HAL Id: tel-01922410
https://tel.archives-ouvertes.fr/tel-01922410
Submitted on 14 Nov 2018
HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Étude des conséquences génomiques et fonctionnelles de
l’instabilité des microsatellites dans le cancer colorectal
Malorie Greene
To cite this version:
Malorie Greene. Étude des conséquences génomiques et fonctionnelles de l’instabilité des microsatel-lites dans le cancer colorectal. Cancer. Université Pierre et Marie Curie - Paris VI, 2017. Français. �NNT : 2017PA066592�. �tel-01922410�
UNIVERSITÉ PIERRE ET MARIE CURIE
École doctorale Physiologie, Physiopathologie et Thérapeutique (ED394) Centre de Recherche Saint-Antoine UMR_S 938, INSERM, UPMC
Équipe « Instabilité des Microsatellites & Cancers »
THÈSE DE DOCTORAT
Spécialité Génétique des CancersÉtude des Conséquences Génomiques et Fonctionnelles de
l’Instabilité des Microsatellites dans le Cancer Colorectal
Par
Malorie GREENE
Dirigée par le Docteur Ada COLLURA
Présentée et soutenue publiquement le 28 novembre 2017 Devant un jury composé de :
Pr. Philippe LE ROUZIC Président
Dr. Dominique BLUTEAU Rapporteure
Dr. Serge ROCHE Rapporteur
Dr. Valérie BORDE Examinatrice
Pr. Pierre AUCOUTURIER Examinateur
Dr. Ada COLLURA Directrice de thèse
UNIVERSITÉ PIERRE ET MARIE CURIE
École doctorale Physiologie, Physiopathologie et Thérapeutique (ED394) Centre de Recherche Saint-Antoine UMR_S 938, INSERM, UPMC
Équipe « Instabilité des Microsatellites & Cancers »
THÈSE DE DOCTORAT
Spécialité Génétique des CancersÉtude des Conséquences Génomiques et Fonctionnelles de
l’Instabilité des Microsatellites dans le Cancer Colorectal
Par
Malorie GREENE
Dirigée par le Docteur Ada COLLURA
Présentée et soutenue publiquement le 28 novembre 2017 Devant un jury composé de :
Pr. Philippe LE ROUZIC Président
Dr. Dominique BLUTEAU Rapporteure
Dr. Serge ROCHE Rapporteur
Dr. Valérie BORDE Examinatrice
Pr. Pierre AUCOUTURIER Examinateur
Dr. Ada COLLURA Directrice de thèse
REMERCIEMENTS
Je tiens à exprimer mes remerciements au Professeur Philippe LE ROUZIC qui m’a fait l’honneur d’accepter de présider mon jury de thèse. J’adresse mes profonds remerciements au Docteur Dominique BLUTEAU ainsi qu’au Docteur Serge ROCHE pour l’intérêt qu’ils ont porté à ce travail en acceptant de le rapporter. Je remercie également le Docteur Valérie
BORDE et le Professeur Pierre AUCOUTURIER pour avoir accepté d’examiner mes travaux de
recherche.
Je remercie Alex DUVAL de m’avoir accueillie au sein de son laboratoire. Merci de m’avoir permis de découvrir le monde captivant de la génétique des cancers ainsi que pour le temps et les moyens accordés à la bonne réalisation des projets.
Je remercie Ada COLLURA de m’avoir guidée tout au long de cette thèse. Merci de m’avoir transmis tes connaissances pratiques et scientifiques et pour ton aide et ta patience.
Je remercie la Ligue Nationale Contre le Cancer pour avoir financé mon doctorat.
Je remercie particulièrement A’dem BOKHARI. Merci mon ‘co-doctorant’ pour ton soutien pendant les heures passées à la paillasse ou à écrire, le partage des chips de crevettes, les sessions de danse et de rires et ton amitié.
Merci Romane BERTRAND pour ton aide technique, ton soutien et ta compagnie en culture cellul-ai. (!), Delphine BOUVET pour tes gâteaux, bon courage pour la fin de ton doctorat et
Kristell WANHERDRICK & Olivier BUHARD pour vos conseils, vos blagues et votre apport
significatif en magnésium.
Je remercie l’ensemble des membres de l’équipe : Martine MULÉRIS, Marie-Annick MARRY,
Anastasia DEMIDOVA, Renato LUPINACCI, Vincent JONCHÈRE et Laetitia MARISA.
Je remercie le service d’anatomopathologie de Saint-Antoine et en particulier le Professeur
Jean-François FLÉJOU, le Docteur Magali SVRCECK ainsi que Sylvie DUMONT pour leur
Merci à Annie MUNIER de la plateforme de cytométrie, Tatiana LEDENT, Laetitia DINARD,
Alizée GUYOMARD, Timothé COULAIS et Quentin POINTOUT de la plateforme
d’hébergement et d’expérimentation animale et Raymond SAMSON & Stephan BILIC de la laverie.
Je tiens à remercier toute l’équipe du Palais de la Découverte et spécialement Philippe
LAVAIVRE, Élodie DUCASSE et Tanguy SCHINDLER.
Mes remerciements vont également à Yannick LERRANT, Valérie SARRAMEGNA, et Yvon
CAVALOC pour leur soutien et leur confiance depuis le début de l’aventure à l’Université de la
Nouvelle-Calédonie.
Un immense merci à Anastasia TCHOUKAEV, Florence SONNEVILLE et Sarah SAGET pour votre présence, votre soutien et votre amitié.
RÉSUMÉ
L’instabilité des séquences répétées microsatellites du génome (courtes répétitions en tandem d’un à cinq nucléotides) est une conséquence de l’inactivation du système MMR (MisMatch Repair), en charge de la réparation des erreurs produites au cours de la réplication de l’ADN. Cette instabilité est associée à un processus de transformation cellulaire original, observé chez l’homme dans des pathologies tumorales fréquentes, nommées MSI (pour Microsatellite Instability). Les localisations primaires les plus fréquentes de ces tumeurs sont le côlon, l’endomètre et l’estomac. Elles peuvent avoir une origine héréditaire (prédisposition familiale ; syndrome de Lynch et apparentés), mais sont dans la majorité des cas de survenue sporadique. La transformation des cellules MMR-déficientes s’observe dans le contexte de l’accumulation de nombreuses mutations somatiques dans l’ADN tumoral. Certaines ont un caractère oncogénique en favorisant la troncature et la perte de fonction de gènes suppresseurs de tumeur ou apparentés, impliqués dans des voies de signalisations diverses et qui contiennent des microsatellites codants (mutations indels d’une à deux paires de base, décalant le cadre de lecture, fréquemment rapportées dans ces tumeurs).
Les travaux présentés dans le cadre de mon doctorat visent à mieux comprendre le rôle de l’instabilité microsatellitaire dans la tumorigenèse MSI. Ils s’inscrivent dans le contexte du décryptage et de l’analyse des données de séquençage d’exome de 47 cancers colorectaux primitifs MSI. Dans le contexte d’un niveau élevé d’instabilité génomique caractérisant ces tumeurs, la mise au point par mon laboratoire d’accueil de modèles probabilistes a permis de dresser une liste restreinte de gènes, remarquables par le fait qu’ils sont affectés par des mutations somatiques dont les fréquences sont exceptionnellement élevées ou basses dans l’ADN tumoral. Sous l’hypothèse que de tels évènements somatiques affectent des gènes clés de la tumorigenèse MSI colique, j’ai focalisé mes recherches sur les gènes dont les altérations sont peu fréquentes. Brièvement, j’ai pu démontrer le caractère délétère d’un petit nombre d’altérations microsatellitaires codantes dont la survenue semble soumise à une pression de sélection négative (N=13). Mes résultats indiquent que ces mutations semblent fragiliser le phénotype tumoral des cellules dans lesquelles elles surviennent, la perte de fonction des gènes qu’elles affectent conduisant à diverses conséquences délétères en fonction du gène candidat (e.g. sensibilisation à la mort cellulaire, perte des capacités proliférative et migratoire, ralentissement de la croissance tumorale).
Ces résultats rapportent pour la première fois et à grande échelle, la sélection négative de mutations dans des tumeurs à forte instabilité génomique MSI. Ils ouvrent de nouvelles voies pour la compréhension de ce mode particulier de transformation cellulaire, et sont potentiellement d’intérêt pour la mise au point de thérapies personnalisées pour les patients.
