193 1," with a patent granted shortly thereafter in 1933.38 It is an acetylcholinesterase inhibitor on the WHO list of essential medicines.39 Neostigmine is commonly used to treat the symptoms of myasthenia gravis, but it is also used to reverse the effects of anaesthetics. The original literature synthesis of neostigmine starts by deprotonation of 3-dimethylaminophenol (1.2) followed by carbamoylation with dimethylcarbamoyl chloride (1.3) to generate an intermediate carbamate (1.4). The carbamate is then methylated with dimethyl sulfate in acetone to afford neostigmine methyl sulfate. The bromide salt of neostigmine is also medicinally relevant, however it was not our desired target. A more recent patent route40 follows this same strategy for synthesis, but utilizes nitroaryl carbonates or phenols to first generate a nitroaryl carbamate followed by carbamate transfer to generate the desired intermediate. The patent route utilizing this carbamate transfer was not considered because selective in-line removal of the nitroaryl byproduct over the desired material was anticipated to be difficult and we preferred to avoid the extra waste.
37 Aeschlimann, J. A.; Reinert, M. J. Pharm. Exp. Ther. 1931, 43 (3), 413-444.
* Aeschlimann, J. A. U.S. Patent 1,905,990, 1933.
* WHO Model Lists of Essential Medicines. http://www.who.int/medicines/publications/essentialmedicines/en/ (accessed April 3, 2019).
Scheme 1-1. Patented routes for the Aeschlimann: OH i. KOH, "alcohol" N CIAN' 1.2 1.3
synthesis of neostigmine methyl sulfate.
0 NN N 1.4 Me2SO4 0 N1 acetone r.t., overnight N MeSO4 1.1
neostigmine methyl sulfate
Dalvi et al: 02N "I LH C1 0 NO2 0 0 M to OOC-NO2 0 KN .0 .31 , Et3N toluene, reflux, 3hr OH N 1.2 02N 0 C O N e2NH luene - rt, 3 hr KOH toluene reflux, 2 hr r.t toluene reflux, 2 hr
Analysis of the routes known to generate neostigmine gave concern due to the solid reagents used. It was envisioned that prudent choice of base and solvent would alleviate any solid formation. Additionally, with this route we suspected minimal problems with reagent incompatibility throughout the synthesis. Another synthesis was envisioned with the steps reversed, which we sought to explore as a novel route to neostigmine.
Scheme 1-2. Concerns with flow translation of patented route to neostigmine.'
Patented route: 0 Cl N KOH, then 1.3 I Toluene, reflux 2 hr 0 Y '- 0 N 1.4 Me2SO4 acetone r.t., overnight ON N+ MeSO4-f N 1.1
-Underlined text indicates suspected flow incompatibilities: solid reagent, solvent switch, and long reaction time.
1.1 Me2SO4 acetone ., overnight N 1
0
N, 1. 4 OH 1.2 4 N,B. Synthetic Route Investigation
Synthesis of neostigmine was first performed in batch by the patent route to assess the compatibility of the route for flow synthesis. This initial test showed that our concerns were valid. Not only was the base insoluble, but the potassium phenolate salt that was generated would rapidly agglomerate into a sticky mass. The subsequent carbamoylation also generated significant quantities of KCl precipitate. Additionally, the disclosed routes both involved a long methylation in acetone after isolation of the material from the carbamoylation step. Looking at alternative syntheses, we still thought that the same general disconnection strategy was the most logical and straightforward. Translation into flow would be possible through proper base and solvent selection in order to afford homogeneity. We also expected that intensification of the methylation step could be safely performed under flow conditions. A novel route with reversed order of synthetic steps was also proposed at this time.
Scheme 1-3. Potential routes to be explored for neostigmine synthesis.
