HAL Id: tel-01077927
https://tel.archives-ouvertes.fr/tel-01077927
Submitted on 27 Oct 2014HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Étude expérimentale de la stabilité d’une bulle unique
de cavitation acoustique : application à la nucléation de
la glace déclenchée par cavitation
William Montes Quiroz
To cite this version:
William Montes Quiroz. Étude expérimentale de la stabilité d’une bulle unique de cavitation acous-tique : application à la nucléation de la glace déclenchée par cavitation. Génie des procédés. Ecole des Mines d’Albi-Carmaux, 2014. Français. �NNT : 2014EMAC0002�. �tel-01077927�
THÈSE
THÈSE
En vue de l’obtention du
DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE
Délivré par :l’École Nationale Supérieure des Mines d’Albi-Carmaux conjointement avec l’INP Toulouse Présentée et soutenue le jeudi 20 février 2014 par :
William Montes Quiroz
Étude expérimentale de la stabilité d’une bulle unique de cavitation acoustique : application à la nucléation de la glace déclenchée par
cavitation JURY
Henri Delmas Professeur, Laboratoire Génie Chimique ENSIACET
Président du Jury Bertrand Dubus Directeur de recherche, Institut
Supérieur d’Électronique du Nord
Rapporteur Frédéric Gruy Professeur, École des Mines de
Saint-Étienne
Rapporteur Fabien Baillon Maître assistant, École des Mines
d’Albi-Carmaux
Examinateur Fabienne Espitalier Professeur, École des Mines
d’Albi-Carmaux
Directeur Loïc Hallez Maître de conférence, Université
de Franche-Comté
Examinateur Stéphane Labouret Ingénieur de recherche, LAGEP
Université de Lyon 1
Examinateur Olivier Louisnard Maître assistant, École des Mines
d’Albi-Carmaux
co-Directeur
École doctorale et spécialité :
MEGEP : Génie des procédés et de l’Environnement Unité de Recherche :
Centre RAPSODEE, CNRS - UMR 5302, École des Mines d’Albi-Carmaux Directeur(s) de Thèse :
Fabienne Espitalier et Olivier Louisnard
En primer lugar quisiera agradecer a mis profesores, por brindarme la oportunidad de formar parte del grupo SONONUCLICE.
Al profesor Olivier L, que compartió conmigo sus bastos conocimientos en física experimental y química, por ser un apoyo en mi desarrollo experimental y por las innumerables “cartas blancas”, para poder realizar todas las ideas locas que no siempre dieron buenos resultados. Mis agradecimientos mas sinceros con el profesor Fabien, por sus siempre oportunas palabras de apoyo, gracias por su completa versión de este template de Latex y por compartir conmigo su experiencia en programación y a la profesora Fabienne muchas gracias por su constante apoyo, por el animo que me transmitió, por su gran paciencia al explicarme un poco de química (que es un tema muy oscuro para mi) no podría imaginar una jefa mas buena que ella.
Quisiera agradecer a mis amistades de Francia, en estos momentos tan difíciles y lejos de casa, en realidad son mucho mas que amistades, son mi familia. . . gracias por compartir conmigo todos esos momentos de felicidad, risas, viajes, tristezas y desesperanza a ratos. Gracias sinceramente por todos esos tan bellos momentos Maria Gracial (MG), Stéphane Griffiths, Jacqueline Azevedo, Nayane Macedo, Wellington Betencurte – Aline Bettencurte (gracias por esos almuerzos de los domongos a los dos), Flavio, Analice Costacurta, Paulo Oliveira, Jérémie y particularmente a Noemie Girot (por esos cafés interminables) y gracias a todos los que no pude nombrar, pero que siempre están en mi corazón.
Finalmente quisiera agradecer a mi Familia, por toda la ayuda que me han brindado en este tiempo, por el amor que me han entregado y el animo que me han transmitido, por ultimo quisiera agradecer a la persona que se la ha siempre jugado por mi, por todo su amor incondicional, por haber cambiado mi vida, por eso digo sin mas gracias Tania Silva, amor de mi vida
Doy gracias a Dios por las personas que puso en mi camino y las oportunidades que me ha dado.
Introduction 1
1 Contexte et objectifs 3
1.1 Contexte : La cristallisation de l’eau . . . 3
1.1.1 Intérêt de l’étude de la sono-congélation . . . 4
1.1.2 Qui sommes-nous ? et notre histoire avec la sono-cristallisation 4 1.2 Lyophilisation . . . 5
1.2.1 Principe de la lyophilisation . . . 5
1.2.2 Les différentes étapes de la lyophilisation . . . 5
1.3 À propos de cette thèse. . . 7
2 Cristallisation 9 2.1 La cristallisation . . . 9 2.2 Nucléation . . . 10 2.2.1 La force motrice. . . 10 2.2.2 La nucléation primaire . . . 11 2.2.3 La nucléation secondaire . . . 14
2.2.4 Facteurs influençant la nucléation secondaire . . . 15
2.3 Cristallisation assistée par ultrasons . . . 15
2.3.1 Les hypothèses de l’effet des ultrasons sur la cristallisation . . 18
2.3.2 Le cas de la glace . . . 23
3 Cavitation acoustique 27
3.1 Introduction . . . 27
3.1.1 L’importance des bulles de cavitation : le puzzle des bulles . . 27
3.1.2 Un peu d’histoire . . . 28
3.2 Dynamique radiale de la bulle de cavitation . . . 29
3.2.1 Tension superficielle . . . 29
3.2.2 Rayon ambiant . . . 31
3.2.3 Seuil de Blake . . . 31
3.2.4 Modélisation de la dynamique radiale de bulle . . . 34
3.2.5 Dynamique d’une bulle . . . 38
3.3 La bulle unique en lévitation . . . 40
3.3.1 Observations expérimentales d’une bulle en lévitation.. . . 40
3.3.2 Conditions de stabilité d’une bulle unique . . . 43
3.3.3 Synthèse sur la bulle unique . . . 56
4 Mise au point du montage expérimental 63 4.1 Principe de la double cellule . . . 65
4.2 Système de refroidissement et de dégazage . . . 65
4.2.1 Système de dégazage et refroidissement des liquides . . . 65
4.2.2 Système de refroidissement des cellules . . . 69
4.3 Système électronique . . . 69
4.3.1 Le transformateur d’impédance . . . 69
4.3.2 Bobine d’adaptation . . . 71
4.3.3 Pilotage et acquisition . . . 72
4.4 Système Acoustique . . . 73
4.4.1 Bibliographie sur les cellules de cavitation acoustique . . . 73
4.4.2 Cellules de cavitation carrées en plastique . . . 78
4.4.3 Cellules de cavitation carrées en verre . . . 81
4.5 Système optique . . . 89
4.5.1 Système d’enregistrement . . . 89
5 Résultats et discussions 97
5.1 La cellule de géométrie sphérique . . . 97
5.1.1 Détection acoustique de la bulle de cavitation . . . 97
5.1.2 Corrélation croisée et caractérisation de la bulle . . . 98
5.1.3 Perturbation de la dynamique de la bulle . . . 102
5.1.4 Images d’une bulle de cavitation acoustique . . . 103
5.1.5 Dynamique de la bulle dans une cellule de géométrie sphérique 106 5.2 Validation de la cellule verre à base carrée . . . 108
5.2.1 Dynamique de la bulle dans une cellule de géométrie cubique . 108 5.2.2 Perturbation d’une bulle dans une cellule cubique . . . 108
5.3 Rampes de pression acoustique. Exploration des régimes de lévitation 110 5.4 Détection acoustique d’objets au voisinage d’une bulle de cavitation . 115 5.5 Dimensionnement de la petite cellule . . . 119
5.5.1 Principe de base . . . 119
5.5.2 Variation de la hauteur de la petite cellule . . . 121
5.5.3 Influence d’une différence de niveau entre les cellules . . . 123
5.5.4 Synthèse . . . 123
6 Conclusion 127
Table des figures 131
Fig. 1 – Photographies de cristaux de glace générés dans une solution de saccharose, par une sonotrode commerciale : (a) Les premiers cristaux de glace apparaissent juste après l’émission de l’onde ultra-sonore, (b) Cristaux observés 5 s plus tard. (Chow, Blindet R. Chivers, 2005)
Différents travaux ont montré l’impact des ultrasons sur la cristallisation ou la précipitation de produits minéraux ou organiques : notamment leur effet sur le temps d’induction, la largeur de la zone métastable, la taille et le faciès des cristaux formés (castro et Priego-capote, 2007; J.Dodds et al.,2007; Lyczko, 2000; Nakagawaet al., 2006). Bien que ces effets présentent un grand intérêt tant sur le plan fondamental que pour des applications industrielles, leur origine reste encore mal connue. Ils sont cependant généralement attribués à la cavitation acoustique, phénomène décrivant l’apparition de bulles de gaz/vapeur subissant des oscillations radiales violentes dans un champ acoustique. Dans ce travail nous nous intéressons à la cristallisation de cristaux de glace, présentant un enjeu majeur dans les opérations de congélation et de lyophilisation. L’application d’ultrasons pendant la congélation de la glace permet par exemple de travailler à des sous-refroidissements plus faibles et d’obtenir des cristaux plus gros (Saclier, Peczalski et Andrieu,2010). Il y a quelques années, plusieurs travaux ont été menés pour étudier la cristallisation de la glace induite par l’immersion d’une sonotrode dans des liquides sous-refroidis, en présence d’un nombre important de bulles. Les nombreux événements de nucléation
induits par les bulles de cavitation acoustique rendent impossible une observation détaillée de la nucléation (figure 1). Une autre difficulté expérimentale majeure de cette méthode est la nucléation hétérogène inévitable à la surface du transducteur plongé dans le liquide sous-refroidi, comme l’indiquent Ohsaka et Trinh (1998). Il est possible de s’affranchir de ces difficultés en se plaçant dans des conditions où une seule bulle de cavitation est générée, et maintenue isolée pendant des heures. Cette technique, initiée par F. Gaitan et al. (1992), consiste à faire léviter une bulle dans une cellule résonante produisant une onde stationnaire.