ABSTRACT
Since the discovery of a link between mismatch repair (MMR) deficiency and cancer, microsatellite instability (MSI) is thought as a process underlying cell transformation and tumour progression and invasion. MSI tumours are a subset of frequent human neoplasms, both inherited and sporadic, associated with several primary locations (colon, stomach, endometrium…). In MMR-deficient cells, MSI generates hundreds of frameshift mutations in genes (MSI Target Genes, MSI-TGs) containing coding microsatellite sequences (e.g. -1/+1 bp, insertions/deletions, i.e. indels). Some of these mutations affect genes with a role in human carcinogenesis and are thus expected to promote the MSI-driven tumorigenic process.
During my PhD, I aimed to decipher the role of MSI in colon tumorigenesis. I exploited exome-sequencing data available in my lab that were generated from the analysis of a series of 47 human MSI primary colorectal cancer (CRC). Through biostatistics analysis and mathematical models that we designed to interpret mutation rates in the context of the high background for instability characterizing MSI in CRC, we identified a few microsatellites containing genes coding mutations that were negatively selected in MSI colon tumours (N=13). Under the hypothesis that these events may have a negative impact in colon tumorigenesis, I demonstrated that the silencing of these MSI target genes (siRNA/shRNA) was deleterious for MSI cancer cells using in vitro and in vivo models (impairment of proliferation and/or migration and/or response to chemotherapy and/or tumour growth) (Jonchère*, Marisa*, Greene* et al., submitted).
TABLE DES MATIÈRES
REVUE BIBLIOGRAPHIQUE ... 1
I. Introduction sur les cancers colorectaux……….……. 3
II. Le phénotype tumoral MSI : historique de la découverte d’un nouveau mécanisme d’oncogenèse………..……….. 5
III. Le système MMR : rôle et fonctionnement………. 6
IV. Les conséquences moléculaires de la déficience du MMR & oncogenèse MSI colique….... 10
1. L’instabilité des microsatellites ... 10
1.a. Mutations dans les régions répétées codantes : gènes cibles de l’instabilité ... 11
1.b. Mutations dans les régions répétées non codantes ... 18
i. Répétitions dans les régions 5’ et 3’ UTR ... 18
ii. Répétitions introniques situées à proximité d’un site accepteur d’épissage ou d’un site régulateur de la transcription ... 20
iii. Répétitions dans les régions intergéniques et les trans-éléments ... 22
1.c. Le séquençage de nouvelle génération (NGS) : un outil indispensable pour l’étude des mutations MSI ... 23
2. Mutations dans les régions non-répétées : altérations ‘hot spot’ ... 24
3. La classification moléculaire des cancers colorectaux ... 26
V. Les cancers de phénotype MSI……….. 29
1. Les cancers MSI héréditaires : syndrome de Lynch ... 29
1.a. Caractéristiques génétiques et moléculaires ... 30
1.b. Spectre clinique et épidémiologie du Syndrome de Lynch ... 31
1.c. Critères d’identification ... 33
2. Les cancers MSI sporadiques ... 35
2.a. Caractéristiques génétiques et moléculaires ... 35
2.b. Spectre clinique et épidémiologie ... 36
3. Caractéristiques cliniques et valeur pronostique de MSI ... 36
4. Néo-antigènes, microenvironnement immun et ICK dans les cancers MSI ... 40
5. Méthodes et marqueurs diagnostiques de MSI dans les CCR ... 43
5.a. Diagnostic par une approche de biologie moléculaire ... 43
6. Méthodes thérapeutiques et valeur prédictive de MSI dans les CCR ... 47
6.a. Chimiothérapie ... 47
6.b. Thérapies ciblées : anticorps monoclonaux et immunothérapies anti-ICK ... 49
i. Anticorps monoclonaux anti-EGF-R et anti-VEGF ... 50
ii. Immunothérapies anti-ICK ... 50
RÉSULTATS ... 53
DISCUSSION & PERSPECTIVES ... 129
ANNEXE ... 141
TABLE DES ILLUSTRATIONS
FIGURES
Figure 1 │ Représentation schématique du fonctionnement du système de réparation MMR . 9 Figure 2 │ Mutation de microsatellites codants et oncogenèse MSI ... 12 Figure 3 │ Représentation non-exhaustive des gènes cibles de l’instabilité microsatellitaire et potentiellement impliqués dans la carcinogenèse colique ... 14 Figure 4 │ Représentation schématique et non-exhaustive de la diversité des mutations génétiques causant la dérégulation de voies de signalisation dans les cancers colorectaux ... 15 Figure 5 │ Modélisation des catégories de gènes cibles de l’instabilité microsatellitaire ... 17 Figure 6 │ Les microsatellites à travers le génome ... 19 Figure 7 │ Classification moléculaire des cancers colorectaux ... 28 Figure 8 │ Mutations germinales hétérozygotes de gènes du MMR, fréquences mutationnelles (%), hétérogénéité phénotypique et risques des tumeurs associées au syndrome de Lynch .. 32 Figure 9 │ Stades & classification TNM histopathologique simplifiée des cancers du côlon et du rectum ... 38 Figure 10 │ Immunité anti-tumorale, ICK et immunothérapies bloquant les ICK ... 42 Figure 11 │ Détermination du statut MSI des tumeurs par PCR-pentaplex et
immunohistochimie ... 45 Figure 12 │ Modèle proposé de progression tumorale MSI ... 56
TABLES
Tableau 1 │ Homologies des gènes MMR chez les procaryotes (E. coli) et les eucaryotes (S. cerevisiae & H. sapiens)………7
ABRÉVIATIONS
5-FU : 5-fluorouracile
ADN : acide désoxyribonucléique
APC : adenomatous polyposis coli
ARN : acide ribonucléique
ARNm : acide ribonucléique messager
BAT : big adenine track
BAX : BCL2-associated X
bp : base pair pour paire de base
CCR : cancer colorectal
CIMP : CpG island methylator phenotype
CIN : chromosomal instability
CMS : consensus molecular subtype CpG : 5’-C-phosphate-G-3’
CTL : cytotoxic T lymphocyte
DFS : disease free survival
E. coli : Escherichia coli
EBV : Epstein-Barr virus
EGF-R : epidermal growth factor receptor
EpCAM : epithelial cell adhesion molecule
FAP : familial adenomatous polyposis
FDA : food and drugs administration
FOLFIRI : 5-FU, LV et IRI
FOLFOX : 5-FU, OXA et LV
Frameshift mutation : mutation décalant le cadre de lecture
GST : gène suppresseur de tumeur
HNPCC : hereditary nonpolyposis colorectal cancer
HSP110 : heat shock protein 110
ICK : immmun chekpoint
IDL : insertion-deletion loop
IHC : immunohistochimie
INCa : Institut National du Cancer
InSiGHT : international society for gastrointestinal hereditary tumors
IRI : irinotécan
KD : knockdown
KO : knockout
LOH : loss of heterozygosity
LS : Lynch syndrome
LV : leucovorine
mAb : monoclonal antibody pour anticorps monoclonal
MAPK : mitogen-activated kinase
MLH : MutL homologue
MMR : mismatch repair
MSH : MutS homologue
MSI : microsatellite instability
MSS : microsatellite stable
NGS : next generation sequencing
OS : overall survival
OXA : oxaliplatine
PAF : polypose adénomateuse familiale
PCNA : proliferating cell nuclear antigen
PCR : polymerase chain reaction
PMS : post-meiotic segregation
q-PCR : quantitative polymerase chain reaction
RFC : replication factor C
RFS : relapse free survival
RLFP : restriction fragment length polymorphism
RNA-seq : RNA-sequencing
S. cerevisae : Saccharomyces cerevisae
shRNA : short hairpin RNA
siRNA : small interfering RNA
TCGA : the cancer genome atlas
TILs : tumor infiltrating lymphocytes
TGi : target genes of instability
TGF-β : transforming growth factor beta
TNM : tumeur primitive – ganglions locorégionaux – métastase(s) à distance
TP53 : tumor protein 53
TS : thymidylate synthase
UTR : untranslated region
VEGF : vascular endothelial growth factor
WES : whole-exome sequencing
WGS : whole-genome sequencing
I. Introduction sur les cancers colorectaux
Le cancer colorectal (CCR) est l’un des cancers les plus représentés au niveau mondial. Il est le troisième cancer le plus fréquent chez l’homme après les cancers de la prostate et du poumon et le deuxième le plus fréquent chez la femme après celui du sein (Torre et al., 2012). Le CCR constitue la deuxième cause de décès liée au cancer avec une incidence élevée ; en France en 2015, plus de 43 000 nouveaux cas étaient estimés dont 45% de femmes et 55% d’hommes (Institut National du Cancer - INCa, 2016). Bien que la mortalité associée à ce cancer décroisse progressivement grâce aux progrès persistants dans le développement des campagnes de dépistage, dans la prise en charge du cancer et du patient et de l’évolution des thérapies, les cancers colorectaux restent un enjeu de santé publique majeur dans le monde occidental (INCa, 2016). Les CCR se déclarent généralement après 50 ans et plusieurs facteurs de risques ont été identifiés. Parmi les facteurs environnementaux et liés au mode de vie, le surpoids, l’alimentation riche en graisses animales, la composition du microbiote, la consommation d’alcool et de tabac, l’inactivité physique, ou encore l’exposition à des substances médicamenteuses sont connus pour influencer le risque de développement de cancer (Haggar and Boushey, 2009; Johnson et al., 2013; Siegel et al., 2017). La prédisposition génétique en cause dans les cancers héréditaires comme le syndrome de Lynch et la polypose adénomateuse familiale est responsable du développement précoce d’un cancer colorectal.
