UNIVERSITE TOULOUSE III–PAUL SABATIER UFR S.V.T
THESE
Pour obtenir le grade deDocteur de L’UNIVERSITE DE TOULOUSE
délivré par l’Université Toulouse III – Paul Sabatier
Discipline : Biologie, Santé et Biotechnologies Spécialité : Cancérologie
Présentée et soutenue par
SKULI NICOLAS
Soutenance Mardi 11 Décembre 2007IMPLICATION DES VOIES DE SIGNALISATION
DE L’ADHERENCE CELLULAIRE DANS LE CONTROLE DE
L’HYPOXIE TUMORALE DES GLIOBLASTOMES
Directrices de Thèse :
Dr TOULAS Christine
Pr COHEN-JONATHAN MOYAL Elizabeth
Jury
Pr Favre Gilles Président
Pr Lambin Philippe Rapporteur
Pr Lartigau Eric Rapporteur
Dr Van Obberghen-Schilling Ellen Examinatrice
Pr Cohen-Jonathan Moyal Elizabeth Directrice de Thèse
Dr Toulas Christine Directrice de Thèse
INSERM U563
Département d’Innovation Thérapeutique et d’Oncologie Moléculaire INSTITUT CLAUDIUS REGAUD
« Remerciements »
A ma famille.
Tout d’abord à mes parents. Merci de m’avoir toujours supporté, encouragé et de m’avoir permis d’atteindre ce niveau. Je vous dois énormément. Je vous aime.
A mon frère Matthieu et ma sœur Hélène, c’est bon de savoir que vous serez toujours à mes côtés. Merci Mat pour ton aide précieuse …
Cinq pour tous et tous pour cinq …
A mes grands-parents, Lucien et Léa, et Marcel et Elizabeth, sans qui non plus je ne serais pas là aujourd’hui et oui forcément...
A toute ma famille, tantes, oncles, cousins et cousines, je pense bien à vous.
A tous mes potes, de Toulouse et d’ailleurs.
En particulier à Lolo, bientôt ça sera la tienne, et Yan, mes acolytes, il y aurait tant à dire mais merci pour les bons délires passés et futurs, viva Espagna …
A la J.S.A.L avec Spit, Julien, Auré (crotte), Youssef, Sim, Yannick, Mathias, Mat A, Maxence, Seb, Mat M, Stephus, Nanus, Alex, Bernard, Claude, Daniel, Mig et tous les autres. Jouer à vos côtés aura été un vrai régal. Je n’oublierai pas l’année de la montée et les soirées qui sont allées avec.
A mes potes de fac qui ont rendu les cours plus supportables. Dans le désordre, p’tit Nico, Patrice, Simon, Mathilde, Seb et Maryline, Mitch, Carlos, Marie, Jérôme, Delphine, Alice, Annick, Jean-Philippe, JF et Marina.
Aux sapeurs pompiers de l’Aude et en particulier à tous les membres (pompiers, mécaniciens ou pilotes) du pélicandrome de Carcassonne que j’ai rencontré au cours de ces 9 ans de service (record à battre …). Merci à Nico, Max, Vincent, Gilles et tous les autres pour l’ambiance, les batailles d’eau (épiques), les coups de bourre, les bons souvenirs et les parties de coinche … Petite pensée pour Régis qui m’a fait piloté mon 1er Tracker.
A la Corse avec Pierre-Jean (on ne se voit peut être pas souvent, mais on se donne des nouvelles régulièrement), Pierre, Pascal et les autres. Que de bonheur, ces moments passés sur cette île magique. A bientôt.
Au laboratoire où j’ai effectué cette thèse.
A Christine et Elizabeth. Je vous dois énormément. J’ai passé 4 années super au sein de l’équipe radibio. C’était énorme. Toujours à fond mais toujours là quand il le faut, que demander de plus … D’ailleurs je sais pas comment vous faites (je crois que Calicéo est dans le coup …) Christine (je te prends quand tu veux au bad), merci pour ton efficacité et ta rigueur surtout sur la fin, et Elizabeth, merci pour tes conseils précieux et ta bonne humeur (j’espère que tu viendras chercher toi-même ta tasse au « black cat » …). S’il fallait vous définir en un mot, ça serait … euh … « pêchu », oui c’est bien ça « pêchu » (me consulter pour la définition exacte). Je vous enverrai des petits poulets voyageurs de l’autre côté de l’océan pour vous donner des nouvelles.
A Caro, ah ma Caro …, depuis le DEA, je suis très fier d’avoir pu travailler avec toi. Tu m’as beaucoup appris (en fait à peu près tout) et tu es la sauveuse des manips perdues. Je ne serais jamais enceinte mais maintenant je suis paré. Je sais tout ce qu’il faut savoir sur le sujet … On a partagé de bons moments quand même. Ca va me manquer. Encore merci. Si j’étais un petit bonobo, je m’appellerais, je m’appelerais C..o.
A Sylvie, j’ai également beaucoup appris à ton contact. Je crois que tu es la postdoc (pardon, maître de conf. maintenant, scuse) la plus gentille que j’ai jamais rencontrée. Toujours de bonne humeur (sauf quand elle a faim, là, il faut pas l’embêter) et prête à rendre service. Merci d’avoir partagé avec moi un peu de ton immense savoir ;). Vive le Yogamasutra…
A Julie M, merci de m’avoir encadré dans mes débuts. Tu es la preuve vivante qu’on peut faire un bébé, rédiger sa thèse, la soutenir et se marier, le tout dans la même année … De quoi je me plains moi …
Au bureau du 3ième, avec les filles : Emilie M (c’est où Limoges ?), Elise (profite bien de ta thèse), Yann (mon remplaçant… y a encore du boulot quand même), Hélène, Olivia, Emilie B du 3ième, merci pour la bonne humeur et la bonne ambiance que vous y faites régner. Au 3ième en général, la team du séquenceur avec Isa G, Sylvia, Domi, Françoise (dit Tigre des Bois), Angèle, Sophie DS, Bettina, Danièle, Alf et aux Jackson Five …
Au 2nd étage, ça en fait du monde … à l’équipe des Wagnériens, Stéphan(ia), Pierre (meilleur au footing qu’au foot, bah c’est presque pareil), Anne V/C (bon courage pour la suite), Gégé (le dieu réunionnais), Sandra et Sam. A Claire G, Jean Charles, Patate (le plus vieux thésard que je connaisse, RDV aux Violettes), Nelly, Laurent. A Anne P, Marie, Anne C, Marie-Ange, Dan T (bonne chance pour ta nouvelle carrière), Valérie, Emilie B du 2ième, Francis, ILM (allez Lagardelle), l’autre Claire, Rémi, Magot (plus connu sous le prénom de Marine), Bubu (le cyclope… non c’est pas drôle).
Au 4ième avec Philou et Mick. A l’équipe des deux immunologistes, Fafita et Guigui (si tu réussis pas dans la recherche, tu pourras toujours monter une agence matrimoniale avec Caro, elle sera contente, elle aura des potins comme ça, merci ;)). A mes copines les petites souris, Christiane et Lourdes.
A la routine et Jeanine, pour les petits « bonjour » au passage.
A la pharmaco, on ne sait pas trop ce qu’ils font mais ils le font… à Ben, Diane (les rats vous se souvenir de nous), Isa H et L, Tihy, Fabienne et Sabrina.
A tous les autres, Arash et Jérôme (deux très bons acteurs, on croit toujours que vous êtes de vrais médecins les gars …), étudiants morts ou vivants passés au labo, Cyril (Allez Brax), Régis, Leïla, Nowel, Pierre B, Hilda, Aurélie, Isa B, Moustapha (un jour, j’irai te voir mais quand ça sera plus calme), Barbie (la néo-bordelaise), Adrien, Adeline, Julie B, Raph, Chantal, Lionel (le chapoteur)…
Aux membres du Jury.
Au Professeur Gilles Favre. Merci de m’avoir accueilli dans votre laboratoire pendant ces 4 années. J’ai passé de très bons moments au sein de votre laboratoire tant au niveau professionnel que relationnel.
Au Professeur Eric Lartigau, merci d’avoir accepté de participer à ce jury de thèse en tant que rapporteur et de m’avoir consacré de votre temps pour évaluer ce travail. Je vous en suis très reconnaissant.
Au Professeur Philippe Lambin, merci également d’avoir accepté de participer à ce jury de thèse en tant que rapporteur. Merci pour votre réactivité, où que vous soyez sur le globe, qui fut très appréciable.
Au Docteur Ellen Van Obberghen-Schilling, merci de faire partie de ce jury en tant qu’examinatrice et d’avoir accepté de juger ce travail de thèse. Merci pour votre gentillesse ainsi que pour votre disponibilité.
A tous ceux que je n’ai pas cité mais qui ont fait partie de cette aventure et que j’ai pu connaître tout au long de ces années.
