T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul Sabatier
Discipline ou spécialité : Physiopathologie Moléculaire et Cellulaire
JURY
Pr Thierry Levade Examinateur Dr Anne Debant Rapporteur Pr Pierre Verrelle Rappoteur Dr Christine Toulas Directrice de Thèse
Dr Elisabeth Cohen-Jonathan Moyal co Directrice de Thèse
Ecole doctorale : Ecole doctorale Botechnologie et Santé
Unité de recherche : Département Innovation Thérapeutique et Oncologie Moléculaire Directeur(s) de Thèse : Dr Christine Toulas et Dr Elisabeth Cohen-Jonathan Moyal
Rapporteurs : Dr Anne Debant et Pr Pierre Verrelle Présentée et soutenue par Julie Milia
Le Le 27 Juin 2005
Titre : IMPLICATION DE RHOB DANS LES MECANISMES CELLULAIRES DE REPONSE AUX RAYONNEMENTS IONISANTS.
2
SOMMAIRE
ABBREVIATIONS_____________________________________________________________5 REVUE BIBLIOGRAPHIQUE ___________________________________________________6
1 Introduction. ______________________________________________________________7 2 Facteurs de radiosensibilité des tumeurs humaines. ______________________________7
2-1 Le nombres de cellules clonogéniques._______________________________________8 2-2 Le facteur oxygène.______________________________________________________8 2-3 La cinétique de prolifération. ______________________________________________8 2-4 La radiosensibilité intrinsèque. _____________________________________________8
3 Mode d’action des rayonnements ionisants (RI). _______________________________13 4 Dommages cellulaires radio-induits __________________________________________16
4-1 les membranes cellulaires : _______________________________________________16 4-2 Lésions de l’ADN : _____________________________________________________17 4-2-1 Type de lésions. ____________________________________________________17 4-2-2 Réparation des lésions de l’ADN radio-induites ___________________________19 4-2-2-1 Réparation par réversion de lésions (directe) __________________________19 4-2-2-2 Réparation par excision de lésion. __________________________________19 4-2-2-3 Réparation des cassures double brin de l’ADN. _______________________20 4-3 Contrôle du cycle cellulaire après IR. _______________________________________22
4-3-1 Progression normale dans le cycle et les points de contrôles dans les cellules de mammifères (Kohn, 1999). ________________________________________________22 4-3-2 Progression dans le cycle cellulaire en réponse aux dommages faits à l’ADN par les RI. ___________________________________________________________________24
4-3-2-1 Mécanismes moléculaires du point de contrôle en G1 (Fig.6). ____________27 4-3-2-2 Mécanismes moléculaires du point de contrôle en S. ___________________30 4-3-2-3Mécanismes moléculaires du point de contrôle en G2.___________________31 4-3-2-4 Mécanismes moléculaires du point de contrôle en M. ___________________37 4-4 Contrôle du cycle centrosomal.____________________________________________40 4-4-1 Structure du centrosome. _____________________________________________40 4-4-2 Cycle de duplication du centrosome.____________________________________42 4-4-3 Anomalies des centrosomes. __________________________________________45 4-5 Mort cellulaire. ________________________________________________________47 4-5-1 Apoptose. _________________________________________________________48 4-5-2 Nécrose. __________________________________________________________51 4-5-3 Sénescence. _______________________________________________________51 4-5-4 Mort mitotique. ____________________________________________________52 4-5-4-1 Caractéristiques morphologiques. __________________________________52 4-5-4-2 Mécanismes cellulaires. __________________________________________52 4-5-4-3 Mécanismes moléculaires de la mort mitotique. _______________________53 4-5-4-4 Les cellules géantes multinuclées : phénotype précurseur de la mort mitotique.
____________________________________________________________________55
5-1 Propriétés générales de la superfamille des petites GTPases Rho. _________________57 5-1-1 Structure. _________________________________________________________57 5-1-2 Cycle d’activation/inactivation des petites GTPases Rho. ___________________58 5-1-3 Régulateurs des GTPases Rho . ________________________________________58 5-1-3-1 Les RhoGEF. __________________________________________________59 5-1-3-2 Les RhoGAP. __________________________________________________59 5-1-3-3 Les RhoGDI. __________________________________________________60 5-1-4 Modifications post-traductionnelles. ____________________________________60 5-1-4-1 Mécanismes généraux. ___________________________________________61 5-1-4-2-Inhibiteurs de Farnésyl Transférase (FTI). ___________________________62 5-1-5 RhoA, RhoB et RhoC : les protéines «Rho ». _____________________________62 5-1-5-1 Structure ______________________________________________________63 5-1-5-2 Fonctions _____________________________________________________65 5-2 RhoB. _______________________________________________________________67 5-2-1 Généralités. _______________________________________________________67 5-2-2-Modifications post-traductionnelles de RhoB. ____________________________67 5-2-3 Localisation cellulaire de la protéine RhoB. ______________________________68 5-2-4 Induction de l’expression de RhoB._____________________________________68 5-2-5 Partenaires protéiques de RhoB. _______________________________________69 5-2-6 Fonctions biologiques de la protéine RhoB. ______________________________69 5-2-6-2 la protéine RhoB et le trafic endocytaire._____________________________69 5-2-6-3 la protéine RhoB et la transformation tumorale. _______________________70 5-2-6-4 la protéine RhoB et la réponse aux stress. ____________________________71 OBJECTIFS _________________________________________________________________73 RESULTATS ________________________________________________________________75 PARTIE 1 ___________________________________________________________________76 1 ARTICLE._______________________________________________________________77 Introduction ______________________________________________________________78 Conclusions ______________________________________________________________80
2-Résultats complémentaires : mécanismes moléculaires de l’effet radioprotecteur de
RhoB-F dans les cellules NIH3T3. _____________________________________________82
2-1 Analyse du rôle de RhoB dans la protection contre les CDB et la réparation des CDB radio-induites .____________________________________________________________82 2-2 Mécanismes moléculaires contrôlant l’accumulation des centrosomes surnuméraires après irradiation dans les cellules NIH3T3. _____________________________________84 2-3 Mécanismes moléculaires contrôlant l’arrêt en G2 induit par l’expression de RhoB après irradiation dans les cellules NIH3T3. __________________________________________87 PARTIE 2 ___________________________________________________________________89
Rôle de RhoB dans la réponse aux RI dans les glioblastomes U87. __________________90
1-RhoB ne contrôle pas la réparation des CDB dans les cellules U87._________________90 2- RhoB contrôle la mort mitotique après irradiation dans les cellules U87. ____________92 3- RhoB et arrêt en G2/M radio-induit dans les cellules U87. _______________________94 4- RhoB et anomalies de duplication des centrosomes radio-induites. _________________96 5-Resultats préliminaires :RhoB et régulation de Cdk1.____________________________96
4
Discussion __________________________________________________________________100 Materiels et méthodes _________________________________________________________105 BIBLIOGRAPHIE ___________________________________________________________109
ABBREVIATIONS
Abl : AbelsonADN : acide désoxyribonucléique ARN : acide ribonucléique
ATM : ataxia telangectasia mutated ATP : adénosine triphosphate
ATR : ataxia telangectasia related gene BER : base excision repair
BRCA : breast cancer susceptibility CAK : cyclin activated kinase CDB : cassures double brin CDK : cyclin dependant kinase Gy : gray
MPF : mitosis promoting factor NER : nucleotide excision repair
PIKK : Phosphatidyl Inositol 3 Kinase related protein kinase PLK : polokinase
RI : rayonnements ionisants TNF : tumor necrosis factor UV : ultrat violet
6
1 Introduction.
La radiothérapie est un outil thérapeutique essentiel dans le traitement des cancers. Son efficacité est cependant parfois limitée par la radiorésistance intrinsèque des tumeurs. Les voies de signalisation qui contrôlent les mécanismes de radiorésistance tumorale ne sont pas encore parfaitement connues. La meilleure connaissance de ces mécanismes est d'un intérêt majeur puisqu'elle permettrait à terme de comprendre l’effet des rayonnements ionisants dans les cellules humaines. Ainsi de nouvelles cibles thérapeutiques pourraient être utilisées pour lever la radiorésistance de certaines tumeurs et par conséquent améliorer l'efficacité de la radiothérapie en clinique humaine. L’équipe du laboratoire d’Innovation Thérapeutique et Oncologie Moléculaire de l’Institut Claudius Regaud a développé un axe de recherche visant à la compréhension de ces mécanismes en s’intéressant en particulier au contrôle de ces voies biologiques par les petites protéines G.
2 Facteurs de radiosensibilité des tumeurs humaines.
Une tumeur est considérée cliniquement comme peu sensible aux rayonnements ionisants, s’il survient une récidive dans le volume irradié après qu’il y a eu régression de la tumeur ou lorsque l’irradiation n’a pu permettre la régression de cette tumeur. Différents travaux ont relié la modulation de la radiosensibilité clinique à certaines caractéristiques liées à la tumeur .
8
2-1 Le nombres de cellules clonogéniques.
Une cellule est dite clonogénique si elle est capable d’établir un nouveau clone de cellules tumorales et de regénérer la tumeur : plus la tumeur contient de cellules clonogéniques et moins elle est radiocurable.
2-2 Le facteur oxygène.
Le manque d’oxygène (hypoxie) est un facteur témoin de la diminution de la radiosensibilité : une tumeur est d’autant moins radiocurable qu’elle contient plus de cellules hypoxiques.
2-3 La cinétique de prolifération.
