Modèles d'analyse simultanée et conditionnelle
pour évaluer les associations entre les haplotypes
des gènes de susceptibilité et les traits des
maladies complexes :
Application aux gènes candidats de l'ostéoporose.
Thèse présentée
à la Faculté des études supérieures de l'Université Laval
dans le cadre du programme de doctorat Médecine Expérimentale
pour l'obtention du grade de Philosophise Doctor (Ph.D.)
FACULTE DE MEDECINE
UNIVERSITÉ LAVAL
QUÉBEC
2010
Les maladies complexes sont des maladies multifactorielles dans lesquelles
plu-sieurs gènes et facteurs environnementaux peuvent intervenir et inter agir. De
nom-breuses études ont identifié des locus (gènes ou régions chromosomiques), avec ou
sans effets marginaux, qui interagissent pour contribuer au risque de la maladie. Pour
les études d'association par polymorphismes, plusieurs méthodes ont été développées
récemment pour évaluer l'interaction gène-gène. Cependant, les études d'association
par haplotypes donnent parfois une meilleure puissance pour détecter l'association.
Mais, la majorité de ces dernières ne permet pas d'évaluer les interactions entre
les haplotypes de deux gènes et celles qui le permettent présentent des restrictions,
comme l'utilisation du phénotype de la maladie en dichotomique (présence ou
ab-sence de la maladie) ou encore n'ajustent pas pour les facteurs environnementaux.
Cette thèse traite cette problématique en deux volets : méthodologique et appliqué.
Au niveau méthodologique, cette thèse rapporte une nouvelle méthode statistique
pour effectuer l'analyse simultanée et l'analyse conditionnelle de deux régions
in-dépendantes (gènes ou régions chromosomiques) dans les études d'associations par
haplotypes des maladies complexes. Une étude de simulation a été effectuée pour
confirmer sa validité. En présence d'un effet d'interaction entre les haplotypes de
deux gènes avec ou sans effets marginaux, les résultats de l'étude de simulation ont
montré que notre modèle d'analyse conditionnelle a plus de puissance pour détecter
l'association et donne une estimation plus précise des effets comparativement aux
méthodes alternatives disponibles actuellement. Au niveau appliqué, l'approche de
la cartographie fine dans un premier échantillon de Québec avec une replication dans un échantillon indépendant de Toronto a été mise à profit pour raffiner l'étude de deux gènes candidats de l'ostéoporose : ESRRG (estrogen receptor-related gamma) et ESRRA (estrogen receptor-related alpha). Pour ESRRG, cette approche combi-née aux deux méthodes d'analyse, par polymorphismes ou par haplotypes, confirma son implication dans l'étiologie de la maladie chez les femmes d'origine européenne, tandis que pour ESRRA, elle a constitué une investigation approfondie révélant une association dans un premier échantillon de femmes préménopausées de Québec, mais sans replication dans un deuxième échantillon indépendant de femmes préméno-pausées de Toronto. Puisque les deux gènes étudiés appartiennent au même sentier métabolique, l'effet conditionnel de ESRRA sachant ESRRG a été évalué par notre méthode. Cette analyse a révélé une association dans un premier échantillon, mais, encore une fois, sans replication dans le deuxième échantillon. Ces résultats sug-gèrent que le premier gène est un gène de susceptibilité de l'ostéoporose. Toutefois, notre étude n'était pas concluante en ce qui concerne l'effet du deuxième gène ainsi que son effet conditionnel sachant l'effet du premier. Ainsi, une replication dans un échantillon indépendant, de même taille ou plus grande que celle de l'échantillon de Québec, s'avère nécessaire pour confirmer ou infirmer les résultats observés chez les femmes provenant de la région métropolitaine de Québec.
Le sujet de ma thèse vise à combler le besoin en méthodologie pour analyser s i m u l t a n é m e n t les effets d e s h a p l o t y p e s d e d e u x gènes sur le t r a i t d e la m a l a d i e e n continu et pour évaluer aussi l'effet conditionnel d u d e u x i è m e gène sachant l'effet d u p r e m i e r . Alors, j'aimerai remercie mon directeur de thèse, Dr François Rousseau, et mes co-directeurs, Dr Alexandre Bureau et Dr Nathalie Laflamme, de m'avoir fait confiance et d'accepter ma supervision pour ce sujet pas-sionnant.
La thèse présentée ici contient deux articles. Le premier de ces articles, présenté au chapitre 3, est rédigé sous la direction de François Rousseau, Alexandre Bureau et Nathalie Laflamme. Cet article est soumis à la revue SAGMB et des commen-taires ont été reçus. Le deuxième, présenté au chapitre 5, est rédigé en collaboration avec François Rousseau, Alexandre Bureau, Nathalie Laflamme, Dr David Cole, co-chercheur sur le projet d'ostéoporose, et Dr Sylvie Giroux, collègue au sien de l'équipe de recherche du laboratoire de François Rousseau. Celui-ci est actuellement publié en ligne dans la revue JBMR. Ces articles sont le fruit de mon travail tant pour la forme que pour le contenu et j'en suis le premier auteur.
Je tiens à remercier mon directeur de thèse et mon ex-patron, François Rousseau, tout d'abord pour ses arrangements concernant les conciliations travail, études et famille, puis pour son ouverture d'esprit, sa compréhension et sa supervision. Mes
scientifique, sa grande disponibilité, ses encouragements et ses conseils précieux aux moments difficiles et Nathalie Laflamme de m'avoir initié au projet de l'ostéoporose, de me donner l'envie et la motivation de continuer à travailler en statistique génétique et aussi pour ses conseils et son support moral tout au long de cette thèse. J'aimerai aussi remercier les deux co-auteurs du deuxième article de cette thèse, David Cole et Sylvie Giroux, pour leurs discussions scientifiques, leurs commentaires ainsi que leurs encouragements. Je remercie également les examinateurs de ma thèse, Dr Yves Giguère, Dr Aurélie Labbe et Dr Mhamed Mesfioui, pour avoir accepté d'évaluer mon travail. Je tiens à vous assurer de l'expression de mon profond respect. Un remerciement spécial va à ma patronne et directrice, Dr Josée Bourdages, dans mon nouvel emploi au sein de la Direction de la surveillance de l'état de santé, Direction générale de la santé publique au ministère de la santé et services sociaux du Québec, de m'avoir accordé le temps et de m'avoir encouragé à compléter la rédaction de cette thèse. Je voudrais aussi remercier tous celles et ceux qui ont contribué de près ou de loin à ce que cette thèse prenne forme.
Toute ma gratitude et mes remerciements spéciaux vont aux membres de ma famille : Ma mère qui m'a soutenue et encouragée tout au long de mes études et non pas seulement pour mes travaux de doctorat, mon mari, mon fils et ma fille pour votre patience et amour et, finalement, ma nièce pour son amitié et son aide. Men-tion d'honneur à mon mari d'avoir absorbé les responsabilités familiales et maintenu l'équilibre dans les moments difficiles. J'aimerais aussi exprimer ma plus vive recon-naissance à mes soeurs et mon frère pour leurs chaleureux mots d'encouragements. Votre soutien m'a permis de mener ce travail de thèse dans de très bonnes conditions. Merci à vous tous !
Finalement, je remercie le Fonds de la recherche en santé du Québec (FRSQ) pour ma bourse de recherche.