Enfin à Cindy, merci pour ton amour et ton soutien de tous les instants. Je n’ai pas la place ici pour écrire tout ce que je te dois. Cette thèse n’aurait pas été la même sans toi, ça c’est sur… Jtm. Doud ;)
SOMMAIRE ... 6
LISTE DES FIGURES... 12
LISTE DES TABLEAUX... 14
RESUME ... 15
ABREVIATIONS ... 16
REVUE BIBLIOGRAPHIQUE... 18
Introduction ... 20
1- Les tumeurs cérébrales... 21
1-1 Généralités ... 21 1-2 Causes ... 23 1-3 La classification histo-pronostique... 23 1-4 Les traitements ... 25 1-5 Le Glioblastome... 25 2- Phénomènes de radiorésistance... 29
2-1 Le nombre de cellules clonogéniques... 29
2-2 La cinétique de prolifération... 29
2-3 La radiorésistance intrinsèque ... 29
2-4 La radiorésistance extrinsèque liée au micro-environnement ... 29
3- L’hypoxie ... 32
3-1 Le facteur inductible par l’hypoxie, Hypoxia Inducible Factor 1 : HIF-1 ... 34
3-2 La sous-unité HIF-1α ... 37
3-3 HIF-1α, HIF-2α, HIF-3α et épissage alternatif ... 39
3-4 Les régulations du facteur inductible par l’hypoxie (HIF-1) ... 41
3-4-1 La principale régulation de l’expression de HIF-1α : le taux d’oxygène... 42
3-4-1-1 L’hydroxylation ... 42
3-4-1-2 L’acétylation ... 45
3-4-2 Les autres régulations de HIF-1 ... 45
3-4-2-1 La phosphorylation ... 45
3-4-2-2 La sumoylation ... 47
3-4-2-3 La nitrosylation ... 48
3-4-3 Les interactions de HIF-1α ... 48
3-4-3-1 Les interactions qui modulent la stabilité et l’activité transcriptionnelle de HIF-1α ... 48
3-4-3-2 Interactions qui régulent préférentiellement la stabilité de HIF-1α ... 51
3-4-3-3 Interactions qui modulent préférentiellement l’activité transcriptionnelle de HIF-1α ... 52
3-6 Survie, prolifération cellulaire et angiogenèse ... 54
4- Les GTPases de la famille des Rho... 57
4-1 Structure des protéines Rho ... 57
4-2 Expression ... 59
4-3 Localisation subcellulaire ... 59
4-4 Les modifications post-traductionnelles ... 61
4-5 Cycle d’activation/inactivation des petites GTPases Rho ... 63
4-5-1 Régulateurs des GTPases Rho ... 63
4-5-2 Les guanine nucleotide exchange factors : GEFs ... 65
4-5-3 Les GTPase activating proteins : GAPs ... 65
4-5-4 Les guanine nucleotide dissociation inhibitors : GDIs... 66
4-6 Fonctions ... 67
4-6-1 GTPases Rho et cytosquelette d’actine ... 67
4-6-2 GTPases Rho et trafic cellulaire ... 69
4-6-3 GTPases Rho, transcription, prolifération et survie cellulaire ... 69
4-6-4 GTPases Rho et oncogenèse ... 70
4-6-5 GTPases Rho, stress et hypoxie ... 73
4-6-5-1 L’hypoxie régule les petites GTPases Rho ... 73
4-6-5-2 Les GTPase Rho peuvent contrôler la réponse et l’adaptation des cellules à l’hypoxie ... 74
4-7 Les inhibiteurs de Farnésyl Transférase (FTIs) ... 75
5- La petite GTPase RhoB ... 77
5-1 Régulation de l’expression de RhoB ... 77
5-1-1 Cycle cellulaire ... 77
5-1-2 Facteurs de croissance ... 79
5-1-3 GTPases Rho ... 79
5-1-4 Stress génotoxiques ... 79
5-1-5 Glucocorticoïdes ... 80
5-2 Modifications post-traductionnelles de RhoB... 80
5-2-1 La prénylation ... 80
5-2-2 La palmitoylation ... 80
5-3 Régulation de l’activation de RhoB (liaison GTP/GDP) ... 81
5-3-1 RhoB et GEFs ... 81
5-3-2 RhoB et GDIs ... 82
5-4 Effecteurs de RhoB ... 82
5-5-1 RhoB et cytosquelette d’actine ... 83
5-5-2 RhoB et trafic intracellulaire ... 84
5-5-3 RhoB et cycle cellulaire ... 85
5-5-4 RhoB et réponse transcriptionnelle ... 86
5-5-5 RhoB, adhérence cellulaire et migration ... 87
5-5-6 RhoB et oncogenèse ... 87
5-5-6-1 RhoB et transformation cellulaire ... 88
5-5-6-2 RhoB et tumeurs ... 90
5-5-6-3 Rôle de RhoB et dans la transformation cellulaire ... 91
5-6 RhoB et réponse au stress ... 93
5-6-1 RhoB et réponse aux stress génotoxiques ... 93
5-6-2 RhoB et chimiotoxiques ... 93
5-6-3 RhoB, UV, radiations ionisantes et hypoxie ... 94
6- Les voies de l’adhérence cellulaire ... 98
6-1 La famille des intégrines ... 98
6-2 La structure des intégrines ... 101
6-3 Les intégrines participent à l’adhérence à la matrice extracellulaire ... 103
6-4 Les voies de transduction du signal dépendantes des intégrines ... 105
6-4-1 La signalisation médiée par l’Integrin Linked Kinase (ILK) ... 107
6-4-2 La signalisation médiée par la Focal Adhesion Kinase (FAK) ... 110
6-4-3 La signalisation médiée par la Particularly Interesting New Cysteine-Histidine rich protein (PINCH) ... 113
6-4-4 La Non-Catalytic (region of) tyrosine Kinase adaptator protein (Nck), un lien entre signalisation des intégrines et facteurs de croissance ... 114
6-5 Expression des intégrines et cellules invasives ... 115
6-6 Intégrines, voies de l’adhérence cellulaire et stress génotoxiques ... 116
6-7 Voies de l’adhérence cellulaire, intégrines et hypoxie ... 119
6-7-1 L’hypoxie régule les protéines des voies de l’adhérence cellulaire ... 119
6-7-1-1 L’hypoxie régule les intégrines ... 119
6-7-1-2 L’hypoxie régule la protéine ILK ... 122
6-7-1-3 L’hypoxie régule la protéine FAK ... 123
6-7-2 Les voies de l’adhérence cellulaire peuvent contrôler la réponse et l’adaptation des cellules à l’hypoxie ... 125
6-7-2-1 Les intégrines modulent la réponse des cellules à l’hypoxie ... 125
6-7-2-2 ILK régule le facteur de transcription HIF-1 ... 126
6-8 Inhibiteurs d’intégrines, d’ILK et de FAK et essais cliniques ... 129 OBJECTIFS DE LA THESE... 132 PUBLICATIONS... 134 ARTICLE 1... 136 ARTICLE 2... 140 ARTICLE 3... 144 CONCLUSION ... 148 BIBLIOGRAPHIE ... 158
LISTE DES FIGURES
Figure 1 : Glioblastome Multiforme observé par IRM au niveau du lobe temporal... 22 Figure 2 : Glioblastome primaire (tranche frontale passant par le pulvinar)... 26 Figure 3 : Schéma représentant les différents types d’altérations ou anomalies
génétiques aboutissant à la formation de glioblastome. ... 28 Figure 4 : Courbe de survie de cellules de mammifères après irradiation en présence ou en absence d’oxygène. ... 31 Figure 5 : Origine de l’hypoxie au sein des tumeurs. ... 33 Figure 6 : Représentation schématique de la structure de HIF-1 et des variants
d’épissage... 38 Figure 7 : Représentation schématique de la structure des différents variants d’épissage de la sous-unité HIF-1α. ... 40 Figure 8 : La sous-unité HIF-1α est soumise à différentes modifications
post-traductionnelles dépendantes ou non des conditions d’oxygénation. ... 44 Figure 9 : La sous-unité HIF-1α peut être régulée par différentes voies
indépendamment des conditions d’oxygénation des cellules et des prolylhydroxylases. ... 50 Figure 10 : Famille des protéines Rho humaines alignées avec les GTPases Ras. ... 58 Figure 11 : Schéma représentant la structure générale des petites GTPases... 60 Figure 12 : Modifications post-traductionnelles des protéines Rho au niveau COOH-terminale. ... 62 Figure 13 : Cycle d’activation des petites GTPases de la famille des protéines Rho.... 64 Figure 14 : Les GTPases Rho sont impliquées dans de nombreuses fonctions
cellulaires. ... 68 Figure 15 : Mutations associées au cancer des protéines Rho et de ses régulateurs. . 72 Figure 16 : Structure primaire et domaines de RhoA, RhoB et RhoC ; RhoA humaine. [gi:10835049], RhoB humaine [gi:51338601], RhoC humaine [gi:132543]. (Base de données NCBI.) ... 78 Figure 17 : Représentation schématique des différentes familles d’intégrines avec les différentes possibilités de dimérisation. (d’après Rüegg et Mariotti, 2003) ... 100 Figure 18 : Modèle de la structure et de l’activation des intégrines... 102
Figure 19 : Structure des différentes molécules retrouvées au niveau des points focaux d’adhérence. ... 106 Figure 20 : Voies de transduction du signal dépendantes des intégrines et médiée par l’Integrin Linked Kinase (ILK) (d’après Helghans et al, 2007). ... 109 Figure 21 : Voies de transduction du signal dépendantes des intégrines et médiée par la Focal Adhesion Kinase (FAK) (issue de http:// www.chuv.ch/cpo_research)... 111 Figure 22 : L’hypoxie tumorale augmente les capacités de métastase, d’invasion et de migration des cellules cancéreuses. ... 118 Figure 23 : L’hypoxie régule les voies de l’adhérence cellulaire. ... 124 Figure 24 : Les voies de l’adhérence cellulaire médiée par les intégrines peuvent
également moduler la réponse et l’adaptation des cellules à l’hypoxie en contrôlant la sous-unité HIF-1α. ... 128 Figure 25 : Schéma récapitulatif des résultats et observations obtenus pendant ces travaux de thèse. ... 147
LISTE DES TABLEAUX
Tableau 1 : Types histologiques des tumeurs astrocytaires selon la classification de l’OMS. ... 24
Tableau 2 : Principaux gènes cibles du facteur de transcription HIF-1. ... 36
Tableau 3 : Tableau récapitulatif des principaux régulateurs (positifs et négatifs) connus de la sous-unité HIF-1α. ... 41 Tableau 4 : Récapitulatif des localisations, modifications post-traductionnelles,
effecteurs, fonctions, implications physiologiques et pathologiques et phénotypes de KO ... 96
Tableau 5 : Récapitulatif bis des localisations, modifications post-traductionnelles, effecteurs, fonctions, implications physiologiques et pathologiques et phénotypes de KO ... 97
Tableau 6 : Les différentes sous-unités d’intégrines humaines (d’après [Takada et al., 2007])... 99
Tableau 7 : Spécificité de liaison au ligand des différentes intégrines humaines (d’après Takada, 2007)... 104
Tableau 8 : L’hypoxie module les voies de l’adhérence cellulaire médiée par les
intégrines ainsi que les protéines de signalisation en aval... 120
Tableau 9 : Liste des différents essais cliniques, en cours ou terminés, ciblant les
RESUME