Le cancer est aujourd’hui reconnu comme étant la conséquence d’un processus complexe d’acquisition de nombreuses altérations génétiques. Ces évènements mutationnels sont susceptibles de conférer, aux clones cellulaires dans lesquels ils surviennent, les caractéristiques nécessaires à la transformation de cellules saines en cellules tumorales (e.g. indépendance des signaux de prolifération, insensibilité aux signaux anti-prolifératifs, résistance à l’apoptose, immortalité, capacité à induire l’angiogenèse, capacités invasives et migratoires…). Il existe deux types distincts d’instabilité génomique :
l’instabilité chromosomique (CIN pour Chromosomal Instability) et l’instabilité des microsatellites (MSI pour Microsatellite Instability).
La prise en compte de cette diversité génétique est cruciale pour le diagnostic des CCR qui devient aujourd’hui systématique en clinique. Cette classification permet la stratification
pronostique de ces cancers et promptement une orientation thérapeutique adaptée (cf. infra : ‘Les cancers de phénotype MSI’).
Dans leur majorité (80-85% des cas), les tumeurs colorectales présentent une instabilité chromosomique et sont nommées tumeurs CIN ou LOH (pour Loss Of Heterozygosity). Elles sont ainsi caractérisées par des pertes alléliques (LOH), un nombre anormal de chromosomes (hypo- ou hyper- diploïdie) et arborent des pertes ou des gains importants de fragments chromosomiques, des translocations chromosomiques ainsi que des amplifications géniques (Vilar and Gruber, 2010). Ces tumeurs étant exemptes de mutations dans les microsatellites, elles sont également appelées tumeurs MSS (pour Microsatellite Stable).
Dans environ 15% des cas, les CCR ont un phénotype MSI, dit aussi phénotype hypermutateur. Les cancers MSI sont caractérisés par une déficience du système MMR (pour Mismatch Repair) de réparation des mésappariements de bases, responsable d’une instabilité nucléotidique générale et d’avantage marquée au niveau des séquences répétées dites microsatellites (Cancer Genome Atlas, 2012). Les CCR MSI ont le plus souvent un caryotype normal diploïde (74% des cas ; Fearon 2011). Des études d’épigénétique ont montré que les tumeurs MSI sont fréquemment associées à un phénotype CIMP (pour CpG Island Methylator Phenotype) se traduisant par une hyperméthylation de nombreux sites CpG (pour Cytosine-Phosphate-Guanine, i.e. séquences riches en résidus C et G) présents dans les promoteurs de gènes comme particulièrement celui du gène MLH1, un des gènes du système MMR, entraînant la répression transcriptionnelle de celui-ci. Les aspects relatifs aux altérations épigénétiques des CCR ne sont pas développés dans cette introduction.
L’émergence des CCR MSS et MSI est consécutive à la dérégulation des principales voies de signalisation impliquées dans la carcinogenèse colique qui sont en partie communes à ces deux types tumoraux ; par exemple les voies WNT/β-caténine et RAS/MAP-Kinases sont activées alors que la voie de signalisation du TGFβ ou de P53 sont inactivées dans ces cancers. Cependant, les mécanismes moléculaires sous-jacents sont responsables d’altérations spécifiques et distinctes aux deux types tumoraux (cf. infra chapitre IV, sections 1 et 2).
II. Le phénotype tumoral MSI : historique de la découverte d’un nouveau mécanisme d’oncogenèse
La notion d’instabilité nucléotidique sur des séquences répétées microsatellites de l’ADN –définies comme des motifs itérés en tandem de 1 à 5 bases (Ellegren, 2004), dans les cancers humains, a été rapportée pour la première fois au début des années 90. En 1993, les groupes de M. Perucho et de S. Thibodeau publient respectivement deux études dans les revues Nature et Science, relatant la présence de mutations somatiques ubiquitaires altérant la taille de microsatellites mononucléotidiques et dinucléotidiques respectivement, dans environ 12 à 15% des cas de CCR (Ionov et al., 1993; Thibodeau, Bren and Schaid, 1993). Ces travaux montrent que dans ces cancers, aussi bien sporadiques qu’héréditaires (syndrome de Lynch appelé à l’époque HNPCC pour Hereditary NonPolyposis Colorectal Cancer), des caractéristiques cliniques spécifiques (cf. infra : section V.3) sont associées à l’instabilité des microsatellites (Aaltonen et al., 1993; Thibodeau, Bren and Schaid, 1993). Ces études, controversées dans un premier temps, révèlent un mécanisme nouveau et unique de carcinogenèse colique.
Par la suite, des travaux ont visé à identifier les causes de cette instabilité. Le phénomène d’instabilité microsatellitaire de ces tumeurs évoque alors le phénotype mutateur identifié dans les années 70 chez la bactérie Escherichia coli (E. coli), dû à un défaut du système de réparation de l’ADN MutHLS en charge de la surveillance de la fidélité de la réplication. Dès la fin de l’année 1993, les homologues humains des gènes MutHLS de E. coli sont localisés sur les chromosomes 2 (MSH2) et 3 (MLH1) et découverts comme mutés de façon constitutionnelle et monoallélique chez les patients Lynch (Fishel et al., 1993; Leach et al., 1993; Bronner et al., 1994). Ces gènes codent pour des protéines essentielles à l’activité du système MMR et permettent de réparer fidèlement les mutations indels (insertions/délétions), fortement représentées au niveau des microsatellites. Ce processus original de transformation cellulaire diffère du modèle d’oncogenèse colique de Fearon et Vogelstein caractérisé par une progression séquentielle spécifique de mutations activatrices d’oncogènes et de l’inactivation biallélique de gènes suppresseurs de tumeurs (Fearon and Vogelstein, 1990). Ce nouveau mécanisme de tumorigenèse appelé ‘RER’ (pour Replicative ERror phenotype) dans un premier temps a ensuite été dénommé ‘MSI’ au cours d’une réunion
III. Le système MMR : rôle et fonctionnement
Le système MMR (pour MisMatch Repair) est le système de réparation de l’ADN en charge de la réparation des erreurs de mésappariement de bases survenant au cours de la phase de réplication de l’ADN. Ce système revêt ainsi un rôle capital dans le maintien de l’intégrité du génome.
Lors de la phase réplicative, l’ADN polymérase peut commettre des erreurs (par l’introduction d’un nucléotide erroné/supplémentaire ou par l’absence d’insertion du nucléotide correspondant au brin matrice) à raison d’une base erronée pour 105 nucléotides incorporés (soient 10-5 mutations par paire de base par génération) (Kunkel and Erie, 2015; Ganai and Johansson, 2016). Ces erreurs sont immédiatement rectifiées par l’activité d’autocorrection exonucléase 3’-5’ de l’ADN polymérase réduisant le taux d’erreur à 10-7. Toutefois, les erreurs échappant à cette activité de relecture sont identifiées, signalées et réparées par le système MMR, faisant chuter le taux d’erreur à 10-9-10-10 et assurant ainsi la fidélité de la réplication à l’échelle du génome. En plus de la réparation des erreurs pendant la réplication, le système MMR est également impliqué dans différents processus cellulaires comme les recombinaisons pendant la méiose et/ou la mitose, la signalisation de dommages de l’ADN et l’apoptose (Jiricny, 2013). Seules les activités de prise en charge des mésappariements, perturbées lors de la tumorigenèse colique, seront développées ci-après. La réparation des erreurs de mésappariement est un processus cellulaire hautement conservé des procaryotes aux eucaryotes. D’abord étudié chez la bactérie E. coli, le système MutHLS est composé de trois protéines principales : MutS, MutL et MutH (Jiricny, 2006). Le système MMR des eucaryotes, plus complexe, comprend dix gènes chez la levure Saccharomyces cerevisiae (S. cerevisiae) dénommés en référence à leurs homologues bactériens (tableau 1). Ces gènes sont pratiquement tous conservés chez les mammifères dont le système MMR est composé de cinq homologues de MutS (MSH 2-6 pour MutS Homologue) et de quatre homologues de MutL (MLH 1 et 3 pour MutL Homologue et PMS1 et 2 pour Post-Meiotic Segregation) (tableau 1). La protéine MutH n’a pas d’homologue connu chez les eucaryotes (Jiricny, 2013).