La technique a largement été utilisée depuis dans le cadre d’études sur la sono-luminescence (M. Brenner, 2002). Ces expériences constituent la base de notre étude pour explorer le phénomène de nucléation de la glace induite par une bulle unique de cavitation acoustique dans de l’eau sous-refroidie. Cette technique devrait permettre de déterminer l’effet d’une bulle de cavitation sur la nucléation de cristaux, et d’apporter des réponses sur les effets observés en situation multi-bulles (telles que celle de la figure 1). Il est à noter que les résultats et les modèles théoriques concernant ces expériences sont à ce jour contradictoires (Hickling,1994; Ohsaka et Trinh,1998; Saclier, Peczalski et Andrieu, 2010).
Cette étude présente la mise au point du dispositif expérimental pour l’étude de la cristallisation de la glace dans de l’eau sous-refroidie en présence d’une bulle unique de cavitation acoustique.
1
Contexte et objectifs
Sommaire
1.1 Contexte : La cristallisation de l’eau . . . . 3
1.2 Lyophilisation. . . . 5
1.3 À propos de cette thèse. . . . 7
1.1 Contexte : La cristallisation de l’eau
La cristallisation est un phénomène thermo-physique important durant lequel une substance précipite en milieu sursaturé. Ce phénomène se compose de deux princi-pales étapes : la nucléation et la croissance de cristaux. La nucléation est la formation d’un nouveau cristal, et dans le cas de l’eau, elle est considérée comme un facteur déterminant pour l’optimisation des procédés industriels liés à la congélation (Naka-gawa et al., 2006). De plus, la morphologie et la distribution de taille des cristaux sont fortement liées à la nucléation. Cependant, la nucléation de la glace se produit de façon spontanée et stochastique dans une vaste gamme de températures. Elle est affectée par plusieurs facteurs comme : les impuretés, les aspérités, les propriétés de surfaces, etc. En général ces facteurs ne peuvent pas être facilement contrôlés (Nakagawa et al., 2006). Autrement dit, la prédiction précise de la température de nucléation est difficile. Par conséquent, une méthode pour contrôler le phénomène de nucléation d’une façon reproductible et prévisible serait prometteuse pour l’industrie de la congélation (Kiani et Sun,2011) . Pour cela, il existe de nouvelles techniques comme : la cristallisation par détente haute pression (Norton et Sun, 2007), par champ électrique (Orlowska, Havent et Le-Bail, 2009), par champ magné-tique (Freitas et al.,1999), par laser pulsé (Soare et al., 2011) et par ultrasons (Ruecroft et al., 2005a). La technique par ultrasons, dite « sono-congélation » ou « sono-cristallisation », est celle que nous avons étudiée dans cette thèse.
1.1.1 Intérêt de l’étude de la sono-congélation
Tant dans le milieu industriel que celui de la recherche, le phénomène de la cristal-lisation de l’eau doit être contrôlé, reproductible et d’un coût relativement faible. Nous nous proposons d’étudier la cristallisation assistée par ultrasons, qui est une méthode relativement nouvelle. La première application à l’échelle du laboratoire a été rapportée en 1927 (Ricards et Loomis,1927). Ensuite, on peut citer des travaux visant à augmenter l’échelle des expériences : échelle du laboratoire (Kapustin,
1963), et échelle industrielle (Ruecroft et al., 2005a).
La sono-congélation joue un rôle important dans les procédés de l’industrie agroali-mentaire et de la lyophilisation. Cette technique n’ajoute pas d’éléments artificiels ou synthétiques à la congélation. Dans le cas de la lyophilisation (expliqué plus loin), cette méthode réduit le coût de l’opération.
Alors que l’utilisation des ultrasons comme méthode de nucléation et de congélation est bien connue, les mécanismes mis en jeu sont complexes et restent non maîtrisés. Plusieurs hypothèses ont déjà été proposées (voir section 2.3.1 page 18) pour les expliquer (Hickling, 1994; Hunt et Jackson,1966; Ohsaka et Trinh, 1998; Saclier, Peczalski et Andrieu, 2010).
1.1.2 Qui sommes-nous ? et notre histoire avec la sono-cristallisation
Le laboratoire RAPSODEE est un centre de recherche basé à ALBI (France), concerné par les processus industriels de traitement des particules solides : poudres, matière granulée, déchets solides et biomasse.
Depuis plusieurs années, RAPSODEE a développé une expertise dans le champ de la sono-cristallisation des solutés, d’un point de vue expérimental et théorique (J.Dodds et al., 2007). Cette expertise est particulièrement développée dans l’acoustique linéaire théorique, la cavitation acoustique et la cristallisation (J.Dodds et al.,2007; Louisnard, 2012a,b; Louisnard et Gonzales-Garcia, 2007).
Différentes expériences réalisées au centre RAPSODEE ont montré l’influence des ultrasons sur la cristallisation/précipitation de produits minéraux ou organiques, leur effet sur le temps d’induction, la largeur de la zone métastable, la taille et le faciès des cristaux formés (Hassen Harzali,2011; Lyczko, 2000). Le centre RAPSODEE a aussi mené des études fondamentales d’une bulle unique de cavitation (Grossier,
2006).
Dans cette thèse, suite à ces recherches, nous nous intéressons à élucider le(s) mécanisme(s) de sono-congélation au travers d’une expérience simplifiée. L’objectif est de concevoir un dispositif expérimental permettant à terme de visualiser et de détecter la formation de cristaux de glace en présence d’une bulle unique de cavitation. Une fois ce dispositif mis au point, il s’agit ensuite de faire varier les paramètres, pression acoustique et température de sous-refroidissement afin d’éclaircir, si possible, les mécanismes par lesquels les ultrasons déclenchent la nucléation.
1.2 Lyophilisation
1.2.1 Principe de la lyophilisation
La lyophilisation, appelée autrefois cryo-dessiccation, est l’opération de déshydrata-tion d’un produit préalablement surgelé, dans une matrice aqueuse (normalement un mélange d’eau et d’alcool), par sublimation. Cette transformation se fait sans passer par une étape de fusion (du solide au liquide), ni par une étape d’évaporation (du liquide au gaz).