L’hypoxie tumorale est un des principaux facteurs de résistance aux thérapeutiques anti-cancéreuses comme la chimiothérapie ou la radiothérapie. Les glioblastomes, tumeurs cérébrales agressives, sont des tumeurs hypoxiques et radiorésistantes qui récidivent dans les champs d’irradiation malgré un traitement bien conduit associant chirurgie et radiothérapie. Les précédents travaux du laboratoire ayant démontréquel’inhibitionde la petite GTPase RhoB induisait une diminution de l’hypoxie tumorale de xénogreffes de glioblastomes. La première partie de ma thèse a consisté à déterminer si, en plus de son effet sur l’angiogenèse, RhoB pouvait contrôler également les voies moléculaires de l’hypoxie cellulaire dans les cellules de glioblastome U87. Nous avons tout d’abord démontré que l’inhibition de RhoB pardeux ARNi dirigés spécifiquement contre RhoB,entraînait une diminution du taux intracellulaire de la sous unité HIF-1α, formant le facteur de transcription inductible par l’hypoxie HIF-1, et également une inhibition de l’activité transcriptionnelle de ce facteur dans des cellules U87 placées en hypoxie. La dégradation de HIF-1α étant médiée par le protéasome, nous avons ensuite montré que la diminution du taux intracellulaire de HIF-1α induite par l’inhibition de RhoB pouvait être réversée par des inhibiteurs du protéasome. Notre travail s’est ensuite orienté vers l’étude des voies de signalisation susceptibles de contrôler le taux de HIF-1α en hypoxie en nous intéressant particulièrement à la voie Akt/Glycogène Synthase Kinase-3. Nos résultats démontrent que l’inhibition spécifique de RhoB induit une diminution de la phosphorylation d’Akt, de la forme inactive de la GSK-3 et, à contrario, une augmentation de la forme active de GSK-3. De plus, l’utilisation d’un inhibiteur de la GSK-3 ainsi que d’ARNi spécifiques permet de réverser l’effet de l’inhibition de RhoB sur le taux intracellulaire de HIF-1α. Enfin, nous avons déterminé que l’hypoxie était capable de stimuler l’activité de RhoB, en favorisant la forme RhoB liée au GTP dans des cellules U87 placées en conditions d’hypoxie. L’ensemble de ces résultats démontre doncque RhoB régule l’hypoxie cellulaire en contrôlant le taux intracellulaire de HIF-1α via GSK-3 et Akt dans les cellules de glioblastomes humains.La deuxième partie de ma thèse s’est ensuite orientée sur la détermination du rôle de l’adhérence cellulaire dans le contrôle de l’hypoxie tumorale. En effet, il est largement démontré que les protéines Rho, Akt et GSK-3 sont impliquées dans la voie de signalisation induite par les récepteurs de la matrice extracellulaire, les intégrines, connues pour avoir un rôle dans l’angiogenèse, la migration, la survie cellulaire et le développement tumoral. Nous avons donc cherché à savoir si les voies biologiques contrôlées par les intégrines étaient impliquées dans le contrôle de l’hypoxie tumorale dans les cellules de glioblastomes. Dans un premier temps, nous avons vérifié l’expression des intégrines αVβ3 et αVβ5 respectivement exprimées par les cellules U87 et SF763 en fonction des conditions d’oxygénation et montré que l’expression des intégrines à la membrane était augmentée ainsi que la phosphorylation de FAK (Focal Adhesion Kinase) en hypoxie. En utilisant des inhibiteurs spécifiques des intégrines et des ARNi, nous avons ensuite démontré que les intégrines αVβ3 pour les cellules U87 et αVβ5 pour les cellules SF763 et la protéine FAK pouvaient réguler le taux intracellulaire de la sous-unité HIF-1α en hypoxie. La diminution du taux intracellulaire de HIF-1α suite à l’inhibition des intégrines est également accompagnée d’une diminution de l’activation de RhoB et de la forme inactive de la GSK-3. Le positionnement en amont de la voie des intégrines par rapport à la voie RhoB/Akt/GSK-3 a pu être confirmé par l’utilisation d’un dominant positif, constitutivement actif, de RhoB (RhoBV14). En présence de RhoBV14, dans des cellules où les intégrines et FAK étaient préalablement inhibées, nous avons pu observer une réversion de la diminution du taux intracellulaire de HIF-1α dans les cellules de glioblastome en hypoxie. Ces différentes observations nous ont permis de mettre en évidence le rôle des voies de l’adhérence dans la réponse cellulaire à l’hypoxie et l’implication de la voie RhoB/Akt/GSK-3, précédemment décrite, dans le contrôle de l’hypoxie cellulaire. Enfin, nous avons étudié l’effet de l’inhibition de cette voie in vivo sur des xénogreffes de cellules U87 implantées en intracrânien et en sous-cutané au niveau de la patte chez la souris immunodéficiente. Grâce au marquage des cellules hypoxiques viables par le 2-nitroimidazole et grâce à l’analyse en immunohistochimie de l’expression de la sous unité HIF-1α et l’observation des phénomènes d’angiogenèse, nous avons confirmé l’effet oxygénant de l’inhibition des voies de signalisation de l’adhérence cellulaire médiée par les intégrines sur ces xénogreffes.
Ces résultats, conjugués à une étude portant sur l’implication des voies d’adhérence cellulaire sur la radiorésistance tumorale, pourraient permettre le développement d’un essai clinique pour tester l’effet réoxygénant et radiosensibilisant de ces inhibiteurs sur les gliomes de haut grade comme l’équipe du laboratoire l’a déjà réalisé, en collaboration avec le Département de Radiothérapie et le Bureau d’Etudes Cliniques de l’Institut Claudius Regaud avec les inhibiteurs de farnésyltransférase testés actuellement dans un essai de phase I-II comme agents radiosensibilisants et réoxygenant des gliomes.
ABREVIATIONS
AA : acide aminéADN : acide désoxyribonucléique ARN : acide ribonucléique
ARNm : acide ribonucléique messager ATP : adénosine triphosphate
bHLH : basic helix loop helix EGF : epidermal growth factor
EGFR : epidermal growth factor receptor EPO : érythropoïétine
FAK : focal adhesion kinase FIH : factor inhibiting HIF FTase : farnésyltransférase FTI : farnesyl transferase inhibitor GAP : GTPase activating protein GDI : GDP dissociation inhibitor GDP : guanosine diphosphate
GEF : guanine nucleotide exchange factor GGTase : géranylgéranyl transférase GGTI : geranylgeranyl transferase inhibitor GSK-3 : glycogen synthase kinase 3 GTP : guanosine triphosphate
Gy : Gray
HIF : hypoxia inducible factor HRE : hypoxia responsive element
HSP : heat shock protein IGF : insulin-like growth factor ILK : integrin linked kinase kDa : kilo Dalton
MAPK : mitogen-activated protein kinase mTOR : mammalian target of rapamycin NLS : nuclear localization signal
NO : nitric oxide
ODDD : oxygen dependent degradation domain PAS : Per-ARNT-Sim motif
PDGF : platelet derived growth factor
PI3K : phosphotidyl inositol triphosphate kinase PKC : protéine kinase C
PTEN : phosphatase and tensin homolog ERO : espèce réactive oxygénée
TAD : transactivation domain TGF : transforming growth factor UV : ultra violet
VEGF : vascular endothelial growth factor VHL : Von Hippel Lindau
Introduction
L’hypoxie tumorale est un des principaux facteurs de résistance aux thérapeutiques anti-cancéreuses comme la chimiothérapie ou la radiothérapie. Ainsi, de nombreuses tumeurs traitées par radiothérapie ont un contrôle local significativement diminué lorsque leur pression partielle d’oxygène, pO2, est réduite. Pour certains
cancers comme les cancers gynécologiques, des voies aéro-digestives supérieures ou des tumeurs cérébrales, la radiothérapie est souvent la base du traitement. Toutefois, certaines de ces tumeurs, en particulier, les glioblastomes, tumeurs cérébrales extrêmement agressives, sont des tumeurs très hypoxiques qui récidivent dans les champs d’irradiation malgré un traitement bien conduit associant chirurgie et radiothérapie [Brat and Mapstone, 2003;Kaur et al., 2005;Stupp et al., 2005] et plus récemment, associant la radiothérapie et la chimiothérapie à visée radiosensibilisante [Stupp et al., 2005]. Ces dernières années, de nombreux travaux de recherche se sont intéressés à l’étude des mécanismes régulant l’hypoxie tumorale. Ceci dans le but de mettre à jour de nouvelles cibles susceptibles de réduire cette hypoxie tumorale, réoxygéner les tumeurs et ainsi augmenter l’efficacité des thérapeutiques anti-cancéreuses.
1- Les tumeurs cérébrales
1-1 Généralités :
Les tumeurs cérébrales se caractérisent par des proliférations anarchiques de cellules au sein du cerveau. Ce sont soit des tumeurs primaires qui apparaissent dans le cerveau ou des métastases issues d'autres tumeurs. Nous focaliserons ici notre attention sur les tumeurs primitives cérébrales. Il existe plusieurs variétés de tumeurs primitives cérébrales mais les gliomes en représentent plus de la moitié dont les astrocytomes et les oligodendrogliomes constituent la majeure partie. Les astrocytomes sont dérivés des astrocytes qui sont des cellules qui activées ont la forme d'étoile. Elles composent avec les oligodendrogliomes et les cellules microgliales la glie. Les astrocytes, véritables passerelles entre le sang et les neurones assurent la nutrition des neurones, gère les connections interneuronales, régulent les neurotransmetteurs [Baudrimont, 1991]. Les astrocytomes sont donc classifiés en quatre grades (tableau 1). Les grades I et II étant considéré comme des astrocytomes de bas grade et les grades III puis IV comme des grades de haute malignité. La transformation en haut grade est notamment associée à l’augmentation de l’index mitotique, des atypies nucléaires et surtout à l’augmentation de la néovascularisation (grade III) puis associée à de la nécrose (grade IV ou glioblastome). Les tumeurs de bas grade évoluent avec le temps vers des astrocytes de haut grade pour aboutir finalement au glioblastome multiforme qui est la forme la plus grave (figure 1). Outre les gliomes, les tumeurs rares du cerveau sont surtout les médulloblastomes et les épendymomes, qui touchent en particulier les enfants, ainsi que les oligodendrogliomes. Les méningiomes, les neurinomes acoustiques, les adénomes de l'hypophyse et les crâniopharyngiomes sont également d'autres tumeurs primaires du cerveau.
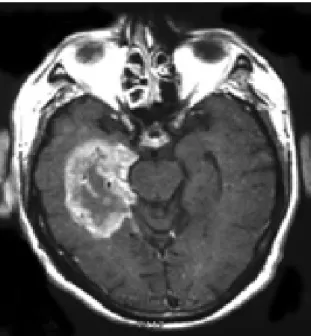
Figure 1 : Glioblastome Multiforme observé par IRM au niveau du lobe temporal.
Le gliobastome est la tumeur la plus commune et la plus grave des tumeurs primaires du cerveau.
1-2 Causes :
Les causes des tumeurs primaires du cerveau sont encore peu connues. Comme pour d'autres types de cancers, il semblerait toutefois que des processus génétiques jouent un rôle important dans leur formation. Ces processus correspondraient à des successions d’anomalies génétiques. En effet, plusieurs séries de mutations sont en général nécessaires avant qu'une cellule ne devienne cancéreuse. Ces mutations vont donc affecter des gènes suppresseurs de tumeur (comme p53, le gardien du génome) en les inactivant, et/ou des oncogènes (comme H-ras, myc …) en les activant. Les mutations des gènes des télomérases ainsi que des gènes impliqués dans les phénomènes de réparation de l’ADN participent à l’instabilité génétique des cellules tumorales et interviennent dans le processus de cancérisation. Il existe également des facteurs de prédisposition liés à une altération constitutionnelle d’un gène du système MMR, MisMatch Repair, (MLH1, MSH2, MSH6). Le système MMR permet de détecter, de réparer les mésappariements et donc de corriger les erreurs survenues lors de la réplication. Par exemple, le syndrome de Lynch ou HNPCC (Hereditary Non Polyposis Colon Cancer) est une prédisposition héréditaire au cancer liée dans 70% des cas à une mutation délétère d’un gène du sytème MMR.