Bactérie E. coli Levure S. cerevisiae Mammifère H. sapiens Fonction MutS
MSH1 - Réparation de l’ADN mitochondrial
MSH2 MSH3 MSH6 MSH2 MSH3 MSH6 Reconnaissance du dommage : MutSα (MSH2-MSH6)
Mésappariement de bases & IDL d’un ou deux nucléotides
MutSβ (MSH2-MSH3)
IDL de plus de deux nucléotides
Liaison à l’ADN et recrutement du complexe MutL
MSH4 MSH5
MSH4 MSH5
Gènes impliqués dans la recombinaison méiotique MutL MLH1 MLH2 MLH3 PMS1 MLH1 PMS2 MLH3 PMS1
Initiation de la réparation par MutLα (MLH1-PMS2) et recrutement des facteurs poursuivant la réparation
MutH - - Endonucléase spécifique du brin d’ADN
néo-synthétisé non méthylé
Tableau 1 │ Homologies des gènes MMR chez les procaryotes (E. coli) et les eucaryotes (S. cerevisiae & H. sapiens)
IDL : insertion-deletion loops Source : Jiricny, 2013.
Les erreurs induites par le glissement de la polymérase conduisent à la formation de boucles d’insertion/délétion (IDL pour insertion/deletion loops) de plusieurs nucléotides extra-hélicoïdaux. Le processus de réparation de ces erreurs nécessite plusieurs complexes protéiques d’hétérodimères en fonction de la nature du dommage et de l’avancement de la réparation. La reconnaissance et l’initiation de la réparation du dommage se font par les complexes MutSα (MSH2-MSH6) et MutSβ (MSH2-MSH3). Ainsi, MutSα est recruté au niveau de mésappariements d’une seule base ainsi que sur les IDL de quelques nucléotides tandis que le complexe MutSβ initie préférentiellement la réparation de grandes IDL de plusieurs bases extra-hélicoïdales. Dans un second temps, les homologues de MutL interagissent en différents complexes d’hétérodimères –MutLα PMS2), MutLβ PMS1) ou MutLγ (MLH1-MLH3), pour recruter le complexe PCNA (pour Proliferating Cell Nuclear Antigen) et l’exonucléase 1 et procéder à l’excision du brin endommagé. Les homologues de MutL sont également en charge de l’activation des évènements ultérieurs, à savoir la resynthèse d’un néo-brin corrigé par l’ADN polymérase δ, relié au brin matrice par l’ADN ligase (figure 1) (Kunkel and Erie, 2015; Ganai and Johansson, 2016; Li and Martin, 2016).
Figure 1 │ Représentation schématique du fonctionnement du système de réparation MMR
Les complexes MutSα (MSH2-MSH6) et MutSβ (MSH2-MSH3) sont en charge de la reconnaissance du dommage (triangle rouge) et interviennent spécifiquement selon la nature de ce dernier. Le complexe MutLα (MLH1-PMS2) est recruté au niveau du dommage par MutSα ou MutSβ. Le complexe PCNA est recruté au niveau de l’ADN par le facteur RFC et interagit avec le complexe MutLα. Ensemble, PCNA et MutLα activent la fonction endonucléase de PMS2 (en jaune) qui amorce l’excision en 3’ du mésappariement. L’excision est complétée par l’exonucléase EXO1. Les protéines RPA (Replication Protein A) stabilisent et protègent la molécule d’ADN monocaténaire de la dégradation et participent à la dernière étape de l’excision. Le néo-brin corrigé est synthétisé par l’ADN polymérase δ et est lié à la molécule d’ADN initiale par l’ADN ligase.
Adaptée de Jiricny et al., 2013 & Li et al., 2016.
Figure 1 │ Représentation schématique du fonctionnement du système de réparation MMR
Les complexes MutSα (MSH2-MSH6) et MutSβ (MSH2-MSH3) sont en charge de la reconnaissance du dommage (triangle rouge) et interviennent spécifiquement selon la nature de ce dernier. Le complexe MutLα (MLH1-PMS2) est recruté au niveau du dommage par MutSα ou MutSβ. Le complexe PCNA (Proliferating Cell Nuclear Antigen) est recrutée au niveau de l’ADN par le facteur RFC (Replication Factor C) et interagit avec le complexe MutLα. Ensemble, PCNA et MutLα activent la fonction endonucléase de PMS2 (en jaune) qui amorce l’excision en 3’ du mésappariement. L’excision est complétée par l’exonucléase EXO1. Les protéines RPA (Replication Protein A) stabilisent et protègent la molécule d’ADN monocaténaire de la dégradation et participent à la dernière étape de l’excision. Le néo-brin corrigé est synthétisé par l’ADN polymérase δ et lié à la molécule d’ADN initiale par l’ADN ligase.
5’ 3’
MSH2 MSH6 MSH2 MSH3
MutSα Base-Base IDL MutSβ
MLH1 MutLα PMS2 ADN ligase ADN polymérase δ Activité endonucléase EXO1 RPA Activité exonucléase
Reconnaissance du mésappariement par MutS
Recrutement de MutL Excision Resynthèse et ligation ADN RFC PCNA 2 3 4 1
IV. Les conséquences moléculaires de la déficience du MMR & l’oncogenèse MSI colique L’oncogenèse MSI est caractérisée par une déficience fonctionnelle du système MMR suite à l’inactivation biallélique d’au moins un des gènes majeurs de ce système de réparation. Le CCR est l’une des premières tumeurs solides où l’hétérogénéité moléculaire fut mise en évidence (Fearon 2011). Il est intéressant de remarquer que dans le modèle de Fearon et Vogelstein, les mutations fréquentes observées dans les tumeurs MSS semblent suffisantes pour initier la progression tumorale (Matano et al., 2015). En revanche, dans les tumeurs MSI (sporadiques ou héréditaires), la première altération est l’inactivation du MMR qui n’est pas per se un évènement transformant. Suite à ce défaut, les clones MMR-déficients (MMR-d) accumulent de nombreuses mutations secondaires (insertions/délétions/mésappariements de bases) susceptibles d’affecter des gènes impliqués dans des processus cellulaires tels que la prolifération cellulaire, l’apoptose ou encore la réponse aux facteurs de croissance, responsables de leur transformation en clones tumoraux. L’instabilité nucléotidique MSI passible de favoriser l’oncogenèse est observée à travers tout le génome et notamment au niveau des régions répétées microsatellites (Ionov et al., 1993; Thibodeau, Bren and Schaid, 1993).
1. L’instabilité des microsatellites
Les microsatellites sont des séquences répétées abondantes réparties tout au long du génome humain. Présents à plus d’un million de loci, ils comptent pour environ 3% du génome humain (Thibodeau, Bren and Schaid, 1993; Ellegren, 2004). Chez les primates, les microsatellites mononucléotidiques sont les plus abondants suivis des dinucléotidiques. Les répétitions de motifs de trois, quatre ou cinq nucléotides sont beaucoup moins représentées et ces dernières ne semblent pas être affectées par l’instabilité causée par la déficience de système MMR (Nadir et al., 1996; Tóth et al., 2000; Subramanian, Mishra and Singh, 2003; Ellegren, 2004).