La lyophilisation est devenue une pratique industrielle dans les années 30, la tech-nologie et les équipements utilisés sont constamment améliorés au fil des décennies, mais le développement de la lyophilisation dans l’industrie est cependant limité par son coût. En fait son développement demeure bien inférieur à celui du séchage convectif, par atomisation ou par contact. La faible productivité en lyophilisation est due au fonctionnement sous vide poussé (de 10 à 200 Pa selon le produit) et à basse température (de −40 à −80◦
C selon le produit), et à la nature le plus souvent discontinue de l’opération, avec des durées de traitement très importantes (d’une dizaine d’heures à plusieurs jours). Tous ces facteurs combinés conduisent aussi à des frais d’exploitation extrêmement élevés (consommation énergétique de 1500 kW h pour éliminer une tonne d’eau). En conséquence, la lyophilisation n’est mise en oeuvre que pour des produits ayant une forte valeur ajoutée, notamment dans les secteurs de la pharmacie (vaccins, sérums, anticorps, hormones) et de la biotechnologie (enzymes, levains) (Butchaiah et Rao,1988; Cherian et Corona, 2006).
Ce procédé présente des avantages. Les produits lyophilisés n’ont pas besoin de réfri-gération, et peuvent être conservés à température ambiante. Le produit se réhydrate « instantanément », lors de son utilisation. Son aspect est conservé, et sa qualité
biochimique est bien supérieure à celle des produits séchés conventionnellement.
1.2.2 Les différentes étapes de la lyophilisation
Le procédé de lyophilisation se décompose en trois étapes : la congélation, la dessic-cation primaire, et la dessicdessic-cation secondaire (LABCONCO,2004) .
Congélation
La congélation (solidification de l’eau) est la première étape de la lyophilisation. Cette phase est essentielle, car elle doit garantir que le produit à lyophiliser ne sera pas altéré. Cette première étape consiste à congeler les produits pour que l’eau qu’ils contiennent soit transformée sous forme de glace (dans le cas où elle se trouvait à l’état liquide). Le procédé de congélation et la température finale du produit congelé peuvent affecter la réussite du processus de lyophilisation.
Il est important de mentionner que la transition liquide-solide ne s’effectue pas à 0◦
C, mais à une température inférieure. L’eau passe donc par un état liquide métastable
lors du refroidissement, et la température à laquelle la transition s’effectue est stochastique, dont la loi de probabilité ne peut être obtenue que par répétition d’un grand nombre d’expériences dans des conditions identiques (Nakagawa et al.,2006). La température de solidification, qui peut descendre jusqu’à −10◦
C dans certains cas, est cruciale, car la taille des cristaux générés, et donc la taille des pores du solide après sublimation de la glace, lui est directement corrélée : plus la température de congélation est basse, plus les cristaux seront petits.
La température de cristallisation dépend de la vitesse de refroidissement. Un re-froidissement rapide conduit à une température de cristallisation plus basse. Ainsi un refroidissement rapide entraîne la formation de petits cristaux de glace, ce qui conduit à un produit plus difficile à lyophiliser, ou parfois dénaturé. Au contraire, si la congélation est lente, les cristaux sont plus gros et la nature du produit est conservée.
Dessiccation primaire
La deuxième étape, la dessiccation primaire permet la sublimation de la glace. La sublimation est un principe physique simple. C’est le passage d’une substance de l’état solide à l’état gazeux directement. On dessèche donc le produit en le mettant sous vide : la glace devient de la vapeur et elle est ensuite récupérée. La vapeur d’eau est condensée par un « piège froid » ou dans un condenseur (appareil dont la fonction principale consiste à transformer un gaz en liquide). La déshydratation du produit se poursuit dans le même temps.
Pour la phase de sublimation, on apporte de la chaleur au produit afin que la glace se sublime. Cette chaleur est apportée principalement par radiation. Pendant cette opération, la température peut varier suivant le produit. Le niveau de vide, la température du produit et le temps de dessiccation sont trois paramètres importants qui permettent de déterminer le flux de sublimation à l’intérieur de la machine (lyophilisateur). Un flux de vapeur trop élevé pourrait altérer le produit que l’on cherche à lyophiliser. Un cycle trop court laisserait trop d’eau dans le produit, qui pourrait alors être dégradé lors de la dessiccation secondaire et un cycle trop long pourrait détériorer certaines molécules du produit. Lorsque la plus grande partie de l’eau a été sublimée, le produit initial a perdu 80 à 90% de son eau.
Dessiccation secondaire
Cette dernière étape débute lorsque toute la glace est sublimée. Les produits sont alors séchés. La phase de dessiccation secondaire consiste à désorber l’eau qui est encore présente dans le produit. Cette eau résiduelle forme des liaisons chimiques avec la matrice solide et elle est donc plus difficile à extraire du milieu. C’est une étape délicate, car poussée trop loin, elle peut dénaturer le produit. Dans cette étape, le vide est poussé jusqu’aux environs de 5 µbar. On maintient ou on augmente la température du produit jusqu’à des valeurs positives. Une température variant entre 20 et 70◦
2 et 8%. Les molécules d’eau qui restent en surface du produit sont arrachées. Le produit est maintenant sec à 95% ou plus.
1.3 À propos de cette thèse
Le but de ce travail de thèse est de concevoir un dispositif expérimental permettant de générer une bulle unique de cavitation acoustique et de la visualiser. Dans une première partie (chapitre2 page 9), nous abordons le phénomène de cristallisation, et plus particulièrement la cristallisation assistée par ultrasons. Dans le chapitre 3 page 27nous présentons les bases théoriques de la cavitation acoustique. Le montage expérimental est abordé dans le chapitre4 page 63, où nous décrivons l’évolution de ce montage étape par étape en indiquant les problèmes rencontrés et les « solutions » proposées. Dans le chapitre 5 page 97nous montrons les résultats de nos expériences de bulle unique. Et pour finir, nous développons les conclusions et perspectives à ce travail (page127).
2
Cristallisation
Sommaire
2.1 La cristallisation . . . . 9
2.2 Nucléation. . . 10
2.3 Cristallisation assistée par ultrasons . . . 15
2.1 La cristallisation
La cristallisation est un important phénomène thermo-physique, qui se produit lorsqu’une espèce initialement en phase liquide passe en phase solide, parce qu’elle y est plus stable. La cristallisation est une transition de phase du premier ordre, ce qui implique que lors de la solidification, le liquide ou la solution est dans un état métastable, elle est aussi dite sursaturée. La cristallisation commence par la nucléation de cristaux, et continue par leur croissance (Kashchiev, 2000; Mullin,
2001). Ce processus est mis en oeuvre dans beaucoup de domaines industriels comme la pharmacie, la chimie de spécialité, la cosmétique ou l’industrie pétrolière. La cristallisation est une étape essentielle, influençant directement les propriétés du produit final comme la distribution de tailles des cristaux, les phases cristallines, la nucléation et la croissance (castro et Priego-capote,2007; Mullin, 2001). Le point de départ pour toute opération de cristallisation est l’obtention d’un état métastable. Cet état de sursaturation peut être atteint de différentes façons, telles que l’évaporation du solvant ou le refroidissement de la solution par exemple. Comme cela vient d’être mentionné, la cristallisation est composée de deux étapes principales, la nucléation et la croissance de cristaux. L’étape de nucléation est définie comme la formation de nouveaux cristaux et se produit, soit dans une solution sans cristaux préexistants (nucléation primaire), soit en présence de cristaux de même nature que le solide qui cristallise (nucléation secondaire). La nucléation primaire
peut être soit homogène (la solution ne contient pas de particules étrangères ou de cristaux), soit hétérogène (des particules ou surfaces étrangères sont présentes). Un résumé des types de nucléation est présenté à la figure (2.1).
Fig. 2.1 – Mécanismes de nucléation (Mersmann, 2001).
Après la formation des germes (nuclei), l’étape suivante du processus de cristallisation est la croissance des cristaux, tant que la solution reste sursaturée. Les germes apparus lors de la nucléation fournissent un modèle structural sur lequel toute la matière se dépose (Mersmann, 2001). Toutefois la nucléation et la croissance de cristaux peuvent se produire simultanément. En outre, les germes initialement formés peuvent être très nombreux, provoquant la limitation de la croissance de chaque cristal. Par conséquent, ces deux étapes vont définir la distribution de taille des cristaux (Mersmann,2001). Par ailleurs, de nombreux facteurs affectent la formation et la croissance, en particulier la concentration, la température, les impuretés, les transferts de masse et de chaleur dans le système et les caractéristiques de la matière à cristalliser. Ces facteurs doivent être considérés lors de l’étude d’un processus de cristallisation.
Nous allons maintenant nous intéresser plus particulièrement à la nucléation.