1-3 La classification histo-pronostique :
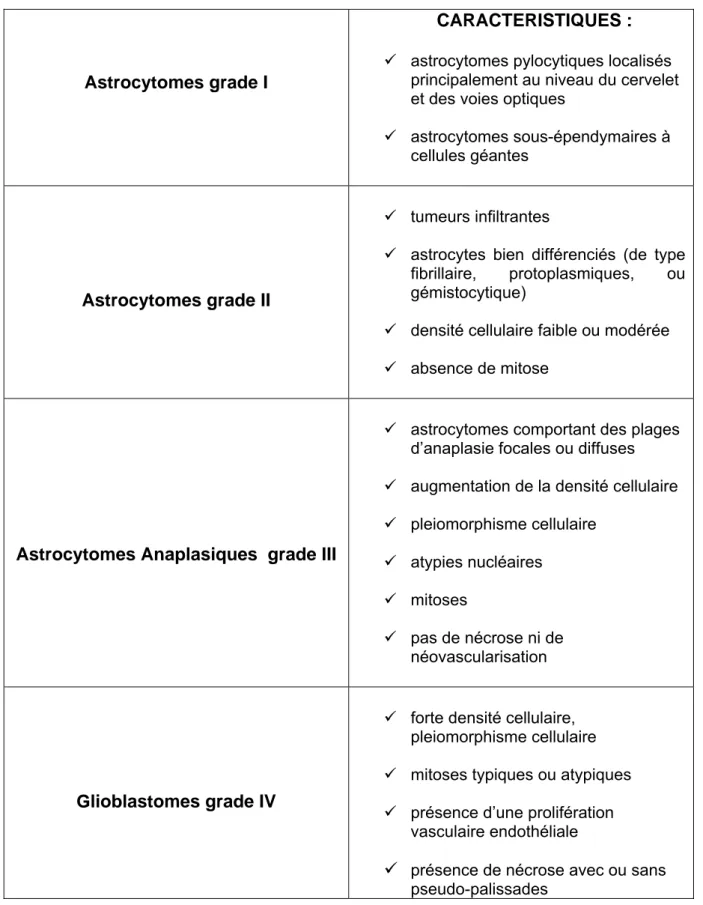
Plusieurs auteurs ont successivement tenté d’établir une classification qui relierait des critères histologiques objectifs, et le pronostic, défini par des grades, des différents types de tumeurs gliales. Selon l’Organisation Mondiale de la santé (OMS), les tumeurs astrocytaires sont classées en quatre grades (tableau 1 suivant) [Darcel, 1996]. Cependant, la classification histo-pronostique des gliomes est encore l’objet de controverses et plusieurs classifications co-existent telle que la classification de l’hôpital Sainte Anne[umas-Duport et al., 1997a;umas-Duport et al., 1997b]. De ce fait, il est possible que des dénominations similaires se rapportent à des lésions différentes [Honnorat, 1999].
Astrocytomes grade I
CARACTERISTIQUES : 9 astrocytomes pylocytiques localisés
principalement au niveau du cervelet et des voies optiques
9 astrocytomes sous-épendymaires à cellules géantes
Astrocytomes grade II
9 tumeurs infiltrantes
9 astrocytes bien différenciés (de type fibrillaire, protoplasmiques, ou gémistocytique)
9 densité cellulaire faible ou modérée 9 absence de mitose
Astrocytomes Anaplasiques grade III
9 astrocytomes comportant des plages d’anaplasie focales ou diffuses 9 augmentation de la densité cellulaire 9 pleiomorphisme cellulaire 9 atypies nucléaires 9 mitoses 9 pas de nécrose ni de néovascularisation Glioblastomes grade IV
9 forte densité cellulaire, pleiomorphisme cellulaire 9 mitoses typiques ou atypiques 9 présence d’une prolifération
vasculaire endothéliale
9 présence de nécrose avec ou sans pseudo-palissades
Tableau 1 : Types histologiques des tumeurs astrocytaires selon la classification de
1-4 Les traitements :
Dans un premier temps, si la localisation de la tumeur le permet, il faut en effet que la tumeur ne soit pas localisée au niveau de zones fonctionnelles, une chirurgie d’exérèse est réalisée puisqu’elle permet un meilleur contrôle de la maladie et une meilleure efficacité des traitements adjuvants. Cependant, bien que les techniques de chirurgie aient évoluée, notamment par la chirurgie éveillée, et permettent de réaliser à présent des exérèses dans des territoires où l’intervention était récusée il y a quelques années, la chirurgie ne consiste encore dans de nombreux cas qu’en une biopsie à visée diagnostique. La chirurgie si elle est réalisable sera la plus large possible en sachant qu'il est en général impossible d'enlever toute la tumeur qui par sa nature infiltre le parenchyme cérébral normal.
En raison de l’envahissement microscopique, une radiothérapie post-opératoire réalisée sur le site d’exérèse ainsi que sur une marge comprenant l’infiltration microscopique est nécessaire. Cette radiothérapie permet d'augmenter la survie des patients quelque soit l’âge [Loiseau, 1996] et d’améliorer la qualité de vie.
Enfin, les chimiothérapies ne se révèlent pas toujours très efficaces à cause de problèmes liés au passage de la barrière hémato-encéphalique, même si cette barrière est souvent altérée dans les tumeurs cérébrales et en particulier dans les glioblastomes. Toutefois, une nouvelle molécule alkylante, le témozolomide (ou Témodal) est depuis quelques années le premier agent chimiothérapeutique oral à traverser facilement la barrière hémato-méningée. Le Témodal associé à la radiothérapie [Stupp et al., 2005] a permis d’obtenir une augmentation de survie moyenne de trois mois en particulier chez les patients opérés.
1-5 Le Glioblastome :
Selon la classification de l'O.M.S., les glioblastomes sont des tumeurs astrocytaires malignes de grade IV. Chez l'adulte, ce sont les tumeurs cérébrales les plus fréquentes avec une incidence de l'ordre de 10 nouveaux cas par an pour 100.000 habitants, ce qui donne pour la France 6000 nouveaux cas chaque année. C'est la deuxième cause de mortalité des cancers chez l'enfant après la leucémie et la troisième chez l'adulte. Les glioblastomes surviennent à tout âge mais dans 70% des cas entre 45 et 70 ans avec un pic à 58 ans. Entre 45 et 50 ans ce sont souvent des glioblastomes secondaires c’est à dire developpés à partir de gliome (astrocytome) de bas grade et entre 50 et 70 ans des glioblastomes primaires. Les causes de cette
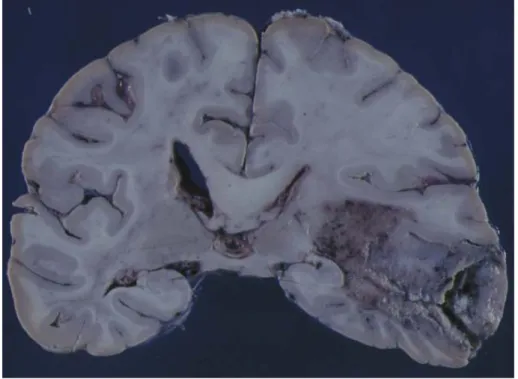
Figure 2 : Glioblastome primaire (tranche frontale passant par le pulvinar).
Volumineuse tumeur temporale droite, dont on reconnaît bien l'aspect nécrotico-hémorragique et le caractère circonscrit. (Photo issue de l’Atlas Interactif de Neuro-Oncologie de l’ANOCEF).
maladie sont encore largement méconnues avec des suspicions d’une origine virale périnatale et de prédispositions génétiques. Par exemple, au niveau des prédispositions génétiques, il a été montré que des mutations des gènes MMR (MisMatch Repair) confèrent un risque accru de tumeurs du système nerveux central [Leung et al., 1998]. Les glioblastomes sont des tumeurs localisées le plus souvent au niveau des hémisphères cérébraux, dans la matière blanche (figure 2). Ce sont des lésions volumineuses, profondes, et donc souvent inopérables.
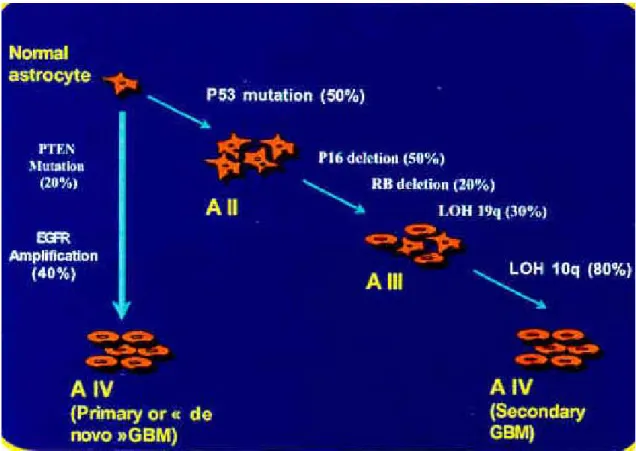
Les voies de progression des glioblastomes, marquées par la survenue de différentes altérations génétiques, inactivation de gènes suppresseurs de tumeur (identifiés comme p53, p16 ou PTEN) ou activation d’oncogènes (EGFR) permettent une classification moléculaire des glioblastomes (figure 3). En effet, les anomalies génétiques dans les tumeurs sont différentes selon qu'il s'agit d'un glioblastome primaire dit « de novo » ou d'un glioblastome secondaire (développé à partir d’une tumeur astrocytaire bénigne). C'est ainsi que les glioblastomes secondaires sont fortement marqués par une mutation ou une délétion du gène suppresseur de tumeur p53 [Nozaki et al., 1999] alors que les glioblastomes primaires dit « de novo » chez les sujets plus âgés présentent une biologie différente avec des altérations multiples, notamment celle d'un autre gène suppresseur de tumeur CDKNA sur le bras court du chromosome 16, et surtout PTEN/MMAC1 sur le chromosome 10. Une mutation de l’EGFR est également retrouvée dans 40% des cas de glioblastome primaire.
Les glioblastomes sont des tumeurs connues pour être très agressives et particulièrement résistantes aux thérapeutiques anti-cancéreuses à savoir chimiothérapie et radiothérapie. En effet, les glioblastomes ont la capacité de récidiver de façon fréquente dans les champs d’irradiation malgré un traitement bien conduit qui peut associer chirurgie, radiothérapie et chimiothérapie. Cette diminution de la radiosensibilité affectant l’efficacité de la radiothérapie fait de la conception et la synthèse d’agents radiosensibilisants un enjeu majeur. Pour une efficacité en clinique, ces radiosensibilisants doivent modifier la radiosensibilité des cellules tumorales sans modifier celle des tissus environnants particulèrement sensibles à l’irradiation. La conception de tels agents ne saurait être réalisée de façon optimale sans acquérir une connaissance approfondie des mécanismes moléculaires impliqués dans la genèse du phénotype « radiorésistant ».