Table des matières
R é s u m é ii
Avant-propos iv
Table des m a t i è r e s x
Liste d e s t a b l e a u x xiii
Liste des figures xv
Liste des a b b r e v i a t i o n s xvi
1 I n t r o d u c t i o n générale 1
1.1 Rappel génétique 1
1.2 Des maladies monogéniques aux maladies complexes 3
1.3 L'hypothèse maladie fréquente/variante fréquente .
1.4 Variations génétiques fréquentes du génome humain 9
1.5 Études d'association et variations génétiques prédisposant aux
mala-dies complexes 10
1.6 Études d'association par polymorphisme : association directe ou
indi-recte 11
1.7 Sélection des SNPs pour les études d'association indirecte 12
1.8.1 Facteurs motivant l'utilisation des haplotypes dans les études
d'association , 14
1.8.2 Détermination des haplotypes : haplotypage moléculaire vs
al-gorithmes de phasage 17
1.8.3 Principe des études d'association par haplotypes 19
1.9 Conclusion 24
Tableaux 27
2 Objectifs d u p r o j e t d e recherche 27
2.1 Résumé de la problématique 27
2.1.1 Problème 1 : sur le plan des méthodes statistiques utilisées . . 27
2.1.2 Problème 2 : sur le plan de la puissance 29
2.2 Hypothèses de travail 29
2.3 Objectifs spécifiques 30
3 S i m u l t a n e o u s a n d Conditional analyzes t o e v a l u a t e associations b e t
-ween complex t r a i t s a n d h a p l o t y p e s in unlinked regions 31
3.1 Introduction 35
3.2 Statistical Method 37
3.3 Simulation study 43
3.4 Discussion 48
References 60
Annexe A : Details on the variance-covariance matrix estimation 67
4 C o m p l é m e n t d e Particle m é t h o d o l o g i q u e 77
4.1 Introduction 77
4.2 Méthode 79
4.2.1 Analyse simultanée 79
4.2.2 Analyse conditionnelle en présence d'une interaction . . . 80
4.3 Résultats 81
4.3.2 Analyse conditionnelle en présence d'une interaction négative . 83
4.3.3 Analyse conditionnelle en présence d'une interaction positive . 86
4.3.4 Synthèse des résultats sur les puissances statistiques 88
4.4 Discussion 91
5 Association w i t h replication b e t w e e n E S R R G v a r i a n t s a n d b o n e
p h é n o t y p e s in w o m e n w i t h E u r o p e a n ancestry. 93
5.1 Introduction 96
5.2 Material & Methods 97
5.3 Results 102
5.4 Discussion 105
References 110
6 E v a l u a t i o n d e l'interaction e n t r e les d e u x gènes E S R R G et E S R R A d u sentier des estrogènes d a n s u n e é t u d e d'association avec les p h é
-n o t y p e s d e l'ostéoporose 123
6.1 Introduction 123
6.2 Matériel et méthodes 125
6.2.1 Échantillons étudiés 125
6.2.2 Sélection des polymorphismes 126
6.2.3 Génotypage 126
6.2.4 Analyse statistique 127
6.3 Résultats 128
6.3.1 Statistiques descriptives des deux échantillons 128
6.3.2 Cartographie fine : association entre les 7 tagSNPs et les
me-sures osseuses dans l'échantillon de Québec 131
6.3.3 Replication dans l'échantillon de Toronto : étude d'association entre les 4 htSNPs et les mesures osseuses chez les femmes
préménopausées de Toronto . 136
6.3.4 Analyse multivariée et évaluation de l'interaction entre les
Annexe B 151
7 Conclusion générale et perspectives
155
7.1 Partie I : Méthode d'analyse développée dans le cadre de cette thèse . 155
7.2 Partie II : Étude d'association entre les mesures osseuses et les
va-riantes de ESRRG et ESRRA chez les femmes d'origine européenne . 158
Liste des tableaux
1.1 Caractéristiques de quelques distributions de GLM 25
1.2 Méthodes de régression pour évaluer l'effet des haplotypes sur les
phé-notypes 26
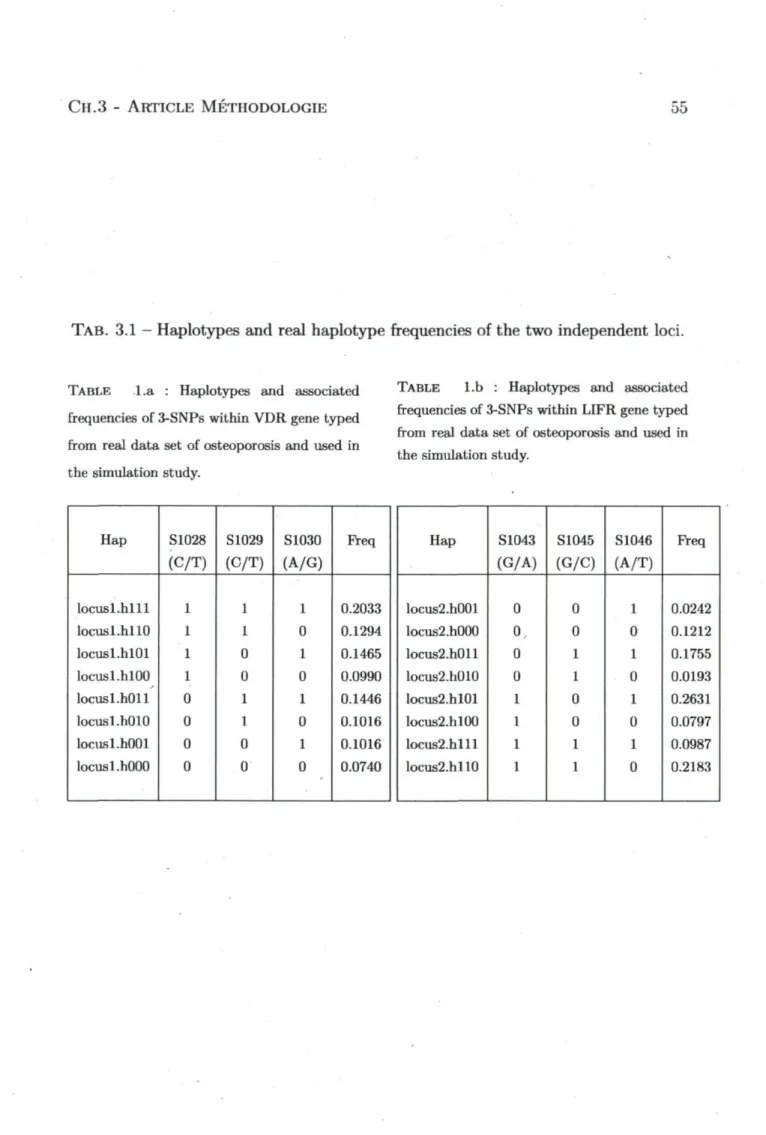
3.1 Haplotypes and real haplotype frequencies of the two independent loci. 55
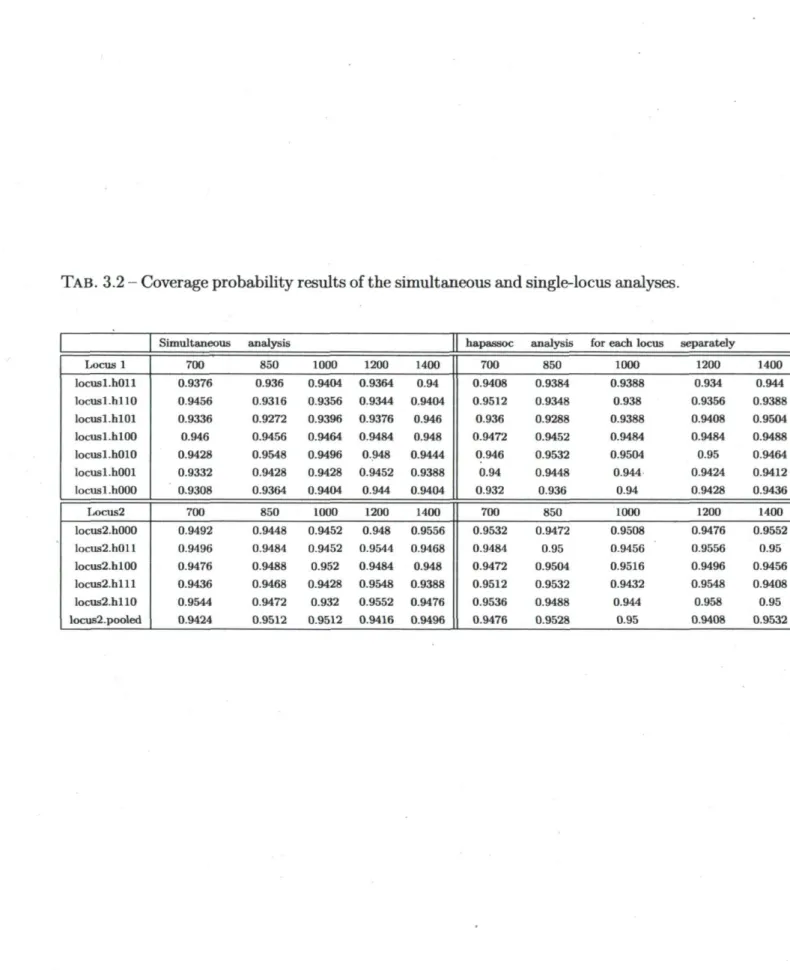
3.2 Coverage probability results of the simultaneous and single-locus
ana-lyses 56
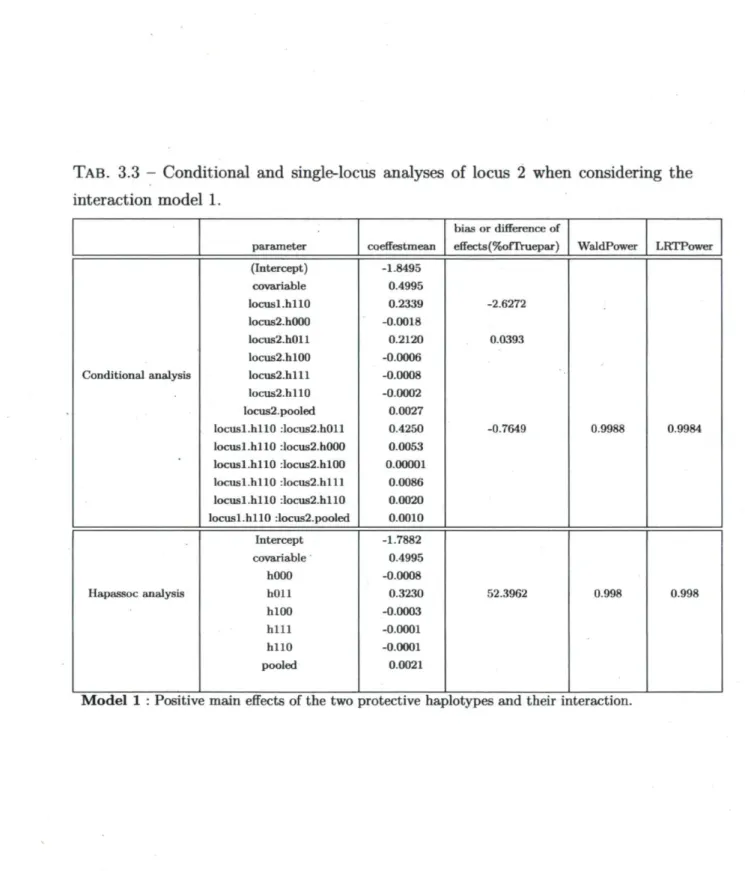
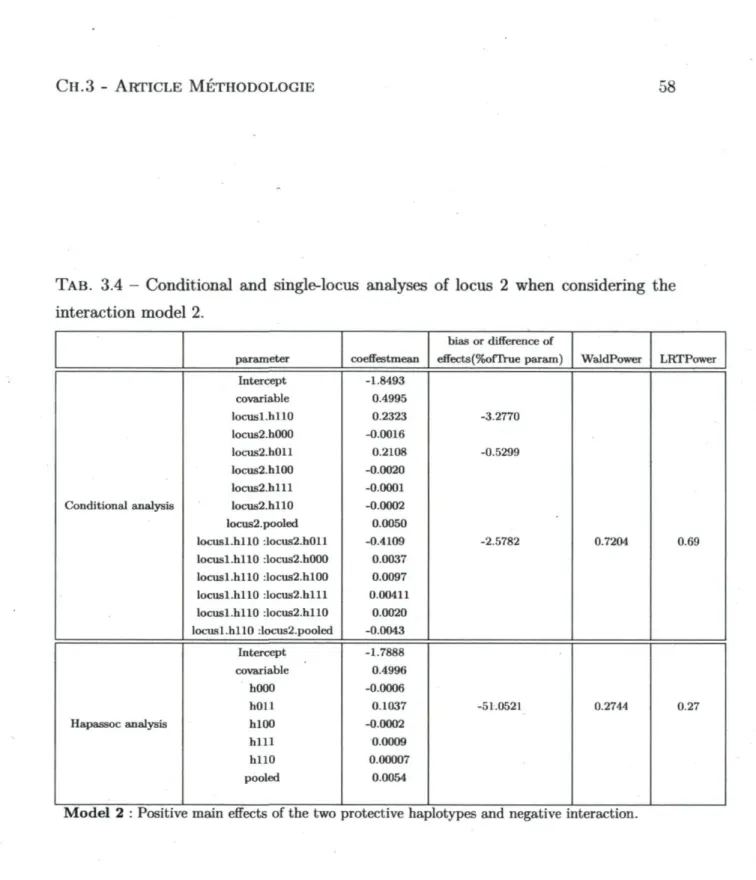
3.3 Conditional and single-locus analyses of locus 2 when considering the
interaction model 1 57
3.4 Conditional and single-locus analyses of locus 2 when considering the
interaction model 2 58
3.5 Conditional and single-locus analyses of locus 2 when considering the
interaction model 3 59
4.1 Résultats de l'analyse simultanée sans interaction au locus 1 82
4.2 Résultats de l'analyse simultanée au locus 2 84
4.3 Résultats de l'analyse conditionnelle, d'un SNP à la fois au locus 2 sachant l'effet des haplotypes au locus 1, en présence d'une interaction
négative 85
4.4 Résultats de l'analyse conditionnelle en présence d'une interaction
po-sitive 87
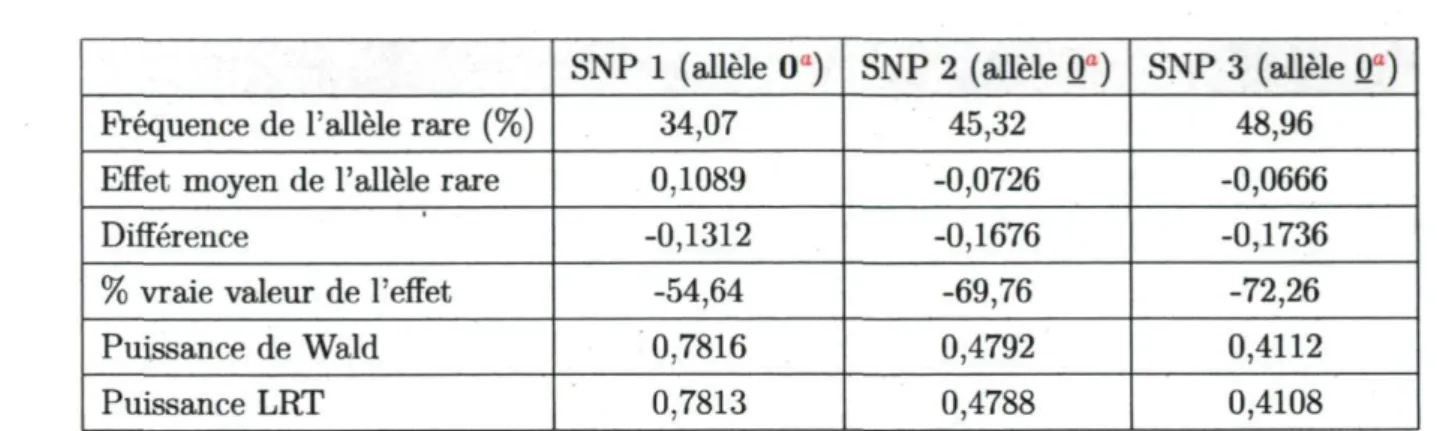
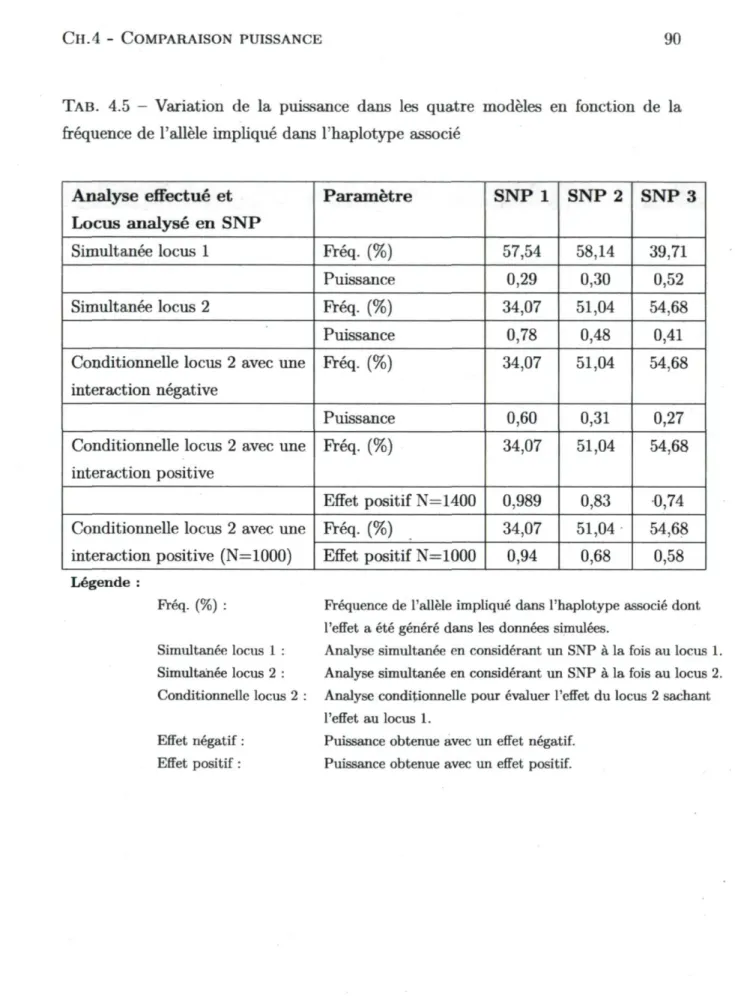
4.5 Variation de la puissance dans les quatre modèles en fonction de la
5.1 Samples characteristics
117
5.2 Minor allele frequency and estimates of the Haplotype frequencies of
the 4 tagSNPs genotyped in the three samples : premenopausal women
from Quebec, premenopausal women from Toronto, and
Perimenopau-sal and postmenopauPerimenopau-sal women from Quebec. These SNPs capture the