Proposed route: Soluble organic A
I
OH base, then CI N 0 N .3 I Me2SO4 t t< 30min qt < 30 min lN N 1.2 1.4 Novel route: OH Me2SO4 OH i.Base ON t < 30 min ii. 1.3 s N+ X- N CI-/X-1.2 1 5 1.1-C I/-X NN Me 0 1.1 alt metathesisInvestigation of both proposed routes allowed the comparison of physical properties of the intermediates generated. Since the known synthesis had already been performed, the novel zwitterionic route was next investigated. The methylation of 3-dimethylaminophenol with methyl iodide41 proceeds smoothly in MeCN to generate quaternary ammonium salt 1.3 in 80%
4 1
(a) Epstein, J.; Plapinger, R. E.; Michel, H. 0.; Cable, J. R.; Stephani, R. A.; Hester, R. J.; Billington, C.; List,
G. R. J Am. Chem. Soc. 1964, 86 (15), 3075-3084; (b) Ortega, P.; Copa-Patiflo, J. L.; Mufloz-Fernandez, M.
A.; Soliveri, J.; Gomez, R.; de la Mata, F. J. Org. Biomol. Chem. 2008, 6 (18), 3264-3269.
yield over 12 hours. Deprotonation of 1.3 with potassium carbonate42 in MeCN over 12 hours grants zwitterion 1.4 in 60% yield. These steps were also found to involve significant heterogeneity, though similar levels as observed in the patent route. Carbamoylation of zwitterion 1.6 proceeded smoothly to generate 1.1-C1 without any formation of solids, however it was noted that there would be significant challenge in attempting salt exchange to the desired methyl sulfate salt. While theoretically possible, an ion exchange in flow would require consumable resins, rendering the synthesis semi-continuous. We also believed that a salt metathesis could be performed between commercially available sodium methyl sulfate and neostigmine chloride to precipitate sodium chloride and generate the desired methyl sulfate salt of neostigmine. While a heterogeneous mixture could be handled by the downstream processing team, the process and purification could be significantly complicated by the other salts present as byproducts from the upstream synthetic steps. Ultimately, this route was predicted to entail more significant complications than a
deprotonation-carbamoylation-methylation sequence analogous to the patent route, and thus it was deprioritized for our development.
Scheme 1-4. Novel route to synthesize neostigmine in batch (up to 1.1-Cl).
OHOH 0- C0 N1O
Mel K2CO3 1.31
I <~<--- --- 1.
MeCN N +- MeCN N THF:DMSO 5:1 . ion exchange N 12h, rt / 12h rt / 60C, 45 min N' Cl
-or-80% yield 60% yield 1-1C salt metathesis
1.2 1.5 1.6 -50% conversion
C. Route Development for Continuous Synthesis of Neostigmine
As suspected from initial route exploration, the synthetic route to neostigmine required significant alteration in order to become flow-compatible. As an overview, these alterations resulted in a series of stepwise improvements of system stability as unit operations were optimized. We began by screening conditions that would allow deprotonation of the phenol starting material homogeneously. Organic bases, such as benzyltrimethylammonium
hours refluxing in solvents like Toluene or DCE. Alkoxide bases were determined to rapidly afford the desired phenolate, and potassium tert-butoxide (1 M in THF) was chosen moving forward into flow. Unfortunately, in toluene the slight turbidity observed in batch translated to immediate clogging at the T-mixer where base and phenol met. Solvents with higher dielectric constants were then screened in batch, where it was observed that no turbidity was present from combinations of the phenol in THF, DMF, DMSO, or DMA treated with the KOtBu solution.
Although the deprotonation heterogeneity was resolved easily with solvent choice, precipitation still occurred after addition of dimethylcarbamoyl chloride, indicating that KCI would be problematic in terms of system stability. Further testing with lithium bases was performed by Dr. Christina Dai as it was thought that the LiCl byproduct may be more soluble.4 3 However, a variety of bases (LiHMDS, n-BuLi, LiOtBu, and LiOEt) in THF or
mixtures of THF and DMSO still generated precipitates. It was also observed that these bases did not consistently drive the reaction to completion, therefore KOtBu remained our base of choice.