Figure 3 : Schéma représentant les différents types d’altérations ou anomalies
2- Phénomènes de radiorésistance
Une tumeur est considérée cliniquement comme peu sensible aux rayonnements ionisants, s’il survient une récidive rapide dans le champ d’irradiation après qu’il y ait eu une régression de la tumeur ou lorsque l’irradiation n’a pu permettre la régression de cette tumeur. Différents travaux ont relié la modulation de la radiosensibilité clinique à certaines caractéristiques liées à la tumeur.
2-1 Le nombre de cellules clonogéniques :
Une cellule est dite clonogénique si elle est capable d’établir un nouveau clone de cellules tumorales et de régénérer la tumeur : plus la tumeur contient de cellules clonogéniques et moins elle est radiocurable.
2-2 La cinétique de prolifération :
La cinétique de prolifération des cellules tumorales influe sur la survie après irradiation. Les tumeurs présentant un pourcentage élevé de cellules en prolifération et un taux de perte cellulaire important sont celles qui sont les plus radiosensibles et les plus radiocurables.
2-3 La radiorésistance intrinsèque :
La radiorésistance intrinsèque correspond à la mise en place de mécanismes moléculaires qui permettent à la cellule de résister aux rayonnements. Le concept de radiorésistance/radiosensibilité intrinsèque introduit par Fertil et Malaise [Fertil and Malaise, 1985] peut être étudié par l’établissement de courbes de survie en fonction de la dose unique délivrée.
2-4 La radiorésistance extrinsèque liée au micro-environnement :
Le facteur oxygène et surtout le manque d’oxygène ou hypoxie est un facteur témoin de la diminution de la radiosensibilité : une tumeur est d’autant moins radiocurable qu’elle contient de cellules hypoxiques [Tannock, 1972;Sutherland et al., 1980;Rockwell and Moulder, 1990;Wouters and Brown, 1997]. En effet, l’implication de l’hypoxie comme facteur de résistance à la radiothérapie est connue depuis de
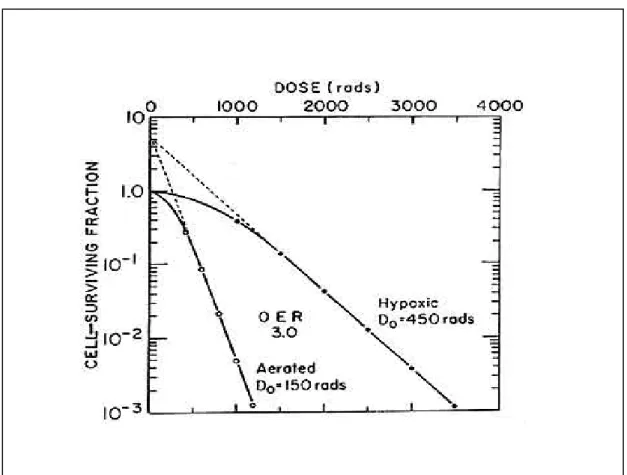
nombreuses années. Considérant l’efficacité des photons, la dose nécessaire à l’obtention d’une survie cellulaire donnée en conditions hypoxiques est trois fois plus importante que la dose nécessaire pour obtenir le même taux de survie en air ambiant. On peut ainsi définir l’OER (oxygen enhancement ratio) (figure 4).
Figure 4 : Courbe de survie de cellules de mammifères après irradiation en présence
ou en absence d’oxygène.
La fraction survivante après irradiation est beaucoup plus importante pour les cellules hypoxiques (A) comparées aux cellules oxygénées (B).
3- L’hypoxie
La diminution de la concentration en oxygène ou hypoxie peut se produire dans de nombreuses pathologies [Hockel and Vaupel, 2001]. C’est le cas pour les ischémies cérébrales et cardiovasculaires, le diabète, l’athérosclérose, mais également les cancers en particulier dans les tumeurs cérébrales comme les glioblastomes. L’hypoxie s’établit lorsque la quantité d’oxygène délivrée aux tissus est insuffisante par rapport aux besoins cellulaires, et en général lorsque la pression partielle en oxygène se situe entre 1 et 10 mmHg. Dans les tissus sains, la tension moyenne en oxygène se situe à environ 7 % [Jiang et al., 1996b] tandis que, au sein des tumeurs solides, elle se situe plutôt à 1,5 % [Adam et al., 1999].
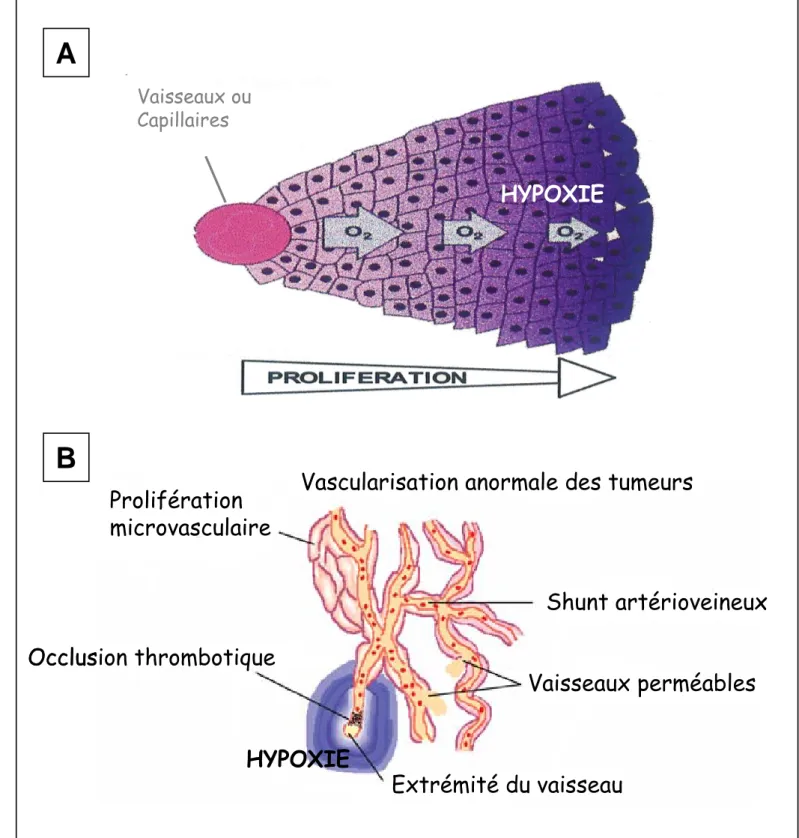
Dans le cas des tumeurs solides, la formation de zones d’hypoxie peut s’expliquer de différentes manières (figure 5). Tout d’abord, les cellules tumorales, en proliférant, vont s’éloigner des vaisseaux ou capillaires qui représentent la source d’oxygène. Cet éloignement est corrélé avec une diminution de la concentration en oxygène présent au niveau des cellules favorisant ainsi la formation de zones hypoxiques. De par leur fort pouvoir de multiplication, les cellules cancéreuses s’expose donc à un environnement appauvri en oxygène. Afin de pallier la faible pression partielle d’oxygène de leur environnement, les cellules tumorales vont, en outre, stimuler leur propre néovascularisation en sécrétant un facteur protéique appelé VEGF (Vascular Endothelial Growth Factor) qui, après s’être lié à des récepteurs spécifiques situés à la surface des cellules endothéliales des vaisseaux sanguins, va inciter ces dernières à proliférer en direction du foyer tumoral [Ferrara, 2002]. Le VEGF est également à l’origine de la perméabilité des vaisseaux tumoraux les rendant ainsi non fonctionnels. La masse tumorale ainsi vascularisée va croître rapidement grâce à un apport plus conséquent de nutriments carbonés et d’oxygène, et les cellules cancéreuses vont également pouvoir se disséminer dans l’organisme via les vaisseaux sanguins ainsi créés.
Vaisseaux ou Capillaires Vaisseaux ou Capillaires
Shunt artérioveineux
Occlusion thrombotique
Prolifération
microvasculaire
Vaisseaux perméables
Extrémité du vaisseau
Vascularisation anormale des tumeurs
Shunt artérioveineux
Occlusion thrombotique
Prolifération
microvasculaire
Vaisseaux perméables
Extrémité du vaisseau
Vascularisation anormale des tumeurs
A
B
HYPOXIE
HYPOXIE
Figure 5 : Origine de l’hypoxie au sein des tumeurs.
A. En proliférant, les cellules s’éloignent des vaisseaux ou capillaires. Cet éloignement
est corrélé avec une diminution de la concentration en oxygène générant ainsi des zones d’hypoxie. B. La vascularisation anormale des tumeurs est également à l’origine de zones d’hypoxie en entraînant une insuffisance dans l’apport en oxygène (d’après [Kaur et al., 2005]).
L’hypoxie peut également se mettre en place grâce au réseau vasculaire même des tumeurs. En effet, la vascularisation des tumeurs et, en général, la néovascularisation donnent naissance à des vaisseaux ou capillaires anormaux, trop petits, des occlusions ou encore des shunts artérioveineux [Shchors and Evan, 2007]. Toutes ces anomalies au niveau de la vascularisation vont engendrer un apport en oxygène insuffisant et donc la création de zones d’hypoxie. On distingue habituellement deux grands types d’hypoxie, l’hypoxie aiguë et l’hypoxie chronique [Coleman, 1988]. L’hypoxie aiguë se caractérise par une exposition des organes, tissus ou cellules à une faible concentration en oxygène pendant des temps relativement courts, pas plus de quelques heures. Au-delà, s’installe une hypoxie dite chronique qui se caractérise donc par des temps d’exposition à une faible concentration en oxygène beaucoup plus long. L’hypoxie chronique fait appel à des phénomènes de réponse et d’adaptation des cellules et est donc un paramètre important dans le processus tumorigéne. Les cellules de mammifères sont justement capables de répondre et de s’adapter à ces conditions d’hypoxie via des voies de signalisation très conservées qui vont aboutir à différents phénomènes physiologiques [Papandreou et al., 2005] comme l’angiogenèse et la formation de néo-vaisseaux. Toutefois, lorsque l’hypoxie est trop sévère, les cellules ne peuvent s’adapter et résister à ces faibles conditions d’oxygénation, ce qui engendre la formation de zones de nécrose au sein des tumeurs.