haplotype diversity of the block "haplotype tagging SNPs' (htSNPs).
118
5.3 Results of the association between the SNP rs945453 and bone
mea-surements in the whole Quebec sample
119
5.4 Marker-by-marker analysis in the two premenopausal samples with
the four htSNPs and the original associated SNP (rs945453)
120
5.5 Associations between rs2818964r (combined genotypes) and bone
mea-surements in premenopausal women from Quebec, premenopausal
wo-men from Toronto, combined data of the two prewo-menopausal samples
(Quebec-I-Toronto), and Perimenopausal and postmenopausal women
from Quebec
121
5.6 Haplotype analysis of the htSNPs (rsll572842, rs2818964, rsll572818,
rsl2087167) in premenopausal women from Quebec, premenopausal
women from Toronto, combined data of the two premenopausal samples
and peri-postmenopausal women
122
6.1 Caractéristiques des 7 tagSNPs génotypes dans l'échantillon des
pré-ménopausées de Québec
133
6.2 Résumé de l'étude d'association (p-values) entre les 7 tagSNPs du
gèene ESRRA et les cinq mesures osseuses dans l'échantillon de Québec
134
6.3 Résultats détaillés des associations significatives entre les
polymor-phismes de ESRRA et les mesures osseuses dans l'échantillon de
Qué-bec. Toutes les analyses sont ajustés pour l'âge, le poids, l'âge à la
6.4 Résultats détaillés des associations significatives entre les 4 htSNPs polymorphismes de ESRRA et les mesures osseuses dans l'échantillon de Québec. Toutes les analyses sont ajustés pour l'âge, le poids, l'âge
à la ménarche, l'activité physique et le statut de tabagisme 135
6.5 Caractéristiques des 4 htSNPs du gène ESRRA génotypes dans
l'échan-tillon des préménopausées de Toronto 137
6.6 Les fréquences des alleles rares et des haplotyopes majeurs des 4 htSNPs (rsl317494, rs660442, rs671976, rs4930702) impliqués dans des associations significatives dans l'échantillon de Québec et génotypes
dans l'échantillon de Toronto '. 138
6.7 Résultats détaillés des analyses d'association entre les SNP impliqués dans des associations significatives dans l'échantillon de Québec et les cinq mesures osseuses chez les femmes préménopausées de Toronto . . 139
6.8 Résultats des analyses d'association entre les haplotypes des htSNPs
et les mesures osseuses dans l'échantillon de Toronto 140 6.9 Analyse simultanée des deux gènes ESRRA et ESRRG chez les femmes
préménopausées de Québec 141
6.10 Analyse simultanée des deux gènes ESRRA et ESRRG chez les
pré-ménopausées de Toronto 142
6.11 Résumé des p-value observés en évaluant les interactions entre rs2818964r du gène ESRRG et les SNPs de ESRRA (rsl317494, rs876064, rs660442, rs671976, rs2276014, rs4930702 et rs499425) dans l'échantillon de
Qué-bec 143
6.12 Résultats de l'analyse conditionnelle par haplotypes chez les femmes
préménopausées de Québec 144
6.13 Résumé des p-values observés en évaluant les interactions entre rs2818964r et les SNP de ESRRA (rsl317494, rs660442 et rs671976) dans
l'échan-tillon de Toronto 145
6.14 Résultats de l'analyse conditionnelle par haplotypes chez les femmes
1.1 Description de la structure du génome, schéma tiré du site web
wiki-pedia (http ://fr.wikipedia.org/wiki/Chromosome) 2
1.2 Les trois étapes de la production de la protéine : la transcription de l'ADN en ARN messager (ARNm), la traduction de l'ARNm en une chaîne d'acides aminés qui détermineront finalement la forme de la
protéine après le repliement 4
1.3 Comparaison des caractéristiques des maladies héréditaires classiques
mendéliennes et des maladies génétiques complexes 6
1.4 L'aspect multifactoriel des maladies complexes
1.5 SNPs, haplotypes et htSNPs 16
3.1 Comparison of power, bias, and dispersion parameter between the hapassoc analysis of locus 1 and locus 2 and the simultaneous analysis
separately according to the sample size 51
3.2 Boxplots of parameter estimate errors (estimate-true value) from 2500 replicates of 700 observations with complex disease trait (gaussian distribution) and two common protective-haplotypes (frequencies : 0.12943,0.17554) within the two loci. True values are the real va-lues used to generate the simulation data and are equal to (0.2401803,
3.3 Boxplots of parameter estimate errors (estimate-true value) from 2500
replicates of 1400 observations with complex disease trait (gaussian
distribution) and two common protective-haplotypes (frequencies :
0.12943,0.17554) within the two loci. True values are the real
va-lues used to generate the simulation data and are equal to (0.2401803,
0.2119258) for the two protective-haplotypes and to 0 for the others. . 53
3.4 Comparison of type I errors, calculated from 2500 replicates with
com-plex disease trait (gaussian distribution), between simultaneous
ana-lysis and hapassoc anaana-lysis 54
5.1 LOCALIZED FINE M A P P I N G : Description of the block with
high linkage disequilibrium around the SNP associated rs945453.