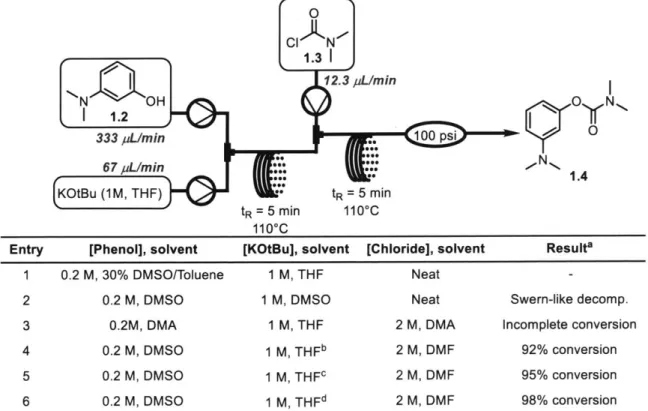
Testing to determine proper combinations of solvents to address this precipitation was then started in flow (Table 1-1). Ultimately, with all of the combinations listed, clogging occurred rapidly and only brief collection of samples after equilibration was possible. Systems involving DMSO (entries 1 and 2) generated the least quantity of precipitate, but use of DMSO as the sole solvent led to significant degradation of the acid chloride through Swern-like reactivity (entry 2). Utilizing a mixture of DMF and DMSO led to near-complete conversion and reduced degradation of the acid chloride, but KCl precipitation remained problematic (entries 4-6). Despite the remaining problems with clogging, we assessed the methylation step with the small amount of crude material collected from these runs.
" Li, M.; Constantinescu, D.; Wang, L.; Mohs, A.; Gmehling, J. Ind Eng. Chem. Res. 2010, 49 (10), 498 1-4988.
Table 1-1. Experiments to determine feasible solvent mixtures to generate carbamate 1.4. qO N CIAN 1.31 12.3 pL/min N' OH 1 N 21.2 M D M 333 pL/min .. 100 psi 0 6 7 pL/M in *0** N
K3 u(1M, DMAF) M, T min 2 Mp5
(K~~ u
1 M THF:) :- tR 5 m in t110 C
1100C
Entry [Phenol], solvent [KOtBu], solvent [Chloride], solvent Resulta
1 0.2 M, 30% DMVSO/Toluene 1 M, THF
Neat-2 0.2 M, IDMVSO 1 M, IDMVSO Neat Swern-like decomp.
3 0.2M, DMA 1 M, THIF 2 M, DMA Incomplete conversion
4 0.2 M, DMSO 1 M, THFb 2 M, DMF 92% conversion 5 0.2 M, DMSO 1 M, THFc 2 M, DMF 95% conversion 6 0.2 M, DMSO 1 M, THFd 2 M, DMF 98% conversion
aAll runs clogged shortly after sample collection. b1 .2 equiv. KOtBu used. c1.3 equiv. KOtBu used. d1 .4 equiv. KOtBu used.
Allowing 1.66 equivalents of Me2SO4 to react over 18 hours at room temperature with crude material from entry 6 (Table 1-1) showed good conversion (approximately 75%), but precipitation of the product was not possible. Addition of acetone to an analogous experiment
(50 v/v%) improved conversion slightly to 85%, but precipitation of the product was still not
possible. In comparison, using pure carbamate with acetone as solvent generated an 80% yield of solid neostigmine methyl sulfate under the same conditions. Given these results, it was predicted that a solvent switch after generation of the carbamate would be advantageous for multiple reasons. First, removal of the polar solvents needed for the first step was predicted to improve our ability to precipitate neostigmine at the end of the process for the downstream processing team to use. Second, a switch into another solvent would allow the identification of a proper solvent in which to intensify the methylation step. Finally, the aqueous wash required for the solvent switch would dissolve any remaining KCI precipitate, and could also be optimized for the removal of any remaining starting materials or byproducts.
consisted of THF with DMSO and/or DMF. We believed this would provide an operationally simpler solvent switch because of the challenges occasionally encountered in fully removing DMF during extractive workup in batch. At this point, however, the stability of the system remained the same, where clogging would occur after approximately 30 minutes of runtime. It was found that a substoichiometric quantity of 18-crown-6 (5-20 mol %) could improve the stability of the system, generating less-cohesive solids, but alone it was not enough to solve the intermittent clogging. Stoichiometric quantities were not used since, as a phase-transfer reagent, 18-crown-6 would further complicate the solvent switch. Because of the improved stability, we began to use 0.12 equivalents of 18-crown-6 from this point forward. This loading corresponds to 0.1 equivalents relative to KOtBu, itself being in slight excess at 1.2 equivalents relative to phenol. The addition of 18-crown-6 did not fully resolve solid formation, so without searching for additional chemical solutions to the problem, we decided to also utilize a tube-in-tube reactor (Figure 1-1) where the precipitate would be generated in a larger cross-section and suspended more effectively.