La cinétique de prolifération des cellules tumorales influe sur la survie après irradiation. Les tumeurs présentant un pourcentage élevé de cellules en prolifération et un taux de perte cellulaire important sont celles qui sont les plus radiosensibles et les plus radiocurables.
2-4 La radiosensibilité intrinsèque.
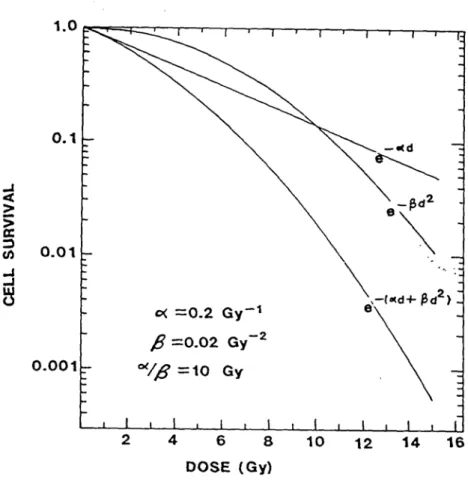
La radiosensibilité intrinsèque correspond à la mise en place de mécanismes moléculaires qui permettent à la cellule de résister aux rayonnements. Le concept de radiosensibilité intrinsèque introduit par Fertil et Malaise (Fertil and Malaise, 1985), peut être étudié par l’établissement de courbes de survie en fonction de la dose unique délivrée.
Courbes de survie de cellules de mammifères irradiées.
La proportion de cellules survivantes, ou taux de survie (S), diminue lorsque la dose (D) augmente. Une étude expérimentale permet d’établir la relation entre le taux de survie, la dose
pour un type cellulaire et des conditions déterminées. La courbe de survie est la représentation graphique de cette relation. Les premières courbes de survie de cellules de mammifère après irradiation ont été obtenues par Puck et Marcus (Puck and Marcus, 1956). Elles sont formées de deux parties linéaires séparées par une incurvation ou un épaulement de la courbe plus ou moins marqué (Fig.1).
L’aspect de cette courbe est expliqué par la théorie de la réparation cellulaire. L’hypothèse étant que toutes les lésions potentiellement réparables le sont à de faibles doses d’irradiation. L’inflexion de la courbe de survie s’explique par le dépassement des mécanismes de réparation lorsque la dose augmente. La pente initiale de la courbe de survie correspond aux doses auxquelles certaines lésions sont d’emblée irréparables. Cette courbe a longtemps été décrite par deux principaux paramètres, l’un appelé n, extrapolation de la deuxième partie rectiligne sur l’axe des ordonnées représentant la partie initiale de la courbe, et l’autre Do (dose létale moyenne) représentant la dose unique qui ne laisse que 37% de cellules survivantes dans la deuxième partie rectiligne. Ce modèle ne présente en fait qu’un aspect descriptif, peu satisfaisant quant au pouvoir prédictif de la radiosensiblité tumorale et n’expliquant pas l’incurvation initiale de la courbe.
Modèles mathématiques
Différents modèles mathématiques décrivant la courbe de survie, et en particulier l’épaulement, ont ensuite été développés pour décrire les mécanismes hypothétiques de cet effet (Tubiana M, 1986).
Le modèle de « une cible à un coup » : une seule lésion d’une cible induit la mort cellulaire.
Le modèle de « n cibles sublétales à un coup » : n cibles doivent être atteintes une fois pour produire la mort cellulaire.
10
Figure 1: Courbe de survie cellulaire de cellules de mammifères après
Le modèle de « une cible à deux coups » (modèle quadratique) : la mort cellulaire résulte de l’addition de deux évènements sublétaux indépendants produits par le passage de deux particules distinctes.
Le modèle conjuguant les deux premiers modèles « Le modèle linéaire quadratique » : la mort
cellulaire peut résulter soit de l’atteinte d’une cible létale, soit de n cibles sublétales.
Ce dernier modèle permet la coexistence de deux évènements létaux, la cellule pouvant être tuée : soit par une lésion directement létale, cet événement constituant la composante linéaire de l’équation (pente à l’origine)
soit par accumulation de lésions sublétales qui constituent la composante quadratique Le taux de survie est donné par : S=e-(αD+βD2)
Les courbes de survies ainsi modélisées se dissocient en deux composantes (Fig. 2) La première, représentée par la partie initiale, correspond à l’équation e-αD La deuxième, correspond à l’équation e-βD2
α : pente à l’origine
β: correspond à la deuxième composante de la courbe D : la dose
Ce modèle reste le plus utilisé pour représenter l’effet biologique de l’irradiation. Ainsi, à partir des données expérimentales, un lissage de courbe est effectué selon ce modèle mathématique et l’aire sous la courbe (MID) est calculée (Fertil and Malaise, 1985). Cette valeur est représentative d’un type cellulaire donné et permet de quantifier la radiosensibilité intrinsèque.
Figure 2: Modèle linéaire quadratique
La courbe de survie est le résultat de deux mécanismes de mort cellulaire : la mort cellulaire induite par une lésion représentée par la première partie de la courbe dont l’équation est e-ad,
et une mort par des lésions cumulées expliquant l’incurvation progressive de la courbe et représentée par le coefficient b. Le rapport a/b représente la dose à laquelle les deux types de mécanismes interviennent de façon égale dans la survenue de la mort cellulaire.
(Issu de Whiters et coll, 1992)
3 Mode d’action des rayonnements ionisants (RI).
Quand ils arrivent au contact du milieu biologique, les photons X et γ (Hν) peuvent interagir avec les noyaux mais surtout avec les électrons des atomes du milieu, en leur cédant une partie de leur énergie
.
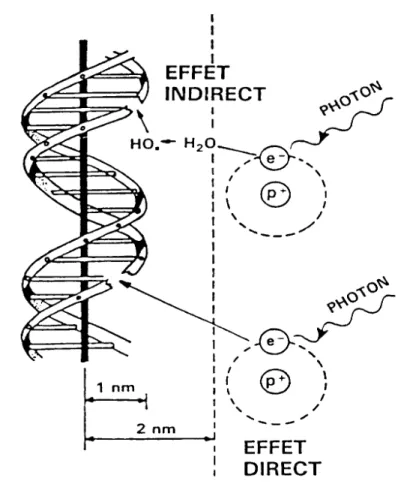
Les photons X ou γ incidents et les électrons arrachés par ces derniers rencontrent, dans le milieu biologique, des atomes appartenant essentiellement aux molécules d’eau ou aux macromolécules membranaires, cytoplasmiques ou nucléaires. De ce fait, les RI vont engendrer deux types de réactions radiochimiques distinctes, chacune aboutissant à la formation de radicaux libres.- Effet direct des RI (Fig.3)
Lorsque le photon ou l’électron libéré secondairement transfère tout ou une partie de son énergie à un atome appartenant à une macromolécule organique de la cellule (protéines, lipides, sucres, acides nucléiques…), celle-ci passe par phénomène d’excitation ou d’ionisation à un niveau énergétique instable et perd l’excèdent d’énergie respectivement :
- par retour à l’état initial avec émission d’un photon de fluorescence.
- par rupture de liaisons chimiques à l’intérieur de la molécule avec formation de radicaux libres.
- Effet indirect des RI : la radiolyse de l’eau (Fig. 3)
Les organes vivants étant très riches en eau (80%), l’interaction entre les rayonnements et les molécules d’eau reste la plus probable. L’interaction rayonnement-eau induit alors rapidement la formation d’espèces réactives de l’oxygène formées à partir des molécules d’eau excitées ou ionisées. Ces réactions radio-chimiques correspondent à la radiolyse de l’eau.
14
Figure 3: Mécanismes d’action des rayonnements ionisants par effet direct
et indirect
.Effet direct : interaction de la molécule d’ADN et d’un électron mis en mouvement à la suite de l’absorption d’un photon.
Effet indirect : un électron mis en mouvement à la suite de l’absorption d’un photon interagit avec une molécule d’eau générant la production d’un radical tel que OH
.
, qui à son tour provoque une lésion au niveau de la molécule d’ADN.Hn + H2O e- + H20+ e
+ H20 H + OH
(H : radical hydrogène) H20+ H+ + OH (OH : radical hydroxyle)
Le photon incident peut, cependant, n’induire qu’une excitation de la molécule d’eau qu’il rencontre.
Hn + H2O H2O* H2O* H + OH
H + H H2 (H2 : dihydrogène) H + OH H2O
OH + OH H2O2 (H2O2 : péroxyde d’hydrogène)
Toutefois, la présence d’oxygène moléculaire tend à amplifier la cascade des réactions chimiques car l’oxygène réagit très facilement sur le radical hydrogène ou sur les électrons libres selon les équations suivantes :
O2 + H OOH
(OOH : radical hydroperoxyde) O2 + e- O2- (O2- : radical superoxyde)
L’eau intracellulaire est finalement décomposée sur le trajet du rayonnement en ions H2O+
, H+,OH-), en radicaux libres (OH, H, OOH, O2-) et en espèces moléculaires (H2O2, H2) hautement réactives, oxydantes ou réductrices.
Les produits de la radiolyse de l’eau vont secondairement attaquer les macromolécules organiques situées à proximité, mécanisme qui aboutit donc à la rupture de liaisons covalentes simples ou doubles, comme dans le cas des effets radio-chimiques directs (Tubiana M, 1986).