Struc-ture of the gene region is shown on top with exon 6 and exon 7.
Tag-SNPs identified with pairwise correlation are encircled 115
5.2 Region covering the 20 kb block in 3' end of ESRRG according to
Uni-versity of California Santa Cruz (UCSC) Genome Browser. At the top
of the figure, every module identified is located. At the bottom of the
figure, homology between species is indicated along with a histogram
of conservation. Module 010170 and module 010175 are encircled. . . 116
6.1 Variation de la densité minérale osseuse chez les femmes selon l'âge
(image extraite de Wasnich, R.D., et al., 1989, p. 179-213) 130
6.2 Description du bloc de déséquilibre de liaison identifié par la
cartogra-phie fine autour du variant * ESRRA Ins/del " en utilisant les données
de HapMap. Nous nous sommes basés sur la méthode de Gabriel pour
définir le bloc et la corrélation allélique r
2entre chacune des paires de
SNPs est donnée dans leur carré d'intersection
132
6.3 Détails sur la tendance d'interaction entre rs2818964r et rs671976 dans
A : adenine
ADN : acide désoxyribonucléique ANCOVA : l'analyse de covariance ANOVA : l'analyse de la variance ARNm : Acide ribonucléique messager
BUA : Mesure de l'atténuation des ultrasons (broadband ultrasound attenuation) C : cytosine
CCL3L1 : le gène codant la chimiokine (Chemokine (C-C motif) ligand 3-like 1) CNV : variations génétiques appelées régions variables (copy number variants) COL : mesure de densité minérale osseuse au niveau du col fémoral (FN=femoral neck)
CP : la probabilité de couverture (coverage probability)
DMO : densité minérale osseuse (BMD=bone mineral density)
DXA : absorptiométrie biénergétique à rayons X (dual energy x-ray absorptiometry) |D'| : la valeur absolue de la mesure du déséquilibre de liaison de Lewontin D'. EM : l'algorithme d'estimation et maximisation (Expectation-Maximisation) ESRl : Gène qui code pour le récepteur des estrogènes a
ESRRA : Les récepteurs reliés aux Estrogènes alpha (Estrogen related receptor al-pha)
ESRRG : Les récepteurs reliés aux Estrogènes gamma (Estrogen related receptor gamma)
G : guanine
GWAS : étude d'association à l'étendue du génome ou pangénomique (genome-wide association study)
HRT : thérapie d'hormone de remplacement (hormone replacememt therapy) htSNPs : les SNPs sélectionnés pour capturer la diversité haplotypique au sein d'un bloc de déséquilibre de liaison (haplotype-tagging SNPs)
HWE : l'équilibre de Hardy-Weinberg (Hardy-Weinberg equilibium) LD : déséquilibre de liaison (Linkage Disequilibrium)
L2L4 : mesure de densité minérale osseuse au niveau des vertèbres lombaires L2-L4 (LS=lumbar spine)
MAF : fréquence de l'allèle rare (minor allele frequency)
MC/VC : Maladie fréquente/Variante fréquente (common disease/common variant) MCMC : l'algorithme de Monte Carlo par chaînes de Markov (Monte Carlo Markov Chain)
QUS : Ultrasons quantitatifs au talon (quantitative ultrasound) r : coefficient de corrélation de Pearson
R2 : coefficient de détermination
SI : l'indice de stiffness (stifness index) caculé en fonction de BUA et SOS SNPs : les polymorphismes nucléotidiques simples (singulier : SNP)
SOS : Vélocité de transmission des ultrasons (speed of sound) T : thymine
tagSNPs : SNPs sélectionnés en fonction de la mesure du LD pour représenter les autres polymorphismes de la région génomique étudiée.
PCR : Amplification en Chaîne par Polymerase (Polymerase Chain Reaction) VIH ou sida : Le virus d'immunodéficience humaine (VIH) est le virus qui cause le syndrome d'immunodéficience acquise (sida).
Introduction générale
1.1 Rappel génétique
Le corps humain, comme tout autre organisme complexe, est composé de plusieurs milliards de cellules de différents états et ayant diverses fonctions. Chaque cellule, que ce soit une cellule cardiaque, une cellule musculaire ou un neurone et à l'exception des globules rouges et des gamètes, renferme une copie diploïde du génome dans son noyau. Le génome humain est l'ensemble de l'information génétique portée par l'ADN (acide désoxyribonucléique), molécule formée de l'assemblage linéaire de quatre bases de nucleotides : adenine (A), thymine (T), cytosine (C) et guanine (G) (figure 1.1). Il est réparti en 23 paires de cliromosomes, transmises par les parents aux enfants, soit 22 paires homologues appelées autosomes, numérotées de 1 à 22 par ordre de taille décroissante, et une paire de chromosomes sexuels appelés gonosomes : XX chez la femme et XY chez l'homme. Chaque chromosome est une suite linéaire de gènes séparés par des régions intergéniques. Le génome humain est composé de plus de trois milliards de bases d'ADN codant entre 25 000 et 30 000 gènes (1; 2; 3).
chromosome
noyaucellule
nucleosomes
paires de bases histories
ADN
double brins
F I G . 1.1 — Description de la structure du génome, schéma tiré du site web wiltipedia (http ://fr.wikipedia.org/wiki/Chromosome)
Un gène représente une séquence d'ADN qui est nécessaire à la synthèse et à la fonction d'une protéine (figure 1.2). L'environnement, le mode de vie, les gènes sont les principaux déterminants du phénotype d'un individu. Celui-ci correspond à l'ensemble des caractéristiques de l'individu, que ce soit son apparence physique (poids, taille, couleur des yeux, ...) ou sa physiologie (groupe sanguin, ...). La contri-bution d'un gène au phénotype d'intérêt est déterminée par la qualité et la quantité
produites d'ARX ou de protéine. Toutefois, la relation quantitative et qualitative entre les gènes et les phénotypes n'est pas simple, comme on l'a longtemps cru. Un simple changement dans la séquence d'ADN d'un gène peut entraîner un change-ment dans son fonctionnechange-ment, une diminution ou une augchange-mentation de la quantité de la protéine ou bien la production d'une protéine dysfonctionnelle et, par consé-quent, occasionner des pathologies génétiques graves. Selon le nombre de gènes de susceptibilité à la maladie, on distingue deux classes de maladies génétiques : les maladies monogénétiques (maladies mendéliennes et anomalies chromosomiques) et les maladies complexes (maladies polygéniques ou multifactorielles) (2).
1.2 Des maladies monogéniques aux maladies
com-plexes
La majorité des maladies humaines ont une composante génétique. Cependant, l'importance de cette composante varie en passant des maladies monogéniques (ou maladies mendéliennes) aux maladies complexes (ou polygéniques) (4) (figure 1.3). Les maladies mendéliennes sont des maladies héréditaires qui suivent les lois de trans-mission de caractères de Mendel (maladies monogéniques). Elles sont caractérisées par des phénotypes bien définis, un ou deux1 locus génétiques avec une forte ou une
complète penetrance, un taux faible de phénocopies2 et des fréquences des alleles
de susceptibilité généralement faibles. Cette relation claire génotype'-phénotype a comme conséquence un modèle de transmission claire d'une génération à l'autre. Les
1 Locus signifie un segment d'ADN. Il est utilisé tout au long de cette thèse pour désigner un gène ou une région génomique d'un chromosome.
2Les phénocopies sont des phénotypes semblables causés par des mutations de différents gènes ou dus à des facteurs environnementaux.
3Le génotype d'un individu est l'information génétique portée par ses gènes, qu'elle soit exprimée ou non sous forme de protéines.
Du gène à la protéine
Brin d'ADN /^ 3 Gène 1 = recette£>5' GATCTGCTGTCCGCAGTATCGGGCTTG 3"
5CGL
ARNm TranscriptionCUAGACGACAGGCS3UCAUAGCCCGAAC
▼ Traduction Acides , e U a SP a SP a r9 a r9 h l S S e r Pr 0 3 S n Aminés(F*
Protéinejf*
Repliement de la protéineFlG. 1.2 Les trois étapes de la production de la protéine : la transcription de l'ADN en ARN messager (ARNm), la traduction de l'ARNm en une chaîne d'acides aminés qui détermineront finalement la forme de la protéine après le repliement
approches classiques d'identification des gènes basées sur l'analyse de liaison ' et de clonage positionnel ont permis des développements spectaculaires dans l'identifica tion des gènes impliqués dans les maladies mendéliennes. Ce succès se résume dans les nombreux désordres mendéliens identifiés (5). A titre d'exemple, on peut citer 4Analyse statistique des recombinaisons chromosomiques au sein des familles (deux ou trois gé nérations) d'individus atteints d'une maladie d'intérêt. Elle consiste à parcourir le génome afin de localiser le gène de la maladie en identifiant la section de chromosome limitée par deux recombi naisons observées dans la famille et partagée par les individus atteints seulement.
la dystrophie musculaire de Duchenne (6), le rétinoblastome (7), la fibrose kystique (8), le syndrome de l'X-Fragile (9) et de la dystrophie myotonique de Steinert (10),
l'anémie de Fanconi (11), l'ataxie télangiectasie (12) et l'hémochromatose (13). Mal-gré ces réalisations remarquables, le problème urgent des maladies complexes qui pose un fardeau beaucoup plus grand du point de vue de la santé publique a attiré l'attention des généticiens.