Figure 1-1. Depiction of a tube-in-tube style reactor.
sySolution A K J4 Mixing occurs over
laer b se-ection 1/8w i f
than in T-mixer
In conjunction with the previous solutions, the implementation of the tube-in-tube reactor resulted in a system that could reliably be run for 2-3 hours at a time. Clogging would still eventually occur from deposition of KCI within the 1/8" i.d. PFA tubing of the tube-in-tube reactor. Knowing further tweaking would later be necessary, we shifted focus toward optimizing the solvent switch parameters. Though the small quantity of phenol 1.2 remaining after carbamoylation (< 1% of total) was found to be fully soluble in neutral water, basic solutions such as 0.5 M NaHCO3 were found to have desirable separation properties in terms of phase resolution.
Scheme 1-5. (a) First telescoped route to generate neostigmine. (b) Throughput (1.4) as a function of toluene flow rate. 261 pLmin OH 1.2 -N -N. (0.23 M, THF) 18-crown-6 (12 mol%) 72 pWmin
(
KOtBu (1 M, THF) -L r(. 600C tR = 3.3 min 90 pLmin 0 Cl N 1.3 1 (1M, THF) tub 250 pL (NaHCO3 (4 e-in-tube -J -U t *R 60*C tR = 2.4 min min 26 pL/min Me2SO4 (3.2 M, toluene) autole neo6 ---- 40 psi aqueous 60*C0 waste tR = 2 .2 min 0 N,% N+ MeSO4 -1.1 10% conversion of (1.4) 1.5 hours collectedb)
Analysis of carbamate (1.4) at varied toluene flow rates[
Carbamate (1.4)] Post-Extraction 25 20 15 E =L L5 0 0 50 100 150 200 250 300 .Toluene flow rate (pL/min)
Carbamate (1.4) Throughput 0 (U M-X a' - ' 0 50 100 150 200 250 300
Toluene flow rate (p1/min)
Percent 100.0% 80.0% 60.0% 40.0% 20.0% 0.0% Recovery of Carbamate (1.4)
-I-Il
75 150 200 250Toluene flow rate (pL/min)
a)
k) 0.125 0.1 0.075 0.05 0.025 0Testing of the telescoped route clarified that the methylation would require further intensification and optimization. From further batch experiments (Table 1-2) we noted that higher polarity solvents had a beneficial effect on the conversion of 1.2 to 1.1. Though the conversion of 1.4 observed in flow was low, and the amount of dimethyl sulfate used was quite high, it showed that toluene was also a viable solvent for methylation.
Table 1-2. Methylation of 1.4 in various solvents.
I
I
0Y N, Me2SO4 (1.4 equiv.) 0 N
0 Solvent, 60 C 0
N N+ MeSO4
-1.4 1.1
Entry Solvent time (min) Conversionb
1a THF/Toluene 2.2 10.0%
2 THF 60 10.3% (5.6%)
3 THF:Acetone (5:1) 60 15.1% (3.9%)
4 THF:iPrOH (5:1) 60 18.8% (8.0%)
aPerformed in flow (scheme 1-5a). After analysis of [1.4] from separator, actual equivalents of Me2SO4 used was determined to be approx.15. bValues in
parentheses are conversion after 20 min.