2.2 Nucléation
C’est l’étape de formation des premiers cristaux. Nous détaillerons successivement la nucléation primaire et la nucléation secondaire.
2.2.1 La force motrice
La nucléation primaire est l’apparition localisée d’une nouvelle phase, ici le solide, au coeur d’une ancienne phase sursaturée, ici le liquide. La sursaturation est nécessaire car elle est la principale force motrice pour la création ou le dépôt d’une phase solide à partir d’une phase liquide (Mullin, 2001).
La sursaturation est calculée à partir de la différence ∆µ entre le potentiel chimique du constituant à cristalliser dans la phase liquide, et le potentiel chimique de ce même constituant dans la phase solide.
Pour une cristallisation en solution, c’est-à-dire la cristallisation d’un soluté initiale-ment dissous dans un solvant, la force motrice, ou écart de sursaturation, est souvent exprimée comme la différence entre la concentration en soluté C et la concentra-tion en soluté à l’équilibre C∗ à la température et à la pression considérée. Cette
sursaturation est appelée sursaturation absolue, et notée ∆C (∆C = C − C∗
). Dans le cas de la cristallisation d’un fondu, ici de l’eau à partir de sa phase liquide, la solution est sursaturée si la phase est à l’état liquide à une température en dessous de son point de fusion. Dans ce cas, la sursaturation dans une eau sous-refroidie peut se calculer par
∆µeau= LS→L
(Tf(P ) − T )
Tf(P )
, (2.2.1)
où Tf(P ) est la température de fusion à la pression considérée P , T la température
de l’eau sous-refroidie et LS→L l’enthalpie de fusion de la glace. 2.2.2 La nucléation primaire
L’apparition d’une nouvelle phase commence par la formation d’une interface construite à partir de germes cristallins. Les germes se forment à partir de fluc-tuations spontanées de la composition ou de la masse volumique au sein de la solution métastable : elles sont les responsables de l’apparition de l’interface. La for-mation des germes est un processus résultant de la concurrence entre deux processus : l’agrégation et la dissociation de molécules ou d’ions constituant le solide.
À température et pression constantes, la théorie classique de la nucléation primaire définit le travail total W nécessaire pour créer un noyau comme étant la somme du travail de formation de surface, Ws, et du travail requis pour former le volume (bulk)
de la particule, Wv (Mullin, 2001; Myerson, 2002).
W = Ws+ Wv, (2.2.2)
Ws est une quantité positive, car les molécules doivent se réorienter pour créer une
nouvelle interface, et Wv est une quantité négative parce que l’état dans le volume
est plus stable, et une certaine quantité d’énergie se libère lors de la fixation de la molécule au cristal. La nucléation implique donc deux phénomènes compétitifs, l’un en volume et l’autre en surface. Le terme de volume traduit un processus exothermique conduisant à une réduction de l’énergie libre de Gibbs. Ce terme favorise donc la nucléation. A contrario, le terme de surface agit contre la nucléation. Il permet à des ensembles moléculaires de se dissoudre ou d’utiliser une certaine quantité d’énergie pour la nucléation. L’équation (2.2.2) peut donc être réécrite, en se basant sur l’énergie de Gibbs selon Kashchiev (2000), Kiani et Sun (2011) et Mullin(2001) :
où ∆G est la variation d’enthalpie libre lors de la transformation de l’espèce considérée de l’état liquide à l’état solide à température et pression constantes et conduisant à une particule de taille L. ∆Gs représente la variation d’enthalpie libre de surface, et
∆Gv est la variation d’enthalpie libre de volume (assemblage des molécules). ∆Gs
est une quantité positive proportionnelle à L2. Dans une solution sursaturée, ∆G v
est une quantité négative proportionnelle à L3, ainsi
∆G = ∆Gs+ ∆Gv = βL2γ −
α ν0
L3∆µ, (2.2.4)
où γ est l’énergie interfaciale cristal/solution, β et α sont respectivement les facteurs de forme en volume et en surface (basés sur la longueur caractéristique L), ν0 le
volume moléculaire du cristal et ∆µ la sursaturation. Pour les noyaux sphériques, L étant le diamètre, le facteur de forme en volume α vaut π
6, et le facteur de forme en
surface β vaut π.
Les deux termes de droite de l’équation (2.2.4) sont de signes opposés et dépendent différemment de L. Le tracé de l’enthalpie libre de formation (∆G) en fonction de L montre que ∆G passe par un maximum (voir figure2.2). Cette valeur maximale, ∆Gc
correspond au noyau critique, Lc, et elle est obtenue en maximisant l’équation (2.2.4),
c’est-à-dire pour ∂∆G ∂L = 0. Lc = 4γν ∆µ0 . (2.2.5)
Pour cette taille critique, le germe est en équilibre instable avec la solution. Si on lui enlève un monomère, c’est-à-dire un élément constitutif du germe, alors L < Lc, et
le germe se dissout ; si on lui ajoute un monomère, alors L > Lc, et le germe croît.
En substituant l’équation (2.2.5) dans (2.2.4), nous obtenons l’énergie libre de formation d’un embryon de taille critique comme :
∆Gc =
16πγ3
ν2 0
3∆µ2 , (2.2.6)
appelé aussi « travail de nucléation ». C’est la barrière d’énergie que les fluctuations thermiques dans le liquide doivent franchir pour pouvoir former un germe de solide. Puisque seuls les noyaux ayant des rayons plus grands que le noyau critique (nucleus) peuvent se développer spontanément jusqu’à la taille macroscopique, le taux de nucléation B est défini comme le nombre de noyaux stables formés dans le système par unité de temps et par unité de volume ou de surface. Ce taux de nucléation dépend généralement de propriétés thermodynamiques et physiques différentes telles que la température, le nombre de noyaux stables formés par unité de temps et par unité de volume, le coefficient de diffusion DAB, la sursaturation ∆µ, la densité du
solide, etc (Rogerson et Cardoso,2003). À sursaturation constante, le nombre de noyaux stables formés suit une distribution de probabilité, et il peut être décrit par le modèle d’Arrhenius. La nucléation primaire, c’est-à-dire la formation d’un nucleus de rayon supérieur au rayon critique, n’arrive pas soudainement, car les
Fig. 2.2 – Variation d’enthalpie libre en fonction de la taille L expliquant l’existence d’un noyau critique.
germes inférieurs à la taille critique se dissolvant spontanément, seul un nombre suffisant de fluctuations peut conduire à la formation d’un nucleus.
Les groupes de molécules sont d’abord créés par des attachements de molécules dus à des collisions par des mouvements aléatoires qu’elles subissent ; ces groupes de molécules forment des embryons. La taille des embryons ne respecte pas les critères de formation du cristal, cependant de nouvelles collisions entre les embryons aboutissent à la formation de noyaux stables (Gunton, 1999; Rogerson et Cardoso,2003). La formule la plus souvent utilisée pour le taux de nucléation B stationnaire provient de la théorie classique (Kashchiev, 2000) et est donnée par :
B = A exp � −∆Gc kBT � , (2.2.7)
avec le terme pré-exponentiel A défini par : A = zf∗
C0, (2.2.8)
où kB est la constante de Boltzman, T est le température, f∗ est la fréquence
coefficient de diffusion des molécules, à la température et à la concentration), C0 est
la concentration de sites de nucléation et z qui traduit la courbure de ∆G(L) pour la taille critique L = Lc. Si l’on note n le nombre de molécules dans un cluster, et nc ce
nombre dans un noyau critique, ce coefficient, appelé facteur de Zeldovich, s’écrit :
z = � −2kTπ d 2∆G dn2 (nc) �1/2 . (2.2.9)
L’équation (2.2.7) implique que la nucléation est un phénomène statistique et que plus la saturation est élevée, plus important est le taux de nucléation. La probabilité de nucléation dépend aussi d’autres paramètres, comme le montre l’équation (2.2.8), via le terme pré-exponentiel A.
L’équation (2.2.7) peut être utilisée pour toutes les sortes de nucléation moyen-nant quelques modifications (Lyczko, 2000; Mersmann, 2001; Mullin, 2001; Rogerson et Cardoso, 2003; Saclier, Peczalski et Andrieu, 2010).
À sursaturation fixée, pour la cristallisation de la glace, on peut par exemple citer pour la vitesse de nucléation stationnaire l’expression donnée par Kashchiev (2000) pour un milieu fondu :
B = 2(γkBT )0.5C0exp � −∆Hcrist kBT � 3πd2 0η exp � ∆µ kBT � exp � − 16πν 2 0γ 3 3kBT ∆µ2 � , (2.2.10) avec d0 le diamètre du monomère moléculaire, et η la viscosité de la solution.