La nature répétée des microsatellites en fait des cibles privilégiées des erreurs de réplication commises par l’ADN polymérase. Leur stabilité durant la réplication dépend notamment de caractéristiques structurales inhérentes aux microsatellites. Il a été observé
que la fréquence des erreurs de l’ADN polymérase augmente avec la longueur de l’unité répétée (Kroutil et al., 1996; Strauss, Sagher and Acharya, 1997). La composition en bases affecte également la mutabilité : les microsatellites composés de C ou G étant plus instables que ceux composés de A ou T aussi bien chez les procaryotes que chez les eucaryotes (Sagher, Hsu and Strauss, 1999; Harfe and Jinks-Robertson, 2000; Boyer et al., 2002; Alhopuro et al., 2012; Kondelin et al., 2017). De plus, la nature des répétitions semble influencer le taux de mutation puisqu’il est rapporté que pour un nombre égal de répétitions, la stabilité augmente avec la taille du motif répété; autrement dit, les répétitions mononucléotidiques sont plus instables que les dinucléotidiques qui sont plus instables que les trinucléotidiques et ainsi de suite (Boyer et al., 2002). La composition des séquences adjacentes rend certains sites plus sensibles au glissement et donc aux erreurs de la polymérase (Duval and Hamelin, 2002). Enfin, il a été montré que le taux de mutation des microsatellites est positivement corrélé à l’état de compaction de la chromatine et à l’activité transcriptionnelle de ces régions de l’ADN : les domaines d’euchromatine et les introns sont enrichis en mutations comparés aux régions d’hétérochromatine et intergéniques respectivement (Kim, Laird and Park, 2013).
De fait, dans un contexte MMR-d, les mutations sont conservées et s’accumulent au fur et à mesure des cycles de réplication au niveau des séquences microsatellites codantes et non codantes. Les conséquences de telles mutations sont détaillées ci-après.
1.a. Mutations dans les régions répétées codantes : gènes cibles de l’instabilité Environ 20% des gènes humains contiennent une répétition microsatellitaire dont la taille est supérieure ou égale à 7 nucléotides au niveau de leur séquence codante (El-Bchiri et al., 2008), rendant le nombre de cibles potentielles de MSI considérablement élevé. Ainsi, les tumeurs MSI accumulent de nombreuses mutations dans des gènes contenant des microsatellites mononucléotidiques au niveau de leur séquence codante. Ces gènes sont nommés gènes cibles de l’instabilité MSI ou TGi pour Target Genes of instability (Duval and Hamelin, 2002). Les erreurs induites par la polymérase sont principalement des délétions et dans une moindre mesure des insertions d’un ou deux nucléotides (Kim, Laird and Park, 2013; Yoon et al., 2013). De telles mutations au niveau de microsatellites codants ont généralement
pour conséquence le décalage du cadre de lecture (mutations dites frameshift) et la génération d’un transcrit porteur d’un codon stop prématuré (figure 2) (Saeterdal et al., 2001).
Figure 2 │ Mutation de microsatellites codants et oncogenèse MSI
Les erreurs commises par l’ADN polymérase lors de la réplication de l’ADN et non réparées directement par celle-ci ne sont pas corrigées dans les cellules ayant un défaut du système MMR donnant lieu à de nombreuses mutations, notamment au niveau des microsatellites. Les conséquences de ces évènements mutationnels sur la transcription des gènes dépendent de la position du microsatellite au sein de la séquence codante du gène (localisation tantôt proximale en 5’ ou distale en 3’ de la phase ouverte de lecture). Dans la majorité des cas et comme présenté dans la figure, ces mutations peuvent causer un décalage du cadre de lecture et l’apparition d’un codon stop prématuré dans le transcrit muté. Ceci résulte en la synthèse d’une protéine tronquée dont le domaine C-terminal est modifié. Les répercussions fonctionnelles associées sont fréquemment des pertes de fonction.
7mG : coiffe 7-méthylguanine ; AAA : queue poly-A ; N-ter : région N-terminale ; ter : région C-terminale ATG AAA 7mG Exon Intron Gène sauvage NNNNNNNNNN (N10) 5’ 3’ NNNNNNNNNN (N9) 5’ 3’
Codon stop prématuré
Figure 3 │ Mutation de microsatellites codants et oncogenèse MSI
Les erreurs commises par l’ADN polymérase lors de la réplication de l’ADN et non réparées directement par celle-ci ne sont pas corrigées dans les cellules ayant un défaut du système MMR donnant lieu à des mutations notamment au niveau des microsatellites. Les conséquences de ces évènements mutationnels sur la transcription des gènes dépendent de la position du microsatellite au sein de la séquence codante du gène (localisation tantôt proximale en 5’ ou distale en 3’ de la phase ouverte de lecture). Dans la majorité des cas et comme présenté dans la figure, ces mutations peuvent causer un décalage du cadre de lecture et l’apparition d’un codon stop prématuré (ou PTC pour premature stop codon) dans le transcrit muté. Ceci résulte en la synthèse d’une protéine tronquée dont le domaine C-terminal est modifié. Les répercussions fonctionnelles associées sont fréquemment des pertes de
N-ter
C-ter Région C-ter modifiée Transcrit muté
Protéine tronquée
Erreur de réplication
MMR déficient: pas de réparation Conservation de la mutation Transcription Traduction Gène muté ARN ADN ADN
Toutefois, en fonction de la localisation proximale (5’) ou distale (3’) de la répétition codante au sein de la phase ouverte de lecture, une mutation peut avoir comme répercussion une perte de fonction de la protéine, totale ou partielle, ou d’autres conséquences comme des effets de dominance négative, de gain de fonction ou d’haplo-insuffisance.
L’apparition de telles mutations dans des gènes impliqués dans la carcinogenèse est susceptible de procurer un avantage sélectif de croissance aux cellules MMR-déficientes (Woerner et al., 2005). Une pléthore de travaux sur les CCR rapporte la présence de mutations fréquentes dans des gènes cibles de MSI impliqués dans la tumorigenèse. Le premier gène cible d’instabilité identifié fût le gène TGFBR2 (pour Transforming Growth Factor β Receptor type 2) dont le microsatellite codant A10 (10 adénines) est muté dans environ 90% des CCR MSI (Markowitz et al., 1995). L’altération de cette région répétée située en début de séquence codante par délétion d’une ou deux bases, engendre la synthèse d’une protéine tronquée non fonctionnelle (Wang et al., 1995). La mutation de ce récepteur abroge son effet suppresseur de tumeur et favorise la prolifération tumorale dans les stades précoces du développement des carcinomes (Lebrun, 2012; Pickup, Novitskiy and Moses, 2013).
Par la suite, grâce à des approches gènes candidats, il a été identifié de nombreux gènes cibles de l’instabilité MSI impliqués dans différents processus cellulaires comme la prolifération (ACVR2A, TCF-4, AXIN2, CDX, IGF2R), le cycle cellulaire et l’apoptose (BAX, CASP-5, RIZ, BCL-10, PTEN, FAS, APAF-1), la régulation épigénétique (ARID1A, HDCA2), la réparation de l’ADN (MSH3, MSH6, MLH3, CHK1, MDB-4, MRE11, BLM, RAD50, ATR) et plus récemment la machinerie des micro-ARN (TARBP2, XPO5) (figures 3 & 4) (Rampino, 1997; Schwartz et al., 1999; Woerner et al., 2001; Duval and Hamelin, 2002; Mori et al., 2002; Thorstensen et al., 2005; Imai and Yamamoto, 2008; Cajuso et al., 2014; Yamamoto and Imai, 2015). De façon intéressante, tous les gènes humains du MMR, à l’exception de MLH1, contiennent une répétition codante mononucléotidique d’au moins sept bases (Chang et al., 2001), illustrant le concept du « mutateur qui mute les autres mutateurs », contribuant à l’instabilité génomique et accélérant l’accumulation de mutations pendant la progression tumorale (Malkhosyan et al., 1996).
Figure 3 │ Représentation non-exhaustive des gènes cibles de l’instabilité microsatellitaire et potentiellement impliqués dans la carcinogenèse colique
De nombreux gènes cibles d’instabilité ont été identifiés dans les CCR MSI. Néanmoins, l’impact sur l’oncogenèse et la pertinence clinique de ces mutations dans la majorité des gènes restent à démontrer.
Adaptée de Yamamoto et al., 2015.