4Les maladies complexes sont des maladies multifactorielles dans lesquelles plu-sieurs gènes et facteurs environnementaux' peuvent intervenir et interagir pour cau-ser la maladie (figure 1.4). Beaucoup plus fréquentes que les maladies génétiques mendéliennes, on retrouve parmi elles entre autres les maladies cardiovasculaires, le cancer, l'asthme, le diabète de type I/II, l'obésité et l'ostéoporose. Compte tenu de l'importante fréquence de ces maladies, on assume qu'il existe une prévalence éle-vée pour l'ensemble des variations génétiques prédisposant à une maladie donnée. Chaque variation contribuera modestement au risque d'être atteint de la maladie. Néanmoins, peu de choses sont connues sur la nature de ces variations génétiques. En outre, la signification de " effet modeste " manque souvent de clarté : est-ce l'impact dans la population qui est faible ou l'effet de la variante sur le phénotype qui est mo-deste? Ainsi, plusieurs possibilités devraient être considérées (14) : (a) une variante fréquente avec un effet modeste, (b) une variante rare avec un grand effet, (c) plu-sieurs variantes rares, chacune avec un effet modeste, (d) une interaction en cis entre deux ou plusieurs variantes fréquentes sur le même gène (ou le même chromosome) ou (e) des combinaisons de toutes ces possibilités. Malheureusement, l'analyse de liai-son a montré sa limite dans l'identification des gènes de susceptibilité des maladies complexes résultant en des effets modestes de plusieurs variantes. Par conséquent, un intérêt particulier a été apporté aux études d'association pour l'identification des
5 Certains auteurs parlent de facteurs environnementaux (ou de covariables environnementales, ou encore tout simplement d'environnement) pour indiquer tous les facteurs de risque non génétiques incluant l'environnement, le mode de vie et les caractéristiques personnelles. Pour simplifier les notations, nous allons adopter cette définition pour le reste de cette thèse.
" P e t i t s " é c h a n t i l l o n » (<IOOO)
ioo°/«
ft
-5 O 1 2 •> O r a m i s é c h a n t i l l o n s I HMMIl N o m b r e d e g è n e s i m p l i q u é sFIG. 1.3 - Comparaison des caractéristiques des maladies héréditaires classiques
mendéliennes et des maladies génétiques complexes
L'abscisse représente le nombre estimé de gènes impliqués dans la maladie, tandis que l'ordonnée indique la proportion de la variance phénotypique expliquée par chacun de ces gènes. Le nombre habituel d'individus à étudier pour identifier ce ou ces gènes est aussi indiqué, montrant que les études visant à identifier les gènes impliqués dans des maladies génétiques complexes nécessitent l'analyse de plus de sujets, étant donné l'effet plus modeste de chaque gène impliqué. Ce schéma est adapté de l'article de Rousseau et Laflamme, MEDECINE/SCIENCES, 2003 (4).
gènes impliqués dans l'étiologie de ces maladies
(15)
pour deux principales raisons :
la taille de l'échantillon qui peut être considérablement réduite comparativement à
celle qui est exigée par l'analyse de liaison ; une puissance suffisante pour détecter
les effets génétiques modestes
(16)
Gènes
Caractéristiques
personnelles
Mode de vie
Environnement
FlG. 1.4 — L'aspect multifactoriel des maladies complexes
Les différents facteurs entrant en ligne de compte pour causer les maladies complexes sont regroupés en quatre principales composantes : les caractéristiques personnelles (poids, âge), le mode de vie (activité physique, statut de tabagisme), l'environnement (air, climat, eau) et les gènes. Toutes ces composantes peuvent interagir les unes avec les autres pour donner la couleur de complexité à ces maladies.
1.3 L'hypothèse maladie fréquente/variante
fré-quente
Selon une hypothèse qui associe une maladie fréquente à une variante génétique fréquente (CD/CV=common disease/common variant), le risque de contracter une maladie fréquente serait associé à des variations génétiques qui sont relativement fré-quentes dans la population. Grâce à son importance du point de vue de l'intervention de la santé publique, cette hypothèse, comparativement à l'autre possibilité qui as-socie une maladie fréquente à plusieurs variantes rares où chacune contribue avec un effet modeste, avait beaucoup de succès dans les dernières années. D'abord, elle était l'idée de base du projet international HapMap (17), un catalogue des variations génétiques les plus fréquentes chez l'humain et décrivant la nature des variantes, leur emplacement dans la séquence d'ADN et leur distribution dans quatre popu-lations de différentes ethnies. Ensuite, combiné aux technologies récentes des puces de génotypage qui permettent de génotyper simultanément des centaines de milliers de SNPs, HapMap a permis d'entreprendre des études d'association à l'étendue du génome (GWAS=genome-wide association studies), menant à la prolifération des découvertes récentes de variantes génétiques fréquentes associées aux maladies com-plexes telles que les maladies cardiovasculaires (18; 19; 20; 21; 22), le cancer du sein
(23; 24; 25), le cancer du poumon (26; 27), le diabète de type II (28; 29; 30; 31; 32; 33),
l'obésité (34) et le cancer de la prostate (35; 36; 37; 38). Malgré les énormes inves-tissements dans les études d'association de criblage du génome, il n'y a pas beau-coup d'évidence à l'appui de la généralité de l'hypothèse de CD/CV. Toutefois, il existe également des exemples de variantes rares influençant les maladies complexes
(39; 40). En conclusion, les variantes rares et fréquentes peuvent contribuer au risque
des maladies complexes, bien que nous ne sachions pas en réalité laquelle des deux possibilités est la plus importante (41).
1.4 Variations génétiques fréquentes du génome
humain
Comme source de variation de l'ADN, les polymorphismes nucléotidiques simples'1
(SNPs= single nucleotide polymorphisms) sont les marqueurs génétiques les plus utilisés par rapport à d'autres, tels que les microsatellites, en raison de leur abondance élevée, de leur taux relativement bas de mutation et de leur adaptabilité facile au génotypage automatique et à haut débit. Des bases de données contenant des millions de SNPs largement distribués à travers le génome humain sont compilées et rendues disponibles via le Web (17; 42; 43; 44; 45; 46). Dans le génome humain, il existe plus de cinq millions de SNPs ayant une fréquence de l'allèle rare (MAF= minor allele frequency) supérieure à 10 % et plus de dix millions ayant une MAF>1 % (47; 48). L'hypothèse maladie fréquente/variante fréquente et l'existence des millions de SNPs fréquents à travers le génome humain motivent leur utilisation dans les études d'association avec les maladies complexes.
Dans le génome humain, il existe aussi d'autres variations génétiques appelées régions variables (CNV= copy number variants) qui peuvent avoir une MAF su-périeure à 1 %. Elles sont alors comprises dans les variantes génétiques fréquentes de la population. Ces régions, qui comportent des séquences de longueurs variables entre deux paire de base à des centaines de milliers de paires de bases d'ADN, sont dupliquées un certain nombre de fois (49; 50). La duplication varie d'un individu à l'autre et la région peut être même absente chez certains individus. En étudiant le génome de 270 personnes aux ancêtres européens, asiatiques et africains, Sebat et al. (50) ont comptabilisé exactement 1447 régions, représentant approximativement 11 % du génome humain. D'un individu à l'autre, seulement une petite proportion de ces 11 % est différente. Un gène au complet, incluant le promoteur, peut être
6Des variations dans l'ADN au niveau desquelles un des quatre nucleotides est substitué à un autre.
compris dans une région variable, ce qui pourra alors avoir un effet de dose sur le
phénotype (49). La duplication du gène CCL3L1 chez les individus fortement
expo-sés au VIH/sida est un exemple concret montrant comment la variabilité du dosage
du gène peut changer la susceptibilité d'être attient de l'infection (51). Le nombre de
copies du CCL3L1 est inversement corrélé à la susceptibilité du VIH. Nous pouvons
donc conclure que les SNPs et les CNV peuvent contribuer à la susceptibilité des
ma-ladies complexes, mais la technologie utilisée pour détecter et analyser les CNV n'a
pas encore attient un niveau d'accessibilité et de polyvalence semblable à celui des
SNPs, ce qui pourra avoir un impact sur leur utilisation dans les études d'association
dans un avenir immédiat (52).
1.5 Etudes d'association et variations génétiques
prédisposant aux maladies complexes
L'utihsation des études d'association dans l'identification des gènes ou des régions
de susceptibilité des maladies complexes se base sur leur capacité à identifier des
va-riantes génétiques fréquentes de risque modeste, et par conséquent sur la présomption
que des variantes génétiques fréquentes sont les principaux déterminants des
mala-dies complexes (CD/CV). Bien que la replication des variantes fréquentes associées
aux maladies complexes soit très courante dans les études récentes, la fréquence de
la variante est inversement corrélée avec la taille de l'effet. En général, des effets
esti-més modestes sont observés avec des variantes fréquentes. Une étude de simulation a
conclu que de telles observations résultent principalement de la puissance des études
d'association menées jusqu'à présent (53). Ces études n'ont donc pas assez de
puis-sance pour détecter les variantes rares. Cependant, avec la collaboration nationale
et internationale, des réseaux sur des sujets de recherche spécifiques se constituent,
ce qui pourra avoir un impact sur l'augmentation des tailles des échantillons utilisés
et, par conséquent, sur l'augmentation de la puissance de l'étude pour détecter les
variantes rares associées aux maladies complexes.
1.6 Etudes d'association par polymorphisme :
as-sociation directe ou indirecte
Le principe des études d'association entre un phénotype d'intérêt et un polymor-phisme est d'identifier une association entre la présence de la maladie et un allele ou un génotype du polymorphisme étudié. Pour ce faire, on a utilisé historiquement des méthodes biostatistiques traditionnelles. Pour un trait dichotomique dans une étude (cas/témoins) et sans ajustement pour les facteurs confondants, l'analyse des tableaux de fréquences avec un test de Chi-deux ou le test de rapport de vraisem-blance sont utilisés pour comparer les fréquences de l'allèle (ou le génotype) entre les cas et les témoins. Dans ce cas, une fréquence significativement plus élevée chez les cas suggérera alors qu'un génotype ou un allele particulier est associé à une suscepti-bilité génétique accrue à développer la maladie. Par ailleurs, des modèles d'analyses multivariées à l'aide des modèles de l'analyse de la variance (ANOVA) ou des modèles d'analyse de covariance (ANCOVA) sont utilisés dans l'ajustement pour les facteurs environnementaux. Le polymorphisme associé peut être directement responsable de la susceptibilité de la maladie, c'est-à-dire en ' association directe ', ou simplement être en déséquilibre de liaison' (LD=Linkage Disequilibrium) avec un polymorphisme causal adjacent, c'est-à-dire en ' association indirecte " (54).