16
4 Dommages cellulaires radio-induits
Parmi les conséquences cellulaires et moléculaires produites par les RI, nous pouvons distinguer : - Les interactions entre les rayonnements et les membranes cellulaires qui sont la conséquence d’une péroxydation par les radicaux libres, libérés lors de la radiolyse de l’eau, des phospholipides des systèmes membranaires. Il s’ensuit des modifications des propriétés biophysiques de la double couche lipidique transformant la structure et la fonction des protéines membranaires (Vit and Rosselli, 2003).
- Les lésions subcellulaires et divers types de lésions de l’ADN notamment l’induction de cassures doubles brins sont responsables des effets létaux des RI (Valerie and Povirk, 2003). - Les dommages induits au niveau cellulaire après exposition aux RI activent les points de
contrôles du cycle cellulaire et inhibent la progression dans les phases G1, S et G2 du cycle cellulaire (Iliakis et al., 2003).
- Les dommages induits au niveau du cytosquelette sur la tubuline alpha, beta et gamma qui sont capables d’induire des changements dans la structure des microtubules (Porter and Lee, 2001). Les radiations peuvent également entraîner une disparition des filaments d’actine modifiant ainsi le cytosquelette d’actine (Somosy et al., 1995).
- La mort cellulaire par apoptose ou par mort post-mitotique.
4-1 les membranes cellulaires :
Certains travaux montrent que les membranes cellulaires sont endommagées par les rayonnements. Ces dommages sont importants dans l’induction de la mort cellulaire radio-induite. Le mécanisme le mieux connu est initié par le clivage radio-induit de la sphingomyéline membranaire par des sphingomyelinases acides ou neutres. Ce clivage donne naissance à un second messager, le céramide. La génération de céramide précède l’apoptose suite à différents
stimuli incluant les RI mais aussi le traitement au TNFα , FasL et l’exposition aux glucocorticoïdes (Kolesnick et al., 1994). Certains travaux montrent que les tissus et les cellules de souris déficientes en sphingomyélinase acide sont beaucoup plus résistants à l’apoptose radio-induite (Santana et al., 1996). De plus, des cellules de lymphone Burkitt (qui sont résistantes à l’apoptose radio-induite) ne sont pas capables d’accumuler le céramide après irradiation (Michael et al., 1997).
4-2 Lésions de l’ADN :
4-2-1 Type de lésions.
Les molécules d’ADN constituent la cible principale des RI. Les anomalies survenant sur l’ADN après irradiation conduisent à la létalité cellulaire. Ces lésions peuvent être de différents types (Fig.4) :
- Altération des bases de l’ADN. Ces dommages incluent les O6
-méthylguanine, les thymines glycols et autres bases de l’ADN réduites, oxydées ou fragmentées qui sont produites par les espèces réactives de l’oxygène ou les RI.
- Pontages intra-brin, inter-brin de l’ADN et ADN-protéine.
Ces lésions ADN-protéines sont produites par la réaction de la forme aldéhyde de sites abasiques avec les protéines.
- Cassures d’un seul ou de deux brins de l’ADN.
Les cassures simple brin sont générées directement par les agents endommageant l’ADN. Les cassures double brin (CDB) peuvent être générées par les radiations ionisantes, les agents radiomimétiques et les espèces réactives de l’oxygène.
Nature de la liaison
Nombres/Gy/cellule diploïde
Lésion de base d’ADN 1000-2000
Lésion de sucre 800-1600
Liaison entre protéine et ADN 150
Liaison entre les deux brins d’ADN 30
Cassure simple brin 500-1000
Cassure double brin 40
4-2-2 Réparation des lésions de l’ADN radio-induites 4-2-2-1 Réparation par réversion de lésions (directe)
Les mécanismes de réversion ont en général une spécificité très étroite pour un type de lésion de l’ADN. Il existe deux types de réparation « directe » dans la majorité des organismes :
- la photoréactivation enzymatique de dimères de pyrimidine (lésions induites par les UV) due à une enzyme, la photolyase qui présente une grande affinité vis-à-vis des dimères de pyrimidine (CDP). Cette chromoprotéine rompt la liaison du dimère pour redonner deux monomères. Son action est contrôlée par la lumière visible.
- le transfert de groupements alkyls qui consiste à « réverser » les lésions formées par la fixation de groupements alkyls sur les sites nucléophiles des bases, mais aussi sur les groupements phosphates. Ce sont des « enzymes suicides » appelées alkyl-transférases qui ôtent le motif alkyl en le liant au niveau des cystéines, ce qui conduit à l’inactivation de la protéine.
4-2-2-2 Réparation par excision de lésion.
- Excision de base (BER).
Ce mécanisme permet d’exciser des bases oxydées ou réduites, alkylées, déaminées ou encore mésappariées. La réparation peut être d’un seul nucléotide (brin court) ou de 2 à 10 nucléotides (long patch) (Sancar et al., 2004).
Brin court: la base endommagée est retirée par la ADN glycosylase constituée de l’AP
20
clive la liaison 5’ du site puis recrute la polymérase β. La coupure est alors reliée par le complexe Lig3(ADN ligase 3)/XRCC1.
Brin long : la base endommagée est retirée par une glycosylase hydrolytique ou une
hydrolyse spontanée. APE1 (endonucléase 1) clive la liaison phosphodiester en 5’ et les complexes RFC/PCNA-Polδ/ε, PCN, puis l’endonucléase FNE1 déplacent le brin en 3’jusqu’à la coupure en produisant un brin flottant de 2 a 10 nucléotides (nt). Un brin de même taille est alors synthétisé par Pold/e avec l’aide de PCNA, la structure flottante est clivée par l’endonucléase FEN1 et la ligation est effectuée par la ligase 1.
- Excision de nucléotides (NER)(Sancar et al., 2004).
Les étapes basiques consistent chronologiquement en une reconnaissance de la lésion, une double incision pour former un oligomère de 24-32 nt, une libération de l’oligomère excisé, une synthèse du brin complémentaire et une ligation. Chez l’homme les protéines impliquées dans ce mécanisme sont RPA, XPA, XPC, TFIIH, XPG et XPF•ERCC1. Il est important de noter qu’une déficience dans ce mécanisme de réparation entraîne le syndrome xeroderma pigmentosum caractérisé par une très grande sensibilité à la lumière entraînant de sévères cancers de la peau (Norgauer et al., 2003).
4-2-2-3 Réparation des cassures double brin de l’ADN.
Il est maintenant admis que l’induction de dommages de type CDB de l’ADN est considérée comme étant à l’origine des effets létaux des RI (Jeggo, 1998). Les CDB sont réparées par deux types de mécanismes : la recombinaison homologue (RH) ou le NHEJ ( Nonhomologous
end-joining) (Khanna and Jackson, 2001) (Gottlieb and Jackson, 1993; Jackson, 2002; Sancar et al., 2004; Valerie and Povirk, 2003).
- Recombinaison homologue (RH).
Ce mécanisme nécessite la présence de séquences homologues qui se trouvent soit sur la chromatide sœur, soit sur le chromosome homologue. La voie de réparation par RH prédomine en phase S et G2 du cycle cellulaire où une chromatide sœur intacte peut servir de modèle (Saleh-Gohari and Helleday, 2004). Les acteurs de la RH chez les cellules de mammifères sont les protéines Rad50, Rad51, Rad52, Rad 54, MRE11 ainsi que d’autres protéines reliées à la recombinase Rad51 comme Rad51B, Rad51C, Rad51D, Brca1, Brca2, XRCC2 et XRCC3 (Khanna and Jackson, 2001). Les CDBs induisent des arrêts du cyle cellulaire médiés par l’activation de la protéine kinase ATM. ATM est une protéine de 350 kDa dont le domaine catalytique présente une forte homologie de séquence avec la famille des PI3K (Phosphoinositide 3 Kinase) mais n’a pas l’activité lipide-kinase. Une fois activée, ATM a pour cible la protéine NBS1 qui appartient au complexe Rad50-MRE11-NBS1. Dès que ce complexe est activé, il stimule l’activité exonucléasique de Rad52, qui au préalable permet la reconnaissance des extrémités de l’ADN. L’action des exonucléases génère des régions d’ADN simple-brin avec une extrémité 3’ libre. Rad51 en coopération avec deux protéines se liant à l’ADN simple brin RPA et Rad52 forme alors un filament nucléoprotéique sur l’ADN simple brin. Ce filament nucléoprotéique composé de Rad51 cherche des régions d’homologie. D’autres protéines telles que Brca1, Brca2, XRCC2, XRCC3, Rad51B,C,D interviennent dans ces étapes. Ce processus de RH est achevé par l’action de polymérases et de ligases (pour revue (Khanna and Jackson, 2001) ; (Dudas and Chovanec, 2004)).
22
- Jonction d’extrémités non homologues (NHEJ).
Dans les cellules de mammifères, le NHEJ joue un rôle fondamental pour la réparation des CDB radio-induites (Jeggo, 1998). Il est admis que la protéine kinase dépendante de l’ADN (DNA-PK) et le complexe XRCC4/Ligase IV sont nécessaires au NHEJ. La DNA-PK est une enzyme hétérodimérique composée d’une sous-unité catalytique de 460 kDa, la DNA-PKcs, sérine/thréonine kinase qui appartient à la famille des phosphatidylinositol 3 kinase (Hartley et al., 1995), et d’une sous-unité régulatrice, l’héterodimère Ku70/Ku86 (Gottlieb and Jackson, 1993). Ku70/Ku86 se lie aux extrémités des CDB et recrute la sous-unité catalytique de la DNA-PK (DNA-DNA-PKcs) (Hartley et al., 1995) qui peut alors phosphoryler ses protéines cibles. La protéine Artémis intéragit avec la DNA-PKcs et permet la dégradation des extrémités de l’ADN libre des CDB grâce à son activité exonucléase 3’ à 5’et endonucléase (Moshous et al., 2001).Le complexe XRCC4/Ligase IV effectue lui, les dernières étapes de la jonction des extrémités de la cassure (Kanaar et al., 1998).