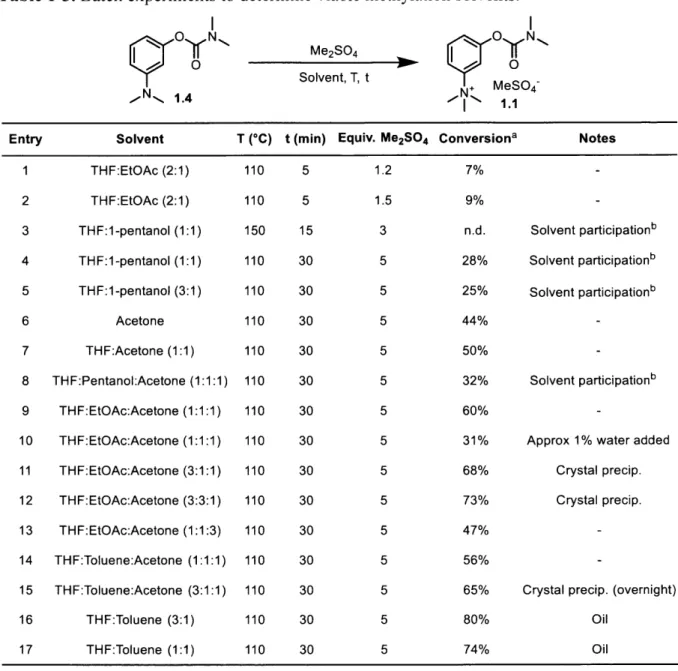
Batch analysis of variety of alternative solvents for methylation was next performed (Table 1-3) in order to better direct our efforts in flow. To narrow down solvent choices, small batch experimentation provided insight on solvents that would provide rapid phase resolution during the solvent switch. The THF/0.5 M NaHCO3 mixture as generated in Scheme 1-5a was found to resolve rapidly with three solvents. In addition to toluene, these were 1 -pentanol and ethyl acetate. It was found that mixtures of THF, acetone, 44 and toluene or ethyl acetate (entries 9, 11-15) resulted in high conversion of carbamate 1.4. The use of 1-pentanol at high temperatures led to carbamate transfer from 1.4 to the solvent. The ratios of solvent were chosen as hypothetical ratios of solvents going in to the solvent switch. In terms of results from a solvent switch in-line, the resultant solvent mixtures were expected to contain little THF
Table 1-3. Batch experiments to determine viable methylation solvents. N N 0 N -,* 1.4 Me2SO4 Solvent, T, t N MeSO4~ Entry Solvent 1 THF:EtOAc (2:1) 2 THF:EtOAc (2:1) 3 THF:1-pentanol (1:1) 4 THF:1-pentanol (1:1) 5 THF:1-pentanol (3:1) 6 Acetone 7 THF:Acetone (1:1) 8 THF:Pentanol:Acetone (1:1:1) 9 THF:EtOAc:Acetone (1:1:1) 10 THF:EtOAc:Acetone (1:1:1) 11 THF:EtOAc:Acetone (3:1:1) 12 THF:EtOAc:Acetone (3:3:1) 13 THF:EtOAc:Acetone (1:1:3) 14 THF:Toluene:Acetone (1:1:1) 15 THF:Toluene:Acetone (3:1:1) 16 THF:Toluene (3:1) 17 THF:Toluene (1:1)
'Based on remaining starting material
carbamate transfer to pentanol.
T (*C) 110 110 150 110 110 110 110 110 110 110 110 110 110 110 110 110 t (min) 5 5 15 30 30 30 30 30 30 30 30 30 30 30 30 30 110 30 Equiv. Me2SO4 1.2 1.5 3 5 5 5 5 5 5 5 5 5 5 5 5 5 5 Conversion' 7% 9% n.d. 28% 25% 44% 50% 32% 60% 31% 68% 73% 47% 56% 65% 80% 74% Notes Solvent participationb Solvent participationb Solvent participationb Solvent participationb
Approx 1% water added
Crystal precip.
Crystal precip.