7Le déséquilibre de liaison (LD) se définit comme une corrélation entre deux alleles plus forte que ce que l'on s'attend à observer selon une distribution aléatoire. Il traduit le fait que les alleles de deux sites polymorphiques donnés situés sur un même chromosome et adjacents ont tendance à être transmis ensemble à la génération suivante, compte tenu de la faible probabilité qu'une recombinaison surgisse entre les deux sites. Ainsi, le LD diminue en fonction de la distance séparant deux polymorphismes.
1.7 Sélection des SNPs pour les études
d'associa-tion indirecte
Afin de réduire de manière significative les coûts de génotypage pour les études d'association génétique, le projet HapMap (17) a été lancé pour décrire la structure du déséquilibre de liaison à travers le génome humain. L'accessibilité de ces données, corpbinée au développement de plusieurs méthodes de sélection du sous-ensemble optimal de SNPs (55), capturant la variabilité génétique au sein de la région (ou de l'ensemble des régions) étudiée, a permis le choix des SNPs à utiliser dans les études d'association. L'avantage de cette approche d" association indirecte * réside dans le fait qu'elle réduit la redondance en exploitant le LD entre les polymorphismes tout en détectant les associations avec les variantes causales par l'intermédiaire de leurs proxys.
Selon la statistique utilisée pour caractériser le LD entre les SNPs de la région d'intérêt, les méthodes de sélection se divisent en trois catégories principales. La première catégorie se base sur le coefficient de corrélation r2 entre les paires de
SNPs situés sur le même gène (ou région génomique) (56; 57). Cette catégorie de méthodes consiste à constituer les sous-groupes de SNPs corrélés à un seuil fixé, par exemple un r2 > 0.80. Pour chacun des sous-groupes, un SNP qui a une corrélation
d'au moins 0,80 avec tous les autres SNPs du sous-groupe en question sera choisi pour représenter les autres. Le SNP ainsi choisi est appelé un tagSNP ; on dit alors que le tagSNP capture à 80 % la variabilité allélique de tous les SNPs du sous-groupe. Par conséquent, l'ensemble de tous les tagSNPs sélectionnés formeront le sous-ensemble optimal à tester dans l'étude d'association portant sur le gène (ou la région génomique) d'intérêt. Cependant, cette première catégorie de méthodes ne tire pas profit de la structure de corrélation entre les tagSNP pour augmenter le nombre de SNP représentés à 100 %.
Pour combler ce besoin, d'autres méthodes, constituant la deuxième catégorie,
ont été développées pour imputer les SNPs non génotypes
(58; 59; 60; 61; 62; 63).
Ces méthodes utilisent une variété de mesures multi-SNPs, notées r
2,, où les
haplo-types
0des tagSNPs sont utilisés pour augmenter la corrélation avec les SNPs non
génotypes et, par conséquent, augmenter la puissance de détecter leurs associations
avec le trait de la maladie.
La troisième catégorie se base sur les méthodes de définition des blocs de
déséqui-libre de liaison
9(62; 63)
pour déterminer tous les blocs de la région étudiée dans un
premier lieu et, dans un second, pour sélectionner les SNPs qui capturent la diversité
de tous les haplotypes au sein de chaque bloc, appelés htSNPs (haplotype-tagging
SNPs). La méthode de Gabriel et al.
(62)
est basée sur la valeur absolue du
coef-ficient de Lewontin |D'| et son intervalle de confiance pour évaluer le LD entre les
paires de SNPs et définir ainsi les blocs de LD. Contrairement aux autres méthodes
qui assument que tous les SNPs doivent être inclus dans des blocks, l'approche de
Gabriel et al.
(62)
est la plus proche de la nature réelle du génome dans le sens
qu'on s'attend à trouver des blocs dans certaines régions du génome, mais pas dans
d'autres et que la proportion du génome sans blocs peut être substantielle
(64; 65).
Finalement, rien ne nous empêche de combiner toutes ces catégories de méthodes
pour tirer plus de profits des corrélations entre les SNPs et de la structure du génome
en blocs de LD, plus spécifiquement pour l'approche des études d'association par
gènes candidats ou à l'étape de la cartographie fine.
8Un haplotype est une combinaison des alleles de deux ou de plusieurs marqueurs adjacents sur un même chromosome.
9Un bloc de déséquilibre de liaison est une région génomique dont les SNPs sont en LD fort ou complet. Donc, il y a peu ou pas d'évidences de recombinaison au sein du bloc.
a»
1.8 Etudes d'association par haplotypes
Plusieurs raisons sont à l'origine de l'utilisation des haplotypes des SNPs
étroite-ment liés dans les études d'association des maladies complexes. Citons entre autres
le rôle fonctionnel des haplotypes dans les études de gènes candidats, la diversité
limitée des haplotypes fréquents au sein des blocs de LD ainsi que son impact
po-sitif sur la réduction du nombre de degrés de liberté dans les modèles statistiques
d'analyse et, par conséquent, l'augmentation de la puissance statistique pour
détec-ter l'association, s'il y en a une, au sein du bloc étudié et, finalement, le fait que
les haplotypes incorporent le déséquilibre de liaison entre plusieurs SNPs adjacents.
Tous ces facteurs sont discutés en détail dans la section suivante.
1.8.1 Facteurs motivant l'utilisation des haplotypes dans les
études d'association
1.8.1.1 Fonction biologique d'un haplotype
Les haplotypes représentent exactement l'organisation des alleles au long du
chro-mosome et reflètent le modèle héréditaire à travers l'évolution. Clark (55) discute du
rôle des haplotypes dans les études de gènes candidats, soulignant que les propriétés
fonctionnelles d'une protéine sont souvent déterminées par sa structure
tridimension-nelle, déterminée elle-même par l'ordre linéaire et les propriétés chimiques des acides
aminés. Cet ordre linéaire est influencé par les variations d'ADN sur un haplotype.
Par ailleurs, il existe des preuves solides que plusieurs mutations sur le même gène
en position cis peuvent agir l'une sur l'autre pour créer un ' super allele ' qui a un
grand effet sur le phénotype observé
(66; 67; 68).
1.8.1.2 S t r u c t u r e d u g é n o m e h u m a i n en blocs d e déséquilibre d e liaison et diversité limitée d ' h a p l o t y p e s d a n s c h a q u e bloc
Les études d'analyse à haute résolution du déséquilibre de liaison (LD) ont mon-tré que le génome humain est découpé en blocs de LD essentiellement complets, de différentes tailles, entrecoupés par de petites régions à faible LD nommées " points
chauds de recombinaisons ' (69; 70). Dans ces blocs de LD fort, la diversité
d'ha-plotypes est limitée et les haplotypes de chaque bloc, appelés blocs d'haplotypes (haplotype blocks), pourraient être différenciés en sélectionnant un nombre relative-ment petit de htSNP (71; 72; 73; 74). En général, 3 à 5 haplotypes majeurs (freq>5 %) repésentent 90 % de tous les haplotypes, quel que soit le bloc ((69) et figure
1.5). Par ailleurs, Clark souligne également que les variations génomiques de la po-pulation sont structurées dans les haplotypes. À partir de ces faits, l'approche basée sur l'utilisation des blocs d'haplotypes dans les gènes candidats a été suggérée pour déterminer le bloc qui est associé à la maladie complexe (55; 75; 76). Cependant, l'identification du bloc associé devrait délimiter la zone requise pour rechercher les variantes causales responsables de la susceptibilité génétique à la maladie.
1.8.1.3 Avantages s t a t i s t i q u e s des é t u d e s d'association p a r h a p l o t y p e s
À cause de la structure héréditaire capturée par la distribution des haplotypes, l'analyse d'association par haplotypes de SNPs, fonctionnels ou non, peut donner une puissance statistique plus grande comparativement à l'analyse des SNPs individuels (77). Plusieurs études ont montré que les méthodes basées sur les haplotypes peuvent donner une puissance élevée et plus de précision sur la cartographie des gènes can-didats que celles qui sont basées sur un SNP à la fois (69; 77; 78; 79). Cependant, la littérature sur l'efficacité relative d'analyser les haplotypes versus les SNPs est compliquée par les différentes hypothèses au sujet du nombre de SNPs étudiés, du nombre de SNPs impliqués dans la susceptibilité de la maladie dans chaque gène (ou
». SNP* . O l O T O l W l A A C A C O C C A . . . Oomo«ome2 A A C A C O C C A . . . Cl-fO-wsomc 3 A A C A T O C C A . . . CtifOmotome* A A C A C O C C A . . . SNP T T C 0 8 0 0 T C AOTCSACCa.... TTCOAOOTC... AOTCA ACCQ.... TTCOOOOTC... AOTCA ACCO.... TTCO OOTC... AOTC ACCO....
b. haplotypn C. htSNPs
L_
Haplotype! C T C A A A O T A C C O T T C A O S C A Haplotype? T T O A T T AC « C A I C A C T A A T â haplotypeJ C C C O A T C T « T O A T A C T O O T • Hapbtype4 T t C A T T C C C C O O T f C A O AC AFIG. 1.5 - SNPs, haplotypes et htSNPs
a) SNPs : Cette partie du schéma montre une courte séquence d'ADN de la même région du chromosome pour des individus différents. La majeure partie de la séquence d'ADN est identique dans cette région, mais les trois bases de couleurs différentes montrent où une variation génétique se produit. Chaque SNP a deux alleles possibles; le premier SNP dans le panneau a les alleles C et T.
b) Haplotypes : Un haplotype est une combinaison linéaire des alleles des SNPs adjacents. La deuxième partie du schéma montre les génotypes observés des 20 SNPs présents à travers une séquence de 6000 bases d'ADN. Seules les bases variables sont montrées, y compris les trois SNPs qui sont montrés en a). Pour cette région, la majorité des chromosomes de la population étudiée ont les haplotypes 1-4.
c) htSNP : Le génotypage des trois htSNPs parmi les 20 SNPs est suffisant pour identifier ces quatre haplotypes uniquement. Par exemple, si un chromosome particulier a l'haplotype A-T-C pour ces trois htSNPs, ceci indique que l'haplotype réel pour ce chromosome est l'haplotype 1. Ce schéma est adapté de ' The international HapMap Project ', Nature, 2003 (76).
région génomique) étudié et du déséquilibre de liaison entre les alleles des SNPs à
tester (80; 81; 82; 83). Le fait que les auteurs de ces comparaisons se soient concentrés
sur deux extrêmes, en utilisant des modèles statistiques bien définis et des ajustement
appropriés pour une analyse des SNPs ou en utilisant un test global pour tous les
haplotypes, limite la portée de ces études (84). Toutefois, la puissance de l'analyse
par SNP dépend toujours du déséquilibre de liaison entre le SNP testé et le locus de
susceptibilité de la maladie. Par ailleurs, l'information sur le LD entre deux SNP
ad-jacents n'est pas incorporée dans les méthodes d'analyse par SNP, ce qui peut avoir
comme impact une diminution de la puissance statistique
(85).