Un grand nombre des mutations de ces gènes a été identifié et leur fréquence mutationnelle analysée, grâce à des approches de criblage à haut débit (Mori et al., 2001; Woerner et al., 2001, 2009). Ainsi, le phénotype MSI implique l’accumulation de nombreuses mutations au sein de gènes, cependant, la présence d’une mutation au sein d’un gène n’implique pas nécessairement son rôle dans le développement tumoral. La difficulté aujourd’hui ne réside plus dans l’identification de nouveaux gènes cibles de MSI mais elle est véritablement dans le fait de déterminer et de distinguer les mutations survenant dans des gènes significativement impliqués dans l’initiation et la progression tumorale MSI, des multiples évènements sans impact significatif dans le processus oncogénique et résultant du bruit de fond de l’instabilité MSI dû au processus d’hyper-mutabilité induit par la déficience du système MMR. MSH6 MSH3 MLH3 MRE11 MDB4 ATM TCF4 HNF1A CDX2 E2F4 ARID1A HDAC2 CHD7/8 SETD2 Réparation de l’ADN Régulations épigénétiques Régulation de la transcription XPO5 TARBP2 TNRC6A AGO2
Biogenèse des micro-ARN
miR-1273C miR-1303 miR-567C Micro-ARN AXIN2 TGFBR2 PTEN ACVR2 RIZ ASTE1 Transduction du signal FAS CASP5 BAX BCL10 Apoptose Autre HSP110 MYB EWSR1 MT1XT20 MMP3 Surveillance immune β2M SEC63 Récepteur Mitochondrie Noyau Cytoplasme
Figure 4 │ Représentation non-exhaustive des gènes cibles de l’instabilité microsatellitaire et potentiellement impliqués dans la carcinogenèse colique
De nombreux gènes cibles d’instabilité ont été identifiés dans les CCR MSI. Néanmoins, l’impact sur l’oncogenèse et la pertinence clinique de ces mutations dans la majorité des gènes restent à démontrer.
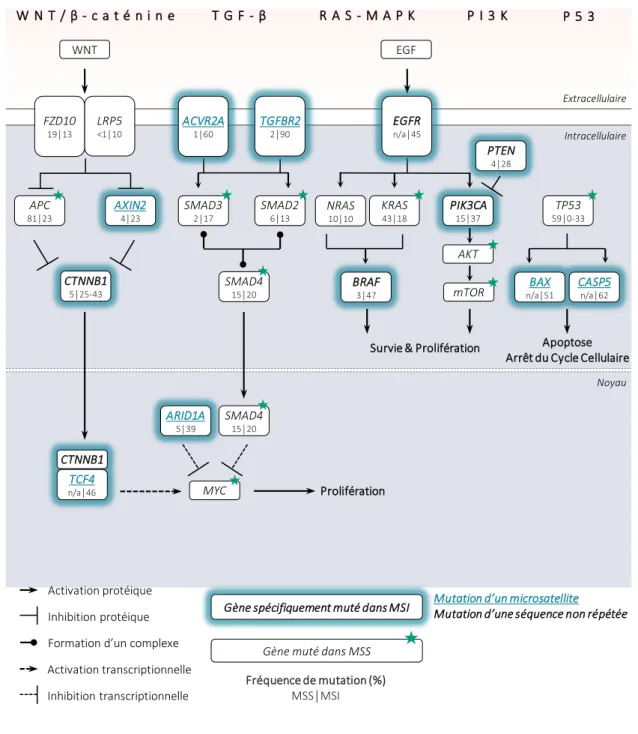
Figure 4 │ Représentation schématique et non-exhaustive de la diversité des mutations génétiques causant la dérégulation de voies de signalisation dans les cancers colorectaux
Bien que les voies de signalisation touchées dans les CCR MSI et MSS convergent, les mutations affectant les différents acteurs de ces voies semblent spécifiques en fonction de l’origine moléculaire du cancer.
n/a : donnée non disponible
Sources : Markovitz et al., 1995 ; Rampino, 1997 ; Schwart et al., 19999 ; Thorstensen et al., 2005 ; Cancer Genome Atlas, 2012 ; Cajuso et al., 2013 ; Tuupanen et al., 2014 ; Lin et al., 2015 ; Dehimi et al.,
2017.
Figure 5 │ Représentation schématique et non exhaustive de la diversité des mutations génétiques causant la dérégulation de voies de signalisation dans les cancers colorectaux
Bien que les voies de signalisation touchées dans les CCR MSI et MSS convergent, les mutations affectant les différents acteurs de ces voies semblent spécifiques en fonction de l’origine moléculaire du cancer.
na: donnée non disponible
Sources: Markovitz, 1995; Rampino, 1997; Schwart et al., 19999; Thorstensen, et al., 2005; Cancer Genome Atlas, 2016; Cajuso, 2013; Tuupanen, 2014; Lin, 2015, Dehimi, 2017
W N T / β - c a t é n i n e T G F - β R A S - M A P K P I 3 K P 5 3 Extracellulaire Intracellulaire Noyau WNT LRP5 <1│10 FZD10 19│13 APC 81│23 SMAD3 2│17 SMAD2 6│13 SMAD4 15│20 NRAS 10│10 KRAS 43│18 AKT mTOR
Survie & Prolifération
TP53
59│0-33
Apoptose Arrêt du Cycle Cellulaire EGF Prolifération MYC EGFR n/a│45 TGFBR2 2│90 ACVR2A 1│60 AXIN2 4│23 CTNNB1 5│25-43 PTEN 4│28 BRAF 3│47 PIK3CA 15│37 CASP5 n/a│62 BAX n/a│51 ARID1A 5│39 TCF4 n/a│46 CTNNB1 SMAD4 15│20 Activation transcriptionnelle Inhibition transcriptionnelle
Gène spécifiquement muté dans MSI
Activation protéique Inhibition protéique Formation d’un complexe
Gène muté dans MSS
Fréquence de mutation (%) MSS│MSI
Mutation d’un microsatellite
En 2002, mon équipe d’accueil a proposé un classement des gènes cibles MSI en fonction de leur implication dans le processus de transformation tumorale (Duval and Hamelin, 2002). Selon cette classification, les gènes dont les mutations participent à la transformation maligne présenteraient un taux de mutation élevé et confèreraient un avantage sélectif pendant la progression tumorale aux clones MSI qui les contiennent ; ces évènements sont soumis à des pressions de sélection positive. À l’inverse, il a été suggéré que des évènements dans des gènes essentiels à la survie de la cellule tumorale seraient contre-sélectionnés (pression de sélection négative) et donc observés à de basses fréquences mutationnelles. Différents modèles statistiques ont été proposés pour l’identification des gènes cibles MSI ayant un réel impact dans la tumorigenèse (figure 5) (Duval et al., 2001; Woerner et al., 2003; Alhopuro et al., 2012). La base de données SelTarbase (pour Selective Targets in human MSI-H tumorigenesis database) répertorie les mutations (et leur fréquence) de microsatellites mononucléotidiques codants, non transcrits, non codants et introniques de CCR à partir de près de 400 publications. À partir de ces données, un modèle de prédiction du potentiel impact de ces mutations sur la tumorigenèse a été proposé (figure 5) (Woerner et al., 2009). Néanmoins, la pertinence fonctionnelle de l’impact sur l’oncogenèse des gènes fréquemment mutés reste à démontrer pour la majorité d’entre eux.
Il est intéressant de noter que le répertoire de gènes cible d’instabilité semble être différent selon la localisation tumorale. En effet, les études comparatives de tumeurs MSI colorectales, gastriques et de l’endomètre ont révélé que les taux de mutations et les gènes affectés varient selon le tissu considéré (Duval et al., 2002; Furlan et al., 2002; Kuismanen et al., 2002). Ces signatures tissu-spécifique ont été confirmées à grande échelle sur un large spectre de tumeurs MSI grâce à des approches de séquençage d’exome à haut débit (Kim, Laird and Park, 2013; Hause et al., 2016). De la sorte, les cancers gastriques et colorectaux semblent partager des ensembles de mutations similaires. En revanche, les tumeurs de l’endomètre arborent des profils mutationnels différents des cancers gastro-intestinaux, suggérant une origine moléculaire distincte en fonction de la localisation tumorale (Duval et al. 2002; Kim et al. 2013). En ce qui concerne une même localisation tumorale, des signatures différentielles d’expression de gènes semblent aussi être retrouvées en fonction de l’origine sporadique ou héréditaire des cancers comme par exemple la mutation de BRAF, quasiment exclusive des CCR MSI sporadiques, ou la mutation de CTNNB1 qui semble être plus fréquente
dans les CCR d’origine héréditaire (Deng et al., 2004; Domingo, 2004; Albuquerque et al., 2010; Cancer Genome Atlas, 2012).
Figure 5 │ Modélisation des catégories de gènes cibles de l’instabilité microsatellitaire
a│ Modèle proposé afin de classifier les gènes cibles de l’instabilité (régions codantes et UTRs) selon le rôle et l’impact de leurs mutations dans la carcinogenèse. Les fréquences mutationnelles sont dépendantes des pressions de sélection. b│ Modèle de régression sigmoïde des répétitions mononucléotidiques non codantes (i.e. régions introniques et UTR) et codantes de CCR MSI. La ligne de régression ajustée est représentée par un trait gris plein et les lignes de prédiction supérieure et inférieure sont représentées par un trait gris en pointillés.