De cette équation, on peut conclure que le taux de nucléation dépend de la sursatu-ration, de la température, de la tension interfaciale liquide-solide, et de la viscosité de la solution. Cependant, malgré l’existence de différentes expressions pour le taux de nucléation, ce dernier doit en pratique être mesuré et corrélé empiriquement pour chaque système.
2.2.3 La nucléation secondaire
Les cristaux déjà créés sont capables de catalyser le phénomène de nucléation, et par conséquent, le taux de nucléation secondaire est généralement plus élevé que celui de la nucléation primaire. De plus, la nucléation secondaire se produit à un rapport de sursaturation inférieur. On trouve sur ce sujet plusieurs théories et recherches dans la littérature, mais les mécanismes ne sont pas entièrement élucidés (Mullin, 2001). Il existe différents types de nucléation secondaire qui dépendent de la source à partir de laquelle elle a été induite : la nucléation secondaire vraie, la nucléation secondaire apparente et la nucléation de contact. Lorsque la nucléation secondaire est provoquée par des interactions cristal-solution, on parle de nucléation secondaire vraie. Lorsque ce sont des interactions cristal-cristal ou cristal-appareillage, on parle de nucléation
secondaire de contact. Lorsque la nucléation résulte de l’ensemencement (des cristaux de même nature que le solide qui cristallise sont volontairement introduits dans la solution), on parle de nucléation secondaire apparente. En effet, à la surface des cristaux utilisés comme semences se trouvent parfois des micro-cristallites qui se détachent dès que les cristaux sont introduits dans le cristallisoir. Une fois détachés, ces fragments présentent des sites de croissance. Cette nucléation est dite apparente, parce qu’il n’y a pas réelle naissance de germes.
2.2.4 Facteurs influençant la nucléation secondaire
La nucléation secondaire dépend de différents facteurs tels que la sursaturation (Bhamidi, Skrzypczak-Jankun et Schall, 2001), la température (Melia et Moffitt, 1963), la pureté des cristaux (Melia et Moffitt, 1963), la vitesse d’agitation de la solution et la force de gravité (Scaife, Cavoli et Suib,1990), la densité en cristaux dans la solution et la rugosité des cristaux.
La sursaturation peut agir sur la nucléation secondaire selon deux modes :
1. La micro-rugosité du cristal parent augmentant avec la sursaturation, plus de micro-cristallites pourront être arrachés par les impacts à sursaturation élevée ; 2. Les micro-cristallites, existant à la surface du cristal et ayant une taille supé-rieure à celle du nucleus critique pour une sursaturation donnée, vont survivre et grossir dans la solution. Quand la sursaturation augmente, le rayon critique diminue, et la fraction de particules qui pourront se développer augmente donc. Les vitesses de nucléation secondaire sont le plus souvent données par des lois empiriques de la forme :
B2 = K2MTjNt∆Cb, (2.2.11)
où B2 est le taux de nucléation secondaire, K2 un constante de vitesse, MT la densité
de la suspension, N l’intensité de l’agitation dans le cristallisoir (dans le cas d’un cristallisoir agité) et ∆C la sursaturation absolue. Les coefficients j, t et b varient en fonction des conditions opératoires.
2.3 Cristallisation assistée par ultrasons
La cristallisation sous ultrasons couple deux phénomènes de nucléation : celui qui donne naissance aux cristaux, et celui à l’origine de l’apparition de bulles de cavitation au sein de la solution. La distribution des bulles dans une expérience de cavitation est inconnue, ainsi que leurs interactions. Ajouté au phénomène de cristallisation, ceci fait de la cristallisation assistée par ultrasons une technique encore difficilement extrapolable au niveau industriel ou même d’une expérimentation à une autre.
Industriellement l’utilisation des ultrasons pourrait pourtant représenter une alter-native à l’ensemencement, méthode classique de contrôle de la nucléation et des propriétés des cristaux produits. Mais comme il n’existe pas d’outil de simulation permettant de dimensionner et d’optimiser des sono-cristallisoirs, cette technique n’est que très peu développée à l’échelle industrielle.
Cependant, la sono-cristallisation semble pouvoir offrir un contrôle accru de certaines caractéristiques des cristaux obtenus : taille, dispersion, structure cristalline, repro-ductibilité comme le montrent des travaux antérieurs (Gatumel, 1997; Hassoun,
2003; Lyczko, 2000; Lyczko et al., 2002).
L’action des ultrasons a été comparée à celle de l’agitation mécanique sur des solutions sursaturées d’alun de potassium. Il a été observé que la distribution de taille obtenue sous ultrasons est plus resserrée. La taille moyenne des cristaux obtenus sous ultrasons est inférieure de 50% à la taille obtenue sous agitation mécanique (A. English et Dole,1950). Amara (Amara,2002) a mené une étude sur l’alun de potassium à une fréquence de 20 kHz et a confirmé que l’uniformité de taille obtenue sous ultrasons était plus importante en comparaison avec une cristallisation sans ultrasons.
Les ultrasons sont aussi reconnus pour assurer une meilleure pureté des cristaux (Ratsimba, Biscans et Delmas, 1999). Par exemple, lors de la cristallisation en milieu fondu, des impuretés peuvent se déposer à la surface du cristal et être redistribuées dans le réseau cristallin par substitutions (Sirinivasan, Shirgaonkar et Pandit, 1995). Les auteurs de cette étude ont proposé un mécanisme rendant compte d’une meilleure pureté du produit cristallisé sous ultrasons. Celui-ci est basé sur l’existence de points chauds au moment de l’implosion des bulles de cavitation, qui, bien qu’ayant une durée de vie éphémère (quelques nanosecondes), permettaient de dissoudre les impuretés présentant des points de fusion plus faibles.
Un des problèmes liés à la sono-cristallisation est l’effet de volume. Des expériences de cristallisation assistée par ultrasons dans des conditions expérimentales similaires (même produit à cristalliser, à même sursaturation, et à température identique), mais réalisées dans des réacteurs de volumes différents ne donneront pas les mêmes résultats (Li et al., 2003). La taille des cristaux obtenus dans le réacteur de volume plus important est plus élevée. Plusieurs explications quant à l’origine de ce phénomène sont proposées :
1. La pénétration de l’onde est moins importante dans un plus grand volume ; 2. Les vibrations acoustiques et le phénomène de cavitation sont plus faibles, car
des zones mortes existent dans un grand réacteur.
Louhi-Kultanen a étudié la cristallisation de la glycine α. Les ultrasons permettent de produire un produit plus pur, plus fin et présentant des grains de morphologie uniforme, avec une distribution granulométrique monomodale (Louhi-Kultanen et al.,2006).
Dans une étude sur la cristallisation de l’hexa-acétate de sorbitol, Price (1997) présente une alternative à l’ensemencement. En effet, grâce à l’application des
ultrasons, l’auteur déclenche la nucléation du produit à des sursaturations plus faibles qu’en l’absence d’ultrasons. L’auteur a montré que l’application des ultrasons, pour initier la nucléation de l’hexa-acétate de sorbitol, permet d’obtenir de simples cristaux non agglomérés, alors que sans ultrasons on obtient de nombreux cristaux agglomérés. Dans cette étude, trois autres points sont abordés : la nucléation, la croissance et la dissolution préférentielle des impuretés. L’auteur rappelle que l’implosion des bulles de cavitation au voisinage des cristaux peut entraîner la formation ou l’arrachement de cristallites, engendrant de la nucléation secondaire. Le mécanisme par lequel les ultrasons affectent la croissance des cristaux reste mal connu.
Une étude de l’effet des ondes ultrasonores sur la taille des particules et la distribu-tion de taille des cristaux, à partir de soludistribu-tions sursaturées d’aluminium refroidies lentement, montre que les ultrasons permettent d’obtenir une meilleure homogénéité des distributions de taille des cristaux par rapport à une agitation mécanique seule, et d’obtenir une taille moyenne plus faible (Enomoto, Sung et Nabagawa, 1992). Une étude sur la cristallisation de solution de saccharose sous ultrasons a permis de montrer la possibilité de former des cristaux répondant aux exigences de l’industrie sucrière (Hassoun,2003; Tai-quin, 1993). Le champ ultrasonore permet de dimi-nuer la barrière énergétique à l’aide des bulles de cavitation, et provoque ainsi la nucléation primaire du saccharose. Cette dernière peut donc être obtenue à de faibles sursaturations en présence d’ultrasons, et la distribution de taille ainsi obtenue est plus resserrée que lorsqu’il n’y a pas d’ultrasons.