4-3 Contrôle du cycle cellulaire après IR.
4-3-1 Progression normale dans le cycle et les points de contrôles dans les cellules de mammifères (Kohn, 1999).
La progression d’une phase du cycle cellulaire à la suivante est contrôlée par l’activation et l’inactivation de protéines appartenant à une famille de serine/thréonine kinases, les kinases dépendantes des cyclines (CDKs). Sous forme de monomères les CDKs sont inactives et sont activées après liaison aux cyclines. Différents complexes cycline/CDK sont assemblés et activés pour contrôler les différents passages d’une phase à l’autre du cycle cellulaire. L’activité de ces complexes est régulées à différents niveaux :
• les CDK peuvent être activées par la phosphorylation d’un résidu thréonine, se trouvant dans leur domaine carboxy terminal, par les kinases CAK (Cyclin Activated Kinase). Le résidu cible est la thréonine 161 pour Cdk1, 160 pour Cdk2 et 172 pour Cdk4 (Solomon and Kaldis, 1998).
• Le complexe cycline-CDK peut être piégé sous forme inactive par phosphorylation d’un ou deux résidus au niveau du site de liaison de l’ATP des CDKs se trouvant dans leur domaine amino terminal. Ces résidus peuvent être un residu thréonine (14) ou tyrosine (15) largement conservé au niveau des CDKs et phosphorylé par exemple par les membres de la famille des kinases Wee1/Mik1/Myt1 (Wee1 et Myt1 chez les vertebrés et Wee1 et Mik1 chez S.pombe)
• Les membres de la famille de la phosphatase Cdc25 déphosphorylent le site de liaison de l’ATP tyrosine et/ou thréonine (14 et 15) des CDKs et donc activent le complexe cycline/CDK.
• Il existe aussi différentes familles d’inhibiteurs des complexes cycline-CDK, les CKIs (Cyclin CDK Inhibitors). Ces protéines sont classées en deux familles en fonction de leur structure et de leur CDK cible :
• Les membres de la famille INK4 (Inhibitors of Cdk4 : p16INK4a
, p15INK4b
, p18INK4c
, p19INK4d ) caractérisés par des motifs ankyrine répétés (Fahraeus et al., 1998). Ils se lient spécifiquement aux kinases Cdk4 et Cdk6 qu’ils inhibent en empêchant la liaison à la cycline D.
• Les membres de la famille Cip/Kip (p21Waf/Cip1/Sid1
, p27Kip1
, p57Kip2
) se lient aux complexes cycline/Cdk en formant des hétérotrimères. p27Kip1 empêche la progression cellulaire en phase S en bloquant les complexes CyclineE/Cdk2 et CyclineA/Cdk2. La p21Waf/Cip1/Sid1
a une spécificité plus large puisqu’elle inhibe l’activité des complexes CyclineA/Cdk2, Cycline E/Cdk2 et CyclineB/Cdk1.
24
4-3-2 Progression dans le cycle cellulaire en réponse aux dommages faits à l’ADN par les RI.
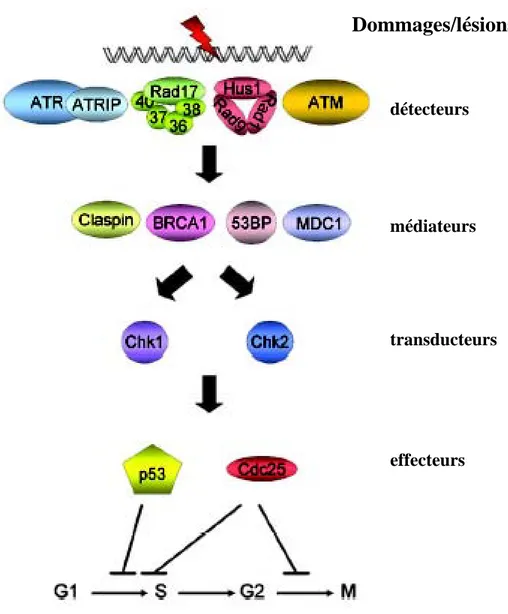
Les dommages à l’ADN induits par les RI induisent des signaux qui peuvent temporairement activer des points de contrôle du cycle cellulaire conduisant soit à la réparation de l’ADN, soit à un arrêt irréversible de la prolifération qui conduit à la mort des cellules (apoptose, nécrose). Il est maintenant admis que l’exposition aux radiations ionisantes entraîne des modifications de la progression normale du cycle cellulaire (Iliakis, 1997) pendant le passage de la phase G1 à la phase S (point de contrôle en G1/S), de la phase S à la phase G2 (point de contrôle en S) et surtout pendant le passage de la phase G2 à la mitose (point de contrôle en G2/M). Les points de contrôle du cycle cellulaire nécessitent des détecteurs (« sensors »), des médiateurs, des transducteurs du signal et des effecteurs (Fig.5).
• Les détecteurs : comme pour la réparation de l’ADN, les points de contrôles du cycle cellulaire nécessitent la reconnaissance du dommage à l’ADN pour initier la voie contrôlant l’arrêt dans une phase du cycle. Des protéines ont été identifiées comme étant des détecteurs spécifiques des point de contrôle du cycle cellulaire en réponse aux RI : il s’agit essentiellement des protéines de la famille des PIKK, ATM et ATR (Durocher and Jackson, 2001), mais également du complexe « RFC/PCNA like », Rad17/9-1-1 (Melo and Toczyski, 2002). Comme nous l’avons décrit précédemment, ATM est une protéine kinase activée par l’apparition de CDBs de l’ADN (Falck et al., 2005). En effet le complexe MRN (Mre11/Rad50/Nbs1) reconnaît les CDB et permet ainsi le recrutement d’ATM et son activation par une autophosphorylation de la serine 1981 (Bakkenist and Kastan, 2003; Falck et al., 2005). Après exposition aux RI, ATM phosphoryle différentes protéines telles que Chk2 (Chan et al., 2000), p53 (Banin et al., 1998), NBS1, BRCA1 (Gatei et al., 2001), et elle même (Bakkenist and Kastan, 2003).
Dommages/lésions
détecteurs
médiateurs
transducteurs
effecteurs
Figure 5: Voies de signalisation des dommages de l’ADN induits par les RI
contrôlant le cycle cellulaire.
26
Ces phosphorylations induisent alors différents arrêts dans le cycle cellulaire. Le complexe hRad17-RFC est formé de la protéine hRAD17 et de 4 sous unités RFC (RFC2, RFC3, RFC4, RFC5). Ce complexe serait responsable de la reconnaissance des cassures simples et double brin de l’ADN et du recrutement de protéines telles que les polymérases d et e mais aussi du complexe 9-1-1 (RAD9, HUS1, RAD1) afin de former une structure permettant le maintien des extrémités d’ADN endommagés (Bermudez et al., 2003; Lindsey-Boltz et al., 2001). Les travaux actuels suggèrent que le complexe Mre11-Rad50-NBS1 et BRCA1 seraient aussi impliqués dans la détection des cassures double brin de l’ADN (Iliakis et al., 2003).
• Les médiateurs : ces protéines s’associent avec les détecteurs et les transducteurs en réponse aux dommages faits à l’ADN. Chez l’homme, trois protéines possèdent le domaine d’interaction protéine-protéine BRCT (domaine répété d’interaction retrouvé sur la protéine BRCA1 et de nombreuses protéines des systèmes de réparation de l’ADN) : 53BP1 (Schultz et al., 2000; Wang et al., 2002), la « topoisomérase binding protéin » TopBP1 et la « Mediator of DNA damage checkpoint 1 » MDC1 (protéine médiatrice du point de contrôle aux dommages à l’ADN 1). Ces protéines interagissent avec des détecteurs comme ATM (Mochan et al., 2004), des protéines de la réparation de l’ADN telles que BRCA1, BRCA2 et des protéines transductrices ou effectrices telles que Chk2 et p53 respectivement. De nombreux travaux montrent qu’elles sont toutes impliquées dans la réponse aux dommages de l’ADN et par conséquent aux RI (Kao et al., 2003; Mochan et al., 2004).
• Les transducteurs : ces protéines transduisent les signaux reçus des protéines médiatrices aux protéines effectrices. Chez l’homme il existe deux kinases, Chk1 et Chk2 (Checkpoint Kinases) qui phosphorylent les protéines cibles sur leurs résidus Serine et Thréonine.