Adaptée de Duval & Hamelin, 2002 & Woerner et al., 2003.
S é l e c t i o n n é e C o n t r e - s é l e c t i o n n é e A n t i - t u m o r a l e P r o - t u m o r a l e b Ta u x d e m u ta ti o n (% ) 0 20 40 60 80 100 2 4 6 8 10 12 14 16 Longueur du microsatellite 18 20 Microsatellites intergéniques Microsatellites non codants Microsatellites codants Cibles MSI Elevé Faible Dépendant de: A/T G/C
Répétition courte Répétition longue Séquences environnantes Fonction présumée de la mutation Taux de mutation a
Figure 6 │ Modélisation des catégories de gènes cibles de l’instabilité microsatellite
a│ Modèle proposé afin de classifier les gènes cibles de l’instabilité (régions codantes et UTRs)
selon le rôle et l’impact de leurs mutations dans la carcinogenèse. Les fréquences mutationnelles sont dépendantes des pressions de sélections. b│ Analyse de régression sigmoïde des répétitions mononucléotidiques non-codantes (i.e. régions introniques et UTR) et codantes de CCR MSI.
Adapté de Duval and Hamelin, 2002 & Woerner et al., 2003
M S I Pression de sélection positive Pression de sélection négative Mutation
1.b. Mutations dans les régions répétées non codantes
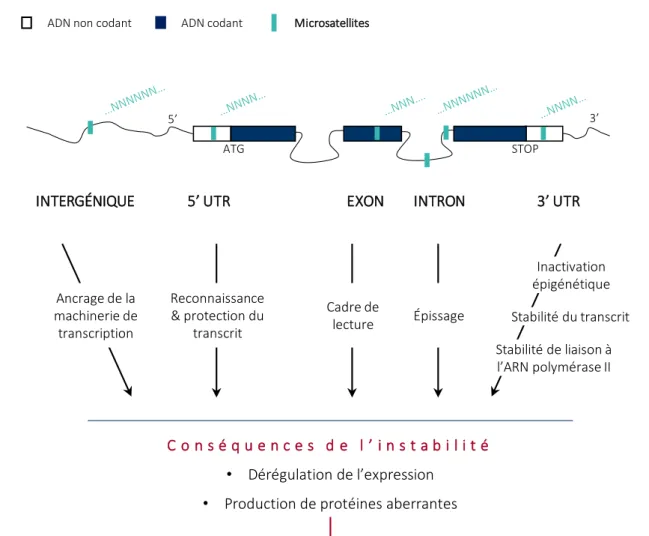
Outre les microsatellites de régions codantes de gènes, la majorité des microsatellites est présente au niveau de séquences répétées non codantes incluant les régions intragéniques et intergéniques (Tóth et al., 2000; Ellegren, 2004). Des mutations dans les régions intragéniques (introns, promoteurs et domaines 5’ et 3’ UTR), non traduites (long ARN non codants et micro-ARN) ou intergéniques (nécessaires à l’amarrage de protéines recrutées pour la transcription et la conformation de la chromatine) sont susceptibles d’influencer les niveaux d’expression de gènes et/ou la fidélité de transcription de l’ARN (figure 6). Cependant, la fonction de répétitions de l’ADN et les conséquences de leur instabilité au niveau de ces régions demeurent majoritairement inconnues. Les quelques travaux rapportant la présence d’altérations dans les microsatellites de ces régions sont décrits dans la section suivante.
i. Répétitions dans les régions 5’ et 3’ UTR
Les microsatellites de régions 5’ et 3’ UTR (pour Untranslated Regions, i.e. régions transcrites et non traduites des gènes) sont susceptibles de contribuer à la régulation de la transcription, à la localisation subcellulaire, à la stabilisation du transcrit ou encore à la fixation de facteurs de reconnaissance du site d’initiation de la traduction (Chatterjee and Pal, 2009). Il a été observé, à la suite d’évènements MSI dans les régions 3’ UTR de certains gènes, des modifications de l’expression de ces derniers (Yoon et al., 2013) ; l’altération de l’activité post-transcriptionnelle d’ARN messagers (ARNm) peut être due à la perturbation de la stabilité de microsatellites présents dans des sites d’ancrage de protéines de liaisons aux ARNm (Ruggiero et al., 2003; Paun et al., 2009). Pour exemples, la mutation d’une séquence répétée A13 (13 adénines) située dans la région 3’ UTR de l’oncogène EGFR (Epidermal Growth Factor Receptor) est présente dans environ 65% de CCR MSI et il a été montré qu’elle est corrélée à une stabilisation du transcrit ainsi qu’à une surexpression de ce facteur. De plus, dans des expériences in vitro sur des lignées cellulaires, cette altération procure un avantage de croissance aux sous-clones tumoraux qui la contiennent (Yuan et al., 2009). Des mutations dans les 3’ UTR des gènes EWSR1 (EWing Sarcoma breakpoint Region 1) ou mPGES-1 (microsomal ProstaGlandin E Synthase-1) ont été rapportées comme fréquemment observées
fonctionnelle n’ait été démontrée sur le rôle de leur instabilité dans le développement tumoral MSI (Kishore et al., 2014; Sasaki, Nakatani and Hara, 2015). Il est à noter que les caractéristiques de faible polymorphisme de ces séquences non codantes dans les populations étudiées indiquent des pressions de sélection au cours de l’évolution sur celles-ci et suggèrent un potentiel rôle fonctionnel dans le contrôle de l’expression génique.
Figure 6 │ Les microsatellites à travers le génome
Les microsatellites sont présents dans les régions intergéniques et intragéniques du génome comme les exons, les introns et les 5’ et 3’ UTR. Des mutations les affectant peuvent avoir des répercussions fonctionnelles sur les ARN et protéines codés par les gènes qui les contiennent.
Source : Li et al., 2004.
Microsatellites ADN codant
ADN non codant
5’ UTR ATG 3’ UTR STOP EXON INTRON INTERGÉNIQUE 5’ 3’ Reconnaissance & protection du transcrit Épissage Cadre de lecture C o n s é q u e n c e s d e l ’ i n s t a b i l i t é • Dérégulation de l’expression
• Production de protéines aberrantes Changements phénotypiques Stabilité du transcrit Inactivation épigénétique Stabilité de liaison à l’ARN polymérase II Ancrage de la machinerie de transcription
Figure 7 │ Les microsatellites à travers le génome
Les microsatellites sont présents dans les régions intergéniques et intragéniques du génome comme les exons, les introns et les 5’ et 3’ UTR. Des mutations les affectant peuvent avoir des répercussions fonctionnelles sur les ARN et protéines codés par les gènes qui les contiennent.
ii. Répétitions introniques situées à proximité d’un site accepteur d’épissage ou d’un site régulateur de la transcription
Jusqu’à présent, peu d’études ont été consacrées aux mutations de microsatellites introniques, nonobstant leurs conséquences éventuelles sur le niveau d’expression et la régulation de l’épissage de leurs cibles. Néanmoins, quelques travaux ont mis en évidence les conséquences fonctionnelles de leur instabilité sur des gènes qui pourraient être associés au développement tumoral MSI.
Un exemple est l’oncogène MYB, qui contient dans son premier intron une séquence répétée de 19 thymidines (T19) ayant pour rôle la régulation négative de la transcription de ce gène. Il a été observé que des mutations au niveau de cette répétition conduisent à une surexpression de MYB dans les tumeurs MSI (Hugo et al., 2006). De manière intéressante, la forte expression de ce gène chez des patients atteints de CCR est associée à un mauvais pronostic de survie en cohérence avec un rôle oncogénique de cette mutation dans les cancers MSI (Biroccio et al., 2001).
Par ailleurs, des mutations peuvent affecter des microsatellites présents dans les séquences non codantes mais ayant un rôle fonctionnel dans l’homéostasie du gène comme les régions à proximité (en amont ou en aval) de sites accepteurs ou donneurs d’épissage. Il a été observé que les bornes introns-exons des gènes sont des régions enrichies en microsatellites d’environ quinze-vingt nucléotides pyrimidiques appelées séquences de poly-pyrimidines (Schellenberg, Ritchie and MacMillan, 2008). Ces éléments cis de l’ADN sont des séquences conservées, généralement de poly-thymidines, reconnues par des éléments trans du spliceosome et déterminants dans la régulation de l’épissage des ARNm précurseurs (Lewandowska, 2013). Ainsi, les gènes MRE11, ATM et HSP110 possèdent des microsatellites mononucléotidiques introniques et sont décrits comme cibles de l’instabilité microsatellitaire dans les CCR MSI.