Une étude sur la précipitation du sulfate de baryum a permis de mettre en évidence les effets des ultrasons sur la nucléation (Gatumel,1997). Les expériences menées en cristallisoir continu ont montré que les ultrasons permettent de s’affranchir des effets de mélange et d’obtenir des cristaux plus petits. La taille et la forme des cristaux obtenues avec ultrasons sont beaucoup moins dispersées que lors de la précipitation sans ultrasons. L’étude de la morphologie des cristaux laisserait à penser à une nucléation proche de la nucléation secondaire de contact. Cette dernière serait liée aux ondes de chocs provoquées par l’implosion des bulles de cavitation, ou les jets de liquide provoqués par une implosion près d’un solide dans le cas d’une étude en cristallisation en continue. L’effet mécanique des ultrasons serait essentiellement un arrachement des agrégats moléculaires sur les cristaux parents.
Les ultrasons permettent d’obtenir des cristaux de K2SO4 de diamètre moyen en
volume plus faible que sans ultrasons. Ils agissent aussi sur la surface des cristaux. En effet, quelle que soit la puissance ultrasonore dissipée dans la solution et le type de sonde ultrasonore utilisée, l’application des ultrasons rend les cristaux de sulfate de potassium poreux. Les ultrasons ont un effet sur la nucléation primaire du sulfate de potassium et sur les caractéristiques des cristaux obtenus (Lyczko,2000). Un autre paramètre important est la réduction drastique du temps d’induction.
Le temps d’induction est défini comme le temps de latence entre le moment où la sursaturation est créée et le moment où les premiers cristaux apparaissent. Ce temps est la somme de trois temps : le temps nécessaire pour obtenir une distribution de clusters stables (quelques nanosecondes), le temps nécessaire pour la formation de nuclei et le temps requis pour que les nuclei formés grossissent jusqu’à une taille
détectable. Dans des conditions classiques de cristallisation, le premier temps est négligé.
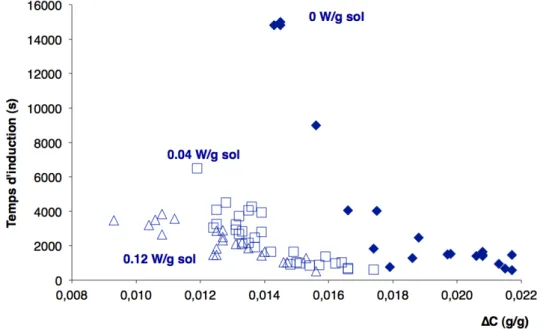
Fig. 2.3 – Courbe sursaturation / temps d’induction pour la cristallisation par refroidissement du sulfate de potassium K2SO4pour différentes puissances ultrasonores (Lyczko et al.,2002).
La figure2.3montre l’évolution du temps d’induction en fonction de la sursaturation absolue dans le cas de la cristallisation du sulfate de potassium par refroidissement (solution aqueuse), avec et sans ultrasons. Elle met en avant un aspect important de la cristallisation : le temps d’induction évolue à l’inverse de la sursaturation, et ce en présence d’ultrasons ou non. Si l’on regarde les points sans ultrasons et les points avec ultrasons, on voit que l’utilisation d’ultrasons est particulièrement appropriée au cas des faibles sursaturations. Dans cette zone, en l’absence d’ultrasons, la naissance d’un cristal est plus qu’improbable dans un temps court. Les ultrasons permettent d’accélérer la nucléation de cristaux dans cette zone (Lyczko et al., 2002). Cette diminution du temps d’induction a aussi été observée dans le cas du saccharose (comme le montre la figure2.4), dans le cas de la glycine (Louisnard et Espitalier,
2007), et dans le cas du sulfate de zinc (H. Harzali, Baillon et al., 2011).
2.3.1 Les hypothèses de l’effet des ultrasons sur la cristallisation
Les ultrasons de puissance engendrent dans un liquide la naissance et l’oscillation très violente de milliards de petites bulles de gaz, phénomène appelé cavitation. Le cycle d’une de ces bulles sur une période acoustique consiste en une phase explosive suivie d’une implosion violente, à la fin de laquelle la température du gaz peut atteindre 10 000 K, et la pression 1 GPa (voir chapitre 3). Ce phénomène très violent pourrait être à l’origine des effets observés, et plusieurs hypothèses sur les mécanismes mis en jeu sont proposées dans la littérature (H. Harzali, Baillon et al., 2011; H.
Fig. 2.4 – Courbe sursaturation / temps d’induction pour la cristallisation par refroidissement du saccharose pour différentes puissances ultrasonores (Hassoun,
2003).
Harzali, Espitalier et al., 2009; J.Dodds et al., 2007; Ruecroft et al., 2005b; Vironeet al., 2006)
1. Refroidissement important au voisinage de la bulle ; 2. Effet de la pression au voisinage de la bulle ;
3. Évaporation rectifiée ;
4. Ségrégation des espèces au voisinage de la bulle. Nous allons maintenant détailler chacune de ces hypothèses.
Refroidissement important au voisinage de la bulle
Les oscillations de volume de la bulle engendrent des compressions et des détentes du gaz qu’elle renferme. On peut donc penser que lors de la longue expansion précédant l’implosion, le gaz se refroidit dans la bulle, et refroidit par conduction une fine couche de liquide au voisinage de l’interface. Ce refroidissement pourrait donc engendrer, dans le cas de produits présentant une solubilité croissante en fonction de la température, des sursaturations locales très importantes (Ruecroft et al.,
2005b). Il est cependant à noter que la couche de diffusion thermique autour de la bulle est très petite (Louisnard,1998; B. Storey et A. Szeri, 2000) et que ces températures extrêmes ne sont atteintes que lors d’une portion réduite du cycle d’oscillations d’une bulle (figure2.5). D’autre part, les simulations de dynamique de
bulle prenant en compte les effets thermiques montrent qu’à 20 kHz cette expansion est quasi-isotherme, car le temps caractéristique de diffusion est bien plus court que le temps caractéristique d’évolution du rayon (B. Storey et A. Szeri,2000), ce qui semblerait infirmer l’hypothèse de refroidissement lors de l’expansion de la bulle. Une variante de cet effet lors des rebonds de la bulle est toutefois à l’étude au laboratoire et conduit à une interprétation alternative plausible de la nucléation de la glace par une bulle de cavitation.
Fig. 2.5 – Effet du refroidissement (H. Harzali, Espitalier et al., 2009).
Effet de la pression
Lors de la fin de la phase d’implosion, la pression dans le liquide au voisinage de la bulle augmente. Cette augmentation de la pression déplace l’équilibre liquide-solide et par conséquent la sursaturation locale (Virone et al., 2006).
Si le soluté est plus dense que la phase cristalline (cas par exemple du saccharose et du sulfate d’ammonium), une augmentation de pression conduira à une augmentation de la sursaturation et donc de la vitesse de nucléation.
Ainsi, l’effet de pression pourrait favoriser la nucléation. C’est le cas pour la sono-cristallisation du sulfate d’ammonium (Virone et al.,2006), pour lequel le temps d’induction est réduit en présence d’ultrasons.
Mais d’autre part, la forte réduction du temps d’induction sous ultrasons a été observée pour d’autres cristaux tels que le sulfate de potassium (Lyczko et al.,
2002), qui à l’inverse, est moins dense en phase solide que sous forme de soluté. Enfin, H. Harzali, Baillon et al. (2011) ont montré que le temps d’induction dans le cas de la cristallisation du ZnSO4 est fortement réduit en présence d’ultrasons, alors que
la solubilité de cette espèce est quasiment indépendante de la pression. Ces derniers résultats démontrent donc que l’effet de la pression ne suffit pas à expliquer tous les phénomènes observés expérimentalement.