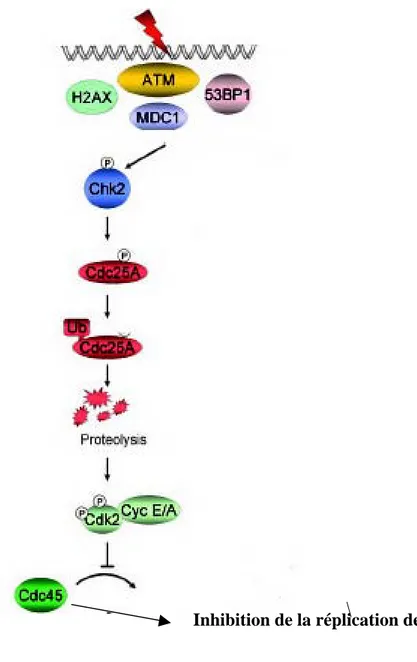
• Les effecteurs : en déphosphorylant les kinases dépendantes des cyclines (CDKs) humaines, les trois phosphotyrosines phosphatases, Cdc25 A, B, C, contrôlent directement des points de contrôle du cycle cellulaire. La phosphorylation des protéines Cdc25 par les Chks, crée un site de liaison pour les protéines adaptatrices 14-3-3. Cette phosphorylation inactive Cdc25 en l’excluant du noyau et/ou en causant sa dégradation protéolytique. Les protéines Cdc25 non phosphorylées induisent la transition G1/S en déphosphorylant Cdk2 et la transition G2/M en déphosphorylant Cdk1 (Cdc2) (Fig.5).
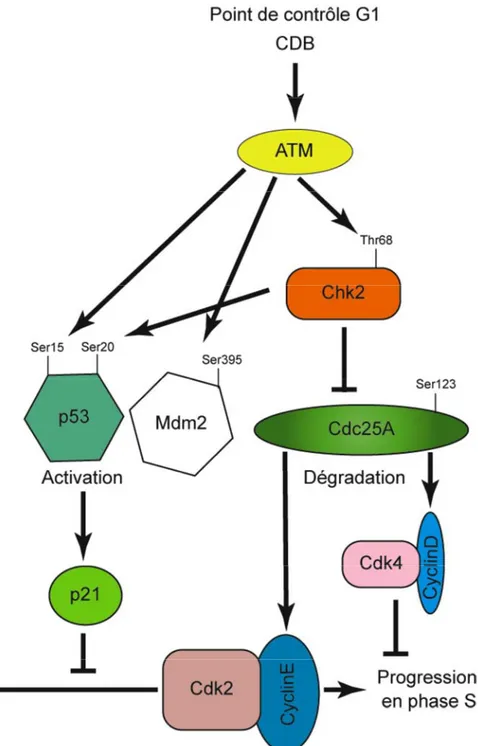
4-3-2-1 Mécanismes moléculaires du point de contrôle en G1 (Fig.6).
Ce point de contrôle prévient l’entrée des cellules en phase S, lorsque l’ADN est endommagé, en inhibant l’initiation de la réplication.
-Mécanisme général.
L’initiation du cycle cellulaire est affectée par l’activation de deux protéines kinases régulatrices, CDK4 et CDK2, en association avec les cyclines D et E respectivement. La phosphorylation de protéines cibles telles que la protéine de rétinoblastome (pRB) ou CDC45 entraîne l’entrée en phase S (Sherr, 1995; Sherr, 1996). Le proto-oncogène c-Myc contribue au passage de la phase G1 à la phase S en activant différents mécanismes, notamment la transcription des cyclines D1, D2, E et de la phosphatase CDC25A. Les voies pRB et c-Myc coopèrent pour augmenter l’activité de CDK2/cyclineE, donc de CDC45 qui favorise alors la réplication.
Figure 6: Signalisation du point de contrôle de la phase G1 en réponse aux
rayonnements ionisants.
-Arrêt en phase G1 induit par les RI.
Si les lésions sont des cassures double brin (CDBs), causées par les RI (Fig6), la protéine ATM est activée et phosphoryle différentes protéines cibles comme p53 ou Chk2. Ces phosphorylations entraînent soit une initiation de l’arrêt, soit un maintien de cet arrêt (Bartek and Lukas, 2001). L’arrêt en G1/S est contrôlé par la phosphorylation de Chk2, qui à son tour phosphoryle la phosphatase Cdc25A, alors inactivée par délocalisation du noyau vers le cytoplasme et dégradée par le système ubiquitine-protéasome (Molinari et al., 2000). La dégradation de Cdc25A activée, entraîne l’accumulation de la forme inactive de Cdk2 (forme phosphorylée) qui est alors incapable de phosphoryler Cdc45 responsable de l’initiation de la réplication. En réponse aux RI, ATM phosphoryle p53 sur la Ser15 directement et la Ser20 via Chk2 (Chehab et al., 2000; Shieh et al., 2000). La phosphorylation de p53 inhibe son export nucléaire et sa dégradation , ce qui entraîne une augmentation du taux intracellulaire de p53 (Chehab et al., 2000). Ce mécanisme semble être complété en réponse aux RI par la phosphorylation de la serine 395 de Mdm2 par ATM, inhibant donc la dégradation de p53 par le protéasome (Maya et al., 2001). p53 active alors ses gènes cibles comme p21WAF-1/CIP1 qui va à son tour lier et inactiver le complexe Cdk2/CyclineE. p21WAF-1/CIP1 se lie alors au complexe Cdk4/CyclineD empêchant ainsi la phosphorylation de pRb (el-Deiry et al., 1994; Harper et al., 1993). Cet événement libère pRb du facteur de transcription E2F qui n’activera pas la transcription des gènes spécifiques de la phase S (Bartek and Lukas, 2001) (Lin et al., 2001).
Il existe un contrôle de cette phase du cycle independante de p53 en réponse aux RI, impliquant c-ABL. En effet ATM phosphoryle c-Abl qui active à son tour p73. Cette cascade aboutit à la transactivation de p21 et Gadd45 donc à l’inhibition de la progression en phase S du cycle cellulaire (Lee and La Thangue, 1999) (Shafman et al., 1997). De plus, certaines voies sont
30
bloquées en réponse aux RI afin de permettre l’arrêt en G1, c’est le cas par exemple de la voie controlée par l’oncogène Myc. Il a été montré qu’en réponse aux RI le taux de cette protéine était fortement diminué dans les cellules MCF7 (Magnet et al., 2001). D’autre part, la surexpression de c-Myc dans des cellules épithéliales mammaires humaines empêche l’arrêt en G1 en réponse aux rayonnements γ (Sheen and Dickson, 2002). La voie de c-Myc semble être régulée négativement en réponse aux rayons afin de permettre l’arrêt en G1 et donc le contrôle de l’entrée en phase S du cycle cellulaire.
En réponse aux RI, l’accumulation de p53 entraîne un arrêt en G1 qui facilite la réparation de l’ADN et/ou en fonction de la lignée, la mort cellulaire. Les travaux concernant le rôle de p53 dans la radiosensibilté des tumeurs est controversé. L’hétérogénéité des résultats est due, soit aux divergences génétiques de chaque lignée cellulaire, soit au type de mutation de la protéine p53 (Bohnke et al., 2004; DiBiase et al., 1999; Matsui et al., 2001; Scott et al., 2003). Certains travaux montrent que quelque soit le statut de p53, la protéine p21 peut aussi jouer un rôle sur la radiosensibilté des tumeurs. Certains glioblastomes connus pour être particulièrement résistants à l’irradiation, présentent une surexpression de la protéine p21. Or, l’utilisation d’ARN antisens déplétant la cellule de p21, radiosensibilise ces glioblastomes indépendamment du statut de p53 (Kokunai et al., 2001).
La modulation des mécanismes de l’arrêt en G1/S est donc intéressante pour la mise en place de protocoles cliniques visant à radiosensibiliser les tumeurs résistantes aux RI.
4-3-2-2 Mécanismes moléculaires du point de contrôle en S.
Lorsque les cellules sont irradiées en phase S du cycle cellulaire, la réplication est bloquée et la cellule tente de réparer les dommages induits par les RI au niveau de l’ADN. Les cellules AT
(Ataxie Telangectasie) par exemple, ne sont plus capables de s’arrêter en phase S du cycle cellulaire, on parle alors de RDS (Radioresistant DNA synthesis).
Après exposition aux RI, la protéine ATM est activée et phosphoryle alors différents substrats comme par exemple Chk2. La cible de Chk2 est la protéine Cdc25A dont la phosphorylation entraîne la dégradation protéolytique par le protéasome. La dégradation de Cdc25A prive alors la cellule d’un activateur essentiel de Cdk2, Cdc45, et bloque ainsi la réplication (Iliakis et al., 2003). Une autre voie connue pour réguler l’arrêt en phase S est la voie ATM/NBS1/BRCA1/SMC1 où le complexe NBS1/BRCA1/SMC est phosphorylé par ATM et affecte alors l’activité de CDK2 (Kitagawa et al., 2004) (Fig.7). Plusieurs pathologies présentent un défaut d’arrêt en phase S qui est associé à une hypersensibilité des cellules aux RI comme par exemple l’Ataxie Telangectasie (Beamish et al., 1996) et l’Anemie de Fanconi (Pichierri and Rosselli, 2004).
4-3-2-3Mécanismes moléculaires du point de contrôle en G2.
Le contrôle de cette phase du cycle cellulaire a été largement décrit et notamment en réponse aux agents endommageant l’ADN. En effet, l’arrêt en G2 des cellules sert à réparer les lésions de l’ADN qui persistent après le contrôle en G1/S, et prévient donc l’entrée en mitose des cellules où l’ADN est lésé.
-Mécanisme général.
La transition G2/M est sous la dépendance d’un complexe protéique appelé MPF (Maturation Promoting Factor). Le complexe MPF est formé de deux protéines, la cycline B1 de 62kDa et la sérine tyrosine kinase Cdk1 de 34kDa (p34cdc2) codée par le gène cdc2. L’activation du complexe MPF permet la phosphorylation des molécules cibles nécessaires à la division cellulaire, telles que les histones, les lamines nucléaires et l’ARN polymérase II.