Les méthodes
d'as-sociation basées sur les haplotypes sont donc généralement considérées comme étant
plus puissantes que les méthodes d'analyse par SNPs individuels
(61; 77; 83),
puisque
les premières exploitent entièrement l'information du LD entre multiples SNPs. Des
études de simulation et empiriques appuient également cette idée
(77;
86).
En plus,
les méthodes d'association par haplotypes ou multi-SNPs sont asymptotiquement
équivalentes et sont plus puissantes que les études d'association par SNP
(61).
1.8.2 Détermination des haplotypes : haplotypage
molécu-laire vs algorithmes de phasage
Pour déterminer les haplotypes des individus, plusieurs méthodes d'haplotypage
moléculaire ont été développées
(87; 88; 89; 90).
Cependant, ces méthodes ne sont
pas largement utilisées, non seulement parce qu'elles sont coûteuses, mais également
parce qu'elles ont un débit très faible et qu'elles ont des limites sur le plan pratique ;
le plus long fragment entier d'ADN purifié qu'on peut obtenir est approximativement
de 10-24 kb
(89; 90)
alors qu'un bloc de déséquilibre de liaison peut couvrir une
ré-gion étendue sur quelques centaines de kb
(91; 92; 93).
En raison de ces limites des
méthodes d'haplotypage direct, les haplotypes ne sont pas généralement observables
et seules les données génotypiques des SNPs sont disponibles en appliquant les
tech-niques de génotypage. En l'absence de données génétiques familiales, les haplotypes
d'un individu ne peuvent être déduits à partir des génotypes, sauf pour les individus
homozygotes pour tous les SNPs ou hétérozygotes pour un seul des SNPS à l'étude ; en général, pour un individu hétérozygote pour k SNPs, il y a 2k~1 combinaisons
d'haplotypes possibles et compatibles avec le génotype observé. Par conséquent, les algorithmes (94; 95; 96; 97; 98; 99) pour inférer les haplotypes des génotypes non pha-ses offrent des solutions pratiques, précipha-ses et rentables (100; 101; 102; 103; 104; 105).
Le but des algorithmes de phasage est d'estimer la fréquence des haplotypes dans l'échantillon de la population tout en attribuant les haplotypes pour chacun des in-dividus. La majorité des méthodes de phasage des haplotypes sont basées sur des approches statistiques. La fonction de vraisemblance combinée avec des hypothèses sur la génétique des populations, tel l'équilibre de Hardy-Weinberg, est à la base de ces méthodes.
Comme dans les autres domaines de la statistique, il existe deux approches pri-maires pour estimer les paramètres : l'approche bayésienne et l'approche fréquentiste. Plusieurs approches bayésiennes ont été proposées pour le phasage des haplotypes
(106; 107; 108; 109; 110; 111; 112; 113). Ces approches calculent la distribution a
posteriori des haplotypes non observés en conditionnant sur les génotypes observés et en incorporant l'information biologique a priori. L'algorithme de Monte Carlo par chaînes de Markov (MCMC ou Monte Carlo Markov Chain), généralement effectué par l'échantillonneur de Gibbs, est utilisé pour échantillonner la paire d'haplotypes pour chaque individu en se basant sur leur distribution a posteriori. Quant à l'ap-proche fréquentiste, elle est généralement basée sur l'algorithme EM (expectation-maximisation) (114). Cet algorithme est couramment utilisé pour le phasage des haplotypes, où les fréquences des haplotypes de la population sont estimées en maxi-misant la fonction de vraisemblance (115; 116; 117; 119). L'algorithme EM donne également la probabilité qu'un individu ait une paire d'haplotypes donnée (120). Ces probabilités peuvent être utilisées soit pour définir les covariables des haplotypes à inclure dans une analyse standard, telle que la régression logistique (60; 121; 122),
ou pour pondérer chaque paire d'haplotypes possible assignée à chaque individu
3.3.3. Étant donné les ressources de laboratoire beaucoup plus élevées requises pour
l'haplotypage direct, le génotypage
est,
en général, plus avantagé, mais ceci doit être
équilibré avec les dépenses liées à la détermination du phénotype et au recrutement
pour augmenter la taille de l'échantillon
(103; 104).
1.8.3 Principe des études d'association par haplotypes
Plusieurs méthodes d'analyse ont été proposées pour évaluer l'association entre
les haplotypes et les traits des maladies complexes. Selon le devis d'étude et les
don-nées utilisées,
ces
méthodes peuvent être classées en deux catégories : les méthodes
populationnelles et les méthodes familiales
(85).
Étant donné l'apparition tardive
caractérisant la majorité des maladies complexes et les difficultés, quand ce n'est pas
l'impossibilité, de recruter un nombre suffisant de membres d'une même famille
(pa-rents et enfants, fratrie) pour répondre aux besoins de l'étude, le devis populationnel
est le plus utilisé dans les études d'association
(102)
et, par conséquent, les méthodes
d'analyse basées sur les données populationnelles sont très demandées. Les méthodes
statistiques développées ont suivi le chemin biostatistique traditionnel, traitant
es-sentiellement les haplotypes en tant que covariables catégorielles. Schaid
(84)
indique
que les méthodes basées sur les modèles linéaires généralisés (GLM=generalized
li-near models) offrent l'avantage d'utiliser une grande variété de méthodes statistiques
déjà établies, avec des extensions nécessaires pour tenir compte de l'incertitude de
la phase des haplotypes. Dans sa revue de la littérature, Schaid
(84)
discute aussi de
plusieurs de ces approches de régression et de leurs propriétés.
1.8.3.1 Rappel quant aux modèles linéaires généralisés .
Dans le cas d'un modèle avec des interactions haplotypes/environnement, soit Y
la variable dépendante et X = (X
e,X
g(H),X
eg) la matrice des variables
indépen-dantes, où X
edénote la matrice des covariables environnementales incluant le vecteur
unité correspondant au paramètre à l'origine, X
g(H) est la matrice des covariables
gé-nétiques, déterminée par la codification numérique des haplotypes et X
egcorrespond
à la matrice des codifications numériques des interactions gène-environnement. Dans
le cadre des modèles linéaires généralisés (GLM), nous supposons que la distribution
de la variable dépendante Y est membre de la famille exponentielle
(125).
La
princi-pale caractéristique du GLM est que la fonction de régression, c'est-à-dire l'espérance
p = E(Y\X) de Y, est une fonction monotone de ô — X
Tf3, où 3 = (3
e, 3
g, 3
eg)est le
paramètre de régression de Y en fonction de X. Soit G la fonction qui relie p et 6 :
E(Y\X) = G(X
T3) <*p = G(0)
Cette fonction G s'appelle la fonction de lien. Notons que McCullagh et Nelder
(125)
dénotent réellement G~
lcomme fonction de lien. La famille exponentielle couvre une
large gamme de distributions, par exemple discrète (Bernoulli ou Poisson) et continue
(gaussienne ou gamma). La fonction de probabilité (si discret) ou sa fonction de
densité (si continu) a la structure suivante :
Pp(y/x) = exp |
V 6~ ^
0 )+ c(y, 4>) j
où a(-), b(-) et c(-) sont des fonctions connues et le paramètre de dispersion.
Un avantage de la formulation de la famille exponentielle est qu'on peut déduire
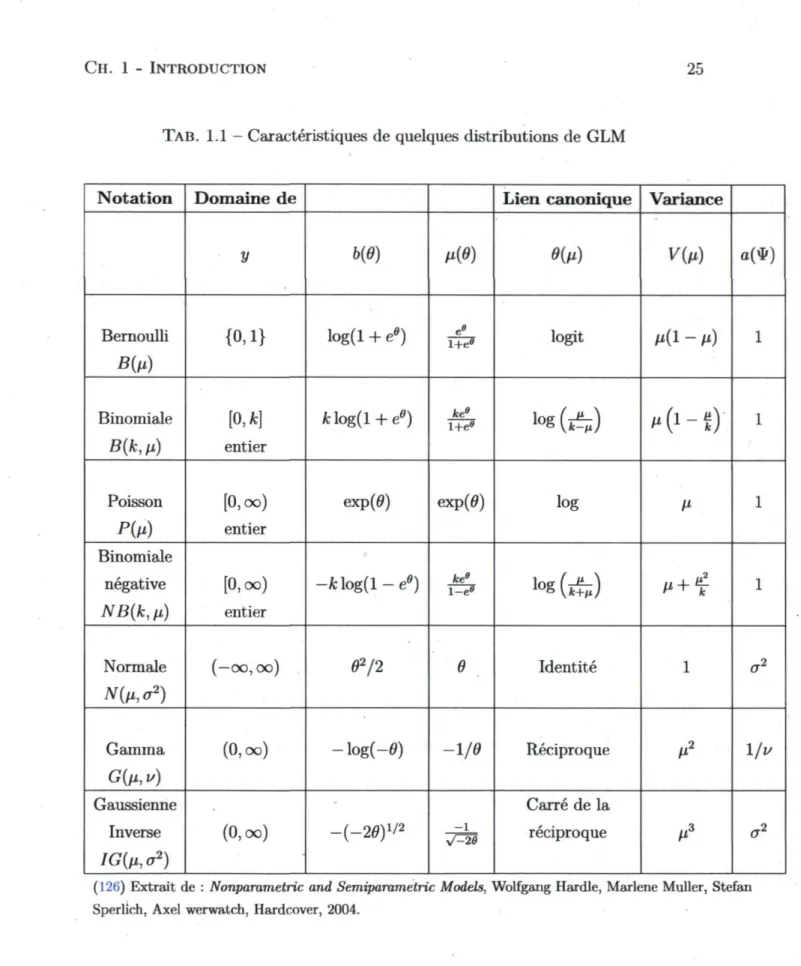
les propriétés de plusieurs distributions au même temps (voir tableau
1.1,
(126))
; de
cet effet, la moyenne et la variance de Y sont données par les formules suivantes :
E(Y) = p = 1/(6) et var(Y) = V(n)a(<fy = b"(0)a(<f>).