Enfin, la diminution intratumorale de la concentration en oxygène est largement associée à une diminution de l’efficacité des thérapeutiques anti-cancéreuses [Wouters and Brown, 1997;Kaur et al., 2005] et une augmentation du pouvoir métastatique des cellules tumorales d’où l’intérêt d’étudier ces phénomènes d’hypoxie pour espérer améliorer l’effet des traitements anti-cancéreux.
3-1 Le facteur inductible par l’hypoxie, Hypoxia Inducible Factor 1 : HIF-1
La réponse et l’adaptation des cellules à l’hypoxie sont contrôlées par différents facteurs de transcription. Parmi ces facteurs de transcription activés par une faible concentration d’oxygène, se trouvent la protéine liant l’élément de réponse à l’AMP cyclique (CREB) [Beitner-Johnson and Millhorn, 1998], l'Activator Protein-1 (AP-1) [Finkenzeller et al., 1995], [Bandyopadhyay et al., 1995], le facteur-KB nucléaire (NFkb) [Koong et al., 1994a;Koong et al., 1994b] et la protéine Early Growth Response-1 (Egr-1) [Yan et al., 1999;Yan et al., 1998;Lo et al., 2001;Nishi et al., 2002]. Cependant, le facteur de transcription inductible par l’hypoxie ou Hypoxia Inducible Factor 1 (HIF-1)
est le régulateur le plus important dans le contrôle de l'homéostasie de l'oxygène. HIF-1, en induisant une batterie de gènes cibles, permet la réponse et l'adaptation des cellules dans un micro-environnement pauvre en oxygène. Ce facteur de transcription a été identifié pour la première fois en 1992 par Gregg Semenza comme régulateur crucial de l’expression du gène de l’érythropoïétine (EPO) en réponse à une faible concentration en oxygène [Semenza and Wang, 1992;Wang and Semenza, 1993]. A l’heure actuelle, plus d’une centaine de gènes ont été identifiés comme cible de HIF-1 (tableau 2). Ces gènes, permettant la réponse et l’adaptation des cellules à l’hypoxie, sont impliqués dans les phénomènes d’angiogenèse, de glycolyse, de prolifération et survie, de régulation du pH, d‘invasion et de migration [Semenza, 2003;Ke and Costa, 2006]. HIF-1 appartient à une classe de facteur de transcription de la famille des protéines basic helix-loop-helix (bHLH)/Per-ARNT-Sim (PAS) et reconnaît des séquences spécifiques sur l’ADN (5'-RCGTG-3') appelées élément de réponse à l’hypoxie (ou Hypoxia Responsive Element (HRE)) [Wang et al., 1995a;Wang et al., 1995b]. HIF-1 est un hétérodimère composé de deux sous-unités : HIF-1 alpha (HIF-1α) et HIF-1 beta (HIF-1β) ou ARNT (aryl hydrocarbon nuclear translocator). Le complexe HIF-1 une fois lié à l'ADN, peut recruter un certain nombre de co-activateurs, tels que p300/CBP, Ref-1, Jab1, SCR-1 et TIF2, facilitant ainsi l'expression des gènes cibles [Wenger, 2002].
Transglutaminase2 Acides aminés
DEC1, DEC2, ETS-1, NUR77 Régulation transcription
KRT14, KRT18, KRT19, VIM Structure du cytosquelette
Anhydrase Carbonique 9 pH
Transferrine, Transferrine-R, Céruloplasmine Fer
Adénylate kinase-3, Ecto-5’-nucléotidase Nucléotide
P53, BNIP3, NIX, Bax, RTP801/REDD1, Ref-1, Bcl-2, NFκB, HSP70, Bid
Apoptose
CXCR4, MMP2, Lox, PAI-1, c-MET, LRP1, MIC2/CD99, FN1, UPAR, Collagène de type V, AMF/GPI, CATHD, ILK
Migration/Invasion Métabolisme cellulaire
P35srj, P21, MDR-1, COX-2, Intestinal trefoil factor
Divers
PFK, PGK, LDHA, GLUT-1/3, Hexokinase-1/2, Enolase-1, GAPDH, ALDA, ALDC, PKM, TPI Glucose
IGF-BP1/2/3, IGF2, CCD1, TGF-α,/-β, P21/WAF-1, Cycline G2, NOS2
Prolifération/Survie
EPO, VEGF, VEGF-R, Ang-2, Tie-2, PDGF,
Leptine, Hème oxygénase-1, Flt-1, Endothéline-1, ADM, récepteur adrénergique α1B, Endogline Angiogenèse
Gènes cibles de HIF-1
Fonctions
Transglutaminase2 Acides aminés
DEC1, DEC2, ETS-1, NUR77 Régulation transcription
KRT14, KRT18, KRT19, VIM Structure du cytosquelette
Anhydrase Carbonique 9 pH
Transferrine, Transferrine-R, Céruloplasmine Fer
Adénylate kinase-3, Ecto-5’-nucléotidase Nucléotide
P53, BNIP3, NIX, Bax, RTP801/REDD1, Ref-1, Bcl-2, NFκB, HSP70, Bid
Apoptose
CXCR4, MMP2, Lox, PAI-1, c-MET, LRP1, MIC2/CD99, FN1, UPAR, Collagène de type V, AMF/GPI, CATHD, ILK
Migration/Invasion Métabolisme cellulaire
P35srj, P21, MDR-1, COX-2, Intestinal trefoil factor
Divers
PFK, PGK, LDHA, GLUT-1/3, Hexokinase-1/2, Enolase-1, GAPDH, ALDA, ALDC, PKM, TPI Glucose
IGF-BP1/2/3, IGF2, CCD1, TGF-α,/-β, P21/WAF-1, Cycline G2, NOS2
Prolifération/Survie
EPO, VEGF, VEGF-R, Ang-2, Tie-2, PDGF,
Leptine, Hème oxygénase-1, Flt-1, Endothéline-1, ADM, récepteur adrénergique α1B, Endogline Angiogenèse
Gènes cibles de HIF-1
Fonctions
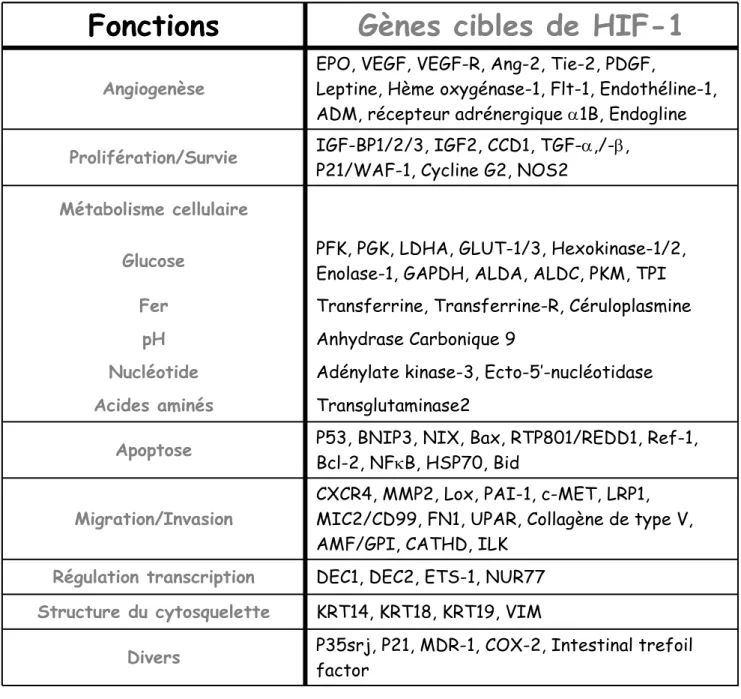
Tableau 2 : Principaux gènes cibles du facteur de transcription HIF-1.
ADM : adrénomédulline ; ALDA : aldolaseA ; ALDC : aldolaseC ; AMF : autocrine motility factor ; Ang-2 : Angiopoïétine 2 ; Bcl-2 : B-cell leukemia/lymphoma 2 ; BNIP3 : Bcl-2 nineteen kilodalton interacting protein 3 ; CATHD : cathepsine D ; CCD1 : Coiled-coil-DIX1 ; COX-2 : cyclo-oxygénase 2 ; CXCR4 : CXC chimiokine récepteur 4 ; DEC1/2 : differentiated embryo-chondrocyte expressed gene 1/2 ; FN1 : fibronectine1 ; Flt-1 : fms-related tyrosine kinase 1 ; GLUT-1/3 : glucose transporteur1/3 ; GAPDH : glycéraldéhyde-3-P-déhydrogénase ; IGF-BP1/2/3 : IGF-factor-binding-protein1/2/3 ; KRT14/18/19 : kératine14/18/19 ; LDHA : lactate déhydrogénase A ; LRP1 : LDL-receptor-related protein 1 ; Lox : lysyl oxydase ; MDR1, multidroguerésistance 1 ; MIC2 : microneme protéine 2 ; MMP2 : matrix métalloprotéinase 2 ; NFκB : nuclear factor kappa B ; NOS2 : nitric oxide synthase 2 ; NUR77 : nuclear receptor77 ; PFK : phosphofructokinase ; PGK : phosphoglycérate kinase ; PAI1 : plasminogen-activator inhibitor 1 ; PKM : pyruvate kinase M ; REDD1 : regulated in development and dna damage responses 1 ; Ref-1 : redox factor-1 ; Tie-2 : endothelium-specific tyrosine kinase-2 ; TPI : triosephosphate isomerase ; UPAR : urokinase plasminogen activator receptor ; VIM : vimentine.