\
Figure 7: Signalisation du point de contrôle de la phase S en réponse aux
rayonnements ionisants.
La modulation de l’activation de ce complexe régule donc le passage du G2 à la mitose. L’activation du complexe Cdk1/cyclineB1 est sous la dépendance de :
- la liaison de la cycline B1 à Cdk1aprés phosphorylation de Cdk1 sur ses résidus Tyr 15 et Thr14 par Wee1 et Myt1 respectivement pour que la liaison se fasse (Liu et al., 1997; McGowan and Russell, 1993; Porter and Donoghue, 2003).
- la phosphorylation sur le résidu thréonine 161 puis la déphosphorylation de Cdk1 sur les résidus Thr14 et Tyr15 par la phosphatase Cdc25C (Smits et al., 2000b) (Draetta and Eckstein, 1997). Cette dernière déphosphorylation de Cdk1 permet alors à la cellule d’entrer en mitose. La phosphatase Cdc25C pour être active doit elle même être phosphorylée juste avant la mitose par l’action du complexe Cdk1/CyclineB1 (Hutchins and Clarke, 2004).
-Arrêt en G2 induit par les RI (Fig.8).
De nombreux travaux montrent que les RI entraînent un arrêt en G2 favorisant ainsi les mécanismes de réparation de l’ADN et permettant donc une entrée en mitose sans lésion radio-induite. Après irradiation, plusieurs mécanismes moléculaires ont été mis en évidence dans le contrôle de l’arrêt en G2. En effet, les RI régule l’activité du complexe Cdk1/CyclineB1 en modifiant sa localisation subcellulaire (Bulavin et al., 2002). La cycline B (avec Cdk1) est initialement localisée dans le cytoplasme pendant la phase S et G2 du cycle cellulaire, puis transloque au noyau au début de la mitose (Pines and Hunter, 1991). Après induction de dommages à l’ADN par les RI, la cycline B reste séquestrée dans le cytoplasme et ce probablement à l’aide de la protéine 14-3-3 (Kao et al., 1999) (Chan et al., 1999). D’autre part, la protéine ATM, dont le gène est muté dans les cellules AT (ataxie telangectasie), est largement impliquée dans le contrôle de l’arrêt en G2 en réponse aux RI.(Xu et al., 2002) (Pandita et al., 2000). L’exposition de cellules AT aux rayonnements g cause des retards dans le cycle cellulaire et notamment dans la phase G2 (Ford et al., 1984) (Beamish et al., 1996).
Figure 8: Signalisation du point de contrôle de la phase G2 en réponse aux
rayonnements ionisants.
Des travaux montrent que cette protéine est activée pendant les phases G2/M (Pandita et al., 2000) et qu’il existe deux arrêts en G2/M distincts dont le plus immédiat, après irradiation, est dépendant d’ATM (Xu et al., 2002). Comme nous l’avons vu précédemment, ATM est une protéine kinase capable de phosphoryler un certain nombre de substrats en réponse aux RI. En général, ATM phosphoryle Chk2 et induit ainsi la cascade permettant l’arrêt en G2 (Melchionna et al., 2000). Cependant des travaux récents montrent qu’elle est aussi capable de phosphoryler Chk1 sur la serine 317 suggérant une implication de Chk1 dans l’arrêt en G2 via ATM après irradiation dans des cellules AT (Gatei et al., 2003). L’histone H2AX et la protéine de liaison à p53, 53BP1( p53 binding protein 1), sont des cibles de phosphorylation d’ATM qui semblent être impliquées dans l’arrêt en G2 radio-induit (Rappold et al., 2001; Rogakou et al., 1998). Les travaux de Fernandez-Capetillo et al., effectués sur des cellules provenant de souris transgéniques dont les gènes ATM ou H2AX ou 53BP1 ont été inactivés, montrent que ATM est requis pour la phosphorylation de H2AX. De plus les cellules déficientes pour H2AX ou 53BP1 présentent un défaut d’arrêt en G2M comme les cellules ATM-/- suggérant l’implication de ces deux protéines dans le contrôle du G2 en réponse aux RI via ATM (Fernandez-Capetillo et al., 2002). Le gène BRCA1 qui code une protéine largement impliquée dans les cancers (notamment les cancer du sein et de l’ovaire) est aussi impliquée dans le contrôle du G2 (Welcsh et al., 2000). En effet des cellules murines déficientes pour ce gène présentent une forte instabilité génétique, une diminution de la réparation par recombinaison homologue et un défaut d’arrêt en G2/M (Xu et al., 1999) (Moynahan et al., 1999). Li et al. montrent que la protéine CtIP, qui interagit avec BRCA1 est hyperphosphorylée en réponse aux RI entraînant sa dissociation de BRCA1, donc la répression de GADD45. Cette phosphorylation est effectuée par ATM suggérant un lien fonctionnel entre ATM et BRCA1 en réponse aux RI (Li et al., 2000). Xu et al., montrent aussi l’implication de BRCA1 dans le contrôle du G2/M en réponse aux RI (Xu et al., 2001a). Ils
36
confirment le lien entre ATM et BRCA1 en mettant en évidence un site de phosphorylation (Ser 1423) de BRCA1 par ATM impliqué dans le contrôle du point de contrôle G2/M post-irradiation. L’ensemble de ces travaux montre une large implication d’ATM dans l’arrêt en G2 induit par les rayons g.
La protéine p53 est décrite dans le contrôle de l’arrêt en phase G2 suggérant donc son rôle dans cet arrêt du cycle en réponse aux RI (Taylor and Stark, 2001). Le rôle de p53 dans le contrôle de l’arrêt en G2/M semble assez redondant ; p53 entraîne une répression de l’activité kinase de Cdk1/Cycline B soit en réprimant la transcription de Cdk1 et de la Cycline B, soit en ciblant des effecteurs qui iront interagir avec le complexe Cdk1/CyclineB afin de l’inhiber. D’autres travaux montrent le rôle de PLK1 et PLK3 qui sont des protéines régulatrices de CDC25C. Elles appartiennent à la famille des Polo Kinases qui jouent un rôle crucial dans différents événements de la mitose (Glover et al., 1998). PLK1 est un régulateur positif de l’activité de Cdc25C dans les cellules non irradiées et permet l’entrée en mitose en phosphorylant Cdc25C. Certaines hypothèses suggèrent que ATM ou ATR phosphorylerait PLK1, privant ainsi la cellule de l’activité de Cdc25C consolidant alors l’arrêt en G2M (Smits et al., 2000a) (van Vugt et al., 2001) en réponse aux RI.
Une autre inhibition de la progression en mitose en présence de lésions sur l’ADN est accompli par l’inhibiteur des CDK : p21 via PCNA (Ando et al., 2001). Il apparaît que la liaison p21/Cdc25 à PCNA/Cdk1-CyclineB exclut l’interaction Cdc25C avec Cdk1, donc la déphosphorylation de Cdk1 nécessaire à l’entrée en mitose. L’implication de p21 dans l’arrêt en G2 implique aussi p53 dans le maintien de l’arrêt et ce par l’activation de trois des protéines cibles de p53 :GADD45, p21, 14-3-3 (Hermeking et al., 1997) (Taylor and Stark, 2001). De plus p21 semble bloquer la phosphorylation activatrice sur le résidu Thr161 de Cdk1/CyclineB1 par CAK (Smits et al., 2000b).
Certaines drogues comme la caféine, la pentoxiftlline ou la staurosporine sont capables d’abolir l’arrêt en G2 et de radiosensibiliser les lignées tumorales . la caféine, molécule appartenant à la famille des méthyl-xanthines, a la propriété de réduire le retard en G2 après irradiation de nombreuses lignées cellulaires, en inhibant entre autre les protéines de la superfamille des PI3K (ATM, ATR) `(Jha et al., 2002) (Zhou et al., 2000). La pentoxifylline appartient aussi à la famille des méthyls-xanthines et a été décrite comme agent radiosensibilisant certaines lignées tumorales (Strunz et al., 2002). UCN-O1, un dérivé de la la staurosporine, est capable de supprimer l’arrêt en G2 et de radiosensibiliser certaines lignées cellulaires de tumeurs colorectales (Playle et al., 2002). De plus cette molécule radiosensibilise des cellules de carcinome du colon humaines probablement en inhibant la kinase Chk2 et donc l’arrêt en G2 radio-induit. (Yu et al., 2002).
4-3-2-4 Mécanismes moléculaires du point de contrôle en M.
L’apparition de cellules géantes multinuclées en réponse aux RI est due à une succession de mitoses abortives dont la cause peut être un défaut du point de contrôle en mitotique.
-Mécanisme général.
L’entrée en mitose est provoquée par l’activation du MPF. Il existe deux points de contrôle importants pour l’entrée mitose. : le premier correspond à la transition Prophase/Prométaphase qui définit la transition G2/M et qui est régulée par les mêmes mécanismes que la fin du G2. Le second correspond à la transition Métaphase/Anaphase qui permet le contrôle de l’alignement des chromosomes avant de poursuivre la division de la cellule. La transition Métaphase/Anaphase (M/A) n’intervient qu’après l’alignement du dernier chromosome : c’est seulement à ce stade que la cohésion entre les chromatides sœurs est rompue (Fig.9).