Crystal precip. (overnight)
Oil
Oil
as determined by HPLC. bOther products were observed, such as
Having observed low conversion of the carbamate from the initial tests (entries 1 and 2) and complete loss of 1.4 at 150 'C (entry 3), we settled on heating the solution to temperatures of 110 'C or below with 5 equivalents of Me2SO4 for 30 minutes. The most
exciting results obtained were from entries 11, 12, and 15-17, where product was obtained either as an oil or as a crystalline precipitate upon standing. From the oil-containing mixture (entry 17), it was found that neostigmine methyl sulfate could be isolated with minimal processing. By heating the mixture to 60 'C, addition of a minimal quantity of acetone to
dissolve the oil, and allowing to cool to room temperature, neostigmine methyl sulfate 1.1 could be isolated as a crystalline solid in 62% yield. Overall, we observed that toluene was the most effective solvent for the methylation.
Notably, the addition of water to the methylation had significant decrease in the conversion of carbamate 1.4. We intuited that the toluene solution exiting the solvent switch would contain water, therefore it was important to quantify the effects of water on methylation ability (Table 1-4). In these trials it was noted that significant water content resulted in further deposition of an oily mixture of 1.4 and 1.1. From entries 3-5, crystallization of 1.1 was not possible under the conditions previously developed.
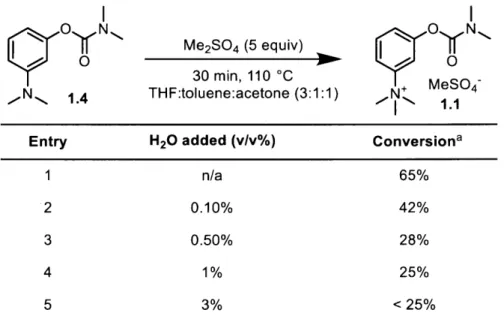
Table 1-4. Effects of water content on methylation step.
YO N, Me2SO4 (5 equiv) N
30 min, 110 C
MeS04-,N 1.4 THF:toluene:acetone (3:1:1) N+
I - 1.1
Entry H20 added (v/v%) Conversiona
1 n/a 65%
2 0.10% 42%
3 0.50% 28%
4 1% 25%
5 3% <25%
aBased on remaining starting material as determined by HPLC.
From this data we determined that water content of the solution of 1.4 was a major factor leading to the low methylation yields previously obtained (Scheme 1-5a). Given this evidence, we focused on reducing water content in the solution prior to methylation. Because we had recently determined that toluene was the optimal solvent for methylation, we focused on adjustments to the aqueous wash or other additives to achieve this goal.
For these tests, the same 3:1 mixture of THF:toluene was used to approximate flow conditions. Under previous conditions, using a 0.5 M solution of NaHCO3, water content observed in the toluene solution was between 4.3-6.7% (wt/wt%).4 5 When a 1:1 mixture of brine:0.5M NaHCO3 was used instead, this water content was lowered to 2.6% (wt/wt%). Despite the decreased water content, this value was still unacceptable. Several attempts to use the methylation reagent itself as a method to dry the reaction were also performed. When dimethyl sulfate was added in large excess-up to 40 equivalents-the conversion of carbamate 1.4
nevertheless remained between 16-30%. It was clear that small adjustments to the originally-telescoped system would not reduce water content to an acceptable level. Thus, we thought it prudent to test other solvent and aqueous base inputs to the solvent switch in order to further
decrease the water content.
Replacement of THF with 2-methyl THF, a green solvent46 with similar properties to THF47 and significantly lower water solubility, granted lowered water content. Where
previously we obtained toluene solutions with water content between 4.3-6.7% (wt/wt%), 2-methyl THF granted reduction to 1.8-2.3% (wt/wt%) (Scheme 1-6). Although a significant reduction, 1.8% water by weight was still higher than the subsequent methylation would tolerate. Under these conditions we also observed approximately 40% loss of product into the aqueous layer during the solvent switch due to the emulsion formed. The emulsion led to sub-optimal performance of the liquid-liquid separator by breakthrough of the aqueous phase. To address the separation performance, bases with higher ionic strength were targeted. We predicted that such a base would improve separation consistency through faster resolution of emulsions and increased interfacial tension.
4' As determined by Karl Fischer titration.
46 Antonucci, V.; Coleman, J.; Ferry, J. B.; Johnson, N.; Mathe, M.; Scott, J. P.; Xu, J. Org. Process Res. Dev.
2011, 15 (4), 939-941.