Évaporation rectifiée
B. Storey et A. Szeri (2000) ont montré que la vapeur d’eau entrant dans la bulle de cavitation est piégée par un blocage diffusionnel lors de l’implosion, et qu’une partie de cette vapeur est décomposée chimiquement. H. Harzali, Baillon et al. (2011) ont mentionné que cet effet pouvait conduire à un appauvrissement en eau
dans la solution au voisinage de la bulle, et donc l’enrichir localement en soluté, ce qui augmenterait localement la sursaturation. Cet effet reste à modéliser et il n’existe aucune vérification expérimentale à ce jour.
Ségrégation au voisinage de la bulle
Les fortes accélérations du liquide au voisinage de la bulle à la fin de la phase d’im-plosion sont susceptibles de ségréger un mélange soluté-solution par un phénomène de diffusion moléculaire forcée suite à un gradient de pression (utilisé notamment en ultracentrifugation). Le calcul de Louisnard, Gomez et Grossier (2007) montre que cette ségrégation est très efficace et peut conduire à des sur-concentrations transitoires. Par conséquent, cet effet serait susceptible d’accélérer la nucléation primaire homogène.
Le chemin d’énergie minimale pour la transition liquide-solide, d’après la théorie classique de la nucléation, est l’agrégation progressive de molécules du soluté, nom-mées « monomères », menant à la formation de clusters. Nous avons vu que le temps d’induction, ou de nucléation, est globalement le temps statistiquement requis pour qu’un cluster parvienne à la taille critique (dès lors dénommé nucleus), au delà de laquelle il ne pourra plus que croître, et former un cristal stable (Kashchiev,2000). Ainsi tout effet microscopique intervenant sur le taux de formation de ces nuclei peut affecter notablement ce temps d’induction.
Ce mécanisme d’agrégation peut être présenté sous forme d’un équilibre chimique entre la nouvelle phase et l’ancienne phase, et deux « chemins réactionnels » sont envisageables :
Cn+ C1 ⇐⇒ Cn+1, (2.3.1)
ainsi que
Cn+ Cm ⇐⇒ Cn+m, (2.3.2)
où C1 est une molécule de soluté, ou « monomère », et Cn est le cluster formé de n
monomères.
L’agrégation directe (Eq. (2.3.2)) de multimères Cn et Cm apparaît comme plus
efficace. Cependant, la probabilité de rencontre de tels clusters est faible, si bien que seul l’équilibre de l’équation (2.3.1) est généralement pris en compte, comme schématisé sur la figure (2.6). La création d’un cluster se fait donc très majoritairement par agrégation d’un monomère C1 et d’un multimère Cn dans des conditions de
En présence d’ultrasons, la forte accélération dans le liquide au voisinage de la bulle à la fin de l’implosion (de l’ordre de 1012
g) est susceptible de ségréger les multimères existant au sein de la solution. Cette ségrégation peut conduire à des surconcentrations transitoires des multimères au voisinage de la bulle favorisant ainsi les agrégations du type de celles exprimées par l’équation 2.3.2 (figure 2.6) (Grossier, Louisnard et Vargas,2007). Cet effet serait susceptible d’accélérer l’attachement des agrégats moléculaires précurseurs des cristaux et donc d’accélérer la nucléation primaire homogène.
Fig. 2.6 – Accroissement de taille d’un cluster par agrégation d’un monomère C1 et d’un n-mère Cn. L’agrégation directe entre n-mères et m-mères présente une très faible probabilité, du fait de leur faible nombre relativement aux monomères présents dans la solution(Grossier, Louisnard et Vargas,2007).
Fig. 2.7 – Accroissement de taille d’un cluster en présence d’une bulle de cavitation : la diffusion de pression concentre les clusters de taille moyenne aux environs de l’interface de bulle à chaque implosion, favorisant ainsi les probabilités d’agrégation décrite par l’équation (2.3.1), et augmentant possiblement l’agrégation directe de clusters de taille raisonnable comme décrit par l’équation (2.3.2). Les clusters les plus larges sont expulsés loin de la bulle (Grossier, Louisnard et Vargas,2007).
Notons que les clusters sont plus qu’un outil conceptuel, et ont été mis en évidence expérimentalement dans des solutions sursaturées (Larson et Garside,1986). Cependant, l’effet de ségrégation n’est pour l’heure supporté que par des calculs théo-riques et n’a jamais été mis en évidence expérimentalement. La thèse de Grossier (2006) avait pour objet de mettre en évidence cet effet dans une cellule de lévitation de bulle unique comparable à celle que nous avons utilisé dans notre étude, mais l’effet n’a pas pu être encore observé. D’autre part, il est important de mentionner que l’effet de ségrégation n’est possible que si la phase solide est plus dense que le liquide, ce qui est le cas en général pour les solutés, mais pas pour certains fondus, et en particulier l’eau.
L’effet de ségrégation ne peut donc être invoqué pour expliquer la nucléation de la glace dans de l’eau sous-refroidie.
2.3.2 Le cas de la glace
Historiquement, deux théories sont susceptibles d’expliquer le déclenchement de la nucléation de la glace par la cavitation acoustique. Les deux théories reposent sur l’effet de pression, dans un cas lors d’une dépression (Hunt et Jackson, 1966), dans l’autre cas lors d’une compression (Hickling,1965, 1994). Les deux théories sont basées sur le fait qu’à la suite de l’implosion de la bulle, une onde de pression divergente se propage dans le liquide, cette onde étant constituée d’un front compressif, suivi d’une forte dépression dans le domaine des pressions négatives.
La température de fusion d’un corps dépend de la pression. De même que dans le cas des solutés, la règle générale est que si le solide est plus dense que le liquide, une augmentation de pression favorise la formation du solide, et à l’inverse si le solide est moins dense que le liquide. On peut exprimer cette règle de façon équivalente en mentionnant que la température de fusion augmente avec la pression si le solide est plus dense que le liquide, et que dans le cas inverse elle diminue avec la pression, ce qui est le cas pour la glace normale, dite glace I. Cette règle n’est en fait que la traduction de la loi de Clausius-Clapeyron, qui spécifie la pente de la courbe d’équilibre : dp dT = LS→L T (vL− vS) , (2.3.3)
où LS→L est l’enthalpie de fusion, vL le volume molaire du liquide, vS le volume
molaire du solide, et dp/dT est la dérivée, sous-entendue le long de la courbe d’équilibre. Cette dernière est bien négative dans le cas de la glace I puisque vL < vS.
Hunt et Jackson (1966) ont mis au point une expérience de cavitation non acoustique dans un tube fermé, et montrent clairement que la glace nuclée pendant ou après l’implosion de la bulle. En accord avec la règle ci-dessus pour la glace I, ils ont postulé que seule la partie dépressive de l’onde émise par l’implosion de la bulle pouvait augmenter la température de fusion de la glace et donc la sursaturation du liquide.
Hickling(1965,1994) considère quant à lui que c’est bien le pic de compression qui est responsable de l’augmentation de la température de fusion, ce qui va à l’encontre de la loi de Clausius-Clapeyron pour la glace I. Pour résoudre le paradoxe, il note que la pente de la courbe d’équilibre p(T ) change de signe pour les glaces à haute pression (figure2.8), dont la formation est envisageable puisque la pression du liquide augmente considérablement dans l’onde de pression émise à la fin du collapse. La glace nucléée serait donc de la glace V, VI ou VII, se retransformant en glace normale une fois les conditions redevenues normales.
Fig. 2.8 – D’après Hickling (1994) : courbe d’équilibre des différentes phases de la glace (courbe en traits pleins). Les courbes en traits pointillés représentent l’évolution de l’état du liquide après une implosion, pour différentes températures initiales de l’eau (0, 30 et 60◦
C).
Les expérimentations en bulle unique de Ohsaka et Trinh, 1998 semblent favoriser l’interprétation de Hickling, et cette dernière fait en général l’unanimité. Des calculs plus récents basés sur ce modèle ont été réalisés par l’équipe du LAGEP, associée à RAPSODEE dans le cadre du projet ANR soutenant la présente étude (Cogné, Peczalskiet Louisnard,2013; Saclier, Peczalski et Andrieu,2010). Le prin-cipal défaut de ce modèle est qu’il doit invoquer la nucléation d’une forme cristalline différente sur des temps très courts, ce qui est difficile à vérifier expérimentalement.