Ainsi, la mutation du microsatellite T11 situé au niveau de la borne d’épissage de l’intron 4 du gène MRE11 a été mise en évidence en 2004 (Giannini et al., 2004). Elle est observée dans 80% des cancers MSI et conduit à la réduction de l’expression de MRE11 ainsi qu’à un défaut d’expression du complexe MRN. Ce dernier, formé par les protéines MRE11, RAD50 et NBS1, est impliqué dans la réparation des cassures doubles brin de l’ADN et sa déficience est
associée à une forte instabilité génomique et à une prédisposition au cancer (Giannini et al., 2004; van der Heijden et al., 2006). La mutation de MRE11 semble par conséquent être oncogénique. En revanche, il a été observé que la mutation biallélique de MRE11 (environ 40% des CCR MSI ; Giannini et al. 2004) sensibilise les cellules CCR MMR-d aux traitements anti-cancéreux utilisant des inhibiteurs de PARP-1 ou les agents de chimiothérapies classiques (Vilar et al., 2008, 2011; McPherson, Shen and Ford, 2014). Ces résultats suggèrent que cette mutation serait un facteur prédictif de réponse positive aux traitements dans les cancers MSI. De même, les mutations de microsatellites poly-thymidiques présents dans les introns 8 et 12 du gène ATM, une sérine-thréonine kinase impliquée dans la transduction du signal des cassures double brin, ont été identifiées. L’instabilité de ces microsatellites est responsable de la synthèse de transcrits aberrants dans des lignées cellulaires de CCR MSI. Les conséquences phénotypiques de telles modifications restent à déterminer, cependant il a été observé dans une lignée cellulaire dérivant d’un patient ayant des antécédents familiaux de cancer, de rares altérations polymorphiques d’ATM semblant être associées à une prédisposition au cancer (Ejima, Yang and Sasaki, 2000). De manière intéressante, la mutation d’ATM dans des lignées cellulaires coliques est corrélée à une résistance aux anticorps monoclonaux anti-EGF-R et constitue un potentiel nouveau marqueur prédictif de la réponse aux thérapies ciblées anti-EGF-R (Geißler et al., 2017).
Enfin, en 2011 mon laboratoire d’accueil a identifié une mutation de la séquence répétée T17 située à proximité du site accepteur d’épissage de l’intron 8 du gène HSP110 (Heat Shock Protein 110), comme une cible d’instabilité universelle, du fait de la présence de mutations dans près de 100% des CCR MSI (Dorard et al., 2011). L’instabilité de ce microsatellite induit un épissage aberrant, le saut de l’exon 9 et a pour conséquence le décalage du cadre de lecture ainsi que la formation d’un codon stop prématuré dans le transcrit mutant généré HSP110DE9. Il a été démontré que la taille de la délétion et l’augmentation du taux d’épissage aberrant pour ce transcrit sont corrélés. D’un point de vue fonctionnel, une grande délétion du T17 (supérieure à 5 bp ; 25% des CCR MSI), induit une perte totale de l’expression de la protéine HSP110. Quant au transcrit mutant HSP110DE9, il est exprimé à un faible taux puisque pris en charge par le système NMD (pour Non-sense Mediated mRNA Decay) de surveillance des transcrits. Celui-ci détecte et induit la dégradation des ARN messagers porteurs d’un codon stop prématuré comme c’est le cas d’HSP110DE9
(Collura et al., 2014). Ce travail rapporte que cette mutation a des effets pro-apoptotiques et qu’elle serait un facteur prédictif de la réponse à la chimiothérapie. En effet, les patients atteints d’un CCR MSI traités par chimiothérapie et porteurs d’une grande délétion de ce microsatellite, présentent un meilleur pronostic en comparaison aux patients avec une petite délétion. Mon laboratoire d’accueil a démontré que la mutation du microsatellite T17 d’HSP110 a pour conséquence une diminution de la prolifération cellulaire et que cet effet anti-tumoral est en partie associé à la diminution du facteur de transcription STAT3 (Berthenet et al., 2016). Une grande mutation du microsatellite T17 est un événement a priori surprenant puisque son action est délétère pour les cellules tumorales (rôle pro-apoptotique et anti-prolifératif). L’hypothèse pour expliquer la survenue fréquente d’un tel événement dans le cancer est que l’instabilité de ce microsatellite est inéluctable dans les cellules tumorales MSI. En effet, ces clones MMR-d dont l’index mitotique est souvent très élevé, ne sont pas en mesure de réparer les erreurs de réplication qui surviennent inévitablement et à haute fréquence au niveau de telles répétitions génomiques introniques de grande taille (Duval et al., 2002).
iii. Répétitions dans les régions intergéniques et les trans-éléments
Peu de travaux sont répertoriés concernant l’instabilité de séquences répétées d’éléments trans-régulateurs, comme les longs ARN non codants et les micro-ARN (miRNA), ou d’éléments cis-régulateurs, comme les régions intergéniques, qui pourraient modifier les niveaux d’expression de gènes impliqués dans les processus de carcinogenèse. Les miRNA sont des petits ARN non codants d’environ vingt-deux nucléotides transcrits mais non traduits qui fonctionnent comme des répresseurs de l’expression de gènes (Earle et al., 2010). Des études ont montré une expression différentielle de miRNA entre les tissus de carcinomes et les tissus sains associés (Lanza et al., 2007). De plus, certains miRNA sont spécifiquement dérégulés dans les CCR MSI versus MSS et une hétérogénéité intra-MSI a été mise en évidence avec des signatures moléculaires de miRNA distinctes entre les CCR MSI sporadiques et héréditaires (Lanza et al., 2007; Earle et al., 2010). En 2012, mon équipe d’accueil a observé que les répétitions microsatellitaires au sein de miRNA sont rares et relativement stables dans les CCR MSI (El-Murr et al., 2012). Parmi les vingt-quatre miRNA étudiés, une instabilité
microsatellitaire fût détectée dans quinze d’entre eux avec des fréquences comprises entre 3 et 100% selon la taille de la répétition. La fréquence mutationnelle importante pour trois de ces miRNA (i.e. hsa-mir-1273c, hsa-mir-567 et hsa-mir-1303), mutés dans plus de 80% des cas de CCR MSI), suggère que ces mutations sont exposées à des phénomènes de pression de sélection au cours de la carcinogenèse MSI. Cependant, bien que les signatures de miRNA soient pertinentes biologiquement et cliniquement et qu’elles participent à la stratification des tumeurs, l’impact fonctionnel et la pathogénicité de ces mutations restent inconnus.
1.c. Le séquençage de nouvelle génération (NGS) : un outil indispensable pour l’étude des mutations MSI
Le développement récent de techniques à haut débit conduit à une révolution de l’appréhension de la complexité du vivant dans son ensemble via l’analyse simultanée d’un grand nombre de variables issues d’approches dites « omiques », à savoir principalement la génomique, l’épigénomique, la transcriptomique, la protéomique et la métabolomique. Les approches de séquençage à haut débit (méthodes de NGS pour Next Generation Sequencing) permettent la détermination des profils génomiques, exomiques ou encore transcriptomiques des tumeurs –grâce respectivement au Whole-Genome Sequencing (WGS), Whole-Exome Sequencing (WES) et au RNA-sequencing (RNA-seq).
Dans un contexte tumoral, et particulièrement dans le contexte MSI dont le phénotype hypermutateur induit une accumulation de mutations cent fois plus importante que dans un contexte non-MSI (soient environ 12 à 40 mutations/mégabase ; Cancer Genome Atlas 2012; Nebot-Bral et al., 2017), ces approches permettent une analyse globale des tumeurs et la détermination du spectre mutationnel spécifique des néoplasmes en comparaison au tissu sain.
En 2013, Kim et ses collaborateurs ont réalisé la première étude offrant une vision globale du profil génomique de tumeurs MSI à partir du séquençage des exomes et génomes de vingt-sept cancers colorectaux et de trente cancers de l’endomètre (Kim, Laird and Park, 2013). De façon intéressante, cette étude met en évidence que l’instabilité des microsatellites est dépendante de l’organisation de la chromatine, celle-ci étant sur-représentée dans les régions euchromatiques et introniques en comparaison des régions hétérochromatiques et