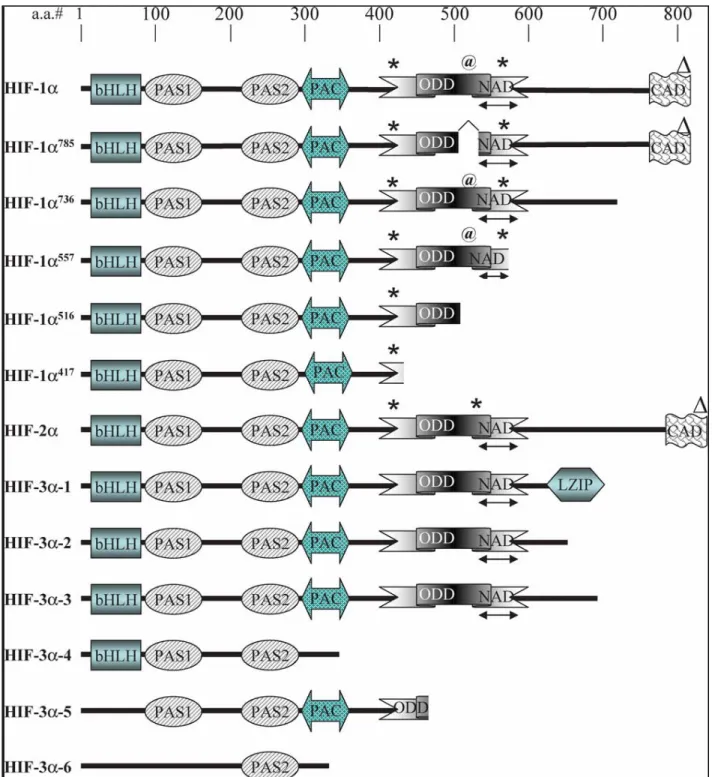
3-2 La sous-unité HIF-1α :
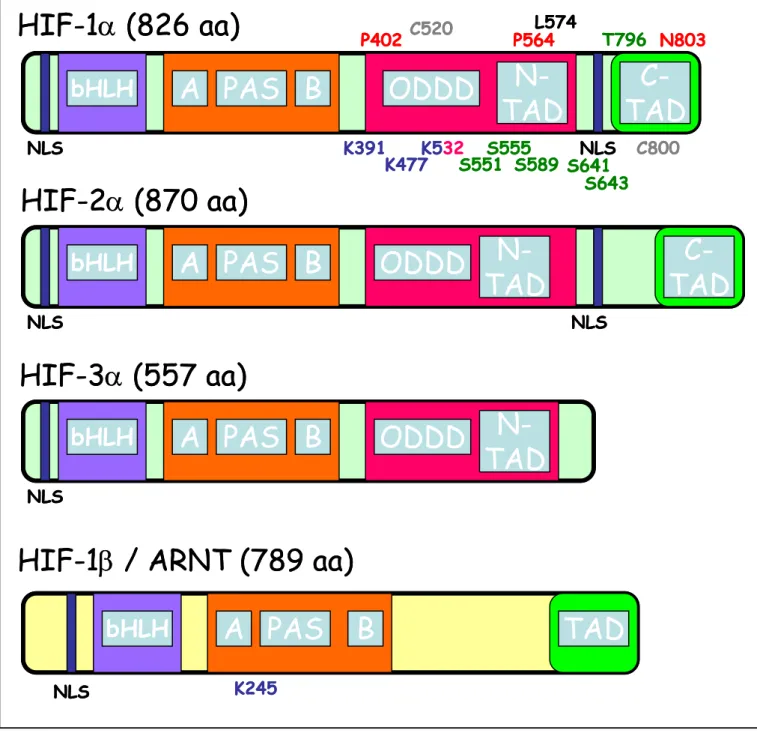
Le gène humain codant pour la sous-unité HIF-1α est situé sur le chromosome 14 (14q21-q24) et est composé de 15 exons donnant, après transcription et traduction, une protéine de 826 acides aminés (figure 6). La protéine HIF-1α est constituée par plusieurs domaines ou régions. La région basique (bHLH) est impliquée dans la dimérisation et dans la liaison directe avec l’ADN. Le domaine Per-ARNT-Sim (PAS) contient des régions hydrophobes nommées PAS A et PAS B. Ce domaine forme une seconde zone de dimérisation entre les sous-unités α et β [Jiang et al., 1996a]. Le domaine PAS est également connu pour lier la protéine chaperone HSP90 qui joue un rôle dans la stabilisation et l'accumulation de la sous-unité HIF-1α. En plus de la région basique N-terminale (bHLH) et des domaines PAS, la protéine HIF-1α possède dans sa moitié C-terminale, deux domaines de transactivation (TADs) impliqués dans la régulation et la stimulation de la transcription : le domaine de transactivation N-teminal (N-TAD) et le domaine de transactivation C-terminal (C-TAD) [Pugh et al., 1997]. Le domaine C-TAD a été montré comme agissant dans l’activation de la transcription via l’intéraction avec des co-activateurs comme p300/CBP [Lando et al., 2002;Hewitson et al., 2002]. En amont du domaine N-TAD, se trouve un domaine de dégradation dépendant de l'oxygène (ODDD), qui confère une certaine instabilité à la sous-unité HIF-1α en présence d’oxygène [Jiang et al., 1997;Huang et al., 1998]. Ce domaine de dégradation dépendant de l’oxygène intervient donc dans la régulation négative de HIF-1α dans des conditions normales d’oxygénation, c’est à dire en normoxie. Le domaine ODD comporte des résidus reconnus et hydroxylés par des prolyl-hydroxylases. Ces hydroxylations permettent la reconnaissance et la liaison de la protéine de von Hippel-Lindau (VHL) identifiée comme suppresseur de tumeur. La protéine VHL fait partie d’un complexe E3 ubiquitine ligase appelé ECV (Elongin/Cullin2/VHL). Ce complexe, en plus de la protéine VHL, est constitué de l'élongineB, l'élongineC, de Rbx1 (également connu sous le nom de ROC1/Hrt1), et de la Culline2 (Cul2). Le complexe ECV a un rôle très important de la stabilité et surtout la dégradation de la sous-unité HIF-1α. En effet, une fois la liaison établie entre le complexe et la sous-unité, la protéine HIF-1α est ubiquitinylée et dirigée vers le protéasome pour la dégradation. En normoxie, le temps de demi-vie de HIF-1α se situe aux alentours de 5 minutes [Maxwell et al., 2001;Maxwell et al., 1999].
HIF-1α (826 aa)
HIF-1β / ARNT (789 aa)
A
B
A
B
PAS
PAS
TAD
bHLH
bHLH
ODDD
HIF-2α (870 aa)
A
PAS
B
bHLH
ODDD
HIF-3α (557 aa)
A
PAS
B
bHLH
ODDD
N-TAD
TAD
C-
N-TAD
TAD
C-
N-TAD
NLS NLS NLS NLSFigure 6 : Représentation schématique de la structure de HIF-1 et des variants
d’épissage.
HIF-1 est un hétérodimère composé de HIF-1α et HIF-1β. La position et les modifications des acides aminés important sont également indiquées : rouge = hydroxylation, vert = phosphorylation, bleu = sumoylation, gris = nitrosylation, rose = acétylation. bHLH : basic helix loop helix ; PAS : Per ARNT Sim domain ; ODDD : oxygen dependant degradation domain ; TAD : transactivation domain ; NLS : nuclear localization signal. NLS NLS P564 P402 N803 K391 K477K532 C800 L574 K245 C520 S551 S589S555 T796 S641 S643
HIF-1α (826 aa)
HIF-1β / ARNT (789 aa)
A
B
A
B
PAS
PAS
TAD
bHLH
bHLH
ODDD
HIF-2α (870 aa)
A
PAS
B
bHLH
ODDD
HIF-3α (557 aa)
A
PAS
B
bHLH
ODDD
N-TAD
TAD
C-
N-TAD
TAD
C-
N-TAD
NLS NLS NLS NLS K391 K477K532 C800 L574 K245 C520 P564 P402 N803 NLS NLS S551 S589S555 T796 S641 S6433-3 HIF-1α, HIF-2α, HIF-3α et épissage alternatif :
L’épissage alternatif de l’ARNm de la sous-unité HIF-1α conduit à la formation de variants d’épissage. Différents variants ont ainsi été décrits : HIF-2α et HIF-3α [Tian et al., 1997;Gu et al., 1998] (figure 6-7). HIF-1α a été initialement identifiée par purification d'affinité en utilisant des oligonucléotides spécifiques du locus du gène de l’érythropïétine [Semenza and Wang, 1992;Wang and Semenza, 1995]. Les sous-unités HIF-2α et HIF-3α ont été identifiées par recherche d’homologie et criblages d'interaction avec HIF-1β. La protéine HIF-2α également appelée EPAS (endothélial PAS domain protein), HLF (HIF-like factor), HRF (HIF-related factor) ou encore MOP2 (member of PAS superfamily 2) est régulée positivement par l'hypoxie et lie HIF-1β pour former le complexe HIF-2. HIF-2, tout comme HIF-1, reconnaît sur l’ADN les séquences HRE. HIF-1α et HIF-2α sont connues pour partager des fonctions communes [Tian et al., 1997;Ema et al., 1997;Flamme et al., 1997;Hogenesch et al., 1997]. Cependant, l'expression de la sous-unité HIF-2α semble être plus limitée comparée à la sous-unité HIF-1α [Lofstedt et al., 2007]. Le variant d’épissage HIF-3α également appelé IPAS (inhibitory PAS domain protein) ou MOP7 (member of PAS superfamily 7), présente de fortes homologies de séquence avec HIF-1α et HIF-2α dans la région basique bHLH et les domaines PAS mais ne possède pas de domaine transactivateur en C-terminal [Gu et al., 1998;Makino et al., 2001]. A ce jour, 6 variants d'épissage de HIF-3α ont été identifiés : HIF-3α1, HIF-3α2, HIF-3α3, HIF-3α4, HIF-3α5, HIF-3α6 (figure 7). HIF-3α1, 2 et 3 partagent un domaine ODD commun qui inclut le motif consensus des prolyl-hydroxylases et lie le complexe VHL/E3 ubiquitine-ligase dans des conditions normales d’oxygénation. La sous-unité HIF-3α est aussi régulée positivement par l'hypoxie et peut interagir avec HIF-1β et les séquences HRE sur l'ADN. Toutefois, au niveau fonctionnel, HIF-3α semble avoir un rôle un peu différent en agissant comme un dominant-négatif de HIF-1α et en inhibant la transcription des gènes cibles induits par l’hypoxie. Cette inhibition passerait par la liaison de HIF-3α à la région N-terminale de HIF-1α empêchant ainsi l’interaction du complexe avec l'ADN.
Figure 7 : Représentation schématique de la structure des différents variants
d’épissage de la sous-unité HIF-1α.
Il existe donc 6 variants HIF-1α, 1 variant HIF-2α et également 6 variants HIF-3α. (d’après Kaur et al, 2005). bHLH : basic helix loop helix ; PAS : Per ARNT Sim domain ; ODDD : oxygen dependant degradation domain ; TAD : transactivation domain N- or C-terminal.
3-4 Les régulations du facteur inductible par l’hypoxie (HIF-1) :
HIF-1, et en particulier la sous-unité HIF-1α, est soumis à de multiples et diverses régulations (tableau 3 et figure 8). Ces régulations de la sous-unité HIF-1α incluent des modifications post-traductionnelles (hydroxylation, acétylation, phosphorylation, sumoylation et nitrosylation) mais également des interactions avec d'autres protéines qui peuvent modifier la stabilité ou l'activité transcriptionelle de HIF-1.