2.3.3 Une nouvelle interprétation
Des calculs menés parallèlement à cette étude dans le cadre de l’ANR SONONU-CLICE montrent que la température au coeur de la bulle chute largement en dessous de 0◦
C lors des rebonds après l’implosion (Fig. 2.9, Louisnard et al., 2014, en pré-paration), sur une échelle de temps de l’ordre de 500 ns, ce qui est court, mais bien plus long que le temps caractéristique d’implosion (typiquement de l’ordre de 1 ns). La vapeur d’eau piégée dans la bulle serait donc susceptible de former des germes de glace, qui une fois re-disséminés dans le liquide sous-refroidi pourraient y déclencher facilement la nucléation.
28
30
32
34
0
20
40
28
30
32
34
−40
−20
0
20
40
R
(µ
m)
a
b
t
(µs)
T
(
◦C
)
t
(µs)
Fig. 2.9 – En haut, zoom sur les rebonds d’une bulle de cavitation inertielle au cours du temps. Le premier minimum de la courbe R(t) correspond à la fin de l’implosion principale. En bas, température calculée au coeur de la bulle. Les points marqués en rouge correspondent aux phases où la température en◦
3
Cavitation acoustique
Sommaire
3.1 Introduction . . . 27
3.2 Dynamique radiale de la bulle de cavitation . . . 29
3.3 La bulle unique en lévitation . . . 40
3.1 Introduction
3.1.1 L’importance des bulles de cavitation : le puzzle des bulles
La cavitation décrit l’oscillation radiale de bulles microscopiques remplies de gaz et/ou de vapeur sous l’influence de variations de pression extérieure. Ces variations de pression peuvent être dues à une onde acoustique, généralement dans le domaines des ultrasons, auquel cas on parle de cavitation acoustique, ou bien résulter d’un effet Bernoulli dans un écoulement accéléré, auquel cas on parle de cavitation hydrodynamique. Bien que présentant des différences notables, les deux types de cavitation partagent un grand nombre de points communs, le principal étant une phase de croissance radiale explosive dans la phase de dépression, suivie d’une phase d’implosion dans la recompresssion.
Les bulles de cavitation sont observables dans beaucoup de domaines. Par exemple : Les bulles sont responsables de la destruction des hélices de bateaux (Rayleigh,
1917) ;
La crevette pistolet est un animal marin de quelques centimètres de longueur, qui fait claquer rapidement une pince spécialisée, produisant une bulle cavitation, qui s’effondre violemment. L’effondrement de cette bulle provoque une onde de
choc que la crevette utilise pour briser la coquille de sa proie, l’étourdir ou la tuer (Versluis et al., 2000) ;
L’apparition de bulles de cavitation limite aussi la vitesse des imprimantes rapides (fabriquées avec des matériaux piezo-électriques) ;
Elles sont présentes dans des applications médicales (elles augmentent le contraste dans des images générées par ultrasons (D. Lohse, 2006) ;
La cavitation trouve de nouvelles applications dans l’industrie pétrolière et du gaz naturel, entre autres pour éliminer l’accumulation de paraffine dans les puits de production et la tuyauterie, sans contact direct (Wilkey, Peters et Furness, 2000).
Dans ce chapitre, nous présenterons les principaux mécanismes physiques régissant la cavitation acoustique, en nous focalisant sur la physique d’une bulle unique stable oscillant de façon périodique pendant plusieurs milliards de périodes dans une cellule de lévitation. L’obtention d’une telle bulle est conditionnée par plusieurs critères de stabilité que nous passerons en revue. Le mécanisme de rectification de l’argon, par lequel une bulle initialement remplie d’air pyrolyse ce dernier pour laisser place à une bulle d’argon pur sera décrit, et les instabilités chimiques liées à cette transition seront mentionnées.
3.1.2 Un peu d’histoire
La cavitation acoustique est un phénomène qui apparaît lorsque des échantillons liquides sont soumis à un champ acoustique. Lorsque la pression de l’onde sonore est plus grande que la pression ambiante, la pression locale devient négative, mettant le liquide sous tension. À tension assez grande, le liquide se brise (cavitation), créant des nuages de bulles instables (Ida,2009; Leighton, 1997). Après ce processus, les bulles peuvent grossir, osciller de façon non linéaire se fragmenter ou bien coalescer, et migrer dans le liquide à des vitesses de l’ordre de 1 m/s sous l’influence des forces de Bjerknes. Ce comportement dynamique dépend fortement des propriétés du liquide (tension superficielle, masse volumique et viscosité), du champ acoustique (intensité et fréquence, ondes continues ou pulsées), et des bulles elles-mêmes (quantité de gaz et de vapeur contenue) (Leighton,1997; Mettin,2005,2007). Dans cette situation, les bulles s’auto-organisent souvent dans des structures dendritiques (Mettin, 2005,
2007). Ces nuages de cavitation s’effondrent avec une énorme force, assez puissante pour faire de graves dommages à la surface de corps solides présents à leur voisinage (Rayleigh, 1917). Les bulles génèrent aussi de la lumière, un phénomène appelé sonoluminescence en multi-bulles (MBSL, multi-bubble sonoluminescence) (Barber et al.,1997; M. Brenner, 2002).
C’est seulement en 1990 qu’on a pu mieux comprendre le phénomène, grâce à Felipe Gaitan (F. Gaitan, 1990; F. Gaitan et al., 1992), alors étudiant à l’université du Mississippi. Ce dernier réussit à produire une seule bulle à la fois, en la piégeant dans un récipient sphérique, dont l’eau a été préalablement dégazée autour de 20% de sa
saturation en oxygène, avec une onde de pression stationnaire imposée. Cette bulle unique a été maintenue pendant des heures en lévitation, dans une configuration acoustique résonante, et elle a pu émettre de la lumière. Depuis lors, ce phénomène a été appelé luminescence par une bulle unique (SBSL, single-bubble sonoluminescence) et est connu pour une gamme de fréquences acoustiques allant de 20 à 50 kHz et une pression acoustique comprise entre 1 et 1.5 bar (Sirvastava et al., 2003). Il est important de dire que dans le cas de SBSL, la bulle doit être nucléée extérieurement soit par l’intermédiaire d’une seringue, d’une micro-pipette, ou à l’aide d’un fil métallique (Ni−Cr) chauffé ou par un laser. En effet, la bulle ne peut pas nucléer spontanément sous l’influence de l’onde acoustique, comme c’est le cas dans les expérimentations classiques en multi-bulles.
L’expérience fondatrice de Gaitan a déclenché une vague de recherche sur la bulle unique, les conditions de son obtention et les propriétés de l’émission lumineuse. L’un des enjeux a été notamment de comprendre pourquoi le spectre lumineux était continu pour la SBSL alors qu’il comporte des raies dans la MBSL. Les différents challenges ont été clairement établis dans la revue préliminaire de Barber et al. (1997), et leur résolution dans la décennie suivante est bien résumée par exemple
dans les revues de M. Brenner (2002), Hammer et Frommhold (2001) et T. J. Matula (2000).
Notons qu’un autre axe de recherche parallèle, plus éloigné de nos travaux, concerne l’auto-organisation des champs multi-bulles en une grande variété de structures (Campos-Pozuelo et al., 2005; Doinikov, 2005; Mettin, 2005, 2007; Mettin
et Doinikov, 2009; Parlitz, Mettin et al., 1999).
3.2 Dynamique radiale de la bulle de cavitation
Une étude détaillée du mécanisme physique montre que de nombreuses propriétés physiques et phénomènes doivent être pris en compte pour décrire la cavitation. Cette liste inclut : la tension superficielle, le gaz dissous, la conductivité thermique du gaz, le rayonnement acoustique, la viscosité, le micro-mélange autour de la bulle, le caractère non linéaire de l’oscillation de la bulle, la fragmentation et coalescence de bulles (Young,2004), ce qui rend ce phénomène difficile à décrire. Cependant l’analyse des résultats expérimentaux et théoriques montre qu’on peut obtenir une information quantitative raisonnablement correcte sur la dynamique radiale d’une bulle à partir de considérations thermo-hydrodynamiques classiques, que nous détaillons dans cette section.
3.2.1 Tension superficielle
La tension superficielle est une propriété liée aux forces d’attraction entre des molécules identiques. Comme telle, elle se manifeste à la surface d’un milieu dense (liquide ou solide) ou à l’interface entre deux milieux denses. Les forces entre molécules dans la masse d’un milieu dense sont toujours égales dans toutes les directions, et par conséquent, aucune force nette n’est exercée sur les molécules. Cependant, à