1.8.3.2 Modèles de régression pour les haplotypes non phases
Pour tenir compte de l'incertitude liée à la phase inconnue des haplotypes,
cer-tains chercheurs utilisent des méthodes statistiques pour inférer la paire d'haplotypes
la plus probable pour chaque individu et s'en servir par la suite comme s'ils étaient
observés. Cette approche ne tient pas compte des autres haplotypes possibles qui ont
été éliminés, et si le LD est faible, cette procédure pourra entraîner une perte
sub-stantielle de l'information (83;
123).
Étant donné ce fait, l'utilisation des haplotypes
les plus probables introduit des erreurs importantes dans la matrice X, ayant pour
résultat des estimateurs biaises pour les effets des haplotypes
(127; 128; 129; 130).
L'approche la plus adéquate est d'utiliser toutes les paires possibles d'haplotypes qui
sont compatibles avec les données génétiques observées. Un résumé des méthodes
uti-lisant cette formulation est donné dans le tableau
1.2
(60; 120;
122;
123; 124; 129; 130;
131; 132; 133; 134; 135; 136; 137; 138; 139;
140;
141; 142),
indiquant aussi le type du
phénotype utilisé et si la méthode développée permet d'ajuster pour les covariables
environnementales ou non. La plupart des méthodes du tableau
1.2
sont limitées aux
traits dichotomiques (cas/témoins). La méthode qui offre plus de flexibilité est la
méthode de Lake et al.
(124). Cette
méthode est applicable à une grande variété de
traits, y compris des traits dichotomiques et continus. Elle permet l'ajustement pour
les covariables environnementales et les interactions haplotype*environnement.
1.8.3.3 Description de la méthode de Lake et al. (2003)
Pour les haplotypes non phases, la méthode de Lake et al. suppose que la
distri-bution de la variable dépendante Y appartient à la famille exponentielle. L'objectif
de cette méthode est d'estimer simultanément les coefficients de régression 3 et les
fréquences des haplotypes, dénotés H, 'y k l'aide de l'algorithme EM. En plus des
notations introduites dans la section précédente, nous utilisons Gi pour dénoter les
génotypes observés de tous les SNPs pour un individu i. Donc, les données observées
sont (Y, Xe,G) et les données complètes recherchées sont (Y,Xe,Xg(H),Xe g,B,')).
Supposons que nous avons un total de K haplotypes distincts dans la population étu-diée et que, pour l'individu i, nous avons un total de J paires d'haplotypes possibles : Htj = (hij^hijz,..., hijK) indique la j e paire d'haplotypes possible pour l'individu i,
où hijk prend des valeurs 0, 1 ou 2 selon le nombre d'haplotypes de type /iiJt inclus
dans la paire Htj. Soit 7^ la fréquence estimée de l'haplotype hk, k = l , . . . , K et
7 = (7i,...,7fl-) le vecteur des fréquences, en assumant une vraisemblance pros-pective et l'indépendance entre les covariables génétiques et environnementales, la contribution de l'individu i à la vraisemblance peut être exprimée de la façon sui-vante :
L i ( 3TyT) = £ P { Yl/ Xe i, Xg( Hl J\ Xg e t i, 3 } P , ( Hi j) (1.1)
où P-r(Hij) est la probabilité a priori de la paire d'haplotypes Hq, calculée en
assu-mant l'équilibre de Hardy Weinberg (HWE) des haplotypes. En général, pour une paire d'haplotypes Hi — ( h u , . . . , hm), cette probabilité est donnée par la formule suivante :
^ . . . M K /
** v
fc=i y
Lorsque le phasage des haplotypes est connu et que l'information complète sur les variables indépendantes est disponible pour tous les individus, la forme GLM est utilisée pour modéliser la probabilité conditionnelle du trait étant donné les cova-riables P {Y/Xei, Xg(Hi), Xg e i, 3 } et, par conséquent, la log-vraisemblance basée sur
les données complètes est donnée par :
log {LÎ(B
T, 7
T)) = ] M i ^ M M _
c(y., J + £
hiklog(
lk)+h
Klog (l - £
7fc) +C
Afin d'ajuster la formule précédente pour l'incertitude du phasage des haplotypes, l'estimation par maximum de vraisemblance des paramètres 3 et 7 est réalisée par l'algorithme estimation-maximisation (EM=expectation-maximisation). En pra-tique, on maximise la fonction log-vraisemblance de tous les individus. L'étape d'es-timation (E) à l'itération (t + 1) consiste à calculer l'espérance conditionnelle de lalog-vraiisemblance des données complètes sachant les données observées et les para-mètres 0t = (/3t)7t) estimés à l'itération t ; l'étape de maximisation (M) maximise la
fonction résultante pour déterminer les nouveaux paramètres 6t+i = (/3t+i,7t+i)- En
appliquant l'algorithme EM, appelé aussi la méthode des poids (143), le problème à l'étape E et à l'itération (t + 1) dans un échantillon de TV individus indépendants se résume comme suit :
l(0/ôt) = ^ { l o g ^ / y , , ^ , ^ } t = i
E( E
-,
t=l {Hij-Gi rK-i^ - ^ 4 , _ i
+ WijiPt) a(<j>)E
hHk
log(7fc) + hiJK log f 1 ~ E T*J |
L * = loù Wij'(6t) coiTespond à la probabilité postérieure de la je paire d'haplotypes possible
pour le ie individu qui est donnée par la formule suivante :
n t . fll P { y i / xe i, xg( Hi r) , xegi j, Bt\ py t( Hij , )
P {xiy/yi, Xi, 0t) = - j — — x (12)
E H ^ G , P { V i / X â X g i H i j l x ^ B t } Pl t(Hij)
L'étape M est équivalente à la maximisation de la fonction 1(6/0t). Cette
fonc-tion est composée de deux termes : le premier terme est en foncfonc-tion de 3 tandis que le deuxième est en fonction de 7. Les deux termes peuvent donc être maximi-sés séparément pour déduire 6t+\ = (/?t+i,7t+i)- Le premier terme correspond à la
log-vraisemblance pondérée du paramètre de régression dans le cadre des GLM. La maximisation de ce terme revient à la résolution d'un problème de régression pondé-rée avec les poids Wij(6t) ; ceci permettra de déduire le paramètre de régression 3t + l.
Pour mettre à jour les fréquences des haplotypes jt , la valeur estimée du nombre
d'haplotypes de type hk est donnée par :
£K
+ 1) = E E ™
t;(0t)M#.;)
t = l H i j - G ,où N est le nombre d'individus dans l'échantillon et Shk(H) compte le nombre
usuelle des fréquences d'une distribution multinomiale est utilisée pour déduire cha-cune des composantes du vecteur jt +i avec la formule suivante :
Tkt + 1 2 N
Une extension de la méthode de Lake et al. (124) est donnée par Burkett et al. (140)
pour augmenter l'information génétique en faisant l'imputation d'un certain nombre des génotypes manquants à l'aide de l'algorithme EM. Cette extension permet aussi d'estimer le paramètre de dispersion. Pour faire les inferences sur les paramètres es-timés, Burkett et al. ont utilisé la formule de Louis (144) pour calculer directement la matrice d'information des données observées, appelée aussi Y information observée pour simplifier la notation, contrairement à Lake et al. qui ont utilisé une approxi-mation de l'inforapproxi-mation observée. Burkett et al. ont aussi développé un test global basé sur le rapport de vraisemblance. Cette méthode est programmée en langage R et librement distribuée dans le package " Hapassoc " (145).
1.9 Conclusion
Ce premier chapitre présente un rappel quant à la génétique humaine et aux maladies complexes ainsi qu'une revue de littérature sur les méthodes d'analyse pour effectuer les études d'association entre les SNPs ou haplotypes et les traits de ces maladies multifactorielles dans les échantillons populationnels. Dans le chapitre 2, nous allons résumer la problématique entourant les méthodes d'association par haplotypes dans le cadre de l'évaluation des gènes de susceptibilité des maladies complexes. Par la suite, les hypothèses de base de ce travail seront énumérées et les objectifs spécifiques de cette thèse seront énoncés.
TAB. 1.1 — Caractéristiques de quelques distributions de GLM
N o t a t i o n D o m a i n e d e L i e n c a n o n i q u e V a r i a n c e
y
m
m
0(p) V(») « ( * )Bernoulli B(y)
{0,1} log(l + ee) 1+e6 logit /*(1 /x) 1
Binomiale
B ( M
[0,fc]
entier ■Uog(l + ee) 1+e» k é l og(A)
" v1" * ) 1 Poisson P & ) [0,oo) entierexp(0) exp(0) log 1* 1
Binomiale négative N B ( k , /x) [0,oo) entier f c l o g ( l ee) l e » ke6 l o
s
(ifc)
»+i
1 Normale ( — 0 0 , 0 0 ) 62/2 6 Identité 1 o2 Gamma G(M, v) (0,oo) l o g ( 0 ) 1 / 6 Réciproquet
l/v
Gaussienne Inverse I G ( p , o2) (0,oo) ( 2 6 )1'2 y / 2 6 1 Carré de la réciproque /*s o2(126) Extrait de : Nonparametric and Semiparametric Models, Wolfgang Hardie, Marlene Mulier, Stefan