IGF/IL-1
PDGF
FOXO4
AngII
HIF-3α/IPAS
Ni
2+/Co
2+P14
arfHSP-90
GSK-3β
mTOR
P53
HDM2
PTEN
PI3K/Akt
Régulateurs de la sous-unité HIF-1α
CITED2/CITED4
CBP/p300
FIH-1
NO
ARD1
MAPK
VHL
ERO
Prolyl-hydroxylases
Hypoxie
Régulateurs négatifs
Régulateurs positifs
IGF/IL-1
PDGF
FOXO4
AngII
HIF-3α/IPAS
Ni
2+/Co
2+P14
arfHSP-90
GSK-3β
mTOR
P53
HDM2
PTEN
PI3K/Akt
Régulateurs de la sous-unité HIF-1α
CITED2/CITED4
CBP/p300
FIH-1
NO
ARD1
MAPK
VHL
ERO
Prolyl-hydroxylases
Hypoxie
Régulateurs négatifs
Régulateurs positifs
Tableau 3 : Tableau récapitulatif des principaux régulateurs (positifs et négatifs) connus
de la sous-unité HIF-1α.
Ces régulateurs agissent au niveau de la transcription, de la traduction ou de la stabilité de la protéine. Les interactions directes entre HIF-1α et régulateurs sont indiquées en grisées.
3-4-1 La principale régulation de l’expression de HIF-1α : le taux d’oxygène.
3-4-1-1 L’hydroxylation :
Contrairement à la sous-unité HIF-1β qui est exprimée constitutivement et de façon ubiquitaire dans les cellules, le niveau intracellulaire de HIF-1α est lui soumis à différentes régulations. La première et la plus importante de ces régulations est celle dépendante de la concentration d’oxygène (figure 8). A un taux normal d’oxygène ou normoxie, la sous-unité HIF-1α est prolyl-hydroxylée sur les résidus 402 et 564 de son domaine ODD [Maxwell et al., 1999;Jaakkola et al., 2001]. Chaque site d’hydroxylation sur la sous-unité présente un motif particulier : LXXLAP [Masson et al., 2001]. Cette prolyl-hydroxylation est dépendante de la quantité d’oxygène et est réalisée par des prolyl-hydroxylases. Pour permettre leur activité et assurer leurs fonctions, ces enzymes ont besoin d’oxygène, d’ions ferreux et de 2-oxoglutarate. Les prolyl-hydroxylases utilisent donc le 2-oxoglutarate comme substrat et les ions ferreux et l’ascorbate comme co-facteurs pour l’hydroxylation. A ce jour, trois prolyl-hydroxylases humaines ont été identifiées (PHD-1/HPH-3/EGLN2 ; PHD-2/HPH-2/EGLN1 ; PHD-3/HPH-1/EGLN3) [Semenza, 2001]. Cependant, un travail récent a montré que la prolyl-hydroxylase 2 semble être senseur principal des variations de la concentration d’oxygène [Berra et al., 2003]. Cette prolyl-hydroxylase régule l’expression de HIF-1α en normoxie en la maintenant à un niveau basal faible. La prolyl-hydroxylation initie l’interaction de la sous-unité HIF-1α avec le complexe ubiquitine-ligase VHL, permettant ainsi l’ubiquitinylation de HIF-1α et sa dégradation rapide par le protéasome 26S. Récemment, le résidu leucine 567 dans le domaine ODD a été montré comme jouant aussi un rôle dans la reconnaissance de HIF-1α par le facteur suppresseur de tumeur VHL [Kageyama et al., 2004]. De plus, la protéine VHL est capable de recruter des histones déacétylases qui vont inhiber la transcription dépendante de HIF-1. En condition d’hypoxie, les prolyl-hydroxylases cessent de fonctionner par le manque d’oxygène et d’ions ferreux. La sous-unité HIF1α peut ainsi échapper à la dégradation et s’accumuler dans le cytoplasme des cellules.
En plus du domaine ODD, le domaine C-TAD de HIF-1α est aussi régulé par les conditions d’oxygénation. En effet, un facteur inhibiteur de HIF-1 ou Factor Inhibiting HIF-1 (FIH-1) a été identifié par criblage double hybride chez la levure [Mahon et al.,
2001]. FIH-1 est également une oxygénase à activité dépendante de la présence de 2-oxoglutarate et de fer. Elle hydroxyle la sous-unité HIF-1α sur l’asparagine 803[McNeill et al., 2002]. Cette asparaginyl-hydroxylase va inhiber l’interaction du domaine C-TAD de HIF-1α avec le cofacteur CBP/p300, et par conséquent l’activité transcriptionnelle HIF-1. De plus, il a été démontré que FIH-1 pouvait également interagir avec le facteur suppresseur de tumeur VHL et faciliter l’ubiquitylation de HIF-1α par VHL.
HIF-1β
HIF-1α
HRE
Gènes ciblesPROTEASOME
VHL
Hypoxie
Normoxie
OH P402 OH P564 Ac K532 Sumo K OH Asp803 PHD1 ,2,3 ARD1 Reconnaissance par VHL et dégradation FIH-1 Inhibition de la liaison avec les co-facteursStabilisation et
augmentation de l’activité transcriptionnelle
SUMO-1
MAPK Augmentation de l’activité transcriptionnelle
GSK-3
Dégradation
HIF-1α
Figure 8 : La sous-unité HIF-1α est soumise à différentes modifications
post-traductionnelles dépendantes ou non des conditions d’oxygénation.
Hydoxylation, acétylation, phosphorylation par la GSK-3 et ubiquitinylation participent à la dégradation de la sous-unité par le protéasome. L’hydroxylation par FIH-1 inhibe l’interaction avec les co-facteurs de transcription. Sumoylation et phosphorylation par les MAPK modulent l’activité transcriptionnelle.
3-4-1-2 L’acétylation :
De récentes études menées sur Saccharomyces cerevisiae ont révélé la présence d’une nouvelle protéine capable d’interagir avec HIF-1, il s’agit d’une acétyltransferase nommée ARD-1 pour ARest Defective-1 [Jeong et al., 2002]. ARD-1 acétyle la sous-unité HIF-1α au niveau de son domaine ODD sur le résidu lysine 532. Cette acétylation constitue alors un deuxième signal de déstabilisation pour HIF-1α [Jeong et al., 2002]. Même s’il a été observé que l’ARN messager de ARD-1 était régulé négativement en conditions d’hypoxie [Chun et al., 2003], l’acétylation de la lysine 532 peut se produire en normoxie comme en hypoxie et ainsi augmenter l’interaction entre HIF-1α et VHL, favorisant l’ubiquitinylation de HIF-1α et sa dégradation par le protéasome. Toutefois, contrairement à l’étude de Jeong et al, un certain nombre d’autres travaux montrent qu’il n’y a pas de régulation en hypoxie de l’ARNm d’ARD-1 ainsi que de la protéine ARD-1 [Fisher et al., 2005;Arnesen et al., 2005]. Ces auteurs montrent également que des modifications d’expression de ARD-1, à savoir surexpression ou extinction, n’ont pas d’effet sur la stabilité de HIF-1α et HIF-2α [Bilton et al., 2005]. Le rôle exact de ARD-1 dans la progression tumorale et dans la régulation de l’expression et de l’activité de HIF-1 reste donc encore à déterminer.
3-4-2 Les autres régulations de HIF-1 :
La sous-unité HIF-1α connaît donc des régulations dépendantes de la concentration en oxygène. Mais un nombre important de régulations a lieu de façon indépendante de la quantité d’oxygène.
3-4-2-1 La phosphorylation :
Une de ces régulations indépendantes de la concentration en oxygène est la phosphorylation (figure 8). Il est maintenant bien connu que la phosphorylation de la sous-unité HIF-1α contribue à des régulations positives et négatives aboutissant à des modifications de la stabilité et de l’activité transcriptionnelle de HIF-1 [Richard et al., 1999;Sodhi et al., 2000]. En particulier, il a été démontré que les Mitogen Activated Protein Kinase (MAPK) ont la capacité de phosphoryler HIF-1α [Richard et al., 1999;Minet et al., 2001]. L’inhibition de p42/44 MAPK (aussi connue sous le nom de Erk1/2 ou de p38 MAPK dans différentes lignées cellulaires) induit une inhibition de
l’activité transcriptionnelle de HIF-1 [Wang et al., 1995b;Hur et al., 2001]. Une autre étude a également montré qu’un fragment recombinant contenant les résidus 531-826 de HIF-1α était phosphorylé in vitro par les MAPK ou la p38 Kinase [Sodhi et al., 2000]. De plus, un récent travail montre que la p42 MAPK modifie le fragment contenant les résidus 531-826 de HIF-1α sur les sérines 641 et 643. Ces résidus ont pu être identifiés par spectrométrie de masse à partir d’un fragment recombinant phosphorylé in
vitro de HIF-1α et confirmés par des analyses de délétion et de mutagenèse dirigée. Ce
travail démontre que l’inhibition de la phosphorylation de la sous-unité HIF-1α sur les serines 641 et/ou 643 induit son exclusion du noyau en inhibant le transport nucléaire dépendant de l’exportine CRM-1/EXPORT-1, ceci sans affecter significativement sa stabilité ou son activité transcriptionnelle [Mylonis et al., 2006]. Ces résultats suggèrent fortement que la phosphorylation de HIF-1α dépendante des MAPK est nécessaire pour une accumulation efficace dans le noyau.
Une autre phosphorylation de la sous-unité HIF-1α par la p38 kinase a également été impliquée dans l’interaction avec le facteur suppresseur de tumeur von Hippel-Lindau (VHL) [Kwon et al., 2005;Emerling et al., 2005]. Enfin, des travaux ont montré que les MAPK ont un rôle activateur de la transcription dépendante de HIF-1 en phosphorylant le co-activateur CBP/p300 permettant ainsi une meilleure interaction entre les deux partenaires [Sang et al., 2003].
La Glycogène synthase kinase 3 (GSK-3), en particulier l’isoforme beta, est aussi impliquée dans une régulation négative de la sous-unité HIF-1α. En effet, la GSK-3 peut phosphoryler le domaine ODD de HIF-1α entraînant ainsi une augmentation de la dégradation protéasome-dépendante de la sous-unité de façon indépendante de la prolyl-hydroxylation et de la liaison à la protéine VHL [Sodhi et al., 2001]. Cette phosphorylation de HIF-1α par la GSK-3 s’effectue sur la sérine 553, la thréonine 555 et la sérine 589 [Flugel et al., 2007].
La thréonine 796 a également été proposée pour être phosphorylée par la Caséine Kinase 2 (CK-2) [Cho et al., 2007]. Cette phosphorylation pourrait augmenter l’activité transcriptionnelle de HIF-1 en favorisant son interaction avec CBP/p300 et en diminuant son affinité pour FIH-1.
Une autre étude très récente [To et al., 2006] a montré que la thréonine 324 de la sous-unité HIF-2α se situait dans une région phosphorylable par la Proteine Kinase D1 (PKD-1), enzyme connue pour appartenir à la famille des PKC en tant que PKCm [Rykx et al., 2003]. En utilisant une forme recombinante de la PDK-1 dans un essai kinase in