Scheme 1-6. Water content obtained with (A) THF and (B) 2-methyl THF as solvent.
261 pUmin 90 pUmin A: 190 pUmin A: 150 ULmin
0 B: 150 p1min B: 141 -165 pUmin
NaHCO3 (aq, 0.5M) toluene
N OH 1.31
| 1.2
(0.23M in A: THF (1M in A:THF
B: 2-MeTHF) B: 2-MeTHF)
+0 N
18-crown-6 (12 mol%) ,tube-in-tube
.... 40 psi0
72Umin ,IN . 1.4
KOtBu (1M in THF)0 aqueous A - H20 content 4.3-6.7%
t = 3.3 min tR = 2.4 min waste B - H20 content 1.8-2.3%
Batch testing of alternative bases showed that K3PO4 (0.4 M, aq.) resulted in
near-identical water content and concentration of 1.4 in the resultant organic phase, but the phases resolved more rapidly. Higher concentration of base led to deposition of solids within the liquid-liquid separator and eventual failure of the unit. Though K3PO4 did not significantly
affect water content of the organic layer, translation into flow did present consistent phase separation when used in the liquid-liquid separator. An additional improvement to separation performance was made by changing the semipermeable PTFE membrane from pore size 0.5 pm to 0.1 ptm. Despite improvements to separation consistency and performance, none of these changes resulted in significant decrease in water content. During these studies, we evaluated drying columns to reduce the water content, but observed that even a large column would not afford the drying capacity required. A packed bed (1/4" i.d, 10 cm length stainless steel tubing) of 600 mesh 4A molecular sieves48 initially showed a large reduction in water content, but its capacity for drying decreased significantly over the course of 1 hour. Additionally, the large void volume of the column meant that the system would not reach equilibrium before requiring replacement of fresh drying agent. The drying column approach was abandoned due to these reasons.
These tests exposed significant stability problems with the system, which could not be operated for much longer than 3 hours without clogging. Changing solvent to DMF and decreasing phenol concentration significantly improved the lifetime of the system (Scheme
1-7a). It was also noted that in DMF the precipitation of KCl in the tube-in-tube reactor was gradual, with solids evenly suspended until agglomeration after approximately 1 minute. Analysis of the reaction showed that carbamoylation was complete in under 30 seconds, and this proved to be the final key to solving system stability. It was found that if the reaction was quenched with the aqueous stream before solid agglomeration had occurred, the system was rendered stable for several hours (Scheme 1-7b). This rapid quench had no negative effects on carbamoylation, as it was complete within the allotted residence time.
Scheme 1-7. (a) Change of solvent and reduced residence time in reactors 1 and 2 results in a stable system. (b) Quench prior to agglomeration prevents system clogging.
a) 6oo pJmin 90 pdJmin
0 1.2 mUmin 300 pUmin N%
)
H.C N.- K3PO4 (aq. 0.4M) toluene 1 .2 ~ 1.3 1 (0.15M in DMF) (2M in THF) 0 N10 mol% 18-crown-6 tube-in-tube
30 psi 0 N 110 110 aqueous 1.4 117= 6 m t = 4 m waste -0.06 M, toluene ( KOtBu (1 M in THF) f tR=2 min tR= 0.5 min b) IL *0 oil -is t t l tR 3 sR R =3-5 min
carbamoylation complete agglomeration starts clogging often occurs aqueous quench here clogging may occur
Once system stability had been addressed, we could address the water content once more. The water content coming from the solvent switch was still too high. NMR analysis of the organic phase obtained from the liquid-liquid separator showed a mixture of 83.3% toluene,
13.7% THF, and 3% DMF. We predicted that the significant quantity of THF and DMF in solution allowed more water to remain within the organic phase. We hypothesized that a
second water wash could afford removal of those solvents and further reduction of water content from the organic phase. Addition of deionized H20 to the toluene stream followed by
a second separator was found to reduce water content from approximately 5500 ppm (0.55 wt/wt%) to 1900 ppm (0.19 wt/wt%). The second separation also provided protection in case of aqueous breakthrough during the first separation. From the combination of DMF, THF, aqueous K3PO4, and toluene prior to the first separation arose a triphasic mixture. The triphasic
mixture presented a remarkable challenge for separator performance, with low interfacial tension between the organic (toluene) phase and middle (DMF, THF, and water) phase. The third phase, concentrated K3PO4, was effectively excluded. The addition of the second
separation provided effective removal for any aqueous breakthrough that occurred and ensured better performance for methylation.