Cdk1 CyclineB APC (ε3) D-Box ε2 Ub Ub Ub Ub Ub Ub Ub Ub Ub Ub Ub MPF Fizzy APC (E3) Fizzy CyclineB securine CyclineB securine ε2 Ubiquitinylation Protéasome 26S Dégradation ε1 ATP ε1 AMP D-Box
Figure 9: Mécanismes moléculaires contrôlant la transition Métaphase/Anaphase
ou point de contrôle mitotique.
La transition M/A coïncide avec la dégradation de la cycline B et l’inactivation de Cdk1. La dégradation de la cycline B nécessite en premier lieu son ubiquitinilation effectuée par l’E3 ubiquitine ligase, APC (anaphase-promoting complex), associée avec son activateur Cdc20/fizzy. APC/Cdc20 fizzy reconnaît et interagit avec la cycline B par son domaine D-box (destruction-box). Elle est ainsi ubiquitinilée et dégradée par la sous unité 26S du protéasome. Cette transition M/A nécessite aussi la dégradation de la sécurine, protéine qui en se liant aux séparases empêche la séparation des chromatides sœurs. A la transition M/A, la sécurine est dégradée et libère la séparase chargée du clivage de la sous-unité Scc1 des complexes cohésines, événement essentiel à la perte de cohésion des chromatides sœurs et à leur ségrégation (Castro et al., 2003). Cette étape est sous la surveillance du point de contrôle mitotique. Expérimentalement ce point de contrôle est révélé par l’arrêt en prométaphase des cellules (par exemple sous l’action du nocodazole). Le mécanisme moléculaire fait intervenir les kinases Bub (Bub1p, Bub2p, Bub3p), Mad (Mad1p, Mad2p, Mad3p), Mps1 (Farr and Cohen-Fix, 1999), la kinase BubR1 qui semblent avoir un rôle dans l’inhibition de APC/Cdc20 fizzy ; la kinésine CENP-E (protéine centromérique). Schématiquement le kinétochore libre émet un signal inhibiteur en se liant à Mad2p via Mad1p. Sous forme de tétramère Mad2p intéragit avec le complexe APC/Cdc20 fizzy qui perd alors son activité ubiquitine ligase. Les cibles d’ APC/Cdc20 fizzy (Cycline B1, sécurine, séparase) ne sont plus dégradées et la transition M/A est alors bloquée (pour revue (Castro et al., 2003; Farr and Cohen-Fix, 1999) (Bharadwaj and Yu, 2004)).
-Point de contrôle mitotique et RI.
Le mécanisme du point de contrôle mitotique en réponse aux RI est encore mal connu chez l’homme. Par contre, chez la levure, de nombreuses équipes se sont intéressées à ces mécanismes. Schématiquement la présence de dommages sur l’ADN de la levure induit l’activation de la kinase Mec1 qui active à son tour Chk1 et Rad53 (Chk2 chez l’homme) (Sanchez et al., 1999).
40
Pds1 (la securine) est directement phosphorylée par Chk1 en présence de lésions sur l’ADN entraînant ainsi l’arrêt en mitose des cellules de levure (Agarwal et al., 2003) (Wang et al., 2001). Il semblerait que le type d’agent génotoxique conditionne l’arrêt en mitose des cellules humaines. En effet des cellules HeLa endommagées par un laser (photoproduits) ou des inhibiteurs de topoisomérase, présentent un arrêt en métaphase caractéristique du point de contrôle mitotique (Mikhailov et al., 2002). Les travaux de Skoufias et al. ne mettent pas en évidence un point de contrôle mitotique en réponse à une irradiation de 12 Gy dans différents types cellulaires humains (Skoufias et al., 2004).
4-4 Contrôle du cycle centrosomal.
L’instabilité génétique de nombreux cancers est due en partie à des défauts de structure et/ou de duplication des centrosomes (Sato et al., 2001). Les RI entraînent aussi des anomalies centrosomales conduisant à la formation de cellules aneuploïdes (Sato et al., 2000a; Sato et al., 2000b).
4-4-1 Structure du centrosome.
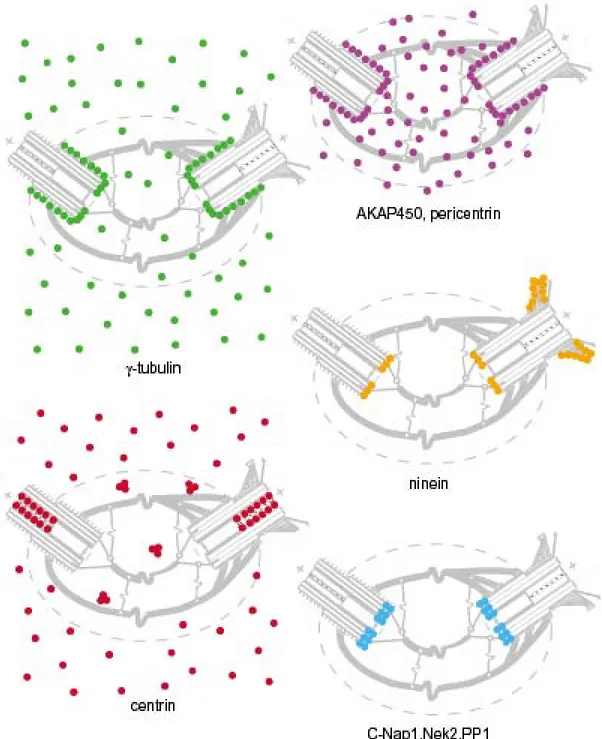
Le centrosome est un organelle formé de deux centrioles et du matériel péricentriolaire. En microscopie électronique, les centrioles ont une forme et une taille relativement constantes. Ils apparaissent comme des structures cylindriques dont les parois sont constituées de neufs triplets de microtubules eux même constitués de tubuline γ. Le matériel péricentriolaire apparaît comme un nuage dense aux électrons. Il est constitué d’un matériel fibrillaire diffus dans lequel peuvent apparaître des agrégats plus denses appelés satellites ou annexes (Paintrand et al., 1992) (Bornens, 2002). Les centrioles sont reliés par un matériel fibrillaire dense ou connexion filamenteuse appelée « matrix »(Bornens, 2002).
Figure 10: Structure schématique du centrosome (Tassin, 1999)
:
•
localisation de AKAP450 et de la péricentrine
•
localisation
tubuline
γ
•
localisation de la ninéine
•
localisation de la centrine
42
Le centrosome sert à la nucléation des microtubules du cytosquelette de la cellule et est composé (Fig10):
-Des complexes protéiques organisés autour de la tubuline γ, les γTuRCs (γ-tubulin-ring-complexes).
-De la pericentrine (Kendrin chez l’homme) qui semble impliquée dans la régulation des γTuRCs (Doxsey et al., 1994).
- De AKAP450, localisée au niveau de la « Matrix ». Elle semble réguler la stabilisation des microtubules naissant (Witczak et al., 1999).
- De la nineine participant, à la capture et à la stabilisation des microtubules naissants (Mogensen, 1999).
- Du complexe C-Nap1/Neck2 (serine/thréonine kinase) localisé au niveau de la « Matrix » à l’extrémité proximale de chaque centriole et participant au maintien de la distance entre les centrioles (Fry et al., 1998).
- De nombreuses kinases localisées au niveau du centrosome participant à la régulation de sa structure : les Polo Kinases (Plks) (Dai et al., 2002), ZYG-1 kinase (O'Connell, 2002), Aurora A kinases (Dutertre et al., 2002)…
- Des protéines Ran, qui sont des petites protéines G, localisées au niveau du centrosome et participant à la nucléation des microtubules (Di Fiore et al., 2004).
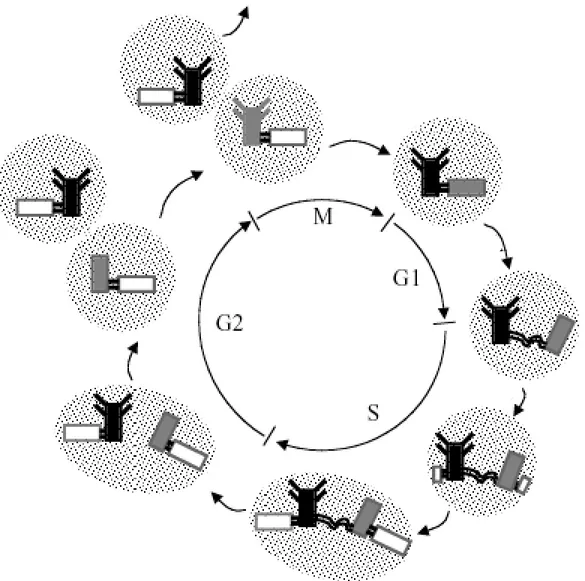
4-4-2 Cycle de duplication du centrosome.
Schématiquement le cycle centrosomal se déroule en trois étapes (Fig.11).
• Les deux centrioles perdent leur orientation orthogonale et se séparent légèrement tout en restant reliés par une connexion flexible ou « matrix » (Bornens, 2002).
44
• Un nouveau centriole se forme perpendiculairement à chaque centriole parental et s’allonge afin d’atteindre la même taille que le parental.
• Enfin les centrioles parentaux se déconnectent complètement l’un de l’autre et le matériel péricentriolaire se divise en deux structures indépendantes identiques à la structure parentale.