With the stability and water content of the system optimized, we moved ahead to finish development of the methylation step prior to transferring the chemistry to the reconfigurable frame developed by our collaborators in the Jensen research group.
D. In-Line Evaporator
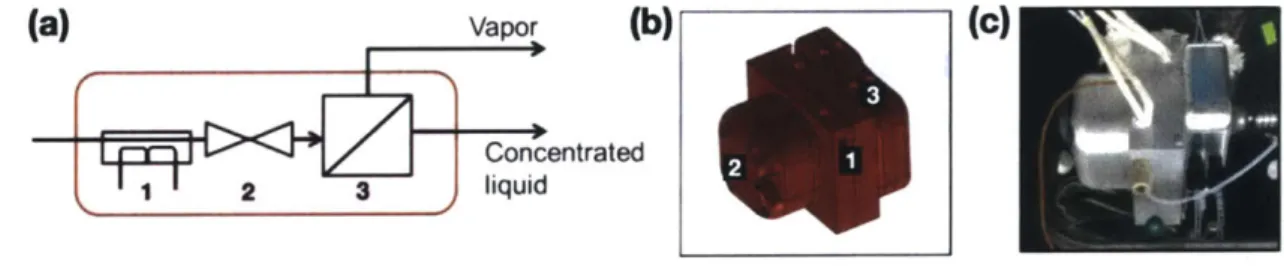
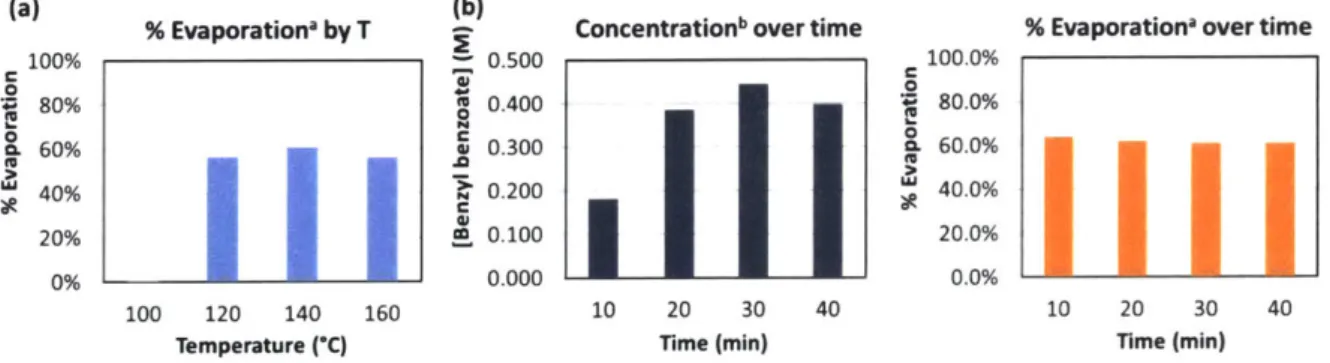
During this time, one of our collaborators in the Jensen lab, Dr. Nopphon Weeranoppanant, had developed a prototype in-line evaporation unit49 that we were eager to
evaluate as an additional means to mitigate the problems encountered above. Several examples of units for continuous small-scale evaporation exist, such as those designed by Hartman et al.50 and Deadman et al.51 Both designs utilize introduction of an inert gas added to the stream
in order to facilitate the evaporation of the desired solvent. The former uses the gas in slug flow in order to achieve rapid equilibrium and afford a consistent distillation through flash evaporation. The latter uses the added gas to nebulize the solution and desolvate, much like a spray chamber functions in ES! instrumentation. The prototype evaporation unit used within