Les cellules entrent en phase G1 du cycle cellulaire avec un centrosome formé de deux centrioles, un centrosome parental et un centrosome fils (généré au cycle précédent) (Fig.11) (Delattre and Gonczy, 2004)). Au moment de la transition G1/S les centrioles se dissocient et deux nouveaux centrioles commencent à se former perpendiculairement au centriole parental. Pendant la phase S, la cellule possède donc deux centrosomes : un centrosome parental, marqué par la présence de l’appendice distal et d’une protéine la cenexine, ainsi que de la tubuline ε au niveau péricentriolaire et un centrosome fils qui n’a pas encore acquis ces caractéristiques. Pendant la phase G2 du cycle cellulaire, le centrosome fils acquiert la tubuline ε du matériel péricentriolaire , l’appendice distal et la cenexine des centrioles. Au début de la mitose, le matériel pericentriolaire augmente. Les deux centrosomes migrent au pôle de la cellule, formant ainsi les deux pôles du fuseau mitotique. La fin de la mitose entraine la formation de deux cellules filles ayant chacune un centrosome.
Comme pour la réplication de l’ADN, la séparation initiale des centrioles (G1/S), dans les cellules de mammifères, nécessite l’activité de la protéine Cdk2 (Matsumoto et al., 1999) et de la cycline E ou A (Meraldi et al., 1999) (Moroy and Geisen, 2004). Deux substrats de Cdk2 sont importants dans la duplication des centrosomes dans les cellules humaines, Mps1 (Fisk et al., 2003) et la nucléophosmine ou NPM (Tokuyama et al., 2001). Il a été montré que la surexpression de Mps1 localisée au niveau des centrosomes, favorise la duplication des
centrosomes alors qu’une inhibition de Mps1 induit une inhibition du cycle centrosomal (Fisk et al., 2003). La NPM est localisée au niveau des centrosomes avant leur duplication et est phosphorylée sur son résidu Thr199, par le complexe Cdk2/CyclineE ce qui permet son détachement du centrosome (Okuda et al., 2000; Tokuyama et al., 2001). Il semblerait que la NPM permette le maintien de la cohésion entre les centrioles et qu’elle soit libérée lorsque les centrioles se séparent (Okuda, 2002). Par ailleurs la duplication du centriole fils est contrôlé par la protéine ZIG-1 (O'Connell, 2002). Deux protéines ont été identifiées comme régulant le mécanisme de séparation des centrosomes dupliqués, les protéines Nek2 et C-Nap1 (Fry, 2002). La phosphorylation de nombreuses protéines joue donc un rôle important dans le maintien et la fonction des centrosomes pendant le cycle cellulaire (D'Assoro et al., 2002b).
4-4-3 Anomalies des centrosomes.
Différents types d’anomalies centrosomales ont été observées dans de nombreux cancers (Salisbury et al., 1999). Les anomalies structurales peuvent être soit une augmentation de la taille du centrosome caractérisée par un marquage plus important de la tubuline γ ou de la pericentrine, soit des modifications morphologiques comme un allongement du centrosome (qui présente normalement une forme arrondie en microscopie à fluorescence), soit une fragmentation de la structure du centrosome(Duensing et al., 2003; Pihan et al., 2001). Les cellules tumorales présentent aussi une amplification du nombre de centrosomes par cellule, ce qui conduit à la formation d’un fuseau mitotique multipolaire et donc à des mitoses aberrantes et à une répartition inégale du matériel génétique (Tarapore and Fukasawa, 2002) (Hollander and Fornace, 2002). Ces défauts peuvent affecter la polarité cellulaire en interphase car l’architecture cytoplasmique et la direction du trafic vésiculaire peuvent être désorganisées. Ils peuvent également augmenter l’incidence de mitoses multipolaires conduisant à des anomalies de ségrégation des chromosomes
46
et donc à l’apparition de cellules aneuploïdes. De nombreux travaux montrent un parallèle entre l’amplification centrosomale et l’instabilité génétique, comme dans les cancers du sein, du pancréas, du colon, (D'Assoro et al., 2002a) (Sato et al., 2001) (Ghadimi et al., 2000).
Les rayonnements ionisants peuvent entraîner une amplification des centrosomes, l’éclatement des centrioles et du matériel péricentriolaire. Sato et al. ont montré que 20 % des cellules U2-OS d’ostéosarcome irradiées à 10 Gy ont plus de deux centrosomes par cellule et que 60% des cellules en mitose présentent des fuseaux multipolaires après irradiation (Sato et al., 2000a). Ce phénotype peut être réversé par la surexpression de p21 dans les cellules d’adénocarcinome pancréatique MIA PaCa2 suggérant donc le rôle de cette protéine dans la surduplication des centrosomes en réponse aux RI (Sato et al., 2000b). De récents travaux tendent à démontrer qu’il existe une voie entre la lésion radio-induite (CDB par exemple) et l’apparition de défauts au niveau de la structure du centrosome et de son cycle de duplication. En effet des travaux visant à comprendre l’embryogénèse de la drosophile (drosophila melanogaster), montrent que seuls les noyaux présentant des lésions sur présentent des défauts sur leurs centrosomes au sein d’un même syncitium (par exemple disparition des centrosomes) et entraînent des défauts de ségrégations des chromosomes (Takada et al., 2003). Les auteurs suggèrent que ces défauts de centrosomes empêchent la division de ces noyaux endommagés et donc la propagation d’une instabilité génétique. Ces mécanismes se font par l’intermédiaire de la protéine Chk2, effecteur connu de la protéine ATM en réponse aux RI. Des travaux effectués sur des cellules de poulet DT40 suggèrent le même processus dans les cellules eucaryotes (Dodson et al., 2004). En effet l’extinction conditionnelle de Rad51, protéine impliquée dans la réparation des CDB, entraîne une amplification des centrosomes associée à un arrêt en G2 en réponse aux RI. Ils suggèrent que les cellules ayant échappé aux points de contrôle de la réparation de l’ADN et de l’assemblage du
fuseau mitotique présentent des défauts de centrosomes ce qui conduit à la mort des cellules par mort mitotique afin d’éviter la propagation des mutations potentielles induites par les RI.
Les mécanismes moléculaires induits par les RI contrôlant l’apparition des anomalies centrosomales sont encore mal connus. Comme nous l’avons vu précédemment il semble que les protéines p21, Chk2, Rad51 soient impliquées dans ces mécanismes. Plus récemment Fletcher et al. ont montré que les RI étaient capables d’inhiber l’activité de Nek2, protéine impliquée dans la séparation des centrosomes. En effet lorsque Nek2 est inhibé par la technique d’interférence d’ARN les cellules irradiées ne sont plus capables de ségréger leurs centrosomes au cours de la division et la croissance cellulaire est inhibée (Fletcher et al., 2004). L’ensemble de ces travaux montre donc un effet des RI sur la structure, la duplication, la séparation des centrosomes au cours du cycle cellulaire.
4-5 Mort cellulaire.
Lorsque la cellule, au cours des différents points de contrôle, n’est pas capable de corriger les erreurs de réparation de l’ADN ou de duplication des centrosomes induits par les RI, elle poursuit son cycle cellulaire vers une mort, programmée ou non programmée (apoptose, catastrophe mitotique et nécrose). Les RI entraînent plusieurs types de mort (cohen-Jonathan et al., 1999; Dikomey et al., 2003):
- l’apoptose voie universelle de mort des cellules hématopoïétique après irradiation. - la mort post-mitotique ou catastrophe mitotique qui semble être la voie majoritaire de réponse aux RI.
- la nécrose. - la sénéscence.
48
4-5-1 Apoptose.
L’apoptose est un mécanisme physiologique de « suicide cellulaire » programmé qui est essentiel pour le développement embryonnaire, le fonctionnement du système immunitaire, le maintien de l’homéostasie tissulaire. La dérégulation de l’apoptose a été impliquée dans différents processus pathologiques : défauts de neurodégénérescence, maladies autoimmunes, cancer (Dewey et al., 1995) (Okada and Mak, 2004).
Elle se caractérise par la condensation de la chromatine nucléaire, par la déstructuration des membranes nucléaires et cytoplasmiques et enfin par la fragmentation des structures nucléaires aboutissant à la formation des corps apoptotiques (cohen-Jonathan et al., 1999).
Après irradiation, le délai d’apparition de l’apoptose varie d’un type cellulaire à l’autre. En effet l’apoptose peut survenir juste après irradiation (apoptose précoce ou interphasique), après l’arrêt en G2 (Merritt et al., 1997) (Radford et al., 1994), ou après plusieurs divisions cellulaires (apoptose tardive) (Dewey et al., 1995) (Yanagihara et al., 1995) (Castedo et al., 2004a).
Au niveau moléculaire l’apoptose se caractérise par une cascade d’évènements contrôlée par la régulation de l’expression de gènes spécifiques de l’apoptose. Les protéines clé de cette cascade appartiennent à la famille des caspases. Il existe deux voies par lesquelles les caspases sont activées. Une activation extrinsèque par l’activation de récepteurs de mort à la surface des cellules. La liaison de ligands tel que FASL (ligand du recepteur de mort FAS) appartenant à la famille des TNF (tumor necrosis factor), sur son récepteur induit la formation d’un complexe protéique de mort (DISC : death induced signalling complexe). Ce complexe recrute alors la caspase 8 qui induit la cascade de clivage des procaspases. Une activation intrinsèque dirigée par différents stress extra ou intra cellulaires (hypoxie, stress genotoxiques, oncogènes…). Les signaux transduits à partir de ces stress convergent alors vers la mitochondrie. Une série d’ événements biochimiques sont induits et entraînent la perméabilisation de la membrane externe